

Abstract
Acute respiratory distress syndrome (ARDS) represents a significant burden to the health care system, with approximately 200,000 cases diagnosed annually in the US. ARDS patients suffer from severe refractory hypoxemia, alveolar-capillary barrier dysfunction, impaired surfactant function, and abnormal upregulation of inflammatory pathways that lead to intensive care unit admission, prolonged hospitalization, and increased disability-adjusted life years. Currently, there is no cure or FDA-approved therapy for ARDS. This work describes the implementation of engineered extracellular vesicle (eEV)-based nanocarriers for targeted non-viral delivery of anti-inflammatory payloads to the inflamed/injured lung. Our results show the ability of surfactant protein A (SPA) functionalized IL-4- and IL-10-loaded eEVs to promote intrapulmonary retention and reduce inflammation, both in vitro and in vivo. We observed significant attenuation in tissue damage, proinflammatory cytokine secretion, macrophage activation, influx of protein-rich fluid and neutrophil infiltration into the alveolar space as early as 6 hours post-eEVs treatment. Additionally, metabolomics analyses showed that eEV treatment caused significant changes in the metabolic profile of inflamed lungs, driving secretion of key anti-inflammatory metabolites. Altogether, these results establish the potential of eEVs derived from dermal fibroblasts to reduce inflammation, tissue damage, and the prevalence/progression of injury during ARDS via non-viral delivery of anti-inflammatory genes/transcripts.
Keywords: Engineered extracellular vesicles, nonviral gene delivery, anti-inflammatory extracellular vesicles, novel nanocarriers, lung injury, pulmonary inflammation
Graphical Abstract
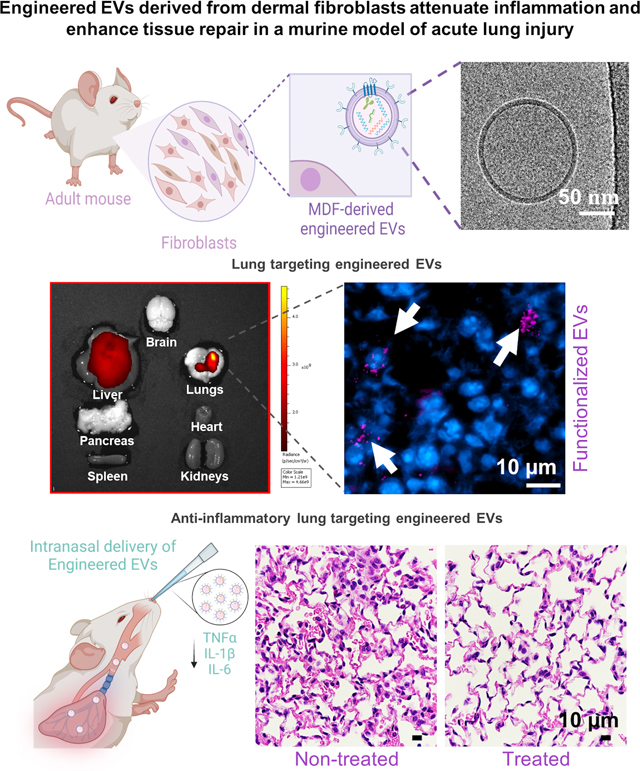
Engineered extracellular vesicles (EVs) derived from adult dermal fibroblasts show enhanced intrapulmonary targeting and retention, and can effectively deliver therapeutic cargo to the inflamed lung to reduce inflammation, mediate tissue repair, and drive metabolic changes that favor further activation of anti-inflammatory pathways. These EVs could potentially represent a powerful tool for the treatment of acute lung injury.
INTRODUCTION
Acute respiratory distress syndrome (ARDS) represents a significant burden to health systems worldwide,[1–4] considering the need for critical care, prolonged hospitalization times, and slow recovery.[1] Pre-COVID-19 there were approximately 200,000 cases of ARDS reported annually in the United States.[5–8] The mortality rate for patients with ARDS is high, ranging between 25% - 40%.[6] ARDS is the most severe form of acute lung injury and is normally caused by direct or indirect injury to the lung (i.e., infections, trauma, pneumonia, hemorrhagic shock, among others).[9–11] Once the lung is injured there is significant upregulation of pro-inflammatory pathways, with marked recruitment of neutrophils, and significant secretion of pro-inflammatory cytokines and other mediators.[5, 6] Although inflammation is critical for the resolution of the underlying condition, hyperinflammation can lead to capillary injury, and further disruption of the alveolar-capillary barrier, followed by a significant influx of protein-rich fluid into the alveolar space.[12–14] This accumulation of edema fluid in the interstitium and alveolar space causes impaired gas exchange and results in poor oxygenation, reduced carbon dioxide excretion, decreased lung compliance, and ultimately acute respiratory failure. For patients with ARDS, mechanical ventilation is used as supportive therapy to maintain adequate oxygenation. However, the mechanical stress imposed during ventilation exacerbates the initial injury and triggers a positive feedback loop that favors inflammation, which could lead to multisystem organ dysfunction/failure and death.[7, 10]
Currently, there is no cure or FDA-approved therapy for ARDS.[15] This unmet clinical need is especially important considering the recent COVID-19 pandemic, where 67% of COVID-19 patients have developed ARDS,[16–19] and in which lung inflammation has played a critical role in determining clinical outcomes. Although progenitor cell-based therapies (e.g., endothelial progenitor cells, and mesenchymal stem cells) have been explored to reduce inflammation and enhance lung repair via trophic and anti-inflammatory mechanisms (e.g., through the secretion of multiple effector molecules, including anti-inflammatory cytokines, growth factors, and antimicrobial peptides),[7, 8, 13, 20–23] significant concerns remain with respect to potential tumorigenesis, high immunogenic responses, limited cell sources, cumbersome and labor-intensive ex vivo processing, and effective delivery methods.[24, 25] On the other hand, while the use of anti-inflammatory approaches to treat the pathophysiology of ARDS has shown significant promise, efficient delivery to the inflamed lung environment remains a major challenge in critically ill patients.[26–28]
Extracellular vesicles (EVs) are cell-derived natural carriers that play a crucial role in mediating short- and long-range cell-cell communication, and share molecular cues with their donor cell.[29–32] EVs have been known to exhibit high stability in biofluids, low immunogenicity, and exceptional biocompatibility. Furthermore, EVs possess an innate ability to cross biological barriers, to transport and deliver a variety of active biomolecules, including proteins, antigens, and nucleic acids, among others, without significant restrictions on size.[33–35] Even though EVs still face some limitations (e.g., heterogeneity,[36, 37] paucity of standardized isolation and purification protocols,[37, 38] and large-scale production for clinical use),[39, 40] they have emerged as promising drug and gene delivery nanocarriers capable of circumventing some of the practical and translational barriers faced by standard nanocarrier systems.[41, 42]
We have previously shown that donor cells can be engineered to stimulate the release of engineered (eEVs) packed with specific molecular cargo for diverse therapeutic applications, and that eEVs can be functionalized with ligands of interest to achieve targeted delivery to cells and tissues.[43–49] Here we report on the implementation of eEVs derived from dermal fibroblasts, loaded with genes, mRNA transcripts, and protein content of anti-inflammatory cytokines interleukin-4 and −10 (IL-4 and IL-10), to dampen lung injury and inflammation, and decorated with Surfactant Protein A (SPA) to promote preferential retention by cell compartments in the injured lung. SPA is an abundant glycoprotein component present in the lung surfactant, which is implicated in the reduction of the surface tension at the pulmonary air-liquid interphase, prevention of alveolar collapse, and is also considered a major inflammatory immunomodulator.[50, 51] In light of this, in addition to enabling targeted delivery to the lung, the presence of mRNA transcripts of SPA in the eEVs could potentially booster the attenuation of lung inflammation due to its role in maintaining immune homeostasis in the lung microenvironment.[52]
These anti-inflammatory eEVs were derived 24 hours after non-viral electrotransfection of adult mouse dermal fibroblasts. Our results highlight the ability of IL-4- and IL-10-loaded eEVs to reduce lipopolysaccharide (LPS)-induced lung inflammation and tissue damage, in vitro and in vivo. In vitro, we observed significant attenuation of proinflammatory cytokine secretion (IL-6, IL-1β, and TNF-α), and macrophage activation. In vivo, we also observed significant reduction in the secretion of pro-inflammatory cytokines (IL-6, IL-1β, and TNF-α), protein-rich fluid influx into the alveolar space, neutrophil infiltration, and tissue damage in mice treated with IL-4- and IL-10-loaded eEVs as early as 6 hours after treatment. In vivo studies suggest that the decoration of eEVs with the SPA targeting ligand improves intrapulmonary eEV retention. Metabolomics analyses also demonstrated that anti-inflammatory eEVs also had a significant impact on the metabolic profile of LPS-challenged mice by inducing the secretion of metabolites related to key anti-inflammatory metabolites. These results establish the potential of using anti-inflammatory eEVs, obtained via non-viral approaches, to reduce inflammation and the prevalence/progression of lung injury during ARDS.
RESULTS
Electrotransfection of dermal fibroblasts with IL-4, IL-10, and SPA leads to the release of SPA-decorated eEVs loaded with IL-4 and IL-10:
eEVs were derived from adult-mouse dermal fibroblast (MDF) cultures electrotransfected with expression plasmids for anti-inflammatory cytokines IL-4 or IL-10 and the targeting ligand SPA (Figure 1A). EVs released from MDF cultures electrotransfected with a sham/empty pCMV6 vector were used as control (i.e., sham eEVs). Western blot analysis confirmed the presence of the EV marker CD63 and the cytoskeletal marker tubulin in the donor cells, IL-10+SPA, IL-4+SPA, and sham eEVs, and a reduced expression of the endoplasmic reticulum protein calnexin detected in eEV formulations compared to donor cells (Figure 1B). Additional EV marker proteins, including ALG-2-interacting protein X (ALIX) and tumor susceptibility gene 101 protein (TSG101) were also found in the eEV formulations and donor cells (Supplemental Figure 1A). Nanoparticle tracking analysis indicated that 24 hours after transfection, adult MDFs release eEVs in the order of 5.7–6.11×1010 eEVs/mL, with an average size ranging between 190 and 226 nm for sham and anti-inflammatory eEVs, respectively (Figure 1C, and Supplemental Figure 1B). Morphological characterization of the eEVs performed via Cryo-electron microscopy (Cryo-EM) revealed discrete eEVs with an intact structure and a characteristic lipid-bilayer (Figure 1D). qRT-PCR analyses confirmed successful transfection of adult-MDF, with significant overexpression of IL-4 and IL-10 after 24 hours (Figure 1E). Adult-MDF also showed significant overexpression of the targeting ligand SPA for cells co-transfected with IL-4+SPA or IL-10+SPA, compared to sham-transfected cells (Figure 1F). We also observed that the co-transfection of IL-10+SPA, had a boosting effect on IL-10 expression in the donor cells, with an increase of approximately two-fold compared to cells electrotransfected only with IL-10 (Figure 1E and 1F) (p-value=0.0140). qRT-PCR characterization of eEV content confirmed robust packing with transcripts for IL-4 and IL-10, 24 hours after electrotransfection of the donor cells with plasmids encoding for IL-4, IL-10, IL-4+SPA, or IL-10+SPA (Figure 1G–H). Additionally, we also verified that there is IL-4 and IL-10 protein content associated with the eEVs as confirmed via ELISA of IL-4+SPA and IL-10+SPA eEV formulations, compared to sham eEVs (Figure 1I–J). Collectively, these results demonstrate the feasibility of packing transcripts of anti-inflammatory cytokines IL-4 and IL-10 in eEVs and functionalizing them with the targeting ligand SPA on their membrane to improve intrapulmonary/alveolar interaction and retention.
Fig. 1. Non-viral electrotransfection of dermal fibroblasts with IL-4, IL-10, and SPA leads to the release of eEVs loaded with IL-4, IL-10, and SPA.

A) Schematic diagram illustrating the transfection of donor cells with plasmids encoding for molecular cargo (IL-4 and IL-10) and targeting ligand (SPA), and the generation of engineered EVs packed and functionalized with episomally expressed plasmids. B) Western blot analysis confirmed the presence of the EV marker, CD63 in IL-10+SPA, IL-4+SPA, sham eEVs, and donor cells. Moreover, the tubulin maker (cell cytoskeleton protein) was found in the donor cells and all eEVs formulations, and the protein calnexin (endoplasmic reticulum related) was detected in the donor cells and to a lesser extent in the eEVs. C) The NanoSight plot shows uniform and narrow distribution across the different groups in terms of concentration and a size, with a peak at ~200nm (n=3). D) Cryo-electron micrograph of IL4+SPA, IL-10+SPA, and sham eEVs revealed an intact structure. E) qRT-PCR analysis displaying the gene expression (mRNA transcripts) of the molecular cargo (IL-4 or IL-10) into adult-MDFs donor cells compared to sham-transfected cells (n=3). F) Donor cells co-transfected with the molecular cargo (IL-4 or IL-10) and the targeting ligand (SPA) showing gene expression (mRNA transcripts) via qRT-PCR compared to sham-transfected cells (n=3). G) qRT-PCR showed that the levels of IL-4 and IL-10 mRNA loaded inside eEVs was ~2–3 order of magnitude higher compared to sham EVs (n=3). H) The levels of the molecular cargo packed inside the eEVs was altered when the donor cells were co-transfected with SPA (IL-4+SPA or IL-10+SPA eEVs), where the expression of IL-4 was found to be one order of magnitude higher with low expression of SPA, and IL-10 was one order of magnitude lower with high expression of SPA. ELISA results of the I) IL-4 and J) IL-10 protein content associated with the eEVs compared to that in sham eEVs. (n=3) All error bars are shown as standard error of the mean (SEM). *p<0.05 and **p<0.001, #significant difference with respect to the control with a p-value<0.05, One-way ANOVA and Two tail t-test when appropriate.
Anti-inflammatory eEVs dampen LPS-induced inflammation in vitro:
Activated monocytes play a major role in the secretion of pro-inflammatory cytokines in response to the activation of pattern recognition receptors by pathogen- or damage-associated molecular patterns.[53, 54] We optimized an in vitro inflammation model where RAW 264.7 macrophage cells were challenged with 100–1000 ng/mL LPS for 3–48 hours to induce significant upregulation of key pro-inflammatory mediators such as IL-6, IL-1β, iNOS, and TNFα (Supplemental Figure 2). To evaluate the therapeutic potential of eEVs in dampening inflammation, macrophages challenged with 100 ng/mL LPS were co- or post-treated with IL-4+SPA, IL-10+SPA, or sham eEVs (Figure 2A and Figure 3A). Cell cultures that were not exposed to LPS (i.e., control) or LPS-challenged without any eEV treatment (i.e., LPS) were included for comparison purposes. For post-treatment experiments, cells that had been LPS-challenged for 12 hours were exposed to eEVs at a concentration of ~2000 eEVs/cell for 3–6 hours. Subsequently, the inflammatory response was evaluated in terms of altered cell morphology, gene expression via qRT-PCR, and protein secretion via ELISA. Early changes in cell morphology were indicative and consistent with successful monocyte/macrophage activation in response to the LPS challenge, where activated cells presented a characteristic spindle shape with reduced roundness compared to unchallenged cells (Figure 2B). LPS-challenged macrophages that were eEV-treated for 3 and 6 hours with IL-4+SPA or IL-10+SPA eEVs showed a significant decrease in the number of activated monocytes compared to control groups (Figure 2B and Supplemental Figure 3A). We also observed a significant decrease in the expression levels of IL-6 and IL-1β for cells post-treated with IL4+SPA eEVs for 3 and 6 hours compared to controls (Figure 2C, and Supplemental Figure 3B). On the other hand, cells treated with IL-10+SPA eEVs only showed a significant reduction in the expression levels of IL-6 and IL-1β at 3 hours post-treatment compared to controls (Figure 2C). Similarly, in terms of protein expression, IL-4+SPA eEV-treated cells showed a significant reduction in the secretion of IL-6, IL-1β, and TNFα at 3 hours post-treatment compared to controls (Figure 2D), while cells treated with IL-10+SPA eEVs showed a significant decrease in IL-6 and TNFα secretion both at 3- and 6-hours post-treatment (Figure 2D and Supplemental Figure 3C).
Fig. 2. Early post-treatment (3 hours) with IL-4+SPA and IL-10+SPA eEVs resolves activation and inflammation in LPS-challenged monocytes.

A) Schematic representation of the inflammatory response driven by RAW 264.7 monocyte cultures in vitro when treated with the endotoxin lipopolysaccharide (LPS), and the therapeutic effect of the post-treatment with anti-inflammatory eEVs to dampen the induced inflammation. B) Representative micrographs illustrating changes in cell morphology revealed that non-treated rounded monocytes get activated by the addition of the LPS, transitioning into a spindled shape typical of an activated or differentiated macrophage. Quantification analyses indicate that cell cultures treated with IL-4+SPA and IL-10+SPA EVs have less activated monocytes compared to the LPS and sham eEV groups. C) qRT-PCR analyses showed increased expression of IL-6, IL1-β, and TNF-α compared to the control (no LPS), and a decreased expression of IL-6, and IL1-β when treated with IL-4+SPA and IL-10+SPA eEVs. D) IL-6 and TNF-α protein expression was reduced when treated with both eEVs, and IL1-β with just IL-4+SPA eEVs. (n=3) All error bars are shown as SEM. *p<0.05 and **p<0.001, #significant difference with respect to the control with a p-value<0.05, One-way ANOVA.
Fig. 3. Early co-treatment (3 hours) with IL-4+SPA and IL-10+SPA eEVs resolves activation and inflammation in LPS-challenged monocytes.

A) Schematic representation of the therapeutic effect of the co-treatment of RAW 264.7 monocytes with LPS and the anti-inflammatory eEVs to dampen inflammation. B) The percentage of activated monocytes (spindle shape) was reduced when treated with IL-4+SPA, IL-10+SPA, and sham eEVs. C) qRT-PCR analysis showing the expression of pro-inflammatory cytokines for the LPS group compared to the control, and the reduction in IL-6, IL1-β, and iNOS expression when treated with IL-10+SPA eEVs, and in IL-6 when treated with IL-4+SPA eEVs. D) At the protein level, IL-6 expression decreased when treated with IL-10+SPA eEVs and IL1-β when treated with IL-4+SPA. (n=3) All error bars are shown as SEM. *p<0.05 and **p<0.001, #significant difference with respect to the control with a p-value<0.05, One-way ANOVA.
For co-treatment experiments, cells treated with LPS + IL-4+SPA eEVs or LPS + IL-10+SPA eEVs showed a significant reduction in IL-6 expression after 3 and 12 hours of co-treatment, and in the expression of IL-1β after 3 hours of co-treatment compared to controls (Figure 3C, Supplemental Figure 4A). Moreover, cells treated with IL-4+SPA eEVs also showed a significant reduction in TNF-α expression after 12 hours of co-treatment compared to controls, while cells treated with IL-10+SPA eEVs showed a reduction in iNOS expression after 12 hours of co-treatment compared to controls (Supplemental Figure 4A). In terms of protein expression, IL-4+SPA eEV-treated cells showed a significant decrease in the secretion of IL-1β after 3 hours of co-treatment compared to controls (i.e., LPS group) (Figure 3D). Cells treated with IL-10+SPA eEVs showed a significant decrease in IL-6 secretion after 3 and 12 hours of co-treatment compared to controls (Figure 3D and Supplemental Figure 4B). In addition, cells treated with IL-10+SPA eEVs also showed a significant decrease in TNFα expression after 12 hours of co-treatment compared to controls (Supplemental Figure 4B). In terms of morphology, LPS-challenged cells co-treated for 3 hours with IL-4+SPA or IL-10+SPA eEVs showed a significant decrease in the percentage of activated monocytes compared to controls (Figure 3B). In addition, we also evaluated the effect of low, medium, and high eEV doses on macrophage responses. We observed that the dose can have an impact in the response of treated macrophages, where lower doses of eEVs seem to have a more beneficial effect on inflammation (Supplemental Figure 5). However, this finding could also be reflecting the effect of the increased nanoparticle load on macrophage activity.[55]
Decoration of eEVs with SPA-targeting ligand leads to enhanced intrapulmonary retention and improved overexpression of the target therapeutic cargo:
To increase the interactions with lung tissue and maximize intrapulmonary retention while minimizing liver clearance, eEVs were functionalized with SPA to promote binding with the P63/CKAP4 receptor on type II alveolar pneumocytes, which are key modulators of inflammation in the lung.[56, 57] The presence of the functionalizing ligand SPA on the membrane of intact eEVs was confirmed via ELISA using intact eEVs (Figure 4A). The ability of the SPA-functionalized eEVs to preferentially accumulate in the lung was assessed in vivo using fluorescently labeled IL-4+SPA or IL-10+SPA eEVs delivered intranasally in 9–15-week-old male mice (Figure 4B). Biodistribution analysis conducted via IVIS at 12 hours after delivery showed improved retention of SPA-functionalized eEVs in the lungs compared to non-functionalized (i.e., sham) EVs (Figures 4C). Fluorescence microscopy analyses of lung tissue sections further revealed robust interaction and retention of the SPA-functionalized eEVs by lung cells (Figure 4D and E) compared to non-functionalized/sham eEVs. Effective transfection and overexpression of the target cargo in recipient cells in the lung was confirmed via 3,3’-diaminobenzidine (DAB) staining, as shown in Figure 4F–G.
Fig. 4. SPA functionalized anti-inflammatory eEVs with improved lung retention and accumulation.

A) Schematic representation of the detection of SPA protein on the surface of the eEVs and data showing the successful transfer of the episomally expressed ligand SPA to the surface of the eEV membrane confirmed via ELISA compared to sham EVs. B) Schematic diagram illustrating the intranasal delivery of fluorescently labeled lung-targeting eEVs and analysis using an in vivo imaging system (IVIS) 12 hours after delivery. C) IVIS images of the major organs, showing the accumulation of the eEVs in the lungs, with a higher retention when treated with SPA-functionalized IL-10+SPA eEVs compared to non-functionalized (sham) EVs and D) comparative analysis of the fluorescence radiant efficiency units to quantify eEV accumulation. E) Immunofluorescence images of lung tissue samples after IVIS of animals treated with fluorescently labeled (Far-Red) sham and IL-0+SPA eEVs co-localized with the nuclei of the cells in blue (DAPI), 12 hours after intranasal delivery (arrows pointing at positive cells). F) Respective fluorescence intensity quantification of 4 lung tissue sections per animal. Scale bar 10μm. G) DAB staining images showing the positive expression of IL-10 in the lungs of animals treated with IL-10+SPA eEVs compared to sham eEVs, this signal was co-localized with the nuclei of the cells. H) Quantification of IL-10 expression (% of positive areas) in the lung tissue showing an overexpression of IL-10 in the IL-10+SPA eEV-treated group compared to the sham eEV-treated group. (n=4) All error bars are shown as SEM. *p<0.05 and **p<0.001, #significant difference with respect to the control with a p-value<0.05, One-way ANOVA or Two tail t-test when appropriate.
SPA-functionalized eEVs loaded with IL-4 and IL-10 dampen LPS-induced lung inflammation in vivo
To assess the efficacy of eEVs in dampening inflammation in vivo, we first established and characterized a model of acute lung injury via intranasal delivery of 0 – 4 mg/Kg LPS to 9–10-week-old male mice (Supplemental Figure 6A). Lung injury parameters, including abnormal BALF (i.e., altered differential cellular counts, and increased protein and cytokine content), and secretion of pro-inflammatory mediators within the lung tissue, were then evaluated in response to the LPS challenge. Total protein content measurements in BALF revealed elevated protein levels for mice challenged with 0.4 or 4 mg/Kg LPS compared to controls without LPS challenge, which potentially correlates with compromised integrity of the alveolar-capillary barrier and associated protein leakage into the alveolar space (Supplemental Figure 6B). Moreover, protein expression analysis conducted via ELISA showed a significant increase in the secretion of key pro-inflammatory mediators (i.e., IL-6, IL-1β, TNF-α) (Supplemental Figure 6C), with a robust increase in the recruitment of neutrophils and decreased macrophage recruitment compared to controls (Supplemental Figure 6D and 6E). Additionally, qRT-PCR analyses of lung tissue showed successful upregulation of the pro-inflammatory mediators IL-6, TNF-α, and iNOS for LPS-challenged mice (Supplemental Figure 6F).
Once we established a model of LPS-mediated acute lung injury, we set to test the therapeutic effect of IL-4+SPA and IL-10+SPA eEVs in mice challenged with 0.4 mg/Kg of LPS. Intranasal delivery of eEVs was conducted 6 hours after the LPS challenge, and the animals were euthanized after 12 hours (Figure 5A). Our results showed a marked reduction in neutrophil recruitment in mice treated with IL-4+SPA or IL-10+SPA eEVs compared to mice treated with sham eEVs (Figures 5B–C). Furthermore, we observed a significant increase in macrophage recruitment for animals treated with IL-10+SPA eEVs in comparison with animals without eEV treatment (i.e., LPS group) (Figure 5B–C). We also found a trend towards decreased BALF protein content compared to LPS-challenged mice without eEV treatment, with robust attenuation in mice treated with IL-10+SPA eEVs (Figure 5D). ELISA analyses revealed a significant decrease in the secretion of the pro-inflammatory mediators IL-6, IL-1β, and TNF-α, in the lungs of mice treated with IL-4+SPA or IL-10+SPA eEVs compared to mice without eEV treatment (Figure 5E).
Fig. 5. SPA-functionalized eEVs loaded with IL-4 and IL-10 dampen LPS-induced lung inflammation and tissue damage in vivo.

A) Schematic diagram depicting the experimental timeline of the intranasal delivery of 0.4 mg/Kg LPS that elicits an inflammatory response (0 hours), treatment with anti-inflammatory eEVs delivered intranasally (6 hours), and subsequent analysis to evaluate lung injury parameters (12 hours). B-C) Differential cellular counts showing reduced neutrophils recruitment in bronchoalveolar lavage fluid (BALF) for animals treated with IL-4+SPA eEVs (~70%), and IL-10+SPA eEVs (~60%) compared to LPS-challenged mice without treatment (~80%). D) BALF analysis shows decreased amount of total protein concentration for animals treated with IL-10+SPA eEVs compared to LPS-challenged mice without treatment. E) Lung tissue protein analysis via ELISA displaying decreased expression of IL-6, IL1-β, and TNF-α for animals treated with IL-4+SPA and IL-10+SPA eEVs with respect to the LPS-challenged mice without treatment. F) Representative micrographs of hematoxylin and eosin-stained lung sections at 90X magnification used for lung tissue morphometric analysis showing G) a significantly higher number of alveoli per hpf (high power field), H) decreased alveolar septal thickness and I) mean linear intercept (MLI) for animals treated with IL-4+SPA or IL-10+SPA eEVs, compared to LPS-challenged mice without treatment and Sham eEV-treated mice (n=4). All error bars are shown as SEM. *p<0.05 and **p<0.001, #significant difference with respect to the control with a p-value<0.05, One-way ANOVA.
Tissue injury in terms of altered lung morphology was evaluated using hematoxylin and eosin-stained lung sections (Figure 5F). This lung tissue morphometric analysis revealed a significant reduction in the degree of tissue injury after LPS-challenge for animals treated with IL-4+SPA and IL-10+SPA eEVs, with an increased number of alveoli per high power field (hpf) (Figure 5G), and a reduced mean septal thickness and mean linear intercept (MLI) (Figure 5H–I), compared to non-treated (i.e., LPS group) and sham eEVs-treated animals. Additionally, measurements including elastance, compliance, resistance of respiratory system, tissue elastance, tissue damping, and inspiratory capacity, are included in Supplemental Figure 7.
Untargeted metabolomics analysis reveals differential metabolic profiles in the BALF of LPS challenged mice treated with IL-4+SPA, IL-10+SPA, and Sham eEVs
An untargeted metabolomics analysis was conducted to identify metabolic changes driven by treatment with the different eEVs formulations in mice with induced acute lung inflammation. For this analysis, BALF samples were collected from LPS-challenged mice (LPS), healthy mice (control), and LPS-challenged mice treated with IL-4+SPA, IL-10+SPA, or sham eEVs (sham). The liquid chromatography with tandem mass spectrometry (LC-MS/MS) in positive and negative ionization modes revealed 3,308 features. Prior to multivariate and univariate statistical analyses, data normalization was performed using auto-scaling (mean-centered, divided by the standard deviation of each variable) and log transformation (Supplemental Figure 8). Unsupervised and supervised data dimensionality reduction approaches were first used to explore all the data (Supplemental Figure 9). Principal component analysis (PCA) was employed to visually discriminate the samples, however, the variance in the dataset explained by PC1 and PC2 showed partial separation (Supplemental Figure 9A–B). Partial least squares-discriminant analysis (PLS-DA) showed better clustering of the four groups (Supplemental Figure 9C–D). Cross validation showed that 5 components were optimal for building the model and the permutation test (n=2000) with a p-value lower than 0.05, indicating that PLS-DA showed a good predictive model (Supplemental Figure 9E–H). After performing a pairwise comparison of the LPS group against all other groups, using the supervised PLS-DA approach, a clear separation between the groups was observed, indicating differential metabolomic profiles between the LPS-challenged mice and the healthy controls, and between the LPS-challenged mice and all the eEVs treatment groups (Figure 6A–D and Supplemental Figure 10A–D). Features with an FDR < 0.1, foldchange > 2, and Variable Importance in the Projection (VIP) score > 1.0 were considered significantly different between groups (Table S5). When samples from healthy controls were compared to samples from mice challenged with LPS, 82 features were upregulated in the control, while 31 were downregulated. Compared to LPS-challenged mice, BALF from the IL-4+SPA eEV-treated mice, showed the downregulation of 33 features, with the same number of features being upregulated; while for the IL-10+SPA eEV-treated mice, 27 features were upregulated and 14 were downregulated. On the other hand, for sham eEV-treated mice, 106 features were upregulated while 15 were downregulated (Figure 6, Supplemental Figure 10, and Table S5). From the set of features that were significantly up/downregulated, only the annotated metabolites that are presented in Figure 7A were further explored. 2’-Deoxycytidine 5’-monophosphate, malic acid, N-phenylacetylglycine, 15-methylpalmitate, heptanoylcarnitine, and fenofibrate, were among the features determined to be statistically increased between LPS-challenged animals and healthy animals (i.e., control group). Interestingly, ascorbic acid and oxoglutarate were both found to be significantly upregulated in the BALF of animals treated with IL-4+SPA and IL-10+SPA eEVs, while tryptophan was significantly downregulated in both groups (Figure 7A). For the IL-4+SPA eEV-treated mice, upregulation of the monosaturated fatty acid 7-heptadecenoic acid, and downregulation of phenylalanine, isoleucine/leucine, and tyrosine were observed. While for the IL-10+SPA eEV-treated mice, phosphodimethylethanolamine, adenosine, inosine, 8E-heptadecenedioic acid, and dodecanoyl-L-carnitine were upregulated, while the triglycerides TG(52:0) and TG(34:0) were downregulated. For the sham eEV-treated group, several other metabolites were upregulated, including uridine, citric acid, vitamin K, glucuronic acid, ubiquinone 6, cortisone, and linoleic acid, among others. The metabolite 9-(2,3-dihydroxypropoxy)-9-oxononanoic acid was increased in BALF samples of animals treated with both, sham and IL-10+SPA eEVs. While 7-heptadecenoic acid was increased in BALF samples from animals treated with sham and IL-4+SPA eEVs.
Fig. 6. Multivariate and univariate statistical analyses of the metabolomics dataset obtained in negative ionization mode.

Two-dimensional score plot of partial squares discriminant analysis (PLS-DA) (left panels) showing the separation of each of the treatment groups and the LPS-challenged mice; heatmap showing hierarchical clustering obtained based on Euclidean distance (middle panels); and volcano plot of univariate statistical analysis showing significant values of the features in each treatment group in comparison with LPS-challenged mice, with fold change values >2 and false discovery rate (FDR) <0.1 (right panels). A) Healthy mice (Control) vs LPS-challenged mice, B) IL-4+SPA eEV-treated mice vs LPS-challenged mice, C) IL-10+SPA eEV-treated mice vs LPS-challenged mice, and D) Sham eEV-treated mice vs LPS-challenged mice.
Fig. 7. Heatmap of annotated metabolites collected from bronchoalveolar lavage fluid (BALF) of mice.

A) Heatmap of the hierarchical cluster analysis showing the detected and annotated metabolites collected from the BALF of mice treated with 0.4 mg/Kg LPS, treated with IL-10+SPA, IL-4+SPA or Sham eEVs. The control treatment group consists of BALF collected from healthy mice that were not exposed to LPS. The metabolites shown in this graph correspond only to the annotated features that had FRD>0.1 and FC>2 resulting from the comparison of each treatment group versus LPS-challenged mice, calculated using one-way ANOVA. The averages of 3 technical, and 3 – 4 biological replicates are presented. Each colored cell on the map corresponds to a concentration value in the data table, with samples in rows and features/compounds in columns. B) Pathway analysis of upregulated pathways after treatment with IL-4+SPA, IL-10+SPA or Sham eEVs. C) Pathway analysis of downregulated pathways after treatment with IL-4+SPA, IL-10+SPA or Sham eEVs.
Pathway analysis comparing BALF samples obtained from LPS-challenged mice vs eEVs-treated mice revealed an increase of the TCA cycle for animals treated with eEVs (IL-4+SPA, IL-10+SPA and sham), while treatment with IL-4+SPA and IL-10+SPA eEVS caused an increase of urea cycle and the metabolism of arginine, proline, glutamate, aspartate, and asparagine. Purine metabolisms was increased upon treatment with IL-10+SPA eEVS. Treatment with sham eEVs caused the upregulation of other distinct pathways including those of linoleate, histidine, vitamin K, ubiquinone, C21-hormone biosynthesis, glycosphingolipid, pyrimidine, de novo fatty acid biosynthesis, among others (Figure 7B). IL-4+SPA and sham eEVs caused an increase in the glycerophospholipid pathway, while treatment with IL-10+SPA eEVs caused a decrease in this pathway. Moreover, tryptophan metabolism was downregulated in IL-4+SPA and IL-10+SPA eEV-treated animals. Tyrosine and biopterin metabolism and the degradation of valine, leucine and isoleucine were decreased in IL-4+SPA eEV-treated animals. Sham eEVs on the other hand, caused a downregulation of the phosphatidylinositol phosphate metabolism (Figure 7B). Our results show that the metabolic profiles in BALF samples of LPS-challenged mice is clearly impacted by treatment with eEVs loaded with IL-4+SPA, IL-10+SPA, but also significantly different than that caused upon treatment with sham eEVs. Overall, we were able to see that different metabolomic responses are elicited depending on the molecular payload of the eEVs.
DISCUSSION
To the best of our knowledge, this is the first report describing the implementation of dermal fibroblasts as donor cells to derive eEVs loaded with anti-inflammatory cargo and functionalized to preferentially target the lung tissue. In recent years, EVs derived from progenitor cells (e.g., endothelial progenitors, and mesenchymal stem/stromal cells) have been postulated as an effective alternative to cell-based therapies.[58] For example, EVs derived from mesenchymal stem cells (MSCs) have been extensively explored for the treatment of pulmonary diseases due to their ability to restore epithelial cell integrity and function and to reduce inflammation during lung injury.[37, 39, 59] Numerous studies have reported that MSC-derived EVs are loaded with multiple effector molecules, including anti-inflammatory cytokines, growth factors (e.g., KGF), and antimicrobial peptides.[22, 58, 60] Progenitor cell derived EVs, however, still face multiple hurdles, including limited cell sources and increased risk for tumorigenicity and immunogenicity. Our results suggest that dermal fibroblasts-derived EVs could potentially circumvent many of these barriers, not only because dermal fibroblasts represent a viable and more abundant and readily available source of EVs, but also due to their reduced immunogenicity likely driven by decreased expression of the major histocompatibility complex (MHC) on their surface.[61] Moreover, dermal fibroblasts are currently being used in clinical applications for dermal grafts, which highlights their potential be a promising cell source for EV-based therapeutics.[62–65]
EVs have been shown to have a therapeutic potential to modulate inflammation in several clinical disorders.[66–68] Their pro- and anti-inflammatory properties can be attributed to the presence of cytokine components exposed on the membrane and/or packed in the lumen, working synergistically.[40, 69] Previous studies have shown that cytokines such as IL-4, IL-10, IL-2, IL-12, and IL-16 are preferentially packed within the EVs, while IL-8, IL-17, and GRO-α are mostly found on the EV surface.[69, 70] Taking advantage of this predisposition to pack cytokine components, we proposed the implementation of IL-4 and IL-10 as model anti-inflammatory cargo. Moreover, IL-4 and IL-10 have well-documented properties to reduce the inflammation and mortality associated with ARDS.[23, 71] IL-10 released by activated macrophages and Th1 cells is known to be involved in the reduction of pro-inflammatory cytokines by downregulation of Th1 cells and inhibiting the production by alveolar macrophages of pro-inflammatory mediators (e.g., TNF-α, IL-1β, IL-6, Interferon ϒ) involved in ARDS.[10, 20, 72, 73] On the other hand, IL-4 possesses immunosuppressive and anti-inflammatory properties mediated by the inhibition of key pro-inflammatory mediators (e.g., TNF-α, IL-1β, IL-6, IL-12, IL-8) and plays an important role on the differentiation of CD4+ T cells to Th2 cells and monocytes into macrophages.[72, 74–76] Additional studies have reported that endogenous and exogenous IL-4 are implicated in the late resolution of lung inflammation by promoting polarization of monocytes towards an anti-inflammatory phenotype (M2) and neutrophil clearance, which dampens inflammation and enhances lung repair.93
Our results highlight the ability of dermal fibroblast-derived eEVs to safely and selectively package mRNA transcripts of the electrotransfected anti-inflammatory cargo, reaching loading levels that are 2–6 orders of magnitude higher compared to eEVs derived from sham-transfected dermal fibroblasts. Additionally, we also verified that besides DNA and mRNA, there is also IL-4 and IL-10 protein content associated with the EVs. Characterization of the eEVs showed that morphology, size distribution, and membrane protein content were consistent with the standards issues by the International Society for Extracellular Vesicle guidelines.[77] Our results also demonstrate the ability of eEVs to effectively transduce recipient cells in the alveolar environment to trigger overexpression and secretion of IL-4 and IL-10, via effective delivery of mRNA transcripts and plasmid DNA encoding for these cytokines. Although our results provide robust pre-clinical data supporting the therapeutic efficacy and safety of IL-4 and IL-10 engineered EVs using a murine model of acute lung inflammation, additional studies are needed to look at toxicology, safety, and efficacy in larger animal models before this technology can be translated into the clinic.
EV-based payload delivery systems offer a promising cell-free therapy, with a highly customizable platform that can potentially be engineered to maximize targeted delivery, of anti-inflammatory therapies for lung injury. This type of targeted delivery enables the implementation of lower doses, with limited off-target systemic effects, which maximizes therapeutic efficacy. While EVs have been shown to have inherent targeting characteristics and display tropism for particular cells or tissues,[78, 79] they can also be modified with ligands (e.g., integrins, protein ligands, peptides, nucleic acids) to facilitate the targeting/recognition of specific receptors for preferential binding and payload delivery, avoiding major off-target effects.[43, 80–82]
Type II pneumocytes constitute 60% of alveolar epithelial cells and makeup 10–15% of all cells present in the lungs.[83] The relevance of this is that alveolar epithelial cells are the first line of cells that secrete pro-inflammatory mediators (e.g., cytokines, chemokines) in response to pathogenic/harmful stimuli, leading and orchestrating recruitment of innate and adaptative immune cells to the site of injury and/or infection.[84, 85] Our results show that the targeting ligand SPA improved intrapulmonary retention of the eEVs with reduced liver clearance and facilitated localized delivery of anti-inflammatory payloads. SPA interacts with the receptor P63/CKAP4 on alveolar pneumocytes[56] and can also bind to Toll-like Receptor – 2 (TLR2),[57] potentially dampening the activation of the NFκβ inflammatory pathway and macrophage activation.[52] SPA has also been shown to decrease pro-inflammatory mediators on LPS-stimulated alveolar macrophages by binding to INF-Y and preventing the interaction between INF-ϒ and its receptor INF-ϒR.[52] Furthermore, SPA/P63 interaction is implicated in the reduction of alveolar surface tension and prevention of the alveoli collapse.[56, 86] In addition, qRT-PCR characterization revealed that eEVs also carry mRNA transcripts of SPA (Figure 1H), which could potentially bolster local SPA expression and may further attenuate lung inflammation due to its role in maintaining immune homeostasis in the lung microenvironment.[52]
The most common models of ARDS included the administration of endotoxin (LPS),[87, 88] acid aspiration,[89, 90] prolonged hyperoxia,[91] mechanical ventilation with low tidal volumes,[92, 93] ischemia/reperfusion,[94] and airway installation of live bacteria or influenza virus,[95] which can cause damage of the pulmonary vascular endothelium and/or bronchoalveolar epithelium.79, 80 Here we ran a comprehensive study looking into the entire process, involving the dynamics, optimal concentration, and treatment method (co- or post-treatment) of the inflammatory response mounted by the LPS in vitro and in vivo expanding on previous reports[87, 88, 96] (Supplemental Figures 2 and 6). Moreover, we confirmed the feasibility of using LPS to simulate an inflammatory response in vivo, to test the therapeutic effect of the eEVs. A suitable in vivo model of ARDS has to meet some basic criteria, including accumulation of neutrophils in the alveolar/interstitial space, increase of protein concentration in BALF representative of the disruption of alveolar-capillary barrier, increase of pro-inflammatory mediators in lung tissue or BALF, and physiological changes consistent with pulmonary dysfunction, in accordance with guidelines from the American Thoracic Society (ATS).[97, 98] Our results show that our in vivo model induces this type of inflammatory response, and it is responsive to treatment via eEVs. The expression of LPS-induced inflammatory mediators (i.e., IL-6, IL-1β, and TNF-α) was consistently reduced in mice treated with IL-4+SPA and IL10+SPA eEVs. The observed differences are consistent with previous reports characterizing cytokine expression in patients with and without ARDS.[99] Additionally, lung tissue morphometric analysis revealed the potential of eEVs to aid tissue repair, where animals treated with IL-4+SPA or IL-10+SPA eEVs showed an increased number of alveoli per high power field (hpf), and a reduced mean septal thickness and mean linear intercept (MLI), compared to non-treated (i.e., LPS group) and sham eEV-treated animals.
In vitro experiments on the other hand show some potential effect of the sham eEVs to reduce some of these markers, but this effect was not consistent across the different treatment options evaluated (i.e., post- or co-treatment for 3–12 hours). Moreover, this effect was not observed when testing the therapeutic effect of the different eEV formulations in vivo.
The untargeted metabolomics approach used in this study aimed at assessing the effect of treating LPS-challenged mice with anti-inflammatory dermal fibroblast-derived eEVs. Cluster separation of healthy controls vs LPS-treated mice indicates a robust change in the metabolomes of the collected BALF samples (Figure 6A, and Supplemental Figure 10A). The accumulation of heptanoylcarnitine in LPS-challenged mice, a derivative of O-acetylcarnitine, had previously been reported as part of the BALF metabolome of patients with acute lung injury.[100] Also, the statistically significant increase of 15-methylpalmitate and N-phenylacetylglycine were a distinguishing characteristic for LPS-challenged mice. The former compound is a long saturated fatty acid, which has been reported to have pro-inflammatory effects on macrophages.[101] Although the biological significance of accumulation of phenylacetylglycine is unclear, its accumulation has been reported in the urine of patients with ischemic heart failure, as well as in plasma of rats subjected to chronic cigarette smoke exposure.[102, 103] Treatment of LPS-challenged mice with IL-4+SPA, IL-10+SPA, and sham eEVs caused changes in the BALF metabolome. However, the metabolic profiles of animals treated with IL-4+SPA and IL-10+SPA eEVs where more similar to each other than to the sham eEV-treated animals; as the sham eEV-treated mice clustered apart from IL-10+SPA, IL-4+SPA, and the healthy control, with some overlapping with the LPS group (Supplemental Figure 9D). Upon treatment with IL-4+SPA and IL-10+SPA eEVs, an upregulation of ascorbic acid and oxoglutarate occurred in the BALF of LPS challenged mice. Ascorbic acid (vitamin C) is a potent antioxidant that has been reported to provide protection against reactive oxygen species, preventing the damaging effects of the oxidative stress, and it is also known for its anti-inflammatory role.[104, 105] Previous studies have shown that ascorbic acid attenuates systemic inflammation induced by acute lung injury.[106] Likewise, oxoglutarate has been reported to have antioxidant properties and to play a key role in macrophage polarization into an M2 anti-inflammatory phenotype.[107, 108] Interestingly, these two metabolites were not significantly upregulated in any of the control groups, suggesting a potential involvement in the reduction of lung inflammation. Moreover, the upregulation of adenosine and inosine in the IL-10+SPA eEV-treated mice, could also be an indication of the therapeutic effect of these eEVs, as these metabolites have also been reported to play a role in anti-inflammatory processes associated with acute lung disease.[109–112] Treatment with IL-4+SPA eEVs also showed to cause a significant reduction of phenylalanine in BALF; the increased accumulation of this amino acid in blood plasma has been described as a potential biomarker and as a mortality predictor for ARDS patients.[113] Furthermore, the metabolomic changes caused by the treatment with sham eEVs also resulted in the upregulation of certain metabolites with antioxidant or anti-inflammatory effects, including ubiquinone, citric acid, cortisone, uridine and linoleic acid.[114–118] We also note that the results obtained from this untargeted metabolomics analysis provide relative abundances of the metabolites, hence the use of standards in future experiments will enable further assessment of the systemic levels of these metabolites and how they compare with known safe levels.[119, 120] Overall, after assessing the effect of IL-4- and IL-10-loaded anti-inflammatory eEVs using an untargeted metabolomics approach, we were able to uncover changes in the metabolic profiles in the BALF of LPS-challenged mice, which paves the way to having a more complete understanding of the unexplored metabolic response of using eEVs as potential therapeutic agents. Our findings shed light into the potential mechanisms involved in the anti-inflammatory response upon treatment with the eEVs.
CONCLUSION
In this study, we describe an effective methodology to generate engineered (eEV)-based nanocarriers derived from primary adult mouse dermal fibroblasts. Our results highlight how SPA-decorated IL-4 or IL-10 loaded eEVs showed enhanced intrapulmonary retention, and can effectively deliver IL-4 and IL-10 transcripts and plasmid DNA to alveolar cells in the inflamed lung environment to boost the levels of these anti-inflammatory cytokines, mitigating the local inflammatory response. Although SPA decoration seems to improve intrapulmonary retention, by likely improved interactions with specific epithelial cell subpopulations in the lung based on ligand-receptor interactions, the main cellular target of the eEVs in the lung microenvironment must be further studied, as their significant therapeutic effect is likely due to the production of IL-4 and IL-10 by multiple types of recipient cells in the lung (e.g., macrophages and epithelial cells). Notably, we report that IL-4+SPA and IL-10+SPA eEVs have the ability not only to reduce the secretion of key proinflammatory mediators such as IL-6, IL-1β, and TNF-α, but also to aid tissue repair and to induce significant metabolomic changes to favor activation of anti-inflammatory pathways mediated in part by the significant upregulation of ascorbic acid, inosine, and oxoglutarate. These results establish the potential of using skin fibroblast-derived anti-inflammatory eEVs, obtained via non-viral approaches, to selectively deliver therapeutic payloads to the inflamed lung to reduce inflammation, tissue damage, and the prevalence/progression of lung injury during ARDS.
MATERIALS AND METHODS
DNA plasmid preparation
Plasmids used in this study (Table S1) were obtained from OriGene Technologies, prepared via bacterial transformation, and isolated using a ZymoPure II kit (Zymo Research) for plasmid DNA isolation, following the procedure described by the manufacturer. DNA concentrations and quality were measured using a Nanodrop 2000c Spectrophotometer (Thermo fisher Scientific).
In vitro cell culture
Adult BALB/c mouse primary dermal fibroblasts (adult-MDFs) were obtained from Cell Biologics (BALB-5067) and cultured using complete fibroblast basal medium with the growth factor supplements provided by the vendor (Cell Biologics, M2267) and incubated at 37°C in humidified air containing 5% CO2 (standard culture conditions). Additionally, monocytes RAW 264.7, a transformed cell line derived from BALB/c mice, were obtained from ATCC (TIB-71) and cultured in RPMI 1640 medium (Gibco), supplemented with 10% heat-inactivated FBS (Corining) at standard culture conditions.
Non-viral in vitro transfection
Non-viral cell transfection of adult-MDFs was performed using a Neon transfection system (Thermo Fisher Scientific). Once the cells (passage <P4) reached >80% confluency, they were detached and resuspended at a final concentration of 1.0×106 cells in 100 μL of electrolytic buffer. Cells were co-transfected with plasmids encoding for IL-4+SPA, and IL-10+SPA in a proportion of 1:1 (at 0.05 μg/μl). pCMV6-GFP (sham/empty vector) was used as a control at a concentration of 0.1 μg/μl. Following the procedure described by the manufacturer, the cells were transfected for 30ms with 1 pulse at 1425 V. This electric field allows for delivery of the molecular cargo into the cells via transient poration of the cell membrane and by driving the genetic material via electrophoresis. After transfection, cells were seeded in 6 wells plates and maintained in fibroblast basal medium with all the growth factor supplements provided by the vendor and supplemented with 10% exosome-depleted FBS (GIBCO, A27208–01). Successful transfection of the molecular cargo into the donor cells was verified via immunofluorescence microscopy (GFP-tag signal) and gene expression of each factor (mRNA) by quantitative reverse transcription polymerase chain reaction (qRT-PCR).
EV isolation and characterization
eEVs were isolated from culture media 24 hours after transfection of the donor cells, as previously reported.[43, 46, 47] The culture media was centrifuged at 2,000g for 30 min at 4 °C to remove dead cells and debris. After centrifugation, Total exosome isolation reagent (Thermo Fisher Scientific, 44–783-59) was added to the supernatant containing the cell-free culture media following manufacturer instructions. Pellets of eEVs were used fresh or stored at −80 °C for subsequent analysis. All eEVs were characterized in solution by measuring their concentration and size distribution via nanoparticle tracking analysis (NTA) and quantitative qRT-PCR was used to verify packing of the molecular cargo inside the eEVs prior to any experiment.
Cryo-electron microscopy (EM):
The intact morphology of the eEVs was evaluated via Cryo-EM. To prepare the Cryo-EM grids, a small aliquot (3μl) of the sample was applied to a Lacey carbon grid or a Lacey carbon grid with a continuous ultrathin carbon layer. After blotting away excess liquid, the grid was immediately plunged into liquid ethane to rapidly form a thin film of amorphous ice using the Vitrobot Mark IV system (Thermo Fisher Scientific, Hillsboro). The Vitrobot was operated at 4°C and 100% humidity. The blotting force was set at +1. The blotting time was 3–5s and the frozen grids were clipped into AutoGrids and transferred into Glacios CryoTEM (Thermo Fisher Scientific, Hillsboro), or stored in a liquid nitrogen tank. Cryo-EM images were captured with Falcon 3EC direct electron detector under linear mode on Glacios Cryo-EM. The microscope was operated at an acceleration voltage of 200kV, and images were collected by using EPU software at 57,000x or 6,700x nominal magnification.
Western Blot:
eEV pellets were resuspended in 20uL of Lysis buffer containing RIPA buffer (Thermo Fisher Scientific, 89900), phosphatase inhibitor cocktail (Sigma-Aldric, P5726–1), protease inhibitor cocktail (Roche, 4693116001), and 1 mM phenylmethylsulfonyl fluoride (PMSF, Thermo Scientific, 36978), vortexed for 15 min at 4°C and incubated overnight at −20°C. The samples were once again vortexed for 15 min at 4°C and centrifuged at 10,000 rpm for 30 min at 4°C, and the cytosolic protein content was recovered and quantified via Bradford assay (Bio-Rad, 500006). 14 ug of total protein was combined with NuPAGE™ LDS Sample Buffer (4X) and NuPAGE™ Sample Reducing Agent (10X) (Thermo Fisher Scientific, NP007, NP009 respectively), heated 2 min at 85°C and loaded into 10–20% Tris-Glycine mini gels (Thermo Fisher Scientific, XP10202BOX). Proteins were electrophoretically transferred to a nitrocellulose or Polyvinylidene difluoride (PVDF) membrane (Thermo Fisher Scientific, LC2001/LC2002). The membranes were washed with TBS-T 1X (Tris-Glycine transfer buffer and tween, Novex and Thermo Fisher Scientific, respectively) and blocked with 5% BSA for 1 hour at room temperature (RT). All antibodies were diluted in TBS-T and 2.5% BSA and incubated overnight at 4°C (primary antibodies), or 1 hour at RT (secondary antibodies). Finally, membranes were visualized using a C-DiGit LI-COR blot scanner. Table S2 contains the list of all antibodies used for this study.
Gene expression analyses
For this type of analysis, donor cells, monocytes, and eEVs were collected using TRizol reagent (Thermo Fisher Scientific, 15596018) to subsequently extract total RNA. Nanodrop 2000c Spectrophotometer (Thermo Fisher Scientific) was used to measure RNA concentrations and quality. Reverse transcription reactions were performed with the superscript VILO cDNA synthesis kit (Thermo Fisher Scientific, 11754050) using 80–2500 ng RNA in a 20μL reaction, preserving equal amounts of cDNA throughout the samples. Taking the cDNA as a template, the mRNA expression levels were verified by qRT-PCR in donor cells and eEV, using predesigned primers (all the primers used in this study are listed in Table S3). qRT-PCR reactions were performed using the QuantStudio 3 Real-Time PCR System with TaqMan fast advance Master Mix (Thermo Fisher Scientific, 4444964). Gene expression was normalized using the housekeeping gene GAPDH and all data presented as fold change relative to sham/control.
After bronchoalveolar lavage fluid (BALF) collection, left lungs were preserved for protein isolation. For this end, lung samples were processed in Lysis buffer (as described in the Western Blot Section above) using a gentleMACS Dissociator (Miltenyi Biotec), with gentleMACS M tubes (Miltenyi Biotec, 130-093-236), subsequently centrifuged at max speed 15 min at 4°C and stored for subsequent analysis.
In vitro assays using Lipopolysaccharide (LPS)
Monocytes RAW 264.7 were seeded overnight in 12-well culture plates at a density of 2.0×105 cells/cm2. All the experiments described below were run using a minimum of 3 biological replicates and 3 technical replicates. Monocytes were challenged in vitro with Lipopolysaccharide (LPS, L4524, Sigma Aldrich) resuspended in RPMI 1640 plain medium at 2 different concentrations (100–1000 ng/mL). Non-treated cells were used as control. 24 hours after LPS challenge, culture plates were imaged using brightfield under a Nikon Ti-2e microscope at 20X to evaluate cell morphology. Moreover, culture media was collected and stored at −80°C for further analysis (e.g., protein analysis). Cells were collected using TRizol reagent, followed by RNA extraction and cDNA synthesis. The inflammatory response was evaluated by the expression of pro-inflammatory mediators (e.g., IL-6, IL-1β, iNOS, TNF-α, and KC) via qRT-PCR as already described. To test the dynamics of secretion of key proinflammatory mediators, monocytes RAW 264.7 cells were seeded overnight and treated with 100 ng/mL of LPS for 3–48 hours. Dose-response experiments were conducted using three different doses of eEVs, including a high dose with 4X109 eEVs, a medium dose with 4X108 eEVs, and low dose with 4X107 eEVs. eEVs were delivered to macrophages challenged with 100 ng/mL LPS (i.e., co-treatment) for 3 hours (Supplemental Figure 5A). The inflammatory response based on the protein expression levels of pro-inflammatory cytokines IL-6, IL-1β, and TNF-α was characterized via U-Plex assay (K15069M-1, Meso Scale Diagnostics LLC) by electrochemiluminesmecence using the MESO QuickPlex SQ 120 instrument (Meso Scale Discovery platform) following manufacturer’s instructions. Analysis was performed using the DISCOVERY WORKBENCH 4.0 Analysis Software.
Morphological analysis
Cell morphology was analyzed using images captured after co- or post-treatment with eEVs. All images were analyzed using the ImageJ-FIJI software, where each cell was categorized and counted as either a non-activated (i.e., circular shape) or an activated monocyte (i.e., spindle shape). A minimum of 3 images per biological and technical replicate were analyzed per condition and the data was presented as the percentage of activated cells based on the total number of counted cells.
Animal Husbandry
All experiments using mice were performed following procedures previously approved by the Animal Care and Use Committee at The Ohio State University (IACUC # 2016A00000074-R1). 8–15-week-old male BALB/c- (000651 strain) and C57BL/6 mice were purchased from Jackson Laboratory. Mice were acclimated for 1-week after arrival. Isoflurane inhalation was used to anesthetize all animals before all experimental procedures.
Biodistribution Analysis
For the biodistribution experiments, eEVs were labeled with a far-red fluorescent cell membrane labeling kit (MIDCLARET-1KT, Sigma Aldrich), according to manufacturer instructions. For this experiment, all mice were grouped and selected randomly. 9–15-week-old male C57BL/6 were anesthetized with inhalation of 5% isoflurane and maintained with 2% while conducting the procedure (based on body weight). For this experiment fluorescently labeled eEVs were intranasally administered to mice. Briefly, mice were positioned on an endotracheal intubation stand (Kent Scientific) to allow delivery of the eEV dose. A 25μL bolus of 1.61X1011 eEVs/mL (IL-10+SPA or sham loaded eEVs) fluorescently labeled and resuspended in saline solution was delivered through the left nostril of the mouse monitoring the respiration rate during the entire procedure to reduce discomfort. Control mice were subjected to delivery of a 25μL bolus of saline solution. Mice were euthanized 12 hours after eEV intranasal delivery, and major organs (i.e., brain, heart, lungs, liver, kidneys, pancreas, and spleen) were collected in 4% FBS (GIBCO) in phosphate buffer solution (PBS, Thermo Fisher Scientific) and stored at 4°C for further characterization. To quantify eEV accumulation, all organs were imaged by using an in vivo imaging system (IVIS) Lumina II (PerkinElmer Inc) right after collection. Images were captured using stage A with an excitation-emission of 640 nm (Cy5.5) and 1s exposure time. All images were analyzed via comparative analysis of the fluorescent radiant efficiency units, and eEV accumulation in each organ was normalized using animals non-treated with eEVs (i.e., control group). Once imaged all major organs (i.e., heart, liver, kidneys, and spleen) were collected and cryopreserved in OCT for further characterization.
Immunohistochemistry
After performing the targeting experiment and imaging the organs with IVIS, lung tissue was embedded in optimal cutting temperature (OCT, Thermo Fisher Scientific) solution and frozen at −80 °C for subsequent analysis. All samples were cryosectioned at 10μm thickness using a CryoStar NX50 cryostat (Thermo Fisher Scientific), and mounted on charged microscope slides (Thermo Fisher Scientific). Tissue samples were fixed with cold Acetone, permeabilized with a solution of 0.05% PBS Tween 20 buffer (PBS-T, Thermo Fisher Scientific), and stained with a DAPI (4’, 6-Diamidino-2-Phenylindole, Dihydrochloride) solution in PBS (1:10000) for 2 min to visualize the nuclei of the cells. Samples were mounted using Vectashield Vibrance Antifade mounting medium (Vector Laboratories), and imaged using an inverted fluorescence microscope (Nikon Ti-2e). All immunohistochemistry imaging was captured at 60X. Finally, mean fluorescence intensity quantification for Far-Red signal (for labeled eEVs) was performed using ImageJ-FIJI software and normalized by the area of the region of interest.
DAB staining:
Right lung samples were fixed with pre-chilled acetone for 3 minutes at RT. After incubation, samples were immersed in methanol containing 10% H2O2 for 15 minutes at RT to inactivate endogenous peroxidases. Then, the samples were rinsed with 0.1% triton-x in PBS-1X for 15 minutes. The slides were blocked with 10% NGS and aviding in PBS-1X solution for 30 minutes at RT. Then primary antibodies (Table S3) were prepared in a solution containing 2.5% NGS with biotin in PBS-1X, and were incubated overnight at 4°C. After primary incubation, the samples were rinsed with 0.1% triton-x in PBS-1X. All samples were subsequently incubated for 1 hour with a biotinylated secondary antibody (Table S3) in a solution containing 2.5% NGS. After incubation, the samples were rinsed and incubated using an ABC (Avidin-Biotin Complex) (Vector laboratories) kit for 20 minutes. The samples were rinsed and a 3,3′-diaminobenzidine (DAB) solution was added to each sample. When the sections turned brown the reaction was stopped with ddH2O. Then, hematoxylin stain (Vector laboratories) was added to each tissue sample for 5 minutes, and the samples were washed with ddH2O. Finally, the samples were dehydrated using an alcohol gradient and then rinsed with xylene before mounting. IL-10 expression in the lung tissue was measured using ImageJ-FIJI software as the percentage of positive area and normalized by the total number of nuclei per section.[121]
In vivo evaluation of the eEVs therapeutic effect
For these experiments 9–10-week-old male BALB/c mice were selected randomly and grouped into 5 experimental groups (with a minimum 4 mice per condition), including a positive (i.e., LPS-challenged mice) and negative (i.e., animals treated with saline solution) control groups, and LPS-challenged mice treated with IL-4+SPA eEVs, IL-10+SPA eEVs, or sham EVs. Animals were anesthetized with inhalation of 5% isoflurane and maintained with 2% while conducting the procedure (based on body weight). Mice were positioned on an endotracheal intubation stand (Kent Scientific) for intranasal delivery (through the left nostril) of a 25μL bolus of 0.4 mg/Kg of LPS resuspended in saline solution. Negative control mice received a 25μL bolus of saline solution. The respiratory rate of the animals was monitored during the entire procedure to reduce discomfort. 6 hours after LPS challenge, mice treated with eEVs were administered a 25μL bolus of 5.82X1010 EVs/mL of IL-10+SPA eEVS, IL-4+SPA eEVs or sham EVs resuspended in saline solution. For lung functional assessments, mice were anesthetized 12 hours after being challenged with LPS, and tracheostomies were initiated with an 18G cannula. Mice were subjected to mechanical ventilation with a FlexiVent FX2 system (SCIREQ Inc) at 150 breathes/min, tidal volume of 10 ml/kg, and a positive end-expiratory pressure (PEEP) of 3 cmH2O prior to baseline measurements. To eliminate breathing efforts and measurement errors, pancuronium bromide (1 mg/kg, Sigma Aldrich) was administered intraperitoneally. Using a “Snapshot-150 perturbation,” respiratory stiffness (Ers) and compliance (Crs) were measured from the forced oscillation technique (FOT) fitted to a single compartment model. Additionally, a broadband FOT maneuver (“Quick Prime-3 perturbation”) was completed to capture the alveolar tissue stiffness/elastance (H). The negative pressure forced expiration (NPFE) extension of the FlexiVent then recorded forced expiratory volume by inflating the lungs to a pressure of +30 cmH2O over 1.2 seconds and then rapidly decreased to a negative pressure of 55 cmH2O. The forced expiratory volume was calculated from the flow-volume loop over 0.1 s (FEV0.1). Mice were euthanized, and lungs were inflated with 10% formalin, collected, and preserved for subsequent histological analysis.
Hematoxylin and eosin (H&E) staining and morphometrical analysis
Lung tissue was fixed in 10% formalin for 72 hours, and subsequently paraffin embedded and sectioned for histologic analysis. Lung sections (n=4 per animal) were stained with H&E to evaluate changes in tissue morphology. For morphometrical analysis, 4 lung fields captured at 90X magnification were randomly selected per section. For each field, a grid with 487.34 μm2 (area per point) was used to count the number of alveolar units represented as alveoli per hpf (high power filed), and the mean septal thickness. Moreover, the mean linear intercept, was calculated as the inverse of the number of air-tissue interfaces to the next measured in microns as previously reported.[122, 123]
Characterization of bronchoalveolar lavage fluid (BALF)
To collect the bronchoalveolar lavage fluid (BALF), the trachea of the mouse was exposed, and a tracheotomy was performed to introduce a 25G gauge needle. 1 mL syringe with 800μL of cold sterile PBS was used to gently wash the lungs twice through the trachea. Once collected, BALF samples (600–700μL) were centrifuged at 400xg for 10 min at 4°C, and stored for downstream analysis. BALF samples were subjected to total protein quantification via Bradford assay compared to a BSA standard curve. For BALF differential cell counts, the pellet containing all free-floating cells from the lung was resuspended in Red Blood Cell Lysis Solution (Miltenyi Biotec), to induce selective lysis of the erythrocytes, following the manufacturer’s protocol. Cell suspensions were subsequently cytocentrifuged onto Epredia Cytoslide microscope slides (Thermo Fisher Scientific) using a Cytospin at 300xg for 3 min. The slides were then air-dried and stained using the Hemacolor Stain Set (Sigma Aldrich, 65044–93) and imaged using a color camera on a Nikon Ti-2e microscope. All images were captured at 40X and analyzed using ImageJ-FIJI software, where the different cell populations present in the BALF samples (e.g., macrophages, neutrophils, lymphocytes) were identified and counted. A minimum of 5 pictures were analyzed per sample.
Enzyme-linked immunosorbent assay (ELISA)
Protein expression levels for pro-inflammatory factors including interleukin 6 (IL-6), interleukin-1 beta (IL-1β), and Tumor necrosis factor-alpha (TNF-α) were quantified via enzyme-linked immunosorbent assay (ELISA), using the commercially available kits DY406, DY401, and DY410 (R&D Systems), respectively. IL-4 and IL-10 protein content associated with the eEVs was evaluated using the commercially available ELISA kits DY404 and DY417 (R&D Systems), respectively. For these assays, protein isolation was performed as previously described. In order to detect the presence of SPA as a ligand on the surface of the eEV, a commercial mouse SPA ELISA kit (Novus Biologicals, NBP2–76693) was used following the protocol described by manufacturer using intact IL-4+SPA, IL-10+SPA, or sham eEVs.
Untargeted metabolomics analysis of BALF
Metabolites from BALF samples were extracted by adding 400 μL of ice cold Methanol:Chloroform LC/MS grade (Optima, Fisher Chemical) (9:1) to 100 mL of clarified BALF, resuspended thoroughly, and filtered using 0.2 μm nylon syringe filters (Thermo Scientific). All samples were normalized by volume to enable relative quantitative comparison between samples. The extracted metabolites were dried on a sample concentrator (Eppendorf® Vacufuge®) at 45°C for 1 hr. The dried samples were reconstituted in 50 μL of LC/MS grade water (Optima, Fisher Chemical) with 0.1% formic acid LC/MS grade (Optima, Fisher Chemical) and 5% methanol (Optima, Fisher Chemical). Samples were mixed by vortexing, sonicated for 15 min, centrifuged at 14,000 g for 1 min, and transferred to liquid chromatography low-volume vials (Waters) for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. The same extraction procedure was performed with PBS (sample blank) and a pooled QC sample was prepared by mixing 2.5 mL of each sample in a single vial. An additional vial sample was prepared as the extraction control, containing the solution used for resuspension of each sample (water with 5% methanol and 0.1% formic acid). No internal standard was used in this experiment. Differences in metabolite abundances in each experimental group were estimated based on relative comparisons between equally treated samples. At least 3 biological replicates were run for each condition.
The untargeted LC-MS/MS analysis was performed on a 6545 Q-TOF LC/MS coupled to a high-performance liquid chromatography (HPLC) system (Agilent 1290 Infinity LC). In each run, 3 mL of sample were injected into the HPLC system for separation, using a SB-C18 column (2.1×100 mm, 2.7 mm particle size, 120 Å pore size, InfinityLab Poroshell 120). The column was maintained at 40°C during the run, using a flow rate of 0.2 mL/min, water with 0.1% formic acid as solvent A and methanol with 0.1% formic acid as solvent B. The gradient used for the chromatographic run started at 2% B for 15 minutes, followed by 90% B for 1 minute, then 98% B for 1 min, and back to 2% B for 1 minute. After each run, the column was equilibrated at 2% B for 8 min. The MS system was calibrated using a standard Agilent ESI Low Concentration tune solution. System stability was checked by analyzing pooled QC regularly in between batches of the run. All samples were analyzed in positive and negative modes using data-dependent acquisition mode. MS spectra (m/z 50–1700. Scan rate 5 spectra/sec) were followed by data-dependent MS/MS spectra (m/z 50–1700. Scan 10 spectra/sec) for the 5 most intense ions per scan with a dynamic exclusion of 30 seconds. Ion selection was based on an intensity threshold of 3,000, fragmented using a collision energy ramp (CE 10–80eV). The injection of samples was randomized, and each sample was analyzed three times, including the extraction control, sample blank and pooled QC samples.
Data processing and analysis included the conversion of raw .d files to .mzXML using MSconvert (ProteoWizard).[124] Feature detection was carried out in MZmine v.2.53[125] using the settings listed in Table S4. Statistical analyses were performed using Metaboanalyst 5.0[126]. Three complementary approaches were integrated for feature annotation: 1) accurate m/z search, based on compounds from the Human Metabolome database (HMDB); 2) spectral search, matching MS/MS fragmentation patterns with mass spectral records from MassBank of North America (MoNA); and 3) de novo molecular structure identification, using SIRIUS (5.5.7) for structure prediction based on MS and MS/MS isotope and fragmentation patterns analysis. KEGG IDs of identified compounds were assigned when possible and pathway analysis was performed in Metascape.[127] The metabolomics datasets were deposited in Massive (https://massive.ucsd.edu/) with the identifiers MSV000089900 for the positive mode data files, and MSV000089807 for the negative mode data files.
Statistical analysis
All data were tested for normality using Shapiro-Wilk test, and outliers were tested using ROUT method with Q=1%. Data that followed a normal distribution were analyzed using one-way ANOVA with post hoc least significant difference test (Fisher LSD method) or two-tailed t-test as pertinent. Data that were not normally distributed were analyzed using nonparametric statistical analysis, i.e., ANOVA on ranks with post hoc Tukey test. All data were graphed as mean ± standard error of the mean (SEM). For in vivo experiments n=4 biological replicates were used and n=3–4 for in vitro experiments. Statistical significance level was defined as P< 0.05 for hypothesis testing. The analyses were run using the statistical software Sigma Plot 14 and GraphPad Prism 9.
Supplementary Material
ACKNOWLEDGEMENTS
This work was partially funded by The Ohio State University Office of Research and the NIH grants 1R01AR079485-01A1 (awarded to NHC), DP1DK126199 and DP2EB028110 (awarded to DGP). Illustrations were created using BioRender.com. The authors would like to thank the Comparative Pathology & Digital Imaging Shared Resource, and Matthew Bernier from the Mass Spec and Proteomics core (project supported by NIH Award Number Grant P30 CA016058) for his support.
REFERENCES
- 1.Moss M. & Thompson BT in Acute Respiratory Distress Syndrome, Edn. Second Edition. (ed. Choi AMK) 1–45 (Informa Healthcare USA, Inc, New York, NY; 2010). [Google Scholar]
- 2.Rubenfeld GD & Herridge MS Epidemiology and outcomes of acute lung injury. Chest 131, 554–562 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Johnson ER & Matthay MA Acute lung injury: epidemiology, pathogenesis, and treatment. J Aerosol Med Pulm Drug Deliv 23, 243–252 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maca J. et al. Past and Present ARDS Mortality Rates: A Systematic Review. Respir Care 62, 113–122 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Kaku S. et al. Acute Respiratory Distress Syndrome: Etiology, Pathogenesis, and Summary on Management. Journal of Intensive Care Medicine 35, 723–737 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Huppert LA, Matthay MA & Ware LB Pathogenesis of Acute Respiratory Distress Syndrome. Semin Respir Crit Care Med 40, 31–39 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthay MA & Zemans RL The acute respiratory distress syndrome: pathogenesis and treatment. Annu Rev Pathol 6, 147–163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Han S. & Mallampalli RK The Acute Respiratory Distress Syndrome: From Mechanism to Translation. The Journal of Immunology 194, 855–860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ragaller M. & Richter T. Acute lung injury and acute respiratory distress syndrome. J Emerg Trauma Shock 3, 43–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bhatia M. & Moochhala S. Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. The Journal of Pathology 202, 145–156 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Rajasekaran S, Pattarayan D, Rajaguru P, Sudhakar Gandhi PS & Thimmulappa RK MicroRNA Regulation of Acute Lung Injury and Acute Respiratory Distress Syndrome. Journal of Cellular Physiology 231, 2097–2106 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Huang Y, Crawford M, Higuita-Castro N, Nana-Sinkam P. & Ghadiali SN miR-146a regulates mechanotransduction and pressure-induced inflammation in small airway epithelium. Faseb j 26, 3351–3364 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthay MA, Ware LB & Zimmerman GA The acute respiratory distress syndrome. J Clin Invest 122, 2731–2740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thompson BT, Chambers RC & Liu KD Acute Respiratory Distress Syndrome. New England Journal of Medicine 377, 562–572 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Cruz FF, Weiss DJ & Rocco PR Prospects and progress in cell therapy for acute respiratory distress syndrome. Expert Opin Biol Ther 16, 1353–1360 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Xu W. et al. Risk factors analysis of COVID-19 patients with ARDS and prediction based on machine learning. Scientific Reports 11, 2933 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gibson PG, Qin L. & Puah SH COVID-19 acute respiratory distress syndrome (ARDS): clinical features and differences from typical pre-COVID-19 ARDS. Med J Aust 213, 54–56.e51 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang D, Comish P. & Kang R. The hallmarks of COVID-19 disease. PLoS Pathog 16, e1008536 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosangi B. et al. COVID-19 ARDS: a review of imaging features and overview of mechanical ventilation and its complications. Emergency Radiology 29, 23–34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J, Li Y. & Li Y. Strategies to Enhance Mesenchymal Stem Cell-Based Therapies for Acute Respiratory Distress Syndrome. Stem Cells International 2019, 5432134 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guillamat-Prats R, Camprubí-Rimblas M, Bringué J, Tantinyà N. & Artigas A. Cell therapy for the treatment of sepsis and acute respiratory distress syndrome. Ann Transl Med 5, 446 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu B. et al. Stem cell derived exosomes-based therapy for acute lung injury and acute respiratory distress syndrome: A novel therapeutic strategy. Life Sciences 254, 117766 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Maron-Gutierrez T, Laffey JG, Pelosi P. & Rocco PRM Cell-based therapies for the acute respiratory distress syndrome. Current Opinion in Critical Care 20, 122–131 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Krause M, Phan TG, Ma H, Sobey CG & Lim R. Cell-Based Therapies for Stroke: Are We There Yet? Front Neurol 10, 656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee AS, Tang C, Rao MS, Weissman IL & Wu JC Tumorigenicity as a clinical hurdle for pluripotent stem cell therapies. Nat Med 19, 998–1004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajasekaran S, Pattarayan D, Rajaguru P, Sudhakar Gandhi PS & Thimmulappa RK MicroRNA Regulation of Acute Lung Injury and Acute Respiratory Distress Syndrome. J Cell Physiol 231, 2097–2106 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Benz F, Roy S, Trautwein C, Roderburg C. & Luedde T. Circulating MicroRNAs as Biomarkers for Sepsis. International journal of molecular sciences 17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Dosari MS & Gao X. Nonviral Gene Delivery: Principle, Limitations, and Recent Progress. The AAPS Journal 11 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hisey CL et al. Investigating the consistency of extracellular vesicle production from breast cancer subtypes using CELLine adherent bioreactors. Journal of Extracellular Biology 1, e60 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamichhane TN et al. Emerging roles for extracellular vesicles in tissue engineering and regenerative medicine. Tissue Eng Part B Rev 21, 45–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stoorvogel W, Kleijmeer MJ, Geuze HJ & Raposo G. The biogenesis and functions of exosomes. Traffic 3, 321–330 (2002). [DOI] [PubMed] [Google Scholar]
- 32.Kalluri R. & LeBleu VS The biology<strong>,</strong> function<strong>,</strong> and biomedical applications of exosomes. Science 367, eaau6977 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kwok ZH, Ni K. & Jin Y. Extracellular Vesicle Associated Non-Coding RNAs in Lung Infections and Injury. Cells 10, 965 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Brien K, Breyne K, Ughetto S, Laurent LC & Breakefield XO RNA delivery by extracellular vesicles in mammalian cells and its applications. Nature Reviews Molecular Cell Biology 21, 585–606 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ha D, Yang N. & Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B 6, 287–296 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalluri R. & LeBleu VS The biology, function, and biomedical applications of exosomes. Science 367 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Khalaj K, Figueira RL, Antounians L, Lauriti G. & Zani A. Systematic review of extracellular vesicle-based treatments for lung injury: are EVs a potential therapy for COVID-19? J Extracell Vesicles 9, 1795365–1795365 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McVey MJ, Maishan M, Blokland KEC, Bartlett N. & Kuebler WM Extracellular vesicles in lung health, disease, and therapy. American Journal of Physiology-Lung Cellular and Molecular Physiology 316, L977–L989 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Worthington EN & Hagood JS Therapeutic Use of Extracellular Vesicles for Acute and Chronic Lung Disease. Int J Mol Sci 21, 2318 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buzas EI The roles of extracellular vesicles in the immune system. Nat Rev Immunol, 1–15 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gantenbein B. et al. Non-viral Gene Delivery Methods for Bone and Joints. Frontiers in Bioengineering and Biotechnology 8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Witwer KW & Wolfram J. Extracellular vesicles versus synthetic nanoparticles for drug delivery. Nature Reviews Materials 6, 103–106 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ortega-Pineda L. et al. Designer Extracellular Vesicles Modulate Pro-Neuronal Cell Responses and Improve Intracranial Retention. Adv Healthc Mater, e2100805 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duarte-Sanmiguel S. et al. In Situ Deployment of Engineered Extracellular Vesicles into the Tumor Niche via Myeloid-Derived Suppressor Cells. Adv Healthc Mater, e2101619 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duarte-Sanmiguel S, Higuita-Castro N. & Gallego-Perez D. Nanoelectroporation and Collection of Genetically Modified Exosomes in Primary Cultures of Dendritic Cells. Methods in molecular biology (Clifton, N.J.) 2050, 79–84 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Tang S. et al. Non-viral reprogramming of human nucleus pulposus cells with FOXF1 via extracellular vesicle delivery: an in vitro and in vivo study. Eur Cell Mater 41, 90–107 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lemmerman LR et al. Nanotransfection-based vasculogenic cell reprogramming drives functional recovery in a mouse model of ischemic stroke. Sci Adv 7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gallego-Perez D. et al. Topical tissue nano-transfection mediates non-viral stroma reprogramming and rescue. Nat Nanotechnol 12, 974–979 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rincon-Benavides MA et al. Engineered Vasculogenic Extracellular Vesicles Drive Nonviral Direct Conversions of Human Dermal Fibroblasts into Induced Endothelial Cells and Improve Wound Closure. Advanced Therapeutics n/a, 2200197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sarker M, Jackman D. & Booth V. Lung Surfactant Protein A (SP-A) Interactions with Model Lung Surfactant Lipids and an SP-B Fragment. Biochemistry 50, 4867–4876 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.King SD & Chen SY Recent progress on surfactant protein A: cellular function in lung and kidney disease development. Am J Physiol Cell Physiol 319, C316–c320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nathan N. et al. Surfactant protein A: A key player in lung homeostasis. The International Journal of Biochemistry & Cell Biology 81, 151–155 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Schutte RJ, Parisi-Amon A. & Reichert WM Cytokine profiling using monocytes/macrophages cultured on common biomaterials with a range of surface chemistries. J Biomed Mater Res A 88, 128–139 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Murray RZ & Stow JL Cytokine Secretion in Macrophages: SNAREs, Rabs, and Membrane Trafficking. Frontiers in Immunology 5 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang L. et al. In vitro inhibition of tumor growth by low-dose iron oxide nanoparticles activating macrophages. Journal of Biomaterials Applications 33, 935–945 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Bates SR et al. Role of P63 (CKAP4) in binding of surfactant protein-A to type II pneumocytes. Am J Physiol Lung Cell Mol Physiol 295, L658–669 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Agrawal V, Smart K, Jilling T. & Hirsch E. Surfactant Protein (SP)-A Suppresses Preterm Delivery and Inflammation via TLR2. PLOS ONE 8, e63990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lanyu Z. & Feilong H. Emerging role of extracellular vesicles in lung injury and inflammation. Biomedicine & Pharmacotherapy 113, 108748 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Mohan A, Agarwal S, Clauss M, Britt NS & Dhillon NK Extracellular vesicles: novel communicators in lung diseases. Respiratory Research 21, 175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Potter DR et al. Mesenchymal stem cell-derived extracellular vesicles attenuate pulmonary vascular permeability and lung injury induced by hemorrhagic shock and trauma. Journal of Trauma and Acute Care Surgery 84 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murphy DE et al. Extracellular vesicle-based therapeutics: natural versus engineered targeting and trafficking. Experimental & Molecular Medicine 51, 1–12 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Morimoto N. et al. An exploratory clinical study on the safety and efficacy of an autologous fibroblast-seeded artificial skin cultured with animal product-free medium in patients with diabetic foot ulcers. Int Wound J 11, 183–189 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thangapazham RL, Darling TN & Meyerle J. Alteration of skin properties with autologous dermal fibroblasts. Int J Mol Sci 15, 8407–8427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sierra-Sánchez Á, Kim KH, Blasco-Morente G. & Arias-Santiago S. Cellular human tissue-engineered skin substitutes investigated for deep and difficult to heal injuries. npj Regenerative Medicine 6, 35 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han S-K in Innovations and Advances in Wound Healing. (ed. Han S-K) 97–126 (Springer Nature Singapore, Singapore; 2023). [Google Scholar]
- 66.Lai P, Weng J, Guo L, Chen X. & Du X. Novel insights into MSC-EVs therapy for immune diseases. Biomarker Research 7, 6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hwang HS et al. Extracellular Vesicles as Potential Therapeutics for Inflammatory Diseases. Int J Mol Sci 22 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Robbins PD, Dorronsoro A. & Booker CN Regulation of chronic inflammatory and immune processes by extracellular vesicles. The Journal of Clinical Investigation 126, 1173–1180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Burgelman M, Vandendriessche C. & Vandenbroucke RE Extracellular Vesicles: A Double-Edged Sword in Sepsis. Pharmaceuticals (Basel) 14 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fitzgerald W. et al. A System of Cytokines Encapsulated in ExtraCellular Vesicles. Scientific Reports 8, 8973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sueblinvong V. & Weiss DJ Cell therapy approaches for lung diseases: current status. Current opinion in pharmacology 9, 268–273 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang P, Wu P, Siegel MI, Egan RW & Billah MM Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. Journal of Biological Chemistry 270, 9558–9563 (1995). [DOI] [PubMed] [Google Scholar]
- 73.Lo C-J, Fu M. & Cryer HG Interleukin 10 Inhibits Alveolar Macrophage Production of Inflammatory Mediators Involved in Adult Respiratory Distress Syndrome. Journal of Surgical Research 79, 179–184 (1998). [DOI] [PubMed] [Google Scholar]
- 74.Huaux F, Liu T, McGarry B, Ullenbruch M. & Phan SH Dual Roles of IL-4 in Lung Injury and Fibrosis. The Journal of Immunology 170, 2083–2092 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Levings MK & Schrader JW IL-4 Inhibits the Production of TNF-α and IL-12 by STAT6-Dependent and -Independent Mechanisms. The Journal of Immunology 162, 5224–5229 (1999). [PubMed] [Google Scholar]
- 76.Hart PH et al. Potential antiinflammatory effects of interleukin 4: suppression of human monocyte tumor necrosis factor alpha, interleukin 1, and prostaglandin E2. Proceedings of the National Academy of Sciences 86, 3803–3807 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lötvall J. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles 3, 26913 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kooijmans SAA, Schiffelers RM, Zarovni N. & Vago R. Modulation of tissue tropism and biological activity of exosomes and other extracellular vesicles: New nanotools for cancer treatment. Pharmacological Research 111, 487–500 (2016). [DOI] [PubMed] [Google Scholar]
- 79.Lara P, Chan AB, Cruz LJ, Quest AFG & Kogan MJ Exploiting the Natural Properties of Extracellular Vesicles in Targeted Delivery towards Specific Cells and Tissues. Pharmaceutics 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim H. et al. Engineered extracellular vesicles and their mimetics for clinical translation. Methods 177, 80–94 (2020). [DOI] [PubMed] [Google Scholar]
- 81.Liu C. & Su C. Design strategies and application progress of therapeutic exosomes. Theranostics 9, 1015–1028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Jong B, Barros ER, Hoenderop JGJ & Rigalli JP Recent Advances in Extracellular Vesicles as Drug Delivery Systems and Their Potential in Precision Medicine. Pharmaceutics 12, 1006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Castranova V, Rabovsky J, Tucker JH & Miles PR The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicol Appl Pharmacol 93, 472–483 (1988). [DOI] [PubMed] [Google Scholar]
- 84.Stanley AC & Lacy P. Pathways for Cytokine Secretion. Physiology 25, 218–229 (2010). [DOI] [PubMed] [Google Scholar]
- 85.Farivar AS, Woolley SM, Fraga CH, Byrne K. & Mulligan MS Proinflammatory Response of Alveolar Type II Pneumocytes to in vitro Hypoxia and Reoxygenation. American Journal of Transplantation 4, 346–351 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Bates SR P63 (CKAP4) as an SP-A Receptor: Implications for Surfactant Turnover. Cellular Physiology and Biochemistry 25, 41–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Domscheit H, Hegeman MA, Carvalho N. & Spieth PM Molecular Dynamics of Lipopolysaccharide-Induced Lung Injury in Rodents. Front Physiol 11, 36–36 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martin I, Cabán-Hernández K, Figueroa-Santiago O. & Espino AM Fasciola hepatica fatty acid binding protein inhibits TLR4 activation and suppresses the inflammatory cytokines induced by lipopolysaccharide in vitro and in vivo. J Immunol 194, 3924–3936 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fukunaga K, Kohli P, Bonnans C, Fredenburgh LE & Levy BD Cyclooxygenase 2 plays a pivotal role in the resolution of acute lung injury. J Immunol 174, 5033–5039 (2005). [DOI] [PubMed] [Google Scholar]
- 90.MODELSKA K, PITTET J-F, FOLKESSON HG, BROADDUS VC & MATTHAY MA Acid-induced Lung Injury. American Journal of Respiratory and Critical Care Medicine 160, 1450–1456 (1999). [DOI] [PubMed] [Google Scholar]
- 91.Frank L, Bucher JR & Roberts RJ Oxygen toxicity in neonatal and adult animals of various species. J Appl Physiol Respir Environ Exerc Physiol 45, 699–704 (1978). [DOI] [PubMed] [Google Scholar]
- 92.Parsons PE et al. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med 33, 1–6; discussion 230–232 (2005). [DOI] [PubMed] [Google Scholar]
- 93.Matthay MA et al. Acute respiratory distress syndrome. Nature Reviews Disease Primers 5, 18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakuma T. et al. Ischemia-reperfusion lung injury in rabbits: mechanisms of injury and protection. Am J Physiol 276, L137–145 (1999). [DOI] [PubMed] [Google Scholar]
- 95.Fox-Dewhurst R. et al. Pulmonary and systemic inflammatory responses in rabbits with gram-negative pneumonia. Am J Respir Crit Care Med 155, 2030–2040 (1997). [DOI] [PubMed] [Google Scholar]
- 96.Vernooy JH, Dentener MA, van Suylen RJ, Buurman WA & Wouters EF Intratracheal instillation of lipopolysaccharide in mice induces apoptosis in bronchial epithelial cells: no role for tumor necrosis factor-alpha and infiltrating neutrophils. Am J Respir Cell Mol Biol 24, 569–576 (2001). [DOI] [PubMed] [Google Scholar]
- 97.Kulkarni HS et al. Update on the Features and Measurements of Experimental Acute Lung Injury in Animals: An Official American Thoracic Society Workshop Report. Am J Respir Cell Mol Biol 66, e1–e14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Aeffner F, Bolon B. & Davis IC Mouse Models of Acute Respiratory Distress Syndrome: A Review of Analytical Approaches, Pathologic Features, and Common Measurements. Toxicol Pathol 43, 1074–1092 (2015). [DOI] [PubMed] [Google Scholar]
- 99.Stukas S. et al. The Association of Inflammatory Cytokines in the Pulmonary Pathophysiology of Respiratory Failure in Critically Ill Patients With Coronavirus Disease 2019. Crit Care Explor 2, e0203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Evans CR et al. Untargeted LC–MS Metabolomics of Bronchoalveolar Lavage Fluid Differentiates Acute Respiratory Distress Syndrome from Health. Journal of Proteome Research 13, 640–649 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hidalgo MA, Carretta MD & Burgos RA Long Chain Fatty Acids as Modulators of Immune Cells Function: Contribution of FFA1 and FFA4 Receptors. Front Physiol 12, 668330 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kang S-M et al. 1H nuclear magnetic resonance based metabolic urinary profiling of patients with ischemic heart failure. Clinical Biochemistry 44, 293–299 (2011). [DOI] [PubMed] [Google Scholar]
- 103.Cruickshank-Quinn CI et al. Transient and Persistent Metabolomic Changes in Plasma following Chronic Cigarette Smoke Exposure in a Mouse Model. PLOS ONE 9, e101855 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lauer A. et al. Ex Vivo Evaluation of the Sepsis Triple Therapy High-Dose Vitamin C in Combination with Vitamin B1 and Hydrocortisone in a Human Peripheral Blood Mononuclear Cells (PBMCs) Model. Nutrients 13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Carr AC & Maggini S. Vitamin C and Immune Function. Nutrients 9, 1211 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fisher BJ et al. Mechanisms of attenuation of abdominal sepsis induced acute lung injury by ascorbic acid. American Journal of Physiology-Lung Cellular and Molecular Physiology 303, L20–L32 (2012). [DOI] [PubMed] [Google Scholar]
- 107.Faulkner A. et al. Dimethyl-2-oxoglutarate improves redox balance and mitochondrial function in muscle pericytes of individuals with diabetes mellitus. Diabetologia 63, 2205–2217 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu S, Yang J. & Wu Z. The Regulatory Role of α-Ketoglutarate Metabolism in Macrophages. Mediators of Inflammation 2021, 5577577 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou Y, Schneider DJ & Blackburn MR Adenosine signaling and the regulation of chronic lung disease. Pharmacol Ther 123, 105–116 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liaudet L. et al. Inosine exerts a broad range of antiinflammatory effects in a murine model of acute lung injury. Ann Surg 235, 568–578 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang N. et al. Inosine: a broad-spectrum anti-inflammatory against SARS-CoV-2 infection-induced acute lung injury via suppressing TBK1 phosphorylation. Journal of Pharmaceutical Analysis (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mao B. et al. Inosine Pretreatment Attenuates LPS-Induced Lung Injury through Regulating the TLR4/MyD88/NF-κB Signaling Pathway In Vivo. Nutrients 14, 2830 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu J. et al. Increased mortality of acute respiratory distress syndrome was associated with high levels of plasma phenylalanine. Respiratory Research 21, 99 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Abdel-Salam OM et al. Citric acid effects on brain and liver oxidative stress in lipopolysaccharide-treated mice. J Med Food 17, 588–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Landolf KM et al. Corticosteroid use in ARDS and its application to evolving therapeutics for coronavirus disease 2019 (COVID-19): A systematic review. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 42, 71–90 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abiri B. & Vafa M. Impact of coenzyme Q10 on inflammatory biomarkers and its role in future therapeutic strategies. Clinical Nutrition ESPEN 43, 25–30 (2021). [DOI] [PubMed] [Google Scholar]
- 117.Zhang L. et al. Uridine alleviates LPS-induced ARDS and improves insulin sensitivity by decreasing oxidative stress and inflammatory processes. Physiol Int 109, 215–229 (2022). [DOI] [PubMed] [Google Scholar]
- 118.Kaveh M, Eftekhar N. & Boskabady MH The effect of alpha linolenic acid on tracheal responsiveness, lung inflammation, and immune markers in sensitized rats. Iran J Basic Med Sci 22, 255–261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sharifi-Rad M. et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front Physiol 11 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pålsson-McDermott EM & O’Neill LAJ Targeting immunometabolism as an anti-inflammatory strategy. Cell Research 30, 300–314 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Crowe AR & Yue W. Semi-quantitative Determination of Protein Expression Using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio-protocol 9, e3465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pua ZJ et al. Histochemical analyses of altered fetal lung development following single vs multiple courses of antenatal steroids. J Histochem Cytochem 53, 1469–1479 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tsikis ST et al. Lipopolysaccharide-induced murine lung injury results in long-term pulmonary changes and downregulation of angiogenic pathways. Scientific Reports 12, 10245 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chambers MC et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology 30, 918–920 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pluskal T, Castillo S, Villar-Briones A. & Orešič M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pang Z. et al. Using MetaboAnalyst 5.0 for LC–HRMS spectra processing, multi-omics integration and covariate adjustment of global metabolomics data. Nature Protocols (2022). [DOI] [PubMed] [Google Scholar]
- 127.Karnovsky A. et al. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics 28, 373–380 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.