

Abstract
A significant proportion of temporal lobe epilepsy (TLE) patients experience drug-resistant seizures associated with mesial temporal sclerosis, in which there is extensive cell loss in the hippocampal CA1 and CA3 subfields, with a relative sparing of dentate gyrus granule cells and CA2 pyramidal neurons (PNs). A role for CA2 in seizure generation was suggested based on findings of a reduction in CA2 synaptic inhibition (Williamson and Spencer, 1994) and the presence of interictal-like spike activity in CA2 in resected hippocampal tissue from TLE patients (Wittner et al., 2009). We recently found that in the pilocarpine-induced status epilepticus (PILO-SE) mouse model of TLE there was an increase in CA2 intrinsic excitability associated with a loss of CA2 synaptic inhibition. Furthermore, chemogenetic silencing of CA2 significantly reduced seizure frequency, consistent with a role of CA2 in promoting seizure generation and/or propagation (Whitebirch et al., 2022). In the present study, we explored the cellular basis of this inhibitory deficit using immunohistochemical and electrophysiological approaches in PILO-SE male and female mice. We report a widespread decrease in the density of pro-cholecystokinin-immunopositive (CCK+) interneurons and a functional impairment of CCK+ interneuron-mediated inhibition of CA2 PNs. We also found a disruption in the perisomatic perineuronal net in the CA2 stratum pyramidale. Such pathologic alterations may contribute to an enhanced excitation of CA2 PNs and CA2-dependent seizure activity in the PILO-SE mouse model.
SIGNIFICANCE STATEMENT Impaired synaptic inhibition in hippocampal circuits has been identified as a key feature that contributes to the emergence and propagation of seizure activity in human patients and animal models of temporal lobe epilepsy (TLE). Among the hippocampal subfields, the CA2 region is particularly resilient to seizure-associated neurodegeneration and has been suggested to play a key role in seizure activity in TLE. Here we report that perisomatic inhibition of CA2 pyramidal neurons mediated by cholecystokinin-expressing interneurons is selectively reduced in acute hippocampal slices from epileptic mice. Parvalbumin-expressing interneurons, in contrast, appear relatively conserved in epileptic mice. These findings advance our understanding of the cellular mechanisms underlying inhibitory disruption in hippocampal circuits in a mouse model of spontaneous recurring seizures.
Keywords: CA2, cholecystokinin, epilepsy, hippocampus, parvalbumin, pilocarpine
Introduction
Temporal lobe epilepsy (TLE) is among the most prevalent neurologic disorders, with a substantial proportion of people living with TLE experiencing refractory seizures that are not effectively controlled by antiseizure medications (Kwan and Brodie, 2000; Asadi-Pooya et al., 2017), deep brain stimulation (Yang et al., 2022), or surgical resection of seizure-generating tissue (Téllez-Zenteno and Hernández-Ronquillo, 2012; Wu et al., 2019). Recurring temporal lobe seizures are often associated with a characteristic pattern of hippocampal neurodegeneration and synaptic reorganization, termed mesial temporal sclerosis, involving a loss of CA1 and CA3 pyramidal neurons (PNs) both in humans and numerous animal models of TLE (Thom et al., 2010; Blümcke et al., 2013; Savitr Sastri et al., 2014; Steve et al., 2014). Particularly notable is the relative resilience of the understudied CA2 region, a narrow area interposed between the larger CA3 and CA1 regions, initially described by Lorente De Nó (1934). A partial loss of CA2 neurons is typically only observed in cases of especially severe hippocampal sclerosis (Wyler et al., 1992). Compared with CA1 and CA3, neurons of the CA2 subfield exhibit distinct gene expression patterns, synaptic connectivity, and electrophysiological properties (Dudek et al., 2016; Lehr et al., 2021). Consistent with a specialized role within the hippocampal circuitry, recent work has demonstrated that CA2 PNs critically regulate social recognition memory and social aggression (Hitti and Siegelbaum, 2014; Leroy et al., 2017, 2018; Meira et al., 2018; Lopez-Rojas et al., 2022). Prior investigations in human TLE tissue have revealed numerous alterations in the CA2 region, including the sprouting of dentate gyrus (DG) mossy fiber axons (Wittner et al., 2009; Freiman et al., 2021), decreases in parvalbumin (PV) expression (Andrioli et al., 2007; Wittner et al., 2009), diminished synaptic inhibition (Williamson and Spencer, 1994), and the appearance of spontaneous interictal-like epileptiform discharges (Wittner et al., 2009).
CA2 PNs are centrally located within the hippocampal network and receive exceptionally strong entorhinal cortex (EC) input, thus acting as the central node in a powerful disynaptic circuit linking the entorhinal cortex directly to CA1 (Chevaleyre and Siegelbaum, 2010; Cui et al., 2013; Hitti and Siegelbaum, 2014; Kohara et al., 2014; Leroy et al., 2017; Sun et al., 2017). CA2 PNs also receive direct input from DG granule cell mossy fibers (Kohara et al., 2014; Whitebirch et al., 2022), and CA2 PN local axonal collaterals contribute to an excitatory recurrent network throughout the CA2 and CA3 subfields (Tamamaki et al., 1988; Ishizuka et al., 1990; Li et al., 1994; Cui et al., 2013; Hitti and Siegelbaum, 2014; Kohara et al., 2014; Okamoto and Ikegaya, 2019; Whitebirch et al., 2022). Among the hippocampal subfields, CA2 contains the greatest density of inhibitory interneurons (INs), including a population of PV+ interneurons with distinct anatomic and physiological properties (Mercer et al., 2007, 2012a; Botcher et al., 2014). CA2 PNs also receive strong inhibitory input from a population of cholecystokinin-expressing (CCK+) interneurons (Botcher et al., 2014; Modi et al., 2019; Loisy et al., 2022).
Accumulating evidence suggests that CA2 functions as a key circuit node regulating both normal and abnormal hippocampal network activity and excitability (Oliva et al., 2023). Sharp-wave ripple (SWR) events, brief bursts of hippocampal population activity, are preceded by the selective recruitment of a subset of CA2 PNs, and optogenetic activation of CA2 neurons triggers SWR-associated population reactivation (Oliva et al., 2016, 2020). Modulation of CA2 PNs influences both the frequency of spontaneous SWRs and the power and coherence of gamma oscillations between the hippocampus and the prefrontal cortex (Boehringer et al., 2017; Alexander et al., 2018). Optogenetic inactivation of CA2 diminishes hippocampal network synchrony and degrades the firing precision of CA1 neurons during both SWRs and behavioral states dominated by theta rhythm (He et al., 2021; MacDonald and Tonegawa, 2021). Most recently, calcium imaging in mice revealed selective recruitment of lateral EC afferents, CA2 PN axons, and CCK+ interneurons in the CA1 subfield during desynchronized network states (Dudok et al., 2021). Together, the unique synaptic connectivity and functional properties of CA2, the resilience of the CA2 subfield to mesial temporal sclerosis, and alterations in CA2 in human TLE tissue suggest that CA2 may have an important role in the epileptic hippocampus.
Indeed, in the pilocarpine-induced status epilepticus (PILO-SE) model of TLE in mice, we recently found that chemogenetic inhibition of CA2 PNs in vivo significantly reduced the frequency of spontaneous recurring convulsive seizures (Whitebirch et al., 2022). Acute hippocampal slice electrophysiology revealed multiple alterations to the cells and circuitry of the CA2 subfield, including a significant impairment to feedforward inhibition that may facilitate the emergence and propagation of epileptiform activity (Whitebirch et al., 2022). Here, we used electrophysiological and immunohistochemical approaches to explore the mechanisms underlying this inhibitory impairment. We found that PILO-SE was associated with alterations to specific interneuronal populations that may compromise inhibitory control of CA2 excitability in epileptic mice.
Materials and Methods
Animals
All procedures were performed in accordance with the Columbia University Institutional Animal Care and Use Committees (IACUC). Adult male and female mice (age range, 8–12 weeks) were housed in a temperature- and humidity-controlled environment with a 12 h light/dark cycle with food and water provided ad libitum. All mice were F1 generation hybrids resulting from a cross between Amigo2-Cre+/− mice (maintained in a C57BL/6J background; The Jackson Laboratory; RRID:IMSR_JAX:030215) and 129S1/SvlmJ mice (stock #002448, The Jackson Laboratory; RRID:IMSR_JAX:002448). A subset of the mice used for experiments (see Fig. 10) were Amigo2-Cre+/− mice maintained on a C57BL/6N background (Whitebirch et al., 2022). As no differences were noted between strains, results were pooled. Genotyping was performed using tail tip samples sent to GeneTyper (Columbia University), and both Cre+/− and Cre−/− animals were used in this study.
Figure 10.
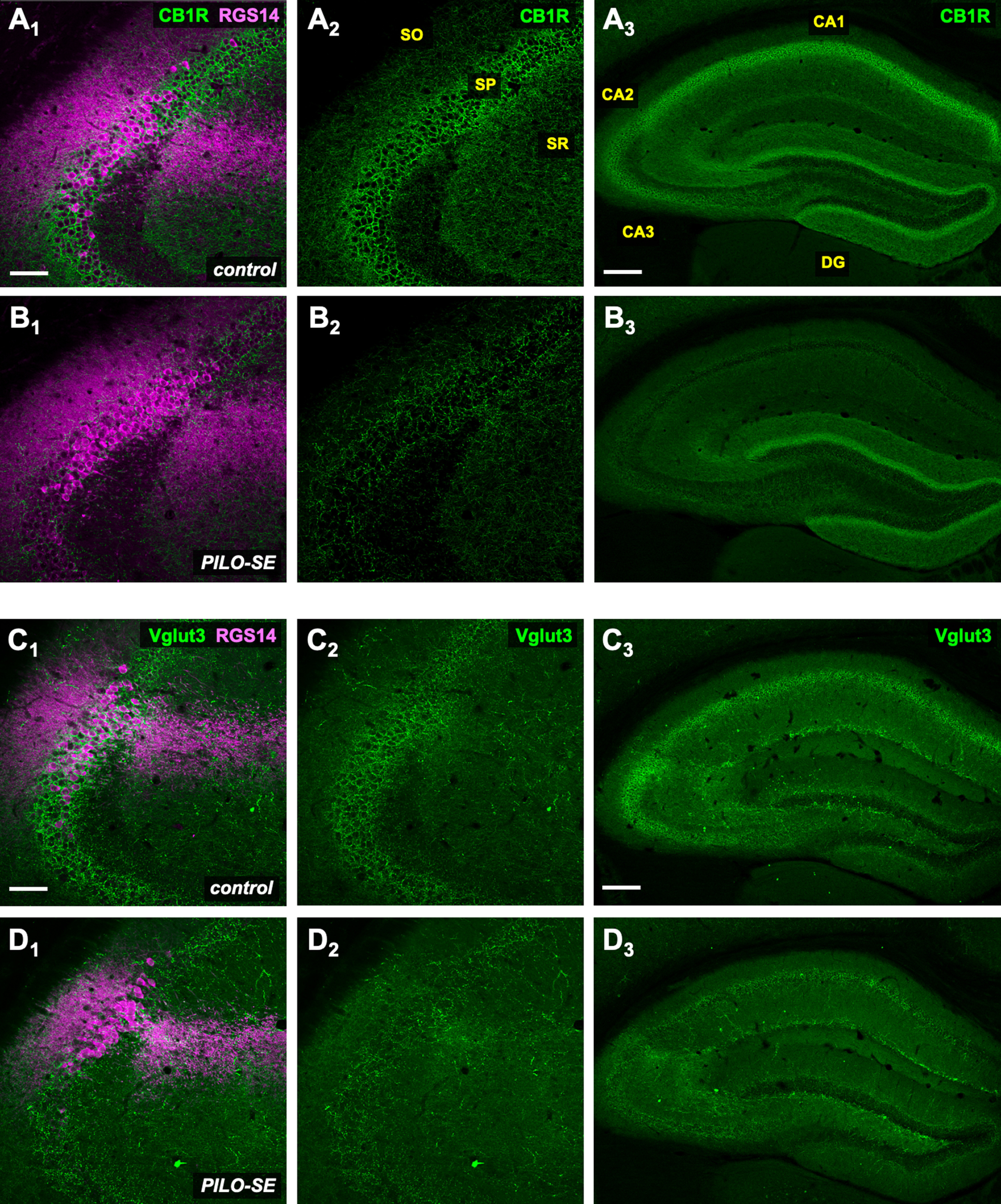
Reduced labeling for molecular markers of CCK+ interneuron axon terminals in PILO-SE mice. A1–D3, Representative hippocampal sections from control (above) and PILO-SE (below) mice, stained for CB1R or VGluT3 (green) and RGS14 (magenta) to delineate the CA2 subfield, as indicated. Scale bars: left and middle panels, 100 µm; right panels, 400 µm.
Pilocarpine-induced status epilepticus
All drugs were administered intraperitoneally. Mice were first administered methylatropine bromide (5 mg/kg, i.p.) to suppress peripheral cholinergic activation from pilocarpine hydrochloride. Pilocarpine was administered 30 min later (350 mg/kg, i.p.), and mice were closely and continually monitored for behavioral indicators of seizures. The onset of SE typically occurred between 30 and 60 min following pilocarpine treatment and was defined as a convulsive seizure (stage 3, 4, or 5 on the Racine Seizure Scale; Racine, 1972) that lasted continually for at least 5 min without resuming normal behavior (e.g., grooming) for several hours. Diazepam (5 mg/kg, i.p.) was administered 1 h after SE onset to curtail seizures. In all cases, diazepam was followed 20 min later by levetiracetam (100 mg/kg, i.p.). Control mice were given an identical time course of drug treatment, except they were not administered pilocarpine. Thus, control animals received methylatropine bromide, then after 150 min were administered diazepam and at 170 min were administered levetiracetam. Immediately after levetiracetam, mice were transferred to a heated and humidified ThermoCare veterinary intensive care unit, where they were closely monitored, provided with dietary supplements in the form of DietGel Recovery (catalog #72-06-5022; Clear H2O), and given subcutaneous hydration twice daily until they showed normal locomotion and feeding (typically within 1–3 d). Control mice were returned to standard housing cages immediately following the drug administration protocol. DietGel was provided, but it should be noted that it was not consumed (H. E. Scharfman, unpublished observations) so that controlling for possible effects of dietary supplementation with DietGel was not possible. After recovery, all mice were kept in standard group housing, except where aggression between cage-mates was observed in which cases aggressors were removed. Taking into account all mice used in this study and a previous series of experiments (Whitebirch et al., 2022), pilocarpine treatment resulted in the following three outcomes: acute mortality during SE because of a generalized tonic-clonic seizure leading to tonic hindlimb extension and death (114 of 411 mice, 27.7%); minor convulsive seizure activity (stage 3 seizures or below the Racine Seizure Scale; Racine, 1972) without convulsive SE (133 of 411 mice, 32.4%); or convulsive SE (164 of 411 mice, 39.9%). Video-electroencephalogram (EEG) recordings were not performed during induction of PILO-SE, and determination of convulsive SE was performed using established behavioral indicators including head nodding, facial automatisms and gnawing, continual bodily tremor, forelimb clonus, and periodic convulsive seizures accompanied by rearing and falling (Turski et al., 1984; Cavalheiro et al., 1996; Lévesque et al., 2016). Pilocarpine treatment for the subset of mice maintained on a C57BL/6N background, used for experiments (see Fig. 10), was performed according to a slightly modified protocol as described (Whitebirch et al., 2022; Lisgaras et al., 2023; Lisgaras and Scharfman, 2023) using a pilocarpine dose of 240 mg/kg, which is sufficient to induce SE in this strain.
Video-electroencephalogram recordings
Video-EEG recordings were performed in a subset of mice to capture spontaneous recurring seizures, the defining feature of experimentally induced epilepsy. Surgeries to implant subdural screw electrodes (stainless steel screws; length, 0.10 inch; catalog #8209, Pinnacle Technology) were performed at least 4 weeks after pilocarpine treatment. For surgery, carprofen was given as an analgesic agent (5 mg/kg, s.c.), and postoperative analgesia was carprofen-supplemented food [either 60 ml of food gel (catalog #CPF-74-05-5022, MediGel) or 2 mg Rodent MD’s carprofen tablets (catalog #MD150-2, Bio-Serv), each estimated to be a dose of ∼5 mg/kg over 24 h of consumption]. Before the commencement of surgery, animals were weighed to record baseline body weight and to calculate carprofen dose. Anesthesia was achieved with isoflurane (NDC 11695-6777-2, Covetrus), with an induction concentration of 4% and a maintenance concentration of ∼2%. Surgical procedures were performed under sterile conditions as approved by the Columbia University IACUC. The stereotactic coordinates used were measured in millimeters relative to bregma [for anterior–posterior (A–P) and medial–lateral (M–L) coordinates]. Subdural screw electrodes were implanted into burr holes located over the left and right dorsal hippocampi (A–P, −1.80; M–L, ±1.30), the left frontal cortex (A–P, −0.30; M–L, −1.50), and the right occipital cortex (A–P, −3.50; M–L, +2.00). An additional two screws were placed over the right olfactory bulb (A–P, +2.30; M–L, +1.80) and the left cerebellum (A–P, −1.5 from λ; M–L, −0.5 from bregma) for ground and reference electrodes, respectively. All screws were wired to an eight-pin connector (prepared from a larger block; catalog #ED90266-ND, DigiKey) and secured using dental cement (C&B Metabond, catalog #S380, Parkell). Following surgery, mice were individually housed and allowed to recover for a minimum of 1 week before video-EEG recordings were initiated. Video-EEG recording was performed with equipment from Pinnacle Technology (four-channel tethered EEG system; catalog #8400-K1). A preamplifier (catalog #8400-SE4, Pinnacle Technology) was connected to the eight-pin connector and wired to the data acquisition system (catalog #8401-HS, Pinnacle Technology) via a multichannel commutator (catalog #8408, Pinnacle Technology) that allowed for uninhibited movement. During long-term continuous recording sessions, mice were housed in circular cages with ad libitum food and water, and synchronous video was recorded using a box camera and an infrared light source (catalog #9000-K10, Pinnacle Technology). Video-EEG recordings were acquired and reviewed using Pinnacle Technology Sirenia software (version 2.2.2). The time–frequency representation of seizure power (Fig. 1F) was generated in MATLAB using the cwt command, which performs a continuous 1-D wavelet transform and outputs a magnitude color scale that represents the L1 normalized power of the frequency components of the input signal.
Figure 1.
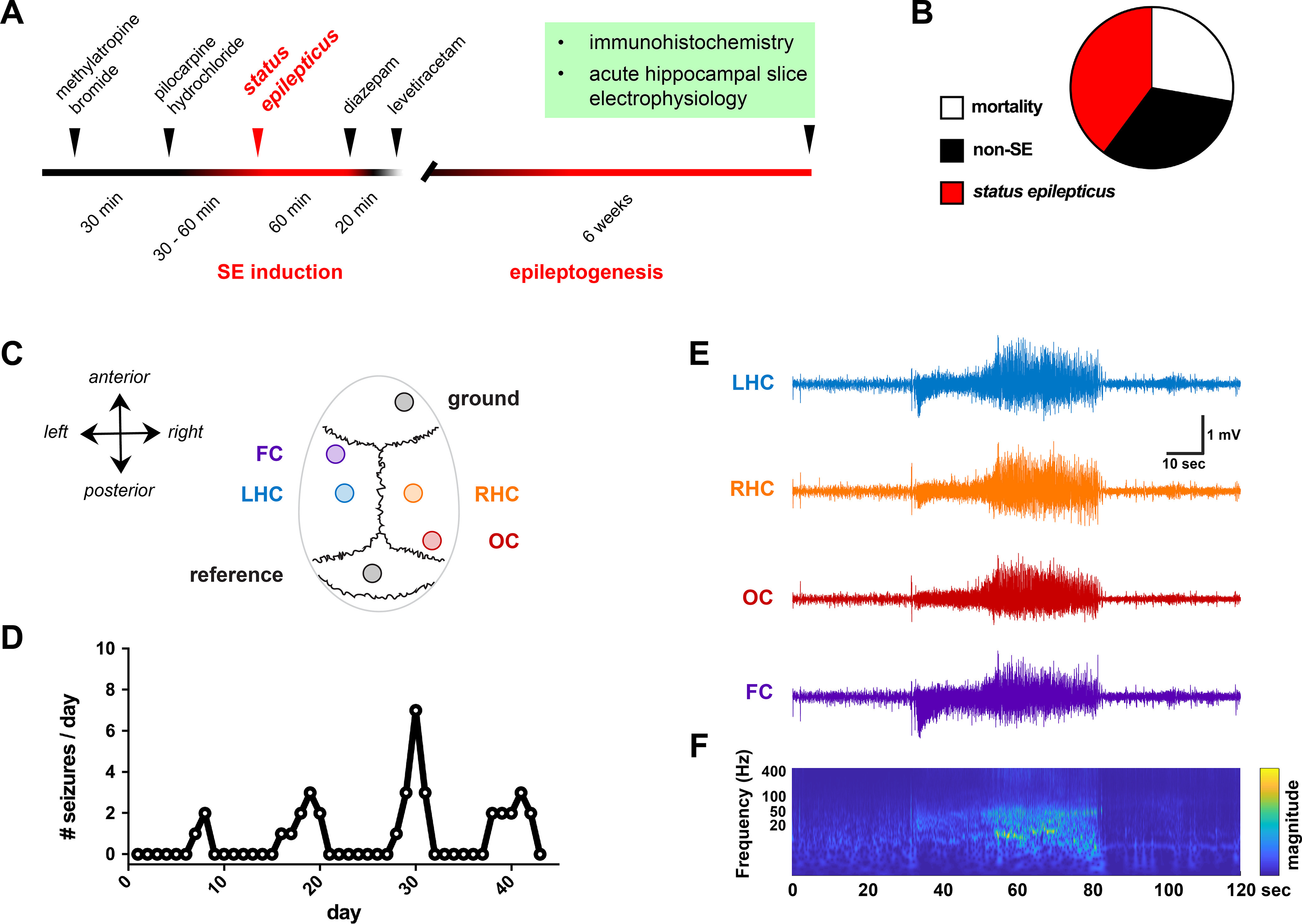
PILO-SE mice experience recurring spontaneous seizures. A, Experimental timeline, in which adult mice are administered PILO to induce acute status epilepticus. Immunohistochemistry and in vitro electrophysiology are performed ∼6 weeks following PILO-SE, when mice exhibit spontaneous, recurring seizures. B, PILO treatment results in acute mortality (114 of 411 mice, 27.7%), minor seizure activity without status epilepticus (133 of 411 mice, 32.4%), or status epilepticus (164 of 411 mice, 39.9%). C, Seizures were captured through continuous video-EEG recordings, with four screw electrodes positioned over the frontal cortex (FC), left hippocampus (LHC), right hippocampus (RHC), and occipital cortex (OC). D, Seizure occurrence frequency for one mouse, illustrating the typical clustered pattern of spontaneous recurring seizures. E, A representative seizure, with highly synchronized and large-amplitude activity visible across all four electrodes. F, A time–frequency representation of the seizure in E, illustrating the increase in power across ∼1–50 Hz.
Slice electrophysiology
Hippocampal slice preparation.
Ex vivo electrophysiology was performed in the chronic phase of PILO-induced epilepsy, ∼6 weeks after status epilepticus, when mice were 14–18 weeks old. Acute hippocampal slices were prepared using artificial CSF (ACSF) and sucrose-substituted ACSF (referred to as sucrose solution). ACSF and sucrose solutions were made with purified water that had been filtered through a 0.22 µm filter. ACSF contained the following (in mM): 22.5 glucose, 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 3 sodium pyruvate, 1 ascorbic acid, 2 CaCl2, and 1 MgCl2. Sucrose solution used for slice preparation contained the following (mM): 195 sucrose, 10 glucose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 sodium pyruvate, 0.5 CaCl2, and 7 MgCl2. ACSF and sucrose cutting solution were prepared fresh before each experiment, and the osmolarity was consistently measured to be between 315 and 325 mOsm. Sucrose solution was chilled on ice and bubbled with carbogen gas (95% O2/5% CO2) for at least 30 min before slice preparation. A recovery beaker was prepared with a 50:50 mixture of ACSF and sucrose solution and was warmed to ∼32°C. Mice were deeply anesthetized by isoflurane inhalation, and immediate incisions were made to sever the diaphragm and access the heart. Transcardial perfusion was performed with ice-cold carbogenated sucrose cutting solution for ∼30–45 s. The mouse was decapitated, and the brain was quickly removed, at which point the hippocampi were dissected free, placed into an agar block, and secured to a vibratome slicing platform with cyanoacrylate adhesive. Hippocampal slices were cut at a thickness of 400 µm, parallel to the transverse plane. Slices were collected from the dorsal and intermediate hippocampus, as CA2 circuits have primarily been characterized within the dorsal hippocampus (Chevaleyre and Siegelbaum, 2010; Hitti and Siegelbaum, 2014; Kohara et al., 2014). Slices were transferred to the warmed recovery beaker and allowed to recover for 30 min, after which the beaker was allowed to come to room temperature and left for an additional 90 min.
Whole-cell recordings.
Recording and stimulation pipettes were prepared from borosilicate glass capillaries using a heated-filament puller programmed to produce pipettes with a resistance of ∼3–5 MΩ. Stimulation pipettes were filled with 1 m NaCl. Recording pipettes were filled with intracellular solution composed of the following (in mm): 135 potassium gluconate (C6H11KO7), 5 KCl, 0.2 EGTA (C14H24N2O10), 10 HEPES (C8H18N2O4S), 2 NaCl, 5 MgATP (C10H16N5O13P3 · Mg2+), 0.4 Na2GTP (C10H16N5O14P3 · Na+), 10 Na2 phosphocreatine (C4H8N3O5PNa2 · H2O), and biocytin (0.2% by weight). Intracellular solution was prepared on ice, with the osmolarity adjusted to ∼295 mOsm and the pH titrated to ∼7.2. Hippocampal slices were individually transferred to the recording chamber of an electrophysiology station where ACSF, warmed to 32°C and continually bubbled with carbogen, was perfused through the chamber at a rate of ∼1–3 ml/min. Whole-cell recordings from CA2 PNs was accomplished primarily through a “blind patching” approach, using the end of the stratum lucidum (SL) as an anatomic landmark. Cells were left unperturbed for several minutes before any recordings to ensure the membrane potential and seal were stable, and cells with a resting membrane potential more depolarized than −50 mV or a series resistance >25 MΩ were discarded. The resting membrane potential was measured by recording for at least 1 min in the current-clamp configuration, without any injected current, and averaging the membrane potential. Current was then applied as needed to hold cells at −70 mV. As previously reported (Chevaleyre and Siegelbaum, 2010; Hitti and Siegelbaum, 2014; Kohara et al., 2014; Whitebirch et al., 2022), CA2 PNs were identifiable based on key intrinsic physiological properties including a relatively small input resistance, delayed action potential firing preceded by a gradual slow depolarizing ramp on depolarizing current injection, a relatively small-amplitude voltage sag on hyperpolarization, a relatively high rheobase current, and a large membrane capacitance. Analysis of intrinsic properties was performed in MATLAB and AxoGraph. Cell identity was confirmed post hoc based on cellular morphology as revealed by biocytin and colocalization with known CA2 markers Purkinje cell protein 4 (PCP4), striatal-enriched protein tyrosine phosphatase (STEP), or regulator of G-protein signaling 14 (RGS14; Hitti and Siegelbaum, 2014).
Synaptic responses and pharmacology.
Electrical stimulation was delivered as a single 0.2 ms pulse (Constant Voltage Isolated Stimulator; model DS2A-Mk.II, Digitimer), ranging in intensity from 8 to 80 V (in 8 V increments). For Figure 2, paired-pulse stimulation consisted of two electrical stimuli separated by 50 ms. Postsynaptic potentials were measured from a holding potential of −70 mV. Fast and slow hyperpolarizing phases in Figure 3 were assessed visually for each cell: the inflection point between the fast and the slow phases of hyperpolarization typically occurred at ∼70 ms after stimulation, and the negative peak was measured both before and after the inflection point. In experiments where inhibitory postsynaptic potentials were pharmacologically isolated, excitatory transmission was blocked with 50 μm D-AP5 (catalog #14539, Cayman Chemical) and 25 μm CNQX (catalog #14618, Cayman Chemical). The CB1 receptor agonist WIN55,212-2 (WIN; catalog #1038, Tocris Bioscience) was applied at a concentration of 1 μm, as indicated. Throughout the course of the experiment, stimulation was applied once every 15 s, and every four evoked IPSPs were averaged to produce a measurement of the IPSP at every minute. In WIN experiments, the baseline IPSP was measured as the mean amplitude from at least 6 min of stable responses. After this baseline, WIN was added to the bath for 15 min and then washed out for an additional 15 min. The post-WIN IPSP amplitudes were measured as the mean amplitude of the IPSPs from minute 11 to 25 (with the application of WIN designated as minute 0). CNQX and WIN55,212-2 were prepared as stock solutions in DMSO, and d-AP5 was prepared as a stock solution in water. Stock solutions were stored at −2°C, and aliquots were thawed and kept on ice on the day of the experiment.
Figure 2.
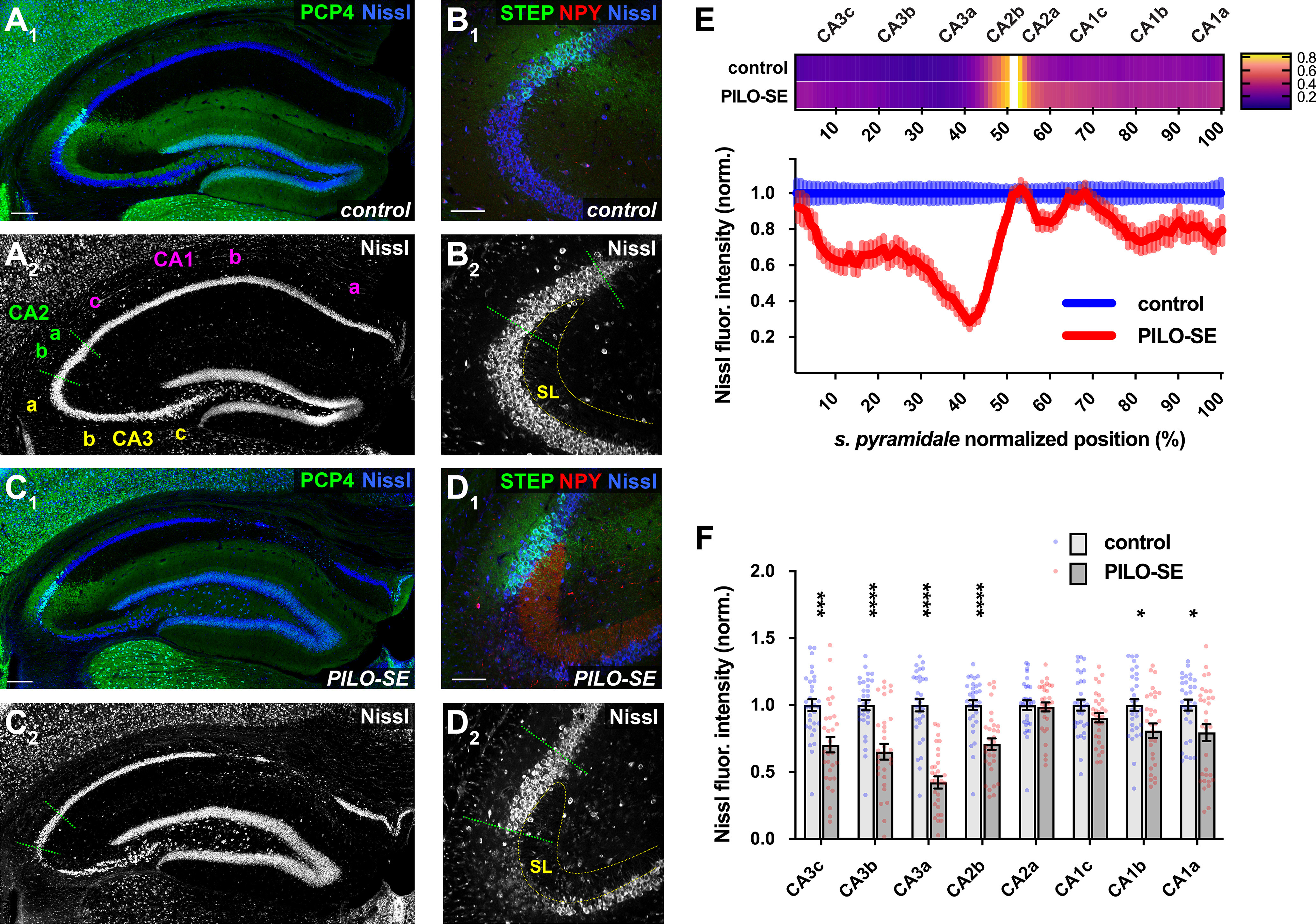
The CA2 subfield was resistant to mesial temporal sclerosis-like neurodegeneration. A1, A2, A representative section from a control mouse stained for PCP4 (green) to label CA2 PNs and for Nissl (blue and white). Scale bar, 200 µm. B1, B2, CA2 PNs (STEP, green) are located adjacent to CA3a at the distal end of the mossy fiber projection in SL. Neuronal somata are visualized with a stain for Nissl (blue and white); labeling for NPY is shown in red. Scale bar, 100 µm. C1, C2, Representative mesial temporal sclerosis-like damage in a section from a PILO-SE mouse stained for PCP4 (green) and Nissl (blue and white). Scale bar, 200 µm. D1, D2, NPY expression in the mossy fibers was visible in all sections from PILO-SE mice. Neuronal somata (Nissl, blue and white) are absent from CA3a and are largely preserved in CA2 (STEP, green). Scale bar, 100 µm. E, Above, a heatmap showing the normalized fluorescence intensity of CA2-specific markers (see Materials and Methods) along the proximal–distal axis of stratum pyramidale in sections from control and PILO-SE mice. Below, normalized Nissl fluorescence intensity across CA3, CA2, and CA1 was decreased significantly in CA3, along with the proximal region of CA2 (CA2b) and the distal portions of CA1 (CA1b, CA1a; n = 45 sections from 31 control mice, n = 56 sections from 31 PILO-SE mice). F, Measurement of the mean normalized Nissl fluorescence in each subregion confirms a characteristic pattern of neurodegeneration in PILO-SE mice with mesial temporal sclerosis-like damage, in which CA2a and CA1c are relatively resilient to neurodegeneration.
Figure 3.
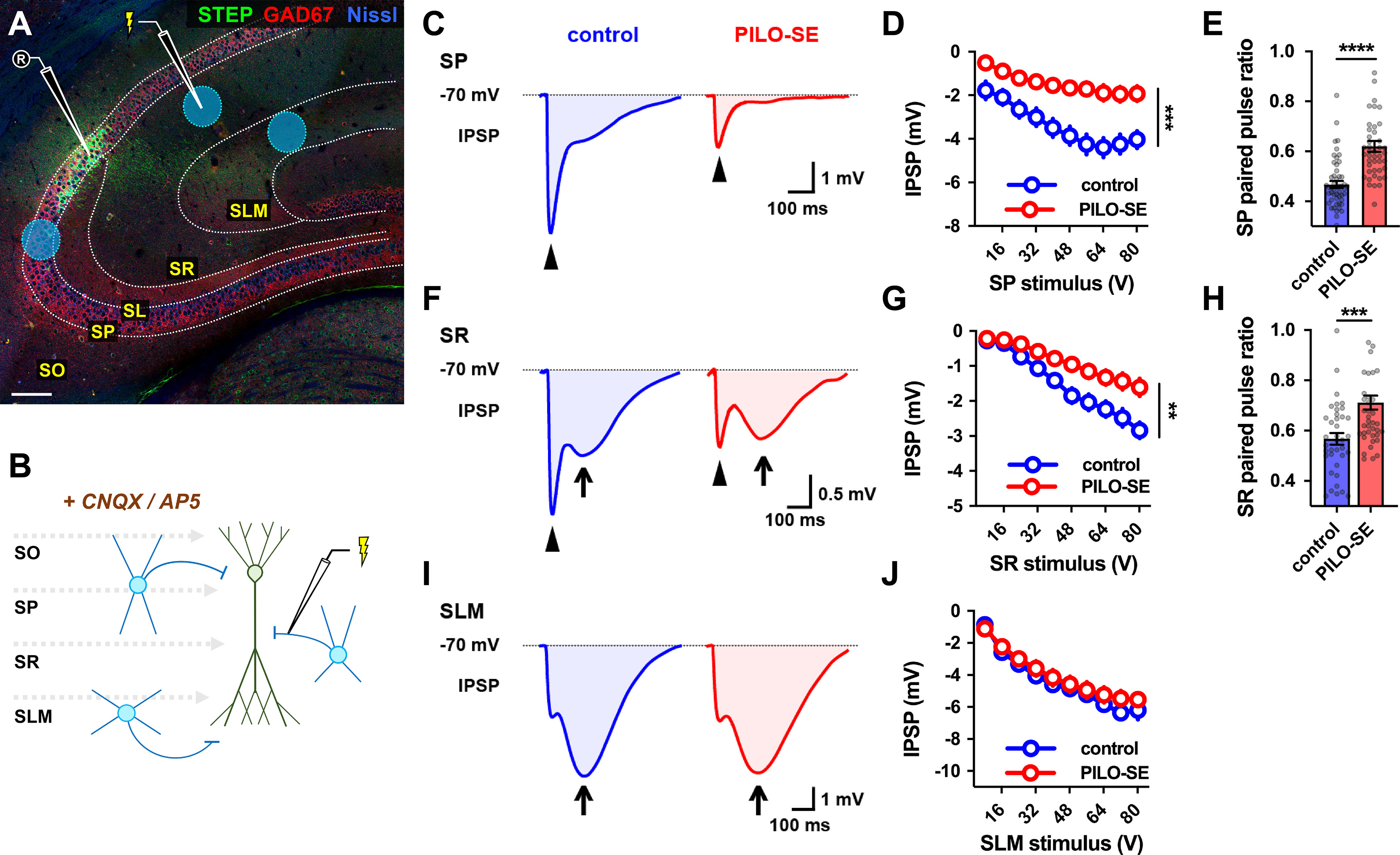
PILO-SE reduced CA2 PN perisomatic and proximal dendritic inhibition but did not alter inhibition at distal dendrites. A, A representative hippocampal section illustrating the stimulation locations for recruitment of monosynaptic inhibition in SP, SR, and SLM (blue circles). Stimulating electrode in SR is shown. CNQX and AP5 were added to block excitatory transmission and isolate IPSPs (see Materials and Methods). Scale bar, 125 µm. B, A circuit diagram illustrating the experimental configuration: a stimulation pipette located in SP, SR, or SLM evokes monosynaptic inhibition by directly activating local interneurons. C, Representative averaged IPSPs evoked in CA2 PNs by 64 V stimulation in SP in slices from control (blue) and PILO-SE (red) mice, with the fast peaks indicated with arrowheads. D, The peak hyperpolarization of the SP stimulation-evoked IPSP was significantly reduced in PILO-SE. E, The PPR of the IPSP evoked in SP was increased in PILO-SE mice. F, Representative averaged IPSPs evoked by 64 V stimulation in SR. The fast peaks are indicated with arrowheads, and the slow peaks with arrows. G, The peak fast hyperpolarization of the SR stimulation-evoked IPSP was significantly reduced in PILO-SE. H, The PPR of the SR stimulation-evoked IPSP was increased in CA2 PNs from PILO-SE mice. I, Representative averaged IPSPs evoked by 64 V stimulation in SLM, with the slow peaks indicated by arrows. J, The peak amplitude of the IPSP evoked by SLM stimulation, taking into account both fast and slow phases, was not significantly different between control and PILO-SE mice.
Immunohistochemistry
Preparation of brain sections.
For immunohistochemistry, mice were placed under deep isoflurane-induced anesthesia and transcardially perfused first with 0.9% NaCl and then with 4% paraformaldehyde (PFA). Brains were extracted and immersed whole in 4% PFA overnight at 4°C. The next day, brains were washed in 0.3% glycine in PBS for 1 h at room temperature on a shaker and were then rinsed three times briefly with PBS. Coronal sections were prepared at a thickness of 60 µm [Figs. 2, 4, 5 (see also Fig. 7)] or 40 µm (see Fig. 9) using a vibratome. Post hoc immunohistochemistry was performed by immersing 400 µm acute hippocampal slices in 4% PFA at the conclusion of recordings and fixing them overnight on a shaker at 4°C. Both 60 µm brain sections and 400 µm hippocampal slices were incubated for 4 h at room temperature in PBS with 0.5% Triton X-100, while 40 µm brain sections were incubated for 2 h at room temperature in PBS with 0.1% Triton X-100. This solution was then exchanged for PBS containing primary antibodies and 0.1% Triton X-100, and the tissue was incubated either overnight (40 and 60 µm brain sections) or for 48 h on a shaker at 4°C. The following antibodies were used: rabbit anti- neuropeptide Y (NPY; 1:1000; catalog #ab30914, Abcam; RRID:AB_1566510); mouse antiparvalbumin (1:1000; catalog #P3088, Sigma-Aldrich; RRID:AB_477329); rabbit anti-proCCK (1:1000; catalog #CCK-pro-Rb-Af350, Frontier Institute; RRID:AB_2571674); rabbit anti-PCP4 (1:500; catalog #HPA005792, Sigma-Aldrich; RRID:AB_1855086); mouse anti-RGS14 IgG2a (1:500; catalog #75-170, NeuroMab; RRID:AB_2179931); mouse anti-STEP IgG1 (1:1000; catalog #4396, Cell Signaling Technology; RRID:AB_1904101); rabbit anti-CB1R (1:1000; catalog #10006590, Cayman Chemical; RRID:AB_10098690); rabbit anti-vesicular glutamate transporter type 3 (VGluT3; 1:500; catalog #135203, Synaptic Systems; RRID:AB_887886); and mouse anti-GAD67 clone 1G10.2 (1:500; catalog #MAB5406, Millipore; RRID:AB_2278725). Primary antibodies were paired with goat anti-mouse and goat anti-rabbit secondary antibodies conjugated to Alexa Fluor 488 (1:500; catalog #A-21121, Thermo Fisher Scientific; RRID:AB_2535764; and 1:500; catalog #A-21131, Thermo Fisher Scientific; RRID:AB_2535771); Alexa Fluor 568 (1:500; catalog #A-11011, Thermo Fisher Scientific; RRID:AB_143157); or Alexa Fluor 647 (1:500; catalog #A-21241, Thermo Fisher Scientific; RRID:AB_2535810). Tissue sections were incubated in the secondary antibody solution for 4 h on a shaker at room temperature or alternatively overnight at 4°C. The Invitrogen NeuroTrace 435/455 fluorescent Nissl stain (1:200; catalog #N21479, Thermo Fisher Scientific) was used to visualize cell bodies in the hippocampus. Perineuronal nets (PNNs) were stained with biotinylated Wisteria floribunda agglutinin (WFA; 1:1000; catalog #L1516, Sigma-Aldrich; RRID:AB_2620171). Biocytin and biotinylated WFA were both visualized using streptavidin conjugated to Alexa Fluor 647 (1:500; catalog #S21374, Thermo Fisher Scientific; RRID:AB_2336066). These antibodies have previously been shown to have high specificity (Hitti and Siegelbaum, 2014; Kohara et al., 2014; Meira et al., 2018).
Figure 4.
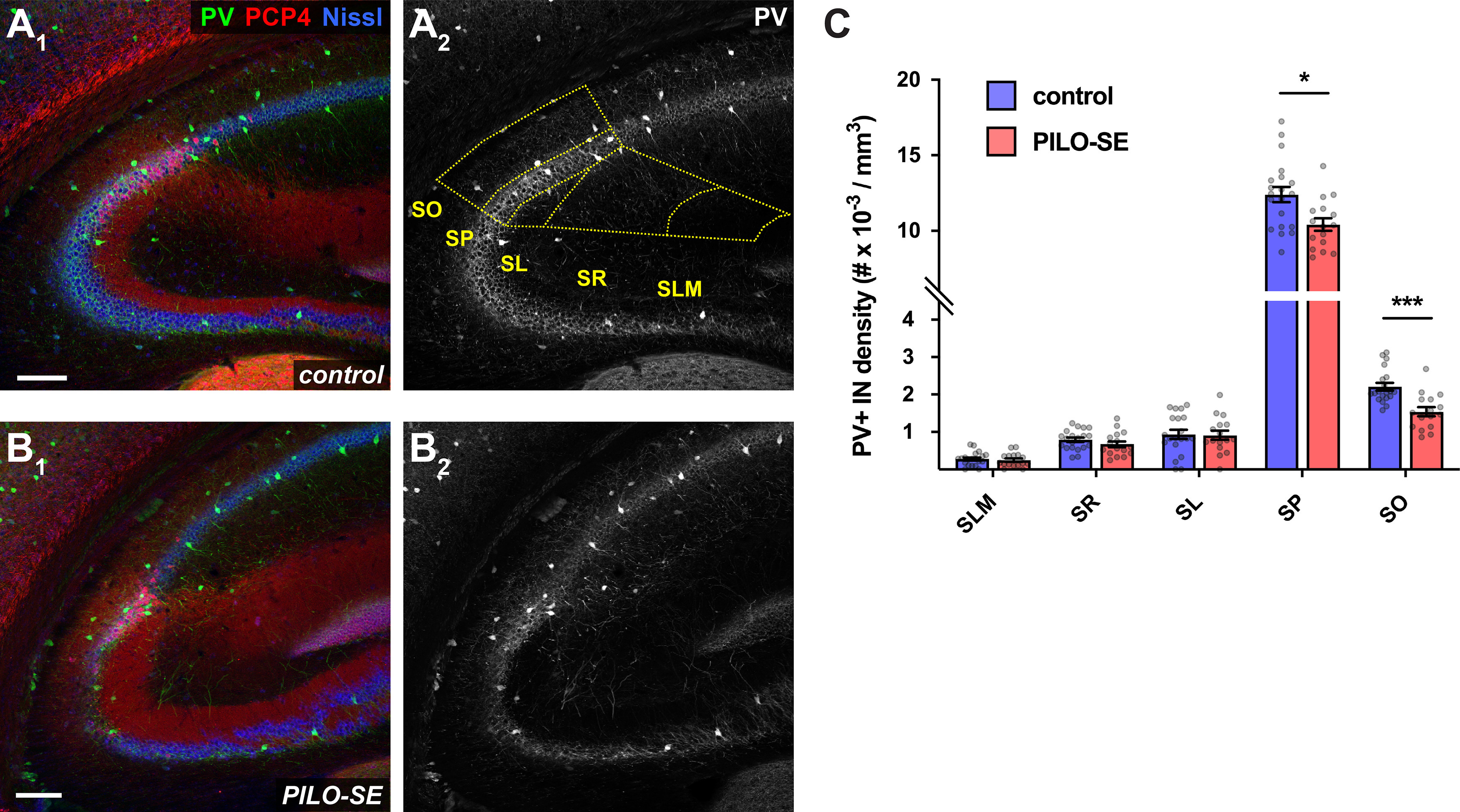
The density of PV+ interneurons was reduced in the CA2 subfield following PILO-SE. A1–B2, Representative hippocampal sections from control (above) and PILO-SE (below) mice, stained for PV (green and white) to visualize PV+ interneurons, Nissl to label neuronal somata (blue), and PCP4 (red) to delineate the CA2 subfield. The bounds of the CA2 region were drawn across all layers of each hippocampal section (A2, yellow dashed lines) and the density of PV+ interneurons was measured for each layer (see Materials and Methods). Scale bars, 150 µm. C, The density of PV+ INs in CA2 was reduced in the SO and SP layers in sections from PILO-SE mice.
Figure 5.
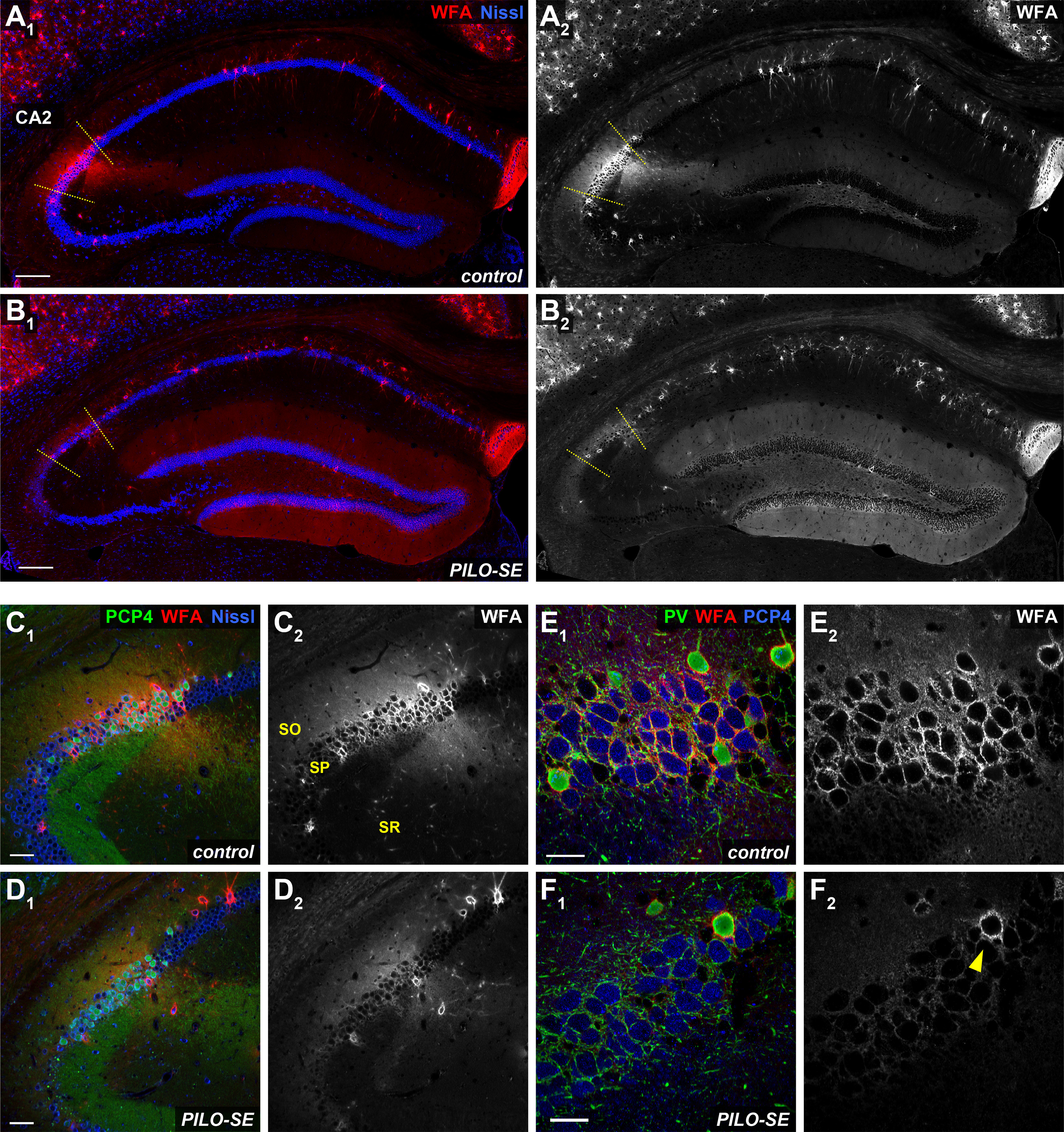
The CA2 pyramidal neuron-associated PNN was diminished in pilocarpine-treated mice. A1, A2, A representative view of hippocampal PNNs stained with WFA (in red and white). Scale bars, 200 µm. B1, B2, In PILO-SE mice the intensity of the WFA stain associated with CA2 PNs was diminished while the intensity of the WFA stain in the DG was enhanced. C1, C2, A representative view of PNNs in CA2. Scale bars, 60 µm. D1, D2, In PILO-SE mice, the intensity of the WFA stain associated with CA2 PNs was diminished. E1, E2, The perisomatic PNN (stained with WFA, in red) surrounds both PV+ interneurons (green) and CA2 PNs (stained with PCP4, in blue). Scale bars, 30 µm. F1, F2, The PNN was degraded around CA2 PNs but was preserved around PV+ interneuron somata in PILO-SE mice (arrowhead).
Figure 7.
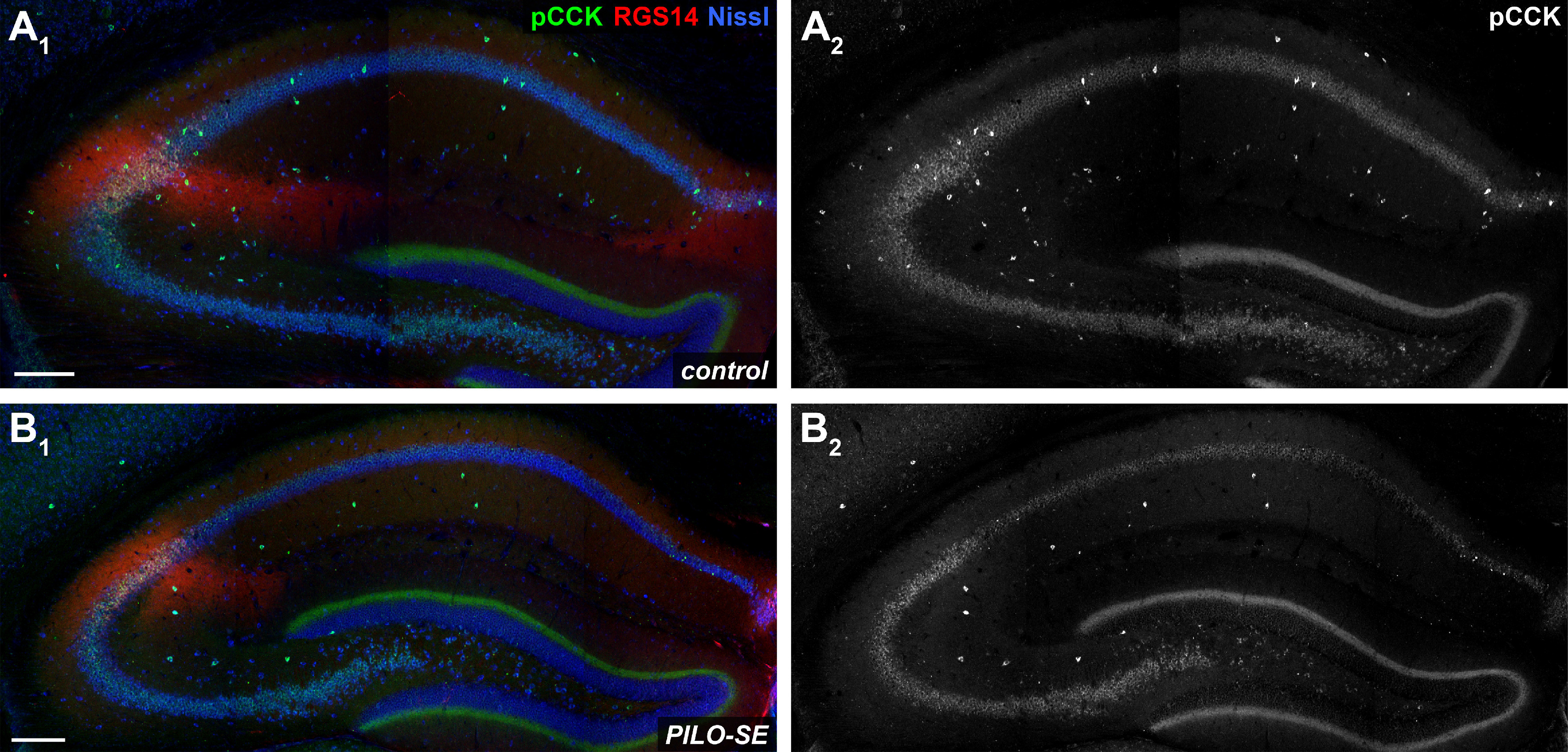
PILO-SE was associated with a widespread decrease of pCCK+ interneurons. A1–B2, Representative hippocampal sections from control (A1, A2) and PILO-SE (B1, B2) mice, stained for pCCK (green and white) to visualize putative cholecystokinin-expressing interneurons, for Nissl to label neuronal somata (blue), and for RGS14 (red) to delineate the CA2 subfield. Scale bars, 200 µm.
Figure 9.
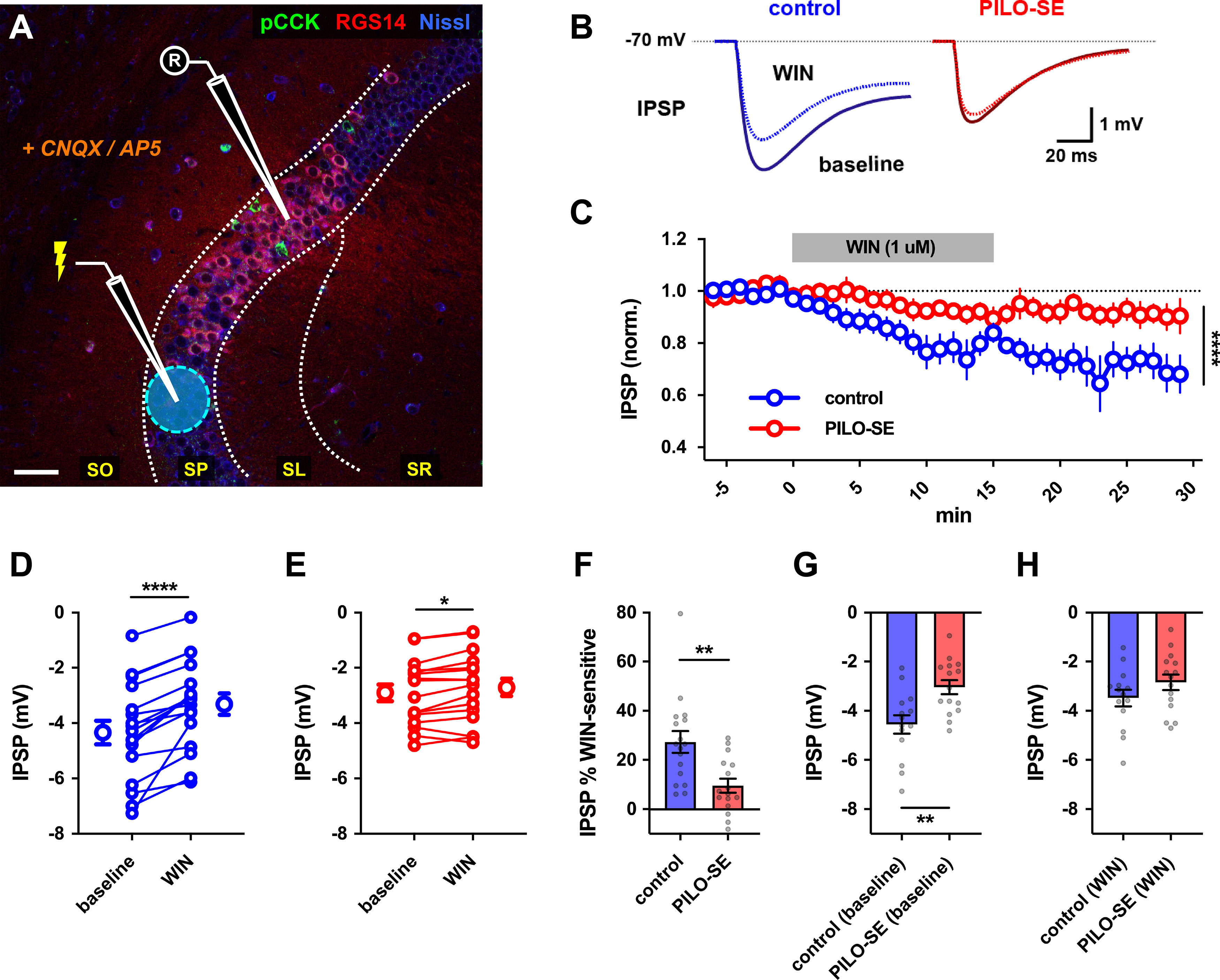
Reduced contribution of CCK+ interneurons to inhibition of CA2 PNs in PILO-SE. A, A representative image illustrating the experimental configuration using electrical stimulation in SP to evoke monosynaptic IPSPs in CA2 PNs (excitation blocked with CNQX and d-APV). Scale bar, 60 µm. B, Representative averaged IPSPs from control (blue) or PILO-SE (red) CA2 PNs before and 11–25 min after application of WIN (1 μm). C, Time course of normalized IPSP amplitude following WIN application in CA2 PNs from control and PILO-SE mice. D, E, IPSPs before and 11–25 min after application of WIN in control (D) and PILO-SE (E) mice. Each pair of points is from one cell in separate slices. F, The percentage reduction of the IPSP (relative to baseline) in control mice was significantly greater than in PILO-SE mice). G, The baseline (pre-WIN) IPSP amplitude in CA2 PNs from control mice was significantly larger in amplitude than the baseline IPSP amplitude in PILO-SE cells. H, There was no significant difference between the post-WIN IPSP amplitude in CA2 PNs from PILO-SE mice and the post-WIN IPSP amplitude in control cells.
The anatomic definition of the CA2 subfield in this study is informed by prior work from our laboratory and others (Chevaleyre and Siegelbaum, 2010; Hitti and Siegelbaum, 2014; Kohara et al., 2014; Meira et al., 2018; Whitebirch et al., 2022). In mice, the dorsal CA2 subfield is ∼300–400 µm in length, and approximately three-quarters of this length overlaps with the stratum lucidum (a portion of distal CA2, near CA1, does not overlap with the stratum lucidum). This delineation of CA2 is clearly seen in the staining patterns of established markers for CA2 pyramidal cells PCP4 (Figs. 1, 3, 4), STEP (Figs. 1, 2), and RGS14 [Figs. 5, 6 (see also Fig. 9)]. All images were acquired with a confocal microscope (model LSM 700, Zeiss) and processed using FIJI software (version 2.0.0-rc-54/1.51 h).
Figure 6.

The CA2 pyramidal neuron-associated PNN was diminished in pilocarpine-treated mice. A, WFA fluorescence intensity in the SO, SP, and SR of CA2 was significantly reduced in PILO-SE. B, WFA staining around CA2 PV+ IN somata was not significantly altered in PILO-SE mice.
Fluorescence and anatomic measurements.
For the normalized stratum pyramidale (SP) Nissl fluorescence intensity profile presented in Figure 2, we first measured the profile of Nissl fluorescence along the length of a line traced along the length of the stratum pyramidale in FIJI (version 2.0.0-rc-54/1.51 h), with the width of the line adjusted to match the thickness of the stratum pyramidale for each image. The fluorescence profile tool in FIJI averages intensity values across the width of the line, at each point along its length. The proximodistal profile was normalized by length centered on the CA2 subfield. To perform the length normalization alongside Nissl fluorescence, a second proximodistal profile was measured of CA2 marker fluorescence intensity. Many tissue slices from the dorsal and intermediate hippocampus (spanning ∼0–3.2 mm from the dorsal pole of the hippocampus) across experiments were combined for this analysis. We used antibodies against a number of markers that are all coexpressed in CA2 PNs, including PCP4, RSG14, WFA, or GFP (the latter based on CA2-selective expression in Amigo2-Cre mice). In the cases where slices were stained for multiple CA2 markers, the marker with the clearest signal-to-noise was used to generate a proximodistal profile and to define the center of the CA2 subfield. A custom MATLAB script was written to transform the full-length proximodistal profiles into vectors 100 values in length, consisting of 50 values from the center of CA2 to the end of CA3, and 50 values from the center of CA2 to the end of CA1. These vectors were then normalized for the intensity of each marker by dividing every value by the maximum, such that for both CA2 markers and Nissl the maximum fluorescence intensity value was 1.0 for every slice measured. Next, to normalize PILO-SE values against the control samples, the control Nissl intensity vectors were averaged across mice and the Nissl vectors from each PILO-SE mouse were divided by the mean control value at each of the 100 positions of the length-normalized vectors, producing the data in Figure 2E. Finally, mean normalized Nissl fluorescence values were generated for each subfield (CA3c, CA3b, CA3a, CA2b, CA2a, CA1c, CA1b, and CA1a) by first defining the bounds of the CA2 subfield based on CA2 marker fluorescence. The CA2a/CA1c border exhibits a distinct transition with little intermingling of CA2 and CA1 PNs and a sharp decline in the fluorescence intensity of CA2 PN markers (Fig. 2E, heatmap). The CA2b/CA3a border exhibits a more gradual transition with significant intermingling of CA3 and CA2 PNs, which is reflected by a gradual decrease in CA2 marker fluorescence (Fig. 2E, heatmap). Therefore, the CA2b/CA3a border was drawn at the approximate halfway point of the intermingled region, so as to balance the exclusion of CA2b neurons against the inclusion of CA3a neurons. Considering recent evidence for proximodistal heterogeneity within the CA2 region (Fernandez-Lamo et al., 2019; Okamoto and Ikegaya, 2019), and our own observations of greater resilience to neurodegeneration in distal CA2, the CA2 subfield was divided into two equal halves designated as CA2a (distal, near CA1) and CA2b (proximal, near CA3). Such a subdivision of the CA2 subfield conforms with reported distinctions between proximal and distal CA2 PNs (Dudek et al., 2016; Okamoto and Ikegaya, 2019; Fernandez-Lamo et al., 2019), and our own observations that many neurons in the distal CA2a do not colocalize with mossy fiber terminals and that neurons in the proximal CA2b are intermingled with CA3a neurons. The CA3 and CA1 regions were each divided into three equal regions by length. CA2-specific markers were used to delineate the distal and proximal boundaries of CA3 and CA1, respectively, such that the distal boundary of CA3 was placed halfway through the intermingled region of CA3a and CA2b neurons, while the proximal boundary of CA1 was placed at the point beyond the mossy fiber terminus at which labeled CA2a neurons were no longer visible. The normalized Nissl fluorescence intensity values were averaged in each region to produce the values represented in Figure 2F.
The areas of the CA2a and CA2b subregions were measured in ImageJ using regions of interest (ROIs) that were manually drawn, with PCP4 used as a CA2-specific molecular marker. Boundaries were defined in the same manner as described above, with the boundary between CA2a and CA2b designated as the midpoint of the entire CA2 subfield as identified by PCP4 immunoreactivity.
PNN integrity was assessed by immunostaining with biotinylated WFA. The bounds of the CA2 subfield were defined based on costaining with PCP4. In FIJI regions of interest were drawn around the CA2 stratum oriens (SO), SP, and stratum radiatum (SR), and the mean WFA fluorescence intensity measured in each region of interest. Regions of interest were drawn to exclude WFA associated with PV+ interneurons. In the dentate gyrus, regions of interest were drawn in a portion of the granule cell layer, molecular layer, and hilus (HIL; measurements were made in the upper blade of the DG). For every animal, background fluorescence intensity was subtracted from the measured WFA fluorescence intensity, and measurements from multiple hippocampal sections were averaged to produce a single set of measurements from each animal. To measure WFA associated with PV+ interneurons, sections were costained for parvalbumin, regions of interest were drawn around several PV+ interneuron soma within the CA2 subfield visualized with PCP4, and the WFA fluorescence intensity from all measured interneurons was averaged for each animal. PV and pro-CCK (pCCK)-expressing interneuron density was measured in hippocampal sections stained with the PV and pCCK antibodies described above, along with a stain against PCP4 or RGS14. The CA2 region was captured in a z-stack series of images, and the bounds of the CA2 subfield were drawn across all layers of the hippocampus using PCP4 or RGS14 immunofluorescence. For pCCK+ interneuron measurements, regions of interest were additionally drawn across all layers of CA3 and CA1, with each subfield divided lengthwise into approximate thirds (CA3c, CA3b, CA3a, CA1c, CA1b, CA1a). As with WFA fluorescence intensity, interneuron density measurements from multiple sections were averaged to produce a single set of measurements for each mouse. All imaging was performed with identical acquisition parameters for samples from control and PILO-SE mice.
For measuring fluorescence intensity values within perisomatic inhibitory axon terminals in SP, images of single optical sections were collected using a Zeiss Plan-Apochromat 20×/0.8 objective (0.5 zoom; pixel size, 0.6252 × 0.6252 μm2). Images displaying GAD67 labeling were converted to a binary format using ImageJ (version 1.53a) after setting the threshold to 3.5% (as we found capturing pixels within the 3.5% highest intensity window allowed for exclusion of the nearby mossy fiber axon signal). Next, a selection was created from the thresholded image to define the ROI. The selected ROI was then applied to the corresponding colabeled images displaying CB1 receptor or VGluT3 labeling. Measurements were obtained for ROI area size and average fluorescence intensity within the ROI for each image (expressed as arbitrary units). Area sizes sampled for control and PILO-SE mice were similar for each of the comparison groups. For PV measurements, the ROI (created as described above) was limited to the CA2 region by excluding portions of the image negative for PCP4 or RGS14 marker labeling, and the threshold was set to 1.5% (to exclude lower-intensity staining in nearby CA1).
Statistical comparisons
All statistical tests were performed in GraphPad Prism. For group comparisons, two-way ANOVA with the Geisser–Greenhouse correction and Holm–Sidak multiple comparisons test was used. For group comparisons with missing data points, GraphPad Prism cannot implement a two-way ANOVA and instead utilizes a mixed-effects model that uses a compound symmetry covariance matrix, which is fit using the restricted maximum likelihood approach. The Mann–Whitney U test was used for comparisons between two datasets, and the Kruskal–Wallis test with Dunn’s multiple-comparisons test was used for comparisons among more than two groups. Comparison of paired datasets was performed using the Wilcoxon matched-pairs signed-rank test. For immunohistochemical and anatomic data, nested t tests were performed to factor both the number of mice (as the sample size n) and measurements from multiple tissue sections from each animal. In the case of electrophysiological measurements, n was defined as the number of individual neurons. In all cases, the cutoff for significance was set to a p value of 0.05. Asterisks are used in figures to designate statistical significance and represent P-values as follows: *, P less than or equal to 0.05; **, P less than or equal to 0.01; ***, P less than or equal to 0.001; ****, P less than or equal to 0.0001.
Results
The CA2a and CA1c subfields are resistant to neurodegeneration following PILO-SE
Our goal here was to provide a detailed quantitative investigation into how the various hippocampal cornu ammonis regions (CA1, CA2, and CA3) are affected in the PILO-SE mouse model of TLE. We were particularly interested in the cellular basis for our previously characterized loss of synaptic inhibitory drive onto CA2 pyramidal cells (Whitebirch et al., 2022). In our previous and current study, we characterized the alterations in the electrophysiological properties of CA2 neurons in acute hippocampal slices from PILO-SE mice that were F1 hybrids from a cross between C57BL/6J and 129S1/SvlmJ mouse lines. We found that this hybrid genetic background provided an optimal compromise between effective induction of SE and mouse survival, which was suitable for subsequent electrophysiological analysis. Although we had previously verified that these mice developed spontaneous seizures, here we first report results from a quantitative analysis of seizure properties in these mice to further validate their use as a TLE model. This is of particular importance as the development of spontaneous seizures in the PILO-SE model is known to be quite sensitive to genetic background, and the commonly used C57BL/6J substrain of mice contains a specific genetic mutation that makes them resistant to seizure generation (Bankstahl et al., 2012; Löscher et al., 2017; Kapur et al., 2020).
Experiments were performed ∼6 weeks following PILO-SE induction (Fig. 1A), at a time when spontaneous seizures have generally started to appear (Mazzuferi et al., 2012; Pitsch et al., 2017; Jain et al., 2019). Pilocarpine treatment led to SE in 39.9% of mice in a large cohort of animals (Fig. 1B). We captured spontaneous seizures in these surviving SE mice using continuous video-EEG recordings and subdural screw electrodes, with recordings taking place at least 4 weeks after SE (Fig. 1C; see Materials and Methods). Video-EEG recordings confirmed that SE reliably led to spontaneous, recurring convulsive seizures in the weeks following pilocarpine treatment (eight of eight mice examined). As previously reported for both human TLE and the PILO-SE model (Haut et al., 2005; Pitsch et al., 2017; Baud et al., 2018, 2019; Whitebirch et al., 2022), spontaneous seizures tended to occur in a clustered pattern across weeks of recording (Fig. 1D). Seizures were large-amplitude and rhythmic events highly synchronized across all four electrodes (Fig. 1E). Examination of the time–frequency representation of the spontaneous seizures revealed an increase in the relative power of frequencies between ∼10 and 50 Hz (Fig. 1F).
As previous studies characterizing cell loss in TLE and its rodent models relied solely on anatomic position to distinguish CA2 from its neighboring CA1 and CA3 regions, we next conducted the first quantitative analysis of cell survival using molecular markers to identify specifically CA2 PNs. Hippocampal sections from PILO-SE mice revealed a characteristic pattern of hippocampal damage, with neuronal depletion most apparent in the CA3 and CA1 subfields (Fig. 2A1,A2,C1,C2) and in the HIL of the DG. In sections from PILO-SE mice, we observed NPY immunoreactivity in DG mossy fibers, consistent with prior reports of NPY upregulation because of recurring seizure activity in the hippocampus (Marksteiner et al., 1990; Kofler et al., 1997; Vezzani et al., 1999). To quantify the neurodegenerative changes in the pyramidal cell layer, we stained hippocampal sections for the Nissl substance along with CA2 molecular markers, such as PCP4 and STEP (Hitti and Siegelbaum, 2014; Kohara et al., 2014). To identify the regions of the stratum pyramidale where seizure-associated neuronal loss most consistently occurred, we first selected hippocampal sections from PILO-SE mice that exhibited overt cell depletion as defined by interruptions in the Nissl stain fluorescence along the entire CA3–CA2–CA1 pyramidal layer in low-magnification images (Fig. 2C1,C2). Next, we measured both Nissl and CA2 marker fluorescence intensity along the proximodistal axis of the stratum pyramidale, normalizing measurements for both fluorescence intensity and proximodistal position (centered on the CA2 subfield; see Materials and Methods). Using this approach, we found clear decreases in normalized Nissl fluorescence in CA1 and CA3, with the most dramatic decrease seen in CA3a (Fig. 2E). In particular, we noted a stark contrast in the vulnerability of CA3 compared with CA2 PNs, with a consistent decrease in Nissl intensity in the CA3a subregion and resilience in the CA2. However, the resilience of CA2 varied across the proximodistal axis. Whereas the half of CA2 closest to CA1 (CA2a; see Materials and Methods) was highly resistant to cell loss, the half of CA2 closest to CA3 (CA2b), was less resilient. These results are consistent with our visual inspection of PILO-SE tissue (Fig. 2B1,B2,D1,D2).
To quantify the neurodegenerative changes in the pyramidal cell layer following PILO-SE, we measured the mean normalized Nissl fluorescence intensity in each subfield (Fig. 2F). This revealed significant decreases in CA3a (p < 0.0001), CA3b (p < 0.0001), CA3c (p = 0.0006), CA2b (p < 0.0001), CA1a (p = 0.0326), and CA1b (p = 0.0326). In contrast, neither CA2a (p = 0.7734) nor CA1c (p = 0.1505) subregions experienced a significant loss of Nissl signal (two-way ANOVA with Holm–Sidak multiple-comparisons test; n = 45 sections from 31 control mice, n = 56 sections from 31 PILO-SE mice).
Thus, our initial anatomic assessment using established molecular markers of the CA2 region confirmed the previously reported relative resilience of CA2 PNs in the epileptic hippocampus (Winawer et al., 2007; Mazzuferi et al., 2012; Blümcke et al., 2013; Steve et al., 2014). Furthermore, our analysis suggested a differentiation between the proximal and distal section of CA2, with the latter showing the highest resilience (CA2a). Finally, our measurements revealed a previously unrecognized resilience of the proximal section of CA1 (CA1c) and highlighted the vulnerability of the CA3 region, in particular the CA3a subfield. However, we note two important limitations of our Nissl stain analysis. First, there is a potential confounding effect of Nissl fluorescence originating from cells other than PNs, including glia. Second, our Nissl stain analysis of total subfield fluorescence intensity lacked cellular level resolution. Thus, while the above analysis identifies regions particularly susceptible to cellular depletion, it cannot rule out the occurrence of more subtle patterns of cell loss.
To address some of these limitations and to characterize in further detail the relative vulnerability of CA2b compared with CA2a, we measured the specific area occupied by the CA2 PNs in each CA2 subregion by drawing ROIs informed by PCP4 immunostaining using higher-magnification images (Fig. 2B,D). We measured the area of CA2a and CA2b in sections from control mice, PILO-SE mice that exhibited overt cell depletion along the pyramidal layer (as defined above), and PILO-SE mice without obvious cell loss. When measurements from both groups of PILO-SE mice were pooled and compared against control measurements, we found a significant decrease in the area of CA2b (nested t test; p = 0.0284; n = 28 sections from 14 control mice, n = 28 sections from 14 PILO-SE mice). Thus, the area of CA2b in control sections was 11,144 ± 339.7 µm2, while in sections from PILO-SE mice the area of CA2b was reduced by 14.93 ± 0.06% to 9481 ± 631.3 µm2. In contrast, the area of CA2a in control sections was 12,133 ± 369.4 µm2, and in sections from PILO-SE mice was 11,637 ± 770.3 µm2, with no significant difference between the groups (nested t test; p = 0.5666; n = 28 sections from 14 control mice, n = 28 sections from 14 PILO-SE mice). When we only considered the subset of PILO-SE mice that exhibited overt cell loss throughout the CA3–CA2–CA1 region; however, we found a significant decrease in the area of both CA2a (nested t test; p = 0.0106; n = 28 sections from 14 control mice, n = 14 sections from 7 PILO-SE mice) and CA2b (nested t test; p = 0.0002; n = 28 sections from 14 control mice, n = 14 sections from 7 PILO-SE mice). In these comparisons, the area of CA2a was decreased by 18.42 ± 0.07% while the area of CA2b was decreased by 26.68 ± 0.05% (t test; p = 0.0154; n = 14 sections from 7 PILO-SE mice). In summary, our measurements of CA2a and CA2b area provide further evidence indicating a difference in vulnerability between the proximal and distal halves of the CA2 subfield, in which the proximal CA2b region exhibited a more dramatic decrease in area. In addition, this analysis revealed that relatively subtle cell loss may also occur in area CA2a, notwithstanding the higher resilience of these neurons to epilepsy-induced damage.
Perisomatic inhibition of CA2 PNs is reduced following pilocarpine administration
Our prior study (Whitebirch et al., 2022) reported diminished inhibition of CA2 PNs following PILO-SE on stimulation of either the CA2/CA3 local recurrent collaterals or the DG granule cell mossy fibers. This loss of inhibition could be explained by a number of mechanisms, including a failure of excitatory axons to sufficiently activate their target interneurons or a reduction in GABA release from recruited interneurons. To explore the basis of this inhibitory deficit, we performed whole-cell recordings from CA2 PNs in slices from control and PILO-SE mice (∼6 weeks after PILO treatment) in the presence of 50 μm d-AP5 and 25 μm CNQX, antagonists of NMDA and AMPA receptors, respectively. Under these conditions, electrical stimulation relatively close to the CA2 subfield directly evoked monosynaptic GABAergic IPSPs (Fig. 3A,B). Electrical stimulation was applied using stimulating electrodes in SP, SR, or stratum lacunosum moleculare (SLM) to activate interneuronal axons targeting the perisomatic region (e.g., those originating from PV+ or CCK+ basket cells), the proximal apical dendrites (where local excitatory axons establish recurrent connections), or the distal apical dendrites (where EC afferents innervate the CA2 region), respectively.
With the stimulating electrode placed in SP, we evoked fast IPSPs in CA2 PNs that were consistent with inhibition mediated by GABAA postsynaptic receptors. On average, PILO-SE caused a reduction in the peak IPSP to 51.7 ± 5.1% of its control value (Mann–Whitney test, p < 0.0001) using a fixed stimulus intensity of 64 V, near the peak of the control IPSP input–output curve (n = 36 cells from 23 control mice, n = 36 cells from 17 PILO-SE mice). This reduction in IPSP amplitude in PILO-SE compared with control cells was observed across the entire IPSP input–output curve (Fig. 3C,D; mixed-effects model, p = 0.0001; n = 17 cells from 10 control mice, n = 20 cells from 7 PILO-SE mice). Pairs of SP stimuli delivered 50 ms apart revealed that PILO-SE also caused a significant increase in the paired-pulse ratio (PPR), consistent with a decreased probability of GABA release (Fig. 3E; Mann–Whitney test; p < 0.0001; n = 51 cells from 22 control mice, n = 44 cells from 18 PILO-SE mice).
We had previously reported that in CA2 PNs from PILO-SE mice, IPSCs decayed more rapidly than the IPSCs in control cells, such that the time constant of decay tau was 52.3 ± 3.8 ms in control cells and 36.9 ± 2.7 ms in cells from PILO-SE mice (Whitebirch et al., 2022). Therefore, here we sought to characterize the temporal characteristics of the SP-evoked IPSP. We additionally measured the 10–90% rise time of the descending slope and the integral of the entire IPSP. We found that IPSPs in CA2 PNs from PILO-SE mice exhibited a faster rise time (Mann–Whitney; p = 0.0008; n = 36 cells from 23 control mice, n = 33 cells from 17 PILO-SE mice). Thus, at a stimulation intensity of 64 V (which typically elicited a maximal response), the 10–90% rise time of the control IPSP was 7.44 ± 0.31 ms while the rise time of the IPSP from PILO-SE mice was reduced to 5.78 ± 0.38 ms. The integral of the SP-evoked IPSP was also greatly reduced: at a stimulation intensity of 64 V the integral was −533.42 ± 61.62 mV × ms in control cells but only −130.05 ± 21.33 mV/ms in cells from PILO-SE mice (mixed-effects model; p < 0.0001; n = 17 cells from 10 control mice, n = 20 cells from 7 PILO-SE mice).
When we placed the stimulating electrode in SR, we evoked IPSPs with clearly separable fast and slow phases, consistent with inhibition mediated by GABAA and GABAB receptors, respectively (Fig. 3F). We found that PILO-SE reduced the peak IPSP to 51.1 ± 4.7% of its control value (Mann–Whitney test; p < 0.0001; n = 27 cells from 17 control mice, n = 31 cells from 16 PILO-SE mice), comparable to the reduction in the SP-evoked IPSP. Comparison of the input–output curves for SR-evoked IPSPs also revealed a decrease in the magnitude of the IPSP amplitudes (Fig. 3F,G; mixed-effects model; p = 0.0055; n = 9 cells from four control mice, n = 12 cells from 5 PILO-SE mice). Separate measurements of the peak hyperpolarization during the fast and slow phases of the IPSP revealed a significant decrease in the amplitude of the fast IPSP (Kruskal–Wallis test with Dunn’s multiple-comparisons test; p = 0.0034; n = 9 cells from four control mice, n = 12 cells from 5 PILO-SE mice) with little or no change in the amplitude of the slow IPSP (Kruskal–Wallis test with Dunn’s multiple-comparisons test; p = 0.7140; n = 9 cells from four control mice, n = 11 cells from 5 PILO-SE mice). Similar to what we observed with SP stimulation, PILO-SE caused a significant increase in the PPR (Fig. 3H; Mann–Whitney; p = 0.0004; n = 38 cells from 16 control mice, n = 42 cells from 18 PILO-SE mice).
In contrast to the loss of inhibition evoked by stimulating the SP or SR layers in PILO-SE mice, we found no change in the size of the IPSP evoked by stimulation in the SLM in the epileptic mice compared with control mice. In both groups, SLM stimulation evoked fast and slow components to the IPSP, with the slow IPSP being dominant (Fig. 3I,J; mixed-effects model; p = 0.5548; n = 5 cells from three control mice, n = 16 cells from 7 PILO-SE mice).
Together, these results suggest that PILO-SE produced a decrease in the magnitude of the monosynaptic fast component of the IPSP evoked by SP or SR stimulation. Furthermore, our PPR data indicate that at least part of the decrease in the IPSP is because of a decrease in the probability of transmitter release, possibly resulting from a functional alteration to inhibitory synapses in CA2. Finally, the preferential loss of the fast component of the IPSP suggests a selective impairment of fast inhibition mediated by GABAA receptors in CA2 PNs.
PILO-SE reduced labeling of CA2 parvalbumin-positive interneurons and the CA2 pyramidal neuron-associated perineuronal net
Perisomatic inhibition in the hippocampus is primarily provided by basket cells that express either CCK or PV (Freund and Katona, 2007; Dudok et al., 2021). In light of past work demonstrating that CA2 contains a particularly dense population of PV+ interneurons (Botcher et al., 2014), including unique PV+ basket cells (Mercer et al., 2007, 2012a), we first used an immunohistochemical approach to examine PV+ interneurons (Fig. 4). We quantified the density of PV+ interneurons and found a significant decrease in the CA2 SP and SO layers of PILO-SE mice compared with controls (Fig. 4C; two-way ANOVA with Holm–Sidak multiple-comparisons test; SP, p = 0.0194; SO, p = 0.0008; n = 98 sections from 20 control mice, n = 81 sections from 16 PILO-SE mice). However, the magnitude of the decrease in density of PV+ interneurons in the SP, the location of the basket cells that provide the main inhibitory drive onto CA2 PN soma, was only 12.9 ± 3.5%, which seems insufficient to fully account for the nearly 50% decrease in the SP-evoked fast IPSP reported above.
A specialized form of extracellular matrix, the PNN, provides a structural scaffold that regulates interneuronal excitability and synaptic plasticity (Slaker et al., 2016; Favuzzi et al., 2017; Wen et al., 2018; Testa et al., 2019). The CA2 subfield exhibits a distinct pattern of PNN expression, with a dense PNN network localized to the perisomatic region of both PV+ interneurons and PNs and a more diffuse PNN extending into the CA2 SO and SR (Noguchi et al., 2017; Domínguez et al., 2019). Recent evidence has implicated the CA2 PNN in activity-dependent regulation of perisomatic inhibition of CA2 PNs (Carstens et al., 2016, 2021; Domínguez et al., 2019). Furthermore, disruption or alteration to the CA2 PNN has been reported in both genetic and chemoconvulsant-based models of epilepsy (McRae et al., 2012; Rankin-Gee et al., 2015; Dubey et al., 2017; Favuzzi et al., 2017; Carstens et al., 2021).
We used the marker WFA to visualize the PNN associated with the CA2 PN perisomatic and proximal dendritic regions, as well as PV+ interneuron soma within the CA2 subfield (Carstens et al., 2016, 2021; Domínguez et al., 2019). In hippocampal sections, we quantified PNN expression in CA2 by measuring the fluorescence intensity of WFA staining in the different layers of each region. We found significant decreases in CA2 WFA intensity (Fig. 6A) in tissue from PILO-SE mice compared with controls, observed in the SP layer (p = 0.0001), SR layer (p = 0.0002), and SO layer (p = 0.0074; all tests using two-way ANOVA with Holm–Sidak multiple-comparisons test; n = 101 sections from 18 control mice, n = 97 sections from 18 PILO-SE mice). Both the dense meshwork of PNNs located around the CA2 PN soma in the SP layer and the diffuse PNN staining in the CA2 SR and SO layers seen in control mice (Fig. 5A1,A2,C1,C2,E1,E2) were markedly diminished in PILO-SE mice (Fig. 5B1,B2,D1,D2,F1,F2). In contrast, when we examined the pattern of PNN distribution in tissue from PILO-SE mice, we noted that the PNN staining around the soma of PV+ interneurons in CA2 appeared intact, even when it was diminished around CA2 PNs (Fig. 5D1,D2,F1,F2). Indeed, the measurement of WFA fluorescence intensity specifically around PV+ interneurons indicated no statistically significant differences between control and PILO-SE cells (Fig. 6B; nested t test; p = 0.8798; n = 65 cells from 15 control mice, n = 79 cells from 15 PILO-SE mice), suggesting a selective loss of the pyramidal cell-associated PNN in the CA2 region of PILO-SE mice. While the mechanisms and functional implications of decreased PNN in CA2 are presently unclear, disruption to the extracellular matrix may be consistent with impairment of inhibitory synaptic function or plasticity in the CA2 region of PILO-SE mice.
PILO-SE is associated with decreased density and functional impairment of CA2 CCK+ interneurons
Given the discrepancy between the substantial impairment of perisomatic inhibition we observed in CA2 PNs from PILO-SE mice and the relatively small decrease of PV+ somata density, we wondered whether the CCK+ interneurons, which comprise another major population of perisomatic-targeting cells, might be impacted following PILO-SE. Similar to the approach taken above for PV+ interneurons, we first assessed the CCK+ interneuron population using an antibody for pCCK and found a substantial decrease in the number of pCCK-immunopositive (pCCK+) interneurons in hippocampal sections from PILO-SE mice (Fig. 7A1–B2). Notably, the magnitude of the decrease in pCCK+ interneuron density was significantly greater than the loss of PV+ IN staining (nested t test; p < 0.0001), with an ∼69.3 ± 4.7% decrease in CA2 pCCK+ neurons (n = 55 sections from 11 PILO-SE mice) compared with a 15.9 ± 3.0% decrease in CA2 PV+ interneuron density (n = 81 sections from 16 PILO-SE mice), when measured across all layers of the hippocampal CA2 region. This suggests that a disproportionate depletion of CCK+ interneurons in PILO-SE mice might contribute to the functional loss of inhibition we observed. Measurement of pCCK+ interneuron density in each layer of CA2 revealed significant decreases in all areas except the SLM (Fig. 8B; two-way ANOVA with Holm–Sidak multiple-comparisons test; SR, p < 0.0001; SL, p < 0.0001; SP, p < 0.0001; SO, p < 0.0001; n = 78 sections from 15 control mice, n = 55 sections from 11 PILO-SE mice), a pattern of loss consistent with the layer-specific pattern of IPSP change seen in CA2 PNs from PILO-SE mice (Fig. 3).
Figure 8.
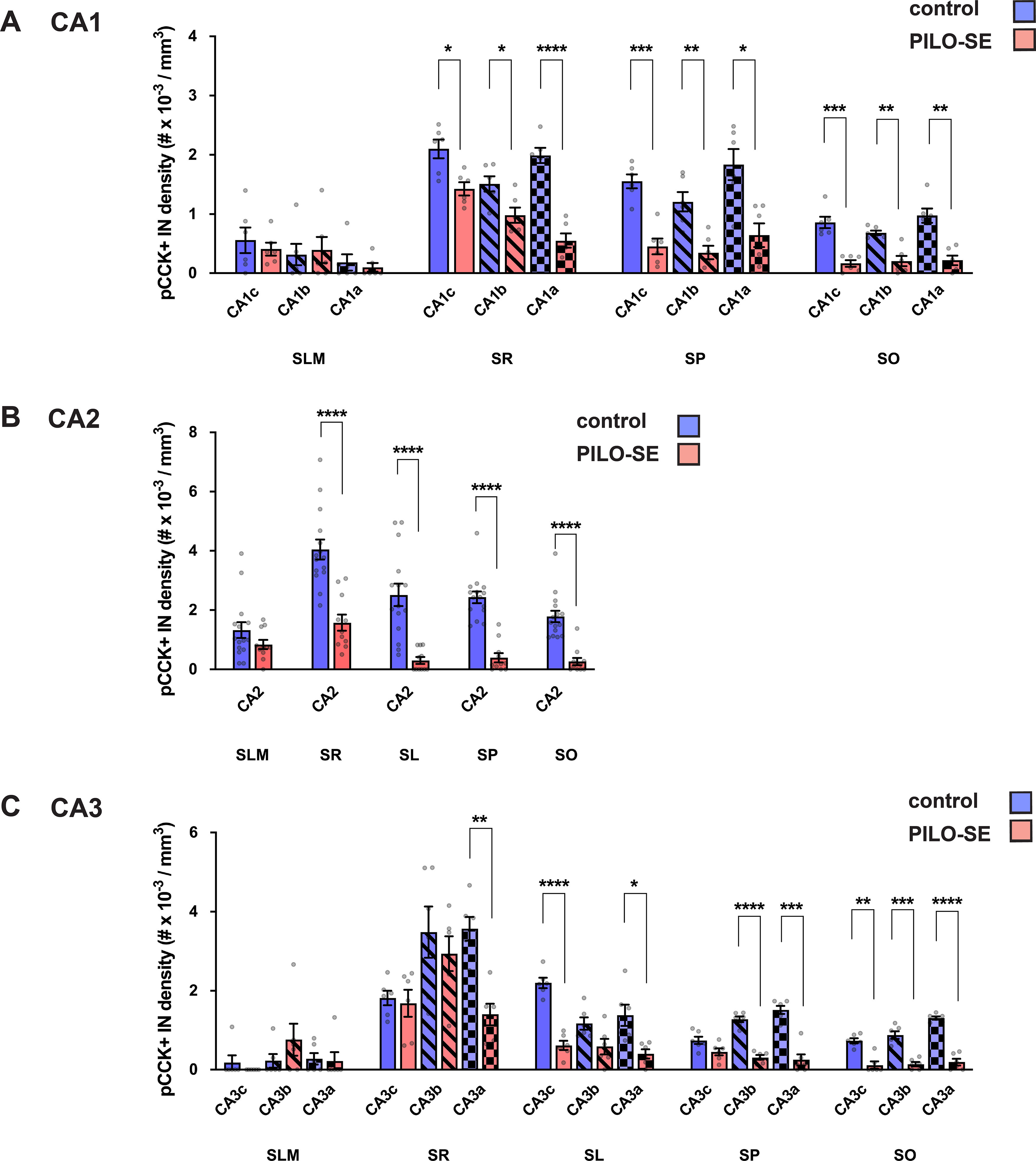
The density of pCCK+ interneurons was reduced in CA2 after PILO-SE. The density of pCCK+ interneurons was significantly reduced across all subfields of the hippocampus. A, pCCK+ interneuron density was reduced in CA1 in SO, SP, and SR. B, pCCK+ interneuron density in CA2 was reduced in PILO-SE mice in SO, SP, SL, and SR. C, pCCK+ interneuron density was reduced in CA3 in SO, SP, SL, and SR, although the statistical significance of the decrease varied in some layers of CA3b and CA3c, as indicated.
Additionally, we observed a significant and widespread decrease of pCCK+ interneuron density in the other hippocampal subfields (Fig. 7). In tissue from PILO-SE mice, pCCK+ interneuron density was reduced across the SR, SP, and SO layers of CA1c (CA1c SR, p = 0.0133; CA1c SP, p = 0.0004; CA1c SO), CA1b (p = 0.0007; CA1b SR, p = 0.0319; CA1b SP, p = 0.0073; CA1b SO), and CA1a subfields (p = 0.0073; CA1a SR, p < 0.0001; CA1a SP, p = 0.0107; CA1a SO, p = 0.0016) when compared with staining in control mice (Fig. 8A; two-way ANOVA with Holm–Sidak multiple-comparisons test; n = 33 sections from 6 control mice, n = 31 sections from 6 PILO-SE mice). Similarly, we found decreases in pCCK+ interneuron density in CA3c (CA3c SR, p = 0.7353; CA3c SL, p < 0.0001; CA3c SP, p = 0.1436; CA3c SO, p = 0.0013), CA3b (CA3b SR, p = 0.5034; CA3b SL, p = 0.1210; CA3b SP, p < 0.0001; CA3b SO, p = 0.0006), and CA3a (CA3a SR, p = 0.0010; CA3a SL, p = 0.0278; CA3a SP, p = 0.0002; CA3a SO, p < 0.0001) compared with control animals (Fig. 8C; two-way ANOVA with Holm–Sidak multiple-comparisons test; n = 33 sections from 6 control mice, n = 31 sections from 6 PILO-SE mice). Thus, overall, pCCK+ interneuron density was reduced across most subregions and layers measured, with the exception of SLM (in all subfields), the SR layer of CA3c and CA3b, the SL layer of CA3b, and the SP layer of CA3c.
As the decrease in pCCK+ interneuron density may reflect a loss of pCCK protein rather than degeneration of pCCK-expressing interneurons, we next examined whether PILO-SE reduced the IPSP evoked by CCK+ interneurons in CA2 PNs. We took a pharmacological approach based on the distinct neuromodulatory profiles of PV+ and CCK+ interneurons, in which CCK+ interneurons, but not PV+ interneurons, express the cannabinoid type-1 (CB1) receptor that on activation by endocannabinoids acts presynaptically to suppress GABA release (Katona et al., 1999; Freund and Katona, 2007). Although CCK+ basket cells are not the only hippocampal interneuronal population that expresses CB1 receptors, by targeting the electrical stimulation to the SP we were able to primarily activate the perisomatic-targeting axons from CCK+ and PV+ basket cells, of which only the former express CB1 receptors (Katona et al., 1999; Wyeth et al., 2010). Because of this anatomic and molecular selectivity, CB1 receptor-mediated depression of perisomatic inhibition has been widely used as a proxy to assess CCK+ basket cell function (Wyeth et al., 2010; Sun et al., 2014; Valero et al., 2015; Drexel et al., 2017). To prevent any modulatory contribution to synaptic transmission of CB1 receptors expressed in excitatory neurons, these experiments were performed in the continual presence of glutamate receptor blockers.
We first used electrical stimulation in the SP layer to evoke direct monosynaptic IPSPs in CA2 PNs. We then applied the CB1 agonist WIN, which selectively suppresses GABA release from CCK+ interneurons, revealing a WIN-insensitive component of inhibition from PV+ interneurons and other interneuronal populations (including any CCK+ interneuron-mediated inhibition not fully suppressed by WIN; Fig. 9A,B). By measuring the difference between the initial baseline IPSP and the remaining component of the IPSP in the presence of WIN, we obtained the WIN-sensitive component of the IPSP, which provided a measure of the CCK+ interneuron component of the baseline IPSP.
In CA2 PNs from control mice, application of WIN induced a gradual depression of the SP-evoked IPSP that stabilized after ∼20 min (Fig. 9B,C), resulting in a significant decrease in IPSP amplitude (Fig. 9D; Wilcoxon test; p < 0.0001; n = 17 cells from 13 control mice). Consistent with the decrease in pCCK+ interneuron density we observed in PILO-SE mice, the absolute magnitude of the WIN-sensitive component of the IPSP was reduced in PILO-SE mice, from −1.1 ± 0.2 mV in control cells to only −0.2 ± 0.1 mV in cells from PILO-SE mice (n = 14 cells from 13 control mice, n = 15 cells from 11 PILO-SE mice). Thus, PILO-SE reduced the magnitude of the WIN-sensitive component of the IPSP by 81.7 ± 7.0% relative to its value in control animals (Mann–Whitney test; p < 0.0001). However, although greatly reduced in amplitude, the effect of WIN on inhibition of the IPSP was still significant in PILO-SE mice (Fig. 9B,C,E; Wilcoxon test; p = 0.0155; n = 16 cells from 12 PILO-SE mice). Whereas the WIN-sensitive component comprised 27.4 ± 4.4% of the total IPSP in control cells, it comprised only 9.6 ± 2.8% of the IPSP in PILO-SE, a decrease to 35% of its value in control animals (Fig. 9F; Mann–Whitney; p = 0.0012; n = 17 cells from 13 control mice, 16 cells from 12 PILO-SE mice).
As reported previously (Whitebirch et al., 2022) and above, the baseline (pre-WIN) amplitude of the IPSP was significantly smaller in PILO-SE mice compared with control mice (Fig. 9H; Mann–Whitney; p = 0.0043; n = 14 cells from 13 control mice, n = 15 cells from 11 PILO-SE mice). Importantly, the amplitude of the IPSP in control mice in the presence of WIN was not significantly different from that in PILO-SE mice, either in the absence or presence of WIN [Fig. 9G,H; Mann–Whitney test; control (WIN) vs PILO-SE (baseline), p = 0.4509; control (WIN) vs PILO-SE (WIN), p = 0.1861; n = 14 cells from 13 control mice, n = 15 cells from 11 PILO-SE mice]. Although the equivalence of the IPSPs could be coincidental, the simplest explanation is that PILO-SE leads to a more or less selective decrease in the CCK+ interneuron-mediated IPSP recorded in CA2 PNs, which is approximately equivalent to the effect of CB1 receptor agonist in control mice, thereby occluding the effect of WIN application.
Selective reduction in the labeling intensity of molecular markers for CCK+ but not PV+ inhibitory axon terminals in CA2 of PILO-SE mice
In principle, the decrease in the WIN-sensitive component of the IPSP in PILO-SE mice reported above could reflect a loss of sensitivity to the CB1 agonist, a loss of CB1 receptors, and/or a decrease in the number of CCK+ interneurons and, thus, the CCK+ interneuron mediated component of the IPSP. To address these possibilities, we performed immunohistochemistry to measure CB1 receptor levels. To limit our measurements to putative inhibitory terminals, we performed double labeling with anti-CB1R and anti-GAD67 antibodies, using the GAD67 signal to design a mask and thus isolate an ROI corresponding to perisomatic axon terminals (see Materials and Methods). Use of a GAD67-defined ROI also allowed for the normalization of any signal decrease because of thinning of the pyramidal layer in PILO-SE tissue. We found a marked decrease in CB1 receptor labeling in tissue from PILO-SE mice compared with control animals (nested t test; p = 0.0004; n = 20 sections from 5 control mice, n = 16 sections from four PILO-SE mice), which was notable throughout the CA1, CA2, and CA3 pyramidal layer (Fig. 10A1–B3; mean ± SEM: 91 ± 3 a.u. in control, and 42 ± 3 a.u. in PILO-SE mice, corresponding to a 54.5% decrease).
As a loss of CB1 receptor labeling may be because of a loss of protein expression rather than an actual loss of axon terminals, we next performed labeling experiments using an antibody directed to VGluT3, a specific marker for CCK+ interneuron terminals in the hippocampus (Somogyi et al., 2004). Using the same double labeling approach described above, we again found a significant decrease in VGluT3 fluorescence intensity (nested t test; p = 0.0002; n = 16 sections from four control mice, n = 12 sections from three PILO-SE mice) within the GAD67-defined ROI in PILO-SE mice compared with control mice (Fig. 10C1–D3; mean ± SEM: 109 ± 4 a.u. in control, and 55 ± 3 a.u. in PILO-SE mice, corresponding to a 49.7% decrease). In stark contrast, measurement of PV fluorescence intensity within the GAD67-defined ROI revealed no significant difference between control and PILO-SE tissue, even when measurements were specifically limited to area CA2 by virtue of triple labeling with the PCP4 or RGS14 CA2 markers (Fig. 4A1–B2; mean ± SEM: 71 ± 4 a.u. in control, and 77 ± 3 a.u. in PILO-SE mice; nested t test; p = 0.4391; n = 28 sections from 7 control mice, n = 28 sections from 7 PILO-SE mice). Together, these data strongly suggest that a selective loss of CCK+ interneurons from the CA2 subfield contributes to the inhibitory impairment in epileptic mice following PILO-SE.
Discussion
Surviving CA2 pyramidal neurons in PILO-SE epileptic mice exhibit impaired perisomatic inhibition
Accumulating evidence indicates that impaired balance and coordination in inhibitory–excitatory networks is a fundamental feature of epilepsy in many different brain regions. Prior reports from human TLE tissue have indicated a loss of inhibitory synaptic transmission in the CA2 subfield (Williamson and Spencer, 1994; Wittner et al., 2009), an area that we found contributes to seizure frequency in the PILO-SE mouse model of TLE (Whitebirch et al., 2022). Consistent with the human findings, our prior study in the PILO-SE mouse model of TLE revealed a widespread loss of inhibition in CA2 circuits. Thus, stimulation of the CA2/CA3 collaterals, CA2 collaterals, or granule cell mossy fibers evoked significantly smaller inhibitory responses in CA2 PNs from PILO-SE animals compared with controls (Whitebirch et al., 2022). Furthermore, feedforward inhibitory responses recorded in CA3 and CA1 PNs evoked by selective photostimulation of CA2 axons were also smaller in PILO-SE mice (Whitebirch et al., 2022). Here we sought to uncover the underlying causes of this inhibitory deficit, reasoning that a decrease in inhibition could result from a number of mechanisms that disrupt distinct components of the feedforward inhibitory circuits, including depletion of specific interneuronal populations and/or functional reduction in synaptic inhibition.
When we pharmacologically abolished excitatory neurotransmission and directly evoked monosynaptic inhibition in CA2 PNs, we found a loss of inhibition in slices from PILO-SE mice that was most apparent in the early phase of the evoked postsynaptic potentials, suggesting a selective impairment in fast GABAA-mediated inhibitory currents (Fig. 3). We previously observed a similar loss of fast inhibition of CA2 PNs recruited by SR stimulation in the absence of excitatory blockade (Whitebirch et al., 2022). A loss of fast inhibition could result from depletion or dysfunction of interneurons that provide synaptic input to PN somata and proximal dendrites, including (though not limited to) perisomatic-targeting interneurons, such as PV+ and CCK+ classes of basket cells (Botcher et al., 2014). We observed an increased paired-pulse ratio in the PILO-SE mice consistent with a decrease in the probability of GABA release from inhibitory cell presynaptic terminals, suggesting that a functional alteration at surviving interneuronal synapses may contribute to the decreased level of inhibition. Alternatively, the increase in the paired-pulse ratio could occur if a surviving population of inhibitory neurons had an inherently lower release probability compared to the neurons that were lost following PILO-SE. It is important to note that our data do not exclude the possibility that an indirect action on disynaptic inhibition resulting from a decrease in the ability of excitatory afferents to effectively recruit interneurons may additionally contribute to the impaired feedforward inhibition of CA2 PNs we previously reported (Whitebirch et al., 2022).
Notably, we did not observe a significant reduction in inhibition evoked by stimulation of the direct entorhinal cortical inputs to CA2 using a stimulating electrode placed in SLM near the PN distal apical dendrites. This preservation of the SLM-evoked IPSP, which was characterized by a predominance of slow, likely GABAB receptor-mediated inhibition (Capogna, 2011), may reflect the preferential survival of interneuronal populations that target PN distal apical dendrites such as the neurogliaform cells (Capogna, 2011). Conversely, the clear reduction in SP- and SR-evoked IPSP amplitudes may reflect a selective loss or impairment of interneuron populations that target those layers of the hippocampus, including SP-SR interneurons (Mercer et al., 2012b), bistratified cells (Mercer et al., 2007), and PV+ or CCK+ basket cells (Mercer et al., 2012a). Although it is possible that some PV+ or CCK+ interneurons may also be recruited by SLM stimulation, relatively few of the somata or dendrites of these interneuron classes are found in the SLM (Botcher et al., 2014).
Cholecystokinin-expressing interneurons are vulnerable in epileptic mice, and their loss may compromise inhibitory control of CA2 excitability
Whereas our immunohistochemistry results revealed a relatively small decrease in the number of PV+ cells in CA1, CA2, or CA3 fields in PILO-SE mice, we observed a striking reduction in pCCK-labeled cell density. In addition, we found a strong decrease in labeling for CB1R and VGluT3, markers of CCK+ inhibitory neuron presynaptic boutons (Katona et al., 1999; Somogyi et al., 2004; Freund and Katona, 2007), throughout the pyramidal layers of the hippocampus, including CA2. In complementary studies, we found a large (threefold), statistically significant decrease in the magnitude of the CCK+ interneuron component of the IPSP recorded from CA2 PNs, based on the magnitude of the effect of the CB1 receptor agonist WIN to inhibit the IPSP evoked by SR stimulation (Fig. 9).
Although the reduced labeling for CB1 receptors and response to WIN could result from a reduction in CB1R expression (Karlócai et al., 2011), rather than a loss of CCK+ INs, our finding of a strong reduction in the CCK+ IN presynaptic marker VGluT3 strongly argues for a true decrease in CCK+ IN density. Moreover, our finding that the magnitude of the CA2 PN IPSP in the presence of WIN in control animals was similar in size to the CA2 PN IPSP in the presence of WIN in PILO-SE animals argues for a decrease in the size of the CCK+ interneuron-dependent component of the IPSP in PILO-SE, rather than just a loss of WIN sensitivity. Furthermore, these electrophysiological results suggest than any effect of PILO-SE to decrease inhibition mediated by PV+ interneurons must be relatively small, otherwise the IPSP in WIN would be smaller in PILO-SE mice than in control mice, consistent with the relatively small change in the intensity of labeling for parvalbumin in the inhibitory axon terminals surrounding PN somas. However, our results do not rule out the possibility that factors other than a loss of CCK+ interneurons may make some contribution to the decreased inhibition in the PILO-SE mice.
Further support for a decrease in magnitude of the inhibition from CCK+ interneurons comes from a consideration of the effect of PILO-SE on the time course of decay of the inhibitory response recorded in CA2. Although both PV+ and CCK+ basket cells target the perisomatic region, CCK+ interneurons evoke an IPSP that exhibits a distinctly slower time course of decay, due in part to slow, asynchronous GABA release, which stands in sharp contrast to the rapid and highly synchronous dynamics of PV+ interneuron-mediated inhibition (Hefft and Jonas, 2005; Basu et al., 2013). A selective loss of CCK+ interneurons is consistent with our recent finding that the IPSCs in CA2 PNs from PILO-SE animals decay more rapidly than the IPSCs in control cells (Whitebirch et al., 2022), and with our present finding that IPSPs evoked by SP stimulation exhibit a more rapid 10–90% rise time in cells from PILO-SE mice. Thus, the most parsimonious interpretation of our combined immunohistochemical and electrophysiological data are that there is a significant reduction in the contribution of CCK+ interneurons to the IPSP, possibly because of the degeneration of CCK+ interneurons, consistent with several recent investigations in chemoconvulsant-treated animals (Wyeth et al., 2010; Sun et al., 2014; Khan et al., 2018; Kang et al., 2021). Notably, Wyeth et al. (2010) reported degeneration of CCK+ and CB1R+ synaptic boutons in the CA1 pyramidal cell layer after PILO-SE in mice, while, in contrast, PV+ perisomatic boutons were conserved (Wyeth et al., 2010). While our measurements of pCCK+ interneuron density are comparable to those previously reported (Botcher et al., 2014), direct comparisons across studies are difficult because of the limited availability of effective antibodies that selectively label CCK+ interneurons and methodological differences across laboratories.
In light of the recent evidence linking CB1R-mediated inhibitory plasticity in CA2 CCK+ interneurons to social memory in mice (Loisy et al., 2022), it is intriguing to consider that a loss of CCK+ interneurons in epileptic mice may contribute to social behavioral comorbidities associated with TLE in human patients and animal models of seizures (Ito et al., 2007; Seo et al., 2013; Lopes et al., 2016; Okruszek et al., 2017; Steiger and Jokeit, 2017; Smolensky et al., 2019). Additionally, recent calcium imaging experiments from Dudok et al. (2021) revealed selective recruitment of CCK+ interneurons and CA2 axons in the CA1 subfield during non-theta, non-SWR desynchronized network activity. These desynchronized network states, which occurred at the transition from locomotion to quiet immobility, were characterized by high levels of CCK+ interneuron activity, widespread inhibition of PN activity, and low levels of PV+ interneuron activity, with the latter fostered in part by reciprocal inhibition between CCK+ interneurons and PV+ interneurons (Caccavano and McBain, 2021; Dudok et al., 2021; Vancura et al., 2023).
The PV+ interneuron network appears relatively preserved in the epileptic hippocampus of our PILO-SE mice, and past reports of PV+ interneuron loss may in fact reflect decreases in PV expression (Wittner and Maglóczky, 2017). Thus, we speculate that a selective loss of CCK+ interneurons in TLE could contribute to disinhibition of surviving PV+ interneurons and permit their excessive activation by PNs, including those in the resilient CA2 subfield. An overactivated PV+ interneuron network may promote epileptiform synchronization among PNs through postinhibitory rebound spiking (de Castro Medeiros et al., 2020) or accumulation of intracellular chloride leading to depolarizing GABAergic signaling (Huberfeld et al., 2007; Ellender et al., 2014; Hamidi and Avoli, 2015). This model is consistent with the reports of increased interneuron activity preceding seizures (Avoli and de Curtis, 2011; Muldoon et al., 2015; Neumann et al., 2017; Rich et al., 2020), including PV+ interneurons specifically (Miri et al., 2018), and the observation that modulation of PV+ interneurons can trigger or sustain seizures (Sessolo et al., 2015; Shiri et al., 2016; Lévesque et al., 2019).
Like CA2 PNs, CA1 PNs in the deep stratum pyramidale sublayer receive strong input from PV+ interneurons (Ribak et al., 1993; Botcher et al., 2014; Lee et al., 2014; Valero et al., 2015). Thus, CA2 and deep CA1 PNs might have the greatest probability of synchronization during periods of intense neural activity. The particularly high density of PV+ interneurons in CA2, including wide-arbor PV+ basket cells and bistratified cells, could promote synchronization of CA2 PNs with nearby CA3a and CA1c neurons (Mercer et al., 2007, 2012a). Synchronous population discharges often originate from distal CA3 and CA2 (Wong and Traub, 1983; Wittner and Miles, 2007; McCloskey and Scharfman, 2011; Oliva et al., 2016), indicating that the CA2 network has an inherent tendency toward population synchronization even in nonepileptic tissue. Thus, CCK+ interneuron impairment and other sequelae of PILO-SE may act in concert with the dense recurrent excitatory circuitry, strong entorhinal cortex input, and PV+ interneuron network in CA2 to create a powerful focus of epileptiform synchronization. Although our pharmacological experiments with WIN suggest a selective decrease in CCK+ interneuron input to CA2 PNs (Fig. 9), in the future it will be important to examine more directly the potential role of CCK+ interneuron function in seizure generation through selective optogenetic or chemogenetic manipulations, as enabled by the recently developed Sncg-Flp transgenic mouse line (Dudok et al., 2021).
CA2 PV+ INs appear relatively resilient in epileptic mice despite PNN disruption following PILO-SE
A previous quantitative analysis of PV+ interneurons in human TLE tissue indicated their widespread loss within the sclerotic hippocampus, including from the CA2 region (Andrioli et al., 2007). Intriguingly, CA2 also showed a significant decrease in PV+ interneuron density even in nonsclerotic tissue (Andrioli et al., 2007). However, when we stained for inhibitory markers to assess the survival of PV+ interneurons in PILO-SE mice, we found only a modest reduction in PV+ interneuron density in CA2 (Fig. 4). Moreover, the small decrease in PV+ interneuron density may reflect a loss of PV immunoreactivity, rather than a true loss of PV+ interneurons, as suggested by various studies in human TLE tissue (Wittner and Maglóczky, 2017). Additionally, our measurements of WIN-sensitive perisomatic inhibition (Fig. 9) suggested that the loss of inhibition in PILO-SE mice may largely be explained by a loss or functional impairment of CCK+ INs. While our PV+ IN density measurements from control mice were lower than in several past reports (Botcher et al., 2014; Piskorowski et al., 2016), this discrepancy may reflect differences in methodology across studies or differences in the particular anti-PV antibodies used.
The CA2 subfield contains a unique PN-associated PNN that has been implicated in the regulation of inhibitory synaptic plasticity at the PV+ interneuron synapses onto CA2 PNs (Carstens et al., 2016, 2021; Domínguez et al., 2019). Additionally, PNNs are negatively regulated by CA2 PN activity levels, such that prolonged chemogenetic activation of CA2 PNs led to a decrease in PNN deposition within the CA2 region (Carstens et al., 2021). Here, we report decreased staining for CA2 PNNs in tissue from epileptic mice (Fig. 5), which may reflect increased levels of CA2 PN activity leading to a downregulation of PNN deposition following PILO-SE. The CA2 PNN may be additionally impacted by seizure-associated degradation of PNN components (McRae et al., 2012; Rankin-Gee et al., 2015; Dubey et al., 2017; Carstens et al., 2021). Intriguingly, recent data indicate that the CA2 PNN is decreased in the Kcna1−/− genetic model of epilepsy in mice (Carstens et al., 2021), which also presents with a pattern of mesial temporal sclerosis-like damage (Wenzel et al., 2007), suggesting similar dysregulation of extracellular matrix components across epilepsy models.
Although our immunohistochemical measures reveal a clear decrease in the CA2 PNN, our electrophysiological data do not suggest a substantial disruption of perisomatic inhibition mediated by PV+ INs. It is possible that, despite recent evidence indicating that the PNN ensheathes PV+ perisomatic synapses onto CA2 PNs (Domínguez et al., 2019), the PNN does not contribute to the baseline magnitude of perisomatic inhibition meditated by PV+ basket cells. Rather, the PNN may be specifically involved in orchestrating inhibitory synaptic plasticity in CA2, notably the δ opioid receptor-mediated inhibitory long-term depression linked to social memory in mice (Carstens et al., 2016, 2021; Domínguez et al., 2019; Cope et al., 2022; Leroy et al., 2022; Rey et al., 2022). The PNN has also been linked to intrinsic physiological properties of ensheathed neurons (Balmer, 2016; Tewari et al., 2018) and may influence the local concentration of extracellular chloride (Glykys et al., 2014). Thus, it is possible that downregulation of CA2 PNNs represents a homeostatic outcome of recurring seizures that may have important implications for neuronal excitability in the CA2 region, such as the decreased resting conductance we observed in CA2 PNs after PILO-SE (Whitebirch, 2022). At present, however, the functional consequences of CA2 PNN loss are unclear and remain an important subject for future investigation.
Footnotes
This work was supported by National Institutes of Health Grants R01-NS-106983 (Principal Inestigators, H.E.S. and S.A.S.) and Grant F31-NS-113466 (A.C.W.).
References
- Alexander GM, Brown LY, Farris S, Lustberg D, Pantazis C, Gloss B, Plummer NW, Jensen P, Dudek SM (2018) CA2 neuronal activity controls hippocampal low gamma and ripple oscillations. Elife 7:1–25. 10.7554/eLife.38052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrioli A, Alonso-Nanclares L, Arellano JI, DeFelipe J (2007) Quantitative analysis of parvalbumin-immunoreactive cells in the human epileptic hippocampus. Neuroscience 149:131–143. 10.1016/j.neuroscience.2007.07.029 [DOI] [PubMed] [Google Scholar]
- Asadi-Pooya AA, Stewart GR, Abrams DJ, Sharan A (2017) Prevalence and incidence of drug-resistant mesial temporal lobe epilepsy in the United States. World Neurosurg 99:662–666. 10.1016/j.wneu.2016.12.074 [DOI] [PubMed] [Google Scholar]
- Avoli M, de Curtis M (2011) GABAergic synchronization in the limbic system and its role in the generation of epileptiform activity. Prog Neurobiol 95:104–132. 10.1016/j.pneurobio.2011.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer TS (2016) Perineuronal nets enhance the excitability of fast-spiking neurons. eNeuro 3:ENEURO.0112-16.2016–13. 10.1523/ENEURO.0112-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankstahl M, Müller CJ, Wilk E, Schughart K, Löscher W (2012) Generation and characterization of pilocarpine-sensitive C57BL/6 mice as a model of temporal lobe epilepsy. Behav Brain Res 230:182–191. 10.1016/j.bbr.2012.02.004 [DOI] [PubMed] [Google Scholar]
- Basu J, Srinivas K, Cheung S, Taniguchi H, Huang ZJ, Siegelbaum S (2013) A cortico-hippocampal learning rule shapes inhibitory microcircuit activity to enhance hippocampal information flow. Neuron 79:1208–1221. 10.1016/j.neuron.2013.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud MO, Kleen JK, Mirro EA, Andrechak JC, King-Stephens D, Chang EF, Rao VR (2018) Multi-day rhythms modulate seizure risk in epilepsy. Nat Commun 9:1–10. 10.1038/s41467-017-02577-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud MO, Ghestem A, Benoliel JJ, Becker C, Bernard C (2019) Endogenous multidien rhythm of epilepsy in rats. Exp Neurol 315:82–87. 10.1016/j.expneurol.2019.02.006 [DOI] [PubMed] [Google Scholar]
- Blümcke I, et al. (2013) International consensus classification of hippocampal sclerosis in temporal lobe epilepsy: a task force report from the ILAE commission on diagnostic methods. Epilepsia 54:1315–1329. 10.1111/epi.12220 [DOI] [PubMed] [Google Scholar]
- Boehringer R, Polygalov D, Huang AJY, Middleton SJ, Robert V, Wintzer ME, Piskorowski RA, Chevaleyre V, McHugh TJ (2017) Chronic loss of CA2 transmission leads to hippocampal hyperexcitability. Neuron 94:642–655.e9. 10.1016/j.neuron.2017.04.014 [DOI] [PubMed] [Google Scholar]
- Botcher NA, Falck JE, Thomson AM, Mercer A (2014) Distribution of interneurons in the CA2 region of the rat hippocampus. Front Neuroanat 8:104. 10.3389/fnana.2014.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccavano AP, McBain CJ (2021) Timing isn’t everything: opposing roles for perisomatic inhibition. Neuron 109:911–913. 10.1016/j.neuron.2021.02.022 [DOI] [PubMed] [Google Scholar]
- Capogna M (2011) Neurogliaform cells and other interneurons of stratum lacunosum-moleculare gate entorhinal-hippocampal dialogue. J Physiol 589:1875–1883. 10.1113/jphysiol.2010.201004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens KE, Phillips ML, Pozzo-Miller L, Weinberg RJ, Dudek SM (2016) Perineuronal nets suppress plasticity of excitatory synapses on CA2 pyramidal neurons. J Neurosci 36:6312–6320. 10.1523/JNEUROSCI.0245-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens KE, Alexander GM, Dudek SM (2021) Perineuronal net degradation rescues CA2 plasticity in Rett syndrome model mice. J Clin Invest 131:e137221. 10.1172/JCI137221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalheiro EA, Santos NF, Priel MR (1996) The pilocarpine model of epilepsy in mice. Epilepsia 37:1015–1019. 10.1111/j.1528-1157.1996.tb00541.x [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Siegelbaum SA (2010) Strong CA2 pyramidal neuron synapses define a powerful disynaptic cortico-hippocampal loop. Neuron 66:560–572. 10.1016/j.neuron.2010.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cope EC, Zych AD, Katchur NJ, Waters RC, Laham BJ, Diethorn EJ, Park CY, Meara WR, Gould E (2022) Atypical perineuronal nets in the CA2 region interfere with social memory in a mouse model of social dysfunction. Mol Psychiatry 27:3520–3531. 10.1038/s41380-021-01174-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Z, Gerfen CR, Young WS (2013) Hypothalamic and other connections with dorsal CA2 area of the mouse hippocampus. J Comp Neurol 521:1844–1866. 10.1002/cne.23263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Castro Medeiros D, Cota VR, Oliveira ACP, Moreira FA, Moraes MFD (2020) The endocannabinoid system activation as a neural network desynchronizing mediator for seizure suppression. Front Behav Neurosci 14:603245. 10.3389/fnbeh.2020.603245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domínguez S, Rey CC, Therreau L, Fanton A, Massotte D, Verret L, Piskorowski RA, Chevaleyre V (2019) Maturation of PNN and ErbB4 signaling in area CA2 during adolescence underlies the emergence of PV interneuron plasticity and social memory. Cell Rep 29:1099–1112.e4. 10.1016/j.celrep.2019.09.044 [DOI] [PubMed] [Google Scholar]
- Drexel M, Romanov RA, Wood J, Weger S, Heilbronn R, Wulff P, Tasan RO, Harkany T, Sperk G (2017) Selective silencing of hippocampal parvalbumin interneurons induces development of recurrent spontaneous limbic seizures in mice. J Neurosci 37:8166–8179. 10.1523/JNEUROSCI.3456-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey D, Mcrae PA, Rankin-gee EK, Baranov E, Wandrey L, Rogers S, Porter BE (2017) Increased metalloproteinase activity in the hippocampus following status epilepticus. Epilepsy Res 132:50–58. 10.1016/j.eplepsyres.2017.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek SM, Alexander GM, Farris S (2016) Rediscovering area CA2: unique properties and functions. Nat Rev Neurosci 17:89–102. 10.1038/nrn.2015.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudok B, Klein PM, Hwaun E, Lee BR, Yao Z, Fong O, Bowler JC, Terada S, Sparks FT, Szabo GG, Farrell JS, Berg J, Daigle TL, Tasic B, Dimidschstein J, Fishell G, Losonczy A, Zeng H, Soltesz I (2021) Alternating sources of perisomatic inhibition during behavior. Neuron 109:997–1012.e9. 10.1016/j.neuron.2021.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellender TJ, Raimondo JV, Irkle A, Lamsa KP, Akerman CJ (2014) Excitatory effects of parvalbumin-expressing interneurons maintain hippocampal epileptiform activity via synchronous afterdischarges. J Neurosci 34:15208–15222. 10.1523/JNEUROSCI.1747-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favuzzi E, Marques-Smith A, Deogracias R, Winterflood CM, Sánchez-Aguilera A, Mantoan L, Maeso P, Fernandes C, Ewers H, Rico B (2017) Activity-dependent gating of parvalbumin interneuron function by the perineuronal net protein brevican. Neuron 95:639–655.e10. 10.1016/j.neuron.2017.06.028 [DOI] [PubMed] [Google Scholar]
- Fernandez-Lamo I, Gomez-Dominguez D, Sanchez-Aguilera A, Oliva A, Morales AV, Valero M, Cid E, Berenyi A, Menendez de la Prida L (2019) Proximodistal organization of the CA2 hippocampal area. Cell Rep 26:1734–1746.e6. 10.1016/j.celrep.2019.01.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiman TM, Häussler U, Zentner J, Doostkam S, Beck J, Scheiwe C, Brandt A, Haas CA, Puhahn‐Schmeiser B (2021) Mossy fiber sprouting into the hippocampal region CA2 in patients with temporal lobe epilepsy. Hippocampus 31:580–592. 10.1002/hipo.23323 [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I (2007) Perisomatic inhibition. Neuron 56:33–42. 10.1016/j.neuron.2007.09.012 [DOI] [PubMed] [Google Scholar]
- Glykys J, Dzhala V, Egawa K, Balena T, Saponjian Y, Kuchibhotla KV, Bacskai BJ, Kahle KT, Zeuthen T, Staley KJ (2014) Local impermeant anions establish the neuronal chloride concentration. Science 343:670–675. 10.1126/science.1245423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidi S, Avoli M (2015) KCC2 function modulates in vitro ictogenesis. Neurobiol Dis 79:51–58. 10.1016/j.nbd.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haut SR, Shinnar S, Moshé SL (2005) Seizure clustering: risks and outcomes. Epilepsia 46:146–149. 10.1111/j.0013-9580.2005.29004.x [DOI] [PubMed] [Google Scholar]
- He H, Boehringer R, Huang AJY, Overton ETN, Polygalov D, Okanoya K, Mchugh TJ (2021) CA2 inhibition reduces the precision of hippocampal assembly reactivation. Neuron 109:3674–3687.e7. 10.1016/j.neuron.2021.08.034 [DOI] [PubMed] [Google Scholar]
- Hefft S, Jonas P (2005) Asynchronous GABA release generates long-lasting inhibition at a hippocampal interneuron-principal neuron synapse. Nat Neurosci 8:1319–1328. 10.1038/nn1542 [DOI] [PubMed] [Google Scholar]
- Hitti FL, Siegelbaum SA (2014) The hippocampal CA2 region is essential for social memory. Nature 508:88–92. 10.1038/nature13028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huberfeld G, Wittner L, Baulac M, Kaila K, Miles R, Rivera C (2007) Perturbed chloride homeostasis and GABAergic signaling in human temporal lobe epilepsy. J Neurosci 27:9866–9873. 10.1523/JNEUROSCI.2761-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka N, Weber J, Amaral D (1990) Organization of intrahippocampal projections originating from CA3 pyramidal cells in the rat. J Comp Neurol 295:580–623. 10.1002/cne.902950407 [DOI] [PubMed] [Google Scholar]
- Ito M, Okazaki M, Takahashi S, Muramatsu R, Kato M, Onuma T (2007) Subacute postictal aggression in patients with epilepsy. Epilepsy Behav 10:611–614. 10.1016/j.yebeh.2007.02.016 [DOI] [PubMed] [Google Scholar]
- Jain S, LaFrancois JJ, Botterill JJ, Alcantara-Gonzalez D, Scharfman HE (2019) Adult neurogenesis in the mouse dentate gyrus protects the hippocampus from neuronal injury following severe seizures. Hippocampus 29:683–709. 10.1002/hipo.23062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YJ, Clement EM, Park IH, Greenfield LJ, Smith BN, Lee SH (2021) Vulnerability of cholecystokinin-expressing GABAergic interneurons in the unilateral intrahippocampal kainate mouse model of temporal lobe epilepsy. Exp Neurol 342:113724. 10.1016/j.expneurol.2021.113724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur M, Ganguly A, Nagy G, Adamson SI, Chuang JH, Frankel WN, Ackerman SL (2020) Expression of the neuronal tRNA n-Tr20 regulates synaptic transmission and seizure susceptibility. Neuron 108:193–208.e9. 10.1016/j.neuron.2020.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlócai MR, Tóth K, Watanabe M, Ledent C, Juhász G, Freund TF, Maglóczky Z (2011) Redistribution of CB1 cannabinoid receptors in the acute and chronic phases of pilocarpine-induced epilepsy. PLoS One 6:e27196. 10.1371/journal.pone.0027196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlágh B, Sík A, Käfalvi A, Vizi ES, Mackie K, Freund TF (1999) Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19:4544–4558. 10.1523/JNEUROSCI.19-11-04544.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Shekh-Ahmad T, Khalil A, Walker MC, Ali AB (2018) Cannabidiol exerts antiepileptic effects by restoring hippocampal interneuron functions in a temporal lobe epilepsy model. Br J Pharmacol 175:2097–2115. 10.1111/bph.14202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler N, Kirchmair E, Schwarzer C, Sperk G (1997) Altered expression of NPY-Y 1 receptors in kainic acid induced epilepsy in rats. Neurosci Lett 230:129–132. 10.1016/s0304-3940(97)00492-8 [DOI] [PubMed] [Google Scholar]
- Kohara K, Pignatelli M, Rivest AJ, Jung H-Y, Kitamura T, Suh J, Frank D, Kajikawa K, Mise N, Obata Y, Wickersham IR, Tonegawa S (2014) Cell type-specific genetic and optogenetic tools reveal hippocampal CA2 circuits. Nat Neurosci 17:269–279. 10.1038/nn.3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan P, Brodie MJ (2000) Early identification of refractory epilepsy. N Engl J Med 342:314–319. 10.1056/NEJM200002033420503 [DOI] [PubMed] [Google Scholar]
- Lee S-H, Marchionni I, Bezaire M, Varga C, Danielson N, Lovett-Barron M, Losonczy A, Soltesz I (2014) Parvalbumin-positive basket cells differentiate among hippocampal pyramidal cells. Neuron 82:1129–1144. 10.1016/j.neuron.2014.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehr AB, Kumar A, Tetzlaff C, Hafting T, Fyhn M, St TM (2021) CA2 beyond social memory: evidence for a fundamental role in hippocampal information processing. Neurosci Biobehav Rev 126:398–412. 10.1016/j.neubiorev.2021.03.020 [DOI] [PubMed] [Google Scholar]
- Leroy F, Brann DH, Meira T, Siegelbaum SA (2017) Input-timing-dependent plasticity in the hippocampal CA2 region and its potential role in social memory. Neuron 95:1089–1102.e5. 10.1016/j.neuron.2017.07.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, Park J, Asok A, Brann DH, Meira T, Boyle LM, Buss EW, Kandel ER, Siegelbaum SA (2018) A circuit from hippocampal CA2 to lateral septum disinhibits social aggression. Nature 564:213–218. 10.1038/s41586-018-0772-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leroy F, de Solis CA, Boyle LM, Bock T, Lofaro OM, Buss EW, Asok A, Kandel ER, Siegelbaum SA (2022) Enkephalin release from VIP interneurons in the hippocampal CA2/3a region mediates heterosynaptic plasticity and social memory. Mol Psychiatry 27:2879–2900. 10.1038/s41380-021-01124-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque M, Avoli M, Bernard C (2016) Animal models of temporal lobe epilepsy following systemic chemoconvulsant administration. J Neurosci Methods 260:45–52. 10.1016/j.jneumeth.2015.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévesque M, Chen LY, Etter G, Shiri Z, Wang S, Williams S, Avoli M (2019) Paradoxical effects of optogenetic stimulation in mesial temporal lobe epilepsy. Ann Neurol 86:714–728. 10.1002/ana.25572 [DOI] [PubMed] [Google Scholar]
- Li XG, Somogyi P, Ylinen A, Buzsáki G (1994) The hippocampal CA3 network: an in vivo intracellular labeling study. J Comp Neurol 339:181–208. 10.1002/cne.903390204 [DOI] [PubMed] [Google Scholar]
- Lisgaras CP, Scharfman HE (2023) High-frequency oscillations (250–500 Hz) in animal models of Alzheimer’s disease and two animal models of epilepsy. Epilepsia 64:231–246. 10.1111/epi.17462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisgaras CP, Oliva A, Mckenzie S, LaFrancois J, Siegelbaum SA, Scharman HE (2023) Hippocampal area CA2 controls seizure dynamics, interictal EEG abnormalities and social comorbidity in mouse models of temporal lobe epilepsy. bioRxiv 524149. 10.1101/2023.01.15.524149. 10.1101/2023.01.15.524149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisy M, Bouisset G, Lopez S, Muller M, Spitsyn A, Duval J, Piskorowski RA, Verret L, Chevaleyre V (2022) Sequential inhibitory plasticities in hippocampal area CA2 and social memory formation. Neuron 110:2854–2866.e4. 10.1016/j.neuron.2022.06.013 [DOI] [PubMed] [Google Scholar]
- Lopes MW, Lopes SC, Santos DB, Costa AP, Gonçalves FM, de Mello N, Prediger RD, Farina M, Walz R, Leal RB (2016) Time course evaluation of behavioral impairments in the pilocarpine model of epilepsy. Epilepsy Behav 55:92–100. 10.1016/j.yebeh.2015.12.001 [DOI] [PubMed] [Google Scholar]
- Lopez-Rojas J, de Solis CA, Leroy F, Kandel ER, Siegelbaum SA (2022) A direct lateral entorhinal cortex to hippocampal CA2 circuit conveys social information required for social memory. Neuron 110:1559–1572.e4. 10.1016/j.neuron.2022.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorente De Nó R (1934) Studies on the structure of the cerebral cortex. II. Continuation of the study of the ammonic system. J Für Psychologie Und Neurologie 46:113–177. [Google Scholar]
- Löscher W, Ferland RJ, Ferraro TN (2017) The relevance of inter- and intrastrain differences in mice and rats and their implications for models of seizures and epilepsy. Epilepsy Behav 73:214–235. 10.1016/j.yebeh.2017.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald CJ, Tonegawa S (2021) Crucial role for CA2 inputs in the sequential organization of CA1 time cells supporting memory. Proc Natl Acad Sci U S A 118:e2020698118. 10.1073/pnas.2020698118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marksteiner J, Ortler M, Bellmann R, Sperk G (1990) Neuropeptide Y biosynthesis is markedly induced in mossy fibers during temporal lobe epilepsy of the rat. Neurosci Lett 112:143–148. 10.1016/0304-3940(90)90193-d [DOI] [PubMed] [Google Scholar]
- Mazzuferi M, Kumar G, Rospo C, Kaminski RM (2012) Rapid epileptogenesis in the mouse pilocarpine model: video-EEG, pharmacokinetic and histopathological characterization. Exp Neurol 238:156–167. 10.1016/j.expneurol.2012.08.022 [DOI] [PubMed] [Google Scholar]
- McCloskey DP, Scharfman HE (2011) Progressive, potassium-sensitive epileptiform activity in hippocampal area CA3 of pilocarpine-treated rats with recurrent seizures. Epilepsy Res 97:92–102. 10.1016/j.eplepsyres.2011.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae PA, Baranov E, Rogers SL, Porter BE (2012) Persistent decrease in multiple components of the perineuronal net following status epilepticus. Eur J Neurosci 36:3471–3482. 10.1111/j.1460-9568.2012.08268.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meira T, Leroy F, Buss EW, Oliva A, Park J, Siegelbaum SA (2018) A hippocampal circuit linking dorsal CA2 to ventral CA1 critical for social memory dynamics. Nat Commun 9:4163. 10.1038/s41467-018-06501-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer A, Trigg HL, Thomson AM (2007) Characterization of neurons in the CA2 subfield of the adult rat hippocampus. J Neurosci 27:7329–7338. 10.1523/JNEUROSCI.1829-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer A, Eastlake K, Trigg HL, Thomson AM (2012a) Local circuitry involving parvalbumin-positive basket cells in the CA2 region of the hippocampus. Hippocampus 22:43–56. 10.1002/hipo.20841 [DOI] [PubMed] [Google Scholar]
- Mercer A, Botcher NA, Eastlake K, Thomson AM (2012b) SP-SR interneurones: a novel class of neurones of the CA2 region of the hippocampus. Hippocampus 22:1758–1769. 10.1002/hipo.22010 [DOI] [PubMed] [Google Scholar]
- Miri M, Vinck M, Pant R, Cardin J (2018) Altered hippocampal interneuron activity precedes ictal onset. Elife 7:e40750. 10.7554/eLife.40750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modi B, Pimpinella D, Pazienti A, Zacchi P, Cherubini E, Griguoli M (2019) Possible implication of the CA2 hippocampal circuit in social cognition deficits observed in the neuroligin 3 knock-out mouse, a non-syndromic animal model of autism. Front Psychiatry 10:1–16. 10.3389/fpsyt.2019.00513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muldoon SF, Villette V, Tressard T, Malvache A, Reichinnek S, Bartolomei F, Cossart R (2015) GABAergic inhibition shapes interictal dynamics in awake epileptic mice. Brain 138:2875–2890. 10.1093/brain/awv227 [DOI] [PubMed] [Google Scholar]
- Neumann AR, Raedt R, Steenland HW, Sprengers M, Bzymek K, Navratilova Z, Mesina L, Xie J, Lapointe V, Kloosterman F, Vonck K, Boon PAJM, Soltesz I, McNaughton BL, Luczak A (2017) Involvement of fast-spiking cells in ictal sequences during spontaneous seizures in rats with chronic temporal lobe epilepsy. Brain 140:2355–2369. 10.1093/brain/awx179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi A, Matsumoto N, Morikawa S, Tamura H, Ikegaya Y (2017) Juvenile hippocampal CA2 region expresses aggrecan. Front Neuroanat 11:41. 10.3389/fnana.2017.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Ikegaya Y (2019) Recurrent connections between CA2 pyramidal cells. Hippocampus 29:305–312. 10.1002/hipo.23064 [DOI] [PubMed] [Google Scholar]
- Okruszek Ł, Bala A, Wordecha M, Jarkiewicz M, Wysokiński A, Szczepocka E, Piejka A, Zaborowska O, Szantroch M, Rysz A, Marchel A (2017) Social cognition in neuropsychiatric populations: a comparison of theory of mind in schizophrenia and mesial temporal lobe epilepsy. Sci Rep 7:484. 10.1038/s41598-017-00565-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva A, Fernández-Ruiz A, Buzsáki G, Berényi A (2016) Role of hippocampal CA2 region in triggering sharp-wave ripples. Neuron 91:1342–1355. 10.1016/j.neuron.2016.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva A, Fernández-Ruiz A, Leroy F, Siegelbaum SA (2020) Hippocampal CA2 sharp-wave ripples reactivate and promote social memory. Nature 587:264–269. 10.1038/s41586-020-2758-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva A, Fernandez-Ruiz A, Karaba LA (2023) CA2 orchestrates hippocampal network dynamics. Hippocampus 33:241–251. 10.1002/hipo.23495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, Siegelbaum SA, Gogos JA, Chevaleyre V (2016) Age-dependent specific changes in area CA2 of the hippocampus and social memory deficit in a mouse model of the 22q11.2 deletion syndrome. Neuron 89:163–176. 10.1016/j.neuron.2015.11.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitsch J, Becker AJ, Schoch S, Müller JA, de Curtis M, Gnatkovsky V (2017) Circadian clustering of spontaneous epileptic seizures emerges after pilocarpine-induced status epilepticus. Epilepsia 58:1159–1171. 10.1111/epi.13795 [DOI] [PubMed] [Google Scholar]
- Racine RJ (1972) Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr Clin Neurophysiol 32:281–294. 10.1016/0013-4694(72)90177-0 [DOI] [PubMed] [Google Scholar]
- Rankin-Gee EK, Mcrae PA, Baranov E, Rogers S, Wandrey L, Porter BE (2015) Perineuronal net degradation in epilepsy. Epilepsia 56:1124–1133. 10.1111/epi.13026 [DOI] [PubMed] [Google Scholar]
- Rey CC, Robert V, Bouisset G, Loisy M, Lopez S, Cattaud V, Lejards C, Piskorowski RA, Rampon C, Chevaleyre V, Verret L (2022) Altered inhibitory function in hippocampal CA2 contributes in social memory deficits in Alzheimer’s mouse model. IScience 25:103895. 10.1016/j.isci.2022.103895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribak CE, Seress L, Leranth C (1993) Electron microscopic immunocytochemical study of the distribution of parvalbumin‐containing neurons and axon terminals in the primate dentate gyrus and Ammon’s horn. J Comp Neurol 327:298–321. 10.1002/cne.903270211 [DOI] [PubMed] [Google Scholar]
- Rich S, Chameh HM, Rafiee M, Ferguson K, Skinner FK, Valiante TA (2020) Inhibitory network bistability explains increased interneuronal activity prior to seizure onset. Front Neural Circuits 13:81. 10.3389/fncir.2019.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitr Sastri BV, Arivazhagan A, Sinha S, Mahadevan A, Bharath RD, Saini J, Jamuna R, Kumar JK, Rao SL, Chandramouli BA, Shankar SK, Satishchandra P (2014) Clinico-pathological factors influencing surgical outcome in drug resistant epilepsy secondary to mesial temporal sclerosis. J Neurol Sci 340:183–190. 10.1016/j.jns.2014.03.026 [DOI] [PubMed] [Google Scholar]
- Seo J, Jung S, Lee S-Y, Yang H, Kim BS, Choi J, Bang M, Shin H-S, Jeon D (2013) Early deficits in social behavior and cortical rhythms in pilocarpine-induced mouse model of temporal lobe epilepsy. Exp Neurol 241:38–44. 10.1016/j.expneurol.2012.11.024 [DOI] [PubMed] [Google Scholar]
- Sessolo M, Marcon I, Bovetti S, Losi G, Cammarota M, Ratto GM, Fellin T, Carmignoto G (2015) Parvalbumin-positive inhibitory interneurons oppose propagation but favor generation of focal epileptiform activity. J Neurosci 35:9544–9557. 10.1523/JNEUROSCI.5117-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiri Z, Manseau F, Lévesque M, Williams S, Avoli M (2016) Activation of specific neuronal networks leads to different seizure onset types. Ann Neurol 79:354–365. 10.1002/ana.24570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaker ML, Harkness JH, Sorg BA (2016) A standardized and automated method of perineuronal net analysis using Wisteria floribunda agglutinin staining intensity. IBRO Rep 1:54–60. 10.1016/j.ibror.2016.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolensky IV, Zubareva OE, Kalemenev SV, Lavrentyeva VV, Dyomina AV, Karepanov AA, Zaitsev AV (2019) Impairments in cognitive functions and emotional and social behaviors in a rat lithium-pilocarpine model of temporal lobe epilepsy. Behav Brain Res 372:112044. 10.1016/j.bbr.2019.112044 [DOI] [PubMed] [Google Scholar]
- Somogyi J, Baude A, Omori Y, Shimizu H, El Mestikawy S, Fukaya M, Shigemoto R, Watanabe M, Somogyi P (2004) GABAergic basket cells expressing cholecystokinin contain vesicular glutamate transporter type 3 (VGluT3) in their synaptic terminals in hippocampus and isocortex of the rat. Eur J Neurosci 19:552–569. 10.1111/j.0953-816x.2003.03091.x [DOI] [PubMed] [Google Scholar]
- Steiger BK, Jokeit H (2017) Why epilepsy challenges social life. Seizure 44:194–198. 10.1016/j.seizure.2016.09.008 [DOI] [PubMed] [Google Scholar]
- Steve TA, Jirsch JD, Gross DW (2014) Quantification of subfield pathology in hippocampal sclerosis: a systematic review and meta-analysis. Epilepsy Res 108:1279–1285. 10.1016/j.eplepsyres.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Sun C, Sun J, Erisir A, Kapur J (2014) Loss of cholecystokinin-containing terminals in temporal lobe epilepsy. Neurobiol Dis 62:44–55. 10.1016/j.nbd.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Sotayo A, Cazzulino AS, Snyder AM, Denny CA, Siegelbaum SA (2017) Proximodistal heterogeneity of hippocampal CA3 pyramidal neuron intrinsic properties, connectivity, and reactivation during memory recall. Neuron 95:656–672.e3. 10.1016/j.neuron.2017.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamamaki N, Abe K, Nojyo Y (1988) Three-dimensional analysis of the whole axonal arbors originating from single CA2 pyramidal neurons in the rat hippocampus with the aid of a computer graphic technique. Brain Res 452:255–272. 10.1016/0006-8993(88)90030-3 [DOI] [PubMed] [Google Scholar]
- Téllez-Zenteno JF, Hernández-Ronquillo L (2012) A review of the epidemiology of temporal lobe epilepsy. Epilepsy Res Treat 2012:1–5. 10.1155/2012/630853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testa D, Prochiantz A, Di Nardo AA (2019) Perineuronal nets in brain physiology and disease. Semin Cell Dev Biol 89:125–135. 10.1016/j.semcdb.2018.09.011 [DOI] [PubMed] [Google Scholar]
- Tewari BP, Chaunsali L, Campbell SL, Patel DC, Goode AE, Sontheimer H (2018) Perineuronal nets decrease membrane capacitance of peritumoral fast spiking interneurons in a model of epilepsy. Nat Commun 9:4724. 10.1038/s41467-018-07113-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M, Liagkouras I, Elliot KJ, Martinian L, Harkness W, McEvoy A, Caboclo LO, Sisodiya SM (2010) Reliability of patterns of hippocampal sclerosis as predictors of postsurgical outcome. Epilepsia 51:1801–1808. 10.1111/j.1528-1167.2010.02681.x [DOI] [PubMed] [Google Scholar]
- Turski WA, Cavalheiro EA, Bortolotto ZA, Mello LM, Schwarz M, Turski LAW (1984) Seizures produced by pilocarpine in mice: a behavioral, electroencephalographic and morphological analysis. Brain Res 321:237–253. 10.1016/0006-8993(84)90177-x [DOI] [PubMed] [Google Scholar]
- Valero M, Cid E, Averkin RG, Aguilar J, Sanchez-Aguilera A, Viney TJ, Gomez-Dominguez D, Bellistri E, de la Prida LM (2015) Determinants of different deep and superficial CA1 pyramidal cell dynamics during sharp-wave ripples. Nat Neurosci 18:1281–1290. 10.1038/nn.4074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vancura B, Geiller T, Grosmark A, Zhao V, Losonczy A (2023) Inhibitory control of sharp-wave ripple duration during learning in hippocampal recurrent networks. Nat Neurosci 26:788–797. 10.1038/s41593-023-01306-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Sperk G, Colmers WF (1999) Neuropeptide Y: emerging evidence for a functional role in seizure modulation. Trends Neurosci 22:25–30. 10.1016/s0166-2236(98)01284-3 [DOI] [PubMed] [Google Scholar]
- Wen TH, Binder DK, Ethell IM, Razak KA (2018) The perineuronal “safety” net? Perineuronal net abnormalities in neurological disorders. Front Mol Neurosci 11:1–17. 10.3389/fnmol.2018.00270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel HJ, Vacher H, Clark E, Trimmer JS, Lee AL, Sapolsky RM, Tempel BL, Schwartzkroin PA (2007) Structural consequences of Kcna1 gene deletion and transfer in the mouse hippocampus. Epilepsia 48:2023–2046. 10.1111/j.1528-1167.2007.01189.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitebirch AC, Lafrancois JJ, Jain S, Leary P, Santoro B, Siegelbaum SA, Scharfman HE (2022) Enhanced excitability of the hippocampal CA2 region and its contribution to seizure activity in a mouse model of temporal lobe epilepsy. Neuron 110:3121–3138.e8. 10.1016/j.neuron.2022.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson A, Spencer DD (1994) Electrophysiological characterization of CA2 pyramidal cells from epileptic humans. Hippocampus 4:226–237. 10.1002/hipo.450040213 [DOI] [PubMed] [Google Scholar]
- Winawer MR, Makarenko N, McCloskey DP, Hintz TM, Nair N, Palmer AA, Scharfman HE (2007) Acute and chronic responses to the convulsant pilocarpine in DBA/2J and A/J mice. Neuroscience 149:465–475. 10.1016/j.neuroscience.2007.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Maglóczky Z (2017) Synaptic reorganization of the perisomatic inhibitory network in hippocampi of temporal lobe epileptic patients. Biomed Res Int 2017:1–13. 10.1155/2017/7154295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Miles R (2007) Factors defining a pacemaker region for synchrony in the hippocampus. J Physiol 584:867–883. 10.1113/jphysiol.2007.138131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L, Huberfeld G, Clémenceau S, Eross L, Dezamis E, Entz L, Ulbert I, Baulac M, Freund TF, Maglóczky Z, Miles R (2009) The epileptic human hippocampal cornu ammonis 2 region generates spontaneous interictal-like activity in vitro. Brain 132:3032–3046. 10.1093/brain/awp238 [DOI] [PubMed] [Google Scholar]
- Wong RK, Traub RD (1983) Synchronized burst discharge in disinhibited hippocampal slice. I. Initiation in CA2-CA3 Region. J Neurophysiol 49:442–458. 10.1152/jn.1983.49.2.442 [DOI] [PubMed] [Google Scholar]
- Wu C, et al. (2019) Effects of surgical targeting in laser interstitial thermal therapy for mesial temporal lobe epilepsy: a multicenter study of 234 patients. Epilepsia 60:1171–1183. 10.1111/epi.15565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyeth MS, Zhang N, Mody I, Houser CR (2010) Selective reduction of cholecystokinin-positive basket cell innervation in a model of temporal lobe epilepsy. J Neurosci 30:8993–9006. 10.1523/JNEUROSCI.1183-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyler AR, Curtis Dohan F, Schweitzer JB, Berry AD (1992) A grading system for mesial temporal pathology (hippocampal sclerosis) from anterior temporal lobectomy. J Epilepsy 5:220–225. 10.1016/S0896-6974(05)80120-3 [DOI] [Google Scholar]
- Yang JC, Bullinger KL, Dickey AS, Karakis I, Alwaki A, Cabaniss BT, Winkel D, Rodriguez-Ruiz A, Willie JT, Gross RE (2022) Anterior nucleus of the thalamus deep brain stimulation vs temporal lobe responsive neurostimulation for temporal lobe epilepsy. Epilepsia 63:2290–2300. 10.1111/epi.17331 [DOI] [PMC free article] [PubMed] [Google Scholar]