

Preface
Scientists have been trying to identify every gene in the human genome since the initial draft was published in 2001. Over the intervening years, much progress has been made in identifying protein-coding genes, and the estimated number has shrunk to fewer than 20,000, although the number of distinct protein-coding isoforms has expanded dramatically. The invention of high-throughput RNA sequencing and other technological breakthroughs have led to an explosion in the number of reported non-coding RNA genes, although most of them do not yet have any known function. A combination of recent advances offers a path forward to identifying these functions and towards eventually completing the human gene catalogue. However, much work remains to be done before we have a universal annotation standard that includes all medically significant genes and maintains their relationships with different reference genomes.
Introduction
The Human Genome Project (HGP) was launched in 1990 with two central goals: “analyzing the structure of human DNA” and “determining the location of all human genes”2. The recent sequencing and assembly of a complete human genome from telomere to telomere3 accomplished the first of these goals: a complete, gap-free DNA sequence. Achieving the second goal, though, has been far more complicated than originally anticipated, despite a vast increase in our knowledge of the location and function of tens of thousands of human genes. Over time, the task of identifying genes and their functions has been augmented with the goal of identifying their regulatory mechanisms. International efforts have been launched to find all functional elements in the genome4,5, including genes as well as transcriptional and post-transcriptional regulatory elements.
Early conceptions of the genome treated it as a repository for genes, most of which were thought to encode a single protein-coding transcript6,7. Today, though, we know that the picture is different, and that human biology can be influenced by thousands of alternative transcripts and transcribed elements that are not translated into proteins8,9, and by hundreds of thousands of regulatory elements4. Further complicating matters, we now know that many transcribed RNA molecules are further processed into smaller RNA fragments that can have functions different from their parent transcripts (Figure 1).
Figure 1:
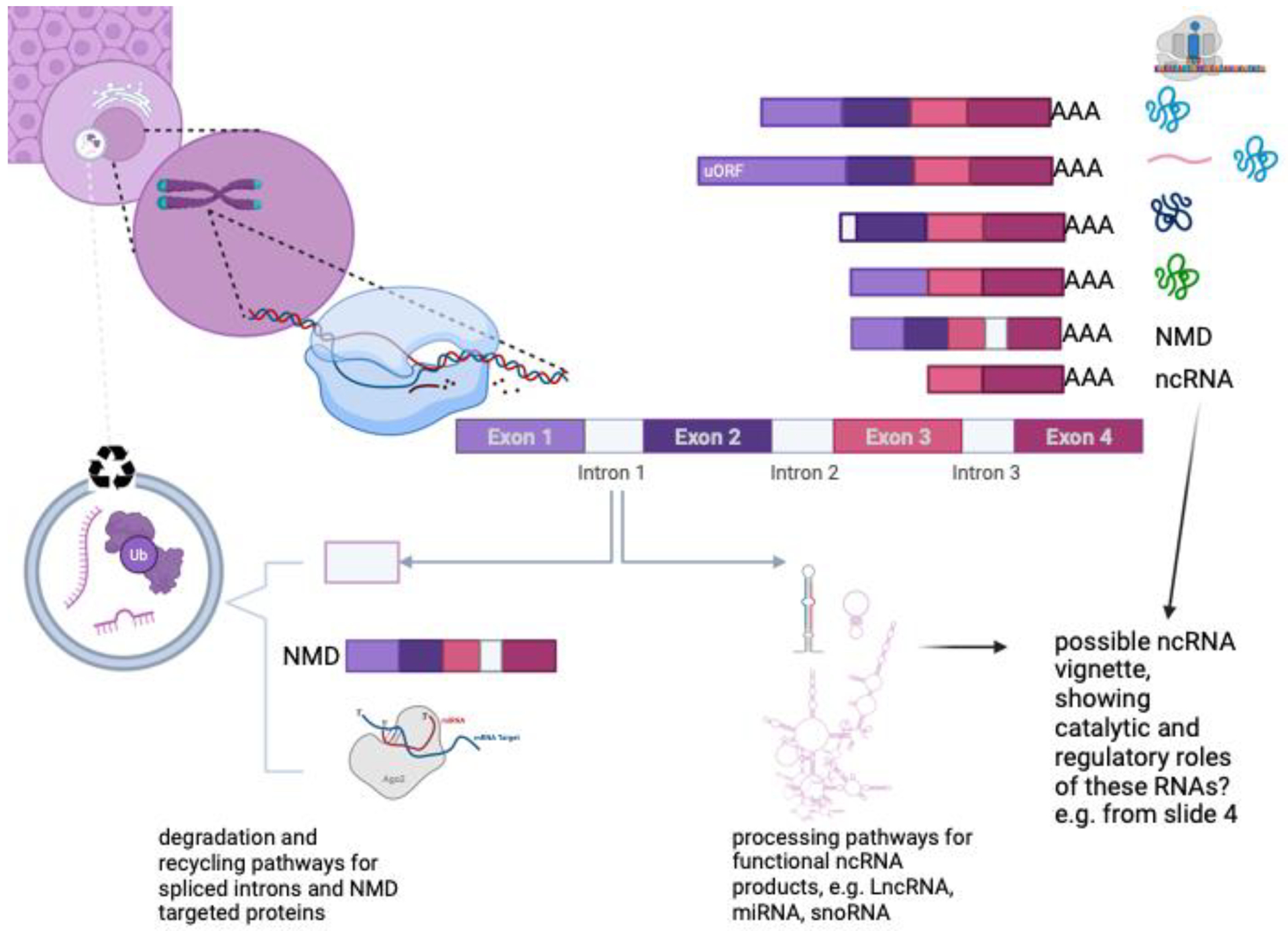
A major challenge for gene annotation is how to capture the diversity of gene products and functions. For example, although the vast majority of protein-coding genes occur on distinct transcripts, a small number of bi-cistronic transcripts encode two distinct open reading frames on the same transcript. Similarly, introns within protein-coding genes may host noncoding RNAs, including miRNAs, snoRNAs or lncRNAs, which may regulate the transcriptional activity of the locus, or may have catalytic roles unrelated to the main protein product. Alternate splicing of transcripts may give rise to proteins that enhance or inhibit each other. Transcripts that are truncated and cannot produce functional proteins are targeted for nonsense-mediated decay (NMD). These products, together with ubiquitinated proteins (Ub) or unwanted intronic material are rapidly recycled by cellular lysozomes. Other seemingly nonproductive transcripts may be repurposed as functional ncRNAs.
The purpose of this perspective is to revisit the goals of the HGP in light of our increased understanding of the diversity of functional elements in the human genome. While the genome contains many different features, this perspective will focus on genes. In the sections that follow, we will consider how we can finish specific aspects of human gene annotation in the years to come. These include (1) completing the list of protein-coding genes and all of their isoforms; (2) compiling a complete list of RNA genes of all lengths and varieties; and (3) identifying medically important genes and gene variants, and linking them to specific disorders. For each of these discussions, we will review where we are today, and what remains to be done, and then finally (4) we discuss technology needed to complete the annotation of human genes.10
Protein-coding genes
Protein-coding genes included in major genome annotation databases–e.g., GENCODE, RefSeq, and CHESS–or captured in reference protein annotation databases such as UniProtKB generally have evidence not just for their translation but also, in many cases, for the function of the protein that they encode11–14. Primary evidence can include the direct biochemical or molecular experiments or inference of function recovered from the scientific literature. The direct observation of function of a gene product or that of a close paralog provides confidence in the assignment of function of the gene and its annotation as protein-coding. In addition, the generation of high-quality genome sequences for a large number of vertebrate species, alongside the development of software (such as PhyloCSF++15, PhastCons16, or PhyloP17) capable of using alignments to identify regions of the genome under purifying selection, as well as indirect evidence of translation from mass spectrometry data, increases our confidence in many protein-coding genes.
Protein-coding gene count
The annotation of protein-coding genes was the primary focus of the Human Genome Project, after capturing the sequence itself, and while this annotation is still incomplete, the scientific community is approaching a consensus on the identities of these genes. From an initial estimate of 50,000–100,000 genes in the 1980s, the estimated number has dropped steadily, falling to 30,000–40,000 with the initial publication of the human genome18,19, and then further to ~20–25,00020,21, 22,0001, and just under 20,000 today3, one recent database release suggests as few as ~19,000 (e.g., 19,370 in GENCODE Release 41).
These refinements came about through a variety of advances, including comprehensive manual review22, improvements in computational annotation methods and analysis, and the generation of ever greater volumes of high-quality experimental transcriptional data. Despite the overall reduction in gene count, novel protein-coding genes continue to be identified, as well as alternative isoforms of known genes.
The Matched Annotation from NCBI and EMBL-EBI (MANE) collaboration23 recently published a near-complete dataset containing one isoform for each protein-coding gene for which two of the leading annotation projects, RefSeq and GENCODE, agree completely. A secondary goal of this project is to converge on an answer to the question of how many protein-coding genes we have. MANE 1.0 contains 19,062 gene loci, which covers ~98% of the curated protein-coding genes in the major human gene catalogs, bringing us closer than ever to one of the central goals of the HGP. An important caveat is that the MANE annotation is provided on the human reference genome known as GRCh38, which still contains gaps, and not on the finished T2T-CHM13 assembly, which was reported to contain 140 additional protein-coding genes3.
We propose a number of future steps to completing the annotation of protein-coding genes in the human genome:
For each protein-coding gene, develop a comprehensive picture of its transcripts and their expression levels in all tissues and cell types available, and determine its conservation in other species.
For all proteins that fold into stable structures, determine their 3-dimensional structure and evaluate their stability.
Determine all alternative sites of transcription initiation and termination, and record how frequently each site is utilized in normal tissues.
Label all reproducible splicing events that lead to non-functional proteins.
Catalog and highlight the many exceptional cases where normal rules appear to be violated. These include (a) bicistronic genes, where two distinct protein-coding genes occur on the same transcript; (b) selenoproteins, where UGA can encode selenocysteine rather than functioning as a stop codon; (c) non-standard splice sites with recognition sites deviating from the most common GT-AG, GC-AG, and AT-AC sites24; (d) coding sequences that use a codon other than ATG as the start codon; and (e) extremely short exons, which are often missed or misplaced by current methods.
Although we are nearing consensus on a protein-coding gene set, the precise set of annotated protein isoforms is still in flux11,25. Determining this number has been challenging for multiple reasons. First, the determination of isoforms today relies primarily on assembly of RNA-seq data, which in turn relies on having a complete sample of all genes in all cell types, including those prevalent during early development. Efforts such as GTEx26 have surveyed a large number of tissues, but still only cover a subset of cell types. Projects such as the Human Cell Atlas aim to identify cell-type-specific RNAs for all human cell types, but much work remains. Second, computational methods do not consistently produce the same splice isoforms from large, complex RNA-seq data sets, in part because short-read RNA-seq sequencing is insufficient to unambiguously determine complete splice structures. Third, even for those isoforms that do appear reproducibly in RNA-seq experiments, many may not encode functional proteins. And fourth, genetic variants in the human population likely introduce splice variants that will only be catalogued as we sequence a greater number of individual humans.
Pseudogenes
Another major challenge, beyond identifying the genes and splice variants themselves, is determining which gene-like elements are pseudogenes. Pseudogenes are sequences that represent defective copies of genes: over 14,000 have been annotated on the human genome. They can be divided into three types: processed (introns removed during retrotransposition), unprocessed (introns retained during duplication), and unitary (pseudogenes without a functioning counterpart in human). Recent evidence using long-read technology suggests that some previously-annotated pseudogenes may in fact be functional27,28, and other reports indicate that some pseudogenes continue to be translated, although the protein products might not be functional29.
Noncoding RNA genes
Non-coding RNA genes (ncRNAs) include a range of different RNA molecules that are transcribed from DNA, that do not encode proteins, and that provide a function in the cell. A variety of subclasses of ncRNAs have been described, including both long ncRNAs (lncRNAs), defined as RNAs ≥200 nt, and many types of shorter ncRNAs such as microRNAs, small nucleolar RNAs, transfer RNAs, piwi-interacting RNAs, and others. We note that although many non-functional RNA sequences might be transcribed in various cells and conditions, our definition will only call them genes if they have a discernable function at the cellular or organismal level. Similarly to protein-coding genes, the functions of lncRNAs need to be determined by primary experimental evidence that reveals their biochemical or molecular function, which can be obtained, e.g., from perturbation of lncRNAs followed by molecular phenotyping30. In contrast to protein-coding genes, though, it is still unclear if function can be inferred by comparing paralogues, due to our limited understanding of the mechanisms of action for most lncRNAs and to the fact that some lncRNAs contain embedded sequences from retrotransposons. In the near term, most annotation efforts will continue to strive to comprehensively catalogue ncRNA transcripts, regardless of their functional status.
Although annotation strategies that search for conserved protein sequences cannot be used for characterizing ncRNAs, high-throughput RNA-seq experiments have provided an abundant source of evidence for transcription of these genes. Compared to protein-coding RNAs, ncRNAs discovered through RNA-seq appear in relatively low abundance, raising questions about whether they encode functional elements or instead represent transcriptional noise. On the larger question of what ncRNA genes do, many possible functions have been described, including regulating expression of other genes, splicing, chromatin architecture, epigenetic regulation, dysregulation in cancer and other diseases, translation, DNA repair, and more31–33. And although tens of thousands of ncRNA transcripts are currently annotated in the human genome, their heterogeneity, poorly understood biology, and other characteristics make the comprehensive discovery of all genes in the ncRNA catalogue an unsolved problem.
A summary of lncRNA gene annotation in current catalogues is shown in Table 1. The two most-widely used are RefSeq and GENCODE, both of which employ human annotators along with large-scale cDNA and RNA sequencing resources12,34,35 to determine which ncRNA genes to include. In parallel, a variety of consortia and individual research laboratories have provided valuable additional resources, including NONCODE, the FANTOM consortium’s CAT resource, LNCipedia, miTranscriptome, CHESS, LncBook, RNAcentral, and others (e.g., see36).
Table 1:
Annotation databases that catalogue long ncRNA genes (figures as of late 2022). Here, “long” refers to loci ≥200 nt.
The overlap between these annotation databases is relatively low35, illustrating how far we are from a consensus on the identification of ncRNA genes. This rather fragmented landscape has nonetheless delivered an impressive achievement in charting the enormous variety of noncoding RNA genes.
Other challenges to ncRNA annotation
A variety of evidence suggests that ncRNA catalogues remain incomplete in a number of ways, and the community is still far from agreement on the true number of ncRNA genes and the true number of transcript isoforms. These issues arise from a variety of sources. First, the transcriptomic datasets from which most ncRNAs are derived originate from a non-exhaustive set of tissues/cell types that are over-represented by adult organs, cell lines and tumors. Rare but important cell types (e.g., tissue stem cells) or difficult-to-access developmental timepoints (e.g., embryonic stages) are poorly represented. This leads to incomplete sampling of existing gene loci and transcript isoforms. Second, the majority of transcriptomic data is produced using oligo-dT reverse transcribed RNA, which largely omits less-studied transcripts such as non-polyA and circular RNAs, although different approaches have been used to circumvent these issues (e.g.42). Third, incomplete reverse transcription of cDNA gives rise to transcript models with inaccurate 5’ ends, and RNA degradation (which affects major organs at different rates post mortem) can lead to fragmented annotations and incorrect transcription start site (TSS) annotation.
The unique biology of ncRNAs also contributes to the challenges of annotating them. Current evidence indicates that they tend to be expressed at low levels43, although this might be explained by technical biases in bulk RNA sequencing44, or in very specific cell types and tissues, leading to relatively infrequent sampling compared to protein-coding RNAs. Their splicing and post-transcriptional processing tends to be as complex as that of protein-coding genes, leading to an ensemble of transcript isoforms that confuses short-read assemblers and human annotators alike45. Note that these same features might also be true of non-functional (noisy) transcripts.
Annotation quality strongly affects our classification of the coding/non-coding biotype of RNAs and interpretation of their biological roles. Non-coding gene annotation efforts are complicated by the fact that some ‘non-coding’ loci in fact encode previously-overlooked protein products. A small but finite fraction of lncRNAs encode ‘micropeptides’ <100 amino acids in length that play diverse and important biological roles25. Their small size confounds conventional open reading frame (ORF) discovery pipelines but may be identified by ribosome profiling or evolutionary signatures of protein conservation using PhyloCSF46. Examples of lncRNAs that have been reclassified in this way include the widely-studied TUG147, whose ORF was only discovered after sequencing of a previously overlooked exon, highlighting how incomplete annotation can lead to misclassification of protein-coding status.
While some small ORF-encoded micropeptides display clear cellular phenotypes, the majority remain functionally uncharacterized and it is conceivable, particularly for those lacking significant evolutionary conservation, that they could represent either non-functional ‘translational noise’ or else early steps in the evolutionary birth of novel proteins. Although evidence of translation from some lncRNAs can be detected by mass spectrometry or ribosome profiling, the assignment of function to any such micropeptides will require further biochemical validation. In a converse fashion, protein-coding loci generate substantial numbers of non-protein-coding transcript isoforms, some of which have been shown to be functional; e.g., MYH7b48. This blurring of the boundary between ‘coding’ and ‘non-coding’ will present a fascinating challenge to future annotation efforts.
Another challenge arises from the dissonance between standard annotation schemas, involving clearly defined, yet arbitrarily defined genes and transcripts, with the messy biological reality of ncRNA transcriptional units. Conventionally, genes are defined as the union of all overlapping transcripts at a locus, and neighboring genes are separated by a clear gap. These definitions worked well in the past. However, with the advent of deep and comprehensive long-read RNA sequencing, annotations are approaching a point at which read-through transcription events will begin to unite nearly all pairs of neighboring genes. Following classical gene definitions, the result could be a single “super gene” on each chromosome49,50, which is clearly not a useful abstraction.
Functional annotation
One of the biggest challenges in ncRNA annotation relates to adding functional labels. For protein-coding genes, we have a rich amount of prior functional evidence, in addition to powerful computational methods for predicting gene function based on primary sequence. For example, DNA-binding transcription factors or membrane-bound receptors can often be predicted from translated amino acid sequences. In contrast, we know little about the vast majority of ncRNAs, and have no validated means of predicting function from sequence. Thus, one near-term goal for annotation of ncRNA genes will be describing the different types of evidence supporting them (e.g., tissue-specific expression levels), even though their function might remain unknown.
The majority of ncRNAs have not been properly assayed for function51: in initial efforts, 10–40% of selected ncRNAs showed some sort of function including effects on cell morphology and proliferation52. Scaling these approaches will require coordinated large efforts, including bulk assays in cell models and single cell sequencing from tissue or organoid models. Even so, the weak phenotypes observed when perturbing some ncRNAs, their tissue and cell type specificity, and the growing number of ncRNAs still being discovered, pose huge challenges that will require new technologies to create genome-scale assays. New methods for large-scale screening for interactions with other cellular compartments (e.g., chromatin) will be needed to address these challenges.
To date, many ncRNAs have been assigned names or biotypes that imply some function53; in particular, ncRNAs are often named after a nearby or overlapping protein-coding gene. For example, FAS-AS1 is an anti-sense (AS) transcript whose name reflects its overlap with the protein-coding gene FAS. This may lead to confusion amongst users, because the lncRNAs in question may not have a function related to that of the neighboring protein-coding gene.
Health and medical annotation
A key application of human gene annotation is its use in diagnosing and treating genetic disease. Over five thousand genes and many thousands of variants of those genes have been associated with single gene disorders and disease risk, as catalogued in OMIM54. For example, the BRCA Exchange database (https://brcaexchange.org/) currently lists over 34,000 variants in the BRCA1 gene alone, of which 2,228 are labeled as pathogenic55.
When assessing variant pathogenicity in a clinical setting, the completeness and accuracy of gene and transcript models is essential. The impacts of variants as determined by programs such as Annovar56 and Variant Effect Predictor57 depend on the predicted open reading frames of transcripts. Further, designs of oligonucleotide baits and PCR primers used in targeted capture sequencing for clinical diagnostic assays depend on the correct annotations of exon boundaries. Even when whole-genome sequencing (WGS) is used for diagnosis, clinicians do not consider unannotated exons as candidates for interpretation.
Flaws in annotation can lead to serious errors in the clinic. Among many examples that might be cited, one case of a false negative diagnosis was caused by missing exons in a transcript of CDKL5, in a proband with seizures who was ultimately diagnosed by WGS after reannotation detected the missing exons58,59. Another striking case led to a new diagnosis of Dravet syndrome after reannotation of an isoform of SCN1A revealed that the original annotation was missing a “poison” exon. In that case, the patient had splicing variants leading to expression of the nonfunctional isoform59.
The need for a clinical standard
Currently, clinical laboratories commonly use RefSeq transcripts as a reference to report variants in well-known disease-linked genes, typically relying on reports from the literature. When the literature is unclear, laboratories tend to choose a transcript using simple criteria such as length or first appearance in annotation databases. This practice is not ideal as the chosen transcript might not reflect the properties needed for clinical diagnosis and leads to inconsistency among different laboratories. To further compound this issue, clinical laboratories commonly still map variant data onto the previous reference genome GRCh37. To realize the full potential of genomic diagnostics, there is a need for a universal transcript reference for every protein-coding gene.
The MANE collaboration, launched in 2018, addressed this need by generating a set of representative reference transcripts (MANE Select) to be used as universal reporting standards. For a small number of genes, the database provides a second transcript labeled as ‘MANE Plus Clinical’ when one transcript alone is not sufficient to report known clinical variants. The MANE set now covers more than 98% of human protein-coding genes and provides a logical starting point for clinically important gene and transcript annotation. However it would be useful in the future to add clinically important annotations of noncoding RNAs and regulatory elements to MANE, at least for those that have been associated with genomic variants linked to disease risk60–62. Clinical interpretation and reporting will also benefit if other databases map their contents to the MANE standard. It is worth noting that the MANE set is anchored on GRCh38, so clinical databases that still use GRCh37 need to make the transition to GRCh38 to get the most out of this set. In addition to using a standard transcript for variant reporting, laboratories and databases also need to use standardized descriptions of genetic variants63 to ensure unambiguous mapping to reference genomes.
Consistent annotation across multiple reference genomes
The hg19 (GRCh37) genome was replaced in 2014 by GRCh38. Although both reference genomes are still in use, they differ in many ways: their coordinates are different, some genes are missing from the older version, gene symbols have changed, and many genes have different exon-intron structures. Even for genes that are unchanged across the two releases, there is no standard way to translate coordinates between genomes without creating artifacts.
The advent of a truly complete human genome sequence, T2T-CHM13, promises to provide much more stability in gene coordinates.3 Looking ahead, we are likely to have many reference genomes for different human sub-populations. We already have annotated reference genomes for Ashkenazi64, Puerto Rican65, and Han Chinese individuals66, and many more are likely to be produced. Another approach is to create a single “pan-genome” representing all populations, and the first draft of this approach, using a sequence graph based on 47 diploid assemblies of a genetically diverse individuals, has just appeared67. Ultimately, we need a gene-centric alternative to gene symbols for referring to the same gene, and a coherent system to denote the same variants on any human reference genome.
Technology to finish the human gene catalogue
Finishing the human gene catalogue will require innovative new technologies to address the challenges ahead, such as resolving the functional relationships between gene products in a diversity of tissues, cells, and developmental stages. Here we touch on a few technologies that are available now or that may be available soon to solve these problems.
Matched long-read sequencing and proteomic analysis of gene products.
Genome-wide measurements of when and where specific isoforms are expressed are currently needed. Measuring gene expression within tissues and at single-cell resolution has already revealed many coordinated patterns of gene expression in cells and tissues68. However, cell-specific splicing estimates from these studies remain problematic69, and the number of splicing events is likely underestimated70.
RNA-seq analysis at the isoform level currently relies on differential expression of exons within a gene69, which is highly dependent on the method of library construction and on sequencing depth71. Even when expression levels are measured accurately, the relative abundance of a transcript does not correlate perfectly with translation72. Ribosomal profiling is a powerful method for measuring the translation of protein-coding isoforms, and it can validate engagement with the translational machinery for many predicted alternate isoforms. Interestingly, transcript analysis from ribosomal scanning and translation complexes at polysome fractions predicts large numbers of unannotated small ORFs73,74, and these need further exploration to determine if they represent valid functional genes. These predictions might be resolved through full-length sequencing, preferably directly from RNA molecules, coupled with further ribosomal profiling or other methods for detecting translation.
While sequencing with single molecule technologies (e.g., Oxford Nanopore Technologies (ONT) and Pacific Biosciences (PacBio)) is capable of providing full-length direct RNA and cDNA sequences, relatively few experiments to date have used these technologies to survey the RNA landscape from each human cell type. Other confounding issues concern sequencing the poly-A terminating RNAs, using ONT oligo-dT reverse transcription primers and oligo-dT linker ligation75. Strategies are being developed to specifically capture total RNA that will use RNA ligases to add a primeable sequence at the true 3’ ends of all RNA transcripts. Another approach uses artificial poly(U) tailing to add a primeable sequence to both capped RNAs and non-capped RNAs76,77. Information on RNA modification, which can be measured from ONT direct RNA sequencing data, will likely provide a powerful new type of functional annotation.
Validation of protein-coding isoforms will ultimately require protein detection through deeper proteomics sequencing as is currently ongoing78 or through other means. Meta-analyses of proteomic data rely heavily on the quality of the transcriptome reference to identify peptides mapping to putative isoforms. However, when coupled with new long-read technologies, dual proteome-transcriptome assemblies are finding evidence of higher isoform diversity than predicted from a representative transcript approach, by resolving peptide fragments that would otherwise fail to map unambiguously to a gene or single isoform. In one recent study, 30% of the gene products identified using a dual PacBio-mass spectrometry approach were distinct isoforms from the same locus, which included thousands of examples where the alternate isoform was not measurable using mass spectrometry alone79. We anticipate that soon, progress in long-read technologies will produce more reliable maps of full-length transcript isoforms, quantifiable isoform switching, and isoform dosage at the resolution of individual cells.
Methods to capture low-expressed transcripts.
Capture sequencing has recently been adapted to target specific RNAs, in order to provide higher sequencing coverage for selected regions of the genome using short- and long-read RNA sequencing in a high-throughput manner34,80. This is particularly useful to enrich for RNA from ultra-low input samples81 and from genes expressed at very low levels. The use of capture technologies, together with recent increases in the throughput of long-read sequencing platforms, could enormously benefit the study of low-expressed transcripts, particularly lncRNAs, which in turn may be vital for the study of gene regulation in both normal and diseased cells.
Conclusions
Over 20 years after the original publication of the human genome, the number of protein-coding genes is stabilizing around 19,500 (Figure 2), although the number of isoforms of these genes is still a subject of intensive study and discussion. The completion of a human genome sequence itself offers the opportunity to map these genes onto a stable, finished sequence and converge to a final number in the next few years. Greater standardization of gene and isoform annotation will improve our ability to apply this knowledge in a clinical setting.
Figure 2:
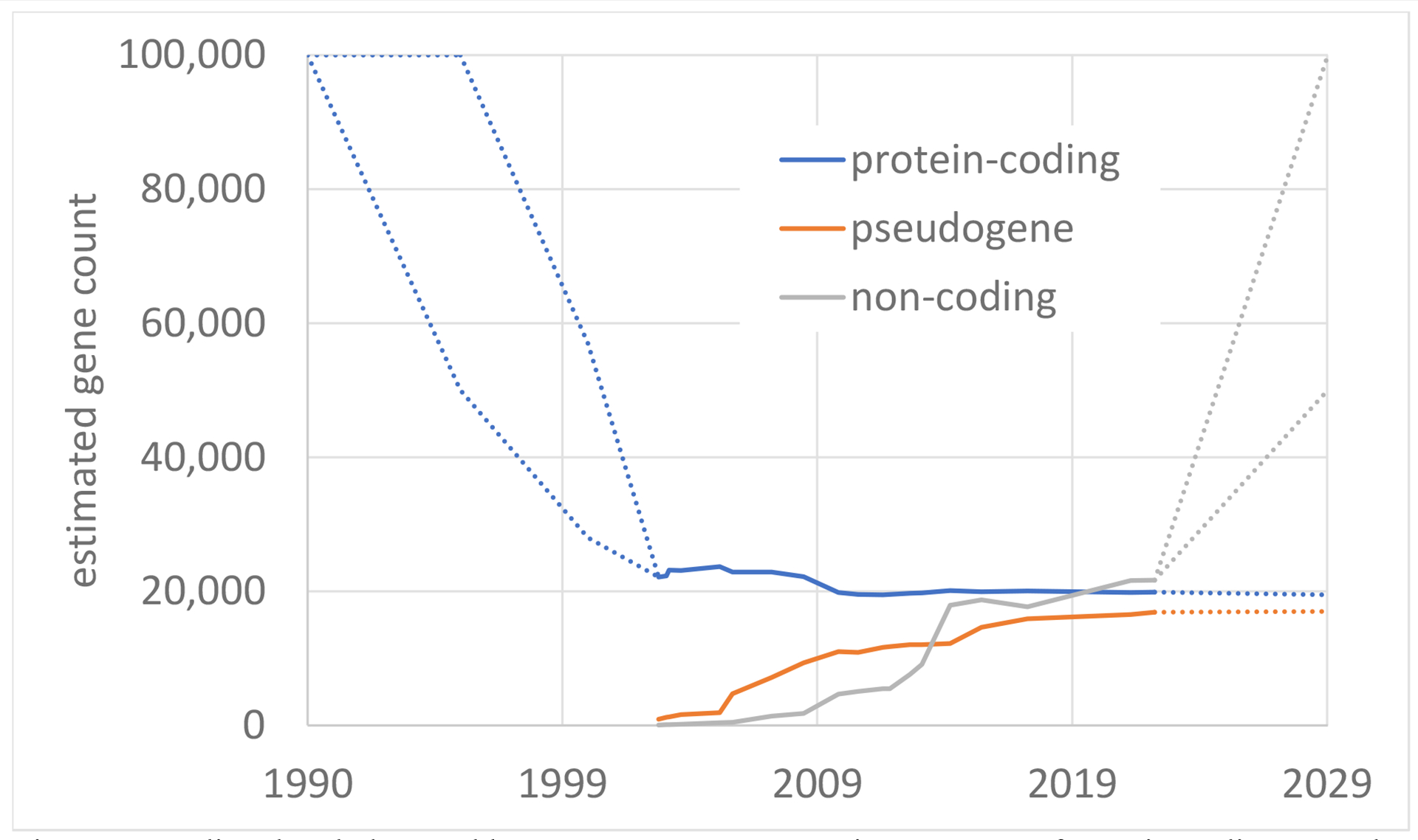
predicted and observed human gene counts over time. Counts of protein-coding, pseudogene, and non-coding genes are shown. Time points before 2003 and after 2023 (dashed lines) represent an average of predictions from the literature1 and extrapolations from this perspective article, respectively. Time points from 2003 to 2023 are based on 20 iterations of the NCBI RefSeq annotation of the human reference genome, including both curated and predicted genes.
In contrast, noncoding RNA genes, particularly lncRNAs, are at an earlier stage of understanding, and are still increasing in number, with current catalogs containing 17,000–20,000 lncRNAs or more. New technologies offer promising avenues to refine this catalog, although a complete functional characterization of lncRNAs is likely many years away. The steady decline in the number of protein-coding genes over the last 20 years makes it only natural to ask if lncRNA numbers may follow a similar trend, as our knowledge of RNA biology and technologies improve.
Finally, we note that even with a complete gene annotation of a finished genome, we will have only one example of the human gene catalogue, one that will not apply to all humans. It is likely that many healthy individuals have more or fewer copies of some genes, and future efforts to survey the diversity of the human population will be an important step towards achieving a more complete view of the gene content of our genome.
Acknowledgements
Thanks to the Banbury Center at Cold Spring Harbor Laboratory and the Cold Spring Harbor Laboratory Corporate Sponsor Program for supporting a workshop that all authors of this work attended. This work was supported in part by: U.S. National Institutes of Health (NIH) under grants R01-HG006677 (MP, SLS, AV), R01-MH123567 (MP, SLS), R35-GM130151 (SLS), U41-HG007234 (AF), and U24-HG007234 (RG, SC); the Wellcome Trust under grant WT222155/Z/20/Z (AF); the European Molecular Biology Laboratory (AF); the U.S. National Science Foundation under grant DBI-1759518 (MP); the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under grant T2EDK-00391 (AH); Science Foundation Ireland through Future Research Leaders award 18/FRL/6194 and the Irish Research Council through Consolidator Laureate award (IRCLA/2022/2500) (RJ); the National Center for Biotechnology Information of the National Library of Medicine, NIH (TDM, KDP, SP); National Health and Medical Research Council (NHMRC) APP1186371 (CAW); Center for Genomic Medicine at the University of Utah Health, and the H.A. & Edna Benning Foundation (MY); Spanish Ministry of Science and Innovation to the EMBL partnership, Centro de Excelencia Severo Ochoa and CERCA Programme / Generalitat de Catalunya (RG, SC); RIKEN Center for Integrative Medical Sciences (PC, HT); and Human Technopole (PC).
References
- 1.Pertea M & Salzberg SL Between a chicken and a grape: estimating the number of human genes. Genome Biol 11, 206, doi: 10.1186/gb-2010-11-5-206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]; Reviews the history of efforts to estimate the human gene count and highlights different computational methods that were used to help with the human gene annotation.
- 2.Understanding our genetic inheritance: The US Human Genome Project, the first five years 1991–1995. (U.S. Department of Health and Human Services and U.S. Department of Energy, 1990). [Google Scholar]
- 3.Nurk S et al. The complete sequence of a human genome. Science 376, 44–53, doi: 10.1126/science.abj6987 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes the first-ever complete, gap-free assembly and annotation of a human genome, which added 140 protein-coding genes and several thousand additional noncoding genes to the human gene catalogue.
- 4.The Encode Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74, doi: 10.1038/nature11247 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kawaji H, Kasukawa T, Forrest A, Carninci P & Hayashizaki Y The FANTOM5 collection, a data series underpinning mammalian transcriptome atlases in diverse cell types. Sci Data 4, 170113, doi: 10.1038/sdata.2017.113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fields C, Adams MD, White O & Venter JC How many genes in the human genome? Nature Genetics 7, 345–346, doi: 10.1038/ng0794-345 (1994). [DOI] [PubMed] [Google Scholar]
- 7.Clamp M et al. Distinguishing protein-coding and noncoding genes in the human genome. Proceedings of the National Academy of Sciences USA 104, 19428–19433 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carninci P et al. The transcriptional landscape of the mammalian genome. Science (New York, N.Y 309, 1559–1563 (2005). [DOI] [PubMed] [Google Scholar]; Demonstrated that transcription is far more complex than previously thought, including large numbers of isoforms and more lncRNAs than protein-coding genes.
- 9.Katayama S et al. Antisense transcription in the mammalian transcriptome. Science (New York, N.Y 309, 1564–1566 (2005). [DOI] [PubMed] [Google Scholar]
- 10.Salzberg SL Next-generation genome annotation: we still struggle to get it right. Genome biology 20, 92, doi: 10.1186/s13059-019-1715-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frankish A et al. GENCODE: reference annotation for the human and mouse genomes in 2023. Nucleic Acids Res 51, D942–D949, doi: 10.1093/nar/gkac1071 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Leary NA et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–745, doi: 10.1093/nar/gkv1189 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pertea M et al. CHESS: a new human gene catalog curated from thousands of large-scale RNA sequencing experiments reveals extensive transcriptional noise. Genome Biol 19, 208, doi: 10.1186/s13059-018-1590-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Presents an enhanced and comprehensive catalog of human genes and transcripts based on very deep RNA sequencing across a broad sample of human tissues.
- 14.UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49, D480–D489, doi: 10.1093/nar/gkaa1100 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pockrandt C, Steinegger M & Salzberg SL PhyloCSF ++: A fast and user-friendly implementation of PhyloCSF with annotation tools. Bioinformatics, doi: 10.1093/bioinformatics/btab756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siepel A et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 15, 1034–1050, doi: 10.1101/gr.3715005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard KS, Hubisz MJ, Rosenbloom KR & Siepel A Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20, 110–121, doi: 10.1101/gr.097857.109 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 409, 860–921, doi: 10.1038/35057062 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Venter JC et al. The sequence of the human genome. Science 291, 1304–1351 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Clamp M et al. Distinguishing protein-coding and noncoding genes in the human genome. Proc Natl Acad Sci U S A 104, 19428–19433, doi: 10.1073/pnas.0709013104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945, doi: 10.1038/nature03001 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Pruitt KD et al. The consensus coding sequence (CCDS) project: Identifying a common protein-coding gene set for the human and mouse genomes. Genome Res 19, 1316–1323 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes a joint effort among three genome annotation centers to converge on coding regions for the annotation of the human and mouse reference genomes.
- 23.Morales J et al. A joint NCBI and EMBL-EBI transcript set for clinical genomics and research. Nature 604, 310–315, doi: 10.1038/s41586-022-04558-8 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes a project to create uniform transcript annotations for every protein-coding gene, thus enhancing the precision of genomic medicine through the accurate identification of genomic variations.
- 24.Alioto TS U12DB: a database of orthologous U12-type spliceosomal introns. Nucleic acids research 35, D110–115, doi: 10.1093/nar/gkl796 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mudge JM et al. Standardized annotation of translated open reading frames. Nat Biotechnol 40, 994–999, doi: 10.1038/s41587-022-01369-0 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]; Outlines a community-led effort to produce a standardized catalog of human open reading frames identified through ribosome profiling.
- 26.Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science (New York, N.Y 369, 1318–1330, doi: 10.1126/science.aaz1776 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Troskie RL et al. Long-read cDNA sequencing identifies functional pseudogenes in the human transcriptome. Genome Biol 22, 146, doi: 10.1186/s13059-021-02369-0 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun M et al. Systematic functional interrogation of human pseudogenes using CRISPRi. Genome Biol 22, 240, doi: 10.1186/s13059-021-02464-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J & Zhang J Are Human Translated Pseudogenes Functional? Mol Biol Evol 33, 755–760, doi: 10.1093/molbev/msv268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ramilowski JA et al. Functional annotation of human long noncoding RNAs via molecular phenotyping. Genome Res 30, 1060–1072, doi: 10.1101/gr.254219.119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cech TR & Steitz JA The noncoding RNA revolution - trashing old rules to forge new ones. Cell 157, 77–94, doi: 10.1016/j.cell.2014.03.008 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Mattick JS et al. Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol, doi: 10.1038/s41580-022-00566-8 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Michelini F et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat Cell Biol 19, 1400–1411, doi: 10.1038/ncb3643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lagarde J et al. High-throughput annotation of full-length long noncoding RNAs with capture long-read sequencing. Nature Genetics 49, 1731–1740, doi: 10.1038/ng.3988 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; Describes a large-scale application of capturing rare RNA species with antisense probes and sequencing them with long-read technology, which revealed a large number of isoforms that were not otherwise detectable.
- 35.Uszczynska-Ratajczak B, Lagarde J, Frankish A, Guigó R & Johnson R Towards a complete map of the human long non-coding RNA transcriptome. Nature Reviews Genetics 19, 535–548, doi: 10.1038/s41576-018-0017-y (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The RNAcentral Consortium. RNAcentral 2021: secondary structure integration, improved sequence search and new member databases. Nucleic acids research 49, D212–220, doi: 10.1093/nar/gkaa921 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L et al. NONCODEV6: an updated database dedicated to long non-coding RNA annotation in both animals and plants. Nucleic Acids Res 49, D165–D171, doi: 10.1093/nar/gkaa1046 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hon C-C et al. An atlas of human long non-coding RNAs with accurate 5′ ends. Nature 543, 199–204, doi: 10.1038/nature21374 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Volders P-J et al. LNCipedia 5: towards a reference set of human long non-coding RNAs. Nucleic acids research 47, D135–139, doi: 10.1093/nar/gky1031 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Iyer MK et al. The landscape of long noncoding RNAs in the human transcriptome. Nature Genetics 47, 199–208, doi: 10.1038/ng.3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ma L et al. LncBook: a curated knowledgebase of human long non-coding RNAs. Nucleic acids research 47, 2699–2699, doi: 10.1093/nar/gkz073 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu Y et al. High-plex protein and whole transcriptome co-mapping at cellular resolution with spatial CITE-seq. Nat Biotechnol, doi: 10.1038/s41587-023-01676-0 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Derrien T et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome research 22, 1775–1789, doi: 10.1101/gr.132159.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stokes T et al. Transcriptomics for Clinical and Experimental Biology Research: Hang on a Seq. Advanced Genetics n/a, 2200024, doi: 10.1002/ggn2.202200024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deveson IW et al. Universal alternative splicing of noncoding exons. Cell Systems 6, 245–255, doi: 10.1016/j.cels.2017.12.005 (2018). [DOI] [PubMed] [Google Scholar]; Describes widespread alternative splicing in noncoding exons, suggesting that noncoding exons are functionally modular and produce a seemingly limitless variety of isoforms.
- 46.Mudge JM et al. Discovery of high-confidence human protein-coding genes and exons by whole-genome PhyloCSF helps elucidate 118 GWAS loci. Genome Res 29, 2073–2087, doi: 10.1101/gr.246462.118 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lewandowski JP et al. The Tug1 lncRNA locus is essential for male fertility. Genome Biol 21, 237, doi: 10.1186/s13059-020-02081-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Broadwell LJ et al. Myosin 7b is a regulatory long noncoding RNA (lncMYH7b) in the human heart. J Biol Chem 296, 100694, doi: 10.1016/j.jbc.2021.100694 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He Y et al. Transcriptional-Readthrough RNAs Reflect the Phenomenon of “A Gene Contains Gene(s)” or “Gene(s) within a Gene” in the Human Genome, and Thus Are Not Chimeric RNAs. Genes 9, doi: 10.3390/genes9010040 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y et al. Identification of the cross-strand chimeric RNAs generated by fusions of bi-directional transcripts. Nature communications 12, 4645, doi: 10.1038/s41467-021-24910-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Hoon M, Shin JW & Carninci P Paradigm shifts in genomics through the FANTOM projects. Mamm Genome 26, 391–402, doi: 10.1007/s00335-015-9593-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yip CW et al. Antisense-oligonucleotide-mediated perturbation of long non-coding RNA reveals functional features in stem cells and across cell types. Cell Rep 41, 111893, doi: 10.1016/j.celrep.2022.111893 (2022). [DOI] [PubMed] [Google Scholar]
- 53.Seal RL et al. A guide to naming human non-coding RNA genes. EMBO Journal 39, e103777, doi: 10.15252/embj.2019103777 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Amberger JS, Bocchini CA, Scott AF & Hamosh A OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 47, D1038–D1043, doi: 10.1093/nar/gky1151 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cline MS et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS genetics 14, e1007752, doi: 10.1371/journal.pgen.1007752 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang K, Li M & Hakonarson H ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38, e164, doi: 10.1093/nar/gkq603 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hunt SE et al. Annotating and prioritizing genomic variants using the Ensembl Variant Effect Predictor-A tutorial. Hum Mutat 43, 986–997, doi: 10.1002/humu.24298 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schoch K et al. Alternative transcripts in variant interpretation: the potential for missed diagnoses and misdiagnoses. Genet Med 22, 1269–1275, doi: 10.1038/s41436-020-0781-x (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; A potent example of the significant impact that precise gene model annotation has on genetic diagnostics, demonstrating how inaccuracies can yield false negatives or positives and potentially compromising the diagnosis of rare disease patients.
- 59.Steward CA et al. Re-annotation of 191 developmental and epileptic encephalopathy-associated genes unmasks de novo variants in SCN1A. NPJ Genom Med 4, 31, doi: 10.1038/s41525-019-0106-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Maurano MT et al. Systematic localization of common disease-associated variation in regulatory DNA. Science (New York, N.Y 337, 1190–1195, doi: 10.1126/science.1222794 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bartonicek N et al. Intergenic disease-associated regions are abundant in novel transcripts. Genome biology 18, article 241, doi: 10.1186/s13059-017-1363-3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aznaourova M, Schmerer N, Schmeck B & Schulte LN Disease-Causing Mutations and Rearrangements in Long Non-coding RNA Gene Loci. Front Genet 11, 527484, doi: 10.3389/fgene.2020.527484 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.den Dunnen JT et al. HGVS Recommendations for the Description of Sequence Variants: 2016 Update. Hum Mutat 37, 564–569, doi: 10.1002/humu.22981 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Shumate A et al. Assembly and annotation of an Ashkenazi human reference genome. Genome Biol 21, 129, doi: 10.1186/s13059-020-02047-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zimin AV et al. A reference-quality, fully annotated genome from a Puerto Rican individual. Genetics 220, doi: 10.1093/genetics/iyab227 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chao KH, Zimin AV, Pertea M & Salzberg SL The first gapless, reference-quality, fully annotated genome from a Southern Han Chinese individual. G3: Genes, Genomes, and Genetics, doi: 10.1093/g3journal/jkac321 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liao WW et al. A draft human pangenome reference. Nature 617, 312–324, doi: 10.1038/s41586-023-05896-x (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Consortium F. et al. A promoter-level mammalian expression atlas. Nature 507, 462–470, doi: 10.1038/nature13182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gonzàlez-Porta M, Frankish A, Rung J, Harrow J & Brazma A Transcriptome analysis of human tissues and cell lines reveals one dominant transcript per gene. Genome biology 14, article R70, doi: 10.1186/gb-2013-14-7-r70 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okazaki Y et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573. (2002). [DOI] [PubMed] [Google Scholar]
- 71.Babarinde IA & Hutchins AP The effects of sequencing depth on the assembly of coding and noncoding transcripts in the human genome. BMC Genomics 23, 487, doi: 10.1186/s12864-022-08717-z (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weatheritt RJ, Sterne-Weiler T & Blencowe BJ The ribosome-engaged landscape of alternative splicing. Nat Struct Mol Biol 23, 1117–1123, doi: 10.1038/nsmb.3317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.van Heesch S et al. The Translational Landscape of the Human Heart. Cell 178, 242–260 e229, doi: 10.1016/j.cell.2019.05.010 (2019). [DOI] [PubMed] [Google Scholar]; Shows that combining ribosome profiling with deep proteomic analysis can detect peptide products translated from a large number of 5’UTRs and annotated lncRNAs.
- 74.Duffy EE et al. Developmental dynamics of RNA translation in the human brain. Nat Neurosci 25, 1353–1365, doi: 10.1038/s41593-022-01164-9 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Workman RE et al. Nanopore native RNA sequencing of a human poly(A) transcriptome. Nature methods 16, 1297–1305, doi: 10.1038/s41592-019-0617-2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mulroney L et al. Identification of high-confidence human poly(A) RNA isoform scaffolds using nanopore sequencing. RNA 28, 162–176, doi: 10.1261/rna.078703.121 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grapotte M et al. Discovery of widespread transcription initiation at microsatellites predictable by sequence-based deep neural network. Nature communications 12, 3297, doi: 10.1038/s41467-021-23143-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sinitcyn P et al. Global detection of human variants and isoforms by deep proteome sequencing. Nat Biotechnol, doi: 10.1038/s41587-023-01714-x (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]; Establishes a valuable resource for identification of isoforms at the proteome level, and provides direct evidence that most frame-preserving alternatively spliced isoforms are translated.
- 79.Glinos DA et al. Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608, 353–359, doi: 10.1038/s41586-022-05035-y (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mercer TR et al. Targeted sequencing for gene discovery and quantification using RNA CaptureSeq. Nature protocols 9, 989–1009, doi: 10.1038/nprot.2014.058 (2014). [DOI] [PubMed] [Google Scholar]
- 81.Curion F et al. Targeted RNA sequencing enhances gene expression profiling of ultra-low input samples. RNA biology 17, 1741–1753, doi: 10.1080/15476286.2020.1777768 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]