

Abstract
Background and Objectives
Accumulation of tau pathology in Alzheimer disease (AD) correlates with cognitive decline. Anti-tau immunotherapies were proposed as potential interventions in AD. While antibodies targeting N-terminal tau failed to demonstrate clinical efficacy in prodromal-to-mild AD, their utility at other disease stages was not evaluated in prior studies. Lauriet is a phase 2 study of an anti-tau monoclonal antibody, semorinemab, in patients with mild-to-moderate AD.
Methods
The phase 2 Lauriet study included a randomized, placebo-controlled, double-blind period, during which participants with mild-to-moderate AD received 4,500 mg of IV semorinemab or placebo every 4 weeks for 48 or 60 weeks. Participants who chose to continue in the subsequent optional open-label extension received 4,500 mg of semorinemab every 4 weeks for up to 96 weeks. Coprimary efficacy endpoints were change from baseline to week 49 or 61 on the 11-item version of the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog11) and the Alzheimer's Disease Cooperative Study-Activities of Daily Living (ADCS-ADL) scale. Secondary efficacy endpoints included change from baseline on the Mini-Mental State Examination (MMSE) and Clinical Dementia Rating-Sum of Boxes (CDR-SB). Safety, pharmacokinetics, and pharmacodynamic effects were also evaluated.
Results
Between December 3, 2018, and February 27, 2020, 624 individuals were screened, 272 participants were randomized, and 238 were included in the modified intent-to-treat population (received ≥1 dose(s) of study medication and underwent baseline and ≥1 postbaseline assessment(s)). Baseline characteristics were well balanced. At week 49, the semorinemab arm demonstrated a 42.2% reduction (−2.89 points, 95% CI −4.56 to −1.21, p = 0.0008) in decline on the ADAS-Cog11 (coprimary endpoint) relative to the placebo arm. However, no treatment effects were observed on the ADCS-ADL scale (coprimary endpoint; absolute difference between the 2 treatment arms in the ADCS-ADL score change from baseline of −0.83 points, 95% CI −3.39 to 1.72, p = 0.52) or on the MMSE or CDR-SB (secondary endpoints). Semorinemab was safe and well tolerated.
Discussion
Based on the results of the prespecified coprimary endpoints, this study was negative. While semorinemab had a significant effect on cognition measured by the ADAS-Cog11, this effect did not extend to improved functional or global outcomes. These results may warrant further exploration of semorinemab or other anti-tau therapies in mild-to-moderate AD.
Classification of Evidence
This study provides Class I evidence that semorinemab does not slow functional decline in patients with mild-to-moderate AD.
Trial Registration Information
The Lauriet study is registered on ClinicalTrials.gov, NCT03828747, and EudraCT 2018-003398-87.
Introduction
Alzheimer disease (AD) is a neurodegenerative disease that is characterized clinically by progressive impairment of cognitive and daily functioning.1 Pathologically, AD is characterized by the accumulation in the brain neocortex of extracellular β-amyloid (Aβ) peptide-containing plaques and intracellular neurofibrillary tangles (NFTs) containing aggregates of the microtubule-associated protein tau.2 Unlike amyloid plaques, the spatial distribution of tau pathology in patients with AD correlates with cognitive decline.3 While most of the Aβ pathology accumulation may have already occurred by the time of symptom onset,4 tau pathology continues to increase with disease progression according to neuropathology,3 CSF and plasma tau levels,5 and tau PET imaging studies.6 Therefore, therapies that target tau in the brain, either by preventing its presumed cell-to-cell spread7 or by reducing the amount of soluble toxic tau species,8 may alleviate cognitive dysfunction and block further synaptic loss, axonal degeneration, and neuronal cell death.
Semorinemab (also known as RO7105705, MTAU9937A, or RG6100) is a humanized monoclonal antibody (immunoglobulin G4) that targets the N-terminal domain of tau (amino acid residues 6–23) and binds to all known isoforms of full-length tau (including hyperphosphorylated and oligomerized species).9 The original therapeutic hypothesis was that semorinemab would prevent cell-to-cell tau spread. Preclinical studies with a murine version of semorinemab reduced tau-related toxicity in cell culture and tau accumulation in tau transgenic mice.9 In a phase 1 study in healthy volunteers and participants with mild-to-moderate AD (NCT02820896), semorinemab exhibited dose-dependent target engagement and a favorable safety profile.9 These encouraging preclinical and phase 1 data triggered 2 phase 2 studies: Tauriel in prodromal (also known as mild cognitive impairment)-to-mild AD (initiated in 2017; NCT03289143) and Lauriet in mild-to-moderate AD (initiated in 2018; NCT03828747). In the Tauriel study, semorinemab failed to demonstrate meaningful efficacy on clinical endpoints at doses up to 8,100 mg.10 Subsequently, phase 2 trials of gosuranemab11 and tilavonemab12 (both N-terminal tau–targeting antibodies) and zagotenemab13 (an antibody targeting an aggregated form of tau) also failed to demonstrate clinical efficacy in prodromal-to-mild AD. The Lauriet study was fully enrolled and ongoing at the time these results became available. Given the uncertain translation of trial results from prodromal-to-mild AD to later disease stages, the study was continued through the end of the double-blind treatment period. The phase 2 Lauriet study was designed to address the primary research question of whether semorinemab administration could slow decline on cognitive and functional endpoints in patients with mild-to-moderate AD.
Methods
Study Design
The Lauriet study was a multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 2 study with an optional open-label extension (OLE) that was conducted at 49 study locations across the United States (n = 31) and Europe (n = 18, including France [n = 5], Poland [n = 7], and Spain [n = 6]). The initial study design comprised a screening period of up to 8 weeks, an extended baseline period (up to 15 days), and a 48-week placebo-controlled treatment period. Due to the coronavirus disease 2019 (COVID-19) pandemic, the protocol was amended in April 2020, creating 2 cohorts. Cohort 1 includes all participants who completed the originally planned 48 weeks of blinded treatment before the protocol amendment implementation and/or who did not miss any blinded study drug doses. Cohort 2 includes all participants who missed at least 1 dose of blinded study drug due to the pandemic. For cohort 2 participants only, the double-blind treatment duration was extended to 60 weeks (see study design schema in eFigure 1, links.lww.com/WNL/D41). The double-blind period of the study was followed by: (1) a 96-week (OLE) period or (2) a treatment completion visit at the end of a 12-week safety follow-up period (for participants who did not wish to continue in the study).
Standard Protocol Approvals, Registrations, and Patient Consents
The Lauriet study has been conducted in accordance with standards of Good Clinical Practice and all applicable federal and local regulations. All participants and care partners signed informed consent before starting any of the screening activities. During the double-blind period of the study, participants, investigators, and the sponsor study team were blinded to treatment allocation. All study documentation was approved by the local or central institutional review board or ethics committee at each site. The study protocol, including statistical considerations and analysis plan, is available online (clinicaltrials.gov/ProvidedDocs/47/NCT03828747/Prot_SAP_000.pdf).
Participants
Adults 50–85 years of age were eligible for participation if they: (1) met National Institute on Aging-Alzheimer's Association core clinical criteria for probable AD dementia14; (2) had a screening MMSE score15 of 16–21, inclusive; (3) had a Clinical Dementia Rating-Global Score (CDR-GS)16 of 1 or 2; (4) had a centralized independent qualitative visual read of amyloid PET scan (new or prior scans) confirming the presence of cerebral amyloid pathology or CSF Aβ (1–42) ≤1,000 pg/mL measured by Elecsys Aβ1–42 CSF immunoassay (Roche Diagnostics, Indianapolis, IN); and (5) had an informant capable of providing accurate information on the participant's cognitive, behavioral, and functional ability. Concurrent treatment with approved symptomatic AD medications (including donepezil, rivastigmine, galantamine, memantine, and Souvenaid) was permitted if dosing was stable for >2 months before screening. The full list of inclusion/exclusion criteria can be found in the eMethods (links.lww.com/WNL/D41).
Randomization and Blinding
Participants were randomly assigned (1:1) through a centralized randomization system to 4,500 mg of semorinemab or placebo IV. The first 3 doses of study drug were administered biweekly to reach steady-state concentrations followed by every 4-week dosing. Participants allocated to cohort 2 who missed 2 or more consecutive study drug doses during the placebo-controlled period resumed dosing with a biweekly regimen for the first 3 doses, with subsequent doses every 4 weeks. Randomization was stratified by both APOE status (presence vs absence of at least 1 APOEε4 allele) and screening MMSE scores (16–18 vs 19–21). For blinding purposes, participants assigned to placebo received the same volume of IV saline as participants receiving semorinemab.
Procedures
During the screening period, participants completed the following procedures: amyloid PET or lumbar puncture to confirm amyloid positivity, brain MRI, MMSE and 11-item version of the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog11),17 physical and neurologic examination, ECG, and laboratory testing (including APOE genotyping). Care partners completed the Alzheimer's Disease Cooperative Study-Activities of Daily Living scale (ADCS-ADL)18 questionnaire, and a qualified rater completed the CDR. Clinical history and results of the clinical outcome assessments were recorded in a diagnostic verification form, which was reviewed by a central vendor. On confirmation that inclusion/exclusion criteria were met, participants were randomized. At the baseline visit, tau PET imaging using [18F]Genentech Tau Probe 1 (GTP1)19 was performed, and clinical outcome assessments including ADAS-Cog11, ADCS-ADL, MMSE, and CDR were repeated.
On day 1, participants underwent a physical and neurologic examination, blood collection, ECG, and the first infusion of blinded study drug (100 mL IV administered over the course of approximately 78 minutes for the first infusion). On weeks 3 and 5, and every 4 weeks thereafter (for a total of 48 weeks for cohort 1 and 60 weeks for cohort 2), participants returned to the site for study drug infusion (100 mL IV administered over the course of approximately 33 minutes). Vital signs, physical and neurologic examination, blood collection (for safety outcomes and semorinemab serum pharmacokinetics and detection of antidrug antibodies), and MRI were performed at different time points. All clinical outcome assessments were repeated at week 25 and week 49 visits (for cohort 1) or at week 25, week 49, and week 61 visits (for cohort 2). The coprimary endpoints (ADAS-Cog11 and ADCS-ADL) were also repeated at week 37 for both cohorts. Because of the COVID-19 pandemic, the option to perform CDR and/or ADCS-ADL assessments through telephone was included. CSF collection by lumbar puncture was performed at baseline in a subset of participants for determination of amyloid positivity, and in a subset of those, it was repeated at week 49 or week 61.
Outcomes
The coprimary efficacy endpoints were the change in the ADAS-Cog11 and ADCS-ADL from baseline to week 49 for both cohort 1 and cohort 2 participants and week 61 for cohort 2 participants. The ADAS-Cog11 is an 11-item cognitive scale administered to the participants and used to assess cognitive domains most often affected in AD (score range 0–70, lower scores indicate better cognitive performance).17 Three different forms of ADAS-Cog11 were used, containing equivalent word recall and word recognition subtests. The 3 alternative forms (list 1, list 2, and list 3) were administered in the same order to all participants (screening—list 1, baseline—list 2, week 25—list 3, week 37—list 1, week 49—list 2, and week 61 [for cohort 2 only]—list 3). The ADCS-ADL scale quantifies the performance of activities of daily living (ADL) in patients with AD and is administered to the care partners (score range 0–78; higher scores indicate better function).18 Secondary efficacy endpoints included change from baseline on the MMSE15 and Clinical Dementia Rating-Sum of Boxes (CDR-SB).20 Assessments were administered in the local language using validated translations. Assessments were administered by separate qualified raters: clinical rater (ADCS-ADL, CDR) and cognitive rater (ADAS-Cog11, MMSE).
The primary safety endpoints were the nature, frequency, severity, and timing of adverse events (AEs) and serious AEs (SAE). Participants were monitored for AEs at all visits. Safety assessments included clinical laboratory testing, clinical examination, and brain MRI (for neuroimaging abnormalities). If study-associated MRIs could not be obtained during the COVID-19 pandemic, a protocol amendment allowed for administration of study drug in the absence of new clinically significant abnormalities in the clinical examination, with a subsequent unscheduled study-associated MRI obtained as permitted by local conditions.
Brain MRI was performed at screening, week 9, week 49 (cohort 1), or week 61 (cohort 2). The imaging sequences and volumetric and safety analyses were performed, as previously described for the Tauriel study.10 Imaging sequences included T1, T2*, and T2 fluid-attenuated inversion recovery. Volumetric and safety analyses were performed by a central MRI vendor (NeuroRx). Changes from baseline to week 49 or 61 in ventricular, whole brain, cerebral cortex, and hippocampal volumes were analyzed from T1 images.
A subset of participants underwent [18F]GTP1 tau PET imaging at baseline, and week 49 (cohort 1) or week 61 (cohort 2), as previously described for the Tauriel study.10 A central vendor (Invicro) performed the analysis of [18F]GTP1 images. Standardized uptake value ratios (SUVR) were calculated using the inferior cerebellar gray matter as reference (without partial volume correction). Change in SUVR from baseline to week 49 or 61 in the whole cortical gray (WCG), cortical lobes, and in vivo Braak regions of interest (ROI)21 were calculated as exploratory biomarker endpoints. The average tau PET accumulation in a 3-month period (between week 49 and week 61) follows a linear trend. Therefore, to use all the observations in these comparisons at once (baseline and week 49 or week 61), we annualized the change in [18F]GTP1 SUVR. Neuroimaging procedures are described in the eMethods (links.lww.com/WNL/D41).
Optional lumbar punctures were performed in a subset of participants at baseline and at the week 49 (cohort 1) or week 61 (cohort 2) visits. Semorinemab concentrations were measured in these CSF samples. In addition, total tau, ptau181, and ptau217 were measured using qualified immunoassays, and N-terminal tau was measured using a targeted mass spectrometry method (amino acids 2–24; includes target epitope for semorinemab). Details of the analytical methods are described in the eMethods (links.lww.com/WNL/D41).
Statistical Analysis
A sample size of 260 participants was determined to provide 80% power to detect a 35% relative reduction in the mean ADAS-Cog11 change from baseline in semorinemab compared with that in placebo. This calculation assumed an average decline of 6 points in ADAS-Cog11 for placebo-treated participants, an SD across participants of 7.5, a dropout rate of 10%, and a 2-sided α = 0.2 significance level.
There were 3 prespecified analysis populations: (1) modified intent-to-treat (mITT) population (all participants who received study drug, had a baseline assessment, and at least 1 valid postbaseline assessment), (2) restricted mITT (all participants included in the mITT population who missed no more than 1 dose of the blinded study drug compared with the total number according to the cohort assignment), and (3) safety evaluable population (all participants who received at least 1 dose of study drug). Between March 2020 and the end of the double-blind treatment period, 110 participants missed a total of 397 doses; in cohort 2 (i.e., participants who missed 1 or more doses during the double-blind portion of the study), 45 participants missed 3 or more doses, and the median number of missed doses was 2. Serum semorinemab concentrations showed similar exposure levels at week 49 in the mITT and restricted mITT populations (eFigure 2, links.lww.com/WNL/D41), suggesting that the effect of missed doses on serum exposures was negligible. For this reason, figures and tables related to efficacy analyses shown in the main manuscript correspond to the mITT population; restricted mITT analyses are found in the eMethods.
All statistical analyses were conducted using SAS statistical software version 9.4 or R software version 3.6.3. For the primary and secondary endpoints, mean changes from baseline were compared across treatment arms using mixed-models for repeated measures (MMRM). The models had terms for intercept, baseline value of the endpoint, APOE4 strata (carrier vs noncarrier of the ε4 allele), baseline disease severity (MMSE scores 16–18 vs 19–21), baseline use of antidementia medications (present or absent), categorical visit terms, treatment group, baseline age, and treatment-by-visit interactions. The unstructured covariance matrix was used to model the within-subject variance-covariance errors. MMRM assumes a missing-at-random data pattern. Sensitivity analyses exploring the effects of missing data included only participants who completed week 49 clinical assessments and yielded similar results to the primary results. The average of the screening score and the day 1 predose score (when available) was used as the baseline score. If either score was missing, a single score was used instead of the average. Prespecified analyses included change in primary and secondary endpoints from baseline to week 49 (for participants in cohorts 1 and 2). For participants in cohort 2, additional analyses focused on the changes in these endpoints from baseline to week 61.
Additional prespecified subgroup analyses included the following: (1) baseline tau burden (with subgroups defined as (i) high: WCG SUVR ≥1.39, the median value for all 264 participants who had a baseline [18F]GTP1 scan or (ii) low: WCG SUVR <1.39), (2) baseline disease severity (defined as MMSE score 16–18 or MMSE score 19–21), and (3) APOEε4 carrier status (defined as 0 or ≥1 APOEε4 allele).
The sponsor formed an unblinded data monitoring committee (independent from the study team) that reviewed and monitored safety data for potential concerns. A prespecified interim analysis was performed when approximately 40% of patients completed the double-blind treatment period; a minimum 10% relative reduction in ADAS-Cog11 progression in the treated arm vs placebo arm was set as the futility threshold.
Data Availability
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (vivli.org/). Further details on Roche criteria for eligible studies are available in: vivli.org/members/ourmembers/. For further details on Roche Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see roche.com/innovation/process/clinical-trials/data-sharing/.
Results
Results have been posted to clinicaltrials.gov and EudraCT. Between December 3, 2018, and February 27, 2020, 624 people were screened, 272 were randomized, and 267 received at least 1 dose of semorinemab or placebo (safety evaluable population) (Figure 1). The mITT population was composed of 238 participants. Approximately one-third of the participants in the mITT population were assigned to cohort 2. The overall discontinuation rate during the placebo-controlled treatment period (excluding safety follow-up) was 24%; reasons for study discontinuation were well balanced across the 2 treatment arms. Of the 208 participants who completed the placebo-controlled treatment period, 8 discontinued during the safety follow-up period, 1 completed the safety follow-up visit and opted out of the optional OLE, and 199 participants enrolled in the optional OLE.
Figure 1. Participant Flow Diagram.

*Two participants who were originally assigned to placebo received semorinemab. OLE = open-label extension.
Baseline demographics and disease characteristics were similar across treatment groups in the mITT population (n = 238, Table 1), in all randomized participants (n = 272, eTable 1, links.lww.com/WNL/D41), in the safety evaluable population (n = 267, eTable 2), and in the restricted mITT population (n = 204, eTable 3). Overall, participants were predominantly non-Hispanic White, with a mean age of 72 years, and two-thirds were women. According to the CDR-GS, 90% of participants in the mITT population were classified as having mild AD (CDR-GS = 1) and 10% as having moderate AD (CDR-GS = 2), while approximately half of the participants had an MMSE score of 16–18 and the other half had an MMSE score of 19–21. Although the presence of tau, measured by [18F]GTP1, was not required as an inclusion criterion in this study, the mean and median WCG SUVR at baseline was 1.4.
Table 1.
Baseline Demographic and Disease Characteristics of the Modified Intention-to-Treat Population
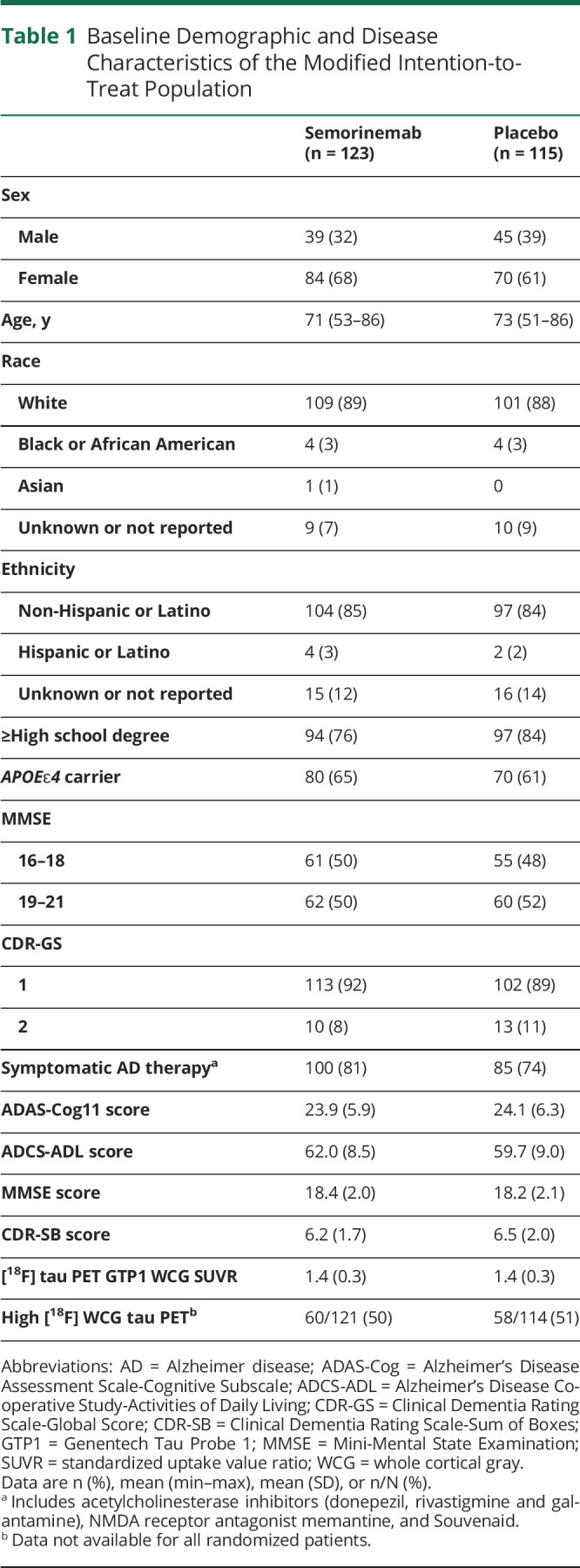
| Semorinemab (n = 123) | Placebo (n = 115) | |
| Sex | ||
| Male | 39 (32) | 45 (39) |
| Female | 84 (68) | 70 (61) |
| Age, y | 71 (53–86) | 73 (51–86) |
| Race | ||
| White | 109 (89) | 101 (88) |
| Black or African American | 4 (3) | 4 (3) |
| Asian | 1 (1) | 0 |
| Unknown or not reported | 9 (7) | 10 (9) |
| Ethnicity | ||
| Non-Hispanic or Latino | 104 (85) | 97 (84) |
| Hispanic or Latino | 4 (3) | 2 (2) |
| Unknown or not reported | 15 (12) | 16 (14) |
| ≥High school degree | 94 (76) | 97 (84) |
| APOEɛ4 carrier | 80 (65) | 70 (61) |
| MMSE | ||
| 16–18 | 61 (50) | 55 (48) |
| 19–21 | 62 (50) | 60 (52) |
| CDR-GS | ||
| 1 | 113 (92) | 102 (89) |
| 2 | 10 (8) | 13 (11) |
| Symptomatic AD therapya | 100 (81) | 85 (74) |
| ADAS-Cog11 score | 23.9 (5.9) | 24.1 (6.3) |
| ADCS-ADL score | 62.0 (8.5) | 59.7 (9.0) |
| MMSE score | 18.4 (2.0) | 18.2 (2.1) |
| CDR-SB score | 6.2 (1.7) | 6.5 (2.0) |
| [18F] tau PET GTP1 WCG SUVR | 1.4 (0.3) | 1.4 (0.3) |
| High [18F] WCG tau PETb | 60/121 (50) | 58/114 (51) |
Abbreviations: AD = Alzheimer disease; ADAS-Cog = Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADCS-ADL = Alzheimer's Disease Cooperative Study-Activities of Daily Living; CDR-GS = Clinical Dementia Rating Scale-Global Score; CDR-SB = Clinical Dementia Rating Scale-Sum of Boxes; GTP1 = Genentech Tau Probe 1; MMSE = Mini-Mental State Examination; SUVR = standardized uptake value ratio; WCG = whole cortical gray.
Data are n (%), mean (min–max), mean (SD), or n/N (%).
Includes acetylcholinesterase inhibitors (donepezil, rivastigmine and galantamine), NMDA receptor antagonist memantine, and Souvenaid.
Data not available for all randomized patients.
The primary and secondary clinical efficacy outcomes are summarized in Table 2. Relative to placebo, semorinemab reduced cognitive decline at 49 weeks (cohorts 1 and 2) by 42.2% based on the ADAS-Cog11, representing an absolute treatment difference in the score change from baseline of 2.89 points (95% CI −4.56 to −1.21, p = 0.0008); however, semorinemab had no effect on other measures of functional (ADCS-ADL), global (CDR-SB), or cognitive (MMSE) decline (Figure 2). These findings were consistent at week 61 (cohort 2 only; change from baseline in the ADAS-Cog11 of −2.75 points, 95% CI −5.31 to −0.20, p = 0.0351; Table 2) and in the restricted mITT population at week 49 (cohorts 1 and 2: change from baseline to week 49 in the ADAS-Cog11 of −2.96 points, 95% CI −4.86 to −1.05, p = 0.0025; eTable 4, links.lww.com/WNL/D41). Prespecified subgroup analyses (i.e., baseline tau burden high vs low, baseline disease severity MMSE score 16–18 vs MMSE score 19–21, and APOEε4 carrier vs noncarrier) for the primary and secondary endpoints were consistent with the overall group results (eFigure 3).
Table 2.
Primary, Secondary, and Exploratory Outcomes in the mITT Population (N = 238)

| Semorinemab | Placebo | Least square mean difference between groups (95% CI) | Relative reduction, %a | p Value | |||
| n | Adjusted mean (95% CI) | n | Adjusted mean (95% CI) | ||||
| Change from baseline to week 49 in the ADAS-Cog11 | 102 | 3.96 (2.66 to 5.26) | 96 | 6.85 (5.58 to 8.11) | −2.89 (−4.56 to −1.21) | 42.2 | 0.0008 |
| Change from baseline to week 61 in the ADAS-Cog11 | 38 | 5.71 (3.92 to 7.51) | 30 | 8.47 (6.55 to 10.38) | −2.75 (−5.31 to −0.20) | 32.5 | 0.0351 |
| Change from baseline to week 49 in the ADCS-ADL | 107 | −7.63 (−9.61 to −5.66) | 101 | −6.80 (−8.72 to −4.88) | −0.83 (−3.39 to 1.72) | −12.3 | 0.5207 |
| Change from baseline to week 61 in the ADCS-ADL | 40 | −9.29 (−11.95 to −6.63) | 33 | −7.57 (−10.47 to −4.68) | −1.72 (−5.50 to 2.07) | −22.7 | 0.3704 |
| Change from baseline to week 49 in the CDR-SB | 109 | 1.80 (1.37 to 2.23) | 101 | 1.54 (1.12 to 1.96) | 0.26 (−0.29 to 0.82) | −17.2 | 0.3501 |
| Change from baseline to week 61 in the CDR-SB | 40 | 2.45 (1.72 to 3.18) | 33 | 2.28 (1.50 to 3.06) | 0.17 (−0.87 to 1.22) | −7.6 | 0.7431 |
| Change from baseline to week 49 in the MMSE | 105 | −2.86 (−3.51 to −2.21) | 97 | −3.12 (−3.76 to −2.48) | 0.27 (−0.58 to 1.11) | 8.5 | 0.5366 |
| Change from baseline to week 61 in the MMSE | 39 | −3.14 (−4.00 to −2.29) | 29 | −4.22 (−5.14 to −3.30) | 1.08 (−0.15 to 2.30) | 25.5 | 0.0851 |
| Annualized change from baseline in WCG [18F]GTP1 tau PETb | 95 | 0.06 (0.04 to 0.08) | 93 | 0.06 (0.04 to 0.08) | 0.00 (−0.02 to 0.02) | 0.2 | 0.9941 |
Abbreviations: AD = Alzheimer disease; ADAS-Cog = Alzheimer's Disease Assessment Scale-Cognitive Subscale; ADCS-ADL = Alzheimer's Disease Cooperative Study-Activities of Daily Living; CDR-GS = Clinical Dementia Rating Scale-Global Score; CDR-SB = Clinical Dementia Rating Scale-Sum of Boxes; GTP1 = Genentech Tau Probe 1; MMSE = Mini Mental State Examination; WCG = whole cortical gray.
Week 49 analyses include patients from both cohorts; week 61 analyses include only cohort 2 patients. n corresponds to the number of patients at each time point.
Positive relative reduction values indicate differences favoring semorinemab, while negative values indicate differences favoring placebo.
Three patients in the mITT population did not have baseline [18F]GTP1 tau PET (2 in the semorinemab arm and 1 in the placebo arm).
Figure 2. Mixed-Models for Repeated Measures-Adjusted Change From Baseline in the Placebo and Semorinemab Treatment Arms, for the Coprimary and Secondary Efficacy Endpoints.

Coprimary efficacy endpoints: (A) ADAS-Cog11 and (B) ADCS-ADL. Secondary efficacy endpoints: (C) CDR-SB and (D) MMSE. Error bars represent 95% CIs. *p < 0.05, *** p < 0.001. In all other time points, the differences observed were not statistically significant. Weeks 0, 25, 37, and 49 time points include participants from cohort 1 and cohort 2, while week 61 time point includes only participants from cohort 2 (shown separate from cohort 1 and cohort 2 analyses). The numbers of participants in each dose arm assessed at each time point on each outcome measure are shown in the data beneath each graph. ADCS-ADL = Alzheimer's Disease Cooperative Study-Activities of Daily Living Scale; ADAS-Cog-11 = Alzheimer's Disease Assessment Scale-Cognitive Subscale; CDR-SB = Clinical Dementia Rating-Sum of Boxes; MMSE = Mini-Mental State Examination.
A post hoc analysis of the ADAS-Cog11 domains and items showed that the effect observed on the ADAS-Cog11 was primarily driven by the memory domain; within the memory domain, the largest effect was observed in the “word recognition” item (eFigure 4, links.lww.com/WNL/D41). Similar post hoc domain analyses in the ADCS-ADL, MMSE, and CDR-SB did not reveal differences between treatment groups (eFigure 5–7).
Semorinemab serum trough concentrations reached steady state around 4 weeks after initiation of treatment (eFigure 8A, links.lww.com/WNL/D41) and were consistent with previously reported pharmacokinetics from healthy volunteers and participants with prodromal-to-mild AD.9,10 CSF samples were available from a subset of participants at baseline and either week 49 or 61 (n = 53) posttreatment (eTable 5, links.lww.com/WNL/D41). In this cohort, there were a larger number of women and APOEε4 carriers in the semorinemab arm; other baseline characteristics were well balanced. Observed semorinemab concentrations in the CSF were consistent with previous studies (eFigure 8B). The mean CSF/serum semorinemab ratio was 0.29% (SD 0.13), consistent with CSF:serum ratios observed in previous studies.9,10 Antidrug antibodies to semorinemab were not detected.
In participants treated with semorinemab, plasma total tau and plasma ptau217 levels increased substantially (>20-fold) compared with those in baseline, consistent with strong peripheral target engagement observed in previous studies (Figure 3A and eFigure 9, links.lww.com/WNL/D41).9,10 Levels for both tau species remained elevated over the course of treatment. A significant reduction in CSF total tau, ptau181, and ptau217 was observed with semorinemab but not with placebo, and no significant change from baseline was observed for N-terminal tau in either treatment group (Figure 3B and eFigure 10).
Figure 3. (A) Plasma Total Tau and (B) CSF Tau Indices.

Error bars represent 95% CIs. **p < 0.05.
The annualized rate of change in [18F]GTP1 WCG SUVR was similar between the treatment arms, suggesting that semorinemab did not alter the rate of tau accumulation and/or spread on this measure (Table 2, eTable 4, and eFigure 11, links.lww.com/WNL/D41). Analyses of frontal, parietal, temporal, and occipital regions (eFigure 11) and in vivo Braak stage ROI21 (eFigure 12) yielded analogous results.
Volumetric MRI analyses demonstrated significant longitudinal declines in whole brain, cerebral cortex, and hippocampal volumes and concomitant increases in ventricular volume across treatment arms. There were no differences between semorinemab and placebo in rates of cerebral atrophy on any measure (eFigure 13, links.lww.com/WNL/D41).
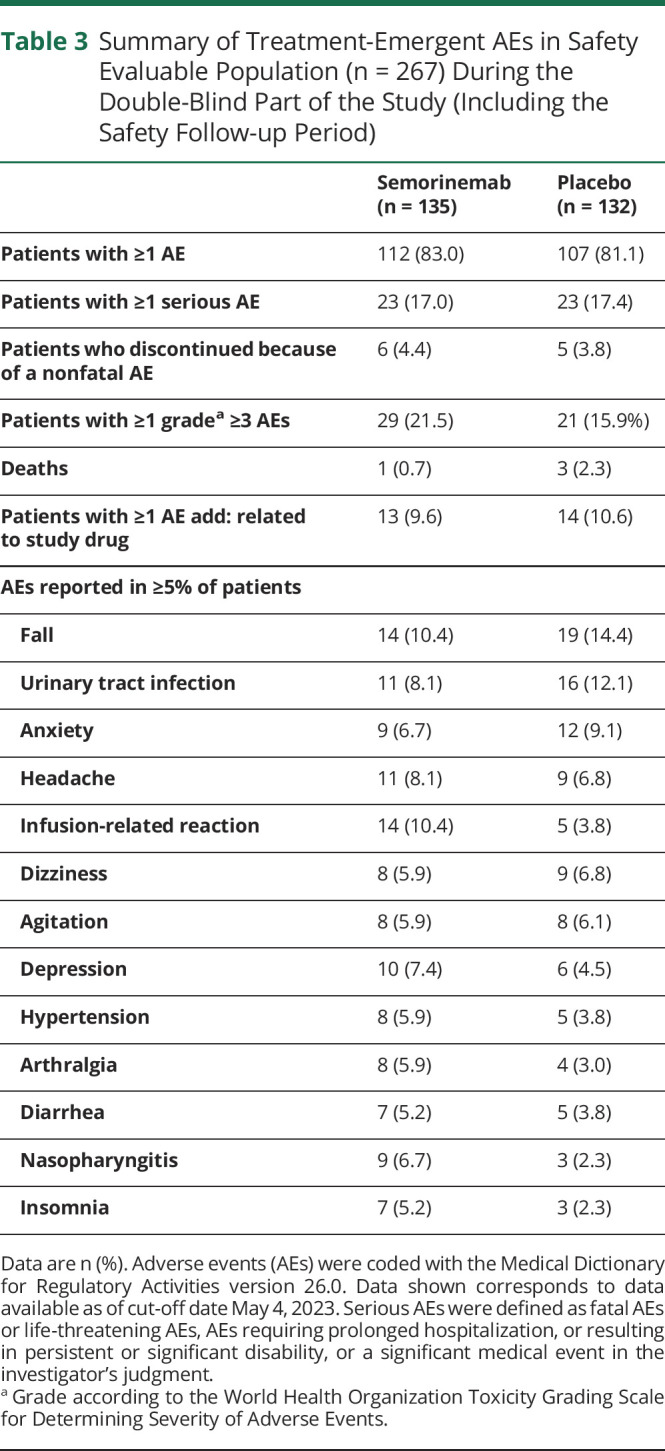
The proportion of participants experiencing at least 1 treatment-emergent AE, regardless of causality assessment, were well balanced between treatment arms (Table 3). The most commonly reported AEs are listed in Table 3. Most AEs were assessed as grade 1 or grade 2 in severity. Grade ≥3 AEs were reported slightly more frequently with the semorinemab arm (22%) relative to the placebo (16%) arm; the most common Grade ≥ 3 AEs were fall (1 [0.8%] patient in placebo, 3 [2.2%] patients in semorinemab), and agitation (3 [2.3%] patients in placebo, 1 [0.7%] patient in semorinemab). Eleven participants (6 in the semorinemab arm, 5 in the placebo arm) discontinued the study because of a nonfatal AE; none were assessed as related to study drug. All infusion-related reactions were grade 1 or 2 in severity, and none were considered serious. The most frequently reported systemic infusion reaction AEs were chills (semorinemab: 3; placebo: 0), dizziness (semorinemab: 2; placebo: 0), headache (semorinemab: 2; placebo: 0), and hypertension/blood pressure increased (semorinemab: 3; placebo: 2).
Table 3.
Summary of Treatment-Emergent AEs in Safety Evaluable Population (n = 267) During the Double-Blind Part of the Study (Including the Safety Follow-up Period)
| Semorinemab (n = 135) | Placebo (n = 132) | |
| Patients with ≥1 AE | 112 (83.0) | 107 (81.1) |
| Patients with ≥1 serious AE | 23 (17.0) | 23 (17.4) |
| Patients who discontinued because of a nonfatal AE | 6 (4.4) | 5 (3.8) |
| Patients with ≥1 gradea ≥3 AEs | 29 (21.5) | 21 (15.9%) |
| Deaths | 1 (0.7) | 3 (2.3) |
| Patients with ≥1 AE add: related to study drug | 13 (9.6) | 14 (10.6) |
| AEs reported in ≥5% of patients | ||
| Fall | 14 (10.4) | 19 (14.4) |
| Urinary tract infection | 11 (8.1) | 16 (12.1) |
| Anxiety | 9 (6.7) | 12 (9.1) |
| Headache | 11 (8.1) | 9 (6.8) |
| Infusion-related reaction | 14 (10.4) | 5 (3.8) |
| Dizziness | 8 (5.9) | 9 (6.8) |
| Agitation | 8 (5.9) | 8 (6.1) |
| Depression | 10 (7.4) | 6 (4.5) |
| Hypertension | 8 (5.9) | 5 (3.8) |
| Arthralgia | 8 (5.9) | 4 (3.0) |
| Diarrhea | 7 (5.2) | 5 (3.8) |
| Nasopharyngitis | 9 (6.7) | 3 (2.3) |
| Insomnia | 7 (5.2) | 3 (2.3) |
Data are n (%). Adverse events (AEs) were coded with the Medical Dictionary for Regulatory Activities version 26.0. Data shown corresponds to data available as of cut-off date May 4, 2023. Serious AEs were defined as fatal AEs or life-threatening AEs, AEs requiring prolonged hospitalization, or resulting in persistent or significant disability, or a significant medical event in the investigator's judgment.
Grade according to the World Health Organization Toxicity Grading Scale for Determining Severity of Adverse Events.
SAEs were well balanced between arms, and none were considered related to study drug (for more details on the most frequently reported SAEs, see eTable 6, links.lww.com/WNL/D41). Four participants died during the double-blind treatment period and/or during the safety follow-up period; none assessed as related to the study drug (1 in the semorinemab arm [unexplained death], 3 in the placebo arm [2 unexplained deaths; 1 because of COVID]).
MRI abnormalities during the double-blind treatment period occurred at slightly higher rates in the placebo arm than in the semorinemab arm, consisting mainly of microhemorrhages, but rates in both arms were within expected background rates.22 There were no reports of vasogenic edema (eTable 7, links.lww.com/WNL/D41).
This study provides Class I evidence that semorinemab does not slow functional decline in patients with mild-to-moderate AD.
Discussion
In this phase 2 study, participants with mild-to-moderate AD (MMSE score 16–21 inclusive) who were treated with 4,500 mg of IV semorinemab for 48 or 60 weeks had a significantly slower cognitive decline relative to those treated with placebo as measured by the coprimary cognitive outcome measure, the ADAS-Cog11; however, no treatment effect was observed on the coprimary functional outcome measure (ADCS-ADL) or on secondary cognitive (MMSE) and global (CDR-SB) outcome measures. The ADAS-Cog11 and ADCS-ADL have previously been used to measure disease progression in other studies of mild-to-moderate AD, and the rates of decline observed in the Lauriet placebo arm on both measures were consistent with those prior reports.23,24 The treatment effect observed in the semorinemab arm on the ADAS-Cog11 was maintained in all 3 prespecified subgroup analyses (i.e., tau burden, disease severity, and APOEε4 status), suggesting that it is unlikely to be driven by a subgroup of participants and/or clinical sites. Scores on the ADAS-Cog and ADCS-ADL are only weakly to moderately correlated in AD,25 which may explain the lack of a corresponding functional benefit in our study.
In addition, post hoc analyses showed that most of the benefits seen with semorinemab on the ADAS-Cog11 appear to be driven by the memory domain, and specifically, the word recognition item. This narrow but significant cognitive effect could contribute to the lack of corresponding treatment effect in other outcome measures, including the ADCS-ADL and CDR-SB, or even in the memory domain of the MMSE, because none directly assess recognition memory. Alternatively, the observed ADAS-Cog11 treatment effect, given the absence of treatment effects on any of the other clinical endpoints, may represent a false-positive result.
In the Tauriel study, participants with prodromal-to-mild AD (MMSE score 20–30) treated with semorinemab in doses up to 8,100 mg over a longer period of time (73 weeks) had no observed clinical treatment effect, including on the ADAS-Cog13.10 Given the different inclusion criteria between the Tauriel and Lauriet studies, the latter study cohort exhibited more severe cognitive and functional deficits at baseline (e.g., mean MMSE 18.3 in Lauriet, 23.3 in Tauriel, CDR-SB 6.5 in Lauriet, 3.9 in Tauriel, and ADCS-ADL 60.3 in Lauriet, 68.4 in Tauriel). These differences in the studies' populations might explain why they yielded different results. Likewise, other 2 N-terminal targeting antibodies (gosuranemab11 and tilavonemab12) and a third targeting aggregated tau with a partial N-terminal epitope (zagotenemab13) reported negative results in their respective phase 2 studies; these were all conducted in prodromal-to-mild AD. None of these other molecules have been studied in phase 2 studies in mild-to-moderate AD. When considered in the context of these prior results, our data raise the possibility that N-terminal tau–targeted therapies might be more effective at later disease stages.
CSF tau indices, including total tau and ptau181, were significantly reduced in the subset of participants with longitudinal CSF collection in both the Lauriet and Tauriel studies.10 Likewise, semorinemab CSF/serum ratios were consistent with prior reports9,10 and concordant with CSF penetration observed with other systemically administered monoclonal antibodies. Together, these data suggest that semorinemab is reaching the brain and modulating tau species that are considered disease relevant. However, semorinemab had no effect on the accumulation of NFTs as measured by tau PET (or on cerebral atrophy measured by MRI, which closely correlates with tau PET).26 While the original therapeutic hypothesis was that semorinemab would slow disease progression by reducing spread and accumulation of NFTs, these results suggest that semorinemab could be alleviating cognitive dysfunction by reducing the amount of soluble tau toxic species. Further analyses of CSF and plasma biomarker data to explore this hypothesis are ongoing.
There are a number of factors that may limit the interpretation of our results. The COVID-19 pandemic contributed to missed doses and/or remote or missed clinical assessments for some participants. The blinded treatment period was extended to account for missed doses, and prespecified analysis of a restricted mITT population yielded results that were very similar to those seen in the mITT population. However, there might be unknown effects of the pandemic on our results that were not assessed by these preplanned sensitivity analyses. The relatively small sample size and lack of racial and ethnic diversity in the study cohort may also restrict the generalizability of our findings.
Data presented in this study suggest that semorinemab may have a modest beneficial cognitive effect in patients with mild-to-moderate AD. A 3-point decline in the ADAS-Cog11 has been described as an appropriate minimal clinical relevant change in patients with AD.27 Although this possible cognitive benefit did not extend to improved functional or global outcomes and therefore may not meet the established regulatory standards for a disease-modifying drug for AD,28 these results may warrant further exploration of semorinemab or other anti-tau therapies in this population. Combining semorinemab or other anti-tau therapies with other disease-modifying drugs such as anti-Aβ antibodies29,30 may be of particular interest.31
Acknowledgment
The authors thank all the study participants and their families and all the site investigators, study coordinators, and staff.
Glossary
- Aβ
β-amyloid
- AD
Alzheimer disease
- ADAS-Cog11
Alzheimer's Disease Assessment Scale-Cognitive Subscale
- ADCS-ADL
Alzheimer's Disease Cooperative Study-Activities of Daily Living
- ADL
activities of daily living
- AE
adverse event
- CDR-GS
Clinical Dementia Rating-Global Score
- CDR-SB
Clinical Dementia Rating-Sum of Boxes
- COVID-19
coronavirus disease 2019
- GTP1
Genentech Tau Probe 1
- mITT
modified intent-to-treat
- MMRM
mixed-models for repeated measures
- MMSE
Mini-Mental State Examination
- NFTs
neurofibrillary tangles
- OLE
open-label extension
- ROI
regions of interest
- SAE
serious adverse event
- SUVR
standardized uptake value ratios
- WCG
whole cortical gray
Appendix 1. Authors

| Name | Location | Contribution |
| Cecilia Monteiro, MD, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and study supervision |
| Balazs Toth, MS | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and statistical analysis |
| Flavia Brunstein, MD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Ashwini Bobbala, MS | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Seema Datta, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; study supervision |
| Ryan Ceniceros, BS | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; study supervision |
| Sandra M. Sanabria Bohorquez, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Veronica G. Anania, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Kristin R. Wildsmith, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and study concept and design |
| Stephen P. Schauer, BA | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Julie Lee, MS | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Michael J. Dolton, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and statistical analysis |
| Vidya Ramakrishnan, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; and statistical analysis |
| Daniel Abramzon, MS | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data |
| Edmond Teng, MD, PhD | Genentech, Inc., South San Francisco, CA | Drafting/revision of the article for content, including medical writing for content; major role in the acquisition of data; study concept and design; and study supervision |
Appendix 2. Coinvestigators

| Coinvestigators are listed at links.lww.com/WNL/D43. |
Footnotes
Editorial, page 593
Class of Evidence: NPub.org/coe
Study Funding
This work was supported by Genentech, Inc.
Disclosure
All authors are employees of Genentech, Inc. and shareholders in F. Hoffmann La Roche, Ltd. Go to Neurology.org/N for full disclosures.
References
- 1.Scheltens P, De Strooper B, Kivipelto M, et al. Alzheimer's disease. Lancet. 2021;397(10284):1577-1590. doi: 10.1016/s0140-6736(20)32205-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimers Dement. 2012;8:1-13. doi: 10.1016/j.jalz.2011.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson PT, Alafuzoff I, Bigio EH, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362-381. doi: 10.1097/nen.0b013e31825018f7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol. 2013;12(4):357-367. doi: 10.1016/s1474-4422(13)70044-9 [DOI] [PubMed] [Google Scholar]
- 5.Janelidze S, Mattsson N, Palmqvist S, et al. Plasma P-tau181 in Alzheimer's disease: relationship to other biomarkers, differential diagnosis, neuropathology and longitudinal progression to Alzheimer's dementia. Nat Med. 2020;26(3):379-386. doi: 10.1038/s41591-020-0755-1 [DOI] [PubMed] [Google Scholar]
- 6.Pascoal TA, Benedet AL, Tudorascu DL, et al. Longitudinal 18F-MK-6240 tau tangles accumulation follows Braak stages. Brain. 2021;144(11):3517-3528. doi: 10.1093/brain/awab248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sanchez JS, Becker JA, Jacobs HIL, et al. The cortical origin and initial spread of medial temporal tauopathy in Alzheimer's disease assessed with positron emission tomography. Sci Transl Med. 2021;13(577):eabc0655. doi: 10.1126/scitranslmed.abc0655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang CW, Shao E, Mucke L. Tau: enabler of diverse brain disorders and target of rapidly evolving therapeutic strategies. Science. 2021;371(6532):eabb8255. doi: 10.1126/science.abb8255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ayalon G, Lee SH, Adolfsson O, et al. Antibody semorinemab reduces tau pathology in a transgenic mouse model and engages tau in patients with Alzheimer's disease. Sci Transl Med. 2021;13(593):eabb2639. doi: 10.1126/scitranslmed.abb2639 [DOI] [PubMed] [Google Scholar]
- 10.Teng E, Manser PT, Pickthorn K, et al. Safety and efficacy of semorinemab in individuals with prodromal to mild Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2022;79(8):758-767. doi: 10.1001/jamaneurol.2022.1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shulman M, Rajagovindan R, Kong J, et al. Top-line results from TANGO, a phase 2 study of gosuranemab in participants with mild cognitive impairment due to Alzheimer's disease and mild Alzheimer's disease. J Prev Alzheimers Dis. 2021;8:S65. [Google Scholar]
- 12.Florian H, Wang D, Guo Q, et al. Phase 2 study of tilavonemab, an antitau antibody, in early Alzheimer's disease. J Prev Alz Dis. 2021;8:S50. [Google Scholar]
- 13.A study of LY3303560 in participants with early symptomatic Alzheimer's disease. ClinicalTrials.gov identifier: NCT03518073. Accessed December 8, 2022. clinicaltrials.gov/ct2/show/NCT03518073.
- 14.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011;7(3):263-269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12(3):189-198. doi: 10.1016/0022-3956(75)90026-6 [DOI] [PubMed] [Google Scholar]
- 16.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43(11):2412-2414. doi: 10.1212/wnl.43.11.2412-a [DOI] [PubMed] [Google Scholar]
- 17.Mohs RC, Knopman D, Petersen RC, et al. Development of cognitive instruments for use in clinical trials of antidementia drugs: additions to the Alzheimer's Disease Assessment Scale that broaden its scope. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11:S13-S21. doi: 10.1097/00002093-199700112-00003 [DOI] [PubMed] [Google Scholar]
- 18.Galasko D, Bennett D, Sano M, et al. An inventory to assess activities of daily living for clinical trials in Alzheimer's disease. The Alzheimer's Disease Cooperative Study. Alzheimer Dis Assoc Disord. 1997;11:S33-S39. doi: 10.1097/00002093-199700112-00005 [DOI] [PubMed] [Google Scholar]
- 19.Sanabria Bohórquez S, Marik J, Ogasawara A, et al. [18F]GTP1 (Genentech Tau Probe 1), a radioligand for detecting neurofibrillary tangle tau pathology in Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2019;46(10):2077-2089. doi: 10.1007/s00259-019-04399-0 [DOI] [PubMed] [Google Scholar]
- 20.Berg L, Miller JP, Storandt M, et al. Mild senile dementia of the Alzheimer type: 2. Longitudinal assessment. Ann Neurol. 1988;23(5):477-484. doi: 10.1002/ana.410230509 [DOI] [PubMed] [Google Scholar]
- 21.Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971-982. doi: 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filippi M, Cecchetti G, Spinelli EG, Vezzulli P, Falini A, Agosta F. Amyloid-related imaging abnormalities and β-amyloid-targeting antibodies: a systematic review. JAMA Neurol. 2022; 79(3):291-304. doi: 10.1001/jamaneurol.2021.5205 [DOI] [PubMed] [Google Scholar]
- 23.Doody RS, Thomas RG, Farlow M, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer's disease. N Engl J Med. 2014;370(4):311-321. doi: 10.1056/nejmoa1312889 [DOI] [PubMed] [Google Scholar]
- 24.Egan MF, Kost J, Tariot PN, et al. Randomized trial of verubecestat for mild-to-moderate Alzheimer's disease. N Engl J Med. 2018; 378(18):1691-1703. doi: 10.1056/nejmoa1706441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu-Seifert H, Siemers E, Selzler K, et al. Correlation between cognition and function across the spectrum of Alzheimer's disease. J Prev Alzheimers Dis. 2016;3:138-144. doi: 10.14283/jpad.2016.99 [DOI] [PubMed] [Google Scholar]
- 26.Ossenkoppele R, Reimand J, Smith R, et al. Tau PET correlates with different Alzheimer's disease-related features compared to CSF and plasma p-tau biomarkers. EMBO Mol Med. 2021;13(8):e14398. doi: 10.15252/emmm.202114398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schrag A, Schott JM; Alzheimer's Disease Neuroimaging Initiative. What is the clinically relevant change on the ADAS-Cog?. J Neurol Neurosurg Psychiatry. 2012;83(2):171-173. doi: 10.1136/jnnp-2011-300881 [DOI] [PubMed] [Google Scholar]
- 28.Leber P. Guidelines for the Clinical Evaluation of Antidementia Drugs (First Draft). 1990. doi: 10.13140/2.1.4164.9445 [DOI] [Google Scholar]
- 29.Van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer's disease. N Engl J Med. 2023;388(1):9-21. doi: 10.1056/nejmoa2212948 [DOI] [PubMed] [Google Scholar]
- 30.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer's disease. N Engl J Med. 2021;384(18):1691-1704. doi: 10.1056/nejmoa2100708 [DOI] [PubMed] [Google Scholar]
- 31.Cummings JL, Tong G, Ballard C. Treatment combinations for Alzheimer's disease: current and future pharmacotherapy options. J Alzheimers Dis. 2019;67(3):779-794. doi: 10.3233/jad-180766 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (vivli.org/). Further details on Roche criteria for eligible studies are available in: vivli.org/members/ourmembers/. For further details on Roche Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see roche.com/innovation/process/clinical-trials/data-sharing/.