

Abstract
Bacteria are ubiquitous lifeforms with important roles in the environment, biotechnology, and human health. Many of the functions that bacteria perform are mediated by proteins and enzymes, which catalyze metabolic transformations of small molecules and modifications of proteins. To better understand these biological processes, chemical proteomic approaches, including activity-based protein profiling, have been developed to interrogate protein function and enzymatic activity in physiologically relevant contexts. Here, chemoproteomic strategies and technological advances for studying bacterial physiology, pathogenesis, and metabolism are discussed. The development of chemoproteomic approaches for characterizing protein function and enzymatic activity within bacteria remains an active area of research, and continued innovations are expected to provide breakthroughs in understanding bacterial biology.
Keywords: Proteomics, Enzymes, Click chemistry, Proteins, Bacteria
Graphical Abstract
1. Introduction
Bacteria are a ubiquitous unicellular lifeform, playing important roles in global nutrient cycling[1] and in causing infectious disease. Bacteria are important industrially in the production of a variety of products, ranging from fermented foods[2] to biologic pharmaceuticals.[3] As such, gaining a deeper understanding of bacterial physiology, pathogenesis, and metabolism are important to improving manufacturing, sustainability, and human health.
Many important bacterial activities in these settings are regulated by bacterial proteins and enzymes. These roles range from essential functions, such as cell envelope biosynthesis, to more specialized processes, including pathogenesis and signal transduction.[4] The importance of bacterial proteins in these numerous contexts warrants novel technologies for understanding their activity.
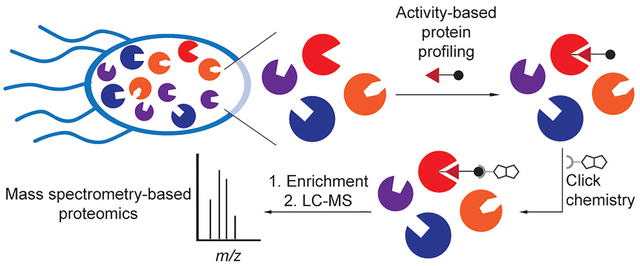
Chemical proteomics has emerged as a powerful technique for profiling protein function and enzymatic activity. Activity-based protein profiling (ABPP) enables the labeling, enrichment, and identification of specific proteins of interest within complex mixtures using an activity-based probe (ABP, Figure 1a). Typically, the ABP contains an electrophilic moiety that reacts with a nucleophilic active site residue, which forms an irreversible covalent adduct with the enzyme.[5–9] The appendage of a fluorophore, followed by resolution by gel electrophoresis, to these protein adducts enables imaging of active enzymes by in-gel fluorescence. Alternatively, an affinity tag, such as biotin, can be appended, which allows for the enrichment of active enzymes that can be further characterized by mass spectrometry-based proteomics. These fluorescent and affinity tags can be appended directly to the probe itself,[5,6,8] or to the protein adduct through a bioorthogonal reaction.[7,9]
Figure 1.
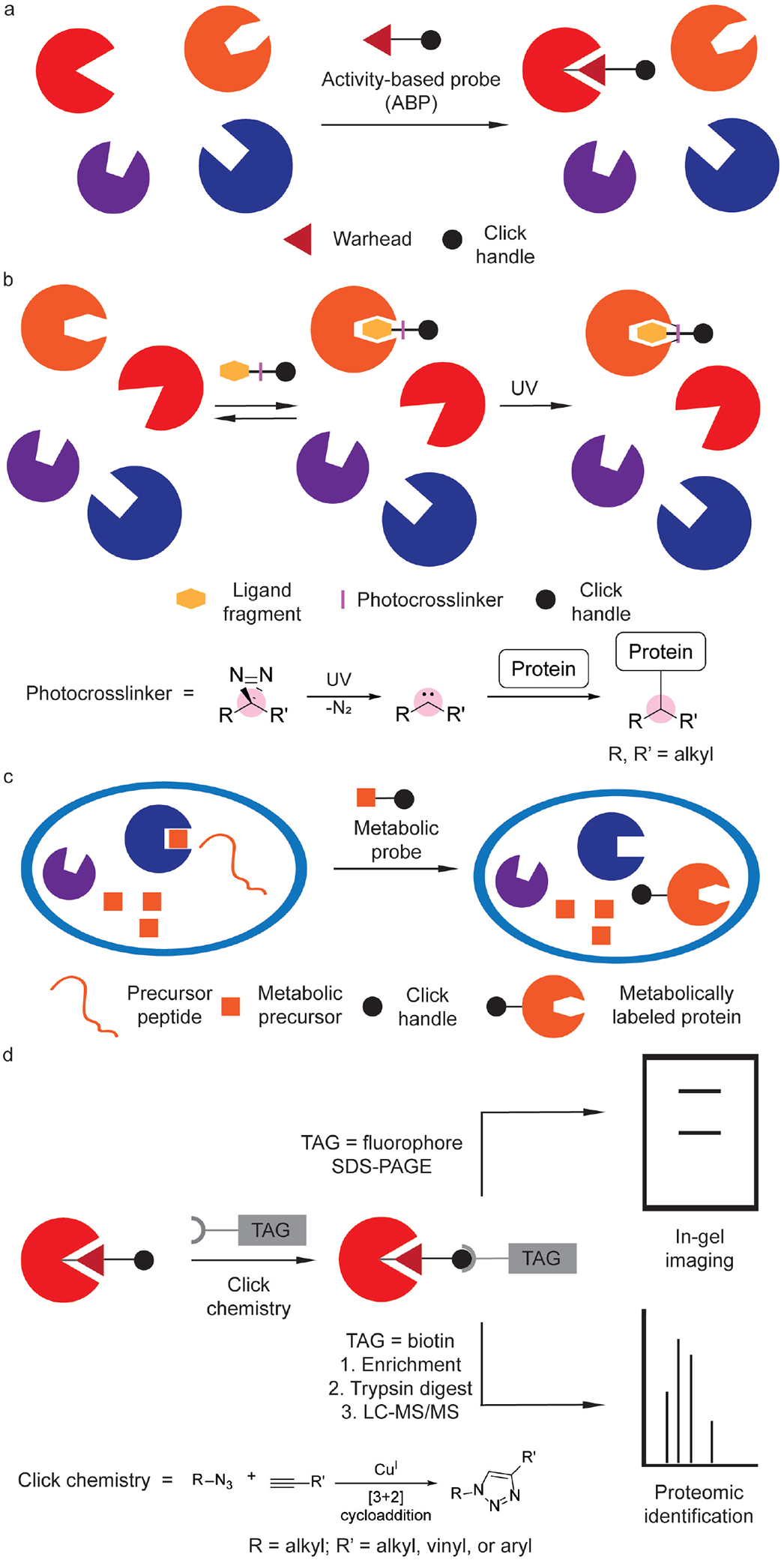
Key chemoproteomic profiling technologies. (a) Labeling of enzyme active sites with activity-based probes (ABPs). (b) Small-molecule protein target labeling with photoaffinity probes, including a chemical schematic of protein labeling by a diazirine photocrosslinker. (c) Metabolic labeling of protein targets using chemical reporters that are metabolically incorporated into newly biosynthesized proteins. (d) Labeled biological targets can be further tagged using click chemistry with an appropriate chemical probe containing an imaging agent or affinity handle for in situ imaging or mass spectrometry-based proteomics, respectively.
Photoaffinity probes are important for determining the cellular targets of compounds that noncovalently bind to proteins (Figure 1b).[10] Whereas ABPP depends on chemoselectivity of the probe warhead, photoaffinity labeling relies on the selective uncaging of a promiscuous photolabile moiety that is generated photochemically and reacts with nearby proteins, enabling their capture and identification. Alternatively, a cell-permeable probe or so-called chemical reporter can be metabolized, wherein it is then incorporated into other biomolecules (Figure 1c). The same analyses and mass spectrometry (MS)-based workflow for ABPP are also used in these cases and typically involves visualization of covalent protein labeling in situ or pull-down and protein identification using MS-based proteomics (Figure 1d). These three strategies – ABPP, photoaffinity labeling, and metabolic labeling – have been key for interrogating bacterial biology using chemical proteomics (Figure 1).
This review highlights specific biological processes in bacteria and contributions of chemical proteomics to understanding these processes. The discussion of works on chemoproteomic profiling of mammalian proteins and its application to studying interactions with bacterial biomolecules,[11] serine hydrolases,[12] and reactive sites in the human proteome[13] have been reviewed elsewhere. Rather, this review highlights seminal technologies for chemoproteomic profiling of bacterial proteins in specific processes and the mechanistic biological studies that these technologies have enabled.
2. Interrogating Prokaryotic Physiology
2.1. Probing Cell Wall Biosynthesis
The bacterial cell envelope is an important barrier for protecting bacteria from the environment. Whereas the cell envelope of Gram-positive bacteria is characterized by a thick peptidoglycan wall surrounded by the cell membrane, Gram-negative bacteria possess inner and outer membranes with a thinner peptidoglycan wall between these membranes.[14] In both cases, peptidoglycan (PG) strands are cross-linked to form a rigid barrier (Figure 2a).[14] Bacterial cell envelope biosynthesis is essential to cell replication. Indeed, numerous clinically important antibiotics act by disrupting cell wall biosynthesis.[15] β-lactam antibiotics are an important class of antibiotics that act as suicide inhibitors of enzymes involved in PG biosynthesis, such as penicillin binding proteins (PBPs).[16] PBPs catalyze the cross-linking of PG strands through transpeptidation, and β-lactam antibiotics inhibit these transpeptidases (Figure 2a).[17] β-lactams specifically disrupt peptidoglycan strand cross-linking by acting as electrophilic substrate analogs.[15] The utility of the β-lactams and related electrophilic scaffolds have enabled numerous ABPP studies of cell wall biosynthesis.
Figure 2.
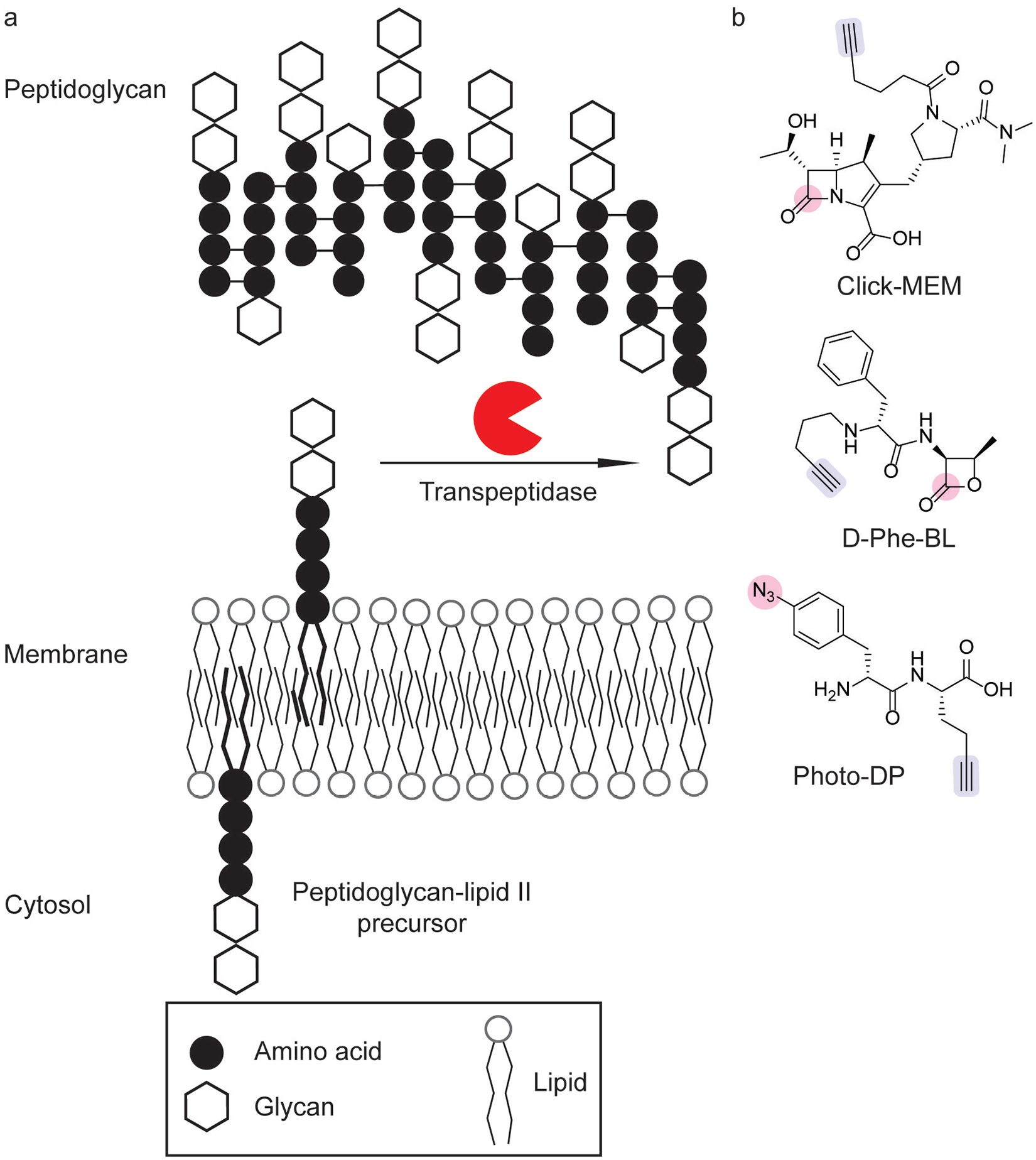
Chemical probes to target bacterial cell wall biosynthesis. (a) Peptidoglycan strand crosslinking by transpeptidases is a key step of cell wall biosynthesis. (b) Chemical probes for profiling peptidoglycan transpeptidases have utilized activity-based labeling[19,20] and photoaffinity labeling modalities[21] (pink), coupled with alkyne click handles (blue) for chemoproteomic identification.
Competitive ABPP has emerged as a useful tool for profiling the selectivity of antibiotics that inhibit cell wall biosynthesis and has led the development of selective probes for probing PBP activity. The fluorescent penicillin analog, BOCILLIN-FL,[18] is a broad-spectrum covalent inhibitor of PBPs that enables ABPP by in-gel fluorescence. Competitive ABPP with BOCILLIN-FL has allowed the rapid characterization of probe selectivity. For example, selectivity profiling of a bioorthogonal analog of meropenem, Click-MEM (Figure 2b), has been investigated in Bacillus subtilis, which selectively labeled PBP3 and PBP5.[19] Clickable β-lactone ABPs have also been evaluated in Streptococcus pneumoniae, in which selectivity depended on linker and click handle identity heavily shaped the selectivity profile, leading to the development of D-Phe-BL (Figure 2b), which selectively labeled PBP2x and PBP2a.[20] In both cases, proteomic analysis of probe-enriched samples corroborated the selectivity profiles of these probes in B. subtilis[19] and S. pneumoniae.[20] Overall, advances in the activity-based profiling of PBPs has led to the development of selective probes to monitor the activity of specific PBPs during bacterial cell growth and division.
More broadly, chemoproteomic profiling of β-lactam-binding proteins has validated additional cellular targets of β-lactams. The characterization of a β-lactam probe library by click chemistry-based chemoproteomics in a panel of bacteria successfully identified differential PBP selectivity based on the β-lactam core.[22] However, proteomic analysis also revealed additional enzymatic targets, including β-lactamases and ClpP, a caseinolytic protease that regulates virulence.[22] This chemoproteomic study unveiled the potential for targeting bacterial virulence with antibiotic scaffolds.
Chemoproteomic profiling with β-lactam probes has also revealed novel antibiotic resistance mechanisms. Chemoproteomic profiling using clickable β-lactam probes in methicillin-susceptible Staphylococcus aureus (SA) and methicillin-resistant S. aureus (MRSA) strains allowed for identification of enzymes that confer resistance to β-lactam antibiotics in MRSA.[23] Specifically, the active enzymes profiled in MRSA included two novel β-lactamases: a novel metallo-β-lactamase and serine hydrolase β-lactamase.[23] Although β-lactamases are a hallmark resistance mechanism,[15] the presence of these enzymes in MRSA contrast the previously characterized model of β-lactam resistance wherein altered PBP binding sites that have a lower affinity for β-lactams.[14]
In addition to forming cross-links that create the rigid cell wall, peptidoglycan precursors are linked to lipid II (Figure 2a), which allows for transport of peptidoglycan from the cytoplasm to exterior side of the cell membrane.[24] The identification of proteins involved in these processes is important for gaining a deeper understanding of bacterial cell wall biosynthesis. Bacteria successfully incorporated peptidoglycan precursor Photo-DP (Figure 2b), which validated the interaction of lipid II with PBP1a/b in live cells.[21]
ABPP has also unveiled novel mechanisms of action for antimicrobial compounds. For example, the investigation of a clickable, photocrosslinking vancomycin analog in SA and Enterococcus faecalis identified novel binding partners.[25] Especially salient in SA, vancomycin was found to inhibit an autolysin domain in a cell wall biosynthetic enzyme.[25] This inhibitory action was identified as a novel resistance mechanism, as autolysin activity is necessary for the bactericidal effects of cell wall biosynthesis inhibitors.[15] Many natural products contain electrophilic scaffolds, such as α,β-saturated carbonyl groups. As a result, these compounds were hypothesized to be protein-reactive electrophiles. Using clickable analogs, chemoproteomic profiling of the nucleoside antibiotic showdowmycin[26] and 3-methylene-γ-lactones[27] were shown to target enzymes involved in cell wall biosynthesis in SA.
Overall, bacterial cell growth and division are essential processes to sustaining bacterial life. The biosynthesis of the cell wall is one aspect of growth that has been extensively examined using ABPP. Strategies for profiling bacterial cell envelope biosynthesis have built on electrophilic scaffolds in antibiotics that inhibit cell wall biosynthesis in actively replicating bacteria. Advances in ABPP of cell wall biosynthesis have illuminated new mechanisms for antimicrobial resistance and previously unidentified cellular targets. Additionally, photoaffinity labeling strategies have also aided in the chemoproteomic identification of enzymes involved in cell wall biosynthesis and antibiotic resistance.
2.2. Probing Lipid Homeostasis
Lipid membranes form an additional protective barrier for cells. Chemoproteomic studies have provided significant insights into the role of serine hydrolases in prokaryotic lipid metabolism. These studies have been important for understanding lipid and membrane homeostasis in mycobacteria.
The membrane composition of mycobacteria, such as Mycobacterium tuberculosis (Mtb), differs in that it contains a distinctive outer membrane outside of its peptidoglycan wall composed of mycolic acid-containing glycolipids, termed the mycomembrane (Figure 3a).[31] Disrupting membrane biosynthesis is an important strategy for treating mycobacterial infections.[32] In order to identify mycomembrane-interacting proteins, clickable diazirine analogs of glycosylated very long-chain fatty acids were synthesized and characterized in Mycobacterium smegmatis.[28] N-x-AlkTMM–C15 (Figure 3b) was successfully incorporated into the membrane, allowing for chemoproteomic profiling of mycolate-interacting proteins.[28] Proteomic analysis identified known mycomembrane proteins and new homologs of membrane remodeling enzymes.[28]
Figure 3.
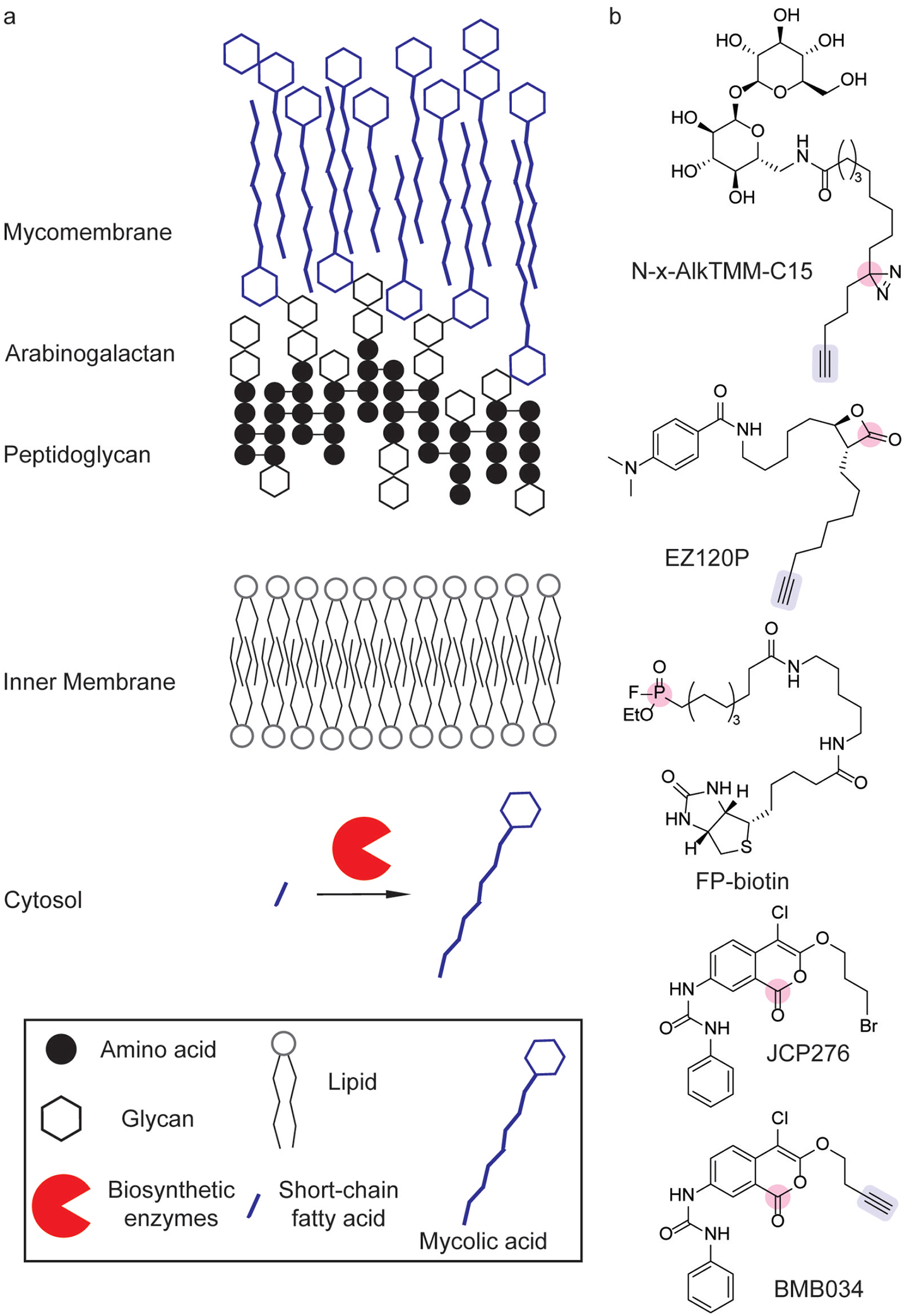
Chemical probes to target mycomembrane biosynthesis. (a) Mycomembrane biosynthesis and composition. (b) Chemical probes for profiling mycomembrane biosynthetic enzymes including photoaffinity[28] and activity-based[6,29,30] labeling strategies (pink) have identified numerous hydrolases involved in lipid metabolism through the use of competitive ABPP and click chemistry handles (blue) for identification.
Target validation of cell wall and membrane biosynthesis inhibitors in Mtb has also benefited from chemoproteomic tools. A β-lactone bearing a long-chain alkyne-containing tail (EZ120P, Figure 3b) has been investigated as a mycolic acid biosynthesis inhibitor.[30] ABPP and subsequent proteomic analysis validated EZ120P as a covalent inhibitor of the Pks13 thioester domain, which is responsible for glycosylation of mycolic acids.[30] Furthermore, this covalent inhibitor synergistically inhibited growth of M. smegmatis when co-administered with vancomycin.[30] Competitive ABPP has also been used to identify the protein targets of antimycobacterial compounds. After screening a triazole urea library for Mtb growth inhibition, competitive ABPP with fluorophosphonatebiotin (FP-biotin, Figure 3b) identified a variety of serine hydrolase targets, including mycolyltransferases, lipases, and a thioesterase, which were inhibited by these compounds.[33] Subsequent biochemical studies validated these targets and induced cell morphologies similar to known cell wall biosynthesis inhibitors.[33] Competitive ABPP with FP-biotin (Figure 3b) and analogs of cyclipostin and cyclophostin similarly identified inhibitors of cell wall biosynthesis in M. abscessus.[34]
A hallmark of Mtb infection is nonreplicative persistence,[35] which can lead to recurrent infection.[36] Owing to the importance of dormancy during infection, chemoproteomic studies have focused on understanding the changes in enzymatic activity during dormancy. Using a clickable tetrahydrolipistatin analog, serine esterase activity was profiled in Mycobacterium bovis undergoing replicative growth and nonreplicative persistence.[37] Proteomic studies identified persistent lipase and thioesterase activity across growth states, although the activity of many lipases decreased during dormancy and increased after recovery from dormancy.[37] Among the lipases downregulated during nonreplicative conditions, ectopic expression of lipase H induced increased levels of long-chain fatty acid triglycerides after recovery from dormancy and decreased susceptibility to the bactericidal effects of tetrahydrolipistatin.[37]
As nonreplicative dormancy occurs under hypoxic conditions,[35] global serine hydrolase ABPP activity in Mtb has also been profiled under hypoxic and aerobic growth conditions.[38] Although overall serine hydrolase activity decreased under hypoxia, global serine hydrolase profiling identified active enzymes involved in membrane biosynthesis during persistence.[38] Subsequent biochemical characterization revealed that these serine hydrolases included novel serine proteases.[38] Esterase profiling using fluorogenic probes identified additional esterases that are active under hypoxia, which were previously unidentified.[39]
ABPP studies on serine hydrolases in Mtb have also identified a subset of serine hydrolases involved in lipid metabolism that are important for Mtb growth.[29] After identifying chloroisocoumarin inhibitors of Mtb growth, the characterization of a resistant strain revealed that the ability to biosynthesize phthiocerol, a cell wall-associated fatty alcohol, conferred resistance to the most potent inhibitor, JCP276 (Figure 3b).[29] Competitive ABPP using FP-biotin and a clickable analog, BMB034, (Figure 3b) revealed numerous lipases and other enzymes involved in lipid metabolism as targets of JCP276 (Figure 3b).[29] Further analysis showed that the inhibitor, along with previously reported antitubercular compounds, targeted a common set of serine hydrolases.[29]
Additionally, ABPP has enabled profiling of active enzymes in extreme environments. Live-cell chemoproteomic profiling of serine hydrolases in thermophilic archaea revealed lipases active at high temperatures and highly acidic conditions.[40] This advance was notable due to previous ABPP studies in archaea being performed under non-native conditions in lysates.[40,41]
Altogether, the lipid membrane is an important protective barrier that exhibits considerable diversity between different prokaryotes. Chemical proteomic characterization of serine hydrolases has identified numerous enzymes involved in lipid metabolism. Further, in Mtb, these studies have revealed potential targets for drug development.
2.3. Probing Protein Homeostasis
The maintenance of protein homeostasis in bacteria is characterized by the precise regulation of protein synthesis and degradation.[42] Chemoproteomic studies of protein homeostasis have yielded insights into these anabolic and catabolic processes. Whereas chemoproteomic methods for studying protein biosynthesis have focused on the use of bioorthogonal noncanonical amino acid tagging (BONCAT), chemoproteomic studies of proteolysis have focused on the ClpXP system.
Chemical proteomic methodologies for studying protein synthesis have relied on the ability to label proteins with noncanonical amino acids (ncAAs). BONCAT is a versatile tool for labeling newly synthesized proteins with unnatural amino acid analogs that can undergo bioorthogonal labeling reactions (Figure 4a).[43] A variety of suitable amino acids have been characterized (Figure 4b). The incorporation of ANL (Figure 4b) in Escherichia coli (E. coli) facilitated selective click chemistry-based imaging and proteomic enrichment in mixed bacterial populations and during bacterial infection.[44] Furthermore, this proteome labeling strategy can be utilized with AHA (Figure 4b) in a variety of environmental bacteria and enabled the study of protein synthesis rates in response to heat shock.[45] Similarly, the incorporation of PEP[46] and AOA[47] (Figure 4b) into Salmonella enterica serovar Typhimurium (STm) have allowed selective proteome labeling and imaging during macrophage co-infection.[47] Continued synthetic efforts[48] towards novel bioorthogonal ncAAs also provide further advances in this area.
Figure 4.
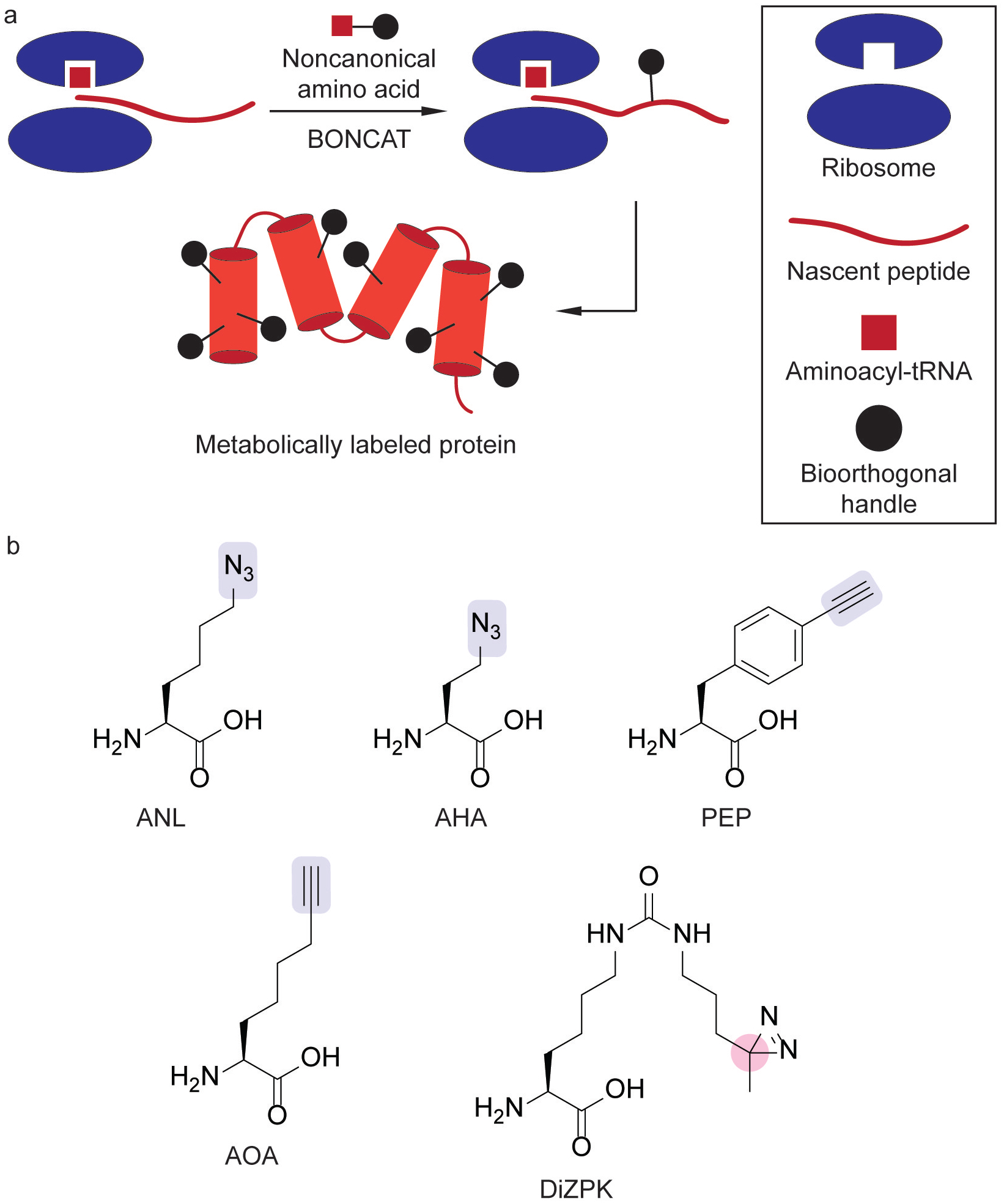
Whole-proteome labeling using bioorthogonal noncanonical amino acid tagging (BONCAT) with noncanonical amino acids (ncAAs). (a) Metabolic labeling of protein biosynthesis using BONCAT in which ncAAs are incorporated into nascent peptides. (b) ncAAs containing click chemistry handles[44,46,47] (blue) or a photo-crosslinker (pink).[49]
Amino acids encoding photoaffinity handles have also been extended to study protein-protein interactions (PPIs). This strategy has provided insight into the roles of protein chaperones in maintaining protein homeostasis by identifying refolding partners. Through the design and incorporation of DiZPK (Figure 4b), which bears a diazirine moiety, into a protein of interest, PPIs were captured through photoaffinity labeling and proteomic analysis.[49] This methodology profiled client proteins of the acid stress chaperone HdeA during E. coli acid stress.[49] In this study, the substitution of DiZPK at different positions of His6-tagged HdeA allowed for the expression of photo-crosslinking HdeA variants in E. coli. The incorporation of a His6-tag into this protein enabled the visualization of photo-crosslinked complexes by Western blotting and enrichment of the PPI partners. Proteomic analysis identified the chaperones SurA and DegP as cooperative partners of HdeA that aid protein refolding during recovery from acid stress. This platform was successfully extended to enteropathogenic E. coli, STm, and Shigella flexneri.[50] Analogous acid stress studies of HdeA in S. flexneri identified novel refolding partners of this protein chaperone.[50]
The ClpP and Lon protease systems are two widely distributed systems in bacteria that regulate cellular protein abundance.[42] Whereas Lon protease is characterized by its recognition and degradation of unfolded proteins, ClpP-mediated proteolysis is characterized by its utilization of specific degrons.[42] Compared to Lon protease, which unfolds its substrates, ClpP relies on complexing with other proteins, such as ClpX, that carry out substrate unfolding.[51] In addition, ClpP protease is an important virulence factor in Listeria monocytogenes (Lm) and SA, making it a potential target for antimicrobial therapies.[52]
ABPP studies of β-lactone probe libraries in Listeria species identified β-lactones as ClpP inhibitors.[55,57] In Lm, VLP (Figure 5a) was identified as a selective inhibitor of ClpP1 and ClpP2.[55] Biochemical characterization found VLP labeling of ClpP1 only occurred in the presence of ClpP2, which led to the identification of a heterooligomeric complex.[55] To understand the mechanism of VLP labeling, additional biochemical and ABPP studies of the oligomeric complex revealed ClpP1 contains a less active catalytic triad that is activated by oligomer formation with ClpP2.[58] Crystallographic analysis of the oligomers provided further insight into the formation of the catalytically active ClpP1/ClpP2 complex.[58]
Figure 5.
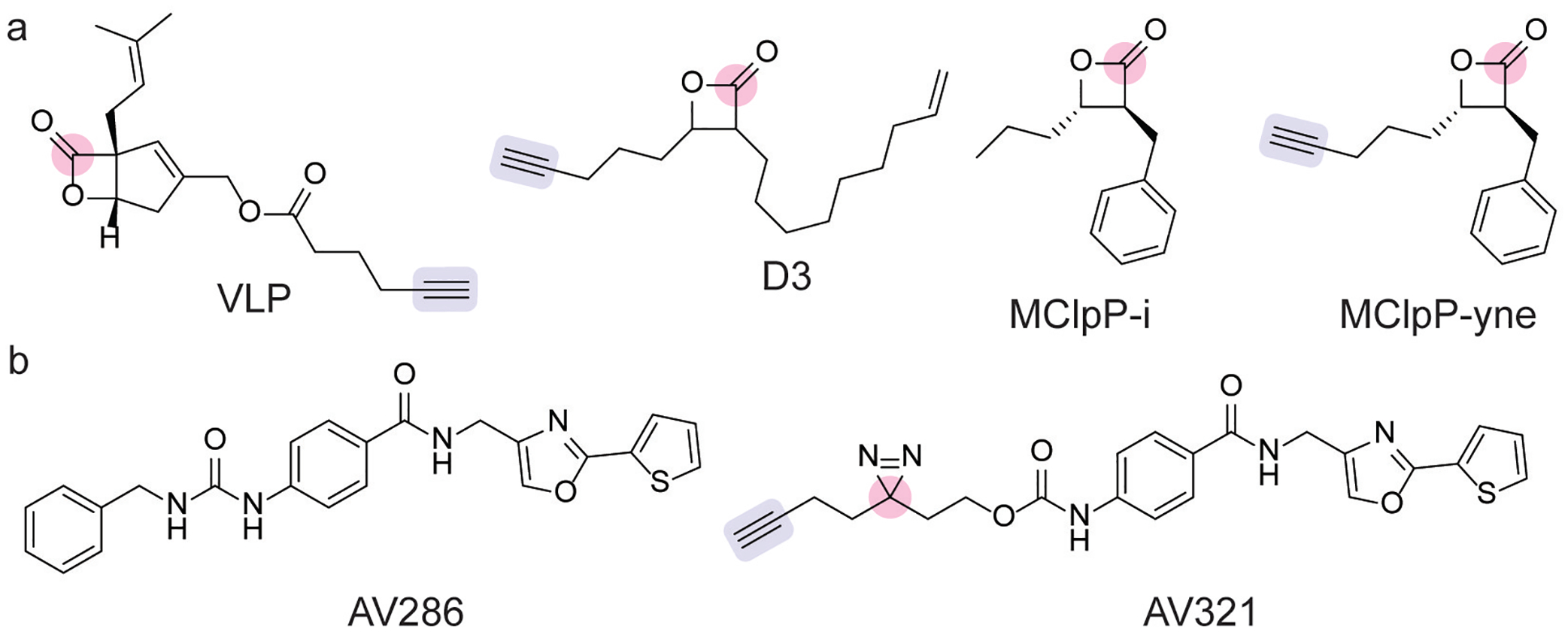
(a) Covalent inhibitors of ClpP proteases, which contain an acylating β-lactone electrophile (pink) and alkyne click chemistry handle (blue).[53–55] (b) Noncovalent ClpP inhibitor AV286 and its photoaffinity profiling analog AV321, which contains a diazirine photocrosslinker (pink) and alkyne click chemistry handle (blue).[56]
Similarly, in SA, the evaluation of a β-lactone ABP library identified a ClpP-selective inhibitor D3 (Figure 5a), which was validated by proteomic identification and inhibited virulence in cellular assays.[54] Further optimization of the β-lactone scaffold led to the development of a highly selective ClpP inhibitor in SA, which had low toxicity in mammalian cells.[59] Noncovalent inhibitors of ClpP have also been developed.[56] The crystal structure of one of these inhibitors with ClpP showed that binding induced active site distortions that moved His123, which prevented participation in the Asp-His-Ser catalytic triad.[56] Further scaffold optimization led to the development of a highly potent inhibitor AV286 (Figure 5b), and chemoproteomic profiling by photoaffinity labeling with AV321 (Figure 5b) validated ClpP as the primary cellular target.[56]
Whereas ClpP proteases are nonessential in Lm and SA, ClpP proteases are essential in Mtb.[53] In search for novel antimycobacterial compounds, a library of β-lactones was screened for antimycobacterial activity in Mtb and M. smegmatis, which identified MClpP-i (Figure 5a) as the most potent inhibitor.[53] The clickable analog of MClpP-i, MClpP-yne (Figure 5a), validated ClpP1 and Clp2 as cellular targets in M. smegmatis. Biochemical and proteomic analysis with recombinant ClpP2 from Mtb corroborated that MClpP-i inhibits ClpP2 by acylating the active site serine residue.
Overall, chemical proteomic strategies for studying bacterial protein homeostasis encompass tools for characterizing protein biosynthesis, PPIs, and protein degradation. BONCAT-based platforms have yielded insights into protein biosynthesis and the role of chaperones during cellular stress. Additionally, chemoproteomic tools to understand protein degradation in bacteria have focused on the ClpP protease system.
2.4. Posttranslational Modifications
Posttranslational modification (PTM) of proteins is an important mechanism for regulating protein activity and localization. Chemical proteomic tools have successfully identified PTMs in bacteria during infection in host and microbial proteins. Clickable fatty acids, such as Alk-14 (Figure 6), first served as metabolic reporters of protein fatty acylation and identified fatty acylated proteins in E. coli.[60] Chemoproteomic profiling of the Clostridium difficile lipoproteome using YnMyr[61] (Figure 6) identified novel signal peptidases involved in surface protein shedding.[62] Genetic and pharmacological inhibition studies showed that these signal peptidases regulated lipoprotein shedding, a necessary process for sporulation.[62]
Figure 6.
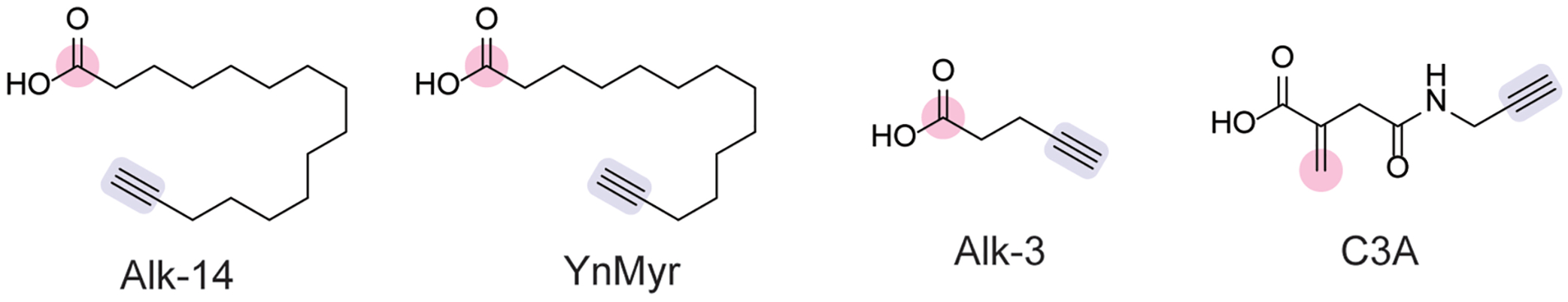
Chemical probes for probing post-translational acylation [60,61] and alkylation[69] in bacteria with moieties enabling protein ligation (pink) and click chemistry-based modification (blue).
STm is an enteric pathogen that causes millions of gastrointestinal illnesses each year.[63] STm encodes two genomic islands, SPI-1 and SPI-2, that regulate invasion and encodes genes important for invasion into the intestinal lumen and host cells.[64] Genetic regulation of SPI-1 is mediated by HilA, a transcription factor.[64] Short-chain fatty acids (SCFAs), such as butyrate, that are produced by the gut microbiota attenuate infection, thereby conferring colonization resistance.[65] Chemoproteomic profiling using Alk-3 (Figure 6) revealed that this colonization resistance mechanism occurs through HilA butyrylation.[66] Butyrylation of HilA downregulated SPI-1 gene expression and attenuated infection in vivo. Furthermore, this mechanism inhibited STm virulence in vitro and in vivo using acylating salicylic acid analogs.[67]
During bacterial infection, the host inflammatory response involves the coordinated production of signaling molecules by immune cells. One such signaling molecule is itaconate, which modulates inflammatory responses in macrophages.[68] Itaconate is an α,β-unsaturated carboxylic acid that is electrophilic at the β position and may act as an alkylating Michael acceptor. Itaconate also attenuates bacterial infection.[68] To examine the role of itaconate in STm infection, chemoproteomic profiling with C3A (Figure 6) in STm characterized alkylated cysteine residues on STm proteins.[69] One notable target of itaconate alkylation was isocitrate lysase, which is important for carbon metabolism in the absence of glucose, and itaconate was shown to inhibit this enzyme in STm.
Inflammation produces a variety of reactive oxygen species that can modify redox-sensitive protein residues. Cysteine is susceptible to numerous oxidation states (Figure 7a). Unoxidized cysteine residues are susceptible to alkylation (Figure 7a), which is quantifiable by click chemistry-based proteomics using IA-alkyne (Figure 7b).[70,71] The attenuation of cysteine alkylation by IA-alkyne can be due to treatment with a cysteine-reactive ligand[72,73] or due to cellular redox changes. To identify redox-sensitive cysteine residues in bacterial pathogens, quantitative proteomics with IA-alkyne was utilized in Pseudomonas aeruginosa (PA) and SA treated with hydrogen peroxide.[74] The identified proteins containing redox-susceptible cysteines included signaling proteins involved in metabolic regulation and quorum sensing, which were validated with additional phenotypic characterization.
Figure 7.
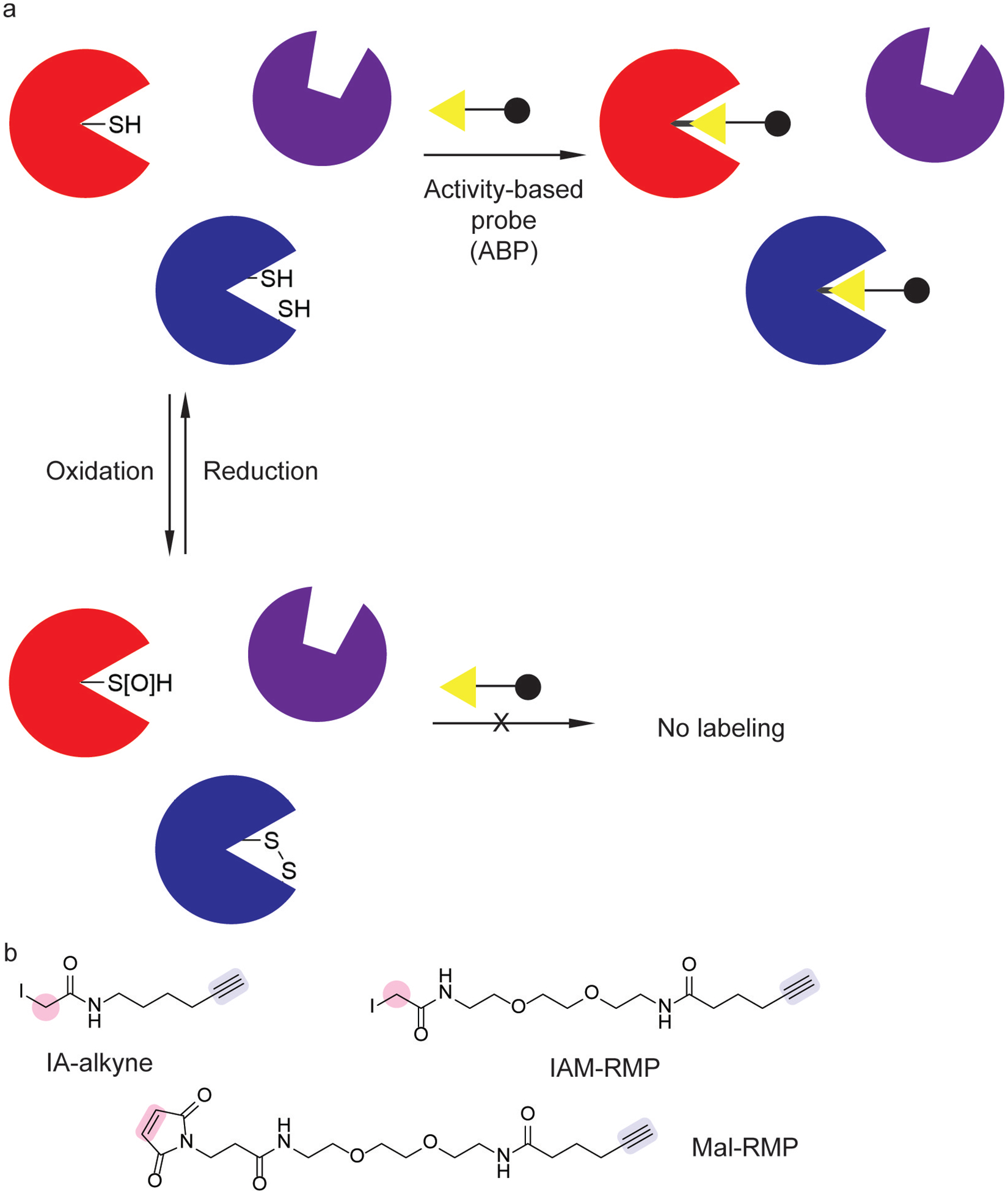
Chemoproteomic tools for profiling cysteine redox changes. (a) Redox-sensitive cysteines can be profiled using activity-based probes (ABPs). (b) ABPs for monitoring reactive cysteine thiols use a cysteine-selective alkylating warhead (pink) and click handle (blue).[71,75]
The role of redox environment changes have also been investigated in photosynthetic bacteria. Changes in redox-sensitive cysteine residues in Synechococcus 7002, a photosynthetic bacterium, was investigated during varying CO2 availability over time.[75] Proteome-wide cysteine labeling with Mal-RMP and IAM-RMP probes (Figure 7b) showed that the temporal dynamics of reduced cysteine residues in proteins cycled upon CO2 replenishment. These temporal dynamics depended on whether the bacterium was growing in carbon-starved conditions. Temporally resolved proteomics showed protein redox changes in response to CO2 varied across processes including transcription, photosynthesis, and secondary metabolism. This platform was extended to study light-to-dark transitions under carbon and nitrogen limitation in Synechococcus 7002, which identified additional redox-sensitive proteins.[76]
In an analogous approach, chemoproteomic profiling of thiol oxidations in Cyanothece sp. strain ATCC 51142 revealed widespread changes in cysteine redox states in response to oxidative stress.[77] This cyanobacterium produces hydrogen gas through nitrogen reduction using dinitrogenase enzymes.[78] Temporally resolved proteomics showed increased cysteine oxidation across numerous proteins during oxygenic photosynthesis, following the formation of reactive oxygen species.[77] Interestingly, however, these global levels of cysteine oxidation subsequently decreased following hydrogen production.[77] These results implicate nitrogen reduction as a mechanism for combating oxidative stress.[77] Altogether, these studies highlight cysteine redox changes as an important mechanism for bacterial protein regulation in response to environmental stimuli.
Studies in bacterial protein phosphorylation have focused on two-component systems, which are important in coordinating bacterial responses to environmental stimuli. Briefly, these systems act through the stimulation of a protein histidine kinase (PHK), which undergoes autophosphorylation at a histidine residue, followed by phosphate transfer to an aspartate residue on a response regulator protein (Figure 8a–b).[79] The response regulator protein then typically activates the transcription of target genes and is dephosphorylated. The phosphorylated residues are highly reactive, electrophilic intermediates, presenting a challenge for reliable monitoring of activity at the protein level.
Figure 8.
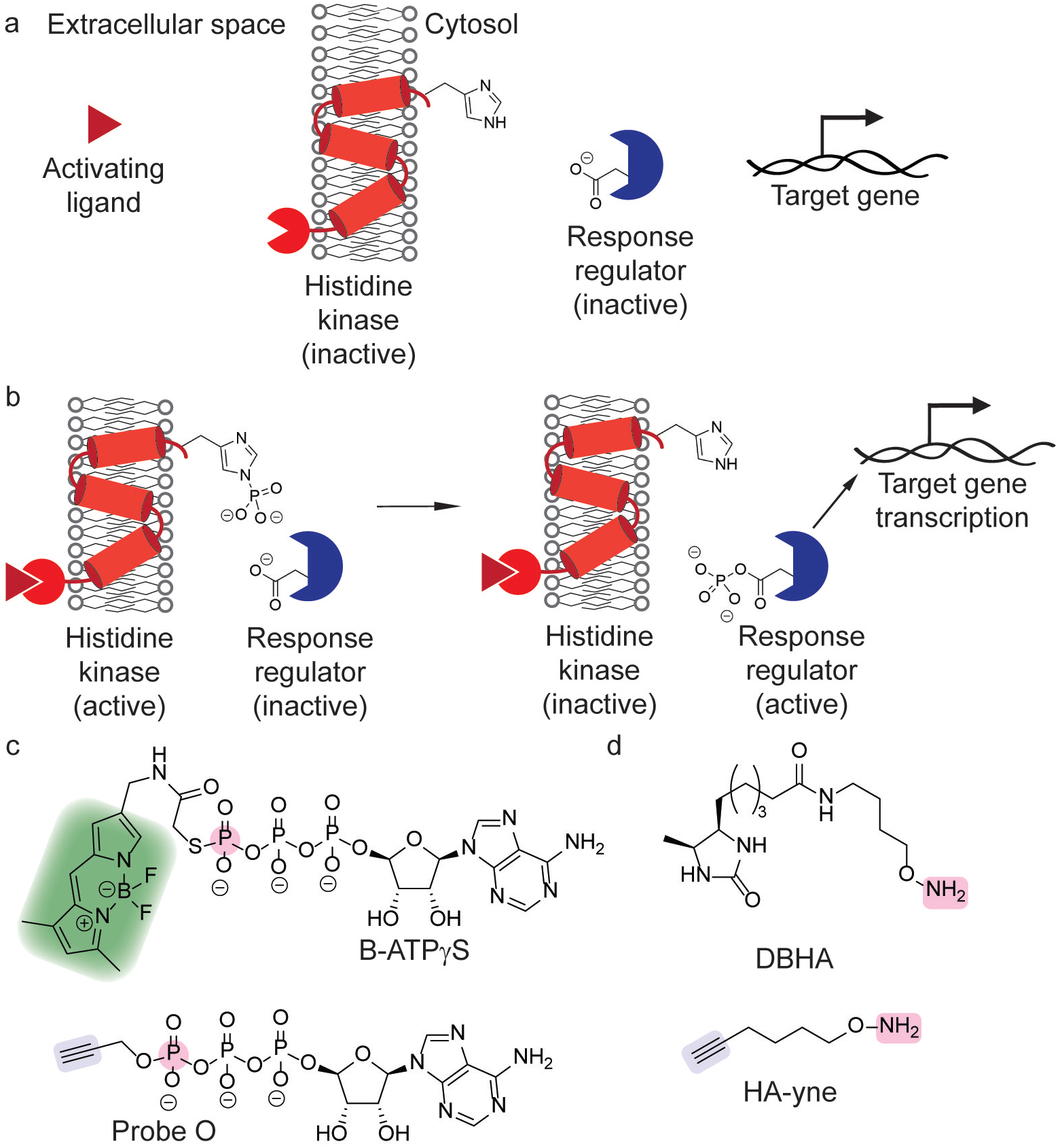
Chemical tools for studying phosphorylation in two-component systems (TCS). (a) Unstimulated two-component system and inactive target genes. (b) Gene activation through the stimulation of a TCS by the activating ligand. (c) ATP analogs for monitoring TCS phosphorylation append a chemical reporter through γ-phosphate (pink) transfer of a fluorophore (green)[80] or alkyne click handle (blue).[81] (d) Nucleophilic hydroxylamine (pink) probes for profiling aspartate phosphorylation utilize a desthiobiotin[82] or alkyne (blue)[83] reporter.
To overcome this challenge, initial proteomic studies for monitoring PHK signaling relied on a less reactive, thiophosphate analog of ATP, whose thiophosphate adduct hydrolyzes much more slowly (B-ATPγS, Figure 8c). The pendant fluorophore of B-ATPγS enabled activity-based profiling of PHK phosphorylation.[80] Furthermore, this activity-based profiling strategy probed phosphate transfer to a response regulator using purified proteins.[80] Biochemical studies showed that thiophosphate analogs are bound similarly and are hydrolyzed more slowly, whereas the introduction of sterically bulkier moieties to the γ-phosphate decreased binding.[84] Additional ATP probes with biorthogonal handles, such as probe O (Figure 8c) expanded on this methodology.[81]
Chemoproteomic profiling of phospho-aspartate modifications has emerged as a complementary strategy for studying two-component systems. While glutamate and aspartate residues are nucleophilic and react with nitrilimines generated from photolyzed 2,5-disubstituted tetrazoles,[85] phospho-aspartate residues are highly electrophilic. Hydroxylamine ethers are suitable nucleophiles for trapping acyl phosphate intermediates, which identified novel phosphorylation sites and quantification of E. coli protein phosphorylation changes in response to osmotic stress using DBHA (Figure 8d).[82]
Chemical proteomic studies in PA also identified a two-component system for PA virulence in response to dynorphin, a mammalian cellular stress hormone. After characterization of dynorphin-induced pyocanin induction, photoaffinity labeling with dynorphin analogs identified ParS, a two-component sensor kinase, as a target PA protein of dynorphin that mediates PA virulence.[86] Subsequent quantitative phospho-aspartate profiling studies then identified the response regulator that relays the ParS signal.[83] Specifically, quantitative proteomics with HA-yne (Figure 8d) identified that dynorphin induces phosphorylation of CprR, cationic peptide resistance regulator, in PA.[83] Taken together, chemoproteomic strategies for studying two-component systems have overcome significant methodological challenges and have successfully been applied to sensory responses in systems relevant to environmental and infection contexts.
Chemoproteomic tools for the characterization of PTMs have facilitated the study of acylation, alkylation, redox modifications, and phosphorylation. These PTM profiling approaches have revealed previously unknown protein modifications that are important for regulating enzymatic and transcriptional activity. These tools have elucidated novel host-pathogen interactions and colonization resistance mechanisms, highlighting their utility.
3. Interrogating Bacterial Pathogenesis
Chemical proteomic strategies have been utilized for identifying proteins involved in bacterial pathogenesis. Specifically, chemoproteomic profiling has identified transcription factors that regulate virulence, aided in the discovery of inhibitors of virulence factors, and led to the discovery of novel virulence factors.
Quorum sensing is an important cell-cell communication process in bacteria, regulating group behaviors including biofilm formation and virulence factor production (Figure 9a).[90] Quorum sensing is regulated by small molecule autoinducers whose detection frequently promotes its own biosynthesis, leading to an amplified group response at high cell densities.[90] Due to its role in regulating virulence, the design of small molecule quorum sensing modulators has emerged as an active area of research.[91] Photoaffinity labeling has been successfully employed as a strategy for the chemoproteomic profiling of quorum sensing receptors, as the probe photo-AHL produced similar phenotypic effects to 3-oxo-C12- AHL and labeled its cognate receptor (Figure 9b).[88] Short-chain AHL analogs have also been developed as photoaffinity probes for quorum sensing receptors.[92] ABPP studies with FP-3 (Figure 9b) validated the role of fimbrolides as inhibitors of autoinducer biosynthesis in Vibrio harveyi.[89]
Figure 9.
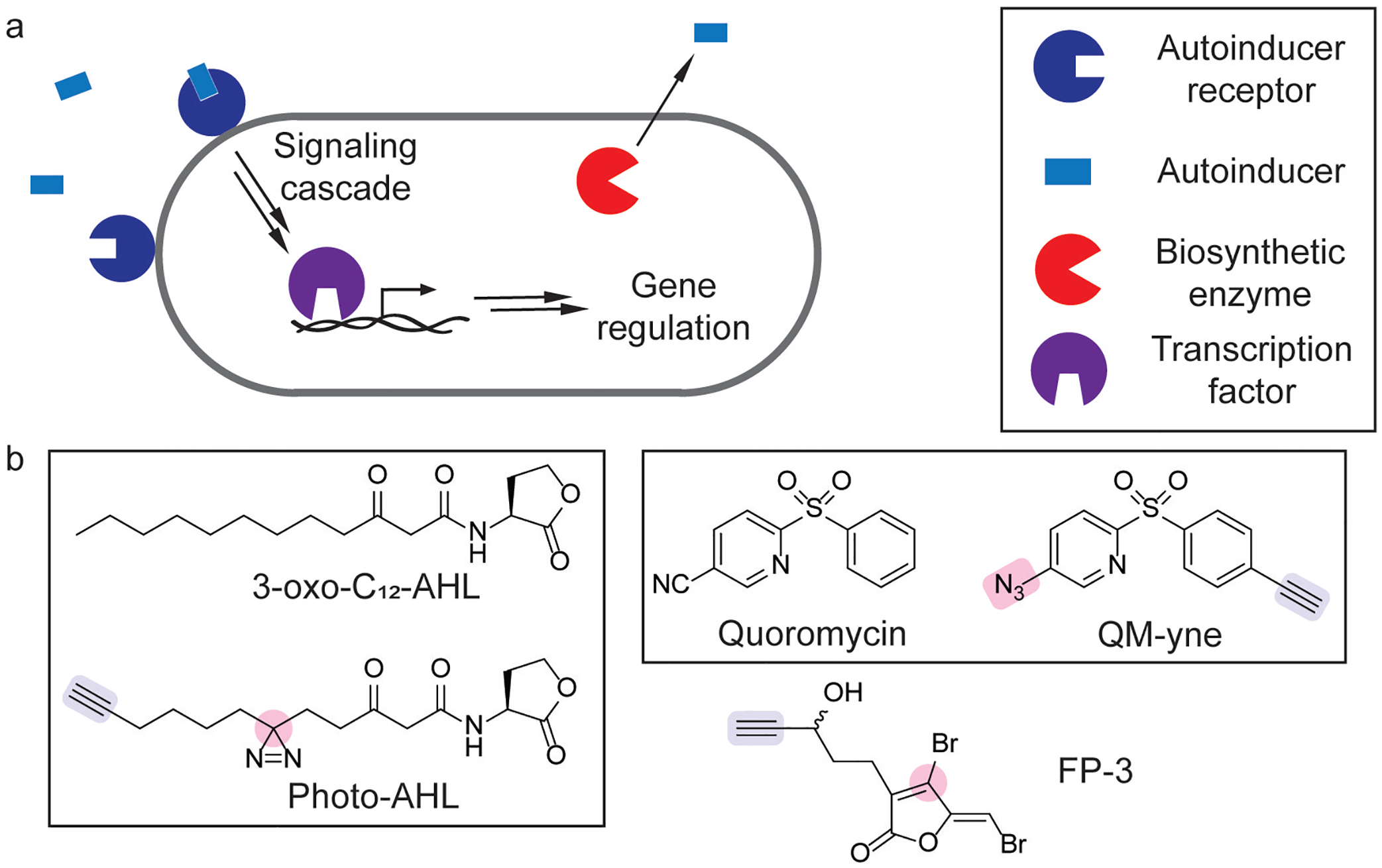
Chemoproteomic tools for studying quorum sensing. (a) Schematic of quorum sensing signaling cascade in a bacterium that is initiated by an autoinducer binding its cognate receptor. (b) Quorum sensing modulators and chemical probes that contain a protein crosslinker (pink) and click handle (blue).[87–89]
Photoaffinity labeling has also been used to validate the cellular target of a quorum sensing inhibitor in Vibrio vulfinicus, an opportunistic bacterial pathogen that causes sepsis.[93] In an effort to attenuate V. vulfinicus virulence, a high-throughput screen of small molecules identified a diarylsulfone, quorumycin, that inhibited collagenase activity (Figure 9b).[87] To identify the target of this diarylsulfone, a photoaffinity analog QM-yne (Figure 9b) identified SmcR, which was validated as a quorum sensing transcription factor.[87] Further studies showed that quorumycin attenuated infection in a rabbit model.[87]
Chemical proteomics has also been important in validating the cellular targets for inhibitors of Mtb virulence. For example, Hip1 is a serine hydrolase that attenuates activation of innate immunity during infection.[94] The development of fluorogenic probes for monitoring Hip1 activity enabled the screening of a small-molecule library to identify inhibitors of Hip1.[95] The selectivity of these inhibitors for Hip1 relative to host proteins was examined by ABPP, with the most promising inhibitors showing few off-target effects on host proteins within macrophages.[95] These inhibitors also successfully inhibited Hip1 in live Mtb cells and in macrophages infected with Mtb.[95]
ABPP with broad-spectrum FP probes has enabled the discovery of novel virulence factors in SA[96] and V. cholerae.[97] These virulence factors, both of which are serine hydrolases, have diverse distribution, biochemical activities, and mechanisms of regulation. Because ABPP identifies catalytically active enzymes, chemoproteomic profiling identified these virulence factors despite their disparate biological characteristics.
Global ABPP of SA serine hydrolases with FP-TMR (Figure 10a)[98] was used in conjunction with competitive ABPP to identify serine hydrolases susceptible to pharmacological inhibition.[96] Proteomic identification using FP-biotin (Figure 3b) and subsequent genetic knockout studies of the enriched proteins identified novel FP-binding serine hydrolases, termed Fph proteins. Biochemical characterization of the most-enriched protein, FphB, demonstrated it is a fatty acid esterase whose activity is stimulated by macrophages.[96] Furthermore, profiling with a FphB-selective fluorescent ABP, JCP251-bT, showed activity in pathogenic SA strains and a non-pathogenic Staphylococcus species (Figure 10b).[96] Lastly, genetic disruption of FphB attenuated SA infection in mice, [96] and the structure of FphB was also characterized.[99]
Figure 10.
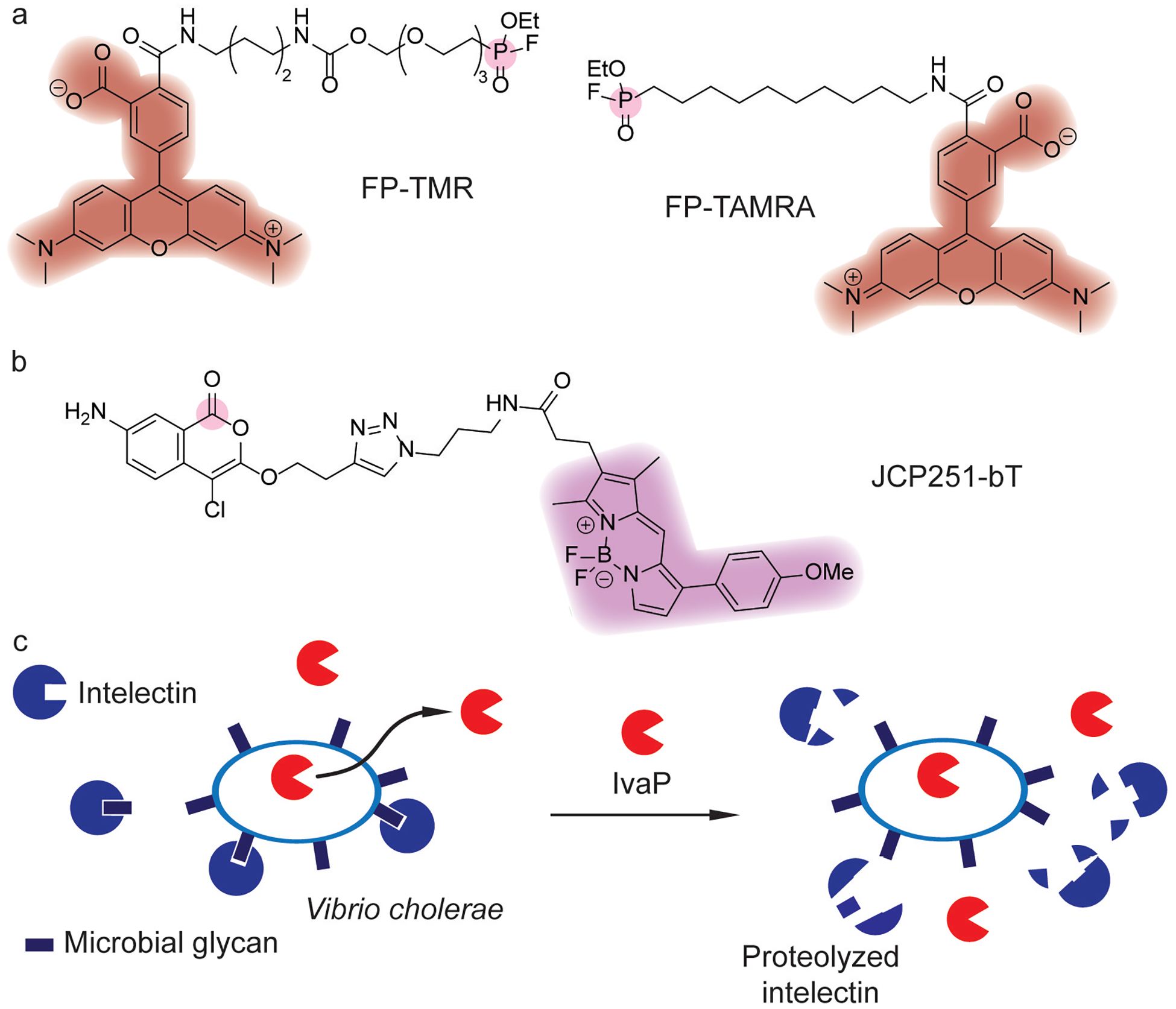
Discovery of bacterial virulence factors using fluorophosphonate (FP) probes.[98] (a) Broad-spectrum fluorophosphonate probes for profiling serine hydrolases utilize a serine-selective fluorophosphonate warhead (pink) coupled with a TAMRA fluorophore (red)[98] for visualization. (b) Activity-based probe for imaging FphB activity using a chloroisocoumarin warhead (pink) and fluorophore (magenta).[96] (c) IvaP in Vibrio cholerae proteolyzes intelectin, which may aid in evading host defense.
Building on this discovery, additional fluorescent triazoleurea ABPs were identified as FphE-selective probes.[100] These FphE-selective probes facilitated single-cell characterization of activity and demonstrated FphE activity in different SA strains depended on growth conditions. FbhE, along with FphB and FphH, have also been identified as the cellular target of oxadiazolones that inhibit the growth of MRSA.[101]
Active Fph homologs are found across numerous Staphylococcus spp., including Staphylococcus epidermis,[102] a commensal skin bacterium that regulates skin barrier integrity.[103] Accordingly, the effects of SA FphB inhibitors on commensal S. epidermis strains were characterized.[102] Although inhibitors of SA FphB also inhibited S. epidermis homologs in commensal skin, further studies showed that genetic deletion and pharmacological inhibition did not affect growth or colonization in vitro and in vivo, respectively. Overall, ABPP in Staphylococcus species has led to the discovery and characterization of FP-binding serine hydrolases. Further biological characterization will provide insights into the role of these enzymes during infection.
In addition, ABPP with FP-TAMRA (Figure 10a) and FP-biotin (Figure 3b) of cecal fluid in a rabbit model of V. cholerae infection uncovered novel serine proteases.[97] Among the V. cholerae proteases identified in the rabbit infection model, an in vivo activated protease (IvaP) was active in clinical choleric stool samples (Figure 10c). IvaP was found to undergo extensive proteolytic processing, decrease the activity of host serine hydrolases, and alter cecal lipid contents. Further biochemical studies investigated the maturation mechanism of IvaP,[104] demonstrating that IvaP undergoes extensive intramolecular proteolytic processing at N- and C-terminal domains. This autoproteolytic processing depended on growth phase and was also regulated by an N-terminal inhibitor peptide domain. These studies also validated that IvaP inhibits intelectin binding to V. cholerae through proteolytic degradation (Figure 10c), which may play a role in intestinal immunity.
Overall, while ABPP has aided in the discovery and validation of modulators of bacterial pathogenesis, it has also uncovered diverse enzymes that drive virulence. Further functional characterization of these enzymes will elucidate their role in pathogenesis, as well as in non-infectious contexts.
4. Interrogating Prokaryotic Metabolism
4.1. Carbohydrate Metabolism
Carbohydrates are an important source of energy for prokaryotes, and the catabolism of simple carbohydrates has been extensively characterized in bacteria at the biochemical and genetic level.[105] Initial chemoproteomic methodologies for profiling bacterial carbohydrate metabolism focused on photoaffinity labeling strategies of enzymes that degrade disaccharides. Specifically, clickable benzophenone photoaffinity probes first enabled chemoproteomic profiling of exo-α-glucosidases in cellular lysates, which was also suitable for labeling yeast exo-α- glucosidases.[106]
Prokaryotes can degrade a variety of complex carbohydrates, which has led to application of bacterial enzymes to convert complex carbohydrates into feedstock chemicals.[107] To explore these applications, ABPP has been utilized to characterize bacterial catabolism of chitin.[107] In Cellvibrio japonica, this chemoproteomic platform allowed for the temporal resolution of chitinase activity and provided insight into how chitin-degrading enzymatic activity changes with carbon source availability. Chemoproteomic profiling of bacterial cellulose metabolism demonstrated the diversity of enzymes involved in this process. In Clostridium thermocellum, a panel of ABPs containing mono- and di-saccharide substrate analogues was used to identify stereospecific glycoside hydrolases and proteins involved in the C. thermocellum cellulosome.[108] In the thermophilic archaeon, Thermococcus sp. strain 2319×1E, ABPP using cyclophellitol aziridine-derived probes identified xylan-degrading enzymes with activities at high temperatures.[109] Collectively, these works highlight how ABPP has provided insight into the complex regulation of carbohydrate degradation by prokaryotes.
Additionally, bacterial metabolism of cellulose is important in mammalian gut microbiomes. The degradation and fermentation of fiber-derived carbohydrates in the gut produces SCFAs.[110] These SCFAs are important gut microbial metabolites because they modulate host epigenetic regulation and mucin production,[110] which contributes to intestinal barrier integrity. Gut microbes are also capable of regulating the mucus layer by hydrolyzing fucose linkages found on these glycans.[111] A probe for α-L-fucosidase activity enabled chemoproteomic profiling of these enzymes in two commensal human gut bacteria, Bacteriodes fragilis and Bifidobacterium bifidum.[112] This platform may enable a deeper understanding of the role of gut microbial α-L-fucosidases in disease.
Overall, advances in chemoproteomic profiling of prokaryotic carbohydrate metabolism span a variety of biological contexts. More recent advances have focused on chemoproteomic platforms for characterizing the degradation of complex carbohydrates by prokaryotes. These chemical tools may provide further insight into prokaryotic carbohydrate metabolism across different biological states that have important implications for human health and biotechnological applications.
4.2. Metabolite-Protein Interactions
In addition to serving as biosynthetic precursors, small molecule metabolites serve many roles in modulating metabolism, such as acting as cofactors and signaling molecules. These metabolites may form noncovalent complexes, preventing the use of traditional ABPs. To address this challenge, chemoproteomic tools for interrogating these transient metabolite-protein interactions have largely relied on photoaffinity labeling strategies.
For example, bifunctionalized thiamine (B1-ABP), riboflavin (B2-ABP), and biotin (B7-ABP) analogs bearing a diazirine photoaffinity handle and clickable alkyne handle have been developed (Figure 11).[113] In addition to labeling known, purified binding proteins, B1-ABP and B2-ABP enabled protein labeling in live Chloroflexus aurantiacus, which scavenges these B vitamins.[113] This live-cell labeling strategy successfully identified transporters and enzymes involved in thiamine and riboflavin metabolism.[113] Furthermore, proteomic analysis identified other proteins that both probes labeled, as well as enzymes for which these B vitamins may be a cofactor.[113] Other photoaffinity probes for folate (Folate-photoclick, Figure 11)[114] and sialic acid (Sialic-photo-click, Figure 11)[115] have been developed and identified bacterial protein binding partners in E. coli and murine gut microbiome isolates, respectively. This strategy has also been applied to identify the cellular targets of the antibacterial natural product, promysalin[116] and cajaninstilbene acid.[117]
Figure 11.
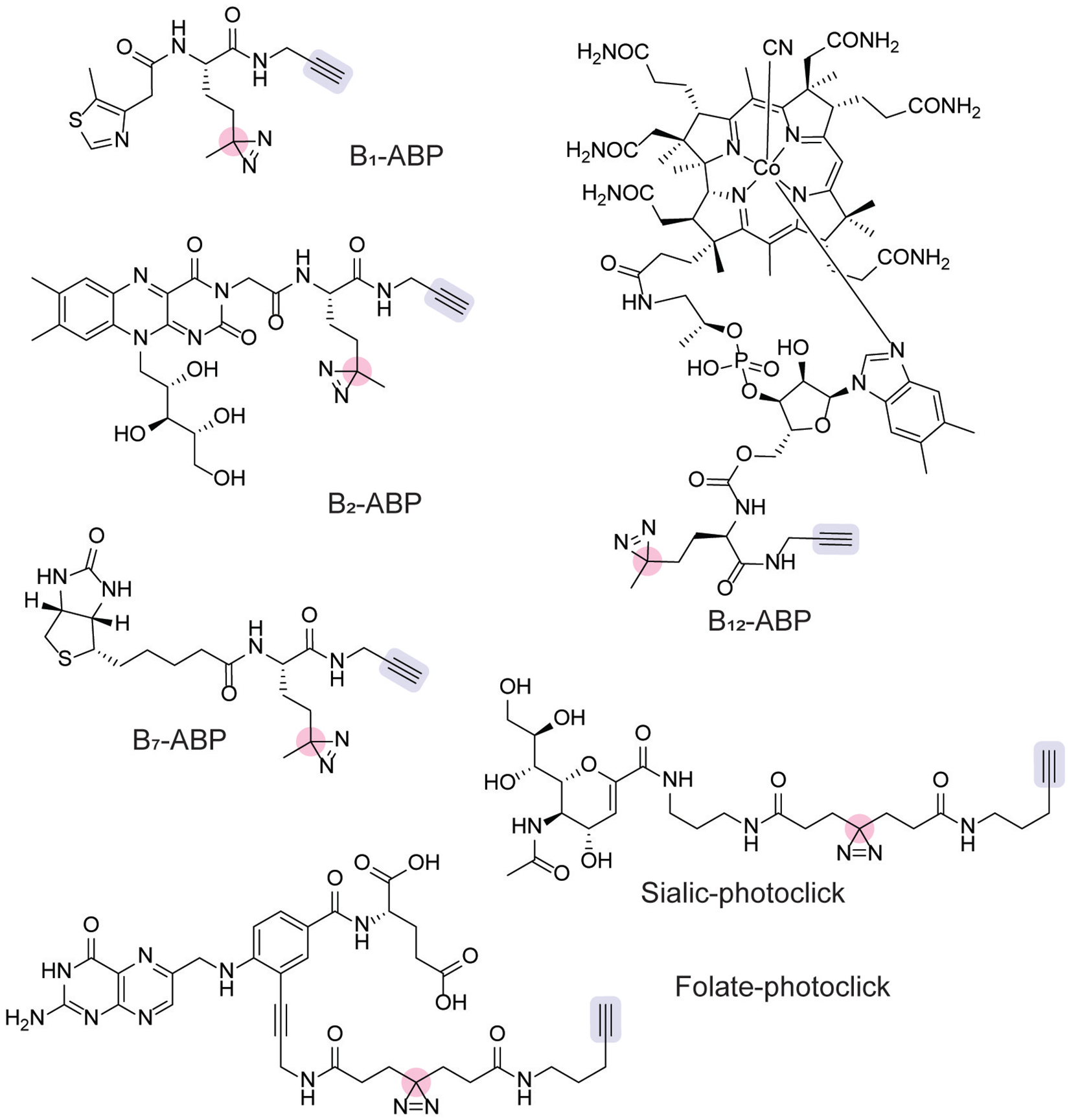
Photoaffinity probes used to identify metabolite-protein interactions [113–115,118] with click chemistry handles (blue) and reactive photocrosslinkers (pink) indicated.
Cobalamin, or vitamin B12, is an important cofactor and dietary nutrient. Photoaffinity labeling has proven fruitful for the identification of vitamin B12-binding proteins. B12-ABP (Figure 11) enabled photoaffinity labeling and click chemistry-based proteomics by the incorporation of a diazirine and alkyne moiety, respectively.[118] B12-ABP recapitulates the effects of vitamin B12 in a variety of bacteria, acting as a surrogate for vitamin B12-dependent growth and regulating gene expression in a similar manner to vitamin B12.[119] Proteomic characterization in E. coli and Rhodobacteracae sp. strain HL-21 identified novel cobalamin-binding proteins, as well as previously known vitamin B12-binding proteins. Notably, the application of this proteomic platform to Halomonas sp. HL-48 uncovered roles for vitamin B12 in folate, methionine, and ubiquinone metabolism.[118] Furthermore, this proteomic study identified the transcriptional regulator, PhrR, as a candidate cobalamin binding protein, and additional mechanistic experiments showed that vitamin B12 enables DNA binding in a light-dependent fashion. The characterization of this probe in the human gut bacterium, Bacteroides thetaiotamicron, identified a novel B12 binding protein, BtuH2, that increased transport of cobalamin precursors.[120] Crystallographic studies validated vitamin B12 binding with BtuH2, and genetic studies showed BtuH2 deletion reduced growth in vitro and colonization in vivo.[120] In summary, the chemoproteomic profiling of cobalamin interactions in bacteria has illuminated novel roles for vitamin B12 in regulating cellular metabolism, gene regulation, and colonization in mammalian hosts.
Additionally, the targeting of reactive lysine residues has been used to identify bacterial ATP binding proteins. The ε-amino group on the side-chain of lysine residues is a nucleophilic moiety susceptible to targeting by electrophilic probes (Figure 12a).[121,122] Leveraging this reactivity, ATP mixed anhydrides have been employed as N-acylating probes for the chemoproteomic identification of nucleotide-binding proteins. Using desthiobiotin-ATP (Figure 12a),[122] the ATP-binding proteome in Mtb was characterized using biotin enrichment during both aerobic and hypoxic growth.[123] This chemoproteomic strategy identified increased ATP-binding signal transduction pathways during hypoxic growth, as well as the decreased abundance of lipid metabolism-associated proteins.
Figure 12.
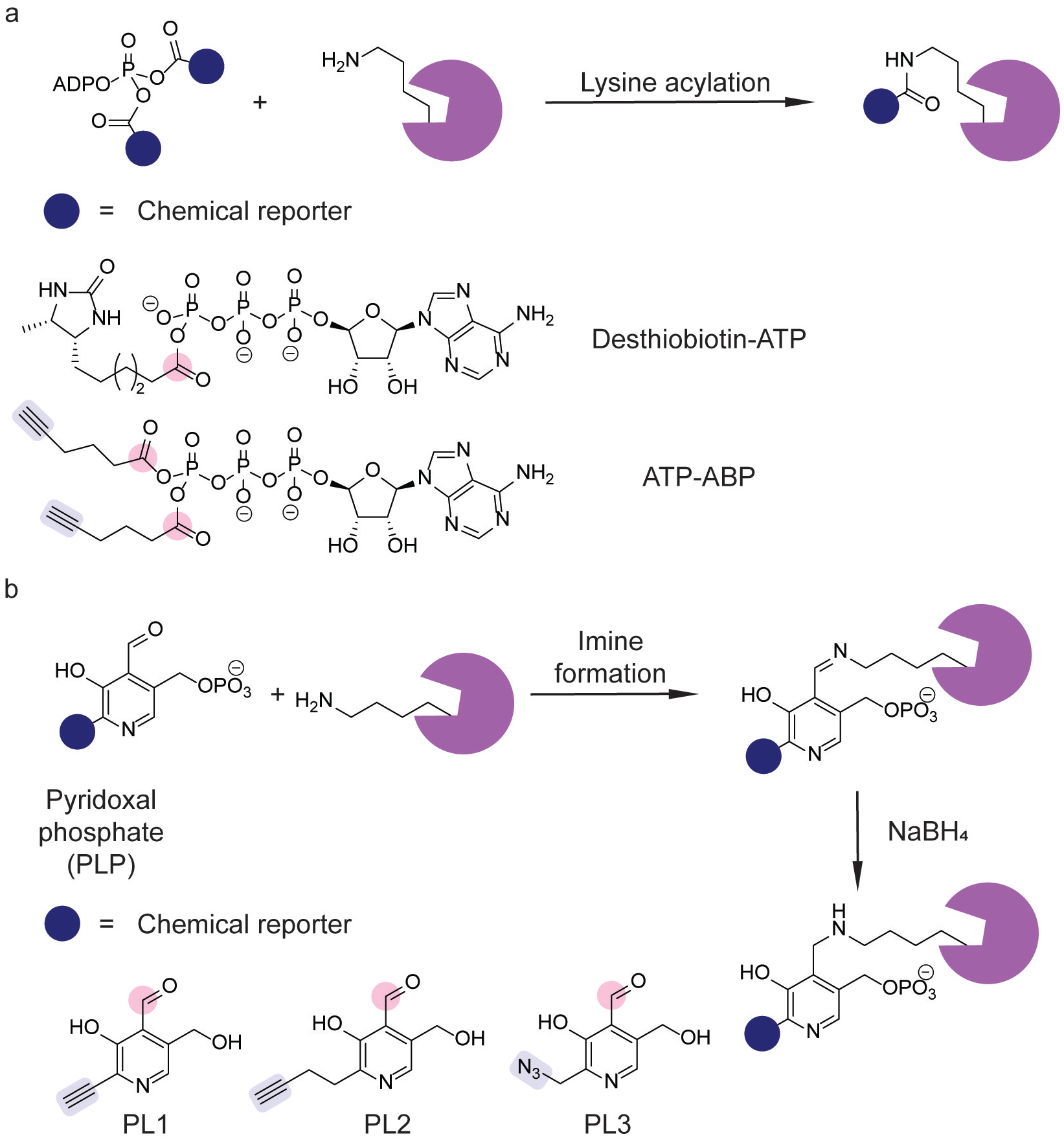
Chemoproteomic platforms exploiting lysine reactivity. (a) Chemical probes for identifying ATP-binding proteins.[122,124] (b) Chemical probes for profiling pyridoxal phosphate (PLP)-interacting proteins.[127] Lysine-reactive electrophiles (pink) and click chemistry handles (blue) are indicated.
The development of a platform using ATP-ABP (Figure 12a) for Mtb ATP-binding proteins identified previously described and novel ATP-binding proteins, and click chemistry-mediated fluorescent labeling allowed for further validation by competitive ABPP.[124] Competitive ABPP using this probe also validated serine/threonine protein kinases as the target of the broad spectrum kinase inhibitor staurosporine, which compromised the viability of Mtb transitioning from hypoxic to aerobic growth.[125] In particular, quantitative proteomics identified decreases of the protein kinases PknB, D, F, and H upon staurosporine treatment. Further genetic studies showed PknB as a regulator of oxygen-dependent replication.[125]
Pyridoxal phosphate (PLP) is an important cofactor for a variety of metabolic transformations.[126] The reactivity of PLP frequently relies on the electrophilic aldehyde moiety, which can form transient aldimine intermediates. These intermediates can form from a primary amine in a metabolite or lysine residue. The formation of lysine-aldimine adducts enabled chemoproteomic profiling of PLP-interacting proteins (Figure 12b).[127] Specifically, pyridoxal analogs PL1–3 (Figure 12b) underwent phosphorylation to form PLP analogs that formed protein-aldimine adducts; subsequent reduction with sodium borohydride formed stable PLP adducts that were characterized by click chemistry-based proteomics.[127] Chemoproteomic profiling in SA using PL2 identified previously uncharacterized PLP-dependent enzymes and characterized the cellular targets of D-cycloserine, a known broad-spectrum inhibitor of these enzymes, through competition experiments.[127] The development of additional pyridoxal analogs identified novel antibiotic compounds.[128]
Metabolite-protein interactions occur through diverse mechanisms, including the formation of noncovalent and transient covalent species. These interactions may be intractable to probe design strategies relying on electrophilic trapping of an active site nucleophile. To overcome this challenge, photoaffinity and reactive lysine labeling strategies have been developed. These chemical proteomic strategies have uncovered novel metabolite-protein interactions involved in gene regulation, cellular signaling, and metabolism, which affect bacterial fitness in diverse contexts.
4.3. Natural Product Biosynthesis
Bacteria produce a wide range of small molecule metabolites.[129] These natural products have a variety of biological effects, including antimicrobial, anticancer, and immunosuppressive activity.[129] As a result, the identification of novel natural products and understanding their biosynthesis has implications for the discovery of new drugs[130] and exploration of chemical space.
Important classes of bacterial natural products include polyketides (PKs) and nonribosomal peptides (NRPs). PK and NRP biosyntheses are carried out by multienzyme complexes that iteratively synthesize these natural products through the activity of specific enzymatic modules, which vary in substrate specificity and catalytic activity (Figure 13a).[131,132] These biosynthetic processes occur through covalently bound intermediates. Whereas PK biosynthesis begins with loading acylcoenzyme A (acyl-CoA) precursors from the cellular pool of acyl-CoA substrates, NRP biosynthesis is initiated by loading an activated carboxylic acid, adenosine monophosphate (AMP) anhydride at an adenylation domain.[131] Acyl carrier proteins, using a pantothenic acid prosthetic group, then tether the intermediates via a thioester linkage.[131] Other domains act on these intermediates, such as dehydratases and reductases, and the final product is cleaved through a thioesterase domain.[131]
Figure 13.
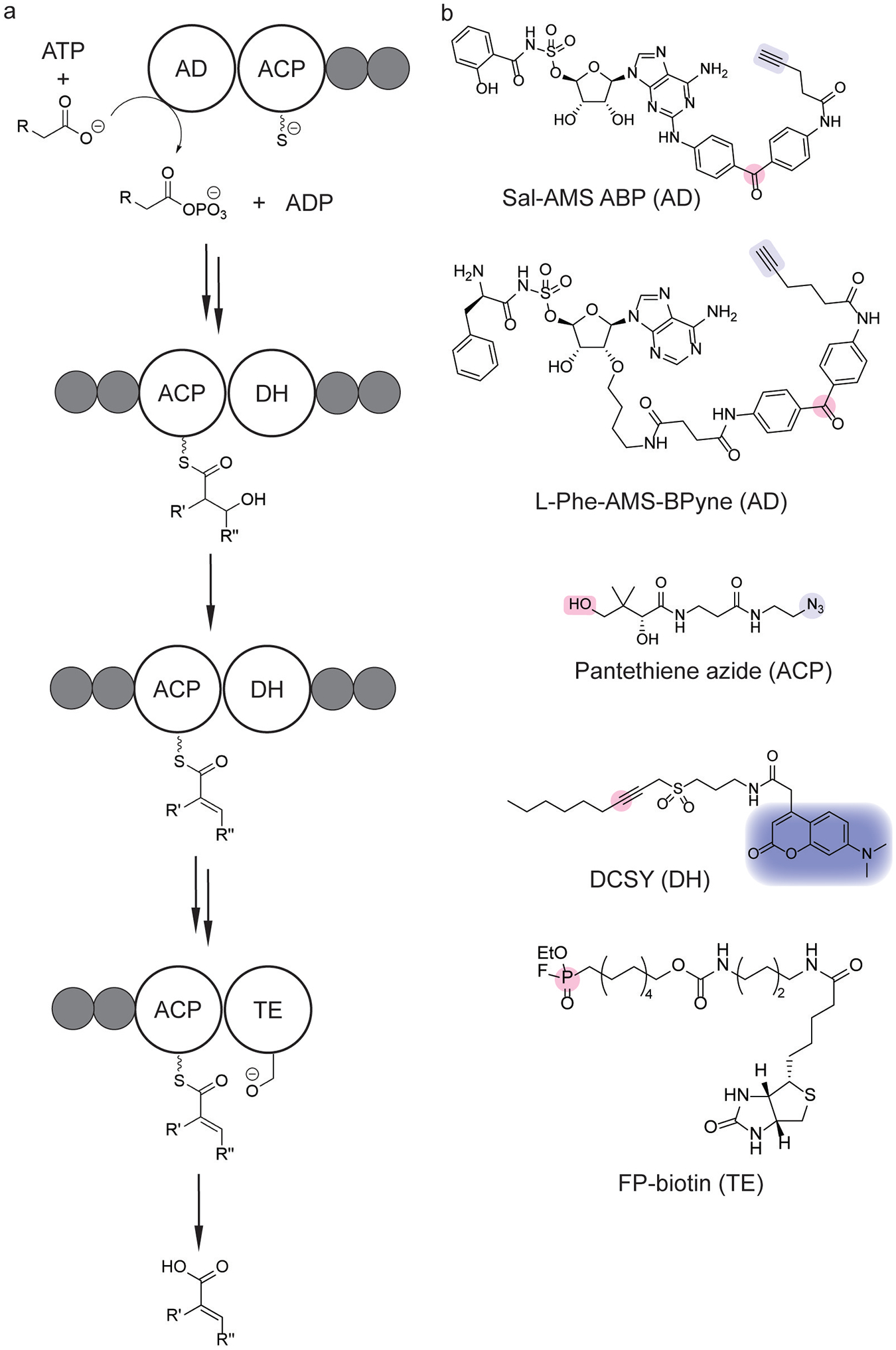
Probing modular nonribosomal peptide (NRP) and polyketide (PK) biosyntheses. (a) Scheme of biosynthetic pathway. (b) Chemical probes for NRP and PK biosynthetic modules utilize photoaffinity,[133,135] metabolic,[140] or activity-based labeling strategies (pink).[6,137] Coupling these labeling moieties with a click handle (light blue),[133–135,139] fluorophore (dark blue),[137] or biotin[6] enables proteomic profiling and identification. The target domain of each probe is indicated in parentheses. AD=adenylation domain; ACP=acyl carrier protein; DH=dehydratase domain; TE=thioesterase domain.
As adenylation is the first step in NRP biosynthesis, the identification of adenylation domains presents a strategy for the identification of enzymes involved in these biosynthetic pathways. Photoaffinity labeling has been one such strategy developed for the chemoproteomic identification of adenylation domains.[133] In particular, the development of L-Phe-SAMS-BPyne (Figure 13b) allowed photoaffinity labeling of adenylation domains involved in the biosynthesis of the antibiotics tyrocidine and gramicidin S.[133] This platform was then extended to label multiple adenylation domains in a gramidicin S synthetase by concurrent labeling with different photoaffinity probes.[134] In a similar strategy, Sal-AMS ABP (Figure 13b) was used to identify proteins involved in the biosynthesis of siderophores derived from salicylic acid precursors.[135] Photoaffinity labeling of adenylation domains has also revealed proteolytic degradation of the NRP synthase of surfactin in live Bacillus subtilis cells.[136] Additional chemoproteomic strategies have been developed to probe alternative domains involved in natural product synthesis. For example, the development of DCSY (Figure 13b) enabled chemoproteomic profiling of dehydratase domains responsible for fatty acid biosynthesis in E. coli and Mtb.[137] FP-biotin (Figure 13b) and pantethiene azide (Figure 13b) have similarly been utilized for chemoproteomic profiling of thioesterase and acyl carrier protein domains, respectively.[138,139]
Overall, numerous chemical proteomic strategies have been developed to probe processes involved in natural product biosynthesis. These complementary approaches highlight the diverse chemistry involved in natural product biosynthesis and wide range of chemoproteomic platforms available.
5. Microbial Metabolism in the Gut Microbiome
The human intestine is colonized by the gut microbiome, a diverse and dynamic community of hundreds of trillions of microbes.[110] The gut microbiota carry out numerous metabolic functions, including the fermentation of otherwise indigestible dietary compounds and the production of molecules that protect against infection and inflammatory disease.[110] In conjunction with genomic and pharmacological strategies, chemical proteomics has aided in the identification of proteins targeted by gut microbial metabolites, as well as the identification of enzymes involved in gut microbial metabolism. These strategies together allow for the elucidation of host-microbe and microbe-microbe interactions at a molecular level.
Inflammatory bowel disease (IBD) is a widespread disease that afflicts millions of people worldwide.[141] Aberrant protease activity in the gastrointestinal tract is an increasingly appreciated aspect of IBD and one target for therapeutic intervention.[141] In a murine model of IBD, quantitative proteomics of fecal samples revealed the increased production of host protease inhibitors and a large number of uncharacterized gut microbial proteins.[142] Activity-based enrichment with chloromethylglycine-biotin led to the proteomic identification of putative microbial cysteine proteases.[142] Along these lines, ABPP of serine hydrolase activity in healthy and ulcerative colitis patient fecal samples led to the enrichment of putative host and microbial serine proteases in ulcerative colitis patients.[143] Together, these results highlight the complexity of the gut metaproteome and utility of ABPP as a proteomic enrichment strategy to understand the role of specific enzyme classes in disease.
Human gut microbiota also produce metabolites that inhibit host protease activity.[144] The elucidation of gut microbial NRPS biosynthetic gene clusters revealed the production of dipeptide aldehydes across numerous human gut bacteria.[144] Subsequent target identification in mammalian cells by competitive ABPP revealed cathepsins B, C, L, and S as host proteases that these dipeptides inhibited.[144] Accordingly, ABPP has elucidated the interplay between gut microbial metabolism and host protease activity.
Gut microbiota produce a wealth of metabolites derived from precursors ingested by the host.[145] These precursors include dietary components, such as amino acids[146] and fiber.[110] Especially salient, however, is the ability of gut microbiota to metabolize pharmaceuticals and their derivatives,[147] which exhibits variability between individuals.[148] Although genome mining and biochemical characterization remain important approaches for characterizing these microbial enzymes,[148] enzymatic activity in biochemical assays may differ in cultures[149] and in vivo. As a result, ABPP methodologies have emerged as a useful strategy for identifying the microbial enzymes actively catalyzing these metabolic transformations in gut microbial communities.
Chemoproteomic tools for profiling xenobiotic metabolism have identified gut microbial enzymes that induce drug toxicity through reactivation. Mammalian drug metabolism involves numerous transformations, including redox modifications, hydrolysis, and conjugation.[150] The conjugation of glucuronic acid onto drugs, glucuronidation, is an important transformation for increasing the water solubility of drugs, thereby promoting excretion in the urine and feces.[150] Glucuronidation is a detoxification pathway for the anticancer drug irinotecan, which can be reactivated in the gut by microbial β-glucuronidases into a toxic metabolite.[151] β-glucuronidase inhibition reverses this reactivation-induced toxicity.[151] The use of GUS-Biotin-ABP and GUS–Cy5-ABP (Figure 14a)[152] identified bacterial β-glucuronidases in human fecal microbiota, which revealed diversity of these enzymes across different genera and variation in enzyme activity between individuals.[153]
Figure 14.
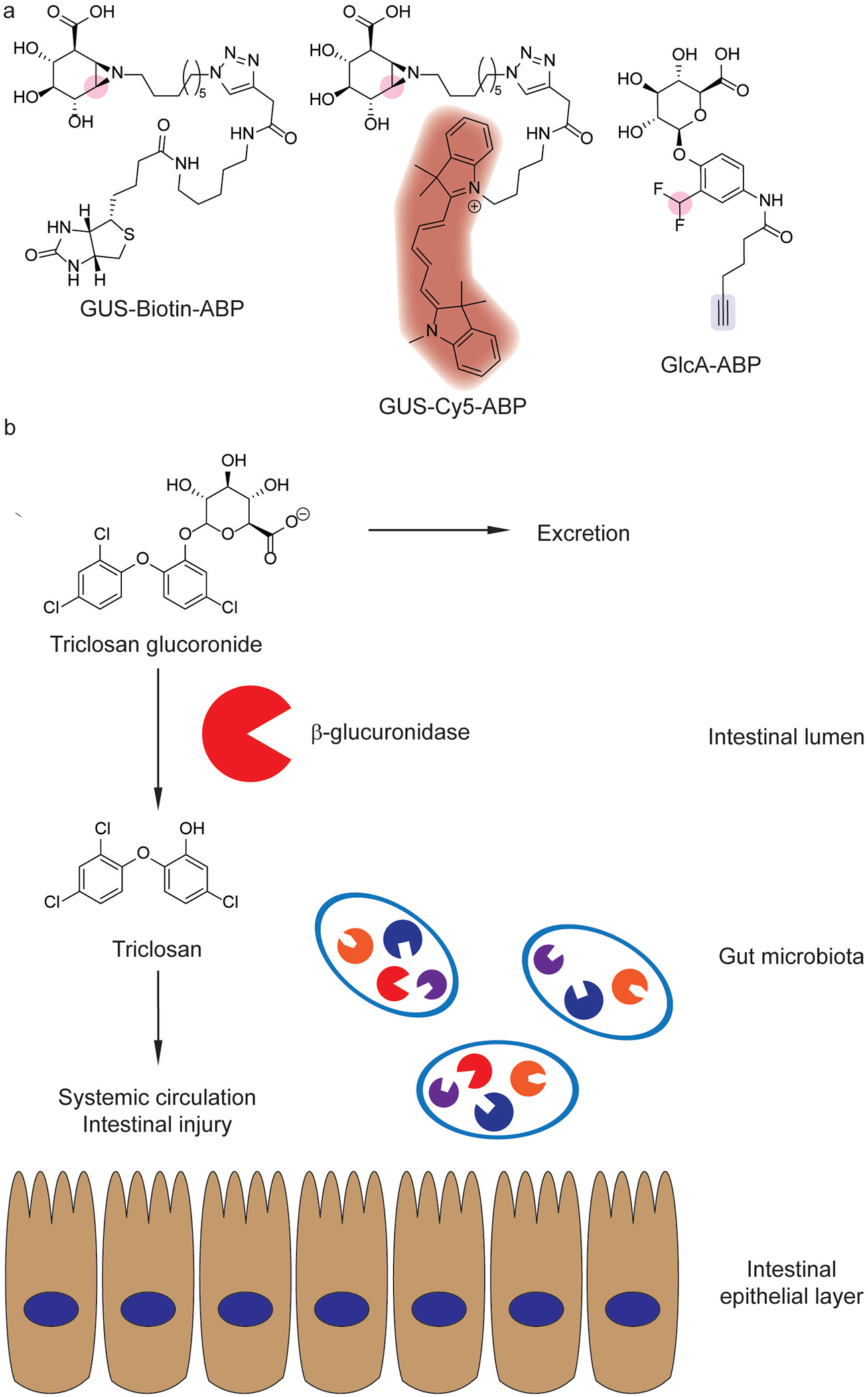
Gut microbial β-glucuronidases in xenobiotic metabolism. (a) Activity-based probes for β- glucuronidases [152,154] with fluorophores (red), enzyme-reactive electrophiles (pink) and click handles (blue) indicated. (b) Triclosan reactivation and reabsorption by gut microbial β-glucuronidases.[155]
Triclosan is an antimicrobial in consumer products whose ubiquitous use is being reevaluated in light of toxicity concerns.[156] Although triclosan is inactivated and secreted as its glucuronide (Figure 14b),[156] further studies show that subclasses of bacterial β-glucuronidases reactivate triclosan in mice.[155] Human fecal isolates reactivated triclosan ex vivo, which correlated with the abundance of active β-glucuronidases identified using GUS-Biotin-ABP.[155] Furthermore, triclosan administration in mice exacerbated dextran sodium sulfate (DSS)-induced colitis, which was reversed upon bacterial β-glucuronidase inhibition.[155]
ABPP has also been extended to the isolation of specific gut microbes with active β-glucuronidase enzymes.[154] GlcA-ABP (Figure 14a) fluorescently labeled cells with active β-glucuronidases, which enabled fluorescence-activated cell sorting for bacterial enrichment.[154] When applied to mouse intestinal contents, this platform characterized the interindividual variability of gut microbial β-glucuronidase activity, which decreased upon antibiotic administration.[154]
Chemoproteomic profiling has also been important in elucidating the roles of gut microbial bile acid metabolism in disease. Bile acids are cholesterol-derived carboxylic acids that are conjugated in the liver to glycine or taurine.[157] When secreted into the intestine, these conjugated bile acids act as detergents and aid in the emulsification of dietary lipids and fat-soluble vitamins.[157] In the intestine, microbial bile salt hydrolases (BSHs) hydrolyze the amide bond, producing unconjugated bile acids that undergo further chemical modifications (Figure 15a).[157] These microbially-produced bile acids are important modulators of host physiology and immunity.[158]
Figure 15.
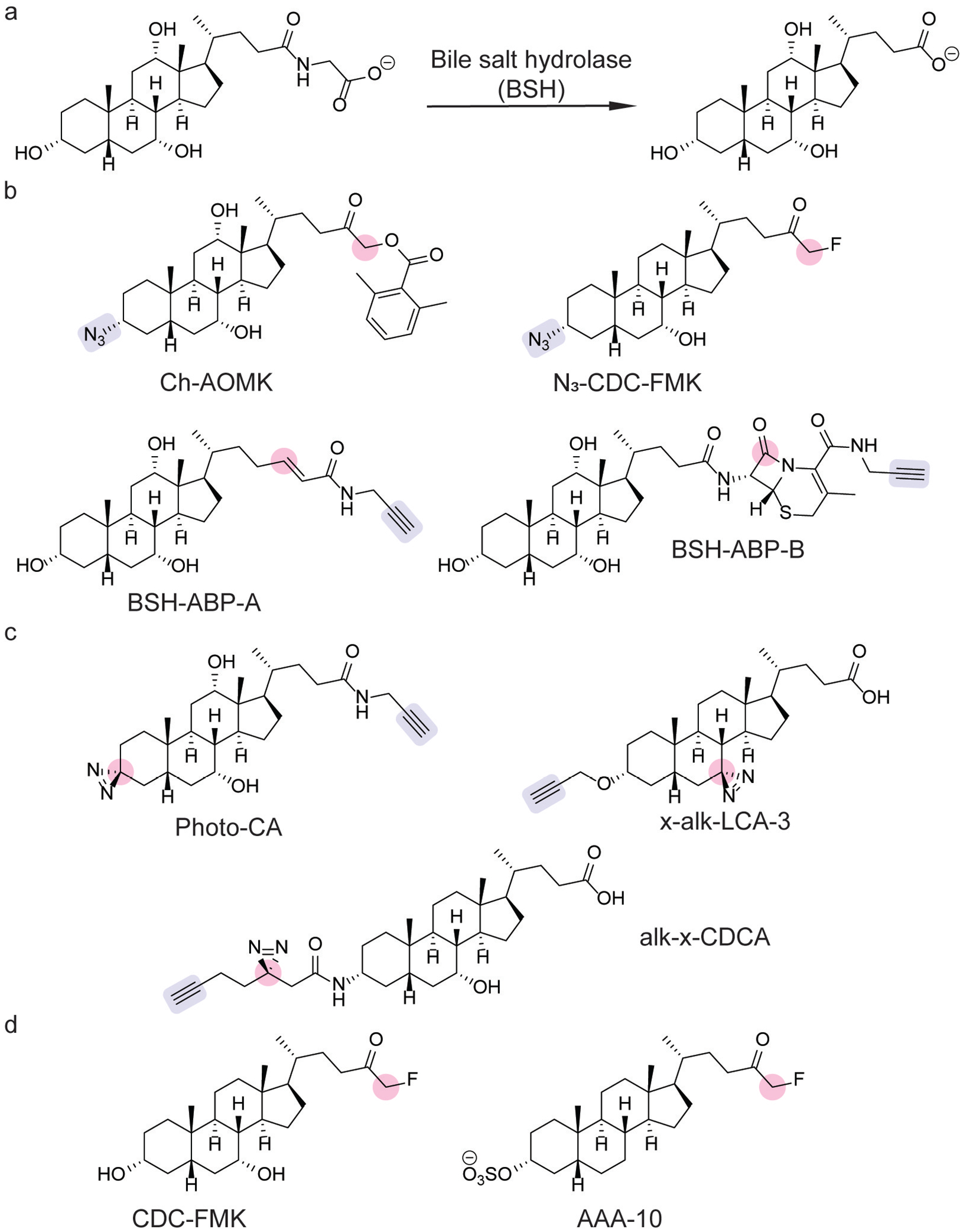
Chemical tools for probing gut microbial bile acid metabolism. (a) Bile salt hydrolase (BSH) deconjugates primary bile acids (shown: cholic acid). (b) Activity-based probes for profiling BSH activity.[159–161] (c) Photoaffinity probes for identifying bile acid-interacting proteins.[162–164] (d) BSH covalent inhibitors.[161,165] Moieties involved in protein labeling and click handles are highlighted in pink and blue, respectively.
Because BSH activity is required for the biosynthesis of secondary or microbially-produced bile acids, BSHs are termed the “gatekeeper” enzymes of microbial bile acid metabolism.[159] The importance of BSHs led to the development of an ABP, consisting of a clickable cholic acid analog bearing an acyloxy methylketone warhead (Ch-AOMK, Figure 15b).[159] Click chemistry-based chemoproteomics with Ch-AOMK detected BSH activity in gut anaerobes and mouse fecal samples.[159] Moreover, this chemoproteomic platform determined that BSH activity increased during intestinal inflammation in a murine model of DSS-induced colitis.[159] Additional ABPs for BSH activity have been developed utilizing acrylamide (BSH-ABP-A, Figure 15b) and β-lactam warheads (BSH-ABP-B, Figure 15b).[160]
ABPP has also been used to validate the specificity of BSH inhibitors. A chenodeoxycholic acid-derived fluoromethyl ketone inhibitor was developed as a covalent BSH inhibitor, which modulated bile acid levels upon oral administration in mice (CDC-FMK, Figure 15 d).[161] Chemoproteomic profiling with a clickable analog (N3-CDC-FMK, Figure 15b) validated its specificity to BSH and showed little off-target effects on host proteins.[161] A second-generation inhibitor (AAA-10, Figure 15d)[165] protected against intestinal barrier erosion and liver inflammation in rats fed a high-fat diet.[166] This protective effect was shown to be caused by the formation of protective micelles by conjugated bile acids, which sequestered the unconjugated bile acids that cause intestinal barrier damage.[166]
Chemoproteomic profiling with photoaffinity labeling has provided insight into bile acid signaling. The development of Photo-CA (Figure 15c) identified bile acid-interacting proteins in HeLa cells.[163] The application of PhotoCA in E. coli revealed a two-component system for bile acid sensing in which EnvZ was identified as a histidine kinase that senses bile acids.[167] The response regulator, OmpR, triggered the production of OmpC, the deletion of which prevented bile acid efflux and survival in the presence of bile acids.[167] The development of x-alk-LCA-3 (Figure 15c) has led to the discovery of novel bile acid-binding proteins, including a previously uncharacterized BSH in Enterococcus faecium.[164]
Chemoproteomic profiling of bile acid-interacting proteins has provided insight into the role of bile acids in regulating genes in bacterial pathogens. For example, chemoproteomic profiling with x-alk-LCA-3 (Figure 15c) identified a transcription factor that is activated by lithocholic acid in Clostridioides difficile.[168] In STm, photoaffinity labeling has elucidated the role of bile acids in colonization resistance. Building on previous work showing that chenodeoxycholic acid (CDCA) inhibits SPI-I expression by destabilizing transcriptional regulator HilD,[169] the development of alk-x-CDCA (Figure 15c) validated this binding interaction and served as a means of generating HilD mutants that were resistant to inactivation by CDCA.[162] These resistant mutants were able to override CDCA-mediated colonization resistance, showing more robust infection in mice and in mice supplemented with CDCA.[162] Thus, the use of chemical proteomic tools has deepened our understanding of how bile acids shape gene expression in enteric pathogens.
Gut microbiota are a complex community of bacteria that carry out a plethora of metabolic transformations. Gut microbial metabolism produces a variety of small molecule metabolites that are important for regulating host physiology and diseases. Chemoproteomic tools for understanding gut microbial metabolism have illuminated ways in which these metabolic transformations change during pathological conditions, contribute to disease, and impact bacterial and host signaling.
6. Conclusion
Prokaryotes are ubiquitous and critical in ecosytems within the environment and biological processes intrinsic to human health. Many of the microbial activities in these contexts are carried out by bacterial proteins and enzymes. As such, gaining a deeper understanding of bacterial protein and enzyme function has led to the development of innovative chemical tools for understanding the roles of bacteria in these important processes. Chemical proteomics has emerged as an important technology for probing bacterial activity, enabling the profiling of microbial proteins and enzymes involved in regulating bacterial physiology, pathogenesis, and metabolism in complex settings, including in live bacteria and microbial communities. Moreover, chemoproteomic approaches have uncovered new functions for bacterial proteins, as well as novel microbial enzymes. These results have led to the characterization of previously unknown virulence factors, mechanistic elucidation of microbe-microbe interactions, and discovery of new host-pathogen interactions. Ultimately, these findings are anticipated to provide significant insights into understanding the role of bacterial proteins and enzymes in the environment and in human health.
Acknowledgements
This work was supported by an NIH R35 Maximizing Investigators’ Research Award for Early Stage Investigators (R35GM133501). K.P.M. is supported by an NIH Chemistry-Biology Interface Predoctoral Training Grant (T32GM138826). Research in the Chang Lab is supported by a Beckman Young Investigator Award (to P.V.C.) from the Arnold and Mabel Beckman Foundation and a Sloan Research Fellowship (to P.V.C.) from the Alfred P. Sloan Foundation.
Biographies

Kien P. Malarney is a Microbiology Ph. D. student in the Department of Microbiology and Immunology at Cornell University. He received his B. S. in Chemistry and Mathematics/Applied Science from the University of California, San Diego. His research is focused on the biochemical and chemoproteomic characterization of microbial enzymes in the gut microbiome.

Professor Pamela V. Chang is an Associate Professor in the Department of Microbiology and Immunology and the Department of Chemistry and Chemical Biology at Cornell University. She completed her undergraduate degree in Chemistry with a minor in Biology from the Massachusetts Institute of Technology (MIT) and her Ph.D. in Chemistry from the University of California, Berkeley. After postdoctoral studies at Yale University in the field of immunology, she joined the Cornell faculty in 2015. Prof. Chang’s research interests span the disciplines of chemistry and biology with a focus on microbiology, immunology, and the gut microbiome.
Footnotes
Dedication: Dedicated to Professor Benjamin F. Cravatt on the occasion of his being awarded the 2022 Wolf Prize in Chemistry.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- [1].Arrigo KR, Nature 2005, 437, 349–355. [DOI] [PubMed] [Google Scholar]
- [2].Dimidi E, Cox S, Rossi M, Whelan K, Nutrients 2019, 11, 1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jozala AF, Geraldes DC, Tundisi LL, Feitosa V. de A., Breyer CA, Cardoso SL, Mazzola PG, Oliveira-Nascimento L. de, Rangel-Yagui C. de O., Magalhães P. de O., Oliveira de MA, Pessoa A, Braz. J. Microbiol 2016, 47, 51–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Needham BD, Trent MS, Nat. Rev. Microbiol 2013, 11, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M, Chem. Biol 2000, 7, 569–581. [DOI] [PubMed] [Google Scholar]
- [6].Liu Y, Patricelli MP, Cravatt BF, Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vocadlo DJ, Bertozzi CR, Angew. Chem. Int. Ed 2004, 43, 5338–5342; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2004, 116, 5452–5456. [Google Scholar]
- [8].Kidd D, Liu Y, Cravatt BF, Biochemistry 2001, 40, 4005–4015. [DOI] [PubMed] [Google Scholar]
- [9].Speers AE, Adam GC, Cravatt BF, J. Am. Chem. Soc 2003, 125, 4686–4687. [DOI] [PubMed] [Google Scholar]
- [10].Smith E, Collins I, Future Med. Chem 2015, 7, 159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhao X, Yang X, Hang HC, Biochemistry 2022, 61, 2822–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Simon GM, Cravatt BF, J. Biol. Chem 2010, 285, 11051–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Roberts AM, Ward CC, Nomura DK, Curr. Opin. Biotechnol 2017, 43, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Silhavy TJ, Kahne D, Walker S, Cold Spring Harbor Perspect. Biol 2010, 2, a000414–a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sarkar P, Yarlagadda V, Ghosh C, Haldar J, MedChemComm 2017, 8, 516–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kong K-F, Schneper L, Mathee K, APMIS 2010, 118, 1–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dörr T, Ann. N. Y. Acad. Sci 2021, 1496, 35–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhao G, Meier TI, Kahl SD, Gee KR, Blaszczak LC, Antimicrob. Agents Chemother 1999, 43, 1124–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Sharifzadeh S, Dempwolff F, Kearns DB, Carlson EE, ACS Chem. Biol 2020, 15, 1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brown NW, Shirley JD, Marshall AP, Carlson EE, ChemBioChem 2021, 22, 193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sarkar S, Libby EA, Pidgeon SE, Dworkin J, Pires MM, Angew. Chem. Int. Ed 2016, 55, 8401–8404; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 8541–8544. [Google Scholar]
- [22].Staub I, Sieber SA, J. Am. Chem. Soc 2008, 130, 13400–13409. [DOI] [PubMed] [Google Scholar]
- [23].Staub I, Sieber SA, J. Am. Chem. Soc 2009, 131, 6271–6276. [DOI] [PubMed] [Google Scholar]
- [24].Kuk ACY, Hao A, Guan Z, Lee S-Y, Nat. Commun 2019, 10, 1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eirich J, Orth R, Sieber SA, J. Am. Chem. Soc 2011, 133, 12144–12153. [DOI] [PubMed] [Google Scholar]
- [26].Böttcher T, Sieber SA, J. Am. Chem. Soc 2010, 132, 6964–6972. [DOI] [PubMed] [Google Scholar]
- [27].Kunzmann MH, Staub I, Böttcher T, Sieber SA, Biochemistry 2011, 50, 910–916. [DOI] [PubMed] [Google Scholar]
- [28].Kavunja HW, Biegas KJ, Banahene N, Stewart JA, Piligian BF, Groenevelt JM, Sein CE, Morita YS, Niederweis M, Siegrist MS, Swarts BM, J. Am. Chem. Soc 2020, 142, 7725–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Babin BM, Keller LJ, Pinto Y, Li VL, Eneim AS, Vance SE, Terrell SM, Bhatt AS, Long JZ, Bogyo M, Cell Chem. Biol 2022, 29, 897–909.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lehmann J, Cheng T-Y, Aggarwal A, Park AS, Zeiler E, Raju RM, Akopian T, Kandror O, Sacchettini JC, Moody DB, Rubin EJ, Sieber SA, Angew. Chem. Int. Ed 2018, 57, 348–353; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 354–359. [Google Scholar]
- [31].Chiaradia L, Lefebvre C, Parra J, Marcoux J, Burlet-Schiltz O, Etienne G, Tropis M, Daffé M, Sci. Rep 2017, 7, 12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Jackson M, Cold Spring Harb. Perspect. Med 2014, 4, a021105–a021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li M, Patel HV, Cognetta AB, Smith TC, Mallick I, Cavalier J-F, Previti ML, Canaan S, Aldridge BB, Cravatt BF, Seeliger JC, Cell Chem. Biol 2022, 29, 883–896.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Madani A, Ridenour JN, Martin BP, Paudel RR, Abdul Basir A, Le Moigne V, Herrmann J-L, Audebert S, Camoin L, Kremer L, Spilling CD, Canaan S, Cavalier J-F, ACS Infect. Dis 2019, 5, 1597–1608. [DOI] [PubMed] [Google Scholar]
- [35].Gengenbacher M, Kaufmann SHE, FEMS Microbiol. Rev 2012, 36, 514–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gollan B, Grabe G, Michaux C, Helaine S, Annu. Rev. Microbiol 2019, 73, 359–385. [DOI] [PubMed] [Google Scholar]
- [37].Ravindran MS, Rao SPS, Cheng X, Shukla A, Cazenave-Gassiot A, Yao SQ, Wenk MR, Mol. Cell. Proteomics 2014, 13, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ortega C, Anderson LN, Frando A, Sadler NC, Brown RW, Smith RD, Wright AT, Grundner C, Cell Chem. Biol 2016, 23, 290–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Tallman KR, Levine SR, Beatty KE, ACS Infect. Dis 2016, 2, 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Zweerink S, Kallnik V, Ninck S, Nickel S, Verheyen J, Blum M, Wagner A, Feldmann I, Sickmann A, Albers S-V, Bräsen C, Kaschani F, Siebers B, Kaiser M, Nat. Commun 2017, 8, 15352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Maaty WS, Steffens JD, Heinemann J, Ortmann AC, Reeves BD, Biswas SK, Dratz EA, Grieco PA, Young MJ, Bothner B, Front. Microbiol 2012, 3, DOI 10.3389/fmicb.2012.00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Mahmoud SA, Chien P, Annu. Rev. Biochem 2018, 87, 677–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lang K, Chin JW, Chem. Rev 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- [44].Ngo JT, Champion JA, Mahdavi A, Tanrikulu IC, Beatty KE, Connor RE, Yoo TH, Dieterich DC, Schuman EM, Tirrell DA, Nat. Chem. Biol 2009, 5, 715–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hatzenpichler R, Scheller S, Tavormina PL, Babin BM, Tirrell DA, Orphan VJ, Environ. Microbiol 2014, 16, 2568–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Grammel M, Dossa PD, Taylor-Salmon E, Hang HC, Chem. Commun 2012, 48, 1473–1474. [DOI] [PubMed] [Google Scholar]
- [47].Grammel M, Zhang MM, Hang HC, Angew. Chem. Int. Ed 2010, 49, 5970–5974; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2010, 122, 6106–6110. [Google Scholar]
- [48].Nair RN, Rosnow JJ, Murphree TA, Bowden ME, Lindemann SR, Wright AT, Org. Chem. Front 2017, 4, 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhang M, Lin S, Song X, Liu J, Fu Y, Ge X, Fu X, Chang Z, Chen PR, Nat. Chem. Biol 2011, 7, 671–677. [DOI] [PubMed] [Google Scholar]
- [50].Lin S, Zhang Z, Xu H, Li L, Chen S, Li J, Hao Z, Chen PR, J. Am. Chem. Soc 2011, 133, 20581–20587. [DOI] [PubMed] [Google Scholar]
- [51].Schramm FD, Schroeder K, Jonas K, FEMS Microbiol. Rev 2020, 44, 54–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Moreno-Cinos C, Goossens K, Salado IG, Van Der Veken P, De Winter H, Augustyns K, IJMS 2019, 20, 2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Compton CL, Schmitz KR, Sauer RT, Sello JK, ACS Chem. Biol 2013, 8, 2669–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Böttcher T, Sieber SA, J. Am. Chem. Soc 2008, 130, 14400–14401. [DOI] [PubMed] [Google Scholar]
- [55].Zeiler E, Braun N, Böttcher T, Kastenmüller A, Weinkauf S, Sieber SA, Angew. Chem. Int. Ed 2011, 50, 11001–11004; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2011, 123, 11193–11197. [Google Scholar]
- [56].Pahl A, Lakemeyer M, Vielberg M, Hackl MW, Vomacka J, Korotkov VS, Stein ML, Fetzer C, Lorenz-Baath K, Richter K, Waldmann H, Groll M, Sieber SA, Angew. Chem. Int. Ed 2015, 54, 15892–15896; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2015, 127, 16121–16126. [Google Scholar]
- [57].Böttcher T, Sieber SA, Angew. Chem. Int. Ed 2008, 47, 4600–4603; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 4677–4680. [Google Scholar]
- [58].Zeiler E, List A, Alte F, Gersch M, Wachtel R, Poreba M, Drag M, Groll M, Sieber SA, Proc. Natl. Acad. Sci. USA 2013, 110, 11302–11307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zeiler E, Korotkov VS, Lorenz-Baath K, Böttcher T, Sieber SA, Bioorg. Med. Chem 2012, 20, 583–591. [DOI] [PubMed] [Google Scholar]
- [60].Rangan KJ, Yang Y-Y, Charron G, Hang HC, J. Am. Chem. Soc 2010, 132, 10628–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Heal WP, Wickramasinghe SR, Leatherbarrow RJ, Tate EW, Org. Biomol. Chem 2008, 6, 2308. [DOI] [PubMed] [Google Scholar]
- [62].Charlton TM, Kovacs-Simon A, Michell SL, Fair-weather NF, Tate EW, Chem. Biol 2015, 22, 1562–1573. [DOI] [PubMed] [Google Scholar]
- [63].Galán JE, Nat. Rev. Microbiol 2021, 19, 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lou L, Zhang P, Piao R, Wang Y, Front. Cell. Infect. Microbiol 2019, 9, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Rivera-Chávez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, Bäumler AJ, Cell Host Microbe 2016, 19, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Zhang ZJ, Pedicord VA, Peng T, Hang HC, Nat. Chem. Biol 2020, 16, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Yang X, Forster ER, Darabedian N, Kim AT, Pratt MR, Shen A, Hang HC, ACS Chem. Biol 2020, 15, 1141–1147. [DOI] [PubMed] [Google Scholar]
- [68].Peace CG, O’Neill LAJ, J. Clin. Invest 2022, 132, e148548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Zhang Y, Qin W, Liu D, Liu Y, Wang C, Chem. Sci 2021, 12, 6059–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zanon PRA, Lewald L, Hacker SM, Angew. Chem. Int. Ed 2020, 59, 2829–2836; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 2851–2858. [Google Scholar]
- [71].Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MBD, Bachovchin DA, Mowen K, Baker D, Cravatt BF, Nature 2010, 468, 790–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Schmidt C, Zollo M, Bonsignore R, Casini A, Hacker SM, Chem. Commun 2022, 58, 5526–5529. [DOI] [PubMed] [Google Scholar]
- [73].Lee KM, Le P, Sieber SA, Hacker SM, Chem. Commun 2020, 56, 2929–2932. [DOI] [PubMed] [Google Scholar]
- [74].Deng X, Weerapana E, Ulanovskaya O, Sun F, Liang H, Ji Q, Ye Y, Fu Y, Zhou L, Li J, Zhang H, Wang C, Alvarez S, Hicks LM, Lan L, Wu M, Cravatt BF, He C, Cell Host Microbe 2013, 13, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sadler NC, Melnicki MR, Serres MH, Merkley ED, Chrisler WB, Hill EA, Romine MF, Kim S, Zink EM, Datta S, Smith RD, Beliaev AS, Konopka A, Wright AT, ACS Chem. Biol 2014, 9, 291–300. [DOI] [PubMed] [Google Scholar]
- [76].Ansong C, Sadler NC, Hill EA, Lewis MP, Zink EM, Smith RD, Beliaev AS, Konopka AE, Wright AT, Front. Microbiol 2014, 5, DOI 10.3389/fmicb.2014.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Sadler NC, Bernstein HC, Melnicki MR, Charania MA, Hill EA, Anderson LN, Monroe ME, Smith RD, Beliaev AS, Wright AT, Appl. Environ. Microbiol 2016, 82, 7227–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Welsh EA, Liberton M, Stöckel J, Loh T, Elvitigala T, Wang C, Wollam A, Fulton RS, Clifton SW, Jacobs JM, Aurora R, Ghosh BK, Sherman LA, Smith RD, Wilson RK, Pakrasi HB, Proc. Natl. Acad. Sci. USA 2008, 105, 15094–15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Stewart RC, Curr. Opin. Microbiol 2010, 13, 133–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Wilke KE, Francis S, Carlson EE, J. Am. Chem. Soc 2012, 134, 9150–9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Espinasse A, Wen X, Goodpaster JD, Carlson EE, ACS Chem. Biol 2020, 15, 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Chang JW, Montgomery JE, Lee G, Moellering RE, Angew. Chem. Int. Ed 2018, 57, 15712–15716; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 15938–15942. [Google Scholar]
- [83].Allihn PWA, Hackl MW, Ludwig C, Hacker SM, Sieber SA, Chem. Sci 2021, 12, 4763–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Chase OM, Espinasse A, Wilke KE, Carlson EE, Biochemistry 2018, 57, 4368–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Bach K, Beerkens BLH, Zanon PRA, Hacker SM, ACS Cent. Sci 2020, 6, 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wright MH, Fetzer C, Sieber SA, J. Am. Chem. Soc 2017, 139, 6152–6159. [DOI] [PubMed] [Google Scholar]
- [87].Kim SM, Park J, Kim MS, Song H, Jo A, Park H, Kim TS, Choi SH, Park SB, ACS Infect. Dis 2020, 6, 3076–3082. [DOI] [PubMed] [Google Scholar]
- [88].Dubinsky L, Jarosz LM, Amara N, Krief P, Kravchenko VV, Krom BP, Meijler MM, Chem. Commun 2009, 7378. [DOI] [PubMed] [Google Scholar]
- [89].Zhao W, Lorenz N, Jung K, Sieber SA, Angew. Chem. Int. Ed 2016, 55, 1187–1191; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 1203–1207. [Google Scholar]
- [90].Mukherjee S, Bassler BL, Nat. Rev. Microbiol 2019, 17, 371–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Welsh MA, Blackwell HE, FEMS Microbiol. Rev 2016, 40, 774–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Yashkin A, Rayo J, Grimm L, Welch M, Meijler MM, Chem. Sci 2021, 12, 4570–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Horseman MA, Surani S, Int. J. Infect. Dis 2011, 15, e157–e166. [DOI] [PubMed] [Google Scholar]
- [94].Madan-Lala R, Peixoto KV, Re F, Rengarajan J, Infect. Immun 2011, 79, 4828–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lentz CS, Ordonez AA, Kasperkiewicz P, La Greca F, O’Donoghue AJ, Schulze CJ, Powers JC, Craik CS, Drag M, Jain SK, Bogyo M, ACS Infect. Dis 2016, 2, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Lentz CS, Sheldon JR, Crawford LA, Cooper R, Garland M, Amieva MR, Weerapana E, Skaar EP, Bogyo M, Nat. Chem. Biol 2018, 14, 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Hatzios SK, Abel S, Martell J, Hubbard T, Sasabe J, Munera D, Clark L, Bachovchin DA, Qadri F, Ryan ET, Davis BM, Weerapana E, Waldor MK, Nat. Chem. Biol 2016, 12, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Patricelli MP, Giang DK, Stamp LM, Burbaum JJ, Proteomics 2001, 1, 1067–1071. [DOI] [PubMed] [Google Scholar]
- [99].Fellner M, Lentz CS, Jamieson SA, Brewster JL, Chen L, Bogyo M, Mace PD, ACS Infect. Dis 2020, 6, 2771–2782. [DOI] [PubMed] [Google Scholar]
- [100].Chen L, Keller LJ, Cordasco E, Bogyo M, Lentz CS, Angew. Chem. Int. Ed 2019, 58, 5643–5647; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 5699–5703. [Google Scholar]
- [101].Bakker AT, Kotsogianni I, Mirenda L, Straub VM, Avalos M, van den Berg RJBHN, Florea BI, van Wezel GP, Janssen APA, Martin NI, van der Stelt M, J. Am. Chem. Soc 2023, 145, 1136–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Keller LJ, Lentz CS, Chen YE, Metivier RJ, Weerapana E, Fischbach MA, Bogyo M, ACS Infect. Dis 2020, 6, 930–938. [DOI] [PubMed] [Google Scholar]
- [103].Zheng Y, Hunt RL, Villaruz AE, Fisher EL, Liu R, Liu Q, Cheung GYC, Li M, Otto M, Cell Host Microbe 2022, 30, 301–313.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Howell M, Dumitrescu DG, Blankenship LR, Herkert D, Hatzios SK, J. Biol. Chem 2019, 294, 9888–9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kaznadzey A, Shelyakin P, Gelfand MS, Biol. Direct 2017,12, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Gandy MN, Debowski AW, Stubbs KA, Chem. Commun 2011, 47, 5037. [DOI] [PubMed] [Google Scholar]
- [107].Zegeye EK, Sadler NC, Lomas GX, Attah IK, Jansson JK, Hofmockel KS, Anderton CR, Wright AT, ChemBioChem 2021, 22, 717–723. [DOI] [PubMed] [Google Scholar]
- [108].Chauvigné-Hines LM, Anderson LN, Weaver HM, Brown JN, Koech PK, Nicora CD, Hofstad BA, Smith RD, Wilkins MJ, Callister SJ, Wright AT, J. Am. Chem. Soc 2012, 134, 20521–20532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Klaus T, Ninck S, Albersmeier A, Busche T, Wibberg D, Jiang J, Elcheninov AG, Zayulina KS, Kaschani F, Bräsen C, Overkleeft HS, Kalinowski J, Kublanov IV, Kaiser M, Siebers B, Front. Microbiol 2022, 12, 734039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Rooks MG, Garrett WS, Nat. Rev. Immunol 2016, 16, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Luijkx YMCA, Bleumink NMC, Jiang J, Overkleeft HS, Wösten MMSM, Strijbis K, Wennekes T, Cell. Microbiol 2020, 22, DOI 10.1111/cmi.13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Luijkx YMCA, Henselijn AJ, Bosman GP, Cramer DAT, Giesbers KCAP, van’t Veld EM, Boons G-J, Heck AJR, Reiding KR, Strijbis K, Wennekes T, Molecules 2022, 27, 1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Anderson LN, Koech PK, Plymale AE, Landorf EV, Konopka A, Collart FR, Lipton MS, Romine MF, Wright AT, ACS Chem. Biol 2016, 11, 345–354. [DOI] [PubMed] [Google Scholar]
- [114].Takamura A, Thuy-Boun PS, Kitamura S, Han Z, Wolan DW, Bioorg. Med. Chem. Lett 2021, 40, 127903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Thuy-Boun PS, Wolan DW, Bioorg. Med. Chem. Lett 2019,29, 2609–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Keohane CE, Steele AD, Fetzer C, Khowsathit J, Van Tyne D, Moynié L, Gilmore MS, Karanicolas J, Sieber SA, Wuest WM, J. Am. Chem. Soc 2018, 140, 1774–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Lu K, Hou W, Xu X-F, Chen Q, Li Z, Lin J, Chen W-M,Eur. J. Med. Chem 2020, 188, 112026. [DOI] [PubMed] [Google Scholar]
- [118].Romine MF, Rodionov DA, Maezato Y, Anderson LN, Nandhikonda P, Rodionova IA, Carre A, Li X, Xu C, Clauss TRW, Kim Y-M, Metz TO, Wright AT, Proc. Natl. Acad. Sci. USA 2017, 114, DOI 10.1073/pnas.1612360114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Rosnow JJ, Hwang S, Killinger BJ, Kim Y-M, Moore RJ, Lindemann SR, Maupin-Furlow JA, Wright AT, Appl. Environ. Microbiol 2018, 84, e00955–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Putnam EE, Abellon-Ruiz J, Killinger BJ, Rosnow JJ, Wexler AG, Folta-Stogniew E, Wright AT, van den Berg B, Goodman AL, mBio 2022, 13, e02845–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Cuesta A, Taunton J, Annu. Rev. Biochem 2019, 88, 365–381. [DOI] [PubMed] [Google Scholar]
- [122].Patricelli MP, Szardenings AK, Liyanage M, Nomanbhoy TK, Wu M, Weissig H, Aban A, Chun D, Tanner S, Kozarich JW, Biochemistry 2007, 46, 350–358. [DOI] [PubMed] [Google Scholar]
- [123].Wolfe LM, Veeraraghavan U, Idicula-Thomas S, Schürer S, Wennerberg K, Reynolds R, Besra GS, Dobos KM, Mol. Cell. Proteomics 2013, 12, 1644–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ansong C, Ortega C, Payne SH, Haft DH, Chauvignè-Hines LM, Lewis MP, Ollodart AR, Purvine SO, Shukla AK, Fortuin S, Smith RD, Adkins JN, Grundner C, Wright AT, Chem. Biol 2013, 20, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Ortega C, Liao R, Anderson LN, Rustad T, Ollodart AR, Wright AT, Sherman DR, Grundner C, PLoS Biol. 2014, 12, e1001746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Du Y-L, Ryan KS, Nat. Prod. Rep 2019, 36, 430–457. [DOI] [PubMed] [Google Scholar]
- [127].Hoegl A, Nodwell MB, Kirsch VC, Bach NC, Pfanzelt M, Stahl M, Schneider S, Sieber SA, Nat. Chem 2018, 10, 1234–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Pfanzelt M, Maher TE, Absmeier RM, Schwarz M, Sieber SA, Angew. Chem. Int. Ed 2022, 61, e202117724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Hug JJ, Krug D, Müller R, Nat. Rev. Chem 2020, 4, 172–193. [DOI] [PubMed] [Google Scholar]
- [130].Trivella DBB, de Felicio R, mSystems 2018, 3, e00160–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Fischbach MA, Walsh CT, Chem. Rev 2006, 106, 3468–3496. [DOI] [PubMed] [Google Scholar]
- [132].Nivina A, Yuet KP, Hsu J, Khosla C, Chem. Rev 2019, 119,12524–12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Konno S, Ishikawa F, Suzuki T, Dohmae N, Burkart MD, Kakeya H, Chem. Commun 2015, 51, 2262–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Ishikawa F, Suzuki T, Dohmae N, Kakeya H, ChemBioChem 2015, 16, 2590–2594. [DOI] [PubMed] [Google Scholar]
- [135].Duckworth BP, Wilson DJ, Nelson KM, Boshoff HI, Barry CE, Aldrich CC, ACS Chem. Biol 2012, 7, 1653–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Ishikawa F, Konno S, Uchida C, Suzuki T, Takashima K, Dohmae N, Kakeya H, Tanabe G, Cell Chem. Biol 2022, 29, 145–156.e8. [DOI] [PubMed] [Google Scholar]
- [137].Ishikawa F, Haushalter RW, Burkart MD, J. Am. Chem. Soc 2012, 134, 769–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Meier JL, Mercer AC, Burkart MD, J. Am. Chem. Soc 2008, 130, 5443–5445. [DOI] [PubMed] [Google Scholar]
- [139].Meier JL, Niessen S, Hoover HS, Foley TL, Cravatt BF, Burkart MD, ACS Chem. Biol 2009, 4, 948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Meier JL, Mercer AC, Rivera H, Burkart MD, J. Am. Chem. Soc 2006, 128, 12174–12184. [DOI] [PubMed] [Google Scholar]
- [141].Mariaule V, Kriaa A, Soussou S, Rhimi S, Boudaya H, Hernandez J, Maguin E, Lesner A, Rhimi M, IJMS 2021, 22, 2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Mayers MD, Moon C, Stupp GS, Su AI, Wolan DW, J. Proteome Res 2017, 16, 1014–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Thuy-Boun PS, Wang AY, Crissien-Martinez A, Xu JH, Chatterjee S, Stupp GS, Su AI, Coyle WJ, Wolan DW, Mol. Cell. Proteomics 2022, 21, 100197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Guo C-J, Chang F-Y, Wyche TP, Backus KM, Acker TM, Funabashi M, Taketani M, Donia MS, Nayfach S, Pollard KS, Craik CS, Cravatt BF, Clardy J, Voigt CA, Fischbach MA, Cell 2017, 168, 517–526.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Donia MS, Fischbach MA, Science 2015, 349, 1254766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Liu Y, Hou Y, Wang G, Zheng X, Hao H, Trends Endocrinol. Metab 2020, 31, 818–834. [DOI] [PubMed] [Google Scholar]
- [147].Zhang J, Zhang J, Wang R, Drug Metab. Rev 2018, 50, 357–368. [DOI] [PubMed] [Google Scholar]
- [148].Javdan B, Lopez JG, Chankhamjon P, Lee Y-CJ, Hull R, Wu Q, Wang X, Chatterjee S, Donia MS, Cell 2020, 181, 1661–1679.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Qian L, Ouyang H, Gordils-Valentin L, Hong J, Jayaraman A, Zhu X, ACS Chem. Biol 2022, 17, 1665–1671. [DOI] [PubMed] [Google Scholar]
- [150].Yang G, Ge S, Singh R, Basu S, Shatzer K, Zen M, Liu J, Tu Y, Zhang C, Wei J, Shi J, Zhu L, Liu Z, Wang Y, Gao S, Hu M, Drug Metab. Rev 2017, 49, 105–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh L-A, Mani S, Redinbo MR, Science 2010, 330, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Wu L, Jiang J, Jin Y, Kallemeijn WW, Kuo C-L, Artola M, Dai W, van Elk C, van Eijk M, van der Marel GA, Codée JDC, Florea BI, Aerts JMFG, Overkleeft HS, Davies GJ, Nat. Chem. Biol 2017, 13, 867–873. [DOI] [PubMed] [Google Scholar]
- [153].Jariwala PB, Pellock SJ, Goldfarb D, Cloer EW, Artola M, Simpson JB, Bhatt AP, Walton WG, Roberts LR, Major MB, Davies GJ, Overkleeft HS, Redinbo MR, ACS Chem. Biol 2020, 15, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Whidbey C, Sadler NC, Nair RN, Volk RF, DeLeon AJ, Bramer LM, Fansler SJ, Hansen JR, Shukla AK, Jansson JK, Thrall BD, Wright AT, J. Am. Chem. Soc 2019, 141, 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Zhang J, Walker ME, Sanidad KZ, Zhang H, Liang Y, Zhao E, Chacon-Vargas K, Yeliseyev V, Parsonnet J, Haggerty TD, Wang G, Simpson JB, Jariwala PB, Beaty VV, Yang J, Yang H, Panigrahy A, Minter LM, Kim D, Gibbons JG, Liu L, Li Z, Xiao H, Borlandelli V, Overkleeft HS, Cloer EW, Major MB, Goldfarb D, Cai Z, Redinbo MR, Zhang G, Nat. Commun 2022, 13, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Weatherly LM, Gosse JA, J. Toxicol. Environ. Health B Crit. Rev 2017, 20, 447–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Ridlon JM, Kang D-J, Hylemon PB, J. Lipid Res 2006, 47, 241–259. [DOI] [PubMed] [Google Scholar]
- [158].Chen ML, Takeda K, Sundrud MS, Mucosal Immunol. 2019, 12, 851–861. [DOI] [PubMed] [Google Scholar]
- [159].Parasar B, Zhou H, Xiao X, Shi Q, Brito IL, Chang PV, ACS Cent. Sci 2019, 5, 867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Brandvold KR, Miller CJ, Volk RF, Killinger BJ, Whidbey C, Wright AT, ChemBioChem 2021, 22, 1448–1455. [DOI] [PubMed] [Google Scholar]
- [161].Adhikari AA, Seegar TCM, Ficarro SB, McCurry MD, Ramachandran D, Yao L, Chaudhari SN, Ndousse-Fetter S, Banks AS, Marto JA, Blacklow SC, Devlin AS, Nat. Chem. Biol 2020, 16, 318–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Yang X, Stein KR, Hang HC, Nat. Chem. Biol 2023, 19, 91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Zhuang S, Li Q, Cai L, Wang C, Lei X, ACS Cent. Sci 2017,3, 501–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Yang X, Zhao X, Chen V, Hang HC, RSC Chem. Biol 2022, 3, 1397–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Adhikari AA, Ramachandran D, Chaudhari SN, Powell CE, Li W, McCurry MD, Banks AS, Devlin AS, ACS Chem. Biol 2021, 16, 1401–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Li DK, Chaudhari SN, Lee Y, Sojoodi M, Adhikari AA, Zukerberg L, Shroff S, Barrett SC, Tanabe K, Chung RT, Devlin AS, Sci. Adv 2022, 8, eabo2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Liu B, Zhuang S, Tian R, Liu Y, Wang Y, Lei X, Wang C, ACS Chem. Biol 2022, 17, 2461–2470. [DOI] [PubMed] [Google Scholar]
- [168].Forster ER, Yang X, Tai AK, Hang HC, Shen A, ACS Chem. Biol 2022, 17, 3086–3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Eade CR, Hung C-C, Bullard B, Gonzalez-Escobedo G, Gunn JS, Altier C, Infect. Immun 2016, 84, 2198–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.