

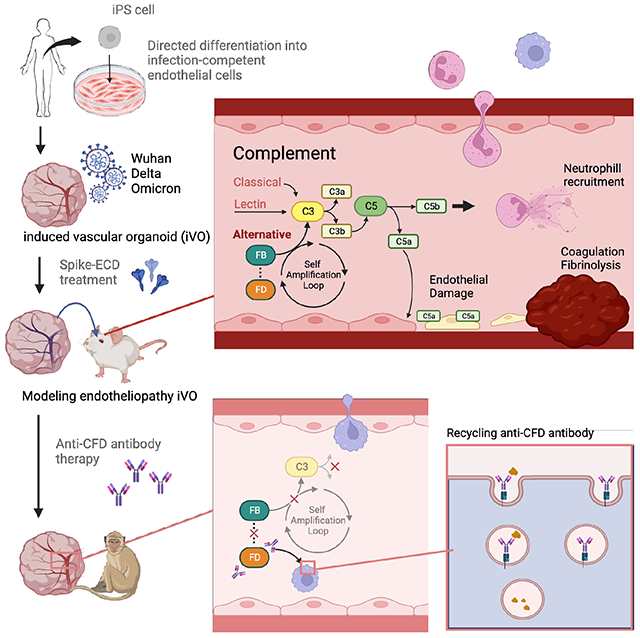
Summary
COVID-19 is linked to endotheliopathy and coagulopathy, which can result in multi-organ failure. The mechanisms causing endothelial damage due to SARS-CoV-2 remain elusive. Here we developed an infection-competent human vascular organoid from pluripotent stem cells for modeling endotheliopathy. Longitudinal serum proteome analysis identified aberrant complement signature in critically ill patients driven by the amplification cycle regulated by complement factor B and D (CFD). This deviant complement pattern initiates endothelial damage, neutrophil activation and thrombosis specific to our organoid-derived human blood vessels, as verified through intravital imaging. We examined a new long-acting, pH-sensitive (acid-switched) antibody targeting CFD. In both human and macaque COVID-19 models, this long-acting anti-CFD monoclonal antibody mitigated abnormal complement activation, protected endothelial cells, and curtailed the innate immune response post-viral exposure. Collectively, our findings suggest the complement alternative pathway exacerbates endothelial injury and inflammation. This underscores the potential of CFD-targeted treatments against severe viral-induced inflammathrombotic outcomes.
Keywords: SARS-CoV-2, iPSC, organoid, endotheliopathy, complement, CFD, thrombopathy, ASHE
eTOC Blurb
COVID-19 can lead to endotheliopathy and multi-organ failure. We developed an infection permissive human vascular organoid to delineate key role of complement pathway causing endothelial damage and inflammathrombosis. In primate model, testing a new CFD targeted antibody mitigated these effects, suggesting its potential against severe COVID-19 complications.
Graphical Abstract
Introduction
The confirmed case count of the global COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is now over seven hundred million, with over six million deaths in Aug, 2023. The typical clinical manifestations of COVID-19 involve fever, cough, and shortness of breath that can progress to pneumonia, which in turn activates immune cells, platelets, and coagulation pathways, leading to a massive cytokine release (cytokine storm), and ultimately, multiple organ failure (MOF) and death1. Mounting clinical and preclinical evidence demonstrates that SARS-CoV-2 infection induces widespread endothelial damage and inflammation and local and systemic coagulopathy2–9. Critically ill COVID-19 patients are prone to both macro- and micro-thrombotic manifestations at various sites, including pulmonary embolisms (20–30% of cases), deep vein thrombosis (DVT), arterial thrombosis as well as microvascular thrombosis affecting lungs, kidneys, and heart10. Of note, micro-thrombotic complications may contribute to acute respiratory distress syndrome (ARDS) and other organ dysfunctions and in the worst case, fatal MOF11. Thus, understanding the diverse thrombotic mechanisms is vital for prioritizing therapeutic pathways in severe COVID-19 cases.
Thrombotic pathophysiology for severe COVID-19 is associated with hypercoagulability and endotheliopathy10. Once SARS-CoV-2 enters host cells by binding the angiotensin-converting enzyme 2 (ACE2), chemokines are released to initiate an inflammatory response12,13. The excessive immune response-induced cytokine storm is thus responsible for hypercoagulability and an endotheliopathy state, leading to macro- and micro-thrombosis, respectively10. While the excessive thrombin formation and the pro- and anti- coagulant imbalance account for a hypercoagulable state14, direct endothelial damage and apoptosis may be critical drivers for micro-thrombotic episodes accompanied by vascular wall oedema, hyaline thrombi, and microhemorrhages15,16. This has been supported by clinically confirmed endothelial damage marker elevation and platelet reduction in patients admitted to the ICU17,18. Interestingly, the pathophysiology for COVID-19-related systemic micro-thrombosis (ultimately complicated by MOF) may be specific and, in particular, different from disseminated intravascular coagulation (DIC)10: indeed, in contrast to sepsis-induced coagulopathy, consumption of platelets, coagulation factors, and fibrinogen, as well as bleeding complications, are atypical in severe COVID-19 patients5,9. The exact pathophysiological mechanisms leading to severe microvascular dysfunction and thrombosis have been unclear.
Preclinical investigative tools have advanced exponentially to interrogate the interplay among COVID-19 severity, endothelial injury, and thrombosis. For instance, animal models that are competent for SARS-CoV-2 entry have been extensively used for infection, disease progression, vaccine, and therapy evaluation, including human ACE-2 transgenic mice19,20, hamsters21–23, ferrets23–25. However, in addition to the fundamental lack of predictive human-relevant inflammatory factors without rodent orthologues of human cytokines26,27, animal models generally present limited inflammatory assaults with modest or absence of significant thrombosis, cytokine storm, and endothelial injury. Given that endothelial dysfunction is involved in acute and post-acute long COVID-1928, it is essential to develop human specific endothelial model permissive for the study of infection-dependent immunothrombosis responses.
Several SARS-CoV-2 permissive in vitro cellular models have been used for studying viral infection and replication. Examples include Caco-229, Calu-330, HEK293T31, Huh732, and Vero-E633,34. However, these immortalized cell lines are devoid of endothelial phenotypes and have been substantially limited in evaluating inflammatory responses. To circumvent this challenge, several ACE-2 dependent or independent cell models31,35,36 are reported to demonstrate viral infection and replication in primary or pluripotent stem cell culture. In contrast, the use of several primary endothelial cells (EC) culture studies suggests that endothelial cells are resistant to SARS-CoV-2 infection36–38 as opposed to alveolar epithelial cells. Thus, despite increasing clinical evidence of COVID-19-related endotheliopathy, the infection competence of SARS-CoV-2 on available human ECs remain inconclusive.
Here, we developed infection-competent venous progenitor-derived vascular organoids (iVO) from human induced pluripotent stem cells (iPSC). In contrast to various commercially available human EC, iVO showed high ACE2 and TMPRSS2 levels, and are efficiently infected by SARS-CoV-2. Human iVO-derived blood vessels can dictate aspects of patients’ phenotypes involving inflammatory cytokine production, neutrophil recruitment, and thrombotic events. Furthermore, longitudinal proteome analysis of critically ill patients’ serum, together with the analysis of published single cell transcriptomic dataset, identify unique complement activation signatures preceded by D-dimer aberration. Differential proteome analysis pointed a complement factor D (CFD, also known as Adipsin) surge during the early phase of severe COVID-19, while their downstream product Factor Ba (Ba) can predict severe and longer-hospitalization outcomes on the initial day of ICU admission. To probe the potential role of complements in thrombotic complications, we investigated a long-acting form of a CFD monoclonal antibody and tested its efficacy in vitro and potency in vivo for alleviating immunothrombosis using a non-human primate COVID-19 model39. Collectively, complement alternative pathways (AP) causes vascular damage, inflammation, and coagulation phenotypes that warrant future preclinical and clinical development of AP targeting in viral infection-associated endotheliopathy.
Results
Endotheliopathy and thrombopathy are hallmarks of severe COVID-19.
In a previous cohort study of COVID-19 patient serum obtained from 62 Japanese individuals (Osaka cohort), inflammatory responses, particularly the release of GDF-15, have been demonstrated to be associated with the severity of the disease40. To further address the pathogenesis of severe illness, we re-analyzed serum samples obtained from 60 patients who were managed on ventilators immediately after ICU admission in the Osaka cohort. Serum levels of the endotheliopathy marker, plasminogen activator inhibitor-1 (PAI-1) in COVID-19 patients at the time of ICU entry (day 1) are markedly higher than 65 septic patients collected in other cohorts and 18 healthy controls (Figure 1A). PAI-1 was significantly decreased in post-recovery serum from non-ICU COVID-19 patients (Figure S1). To test if the PAI-1 level was associated with a more severe outcome, all the patients were assigned into two groups, PAI-1 high and low, with the threshold defined by the maximum value in the control group and compared the number of days required to wean off the ventilator. The PAI-1 low group had faster recovery with a median of approximately 10 days, while the PAI-1 high group required significantly more days to recover from ventilator management (Figure 1B). We also found that PAI-1 levels positively correlated with inflammatory cytokines (GDF-15 and IL-6) on day 1 but not with fibrinolysis/thrombosis markers (D-dimer and FDP) (Figure 1C)41,42. These results indicate endotheliopathy and inflammation are early hallmarks of severe COVID-19.
Figure 1. Endotheliopathy and inflammathrombosis is a hallmark for severe COVID-19.
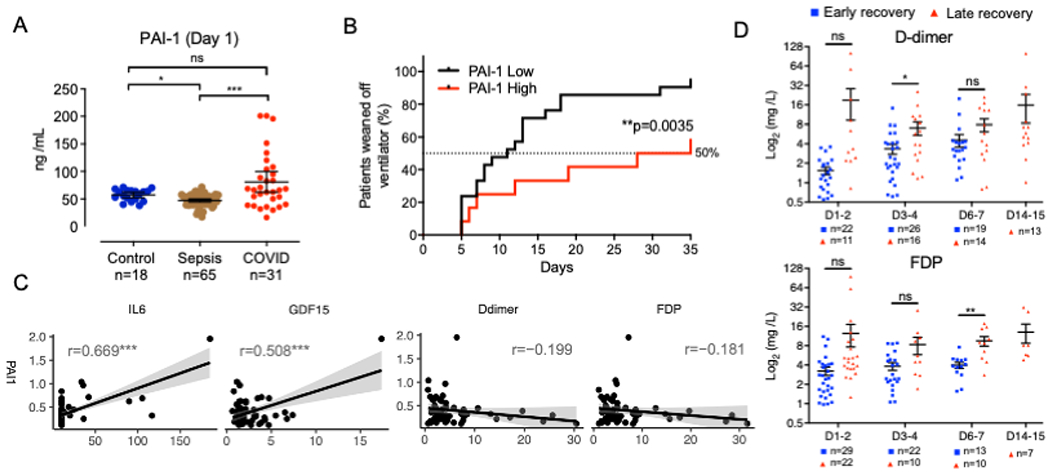
(A) Quantification of PAI-1 in healthy control, sepsis patient and COVID-19 patient plasma (mean ± SE; Kruskal-Wallis test with Dunn’s post-hoc test; *p < 0.05; ***p < 0.001; ns: not significant). The number of samples is as indicated.
(B) Kaplan-Meier survival curves for time to weaning from ventilator in COVID-19 patients separated into PAI-1 low and high groups (Log-rank test; **p<0.01).
(C) Correlation plots of PAI-1 with IL-6, GDF-15, D-dimer and FDP with linear regression. r means Pearson correlation coefficient (***p < 0.001).
(D) Time-series comparison of plasma D-dimer and FDP levels between the early recovery (blue dot) and late recovery (red dot) groups. The mean ± SE are shown together in each dot plot with a black line (Welch’s t-test; *p< 0.05; ns: not significant). The number of samples is as indicated.
To further study the temporal dynamics of published prognostic markers, we re-analyzed longitudinally collected serum samples grouped into early or late recovery phases based on the published reports40,43. We assessed the temporal dynamics of reported prognostic markers of fibrinolysis/thrombosis (D-dimer and FDP)41,42. In the late recovery group, D-dimer and FDP were significantly higher on day 3-4 and 5-6, respectively, demonstrating that the thrombotic state became apparent later than the GDF-15 increase observed on day 1-2 as previously reported40 (Figure 1D). Our cohort data indicate that the level of endothelial damage and associated inflammation precedes the manifestation of life-threatening thrombotic states.
In pulmonary endothelial cells, SARS-CoV-2 RNA is enriched, and gene expression levels of cytokine, inflammation, and chemotaxis genes are increased in response to the infection44,45. To dissect the key molecular signature in the damaged and shed endothelial cells, we analyzed publicly available single-cell RNA-seq data derived from bronchoalveolar lavage fluid (BALF)46 and extracted populations expressing VWF, PECAM1, ENG, and FLT1 (Figure S1B). hierarchical clustering identified a set of genes with altered expression in endothelial cells from infected patients (Figure S1C). Gene ontology analysis revealed the significant upregulation of the complement pathway and inflammatory response-related gene sets (Figure S1D). Notably, molecular alterations in severely inflamed endothelial cells involve complement alternative pathway (AP) genes such as CFD, CFB, and CFP, which are highly expressed in severe COVID-19 patients relative to mild COVID-19 or healthy groups (Figure S1E and S1F). Heatmaps showing the predicted ligand-receptor interactions between sender (innate immune cells (macrophages (Mφ), monocyte, and neutrophil) and receiver (endothelial cells), indicated important cytokines and chemokines crosstalk (Figure S1G). Consistent with the vast literature, these results suggest that the progression of the inflammatory process is accompanied by the activation of the innate immunity program in impaired endothelial cells.
Development of SARS-CoV-2 infection competent vascular organoids using iPSC
To further understand the molecular alterations that interlink endotheliopathy and hypercoagulability, appropriate in vitro infection models are essential for deciphering the causative biological processes leading to vascular injury and thrombosis. However, current experimental systems using primary ECs37,47 or human pluripotent stem cell (PSC)-derived ECs and organoids38, have demonstrated minimal viral infectivity and amplification even upon transgene overexpression48. More importantly, their downstream effects, such as inflammatory and thrombotic events, could not be modeled.
For the PSC-derived systems, past differentiation protocols require continuous exposure of relatively high VEGF-A (30-200 ng/mL) for EC induction49–52. However, this iteration led to proliferative and angiogenic ECs53, as opposed to the native and intact vascular state. Here, we adapted our VEGF-weaning induction protocol to generate infection-competent naïve endothelial cells (Figure 2A). Briefly, the mesodermal lineage cell population was formed from iPSC colonies by adding BMP4, VEGF-A, and GSK3 inhibitor, and induced to angioblast by VEGF-A (80 ng/ml), SCF, bFGF, and TGFb inhibitor (Figure S2A). Subsequently, CD34+ (endothelial marker) CD73+ (venous marker) CXCR4dim (arterial marker) venous endothelial cells (iVEC) were gained by stepwise reduction of VEGF-A concentration from 80ng/ml, 50 ng/ml to 5 ng/ml in the medium containing SCF, Flt-3, IL-3, and TPO (Figure 2B, S2B, and S2C). Immunostaining confirmed that another venous endothelial marker, COUP-TFII, was also expressed in CD31+ iVEC (Figure 2C).
Figure 2. Human vascular organoids are permissive for SARS-CoV-2 infection to induce inflammation.
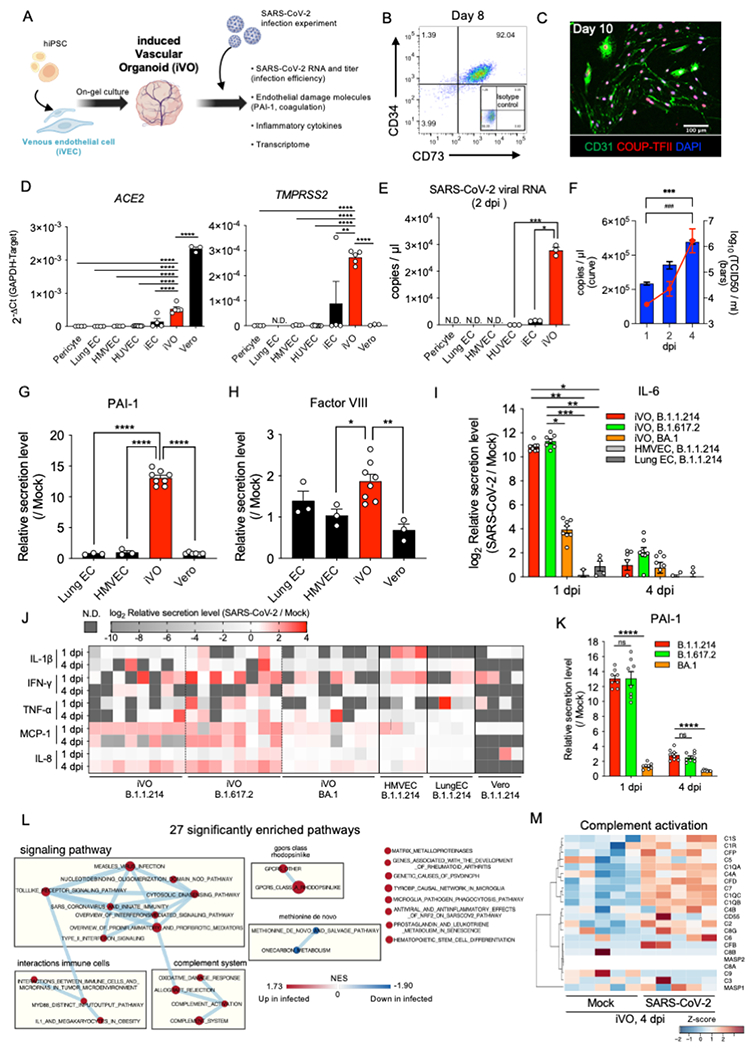
(A) The schema of SARS-CoV-2 infection and phenotypic analysis for induced vascular organoid (iVO).
(B) Scatter dot-plots of the flow cytometry analysis of CD34 and CD73 expression at day 8 in CD34+ purified iVEC cultured with 5 ng/ml VEGF-A at days 6 to 8.
(C) Immunofluorescent staining of iVEC for CD31/COUP-TFII/DAPI on day 10 after CD34+ purification.
(D) qRT-PCR analysis of ACE2 (left) and TMPRSS2 (right) (n=3-8; mean ± SE; One-Way ANOVA with Tukey’s post-hoc test; **p<0.01; ****p<0.0001). HMVEC, human microvascular endothelial cell; HUVEC, human umbilical vein endothelial cell; iEC, iPSC-derived endothelial cell.
(E) qRT-PCR analysis of SARS-CoV-2 viral copy number in cell culture supernatant at 2 dpi (n=3; mean ± SE; One-Way ANOVA with Tukey’s post-hoc test; *p < 0.05; ***p < 0.001).
(F) Time-series fold change in SARS-CoV-2 viral RNA copy number (red curve) and titer (blue bars) in cell culture supernatant expression of iVOs (n=4, RNA copy number; n=3, titer; mean ± SE; Two-Way ANOVA; ***p < 0.001, RNA copy number; ###p < 0.001, titer).
(G) Quantification of PAI-1 in culture supernatant at 1 dpi. Each value was normalized by the quantitative value in the mock control of the same cell type (n=3-8; mean ± SE; One-Way ANOVA with Dunnet’s post-hoc test compared to iVO; ****p < 0.0001).
(H) Quantification of Factor VIII in culture supernatant at 1 dpi. (n=3-8; mean ± SE; One-Way ANOVA with Dunnet’s post-hoc test compared to iVO; *p < 0.05; **p < 0.01).
(I) Quantification of IL-6 in culture supernatant of iVO infected with different strains and primary endothelial cells infected with B. 1.1.214 strain at 1 dpi and 4 dpi. Each value was normalized by the quantitative value in the mock control of the same cell type (n=4-8; mean ± SE; One-Way ANOVA Kruskal-Wallis test with Dunn’s post-hoc test; *p < 0.05; **p < 0.01; ***p < 0.001).
(J) The heatmap showing inflammatory cytokines and chemokines at 1 dpi and 4 dpi. Each value was log2 transformed fold change of infection / mock.
(K) Quantification of PAI-1 in culture supernatant of iVO infected with different strains at 1 dpi and 4 dpi. Each value was normalized by the quantitative value in the mock control of the same cell type (n=8; mean ± SE; One-Way ANOVA with Dunnet’s post-hoc test compared to B. 1.1.214 infected iVO; ****p < 0.0001).
(L) Network map of connections among significantly enriched pathways by GSEA of SARS-CoV-2 infected and mock (FDR<20%, p<0.05). Each circle indicates the size of the gene set, and the color scale indicates the normalized enrichment score (NES).
(M) The heatmap showing genes in the complement activation pathway. Each value was standardized to z-score.
Given that the surface polarity of endothelial cells is an essential factor to the success of infection54,55, we induced vascular organoids (iVOs) from iVEC on a gel plug (Figure 2A). iVEC quickly developed into a branched endothelial structure composed of CD31+COUP-TFII+ venous endothelial cells (Figure S2D and S2E), surrounded by PDGFRb+ pericytes (Figure S2F). The outer layer of the endothelium produced collagen IV basement membrane (Figure S2G). Generated iVO expressed higher levels of viral entry receptor and protease, ACE2 and TMPRSS2 than the other available primary- or stem cell-derived ECs and primary pericyte (Figure 2D). iVOs and other available ECs were treated with SARS-CoV-2 at 1x105 TCID50/ml for 2 hrs. Higher SARS-CoV-2 RNA copies were observed in iVOs supernatant at 2 days post-infection (dpi) than other ECs (Figure 2E). Tracking the kinetics of viral RNA and titer (TCID50/ml) after infection showed approximate 4- and 41-fold amplification in 4 days, respectively (Figure 2F). Immunostaining confirmed the localization of SARS-CoV-2-N protein, which was co-localized on iVOs (Figure S2H). Notably, our infection model induces endotheliopathy-associated phenotypes as confirmed by secreted protein measurements: e.g., the release of PAI-1 (Figure 2G), coagulation factor VIII (FVIII) (Figure 2H), and known endothelium-derived inflammatory cytokine (IL-6) (Figure 2I) and chemokine (IL-8, MCP1) production (Figure 2J). Interestingly, in all tested SARS-CoV-2 strains, B.1.1.214, B.1.617.2 (Delta variant), BA.1 (Omicron variant), viral RNA detected at 1 dpi showed evidence of infection with iVO, but for the BA.1 infection, viral replication up to 4 dpi was modest (Figure S2I). Consistently, B.1.1.214 and B.1.617.2 strains elicit inflammatory cytokine responses in iVOs, but such inflammatory responses were lower in BA.1 strain as was observed in primary ECs and infection-competent Vero cells (Figure 2J). Endothelial damage as measured by PAI-1 increase, was more prominent in B.1.1.214 or B.1.617.2 -infected iVO as early as 1 dpi (Figure 2K).
We further performed transcriptome analysis at 4 dpi to probe infected endothelial cell-specific signatures. Pathway enrichment analysis showed that activation of signaling cascades mediated by inflammatory responses after infection (toll-like receptor signaling and IFN signaling pathway) increased expression of the complement pathway and immune cell interaction-related molecules (Figure 2L). In particular, we noted a marked increase in the expression of an intermediate and final product of the complement pathway that is part of the membrane attack complex, and molecules that catalyze upstream reactions (classical; C1QA, C1QB, C1QC, and alternative; CFD) (Figure 2M). Together, we have successfully established an in vitro endothelial organoid model that can reproduce the vascular injury and inflammatory phenotype associated with the viral strain dependent response.
Inflammathrombosis in iVO-derived engineered human blood vessels
We then evaluated virus-induced damages in perfused human blood vessels from iVO. By taking advantage of the cranial window model56,57, iVOs were transplanted to develop perfused human blood vessels by anastomosing the recipient mouse circulatory system (Figure 3A). We infused SARS-CoV-2 spike protein (extracellular domain, ECD) in the mouse to determine the impact on chimeric human-mouse blood vessels formed under the cranial window. Intravital immunofluorescence imaging with human-specific CD31 antibody confirmed the development of fully patent vessels from transplanted iVO. At multiple sites in human CD31+ vessels, perfused fluorescent dextran signals were lost within 24 hrs of ECD administration, whereas mouse blood vessels remain intact (Figure 3B). Quantification of blood flow perfused into the engineered human vessels indicated a significant decrease at 24 hrs only after ECD administration but not in control peptide or saline group (Figure 3C). Progressive acquisition of fibrin/fibrinogen was observed in the obstructed human blood vessels (Figure 3D). These results indicate iVO-derived human vessels recapitulate thrombotic episodes in response to ECD exposure in mice.
Figure 3. Modeling inflammathrombosis specific to engineered human blood vessels.
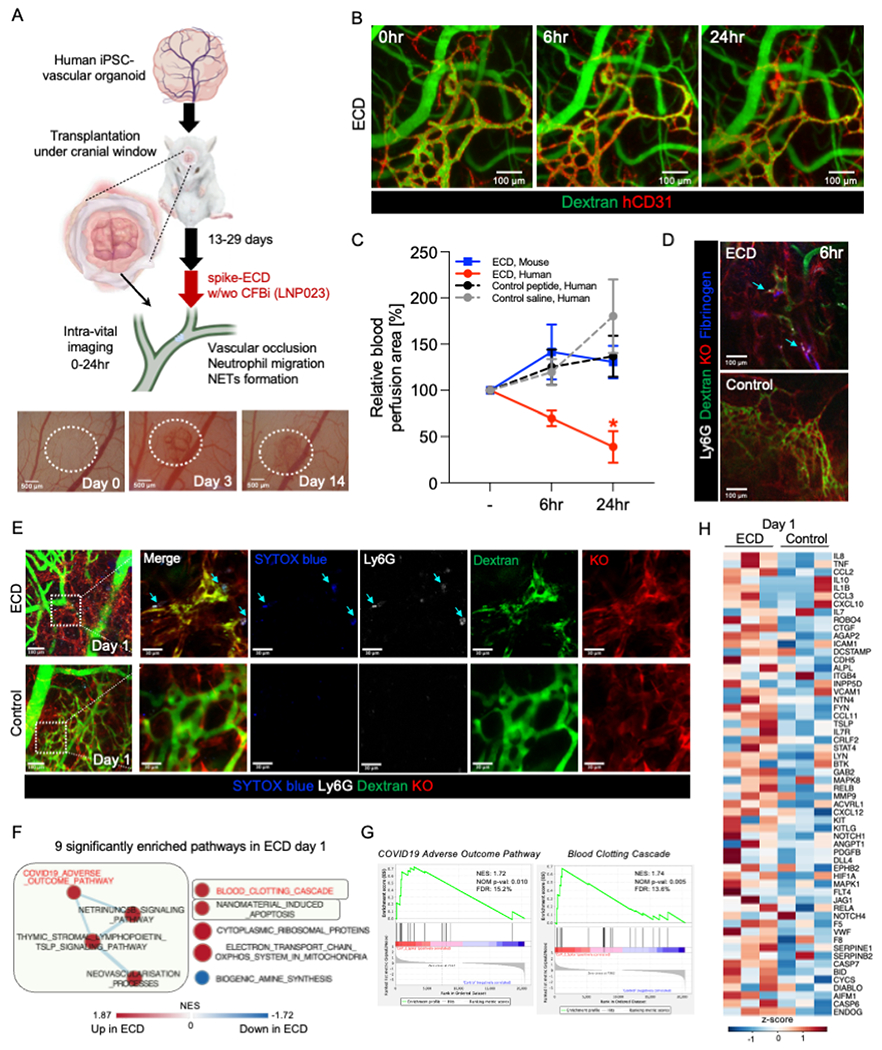
(A) Top, schematic representation of our transplantation strategy. Bottom, macroscopic image of transplanted human iPSC-vascular organoid under cranial window. Dotted area indicates the transplanted human iPSC-vascular organoid.
(B) Intravital fluorescence microscopy imaging of the organoid-transplanted mice treated with spike-ECD via tail vein. The organoid-derived human vascular and blood flow are visualized using a human-specific anti-CD31 antibody (hCD31) and FITC-dextran, respectively.
(C) Quantification of relative blood perfusion area in human and mouse vessels after spike-ECD infusion and in human vessels after control peptide or saline infusion to 0hr (mean ± SD; n = 3; *p < 0.05; ns: not significant; compared to 0hr [-] by ANOVA followed by Dunnett’s test.).
(D) Intravital fluorescence microscopy thrombus imaging of transplanted human iPSC-vascular organoid (Kusabira orange (KO)) injected with FITC-dextran, anti-mouse Ly6G antibody (neutrophil), and fluorescence-conjugated fibrinogen 6 hrs after spike-ECD or control peptide infusion.
(E) Intravital fluorescence microscopy NETs imaging of transplanted human iPSC-vascular organoid (Kusabira orange (KO)) injected with FITC-dextran, anti-mouse Ly6G antibody (neutrophil), and SYTOX blue (extracellular DNA) 24 hrs after spike-ECD or control peptide infusion.
(F) Network map of connections among significantly enriched pathways by GSEA of spike-ECD infusion (FDR<20%, p<0.05). Each circle indicates the size of the gene set, and the color scale indicates the normalized enrichment score (NES).
(G) Enrichment plots of selected significantly enriched pathways.
(H) The heatmap showing core enrichment genes in pathways highlighted in green in Figure 3F. Each value was standardized to z-score.
Next, we sought to examine pre-thrombotic innate immune response towards engineered human blood vessels. Serial intravital imaging analysis showed that Ly6G labeled neutrophil migration was initiated as early as 6 hrs post ECD infusion, whereas vascular occlusion occurs within 24 hrs (Video S1). Notably, these neutrophil trapping and subsequent thrombotic events are highly specific to human-, but not mouse-, derived vasculatures (Figure S3A and S3B). Tracking analysis of neutrophils across time-lapse observation revealed that in human vessels, trapped neutrophils emerged 6 hrs post-ECD fusion and most migrating neutrophils were found on day 1 compared to those observed in mouse vessels (Figure S3C). In trapped neutrophils after 1 day post ECD infusion, intravital co-staining of SYTOX blue and Ly6G found neutrophil extracellular traps (NETs)-like DNA release (Figure 3E). Transcriptome analysis of transplanted iVO grafts at the time of thrombosis (day 1) revealed that blood clotting pathway genes (e.g., F5 and VWF) and chemokines with direct evidence of association with and COVID-19 pathogenesis (e.g., IL8 and MCP-1) were increased in response to local vascular disturbances involving neutrophil migration and occlusion (Figure 3F–3H). Thus, in vitro and in vivo human blood vessel models are permissive for modeling different aspects of inflammathrombosis associated with a viral infection.
Proteome analysis identified early complement activation preceding endotheliopathy and hypercoagulable states.
Longitudinal proteome analysis was conducted to gain further insights into molecular mediators that drive vascular complications (Table S1). Differential signatures were identified by comparing late- and early- recovery groups. In the late recovery group, there were significant increases in FCGBP (anti-inflammation)58, SFTPB (lung surfactant metabolism)59, coagulation system (A2M and SERPINA1), and complement proteins (C7, CFHR5, and CFD) on day 1-2 (Figure 4A and S4A). In particular, CFD, a serine protease of the AP, remained elevated after day 3-4 (Figure S4B). Ba, a CFD cleavage product, showed a positive correlation not only with cytokines but also with thrombosis/fibrinolysis markers (PAI-1, D-dimer and FDP) and tissue damage markers such as creatinine (Cr) and LDH41,42, which showed a negative correlation with PLT (Figure 4B). The end-product of the complement pathway (terminal complement complex; TCC) was higher in COVID-19 than in sepsis (Figure 4C). TCC and Ba significantly decreased after recovery of patient symptoms (Figure S4C). We also compared the chrono-dynamics of C4d (classical pathway; CP) and Ba, relative to the COVID-19 recovery time. In late recovery, the alternative pathway Ba was continuously and significantly elevated from the beginning of ICU admission (Figure 4D and 4E). These are consistent with scRNAseq and in vitro RNAseq analysis models, wherein the complement AP pathway was activated in endothelial cells after infection, along with inflammation-related signaling pathways. To determine the involvement of complement amplification in inflammation and thrombosis, we challenged our in vivo transplanted human vascular organoid model with LNP023, a factor B inhibitor (Figure 4F). LNP023 treatment prevented thrombus formation against engineered human blood vessels (Figure 4G). Collectively, we posit that in severe COVID-19, the persistent activation of the complement amplification loop potentially drives vascular damage and clot formation (Figure 4H).
Figure 4. Complement amplification precedes inflammathrombosis.
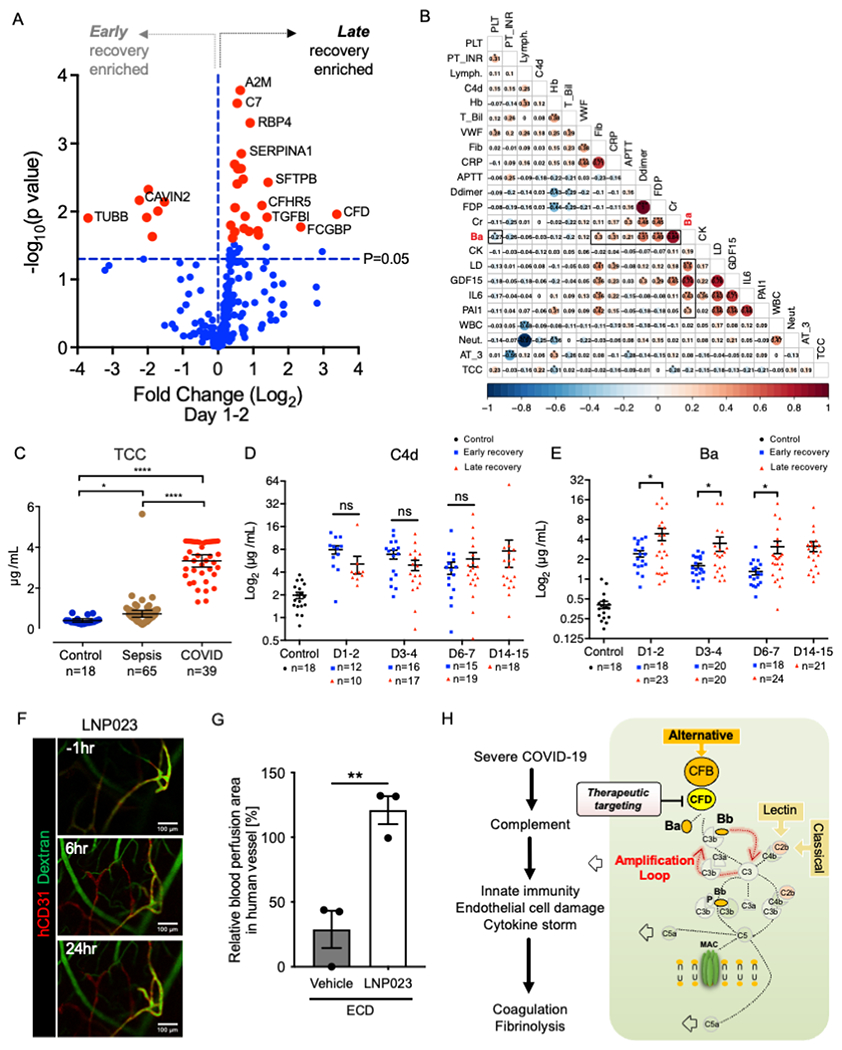
(A) Volcano plots showing differentially expressed protein profile between late recovery and early recovery at 1-2 day post ICU admission. Red dots indicate proteins that satisfied FDR cut off (20%). The blue dashed line marks a threshold of p=0.05.
(B) Pairwise correlation matrix of complement (Ba, C4d, TCC), PAI-1, cytokines (IL-6, GDF-15) and clinical laboratory parameters in patients with COVID-19. Each value represents a Pearson correlation coefficient (*p < 0.05; **p < 0.01; ***p < 0.001).
(C) Quantification of TCC in healthy control, sepsis patient and COVID-19 patient plasmas (mean ± SE; Kruskal-Wallis test with Dunn’s post-hoc test; *p < 0.05; ****p < 0.0001). The number of samples is given under the group name.
(D-E) Time-series comparison of plasma C4d and Ba levels between the early recovery (blue dot) and late recovery (red dot) groups. The mean ± SE are shown together in each dot plot with a black line (Welch’s t-test; *p < 0.05, ns: not significant).
(F) Intravital imaging of the organoid-transplanted mice treated with complement factor B inhibitor LNP023 and spike-ECD. LNP023 was orally administrated 1 hr before spike-ECD treatment. The organoid-derived human vascular and blood flow are visualized using a hCD31 and FITC-dextran, respectively.
(G) Relative quantification of blood perfusion area in human vessels after LNP023 and spike-ECD or sham (ECD+DMSO) treatment to before treatment (mean ± SE; n = 3; Welch’s t-test; **p < 0.01).
(H) Aberrant activation of complement amplification loop in endotheliopathy.
A long-acting ASHE (Acid Switched Half-Life Extended) anti-CFD antibody.
The AP is part of the innate immune system and is triggered by the cleavage of factor B into Ba and Bb by CFD. This is followed by C3 degradation via C3 convertase, which forms the terminal complement complex (TCC) with cell lysis and death activity60. To efficiently neutralize CFD, a human CFD antibody (Ab) was engineered with Acid-Switch Half-life Extension (ASHE) feature. An acid-switched antibody can bind its target at physiological pH but is released at acidic pH in the lysosome allowing the target free antibody to recycle into the bloodstream while the target itself is degraded, thereby enhancing the pharmacodynamic effect of the antibody (Figure 5A). This antibody has enhanced binding feature to FcRn that increase FcRn mediated recycling and enhance half-life. ASHE-CFD Ab showed significantly prolonged Ab turnover kinetics compared to conventional CFD antibodies61 in non-human primate serum (Figure 5B). Subcutaneous ASHE-CFD Ab injection totally inhibited TCC formation for an extended period compared to intravenous injection: less than 5 days (data not shown) and more than 8 days, respectively (Figure 5B). To further determine target engagement in the human model, ASHE-CFD Ab was exposed to our in vitro iPSC-endothelial cell simultaneously with ECD exposure. The deposition of membrane attack complex (MAC) and the high level of Ba was induced by treatment with SARS-CoV-2 spike-ECD was detected (Figure S4D and S4E). The CFD Ab inhibited MAC deposition with a lower IC50 of 2.581 μg/mL than that of Eculizumab (anti-C5), which has an IC50 of 9.283 μg/mL (Figure S4F). For Ba levels, CFD inhibition effectively reduced Ba even at low concentration ranges (Figure S4G). Together, ASHE-CFD Ab is a potent and durable inhibitor of complement factor D.
Figure 5. Factor D monoclonal antibody alleviates inflammation and coagulation responses in SARS-CoV-2 infected non-human primates.
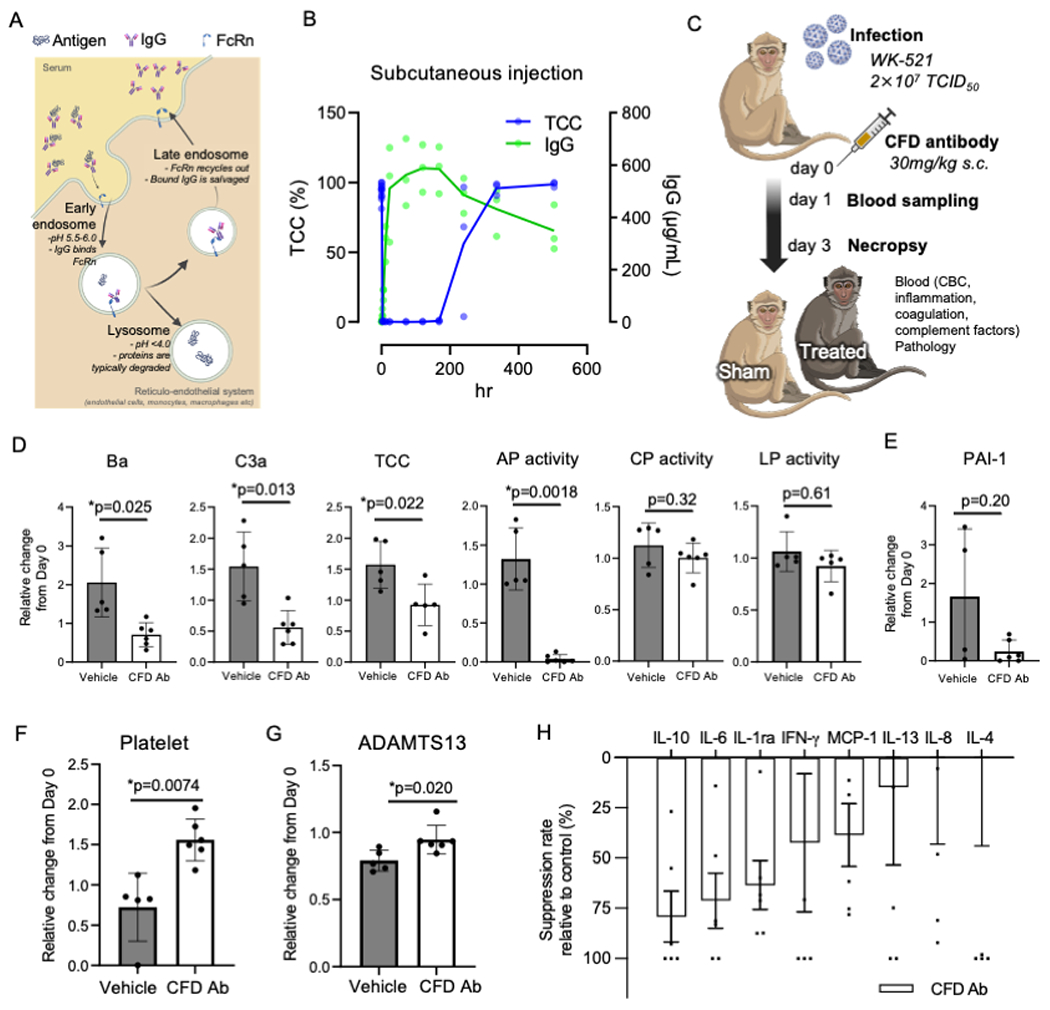
(A) Schematic diagram for ASHE concept for CFD monoclonal antibody.
(B) Time-series MAC inhibition activity and CFD Ab concentration in cynomolgus macaques (CMs) serum after subcutaneous injection.
(C) Schematic diagram for potency of CFD Ab; CMs were injected vehicle (n = 5) and anti-Factor D (n = 6) treated simultaneously with SARS-CoV-2 infection.
(D) Complement factor Ba, complement C3a, TCC, alternative pathway activity, classical pathway activity and lectin pathway activity in serum of experimental CMs (mean ± SD; n=5 or 6 per group; Welch’s t-test; *p< 0.05).
(E) The concentration of PAI-1 in plasma (mean ± SD; n=5 or 6 per group).
(F) The count of platelet in peripheral blood cells (mean ± SD; n=5 or 6 per group; Welch’s t-test; *p<0.01).
(G) The concentration of ADAMTS13 in plasma (mean ± SD; n=5 or 6 per group; Welch’s t-test; *p< 0.05).
(H) The level of the inflammatory cytokines and chemokines in day 1 plasma by CFD Ab. Each value was normalized by those of the same individual on day0 and then the percentage of suppression to vehicle was calculated. (mean ± SE; n=6). Statistical evaluation was performed by Student’s t-test (p=0.24, 0.09, 0.23, 0.53, 0.20, 0.75, 0.78, 0.56).
CFD inhibition on endothelial injury, cytokine, and coagulation in SARS-CoV-2-infected macaques.
Human complement factor D has high homology with cynomolgus macaque (96.6%) when determined by Blastp and has low protein sequence homology with rodents (66.3% with mice and 69.0% with hamsters) (https://blast.ncbi.nlm.nih.gov/Blast.cgi). To further explored the mechanistic hypothesis using human CFD Ab on thrombosis-related pathology caused by SARS-CoV-2, macaques were utilized as an in vivo model due to a lack of the antibody cross-reactivity to rodents. SARS-CoV-2 JPN/TY/WK-521/2020 (WK-521) was inoculated into the conjunctiva, nasal cavity, oral cavity, and trachea of cynomolgus macaques following previously published methodology62. Ground glass appearance indicating pneumonia in X-ray analysis was observed on day 7 after inoculation compared to pre-infection (Figure S5A, Left). On day 7, dark red lesions but not fresh red lesions were macroscopically observed on the surface of lungs, indicating that inflammation was beginning to improve (Figure S5A, Right). Histological analysis using hematoxylin-eosin staining revealed that not only were there neutrophil accumulation, but vasculitis and thrombosis were present in the lungs 3 days post-infection (Figure S5B). To optimize the timing of antibody administration, we first investigated the dynamics of endothelial injury, coagulation, immune-related and complement factors up to day 7 after infection. sICAM-1 and sVCAM-1 were significantly upregulated on day 3, and PAI-1 elevation and thrombocytopenia were observed on day 1 of infection (Figure S5C). In addition, vWF and D-dimer were increased on day 3 and 5, whereas ADAMTS13 which cleaves vWF multimer, was decreased by day7 (Figure S5D). Multiplexed cytokine assays showed that inflammation markers such as IL-1ra, MCP-1, and CCL4 levels were increased on day 1 post-infection (Figure S5E). Furthermore, SARS-CoV-2 infection increased plasma complement D, B, and Ba (components of the alternative pathway). Of the classical and lectin pathway components, complement C4d was slightly elevated, but C1q was unchanged (Figure S5F). Immunohistological analyses, furthermore, showed that vWF and C3 were detected in multiple organs, including lung, liver, and kidney of infected macaque at day 7 (Figure S5G). These are in line with published SARS-CoV-2-infected rhesus monkey models that show complement activation and signs of thrombosis peaking on the second day of infection39. Collectively, complement, coagulation, and endothelial damage markers increase from day 1 to day 3 in SARS-CoV-2-infected macaques, whereas cytokines and chemokines transiently increased on day 1.
To investigate the in vivo involvement of CFD in the physiopathology of COVID infection, 11 macaques were randomly assigned to two arms (Table S2). SARS-CoV-2 WK521 were inoculated into all macaques, and PBS (n = 5) or CFD antibody (n = 6) were administered subcutaneously at the same time (Figure 5C and Figure S5H). Complement antibody treatment with anti-CFD Ab significantly reduced the formation of complement C3a, complement factor Ba, and soluble terminal complement complex (TCC) in the serum of SARS-CoV-2-infected macaques on day 1 after virus inoculation (Figure 5D). In addition, the alternative pathway but not the classical and lectin pathways was specifically inactivated in anti-CFD Ab-treated macaques (Figure 5D). Endotheliopathy marker PAI-1 was decreased in plasma of CFD Ab treated group on day3 after treatment (Figure 5E). Anti-CFD Ab treatment also resolved coagulation abnormalities as evidenced by improvements in thrombocytopenia and significant increase in plasma ADAMTS13 concentration on day3 (Figure 5F and 5G). IFN-gamma, IL-10, IL-6, IL1ra, and MCP-1 are known to be acutely induced around day 1 62 (Figure S5E), whereas IL-4, IL-8, and IL-13 are not till day10. Plasma protein quantification revealed the reduction of such acute pro-inflammatory cytokine and chemokine responses by anti-CFD Ab treatment (Figure 5H). Together, ASHE-CFD Ab normalized the early complement alternative pathway aberration and suppressed inflammathrombotic responses induced by SARS-CoV-2 in non-human primate model.
DISCUSSION
Endotheliopathy and hypercoagulable states are recognized as key hallmarks of severe COVID-19 patients. Whether complement activation is driving the features of hypercoagulability and microthromboses in COVID-19 is currently an area of immense interest 63, in part because, in stark contrast to DIC, the systemic microangiopathy of COVID-19 shares clinical presentation with complement-driven thrombotic microvascular disease (TMA). However, the evolutionary conservation of crosstalk between the complement and coagulation systems, has complicated delineation and understanding temporal dynamics and perturbation responses of this crosstalk but is crucial in defining causative mechanisms where complement exacerbates endothelial injury and thrombosis in human systems. Here, we establish a traceable and manipulatable human iPSC-derived vascular organoids (iVO) that are permissive for SARS-CoV-2 infection and subsequent complement-driven immunothrombotic events. Unlike conventional primary endothelial cell models or iPSC-derived endothelial cells, iVO expresses both ACE2 and TMPRSS2, and generates a high titer of viral particles comparable to Vero cells. Importantly, infected iVO exhibited interferon (IFN) response and endothelial damages that were poorly represented in immortal cell lines. Newly developed iVO models display arrays of key endotheliopathy-associated phenotypes ranging from the neutrophil migratory response, endothelial injury, PAI-1 secretion, complement activation, and inflammation, to fibrin clotting. Interestingly, evidence of inflammatory inducibility of the SARS-CoV-2 Spike protein in human endothelial cell models was controversial64,65. Our iVO transplantation studies indicated that spike protein could specifically target human endothelial cells, but not murine endothelial cells, which leads to neutrophil migration by creating a coagulopathic milieu and therein generating microthrombi, a process which has been impossible to model in animal infection models 66. Conventional cell-based assays lacked several complement regulatory proteins and, as isolated systems, does not represent all cell populations, including immune cell or tissues, associated with pathogenesis in vivo65,67. The deployment of human tissue model could inform personalized insight into the risk assessment of post-infection severity by reflecting environmental factors such as an increased sugar (hyperglycemia)51 and hypoxia (oxidative stress)68, and genetic factors using multi-donor iPSC libraries69. Thus, our human vascular organoid based infectious disease modeling allows for interrogating viral infection-related endotheliopathy.
Complement is a crucial player in protective immunity against pathogens, but in excess or deregulated activation may result in collateral tissue injury, as often seen in severe COVID-19 cases 70. Of the three complement pathways, the classical pathway, the mannose-binding lectin pathway, and the alternative pathway, the AP is of central importance as it directly feeds into the amplification loop which serves to augment activation of all three complement pathways via factor D (CFD), a rate-limiting component for propagating amplification signals and producing large amounts of C3b. CFD, also known as Adipsin, is a 24 kDa serine protease comprising 228 amino acids71. Unlike most proteins of the complement system, which are synthesized by the liver and immune cells72, CFD is predominantly produced by adipocytes73. The normal level of CFD in serum is within the range of 1–2 μg/mL, which are concentrations lower than those of other complement proteins. While there are controversies about levels of CFD and other AP components in patients with severe SARS-CoV-2 infection9,74,75, our unbiased proteomic analysis highlighted prominent CFD increases at the early stage of severe COVID-19 in the late recovery group, followed by fibrinolysis/thrombosis marker aberration, such as D-dimer and FDP. Furthermore, our results support the hypothesis that the activation of the amplification pathway is more significant, as confirmed by increased cleaved product Ba and terminal sC5b-9 components9,74 at the time of ICU entry. Complement contributions in COVID-19 are envisioned as both protective and hostile. The recent “friend and foe” hypothesis 70 proposed that complement intervention could become a “foe” around the second or third weeks of infection, leading to acute respiratory distress syndrome4. Typically, ICU entry occurs around this stage; therefore, factor Ba and CFD might be potential prognostic indicators for severe COVID-19 after ICU entry. Further clinical interrogation on AP factors and other confounders will provide much-conclusive answers.
The local contribution of elevated CFD, both protein and RNA, is increasingly accepted44,76,77. In line with emerging studies, COVID-19 lungs contain more CFD transcripts in macrophage, endothelial, and alveolar epithelial cells than control lungs. Interestingly, recent in situ transcriptomes analysis of COVID-19 lung samples at the late-phase pneumonia stage (between 13-17 days post-infection) indicated that certain complement factors (CFD, C1QB, C7) and interferon response genes (IFI6, ISG15) are upregulated in the infected COVID-19 lungs but mainly at the SARS-CoV-2− sites within the lungs 77. These results support our hypothesis that subsequent local activation of the complement AP system, beyond a viral defense mechanism, potentially acts as an inflammatory mechanism of disease during the pneumonia stage.
Some small-molecule CFD inhibitors (ALXN2040, ALXN2050, and BCX9930) or CFD antibody (lampalizumab) have been explored as a therapy against complement-associated diseases, such as PNH60. However, because of the rapid turnover of complement factors, small molecules have to be administered at least two to three times daily. ASHE-CFD antibody has properties with significantly prolonged CFD turnover kinetics and inhibition of MAC formation due to its ASHE property, i.e., exhibits high affinity in neutral conditions and dissociates from the antigen in acidic (endosomal) conditions to allow for enhanced removal of CFD from circulation. The potential of CFD targeting in COVID-19 has been indicated by the fact that SARS-CoV-2 spike protein-derived complement activation was inhibited by the CFD inhibitor in the gene-modified human erythroblast cell line, TF1PIGAnull cells65. In this present study, we provide support that the activation of CFD is accompanied by endothelial damage and its deactivation protects against complement activation and endothelial damage. The IC50 value of CFD inhibition via our antibody on MAC deposition was about 4 times lower than that of a clinical-stage C5 inhibitor, Eculizumab. These results suggest that CFD overactivation inflicts endothelial damage, rationalizing CFD inhibition as a potential target for endotheliopathy induced by SARS-CoV-2 infection.
In summary, our combinatorial investigations using human organoids, patients, and the cynomolgus macaque model illustrates the causative linkage amongst the complement alternative pathway, endotheliopathy and inflammathrombosis by SARS-CoV-2 infection. Complement inhibition rectifies inflammation, coagulation markers, and associated phenotypes. These findings support CFD targeting as a strategy to counter severe inflammathrombotic complications in COVID-19 and possibly other infections.
Limitations of the study
In our in vitro model of the SARS-CoV-2 infection-competent induced vascular organoid (iVO), we observe a nascent branching architecture characterized by venous endothelial cells in proximity to pericytes. However, the modest deposition of collagen IV suggests that the organoid is yet to reach full maturity. Extending the culture period for over 10 days might promote further maturation, offering richer mechanistic insights into potential changes in endothelial cell infectivity51.
Utilizing in vivo transplantation, vascular organoids can mature into a fully perfused vascular network, enabling the study of physiological phenomena like immune cell interactions and thrombus formation in human vessels. While iVECs display venous markers, the acquisition of organ-specific attributes—ranging from surface markers and genetic profiles to morphological and functional characteristics—remains uncertain. The debate over SARS-CoV-2 infectivity across endothelial cells from different organs necessitates further exploration into organ-specific endothelial cell induction47,78–81. This will enhance our understanding of virus tropism and related endotheliopathies. Spike-ECD injection studies, for now, provide a glimpse into the ACE2-targeted cell damage pathway, prominently observed in iVO. Comprehensive viral infection experiments are essential for a deeper understanding of the severe infection pathogenesis.
Our research in non-human primates primarily assessed the efficacy and longevity of the CFD antibody, aiming to elucidate the role of the amplification pathway. Yet, consistent with other studies62, our observations suggest modest clinical outcomes, limiting its implications for severe COVID-19 cases. Considering CFD’s primary synthesis in adipose tissues, an ideal approach would involve assessing anti-CFD antibody treatment in CFD-humanized animals displaying more pronounced SARS-CoV-2 symptoms.
STAR★Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to the Lead Contact, Takanori Takebe (takanori.takebe@cchmc.org)
Material Availability
This study did not generate new unique reagents.
Data and Code Availability
The raw and processed gene expression data of in vitro iVO with SARS-CoV-2 infection and in vivo transplanted iVO with spike-ECD infusion have been deposited into the NCBI GEO database under accession GSE214694 and GSE 235232, respectively.
The raw mass spectrometry proteomics data have been deposited into the PRIDE database with the dataset identifier PXD044386.
The processed proteomics data in this paper are available in Table S1.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
EXPERIMENTAL MODELS AND STUDY PARTICIPANT DETAILS
Human subjects
Human specimen study was conducted according to the principles of the Declaration of Helsinki and was approved by the institutional review board of Osaka University Hospital [Approval numbers: 885 (Osaka University Critical Care Consortium Novel Omix Project; Occonomix Project)]. Informed consent was obtained from the patients or their relatives and the healthy volunteers for the collection of all blood samples, which was conducted at the Department of Traumatology and Acute Critical Care Medicine, Osaka University Graduate School of Medicine and Osaka Prefectural Nakakawachi Emergency and Critical Care Center from August 2020 to December 2020. All patients were diagnosed as having RT-PCR-confirmed SARS CoV-2 and pneumonia based on computed tomography (Osaka cohort). To compare with the sepsis pathogenesis, patients with sepsis in a retrospective cohort managed at the Department of Traumatology and Acute Critical Care Medicine, Osaka University Graduate School of Medicine between December 2020 to November 2021 were used. All sepsis patients were >18 years old and fulfilled the Sepsis-3 criteria. The healthy control population comprised outpatients recruited via public poster advertisements.
The blood samples from the patients were systematically measured by the central laboratory at each hospital to obtain the laboratory data. Demographic variables [age, sex, body mass index (BMI)], comorbid conditions (hypertension, diabetes, hyperlipidemia), and clinical variables [laboratory data, Acute Physiology and Chronic Health Evaluation (APACHE) II score, Sequential Organ Failure Assessment (SOFA) score, the day of weaning off ventilator, and mortality] were extracted from electronic medical records by the investigators. All blood samples were centrifuged after addition of sodium citrate to separate plasma. Plasma samples were stored at −30 °C until use.
The serum samples from pre- and post- recovery non-ICU COVID19 patients were purchased from Reprocell Inc.. All samples are ethically collected according to HIPAA regulations and IRB approved protocols in Reprocell’s Bioserve Global Repository.
iPSCs
Human iPSC lines, 1383D2, 1383D682 and 625-A483 were established by Kyoto University and derived from ePBMC® (Cellular Technology Limited, OH; https://immunospot.com/). Kusabira orange (KO)-expressing iPSC line was generated by knocking in the KO reporter into the AAVS1 in 1383D6 iPSC line 82. For the LifeAct reporter, the sequence of a short F-actin-binding peptide (GVADLIKKFESISKEE) was fused to the N-terminus of mCherry in the donor construct pAAVS1-P-CAG-mCh (Addgene #80492). This reporter was knocked in the AAVS1 locus by co-electroporating with the AAVS1-targeting Cas9-gRNA expression plasmid pXAT2 (Addgene #80494). The electroporated cells was selected with 0.5 mg/ml of puromycin, and single clones were obtained by picking the isolated colonies with CELL PICKER (Shimazu Corp.) The isolated, homologously knocked-in clones were validated by PCR genotyping and Southern blotting. All iPSCs were cultured on laminin 511 E8-fragment-coated (iMatrix-511, Nippi) dishes in StemFit AK02N (Ajinomoto).
Animals
Pharmacokinetics study of CFD antibody using cynomolgus macaques was conducted in accordance with the applicable Covance Laboratories Inc. (Covance) standard operating procedures (SOPs). All Protocol procedures comply with the Animal Welfare Act Regulations (9 CFR 3). Potency of CFD Ab using cynomolgus macaques were carried out in strict accordance with the Guidelines for the Husbandry and Management of Laboratory Animals of the Research Center for Animal Life Science at Shiga University of Medical Science and Standards Relating to the Care and Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology, Japan. The protocols were approved by the Shiga University of Medical Science Animal Experiment Committee. Regular veterinary care and monitoring, balanced nutrition and environmental enrichment were provided by the Research Center for Animal Life Science at Shiga University of Medical Science. Experiments using the virus were performed in the biosafety level 3 facility of the Research Center for Animal Life Science, Shiga University of Medical Science. All treatments within the experimental period were performed under ketamine and xylazine anesthesia and all individuals were euthanized at the endpoint using ketamine/xylazine followed by intravenous injection of pentobarbital (200 mg/kg). Macaque monkeys were monitored every day during the study to be clinically scored as shown and to undergo veterinary examinations to help alleviate suffering. Animal studies using mice were bred and maintained following institutional guidelines by Tokyo Medical and Dental University for the use of laboratory animals (approval numbers: A2022-133 and A2022-138).
METHOD DETAILS
Clinical outcomes on coagulation
Clinical blood data on coagulation (D-dimer and FDP) were obtained by CS-5100 (Sysmex) according to the manufacturer’s protocols.
Proteomics for COVID-19 patient plasma and differentially expression analysis
Mass spectrometry, processing of raw mass spectrometry spectra, peptide identification, and summarizing peptide search results were conducted as previously described84. The summarized results were quantified using the exponentially modified protein abundance index (emPAI) 85 and exported in the CSV format for further analysis. DECOY proteins and immunoglobulins were removed, and the top 256 proteins of the total emPAI summation were used. Differential expression analysis between early and late recovery were performed by using GraphPad Prism software ver.9 (GraphPad Software, Inc.). The differentially expressed proteins were partitioned into upregulated and downregulated groups based on the log2-fold change criteria between −5 and 4. A cutoff threshold by false discovery rate (FDR) was provided for the evaluation of significance (FDR<20%).
scRNA-seq data analysis
The public scRNA-seq data of the lung immune microenvironment in the bronchoalveolar lavage fluid (BALF) from 6 severe and 3 moderate COVID-19 patients and 3 healthy controls were obtained GSE115469 of Gene Expression Omnibus46. All UMI count matrices were subjected to cell and gene filtering and data scaling using the Seurat v3 package implemented in R86. Cells which have unique feature counts less than 500 or mitochondrial counts more than 20% were removed. Genes which were expressed in fewer than 3 cells were also removed. The data was normalized using default global-scaling method. Subsequently, cell populations annotated as endothelial cells were extracted as input into downstream analysis.
Data integration, clustering, visualization and differentially expression analysis were performed in same environment (Seurat v3). Scaled data of each patient were integrated through batch correction using mutual nearest neighbor alignment with dimensionally reduction loaded 50 PCs for UMAP visualization. Pathway enrichment analysis of the identified gene sets was performed using g:Profiler. Annotations for each cluster were assigned using automated searches with scCATCH 87 and marker information available in the Human protein atlas (https://www.proteinatlas.org). Prediction of ligand-receptor interactions was performed by NicheNet package implemented in R88.
Venous endothelial cells (iVEC)
625-A4 iPSCs expressing GFP-Luciferase83 or LifeAct-mCherry reporter iPSCs were pre-cultured by seeding 300 cells on laminin 511 E8-fragment-coated 10 cm dish with StemFit AK02N (Ajinomoto) and changing medium every other day for 7 days. The medium was changed to Essential 8 medium (Thermo Fisher Scientific) supplemented with 80 ng/mL BMP4 (R&D Systems), 80 ng/mL VEGF-A (Invitrogen), and 2 μM CHIR99021 (Cayman) on day 0. 2 days later, cells were replaced in Essential 6 medium (Thermo Fisher Scientific) with 5 ng/mL bFGF (Wako), 50 ng/mL SCF (R&D Systems), 80 ng/mL VEGF-A, and 2 μM SB431542 (STEMCELL Technologies). To induce hemangioblasts, the medium was changed to StemPro 34 SFM (Thermo Fisher Scientific) supplemented with 50 ng/mL VEGF-A, 50 ng/mL SCF, 50 ng/mL Flt-3 (PeproTech), 50 ng/mL IL-3 (R&D Systems), and 5 ng/mL TPO (R&D Systems) on day 4, and then to day 4 medium with VEGF-A concentration switched to 5 ng/ml on day 6. On day 8, cells were dissociated by TrypLE Select (Invitrogen), and CD34+ endothelium cell fractions were isolated using CD34 immunomagnetic beads (Miltenyi Biotec) and Quadro MACS Separator with LS Columns (Milteny Biotec) according to the manufacturer’s protocol.
Flow cytometry
Flow cytometric analyses was performed using the BD LSRFortessaTM cell analyzer (BD Bioscience). Antibodies and their dilution rates are as follows: anti-CD34 (BD Biosciences, 562577, 1:50), anti-CD73 (Milteny Biotec, 130-095-183, 1:50), anti-CXCR4 (BioLegend, 306506, 1:50).
Induced vascular organoid (iVO) generation
For SARS-CoV-2 infection experiment, the 8-d-differentiated cells for iVEC from 625-A4 iPSCs or LifeAct-mCherry reporter iPSC were seeded on growth factor-reduced matrigel (Corning) pre-coated 48-well culture plates at 8.0 × 104 cells / well and cultured with StemPro 34 SFM supplemented with 5 ng/mL VEGF-A, 30 ng/mL bFGF, and 10 μM ROCK inhibitor (Y276932). On the next day, half of the medium was replaced with a same medium minus the ROCK inhibitor. After capillary-like structures were confirmed (at least 24 hrs later), iVOs were used for SARS-CoV-2 infection assay.
SARS-CoV-2 infection assay using iVO
The SARS-CoV-2 strain B. 1.1.214 (GISAID accession number: EPI_ISL_2897162), B.1.617.2 (GISAID accession number: EPI_ISL_9636792), and BA.1 (GISAID accession number: EPI_ISL_9638489) were isolated from a nasopharyngeal swab sample of a COVID-19 patient89. This study has been approved by the research ethics committee of Kyoto University. The virus was plaque-purified and propagated in TMPRSS2/Vero cells (JCRB1818, JCRB Cell Bank)90. SARS-CoV-2 was stored at −80 °C. All experiments including virus infections were done in biosafety level 3 facilities at Kyoto University strictly following regulations. iVOs were infected with SARS-CoV-2 at 1x105 TCID50/mL for 2 hr. Culture supernatants were collected and media were changed every day. iVO was fixed for immunostaining or lysed for RNA extraction at the endpoint (4 dpi).
Viral titration of SARS-CoV-2
Viral titers were measured using median tissue culture infectious dose (TCID50) assays. TMPRSS2/Vero cells (JCRB1818, JCRB Cell Bank)90, which were cultured with Minimum Essential Media (MEM, Sigma-Aldrich) supplemented with 5% fetal bovine serum (FBS), and 1% penicillin/streptomycin (Invitrogen) were seeded into 96-well cell-culture plates (Thermo Fisher Scientific). The samples were serially diluted tenfold from 10−1 to 10−8 in cell-culture medium. Dilutions were placed onto TMPRSS2/Vero cells in triplicate and incubated at 37 °C for 96 hr. Cytopathic effects were evaluated under a microscope. TCID50/mL was calculated using the Reed-Muench method.
Intravital imaging of human iPSC-vascular organoids under a cranial window
Female non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice were purchased from CLEA Japan, Inc.. In preparation of the cranial window, NOD/SCID mouse was anesthetized by an intraperitoneal injection (0.2 ml/20 g body weight) of 20 mg/ml ketamine and 3 mg/ml xylazine in 0.9% NaCl. After removing the 7-mm diameter circular hole of the skull, we removed the dura on the right and left sides of the brain and gently place a circular 8-mm coverslip (Matsunami Glass Inc., Japan) in diameter to cover the open skull enclosed with a biocompatible glue. After allowing the mice to recover for 7 days, the coverslip was removed, and the transplantation material was placed on the cortex, followed by sealing of the glass-covered window with a biocompatible glue56. 8- to 14-week-old mice were used for preparing the cranial window. For transplantation, the 8-d-differentiated cells for iVEC (from Kusabira orange [KO]-expressing iPSCs) were isolated using CD34 immunomagnetic beads (Miltenyi Biotec) according to the manufacturer’s protocol. 8.75 × 104 CD34-positive cells mixed with 1.25 × 104 CD34-negative cells (7:1) were suspended in 100 μl of a mixture of endothelial cell growth medium (EGM, Lonza) and hepatocyte culture media (HCM, Lonza) with 5 ng/mL VEGF-A and 10 ng/mL Oncostatin M (PeproTech). The cell suspension was pipetted into a well (1 × 105 cells per well) of Ultra-Low Attachment 96-well plates and incubated at 37 °C in a humidified atmosphere containing 5% CO2 for 4 days. The 4-d-cultured iPSC-vascular organoids were transplanted on the mouse cortex under a cranial window of 9- to 16-week-old mice56.
For ECD treatment, mice were tail vein injected with recombinant SARS-CoV-2 spike-ECD protein (1 μg/mouse; GenScript) solution in saline. An equal volume of poly-His and FLAG control peptide (GenScript) or vehicle (saline) was used for controls. For treatment with ECD and LNP023, mice were pretreated with LNP023 (25 mg/kg; MedChemExpress) solution in 1% DMSO diluted in saline by oral administration 1 h before ECD injection. Intravital imaging was performed using an Olympus FV3000 confocal laser scanning microscope. The perfused vessels were highlighted by tail vein injection of Fluorescein isothiocyanate (FITC)-labeled dextran (MW 2 000 000; Sigma-Aldrich). For immunostaining, Alexa Fluor 647-conjugated anti-human-specific CD31 antibody (human vascular endothelial cells; WM59; Biolegend) and Brilliant Violet 421-conjugated anti-mouse Ly6G antibody (mouse neutrophils; 1A8; Biolegend) were tail vein injected. SYTOX blue (Thermo Fisher Scientific) for extracellular DNA staining was co-injected with Ly6G antibody. Alexa Fluor 647-fibrinogen (Thermo Fisher Scientific) was co-injected with ECD. The excitation wavelength or laser power must be adjusted to each fluorescent sample at every experimental setting.
Intravital imaging Analysis
Detecting blood perfusion area (dextran-positive area) and transplanted human blood vessel area (human CD31-positive or KO-positive area) were analyzed using NoviSight software (Evident, Tokyo, Japan). For detecting blood area, the xyz images of vessels were projected and used for analysis. For tracking analysis of neutrophils, the neutrophils were extracted from each frame of time lapse movie and overlayed for heatmap. The Dice coefficient for neutrophil-occupying area between the first frame and the subsequent 49 frames in representative time-lapse movies.
In vitro MAC deposition on iPSC-derived endothelial cells
iPSC was seeded into on laminin 511 E8-fragment-coated dish in StemFit AK02N for 24 hrs (Day 0). On day 1, the medium was changed to DMEM/F-12 (Invitrogen) with 1% Penicillin/Streptomycin, 1% Glutamax (Invitrogen), 1% B27 Supplement (Invitrogen), 25 ng/mL BMP-4 (R&D), 8 μM CHIR99021 to induce mesoderm for 3 days. On day 4, the medium was changed into StemPro-34 SFM with 1% Penicillin/Streptomycin, 2 μM Forskolin (Cayman) and 200 ng/mL VEGF-A (Invitrogen) to differentiate into endothelial cells at 37 °C in 5% CO2 for 3 days. On day 7, cell aggregates were removed by PBS wash and endothelial cells were passaged at appropriate cell numbers and maintained with same medium for 72 hrs.
Complement activation via SARS-CoV-2 spike protein stimuli was demonstrated at culture day 10. SARS-CoV-2 were used to recapitulate complement pathway activation via SARS-CoV-2 spike-ECD protein (GenScript) infection. First, 40% normal human serum (NHS) (Biopredic) were pre-incubated with 10 μg/mL SARS-CoV-2 spike-ECD and multiple concentration of test compounds (anti-CFD Ab and Eculizumab) in StemPro-34 SFM with 1% Penicillin/Streptomycin and 2 μM Forskolin and 200 ng/mL VEGF-A medium at 37 °C for 1 hr. Then, cell cultured medium was changed to this medium after treatment of final concentration of 1 mg/ml anti-human CD59 antibody (BIO RAD) at 37 °C for 0.5 hr. 24 hrs later, culture supernatant was collected for ELISA and fixed by 4% paraformaldehyde solution for immunocytochemistry.
Virus infection with cynomolgus monkeys
Total 7 individuals were assigned for phenotypic analysis by SARS-CoV2 infection. For drug efficacy evaluation, total 11 subjects were randomized into control and drug administration group, based on age, birth country, body weight. SARS-CoV-2 JP/TY/WK-521/2020 (WK-521) was used as previously reported62. All macaques were challenged with the WK-521 virus (2 × 107 TCID50) inoculated into the conjunctiva (0.05 mL × 2), nostrils (0.5 mL× 2), oral cavity (0.9 mL), and trachea (5 mL) with pipettes and catheters under ketamine/xylazine anesthesia. PBS (−) or complement factor D antibody (30 mg/kg) were subcutaneously injected simultaneously with SARS-CoV-2 inoculation. Chest X-ray radiographs were taken using the I-PACS system (Konica Minolta Inc.) and PX-20BT mini (Kenko Tokina Corporation). EDTA plasma and serum were collected on pre-inoculation and on 1, 3, 5, and 7 days after infection for hematological testing, blood counting (Vetscan), ELISA and Milliplex assay. Lung, liver and kidney tissues were collected at 3 or 7 days after infection. At day 3 or 7, macaques were anesthetized and dissected after whole blood collection. Their tissues were immersed and fixed in 10% formaldehyde for histological analysis.
RNA isolation and sequencing.
Total RNA was extracted from cultured cells, using a PureLink RNA Mini Kit (Thermo Fisher Scientific). For transplanted iVO, graft was stabilized by immersion in RNAlater and then I to beads pre-filled tube and added lysis buffer of NucleoSpin RNA Plus XS (MACHEREY-NAGEL). Tissue homogenization was performed by SHAKE MASTER tissue disrupter (BMS Japan) for 2.5 min and total RNA was extracted by NucleoSpin RNA Plus XS. A cDNA library was generated from 10 ng of total RNA, using a Superscript VILO cDNA Synthesis Kit (Thermo Fisher Scientific). Amplification, primer digestion, and adapter ligation were performed using an Ion AmpliSeq Transcriptome Human Gene Expression Kit (Thermo Fisher Scientific). The cDNA library was purified using Agencourt AMPure XP Reagent (Beckman Coulter) and quantified using Ion Library TaqMan Quantitation Kit (Thermo Fisher Scientific), followed by dilution to 75 pM with water and pooled equally. Eight samples per pool were sequenced using an Ion 540 Chip Kit (Thermo Fisher Scientific) simultaneously, using IonS5 XL (Life technologies) and Ion Chef instrument systems (Life technologies) with Ion 540 Kit-Chef (Life technologies). All procedures were performed according to the manufacturers’ protocols.
Bulk RNA sequencing data analyses.
From the sequencing output, quality control, alignment reads on hg19 (hg19_AmpliSeq_Transcriptome_21K_v1.bed), read counts and the normalization (reads per million, RPM) for each gene were obtained using the Torrent ampliSeqRNA plugin (hg19 AmpliSeq Transcriptome ERCC v1) in Torrent Suite Software v5.2.1. For transplanted iVO, to exclude mouse tissue-derived reads, mapping and alignment was performed using a custom reference sequence with mouse Refseq. Data scaling of AmpliSeq count data were performed by converting into log2(RPM+1) or z-score. Log2(RPM+1) scaled dataset were first subjected to gene functional classification and pathway enrichment analysis by GSEA ver.4.3.1. Gene set for each pathway derived from the Molecular Signatures Database (MsigDB) ver.7. Network map of connections among pathways were generated by Enrichment Map implemented in Cytoscape ver.3.9.1 using significantly enriched geneset with a nominal p value <0.05 and FDR <25%. Unsupervised clustering using Euclidean distance and average linkage method and visualization of heatmap and dendrogram were performed by heatmap.2 in ggplot2 package implemented in R using a z-score scaled data set.
Quantification of SARS-CoV-2 viral RNA copy number in vitro
The cell culture supernatant was mixed with an equal volume of 2×RNA lysis buffer (distilled water containing 0.4 U/μL SUPERase•In Rnase Inhibitor (Thermo Fisher Scientific), 2% Triton X-100, 50 mM KCl, 100mM Tris-HCl (pH 7.4), and 40% glycerol) and incubated at room temperature for 10 min. The mixture was diluted 10 times with distilled water. Viral RNA was quantified using a One Step TB Green Prime Script PLUS RT-PCR Kit (Perfect Real Time) (Takara Bio) on a QuantStudio 1 Real-Time PCR System (Thermo Fisher Scientific). The PCR primer sequences are shown in Key resources table.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD34 MicroBead Kit, human | Miltenyi Biotec | Cat# 130-046-702; RRID:AB_2848167 |
| anti-human CD59 antibody | Bio-Rad Laboratories, Inc | Cat# MCA715G; RRID:AB_566852 |
| Alexa Fluor 647-conjugated anti-human-specific CD31 antibody (Clone; WM59) for intravital imaging | BioLegend | Cat# 303111; RRID:AB_493077 |
| Brilliant Violet 421-conjugated anti-mouse Ly6G antibody (Clone; 1A8) | BioLegend | Cat# 127627; RRID:AB_10897944 |
| Anti-CD31 for immunocytochemistry | Abcam | Cat# ab28364; RRID:AB_726362 |
| Anti-human terminal complement complex MAC | Hycult Biotech | Cat# HM2167; RRID:AB_533189 |
| Anti-SARS-CoV-2-NP | Sino Biological | Cat# 40588-T62 |
| Anti-COUPTF-II | R&D systems | Cat# PP-H7147-00; RRID:AB_2155627 |
| Anti-Collagen IV | Abcam | Cat# ab6586 RRID:AB_305584 |
| Anti-PDGFRb | R&D systems | Cat# AF385 RRID:AB_355339 |
| CD73 Antibody, anti-human, APC | Milteny Biotec | Cat# 130-095-183; RRID:AB_10828447 |
| BD Horizon™ BV421 Mouse Anti-Human CD34 | BD Biosciences | Cat# 562577; RRID:AB_2687922 |
| PE anti-human CD184 (CXCR4) Antibody | BioLegend | Cat# 306506; RRID:AB_314612 |
| Sulfo-tagged goat anti-Human IgG antibody | Meso Scale Discovery | Cat# R32AJ; RRID:AB_2905663 |
| Anti-human vWF | Abcam | Cat# ab179451; RRID:AB_2890242 |
| Anti-human C3 | Sigma-Aldrich | Cat# HPA020432; RRID:AB_1847118 |
| Anti-human IgG | Abcam | Cat# ab124055; RRID:AB_10949474 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 488 | Thermo Fisher Scientific | Cat# A21206; AB_2535792 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 555 | Thermo Fisher Scientific | Cat# A-31572; RRID:AB_162543 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 594 | Thermo Fisher Scientific | Cat# A-21207, RRID:AB_141637 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 555 | Thermo Fisher Scientific | Cat# A-31570, RRID:AB_2536180 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | Cat# A-31571; RRID:AB_162542 |
| Donkey anti-Goat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Thermo Fisher Scientific | Cat# A-21447, RRID:AB_141844 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 555 | Thermo Fisher Scientific | Cat# A21137; AB_2535776 |
| Peroxidase-AffiniPure F(ab’)2 Fragment Donkey Anti-Rabbit IgG (H+L) | Jackson ImmunoResearch Labs | Cat# 711-036-152; RRID:AB_2340590 |
| -Cellstain®- DAPI solution | DOJINDO | 340-07971 |
| Bacterial and virus strains | ||
| SARS-CoV-2 JP/TY/WK-521/2020 | National Institute of Infectious Disease, Japan | N/A |
| SARS-CoV-2 strain B.1.1.214 | Hashimoto et al., 202389 | GISAID: EPI_ISL_2897162 |
| SARS-CoV-2 strain B.1.617.2 Delta | Hashimoto et al., 202389 | GISAID: EPI_ISL_9636792 |
| SARS-CoV-2 B.1.1.529 BA.1 Omicron | Hashimoto et al., 202389 | GISAID: EPI_ISL_9638489 |
| Biological samples | ||
| COVID-19 patient serums collected in Osaka cohort | Ebihara et al., 202140 | N/A |
| Sepsis patient serums collected in Osaka cohort | This study | N/A |
| Healthy control serums collected Osaka cohort | Ebihara et al., 202140 | N/A |
| Pre- and Post- recovery COVID-19 patient serums | Reprocell Inc. | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| StemFit AK02N | Ajinomoto | Cat# AJ100 |
| Essential 8 medium | Thermo Fisher Scientific | Cat# A1517001 |
| Essential 6 medium | Thermo Fisher Scientific | Cat# A1516401 |
| StemPro 34 SFM | Thermo Fisher Scientific | Cat# 10639011 |
| Hepatocyte Culture Medium | Lonza | Cat# CC-3198 |
| Endothelial Growth Medium | Lonza | Cat# CC-3162 |
| DMEM/F-12 | Invitrogen Corp. | Cat# 11330057 |
| Minimum Essential Media (MEM, Sigma-Aldrich) | Sigma-Aldrich | Cat# M4655 |
| Recombinant Human BMP-4 | R&D Systems | Cat# 314-BP-01M |
| Recombinant Human VEGF | Invitrogen Corp. | Cat# PHC9393 |
| CHIR99021 | Cayman | Cat# 13122 |
| SB431542 | Wako | Cat# 033-24631 |
| Recombinant Human bFGF | Wako | Cat# 060-04543 |
| Recombinant Human SCF | R&D Systems | Cat# 255-SC-001MG/CF |
| Recombinant Human Flt3-Ligand | PeproTech Inc. | Cat# 30019250UG |
| Recombinant Human IL-3 | R&D Systems | Cat# 203-GMP-050 |
| Recombinant Human Thrombopoietin | R&D Systems | Cat# 288-TPN-100/CF |
| Forskolin | Cayman Chemical Co. | Cat# 11018 |
| ROCK inhibitor (Y276932) | CultureSure | Cat# 034-24024 |
| Oncostatin M | PeproTech Inc. | Cat# 300-10 |
| GlutaMAX Supplement | Invitrogen Corp. | Cat# 35050061 |
| B27 Supplement | Invitrogen Corp. | Cat# 17504001 |
| TrypLE Select | Invitrogen Corp. | Cat# 12563029 |
| Penicillin/Streptomycin | Invitrogen Corp. | Cat# 15140163 |
| Laminin 511E8-fragment | Nippi | Cat# 892011 |
| Matrigel® Basement Membrane Matrix | Corning | Cat# 356237 |
| Complement factor D antibody | Takeda Pharmaceutical Company, Ltd | N/A |
| LNP023 | MedChemExpress | Cat# HY-127105 |
| Eculizmab | Absolute Antibody Ltd. | Cat# Ab00296-1.1 |
| Recombinant SARS-CoV-2 Spike-ECD protein | GenScript | Cat# Z03481 |
| poly-His and FLAG control peptide | GenScript | N/A |
| Normal human serum (NHS) | Biopredic | Cat# SER018050 |
| Dextran, Fluorescein isothiocyanate (FITC), 2,000,000 MW | Sigma-Aldrich | Cat# FD2000S |
| Alexa Fluor 647-fibrinogen | Thermo Fisher Scientific | Cat# F35200 |
| SYTOX blue | Thermo Fisher Scientific | Cat# S11348 |
| Critical commercial assays | ||
| Alternative pathway ELISA kit | Svar Life Science | Cat# COMPLAP330 |
| Classical pathway ELISA kit | Svar Life Science | Cat# COMPLCP310 |
| Lectin pathway ELISA kit | Svar Life Science | Cat# COMPLMP320 |
| human Ba ELISA kit | QUIDEL | Cat# A033 |
| human C3a ELISA | BD biosciences | Cat# 550499 |
| human TCC ELISA kit | Svar Life Science | Cat# COMPLTCCRUO |
| human C4d ELISA kit | Svar Life Science | Cat# COMPLC4DRUO |
| human FVlll ELISA kit | Biomatik Corporation | Cat# EKU03270 |
| human vWF ELISA kit | Abcam | Cat# ab223864 |
| human PAI-1 ELISA kit | Abcam | Cat# ab269373 |
| LEGENDplex™ Human Inflammation Panel 1 (13-plex) | BioLegend | Cat# 740809 |
| Non-human Primate Cytokine Magnetic Bead Panel (Millipore) | Millipore | Cat# PRCYTOMAG-40K |
| Human Cardiovascular Disease (CVD) Panel 2 | Millipore | Cat# HCVD2MAG-67K |
| human Complement Magnetic Bead Panel 1 | Millipore | Cat# HCMP1MAG-19K |
| human Complement Magnetic Bead Panel 2 | Millipore | Cat# HCMP2MAG-19K |
| Deposited data | ||
| scRNA sequence data of the lung immune microenvironment in the bronchoalveolar lavage fluid (BALF) | Liao et al., 202046 | GSE115469 |
| RAW bulk RNA sequence data (in vitro iVO with SARS-CoV-2 infection) | This study | GSE214694 |
| RAW bulk RNA sequence data (in vivo transplanted iVO with spike-ECD infusion) | This study | GSE235232 |
| Raw mass spectrometry proteomics data | This study | PXD044386 |
| Processed proteomics data | This study | Table S1 |
| Experimental models: Cell lines | ||
| human iPSC line, 1383D2 | Nakagawa et al., 201482 | N/A |
| human iPSC line, 1383D6 | Nakagawa et al., 201482 | N/A |
| human iPSC line, 625-A4 | Oceguera et al., 201583 | N/A |
| Kusabira orange [KO]-expressing iPSC line | This study | N/A |
| TMPRSS2/Vero cells | JCRB Cell Bank | Cat# JCRB1818; RRID:CVCL_YQ48 |
| LifeAct reporter iPSC line | This study | N/A |
| Experimental models: Organisms/strains | ||
| non-obese diabetic/severe combined immunodeficient (NOD/SCID) mice | CLEA Japan, Inc. | NOD/ShiJic-scidJcl |
| Cynomolgus macaques for SARS-CoV-2 infection experiment | Research Center for Animal Life Science at Shiga University of Medical Science | N/A |
| Cynomolgus macaques for pharmacokinetics study of CFD antibody | Covance Research Products Inc., Alice, Texas | N/A |
| Oligonucleotides | ||
| SARS-CoV-2 viral RNA forward primer (in vitro): AGCCTCTTCTCGTTCCTCATCAC | This study | N/A |
| SARS-CoV-2 viral RNA reverse primer (in vitro): CCGCCATTGCCAGCCATTC | This study | N/A |
| Human ACE2 forward primer: ACAGTCCACACTTGCCCAAAT | This study | N/A |
| Human ACE2 reverse primer: TGAGAGCACTGAAGACCCATT | This study | N/A |
| Human TMPRSS2 forward primer: GTCCCCACTGTCTACGAGGT | This study | N/A |
| Human TMPRSS2 reverse primer: CAGACGACGGGGTTGGAAG | This study | N/A |
| Human GAPDH forward primer: GGAGCGAGATCCCTCCAAAAT | This study | N/A |
| Human GAPDH reverse primer: GGCTGTTGTCATACTTCTCATGG | This study | N/A |
| Chlorocebus sabaeus ACE2 forward primer for Vero cells: GTGCACAAAGGTGACAATGG | This study | N/A |
| Chlorocebus sabaeus ACE2 reverse primer for Vero cells: GGCTGCAGAAAGTGACATGA | This study | N/A |
| Chlorocebus sabaeus TMPRSS2 forward primer for Vero cells: GTGCTCCGACTCTGGGATAG | This study | N/A |
| Chlorocebus sabaeus TMPRSS2 reverse primer for Vero cells: CATACAGGGTGCCAGGACTT | This study | N/A |
| Chlorocebus sabaeus GAPDH forward primer: CGAGATCCCTCCAAAATCAA | This study | N/A |
| Chlorocebus sabaeus GAPDH reverse primer: TGACGATCTTGAGGCTGTTG | This study | N/A |
| Software and algorithms | ||
| R | CRAN | Version 4.0.5; https://cran.r-project.org/ |
| RStudio | RStudio | Version 1.3.1073; https://www.rstudio.com; RRID:SCR_000432 |
| Seurat R package | Satija Lab | Version 4.1.1; http://satijalab.org/seurat/; RRID:SCR_016341 |
| scCATCH | Shao et al., 2020 87 | Version 3.2; https://github.com/ZJUFanLab/scCATCH |
| NicheNet | Browaeys et al., 2020 88 | Version 1.0.0; https://github.com/saeyslab/nichenetr |
| g:Profiler | University of Tartu | https://biit.cs.ut.ee/gprofiler/gost; RRID:SCR_006809 |
| Torrent Suite Software | Thermo Fisher Scientific | Version.5.2.1; https://tools.thermofisher.com/content/sfs/manuals/TorrentSuite_v521_ReleaseNotes.pdf |
| GSEA | Broad Institute, Inc. | Version 4.2.3; http://www.gsea-msigdb.org/gsea/index.jsp; RRID:SCR_016863 |
| Cytoscape | cytoscape.org | Version 3.9.1; https://cytoscape.org; RRID:SCR_003032 |
| NoviSight | Evident | Version 1.2.3.9; https://www.olympus-lifescience.com/en/software/novisight/ |
| INCell Developer Toolbox | GE healthcare | version 1.9.2; https://download.cytivalifesciences.com/cellanalysis/download_data/incell/6500/incell_6500_download_page.htm; RRID:SCR_015790 |
| Prism | GraphPad Software | Version 9.3.1; https://www.graphpad.com; RRID:SCR_002798 |
| Image J | NIH | Version 2.9.0; https://imagej.nih.gov/ij/ |
Quantitative PCR for ACE2 and TMPRSS2
Total RNA was isolated using ISOGENE II (NIPPON GENE). cDNA was synthesized using 500 ng of total RNA with a Superscript VILO cDNA synthesis kit (Thermo Fisher Scientific). Real-time RT-PCR was performed with the SYBR Green PCR Master Mix (Thermo Fisher Scientific) using a QuantStudio 1 Real-Time PCR System (Thermo Fisher Scientific). The relative quantitation of target mRNA levels was performed using the 2-ΔΔCT method. The values were normalized by those of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The PCR primer sequences are shown in Key resources table.
Immunocytochemistry
Fixed cells were blocked by protein block serum free blocking solution (DAKO, X909) or donkey serum (Millipore) and the primary antibody was added and incubated at 4 °C overnight. Next day, wells were washed by PBS and species-specific secondary antibody conjugated with Alexa Flour 488, 555 or 647 (Life Technologies) and DAPI (DOJINDO) with 1% blocking solution was incubated at RT for 1 hr. Then, wells were washed by PBS and the stained images were observed with BZ-X710 (Keyence). The primary antibodies and their dilution rates were used as below; anti-terminal complement complex MAC (HCB Hycult Biotech, 1/200), CD31 (Abcam, 1/50), and COUPTF-II (R&D, 1/100). MAC deposition was observed and counted by IN Cell Analyzer 6500HS and IN Cell Developer Toolbox (GE healthcare). Data was calculated and standardized by fluorescence of DAPI value.
Whole mount clearing and immunocytochemistry.
Organoids were fixed in 4% paraformaldehyde in PBS for 2 hours at 4 °C. Clearing and immunofluorescent staining were performed by using SCALEVIEW-S (Wako) according to AbScale protocol supplied from manufacturer. Primary antibodies and their dilution rates are as follows: CD31 (Abcam, 1/50), SARS-CoV-2-NP (Sino Biological, 1/100), Collagen IV (Abcam, 1/100), and PDGFRb (R&D, 1/100). Species-specific secondary antibodies conjugated with Alexa Fluor 555 or 647 (Life Technologies) were used at 1:500 dilution. DAPI (Sigma) for nuclear staining was diluted 1:1000. z-stack images were acquired using LSM 880 with Airy scan (Zeiss). 3-D image processing and volume rendering were managed by Imaris software (Oxford Instruments).
Enzyme-linked immunosorbent assay (ELISA)
Human serum, culture supernatant, animal EDTA plasma and animal serum were used for protein qualification. Complement factor-related proteins were quantified using alternative pathway ELISA kit (Svar Life Science), classical pathway ELISA kit (Svar Life Science), lectin pathway ELISA kit (Svar Life Science), human Ba ELISA kit (QUIDEL), human C3a ELISA kit (BD biosciences), human TCC ELISA kit (Svar Life Science) and human C4d ELISA kit (Svar Life Science). Coagulopathy and endotheliopathy related proteins were measured by human FVIII ELISA kit (Abcam), human vWF ELISA kit (Abcam), human D-dimer ELISA kit (Abcam), human PAI-1 ELISA kit (Abcam). All assays were conducted according to the manufacturer’s protocols.
Human IgG quantification
For pharmacokinetics of complement factor D antibody in macaque, human IgG was measured by electrochemiluminescence immunoassay. High bind MSD plates (Meso Scale Discovery) were coated with 2 μg/ml mouse anti-human IgG (Abcam) in 50mM sodium bicarbonate solution at pH 9.6, shaking at room temperature. After one hr, the plates were washed three times with PBS/0.05% Tween 20 buffer. Plates were blocked for 1 hr with 5% MSD Blocker A (Meso Scale Discovery) shaking at ambient temperature. Blocked plates were washed three times with 1X PBS/0.05% Tween 20 buffer. Immediately after the wash, the standards, samples, and controls with the drug of interest diluted in 5% MSD Block A solution were dispensed on the plate and incubated at room temperature, shaking for 1 hr. Plates with captured drug were washed as specified in the previous wash steps. A solution of Sulfo-tagged goat anti-Human IgG antibody (1 μg/ml; Meso Scale Discovery) in 5% MSD Block A was pipetted into the plate. After one hr of incubation at room temperature, any unbound antibody was washed off. 1X MSD Read Buffer T (Meso Scale Discovery) was dispensed in each well and plates were immediately read on the SECTOR Imager 2400 plate reader (Meso Scale Discovery).
Multiplex assay
The cytokine and chemokine concentrations were measured by the Non-human Primate Cytokine Magnetic Bead Panel (Millipore) in macaque EDTA plasma and by LEGENDplex human inflammation panel 1 (13-plex; BioLegend) in human cell culture supernatants. Complement factors in macaque EDTA plasma were qualified by human Complement Magnetic Bead Panel 1 and 2 (Millipore). Coagulation, endothelial activation, and thrombosis associated factors in macaque EDTA plasma were analyzed by Human Cardiovascular Disease (CVD) Panel 2 (Millipore). All assays were performed in accordance with the manufacturer’s instruction, followed by plate reading on Luminex 200 (Thermo Fisher Scientific) for macaque EDTA plasma, and by the BD LSRFortessaTM cell analyzer (BD Bioscience) for human cell culture supernatants.
Immunohistochemistry
For histopathological analysis, tissues obtained at autopsy were immersed in 10% neutral buffered formalin for fixation, embedded in paraffin, and cut to 3-μm-thick sections on glass slides. Sections were stained with hematoxylin and eosin (H&E) or Carstairs’ stain using standard protocols.
For immunohistochemistry, antigen retrieval was performed with pH6.0 or pH9.0 Target Retrieval Solution (DAKO) in the slide chamber, at 115 °C for 15 min. The slides were cooled, washed, and endogenous peroxidases were blocked with POD blocking solution (DAKO) for 10 minutes. And then samples were blocked by 10% donkey serum (Millipore) in TBS (DAKO) for 30 minutes at room temperature and incubated at 4 °C overnight with antibodies against vWF (Abcam) and C3 (Sigma). After washing, slides were then incubated with anti-rabbit labelled with HRP (The Jackson Laboratory). Signals were detected using DAB substrate buffer (DAKO), counterstained with hematoxylin. After washing, slides were mounted in mount quick. All procedures were required wash step with TBST. Images were scanned and stored on a NanoZoomer (NIKON).
Quantification and Statistical Analysis
In all patient serum data, the dots refer single patient specimens at each time point and does not include technical replication. In all in vitro and mouse iVO transplantation experimental data are representative or pooled of values from at least three or more, different differentiation batches. In cynomolgus macaque experiment, due to the death of one control macaque from unexpected causes 2 hours after virus inoculation, the control group (n=5) and the FD Ab group (n=6) were finalized as the data set. Statistical analysis was performed using GraphPad Prism 9. Data were statistically evaluated from biological independent replicates. The details of statistics and exact number of replicate (n) are shown in Figure panel or legend. Briefly, for two-samples comparison, unpaired two-tailed Welch’s t-test, Student’s t-test at equal variances assumed, Mann-Whitney’s U-test, Wilcoxon’s rank-sum test was used. If repeated tests were performed, p-values were adjusted by Holm-Sidak method or cutoff by the false discovery rate (FDR) < 20%. For comparisons between more than two samples, one-way or two-way analysis of variance (ANOVA) with Dunnett’s or Tukey’s multiple comparisons test, p-values or adjusted p-values < 0.05 were considered statistically significant.
Supplementary Material
Table S2. Grouping of monkeys recruited in SARS-CoV-2 infection experiments, Related to Figure 5
Video S1. Intravital imaging of neutrophils within human (transplant) blood vessels 6 hrs after spike-ECD treatment, Related to Figure 3 and S3
Highlights.
Longitudinal proteomics reveals aberrant complement patterns in severe COVID-19
Human PSC-vascular organoid models endotheliopathy by SARS-CoV-2 infection
SARS-CoV-2 spike-ECD triggers inflammathrombosis in engineered human vessels
Complement Factor D antibody reduces inflammathrombosis in primate COVID-19 model
ACKNOWLEDGEMENTS
We thank Shinya Yamanaka, Yasushi Kajii, Yoshiaki Kassai and Vivian Hwa for their critical comments. We thank Kanae Otsu, Yoshihiro Shimada, Rio Ohmura (Tokyo Medical and Dental University), Natsumi Mimura, Ayaka Sakamoto, Emi Sano, and Yusuke Torisawa (Kyoto University) for technical assistance with in vitro SARS-CoV-2 infection experiment and analyses, Yoshio Koyanagi, Takeshi Noda, Kazuya Shimura, and Naoko Misawa (Kyoto University) for setup and operation of the BSL-3 laboratory at Kyoto University, Miki Nagao and Yasufumi Matsumura (Kyoto University) for preparing SARS-CoV-2 isolates and Naoko Kitagawa (Shiga university of medical science) for technical assistance with in vivo SARS-CoV-2 infection study. We are thankful to Aki Kuwano, Shika Inoue, Yui Noguchi, Aya Takino, Tsuyoshi Sakairi, Akihiro Nukuda, Megumi Fukui and Masazumi Waseda for their technical support, Midori Yamasaki, Hirokazu Matsumoto, Hiroshi Yukitake, Maria Yogo, Kae Matsuda, Takanori Matsuo, Momoko Oohori, Saori Minami and Shinji Ogawa for valuable discussions and helpful support. For SARS-CoV-2 in vitro experiments, 1383D2 and 1383D6 iPS cells were kindly provided by Masato Nakagawa (CiRA, Kyoto University), and 625A-4 iPS cells were kindly provided by Knut Woltjen (CiRA, Kyoto University). Figures were partly created with BioRender.com. This work was supported by Japan Agency for Medical Research and Development (AMED) under Grant Number JP21bm0404045, JP20fk0108280, 20fk0108404, 23gm1610005, 23gm1210012, 19fk0108172, 20fk0108410, 20fk0108538, and 223fa627008, JSPS 18H02800, 19K22416, and JST Moonshot R&D, Grant Number JPMJPS2033, JPMJMS2022. This study was also supported by Takeda-CiRA (T-CiRA) Joint Program from Takeda Pharmaceutical Co., Ltd. by Cincinnati Children’s Research Foundation grant, CURE award, NIH Director’s New Innovator Award (DP2 DK128799-01), the Falk Transformational Awards Program and by World Premier International Research Center Initiative (WPI), MEXT, Japan.
Declaration of interests
This project is partly funded by Takeda Pharmaceuticals. N.S. and T.T. are patent holder associated with the technology described in this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
References
- 1.Datta PK, Liu F, Fischer T, Rappaport J, and Qin X (2020). SARS-CoV-2 pandemic and research gaps: Understanding SARS-CoV-2 interaction with the ACE2 receptor and implications for therapy. Theranostics 10, 7448–7464. 10.7150/thno.48076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, and Remuzzi G (2021). Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nat Rev Nephrol 17, 46–64. 10.1038/s41581-020-00357-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Merrill JT, Erkan D, Winakur J, and James JA (2020). Emerging evidence of a COVID-19 thrombotic syndrome has treatment implications. Nat Rev Rheumatol 16, 581–589. 10.1038/s41584-020-0474-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, and Lambris JD (2020). Complement as a target in COVID-19? Nat Rev Immunol 20, 343–344. 10.1038/s41577-020-0320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Campbell CM, and Kahwash R (2020). Will Complement Inhibition Be the New Target in Treating COVID-19-Related Systemic Thrombosis? Circulation 141, 1739–1741. 10.1161/CIRCULATIONAHA.120.047419. [DOI] [PubMed] [Google Scholar]
- 6.O’Sullivan JM, Gonagle DM, Ward SE, Preston RJS, and O’Donnell JS (2020). Endothelial cells orchestrate COVID-19 coagulopathy. Lancet Haematol 7, e553–e555. 10.1016/S2352-3026(20)30215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, et al. (2020). Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N Engl J Med 383, 120–128. 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Costanzo M, Caterino M, Fedele R, Cevenini A, Pontillo M, Barra L, and Ruoppolo M (2022). COVIDomics: The Proteomic and Metabolomic Signatures of COVID-19. Int J Mol Sci 23. 10.3390/ijms23052414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Messner CB, Demichev V, Wendisch D, Michalick L, White M, Freiwald A, Textoris-Taube K, Vernardis SI, Egger AS, Kreidl M, et al. (2020). Ultra-High-Throughput Clinical Proteomics Reveals Classifiers of COVID-19 Infection. Cell Syst 11, 11–24.e14. 10.1016/j.cels.2020.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Joly BS, Siguret V, and Veyradier A (2020). Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensive Care Med 46, 1603–1606. 10.1007/s00134-020-06088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonaventura A, Vecchie A, Dagna L, Martinod K, Dixon DL, Van Tassell BW, Dentali F, Montecucco F, Massberg S, Levi M, and Abbate A (2021). Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat Rev Immunol 21, 319–329. 10.1038/s41577-021-00536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, Zhang L, Fan G, Xu J, Gu X, et al. (2020). Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sa Ribero M, Jouvenet N, Dreux M, and Nisole S (2020). Interplay between SARS-CoV-2 and the type I interferon response. PLoS Pathog 16, e1008737. 10.1371/journal.ppat.1008737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang N, Bai H, Chen X, Gong J, Li D, and Sun Z (2020). Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 18, 1094–1099. 10.1111/jth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox SE, Akmatbekov A, Harbert JL, Li G, Quincy Brown J, and Vander Heide RS (2020). Pulmonary and cardiac pathology in African American patients with COVID-19: an autopsy series from New Orleans. Lancet Respir Med 8, 681–686. 10.1016/S2213-2600(20)30243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carsana L, Sonzogni A, Nasr A, Rossi RS, Pellegrinelli A, Zerbi P, Rech R, Colombo R, Antinori S, Corbellino M, et al. (2020). Pulmonary post-mortem findings in a series of COVID-19 cases from northern Italy: a two-centre descriptive study. Lancet Infect Dis 20, 1135–1140. 10.1016/S1473-3099(20)30434-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goshua G, Pine AB, Meizlish ML, Chang CH, Zhang H, Bahel P, Baluha A, Bar N, Bona RD, Burns AJ, et al. (2020). Endotheliopathy in COVID-19-associated coagulopathy: evidence from a single-centre, cross-sectional study. Lancet Haematol 7, e575–e582. 10.1016/S2352-3026(20)30216-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falcinelli E, Petito E, Becattini C, De Robertis E, Paliani U, Sebastiano M, Vaudo G, Guglielmini G, Paciullo F, Cerotto V, et al. (2021). Role of endothelial dysfunction in the thrombotic complications of COVID-19 patients. J Infect 82, 186–230. 10.1016/j.jinf.2020.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, Alfajaro MM, Wei J, Dong H, Homer RJ, et al. (2020). Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J Exp Med 217. 10.1084/jem.20201241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rathnasinghe R, Strohmeier S, Amanat F, Gillespie VL, Krammer F, Garcia-Sastre A, Coughlan L, Schotsaert M, and Uccellini MB (2020). Comparison of transgenic and adenovirus hACE2 mouse models for SARS-CoV-2 infection. Emerg Microbes Infect 9, 2433–2445. 10.1080/22221751.2020.1838955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun W, McCroskery S, Liu WC, Leist SR, Liu Y, Albrecht RA, Slamanig S, Oliva J, Amanat F, Schaefer A, et al. (2020). A Newcastle disease virus (NDV) expressing membrane-anchored spike as a cost-effective inactivated SARS-CoV-2 vaccine. bioRxiv. 10.1101/2020.07.30.229120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tostanoski LH, Wegmann F, Martinot AJ, Loos C, McMahan K, Mercado NB, Yu J, Chan CN, Bondoc S, Starke CE, et al. (2020). Ad26 vaccine protects against SARS-CoV-2 severe clinical disease in hamsters. Nat Med 26, 1694–1700. 10.1038/s41591-020-1070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cleary SJ, Pitchford SC, Amison RT, Carrington R, Robaina Cabrera CL, Magnen M, Looney MR, Gray E, and Page CP (2020). Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. Br J Pharmacol 177, 4851–4865. 10.1111/bph.15143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim YI, Kim SG, Kim SM, Kim EH, Park SJ, Yu KM, Chang JH, Kim EJ, Lee S, Casel MAB, et al. (2020). Infection and Rapid Transmission of SARS-CoV-2 in Ferrets. Cell Host Microbe 27, 704–709 e702. 10.1016/j.chom.2020.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi J, Wen Z, Zhong G, Yang H, Wang C, Huang B, Liu R, He X, Shuai L, Sun Z, et al. (2020). Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 368, 1016–1020. 10.1126/science.abb7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Li J, Gao M, Fan H, Wang Y, Xu X, Chen C, Liu J, Kim J, Aliyari R, et al. (2020). Interleukin-8 as a Biomarker for Disease Prognosis of Coronavirus Disease-2019 Patients. Front Immunol 11, 602395. 10.3389/fimmu.2020.602395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, Lavin Y, Swartz TH, Madduri D, Stock A, et al. (2020). An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med 26, 1636–1643. 10.1038/s41591-020-1051-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Rooij L, Becker LM, and Carmeliet P (2022). A Role for the Vascular Endothelium in Post-Acute COVID-19? Circulation 145, 1503–1505. 10.1161/CIRCULATIONAHA.122.059231. [DOI] [PubMed] [Google Scholar]
- 29.Bojkova D, Klann K, Koch B, Widera M, Krause D, Ciesek S, Cinatl J, and Munch C (2020). Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 583, 469–472. 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swaim CD, Dwivedi V, Perng YC, Zhao X, Canadeo LA, Harastani HH, Darling TL, Boon ACM, Lenschow DJ, Kulkarni V, and Huibregtse JM (2021). 6-Thioguanine blocks SARS-CoV-2 replication by inhibition of PLpro. iScience 24, 103213. 10.1016/j.isci.2021.103213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, Tang X, Zhu J, Zhao Z, Jaffre F, et al. (2020). A Human Pluripotent Stem Cell-based Platform to Study SARS-CoV-2 Tropism and Model Virus Infection in Human Cells and Organoids. Cell Stem Cell 27, 125–136 e127. 10.1016/j.stem.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, et al. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273. 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma C, Sacco MD, Hurst B, Townsend JA, Hu Y, Szeto T, Zhang X, Tarbet B, Marty MT, Chen Y, and Wang J (2020). Boceprevir, GC-376, and calpain inhibitors II, XII inhibit SARS-CoV-2 viral replication by targeting the viral main protease. Cell Res 30, 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caly L, Druce JD, Catton MG, Jans DA, and Wagstaff KM (2020). The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Res 178, 104787. 10.1016/j.antiviral.2020.104787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Han Y, Yang L, Lacko LA, and Chen S (2022). Human organoid models to study SARS-CoV-2 infection. Nat Methods 19, 418–428. 10.1038/s41592-022-01453-y. [DOI] [PubMed] [Google Scholar]
- 36.Rosa RB, Dantas WM, do Nascimento JCF, da Silva MV, de Oliveira RN, and Pena LJ (2021). In Vitro and In Vivo Models for Studying SARS-CoV-2, the Etiological Agent Responsible for COVID-19 Pandemic. Viruses 13. 10.3390/v13030379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schimmel L, Chew KY, Stocks CJ, Yordanov TE, Essebier P, Kulasinghe A, Monkman J, Dos Santos Miggiolaro AFR, Cooper C, de Noronha L, et al. (2021). Endothelial cells are not productively infected by SARS-CoV-2. Clin Transl Immunology 10, e1350. 10.1002/cti2.1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ma Z, Li X, Fan RLY, Yang KY, Ng CSH, Lau RWH, Wong RHL, Ng KK, Wang CC, Ye P, et al. (2022). A human pluripotent stem cell-based model of SARS-CoV-2 infection reveals an ACE2-independent inflammatory activation of vascular endothelial cells through TLR4. Stem Cell Reports 17, 538–555. 10.1016/j.stemcr.2022.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aid M, Busman-Sahay K, Vidal SJ, Maliga Z, Bondoc S, Starke C, Terry M, Jacobson CA, Wrijil L, Ducat S, et al. (2020). Vascular Disease and Thrombosis in SARS-CoV-2-Infected Rhesus Macaques. Cell 183, 1354–1366 e1313. 10.1016/j.cell.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ebihara T, Matsumoto H, Matsubara T, Togami Y, Nakao S, Matsuura H, Kojima T, Sugihara F, Okuzaki D, Hirata H, et al. (2021). Cytokine Elevation in Severe COVID-19 From Longitudinal Proteomics Analysis: Comparison With Sepsis. Front Immunol 12, 798338. 10.3389/fimmu.2021.798338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghaith MM, Albanghali MA, Aldairi AF, Iqbal MS, Almaimani RA, AlQuthami K, Alqasmi MH, Almaimani W, El-Readi MZ, Alghamdi A, and Almasmoum HA (2021). Potential Predictors of Poor Prognosis among Severe COVID-19 Patients: A Single-Center Study. Can J Infect Dis Med Microbiol 2021, 6656092. 10.1155/2021/6656092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaftan AN, Hussain MK, Algenabi AA, Naser FH, and Enaya MA (2021). Predictive Value of C-reactive Protein, Lactate Dehydrogenase, Ferritin and D-dimer Levels in Diagnosing COVID-19 Patients: a Retrospective Study. Acta Inform Med 29, 45–50. 10.5455/aim.2021.29.45-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Martin-Villares C, Perez Molina-Ramirez C, Bartolome-Benito M, Bernal-Sprekelsen M, and Group COEC (2021). Outcome of 1890 tracheostomies for critical COVID-19 patients: a national cohort study in Spain. Eur Arch Otorhinolaryngol 278, 1605–1612. 10.1007/s00405-020-06220-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delorey TM, Ziegler CGK, Heimberg G, Normand R, Yang Y, Segerstolpe A, Abbondanza D, Fleming SJ, Subramanian A, Montoro DT, et al. (2021). COVID-19 tissue atlases reveal SARS-CoV-2 pathology and cellular targets. Nature 595, 107–113. 10.1038/s41586-021-03570-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang S, Yao X, Ma S, Ping Y, Fan Y, Sun S, He Z, Shi Y, Sun L, Xiao S, et al. (2021). A single-cell transcriptomic landscape of the lungs of patients with COVID-19. Nat Cell Biol 23, 1314–1328. 10.1038/s41556-021-00796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, Wang F, et al. (2020). Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med 26, 842–844. 10.1038/s41591-020-0901-9. [DOI] [PubMed] [Google Scholar]
- 47.Nascimento Conde J, Schutt WR, Gorbunova EE, and Mackow ER (2020). Recombinant ACE2 Expression Is Required for SARS-CoV-2 To Infect Primary Human Endothelial Cells and Induce Inflammatory and Procoagulative Responses. mBio 11. 10.1128/mBio.03185-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dolci M, Signorini L, D’Alessandro S, Perego F, Parapini S, Sommariva M, Taramelli D, Ferrante P, Basilico N, and Delbue S (2022). In Vitro SARS-CoV-2 Infection of Microvascular Endothelial Cells: Effect on Pro-Inflammatory Cytokine and Chemokine Release. Int J Mol Sci 23. 10.3390/ijms23074063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dailamy A, Parekh U, Katrekar D, Kumar A, McDonald D, Moreno A, Bagheri P, Ng TN, and Mali P (2021). Programmatic introduction of parenchymal cell types into blood vessel organoids. Stem Cell Reports 16, 2432–2441. 10.1016/j.stemcr.2021.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patsch C, Challet-Meylan L, Thoma EC, Urich E, Heckel T, O’Sullivan JF, Grainger SJ, Kapp FG, Sun L, Christensen K, et al. (2015). Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol 17, 994–1003. 10.1038/ncb3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wimmer RA, Leopoldi A, Aichinger M, Wick N, Hantusch B, Novatchkova M, Taubenschmid J, Hämmerle M, Esk C, Bagley JA, et al. (2019). Human blood vessel organoids as a model of diabetic vasculopathy. Nature 565, 505–510. 10.1038/s41586-018-0858-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wörsdörfer P, Dalda N, Kern A, Krüger S, Wagner N, Kwok CK, Henke E, and Ergün S (2019). Generation of complex human organoid models including vascular networks by incorporation of mesodermal progenitor cells. Sci Rep 9, 15663. 10.1038/s41598-019-52204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, and Ferrara N (1998). Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem 273, 30336–30343. 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 54.Wazny V, Siau A, Wu KX, and Cheung C (2020). Vascular underpinning of COVID-19. Open Biol 10, 200208. 10.1098/rsob.200208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, et al. (2020). Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 181, 905–913.e907. 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takebe T, Zhang RR, Koike H, Kimura M, Yoshizawa E, Enomura M, Koike N, Sekine K, and Taniguchi H (2014). Generation of a vascularized and functional human liver from an iPSC-derived organ bud transplant. Nat Protoc 9, 396–409. 10.1038/nprot.2014.020. [DOI] [PubMed] [Google Scholar]
- 57.Takebe T, Sekine K, Enomura M, Koike H, Kimura M, Ogaeri T, Zhang RR, Ueno Y, Zheng YW, Koike N, et al. (2013). Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 499, 481–484. 10.1038/nature12271. [DOI] [PubMed] [Google Scholar]
- 58.Yuan Z, Zhao Z, Hu H, Zhu Y, Zhang W, Tang Q, Huang R, Gao F, Zou C, Wang G, and Wang X (2021). IgG Fc Binding Protein (FCGBP) is Down-Regulated in Metastatic Lesions and Predicts Survival in Metastatic Colorectal Cancer Patients. Onco Targets Ther 14, 967–977. 10.2147/ott.S285171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Islam A, and Khan MA (2020). Lung transcriptome of a COVID-19 patient and systems biology predictions suggest impaired surfactant production which may be druggable by surfactant therapy. Sci Rep 10, 19395. 10.1038/s41598-020-76404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barratt J, and Weitz I (2021). Complement Factor D as a Strategic Target for Regulating the Alternative Complement Pathway. Front Immunol 12, 712572. 10.3389/fimmu.2021.712572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Loyet KM, Good J, Davancaze T, Sturgeon L, Wang X, Yang J, Le KN, Wong M, Hass PE, van Lookeren Campagne M, et al. (2014). Complement inhibition in cynomolgus monkeys by anti-factor d antigen-binding fragment for the treatment of an advanced form of dry age-related macular degeneration. J Pharmacol Exp Ther 351, 527–537. 10.1124/jpet.114.215921. [DOI] [PubMed] [Google Scholar]
- 62.Ishigaki H, Nakayama M, Kitagawa Y, Nguyen CT, Hayashi K, Shiohara M, Gotoh B, and Itoh Y (2021). Neutralizing antibody-dependent and -independent immune responses against SARS-CoV-2 in cynomolgus macaques. Virology 554, 97–105. 10.1016/j.virol.2020.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gavriilaki E, and Brodsky RA (2020). Severe COVID-19 infection and thrombotic microangiopathy: success does not come easily. Br J Haematol 189, e227–e230. 10.1111/bjh.16783. [DOI] [PubMed] [Google Scholar]
- 64.Joffre J, Rodriguez L, Matthay ZA, Lloyd E, Fields AT, Bainton RJ, Kurien P, Sil A, Calfee CS, Woodruff PG, et al. (2022). COVID-19-Associated Lung Microvascular Endotheliopathy: A “From the Bench” Perspective. Am J Respir Crit Care Med. 10.1164/rccm.202107-1774OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu J, Yuan X, Chen H, Chaturvedi S, Braunstein EM, and Brodsky RA (2020). Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood 136, 2080–2089. 10.1182/blood.2020008248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodrigues LM, Midwood CJ, Coombes RC, Stevens AN, Stubbs M, and Griffiths JR (1988). 31P-nuclear magnetic resonance spectroscopy studies of the response of rat mammary tumors to endocrine therapy. Cancer Res 48, 89–93. [PubMed] [Google Scholar]
- 67.Kulkarni HS, and Atkinson JP (2020). Targeting complement activation in COVID-19. Blood 136, 2000–2001. 10.1182/blood.2020008925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nzou G, Wicks RT, VanOstrand NR, Mekky GA, Seale SA, El-Taibany A, Wicks EE, Nechtman CM, Marrotte EJ, Makani VS, et al. (2020). Multicellular 3D Neurovascular Unit Model for Assessing Hypoxia and Neuroinflammation Induced Blood-Brain Barrier Dysfunction. Sci Rep 10, 9766. 10.1038/s41598-020-66487-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kimura M, Iguchi T, Iwasawa K, Dunn A, Thompson WL, Yoneyama Y, Chaturvedi P, Zorn AM, Wintzinger M, Quattrocelli M, et al. (2022). En masse organoid phenotyping informs metabolic-associated genetic susceptibility to NASH. Cell 185, 4216–4232.e4216. 10.1016/j.cell.2022.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Java A, Apicelli AJ, Liszewski MK, Coler-Reilly A, Atkinson JP, Kim AH, and Kulkarni HS (2020). The complement system in COVID-19: friend and foe? JCI Insight 5. 10.1172/jci.insight.140711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White RT, Damm D, Hancock N, Rosen BS, Lowell BB, Usher P, Flier JS, and Spiegelman BM (1992). Human adipsin is identical to complement factor D and is expressed at high levels in adipose tissue. J Biol Chem 267, 9210–9213. [PubMed] [Google Scholar]
- 72.Choy LN, Rosen BS, and Spiegelman BM (1992). Adipsin and an endogenous pathway of complement from adipose cells. J Biol Chem 267, 12736–12741. [PubMed] [Google Scholar]
- 73.Gomez-Banoy N, Guseh JS, Li G, Rubio-Navarro A, Chen T, Poirier B, Putzel G, Rosselot C, Pabon MA, Camporez JP, et al. (2019). Adipsin preserves beta cells in diabetic mice and associates with protection from type 2 diabetes in humans. Nat Med 25, 1739–1747. 10.1038/s41591-019-0610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma L, Sahu SK, Cano M, Kuppuswamy V, Bajwa J, McPhatter J, Pine A, Meizlish M, Goshua G, Chang CH, et al. (2021). Increased complement activation is a distinctive feature of severe SARS-CoV-2 infection. bioRxiv. 10.1101/2021.02.22.432177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Reiterer M, Rajan M, Gómez-Banoy N, Lau JD, Gomez-Escobar LG, Ma L, Gilani A, Alvarez-Mulett S, Sholle ET, Chandar V, et al. (2021). Hyperglycemia in acute COVID-19 is characterized by insulin resistance and adipose tissue infectivity by SARS-CoV-2. Cell Metab 33, 2174–2188.e2175. 10.1016/j.cmet.2021.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Melms JC, Biermann J, Huang H, Wang Y, Nair A, Tagore S, Katsyv I, Rendeiro AF, Amin AD, Schapiro D, et al. (2021). A molecular single-cell lung atlas of lethal COVID-19. Nature 595, 114–119. 10.1038/s41586-021-03569-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sounart H, Lázár E, Masarapu Y, Wu J, Várkonyi T, Glasz T, Kiss A, Borgström E, Hill A, Jurek A, et al. (2022). Dual spatially resolved transcriptomics for SARS-CoV-2 host-pathogen colocalization studies in humans. bioRxiv, 2022.2003.2014.484288. 10.1101/2022.03.14.484288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bhatnagar J, Gary J, Reagan-Steiner S, Estetter LB, Tong S, Tao Y, Denison AM, Lee E, DeLeon-Carnes M, Li Y, et al. (2021). Evidence of Severe Acute Respiratory Syndrome Coronavirus 2 Replication and Tropism in the Lungs, Airways, and Vascular Endothelium of Patients With Fatal Coronavirus Disease 2019: An Autopsy Case Series. J Infect Dis 223, 752–764. 10.1093/infdis/jiab039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, and Lindskog C (2020). The protein expression profile of ACE2 in human tissues. Mol Syst Biol 16, e9610. 10.15252/msb.20209610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu F, Han K, Blair R, Kenst K, Qin Z, Upcin B, Wörsdörfer P, Midkiff CC, Mudd J, Belyaeva E, et al. (2021). SARS-CoV-2 Infects Endothelial Cells In Vivo and In Vitro. Front Cell Infect Microbiol 11, 701278. 10.3389/fcimb.2021.701278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McCracken IR, Saginc G, He L, Huseynov A, Daniels A, Fletcher S, Peghaire C, Kalna V, Andaloussi-Mäe M, Muhl L, et al. (2021). Lack of Evidence of Angiotensin-Converting Enzyme 2 Expression and Replicative Infection by SARS-CoV-2 in Human Endothelial Cells. Circulation 143, 865–868. 10.1161/circulationaha.120.052824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nakagawa M, Taniguchi Y, Senda S, Takizawa N, Ichisaka T, Asano K, Morizane A, Doi D, Takahashi J, Nishizawa M, et al. (2014). A novel efficient feeder-free culture system for the derivation of human induced pluripotent stem cells. Sci Rep 4, 3594. 10.1038/srep03594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Oceguera-Yanez F, Kim SI, Matsumoto T, Tan GW, Xiang L, Hatani T, Kondo T, Ikeya M, Yoshida Y, Inoue H, and Woltjen K (2016). Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 101, 43–55. 10.1016/j.ymeth.2015.12.012. [DOI] [PubMed] [Google Scholar]
- 84.Tachino J, Matsumoto H, Sugihara F, Seno S, Okuzaki D, Kitamura T, Komukai S, Kido Y, Kojima T, Togami Y, et al. (2022). Development of clinical phenotypes and biological profiles via proteomic analysis of trauma patients. Crit Care 26, 241. 10.1186/s13054-022-04103-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Masuda T, Tomita M, and Ishihama Y (2008). Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J Proteome Res 7, 731–740. 10.1021/pr700658q. [DOI] [PubMed] [Google Scholar]
- 86.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821. 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shao X, Liao J, Lu X, Xue R, Ai N, and Fan X (2020). scCATCH: Automatic Annotation on Cell Types of Clusters from Single-Cell RNA Sequencing Data. iScience 23, 100882. 10.1016/j.isci.2020.100882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Browaeys R, Saelens W, and Saeys Y (2020). NicheNet: modeling intercellular communication by linking ligands to target genes. Nat Methods 17, 159–162. 10.1038/s41592-019-0667-5. [DOI] [PubMed] [Google Scholar]
- 89.Hashimoto R, Tamura T, Watanabe Y, Sakamoto A, Yasuhara N, Ito H, Nakano M, Fuse H, Ohta A, Noda T, et al. (2023). Evaluation of Broad Anti-Coronavirus Activity of Autophagy-Related Compounds Using Human Airway Organoids. Mol Pharm 20, 2276–2287. 10.1021/acs.molpharmaceut.3c00114. [DOI] [PubMed] [Google Scholar]
- 90.Matsuyama S, Nao N, Shirato K, Kawase M, Saito S, Takayama I, Nagata N, Sekizuka T, Katoh H, Kato F, et al. (2020). Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc Natl Acad Sci U S A 117, 7001–7003. 10.1073/pnas.2002589117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Grouping of monkeys recruited in SARS-CoV-2 infection experiments, Related to Figure 5
Video S1. Intravital imaging of neutrophils within human (transplant) blood vessels 6 hrs after spike-ECD treatment, Related to Figure 3 and S3
Data Availability Statement
The raw and processed gene expression data of in vitro iVO with SARS-CoV-2 infection and in vivo transplanted iVO with spike-ECD infusion have been deposited into the NCBI GEO database under accession GSE214694 and GSE 235232, respectively.
The raw mass spectrometry proteomics data have been deposited into the PRIDE database with the dataset identifier PXD044386.
The processed proteomics data in this paper are available in Table S1.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.