

Abstract
Purpose To assess the spectrum of associated cardiac anomalies, the intrauterine course, and postnatal outcome of fetuses with double inlet ventricle (DIV).
Methods Retrospective analysis of prenatal ultrasound of 35 patients with DIV diagnosed between 2003 and 2021 in two tertiary referral centers in Germany. All fetuses underwent fetal echocardiography and a detailed anomaly scan. Postnatal outcome and follow-up data were retrieved from pediatric reports.
Results 33 cases of DIV were correctly diagnosed prenatally. 24 fetuses (72.7%) had a double inlet ventricle with dominant left (DILV), 7 (21.2%) with dominant right ventricular morphology (DIRV), and 2 cases (6%) with indeterminate morphology (DIIV). 4 (16.6%) were Holmes hearts. 5 of the 7 fetuses (71.4%) with DIRV had a double outlet right ventricle (DORV). Malposition of the great arteries was present in 84.8%. Chromosomal abnormalities were absent. Termination of pregnancy was performed in 8 cases (24.2%). 24 fetuses (72.7%) were live-born. 5 (20.8%) were female and 19 (79.2%) were male. The median gestational age at birth was 38+2.5 weeks. All but one child received univentricular palliation. The median follow-up time was 5.83 years with an adjusted survival rate of 91.6% (22 of 24 live-born children). There was one case of Fontan failure at 15.7 years.
Conclusion DIV remains a major cardiac malformation although both prenatal diagnostics and cardiac surgery have improved over the years. The course of pregnancy is commonly uneventful. All children need univentricular palliation. The children are slightly physically limited, develop a normal intellect, and attend school regularly.
Keywords: Echocardiography, Fetal cardiology, Pediatric cardiology, Double inlet ventricle, Prenatal diagnosis
Zusammenfassung
Ziel Beurteilung der assoziierten kardialen Anomalien, des intrauterinen und postnatalen Verlaufs von Feten mit doppeltem Einlassventrikel (DIV).
Methoden Retrospektive Analyse der pränatalen Befunde von 35 Patienten mit DIV zwischen 2003 und 2021 in 2 tertiären Referenzzentren in Deutschland. Bei allen Feten wurden eine fetale Echokardiografie und eine Feindiagnostik durchgeführt. Postnatale Follow-up-Daten wurden aus pädiatrischen Berichten entnommen.
Ergebnisse 33 Fälle von DIV wurden pränatal korrekt diagnostiziert. 24 Feten (72,7%) hatten einen DIV mit dominanter linker (DILV), 7 (21,2%) mit dominanter rechtsventrikulärer (DIRV) und 2 Fälle (6%) mit unbestimmter Morphologie (DIIV). 4 (16,6%) waren Holmes-Herzen. 5 der 7 Feten (71,4%) mit DIRV hatten einen DORV. Eine Malposition der großen Arterien lag in 84,8% vor. Aneuploidien waren nicht vorhanden. Ein Abbruch wurde in 8 Fällen (24,2%) durchgeführt. 24 Feten (72,7%) wurden lebend geboren, davon waren 5 (20,8%) weiblich und 19 (79,2%) männlich. Medianes Gestationsalter bei der Geburt betrug 38+2,5 Wochen. Bis auf ein Kind erhielten alle eine univentrikuläre Palliation. Das mediane Follow-up betrug 5,83 Jahre mit einer adjustierten Überlebensrate von 91,6% (22 von 24 Lebendgeborenen). Ein Fall von Fontan-Versagen mit 15,7 Jahren.
Schlussfolgerung DIV bleibt ein schwerer Herzfehler, auch wenn sich die pränatale Diagnostik und kardiochirurgische Palliation im Laufe der Jahre verbessert haben. Der Verlauf der Schwangerschaft ist meist problemlos. Alle Kinder erwartet eine univentrikuläre Palliation. Die Kinder sind körperlich leicht eingeschränkt, bei normalem Intellekt und gehen regelmäßig zur Schule.
Schlüsselwörter: Echokardiografie, Fetale Kardiologie, Pädiatrische Kardiologie, Double inlet ventricle, Pränatale Diagnostik
Introduction
Double inlet ventricle (DIV) is present when both atria drain into the same ventricle via a right and left atrioventricular valve. DIV has a prevalence of less than 1 per 10,000 live births and accounts for about 1% of all congenital heart defects 1 2 . Maternal pregestational diabetes is associated with an increased risk of DIV 3 . DIV, considered the most common form of univentricular atrioventricular junction, can be complex. The nomenclature of univentricular hearts is still controversial. First described by van Praagh as a single ventricular heart 4 , it is now clear that DIV has a big and a second small incomplete chamber within the ventricular mass. In normal hearts, there is a single left ventricle coexisting with a single right ventricle with each containing three segments: inlet, apex, and outlet with the muscular ventricular septum interposing between the right and left ventricular apical component carrying the atrioventricular conduction axis 5 6 . In double inlet ventricular hearts, one chamber is incomplete, meaning it no longer possesses all of its components. These hearts are differentiated into three types: left ventricle type with incomplete right ventricle (DILV, most common), right ventricle type with incomplete left chamber (DIRV, sometimes), and solitary ventricle of indeterminate morphology (DIIV, rare). The latter type is frequently seen in association with heterotaxy (mostly right isomerism). DIRV is often associated with DORV. The interventricular communication between the dominant and the incomplete chamber (rudimentary outlet chamber) is often through a ventricular septal defect formerly known as the bulboventricular foramen 6 . A discordant ventriculoarterial connection (malposition of the great arteries (MGA)) and obstruction of the outflow tracts are common. The more uncommon case of a DILV with concordant ventriculoarterial connection is called Holmes heart 7 8 .
Differential diagnosis of DIV and DILV include tricuspid atresia (TA), hypoplastic left heart and unbalanced atrioventricular septal defect (AVSD).
The aim of this study was to assess the spectrum of associated cardiac anomalies, the intrauterine course, and postnatal outcome of fetuses with DIV. The morphological variants and the accuracy of prenatal echocardiographic diagnosis of DIV were assessed.
Patients and Methods
All prenatally diagnosed cases of DIV between 2003 and 2021 in two tertiary referral centers were retrospectively reviewed with regard to intrauterine course and outcome. 35 pregnant women were enrolled in this study. Two cases had to be excluded because DILV was not confirmed postnatally; instead TA was present.
All fetuses underwent a full anatomic scan and echocardiography in a standardized fashion including color and pulsed wave Doppler examinations. Ultrasound examinations were carried out with 5-MHz, 7.5-MHz or 9-MHz probes (IU 22 and EPIQ7, Philips, Hamburg, Germany; Aplio i900 Canon, Tokyo, Japan; Voluson E8 and E10, GE Healthcare, Solingen, Germany). All patients received consultation from a pediatric cardiologist at least once. If the patient decided to continue with the pregnancy, repeated sonographic scans were scheduled. In the early postnatal period, all patients underwent echocardiography performed by a pediatric cardiologist.
As far as retrospectively achievable, prenatal databases were analyzed regarding the type of double inlet ventricle (DILV/DIRV/DIIV), the great arteries, the incomplete outlet chamber, the patency of both atrioventricular valves, the presence of outflow tract obstructions, and additional cardiac and extracardiac anomalies. The following variables were assessed: maternal age, gestational age at diagnosis and at delivery, mode of delivery, birth weight, and Apgar score. Karyotype analysis including 22q11 was evaluated when available. Pediatric charts and autopsy results were analyzed and compared to prenatal findings. All data were retrieved from medical files, stored images, and video recordings. Structured telephone interviews were conducted with each family regarding quality of life (QOL). Pre- and postnatal diagnoses of all live-born children were compared to assess the accuracy of prenatal diagnosis. There was no autopsy conducted in six cases of termination of pregnancy due to refusal of the parents.
In this study DIV was defined by the presence of a dominant and an incomplete chamber based on the respective morphological appearance and presence or absence of the inlet, apex, and outlet component. Cases of TA where the atretic tricuspid valve is in communication with the dominant chamber were defined as DIV with TA. Cases in which the atretic AV valve belongs to the incomplete ventricle were defined as true TA. In addition, we defined cases with AVSD as DIV with AVSD if more than 2/3 of the common AV valve drained into the dominant chamber.
All cases were classified into five groups according to the pregnancy outcome: termination of pregnancy (TOP), neonatal death (NND), death in infancy (INFD), and survivors. Neonatal death was defined as death within the first 28 days of life, and INFD as any death after 28 days of life. Postnatal medical files of echocardiography, cardiac catheterization, surgery, or autopsy were available for confirmation of the prenatal diagnosis in all live-born children.
Data analyses were performed using the Statistical Package for Social Sciences (SPSS 20.0 INC. Chicago, III USA) but were limited to descriptive analysis.
Approval from the ethics committee was obtained.
Results
Between 2003 and 2021, 33 cases of DIV were diagnosed at our centers with two pregnancies currently ongoing. The median gestational age at first presentation in our clinic was 23+3 weeks (range: 14–37) ( Table 1 ).
Table 1 Patient population.
| Total cases | 33 |
| Lost to follow-up | 3% (1/33) |
| Termination of pregnancy | 24.2% (8/33) |
| Live birth | 72.7% (24/33) |
| Overall survival rate | 66.6% (22/33) |
| Adjusted survival rate (live births) | 91.6% (22/24) |
| Neonatal death (NND) | 4.1% (1/24) |
| Infant death (INFD) | 4.1% (1/24) |
| Median maternal age at first presentation | 30 years (20–43) |
| Median gestational age at first presentation in our clinic | 23 + 3 weeks (14–27) |
| Median gestational age (GA) at delivery | 38 + 2.5 weeks (34–41) |
| Fetal sex live born | 5 female (20.8%) |
| 19 male (79.2%) | |
| Median birth weight | 3220g |
| Median follow-up time | 5.83 years (1 day to 15.7 years) |
Cardiac anomalies
24 of the 33 fetuses (72.7%) had a DILV, 7 (21.2%) had a DIRV, and 2 (6%) had a DIV of indeterminate type. Of the 24 cases with DILV, one (4%) had a double outlet left ventricle (DOLV) and four (16.6%) had a Holmes heart. In addition, normal position of the great vessels was present in two further cases. In one of the cases, there was pulmonary atresia (PA), while the aorta was normally coming from the left ventricle (LV) and in the other case, DIRV was present. 71.4% of cases of DIRV (5/7) had a double outlet right ventricle (DORV). There were no cases of DIV with TA or with AVSD.
With regard to the position of the great arteries, there was malposition in 28 cases (84.8%), 16 cases of dextro-MGA (57.1%), 10 cases of levo-MGA (35.7%). Two fetuses (7.1%) had an undefined malposition, including one case of DILV with mirror image dextrocardia. In this case, the outflow was left-sided via a rudimentary outlet chamber into the aorta and a hypoplastic pulmonary trunk right parallel to the aorta. There were no cases of isomerism (left or right) in our cohort, but one child showed an interrupted inferior caval vein and azygos continuation. Pulmonary outflow tract obstruction was present in 13 cases (39.4%), of which 8 were pulmonary stenosis (PS) (61.5%) and 5 were pulmonary atresia (PA) (38.5%). Anomalies of the aortic arch were seen in 36.4% (12/33). These include right aortic arch (25%), hypoplastic aortic arch (16.7%), and aortic coarctation (CoA) (58.3%); CoA was severe in 71.4% (5/7). Three fetuses had a left persistent superior caval vein and one had an aberrant left subclavian artery. One fetus with DILV had third-degree AV block.
Genetic testing and additional extracardiac anomalies
Chromosomal analysis was offered to all patients and was carried out at the patient’s request in 19 cases (57.6%) all having a euploid karyotype. In addition, analysis for microdeletion syndrome 22q11 was performed in 15 cases. All showed normal results.
Extracardiac anomalies were present in 6 of 33 cases (18.2%) including three cases with a single umbilical artery, and one case each with a left-sided renal agenesis, azygos continuation, and gallbladder duplication.
Outcome
In 8 of 33 cases (24.2%), the parents-to-be opted for TOP. One case was lost to follow-up (3%); lastly this patient considered TOP. There was no IUFD. Overall 24.2% (8/33) were female and 75.8% (25/33) were male. 24 neonates (72.7%) were live-born of which 5 (20.8%) were female and 19 (79.2%) male. 11 (45.8%) of 24 pregnancies were delivered vaginally. In 13 cases (54.2%), a caesarean delivery (CD) was performed due to obstetrical indications, with 30% being unplanned. The median gestational age at birth was 38+2.5 weeks (range: 34–41). One fetus had CD at 34+0 weeks due to a pathological CTG during induction of labor after preterm premature rupture of membranes. The median birth weight was 3220 g (range: 1980–4570 g). IUGR was not present. The median 5-minute Apgar score was 9 (range: 5–10).
NND and INFD occurred in one of 24 cases each. One neonate who received Norwood I on day 13 of life with the Damus-Kaye-Stansel procedure, Sano shunt, and aortic arch reconstruction by patch-plasty died three days after surgery due to heart and acute renal failure on ECMO therapy. The other neonate underwent a modified Norwood procedure with patch extension of the aortic arch into the descending aorta, a BT shunt to the right pulmonary artery, and an atrioseptectomy on day 20 and died at 3 months of age on ECMO therapy prior to bidirectional Glenn (PCPC) also due to heart failure after a BT shunt closure.
The remaining 22 children were alive (5 patients > 10 years of age included) at the time this manuscript was written, resulting in an adjusted survival rate of 91.6%. Follow-up was available between 1 day and 15.7 years after birth. The median follow-up time was 5.83 years ( Fig. 1 ).
Fig. 1.
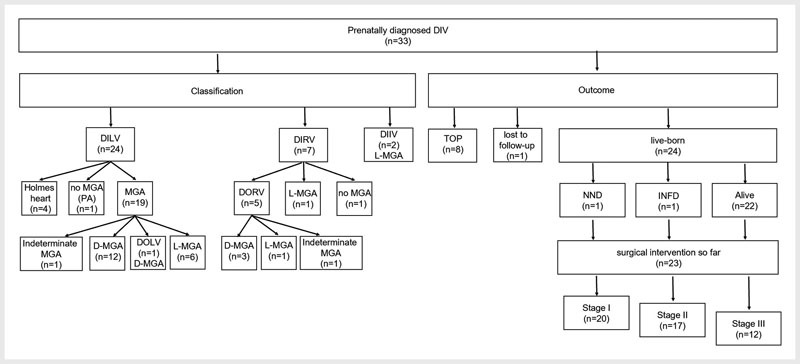
Classification and outcome: Abbreviations: DIIV: double inlet indeterminate ventricle; DILV: double inlet left ventricle; DIRV: double inlet right ventricle; DIV: double inlet ventricle; D-MGA: dextro-malposition of the great arteries; DOLV: double outlet left ventricle; DORV: double outlet right ventricle; INFD: infant death; L-MGA: levo-malposition of the great arteries; NND: neonatal death; PA: pulmonary atresia; TOP: termination of pregnancy.
Postnatal cardiac surgery
All but one child underwent surgery with the aim of univentricular palliation. This 11-month-old child has a DILV with PA with centrally located small pulmonary arteries and multiple aorto-pulmonary collaterals. In this case, the central pulmonary arteries were too small for a Fontan circulation, so compassionate care was requested by the parents. Excluding this case, 86.9% (20 of 23) of the neonates received a first stage of univentricular palliation in the form of a (modified) Norwood (-Sano) procedure, BT (Blalock-Taussig) shunt, DKS (Damus-Kaye-Stansel procedure), pulmonary artery banding (PAB), and/or atrioseptectomy with a mean age at the time of operation of 0.65 months with 16 of 18 (88.9%) being performed within the first three weeks of life. Minus the two cases of NND and INFD, respectively, the bidirectional Glenn procedure (stage II; PCPC) was performed in 94.4% (17/18) of cases (mean age at operation 6.76 months), including one Kawashima procedure due to an interrupted inferior caval vein with azygos vein continuity into the superior caval vein. The Fontan procedure (stage III; TCPC) was performed in 12 of 20 children (60%). The mean age at Fontan completion was 39 months (3 3/12 years). At this point, it should be considered that, at the time the manuscript was written, seven children were younger than three years. Univentricular palliation is the common final course of action in all children of our collective. 5 of 22 children (22.7%) received PAB. Four children (18.2%) had a pacemaker implanted. The reason for this was a third-degree atrioventricular block in two cases (in one case congenitally, in the other postoperatively) and postoperative sinus bradycardia in two cases. Postoperative diaphragmatic paresis was seen three times. Failing Fontan occurred once (8.3%) after 15.7 years. None of our patients underwent heart transplantation ( Table 2 ).
Table 2 Prenatal findings and outcome.
| GA at first presentation w+d | Type of DIV | Malposition of the great arteries | Out-flow tract obstruction | Outlet chamber/position | Additional cardiac anomalies | Additional extra-cardiac anomalies | Diagnosis/obduction | Deliv-ery | GA at birth w+d | Birth weight (g) | Percen-tile weight/length | Out-come | Surgery | Age at death or last follow-up (years/months) | Long-term outcome | |
| Abbreviations: ALSA: aberrant left subclavian artery; BT shunt: Blalock-Taussig shunt; CoA: aortic coarctation; CD: caesarean delivery; DCDA twins: dichorionic diamniotic twins; DILV: double inlet left ventricle; DIRV: double inlet right ventricle; DIIV: double inlet indeterminate ventricle; DKS: Damus-Kaye-Stansel procedure; D-MGA: dextro-malposition of the great arteries; INFD: infant death; ICV: interrupted inferior caval vein; IVC: interventricular communication; L-MGA: levo-malposition of the great arteries; LPSCV: left persistent superior caval vein; LV: left ventricle; MAPCA: major aorto-pulmonary collateral artery; NND: neonatal death; Pa: pulmonary artery; PA: pulmonary atresia; PAB: pulmonary artery banding; PCPC: partial cavo-pulmonary connection; PM: pacemaker; RAA: right aortic arch; RPA: right pulmonary artery; RV: right ventricle; SUA: single umbilical artery; TCPC: total cavo-pulmonary connection; TOP: termination of pregnancy; TP: pulmonary trunk; VB: vaginal birth; valv. PS: valvular pulmonary stenosis | ||||||||||||||||
| 1 | 16+3 | DILV | D-MGA | PS | RV/right | CoA | – | DILV, D-MGA, peripheral PS, preductal CoA | VB | 18+4 | – | TOP | – | – | – | |
| 2 | 24+5 | DIRV | D-MGA | PS | DORV/right | Severe CoA | – | DIRV+DORV, D-MGA, PS, subpulmonary VSD, aortic arch atresia | VB | 26+0 | – | TOP | – | – | – | |
| 3 | 21+6 | DIRV | L-MGA | – | DORV/right | – | – | DIRV+DORV, L-MGA | VB | 23+5 | – | TOP | – | – | – | |
| 4 | 14+1 | DILV | no | – | RV/right | – | Left-sided renal agenesis | DILV – Holmes heart, left-sided renal agenesis | VB | 17+2 | – | TOP | – | – | – | |
| 5 | 20+1 | DILV | L-MGA | – | LV/left anterior | Narrow aortic arch | – | DILV, L-MGA, narrow aortic arch | VB | 20+2 | – | TOP | – | – | – | |
| 6 | 18+4 | DIRV | L-MGA | valv. PS | LV/left | LPSCV | – | DIRV, L-MGA, valv. PS, LPSCV | VB | 19+4 | – | TOP | – | – | – | |
| 7 | 18+1 | DIRV | D-MGA | – | DORV/right | – | – | DIRV+DORV, D-MGA | VB | 21+1 | – | TOP | – | – | – | |
| 8 | 26+1 | DILV | no | PA | RV/right | – | SUA | DILV – Holmes heart, PA | VB | 28+1 | – | TOP | – | – | – | |
| 9 | 29+2 | DILV | D-MGA (not side-by-side) | PS | DOLV/left | – | – | DILV+DOLV, D-MGA, PS | – | – | – | – |
Lost to follow-up |
– | – | – |
| 10 | 22+5 | DILV | D-MGA | – | RV/right | – | – | DILV, D-MGA | VB | 39+3 | 4260 | 95/64 | NND | Mod. Norwood (aortic arch reconstruction by patch-plasty, DKS, Sano-shunt) | 16 days | |
| 11 | 20+5 | DILV | L-MGA | – | RV/left anterior | Hypo-plastic aortic arch | – | DILV, L-MGA, hypo-plastic aortic arch | VB | 36+5 | 3180 | 65/38 | INFD | Mod. Norwood with patch extension of the aortic arch into the desc. Ao, BT shunt to the RPA, atrioseptectomy |
3 months | |
| 12 | 28+0 | DIIV | L-MGA | – | Left | – | – | DIIV with L-MGA, mild subaortic obstruction | CD | 38+0 | 2900 | 26/27 | Alive | BT shunt, atrioseptectomy, ligation of DA, DDD-PM | 0 3/12 | Postoperative AV block III; next check-up in 2 weeks |
| 13 | 22+1 | DIRV | no | – | LV/left | – | – | DIRV with normally related great arteries | CD | 35+4 | 3900 | >97/93 | Alive | BT shunt | 0 4/12 | 70cm, 7,4kg; PCPC planned |
| 14 | 27+4 | DILV | no | – | RV/right | LPSCV | Gallbladder duplication | DILV – Holmes heart – with narrow (antegrade) TP from the rud. outlet chamber | CD | 39+3 | 3534 | 50/79 | Alive | Bilateral PAB, epimyocardial PM | 0 4/12 | 63cm, 5,6kg; post-interventional sinus-bradycardia; next check-up in 4 weeks |
| 15 | 23+5 | DILV | MGA | PA | Left | Dextro-cardia with mirror imaging | DCDA twins, SUA | Mirror image dextrocardia; DILV; outflow tract lying on left via rud. outlet chamber into aorta; PA with hypopl. TP right parallel to aorta; native Pa´s and MAPCAs | VB | 38+4 | 2315 | 7/22 | Alive | None so far | 0 11/12 | 52cm, 3kg; central pulmonary arteries too small for Fontan circulation, so compassionate care requested by parents |
| 16 | 30+5 | DILV | D-MGA | PS | Right | – | SUA | DILV, D-MGA, PS, right AV insufficiency | CD | 38+1 | 3130 | 32/62 | Alive | AV valve reconstruction, PAB, PCPC | 1 1/12 | 82cm, 11,4kg, BMI 17, normal development, no signs of cardiac insufficiency |
| 17 | 25+6 | DILV | L-MGA | – | RV/left anterior | – | – | DILV- L-MGA | CD | 38+5 | 3100 | 32/52 | Alive | Norwood-Sano, DKS, PCPC | 1 3/12 | 74cm, 11kg, BMI 20 |
| 18 | 28+1 | DILV | L-MGA | – | RV/left anterior | – | – | DILV- L-MGA | VB | 39+3 | 3500 | 46/64 | Alive | Norwood-Sano, DKS; atrioseptectomy, PCPC | 1 7/12 | Lean nutritional condition, no signs of cardiac insufficiency |
| 19 | 25+6 | DIIV | L-MGA | PA | Left | – | Azygos contin-uation, interrupted ICV | DIIV, L-MGA, PA, Azygos continuation, interrupted ICV | CD | 39+3 | 3050 | 20/60 | Alive | BT shunt, Kawashima | 1 10/12 | 85cm, 10,8kg, BMI 15, good development. Heterotaxy excluded. She is thriving, runs freely, and speaks well |
| 20 | 24+2 | DILV | D-MGA | – | RV/right anterior | Severe CoA, AV block III | – | DILV, D-MGA, severe CoA, AV block III | CD | 38+0 | 3150 | 36/18 | Alive | Norwood-Sano, PAB; atrioseptectomy, PM implantation | 1 11/12 | 84cm, 11kg, BMI 15,6, normal development, no signs of cardiac insufficiency |
| 21 | 25+4 | DILV | D-MGA | – | RV/right | CoA | – | DILV, D-MGA, CoA | VB | 40+0 | 3910 | 74/86 | Alive | Norwood-Sano with aortic arch reconstruction and atrioseptectomy, PCPC | 4 8/12 | 104cm (44. Perc), 16kg, BMI 14.8; age-appropriate resilience; poor tooth status; Fontan planned, after dental restoration |
| 22 | 21+6 | DILV | D-MGA | – | RV/right anterior | Hypoplastic aortic arch, LPSCV | – | DILV, D-MGA, hypoplastic aortic arch, LPSCV | VB | 37+2 | 3650 | 91/40 | Alive | Norwood-Sano, atrioseptectomy, PCPC + ligation of LPSCV, TCPC | 5 0/12 | 109cm (31. Perc), 18.6kg; BMI 15.7; slight physical limitations, speech development delay |
| 23 | 22+6 | DILV | D-MGA | PA | RV/left anterior | RAA, ALSA | – | DILV, D-MGA, PA, MAPCAs, RAA, ALSA | VB | 37+0 | 3210 | 59/83 | Alive | Modif. BT shunt, PCPC, TCPC | 6 8/12 | 121cm (49. Perc), 21kg, BMI 15.71, no physical limitations, asymptomatic SarsCoV2 infection |
| 24 | 25+1 | DILV | D-MGA | – | RV/right anterior | Severe CoA | – | DILV, D-MGA, severe CoA | CD | 38+1 | 2820 | 12/8 | Alive | Modif. BT shunt, PCPC, TCPC | 8 1/12 | Normal development, slight physical limitations |
| 25 | 20+1 | DILV | L-MGA | – | RV/right anterior | Severe CoA | – | DILV, L-MGA, severe CoA | VB | 39+2 | 3450 | 43/39 | Alive | Modif. Norwood + BT shunt, PAB, PCPC, TCPC, DDD-PM |
8 6/12 | 122cm (7. Perc), 29kg, BMI 19,5, AV block III, likes to go to school, slight physical limitations |
| 26 | 36+4 | DIRV | MGA | – | DORV/right | Mitral dysplasia | – | DIRV+DORV, MGA | CD | 38+5 | 3400 | 60/34 | Alive | Norwood-Sano, DKS, PCPC, TCPC | 8 9/12 | 142cm (86. Perc), 35.1kg; BMI 17.4; started school 1 year later, now in 2nd grade primary school, almost normal everyday resilience, ADHD |
| 27 | 22+0 | DILV | No | – | RV/right | – | – | DILV – Holmes heart | CD | 34+0 | 1980 | 20/92 | Alive | PCPC, TCPC | 9 4/12 | 132cm (13. Perc), 28.9kg; BMI 16.6, normal intelligence, slight physical limitations |
| 28 | 22+0 | DILV | D-MGA | PS | RV/right anterior | RAA | – | DILV, D-MGA, PS, RAA | CD | 38+1 | 3080 | 39/6 | Alive | PCPC, TCPC | 9 10/12 | The basic development, including the school situation (high school), is positive; slight physical limitations |
| 29 | 23+1 | DILV | D-MGA | – | RV/right anterior | RAA, ALSA | – | DILV, L-MGA, PS | CD | 37+5 | 3420 | 65/36 | Alive | BT shunt, PAB, PCPC, TCPC | 10 1/12 | 134cm (12. Perc), 25kg; BMI 13.9, asymptomatic SarsCov2 infection, normal intelligence, slight physical limitations, delayed speech development (his parents are both deaf) |
| 30 | 20+1 | DILV | D-MGA | PA | RV/left anterior | Dextro-cardia | – | DILV, D-MGA, PA, dextrocardia in situs solitus, MAPCA | CD | 38+0 | 3220 | 14/71 | Alive | BT shunt, closure of MAPCA, PCPC, TCPC with atrial PM | 12 3/12 | 32.7kg, sinus bradycardia after PCPC; slight physical limitations, 7th grade secondary school, hereditary spherocytosis (asymptomatic) |
| 31 | 26+5 | DILV | L-MGA | valv. PS | RV/left anterior | Dextro-cardia | – | DILV with anterior outlet chamber, dextrocardia, L-MGA, valvular PS, hypoplastic IVC | VB | 38+6 | 3220 | 29/<3 | Alive | BT shunt, PAB, atrioseptectomy, PCPC, TCPC | 13 2/12 | 148cm (9. Perc.), 50.8kg, BMI 23.19, bilateral diaphragm paresis, slight physical limitations, 7th grade secondary school |
| 32 | 23+0 | DIRV | D-MGA | valv. PS | DORV/right | – | – | DIRV+DORV, D-MGA, valv. PS | VB | 40+6 | 4570 | >97/80 | Alive | PCPC, TCPC | 14 0/12 | 164cm (36. Perc), 55kg, BMI 20.4, is now in 8th grade, dyslexia, IQ129, depressive episode with school refusal during Covid pandemic, physically well |
| 33 | 31+3 | DILV | D-MGA | – | RV/left anterior | Severe CoA | – | DILV, D-MGA, severe CoA | VB | 39+2 | 3630 | 60/72 | Alive | Norwood-Sano, PCPC, TCPC | 15 8/12 | 152cm (<3. Perc), 37kg, BMI 16; behind in growth, slight physical limitations, no pubertal development so far, currently probably failing Fontan with progressive edema |
Quality of life
Structured telephone interview analysis with each family showed that on average, a satisfactory overall quality of life can be recorded over a period of more than 15 years. According to the parents, the children attend school regularly and have an age-appropriate intellect. One child started school one year late. Another child had delayed language development (parents are deaf). Furthermore, all children participate normally in social life. The majority enjoy sports, but almost all children have limited physical capacity. All children were checked by pediatric cardiologists every 6 months. Of the children aged 4 years and older (13 children), current height and weight values were available in 10 cases. In terms of length growth, 5 of the 10 children were below the 14th percentile and one of them below the 3rd percentile. Here, pubertal development has not yet occurred. This is the oldest child in our cohort (15.7 years), who currently appears to be developing a failing Fontan. One child has an asymptomatic spherocytosis, another child has attention deficit hyperactivity disorder, and two children have had an asymptomatic SARS-CoV-2 infection. Anti-aggregative or anticoagulant therapy had to be continued in 8 cases. Five of these were treated with phenprocoumon and three with acetylsalicylic acid. Antihypertensive or vasodilator therapy was continued in 7 of the children.
Discussion
Prenatal diagnosis has improved over the last few years. The four-chamber view (4CV) shows only one ventricle without a ventricular septum. So, the connection between two atria and a single ventricle is mandatory for the diagnosis of DIV. The small, incomplete chamber typically cannot be visualized in the 4CV, as is usually possible in the hypoplastic left or right heart, but in a more cranial plane on the left or right side of the dominant ventricle ( Fig. 2 ). In cases of a dominant LV, the rudimentary outlet chamber is usually located antero-superiorly to the right of the dominant ventricle, whereas in a dominant right ventricle it is mainly located postero-inferiorly to the left side of the dominant ventricle 9 . Recognizing the dominant ventricle in terms of its morphological nature can be difficult prenatally. Histoanatomically, this differentiation is usually quite possible. Here, attention should be paid to the apical trabeculation (left ventricle with fine crisscross, right ventricle with coarse trabeculations). However, both fine and coarse trabeculation can be visualized sonographically ( Fig. 3 ). Furthermore, in DILV, both valves usually resemble mitral valves, where both of them are not in contact with the antero-superiorly located ventricular septum (septophobic), whereas in DIRV, the valves seem to be truly tricuspid (septophilic) and mitral valves 9 . However, such a prenatal distinction is often not possible due to the difficult to delineate attachment of the papillary muscles to the rudimentary outlet chamber.
Fig. 2.
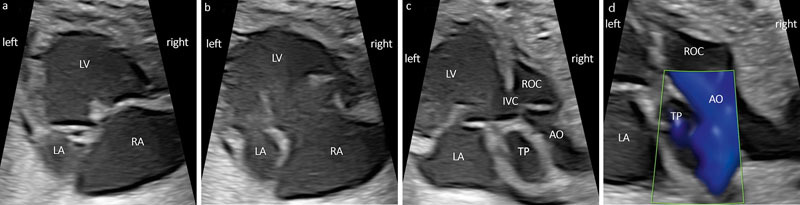
a DILV (30 + 5 weeks): a) Four-chamber view (4CV) in systole with left and right atrium (LA, RA) and the dominant (left) ventricle (LV). b 4CV in diastole showing both AV valves opening into the same ventricle; note the unusual difference in size regarding the two atria. After birth, the AV valve had to be reconstructed due to a tricuspid insufficiency. c More cranial plane showing the interventricular communication (IVC) to the rudimentary outlet chamber (ROC) on the right side and the aorta (Ao) coming from the ROC in dextro-malposition. d Color Doppler in systole with the pulmonary trunk (TP) originating from the LV and the aorta coming from the ROC.
Fig. 3.
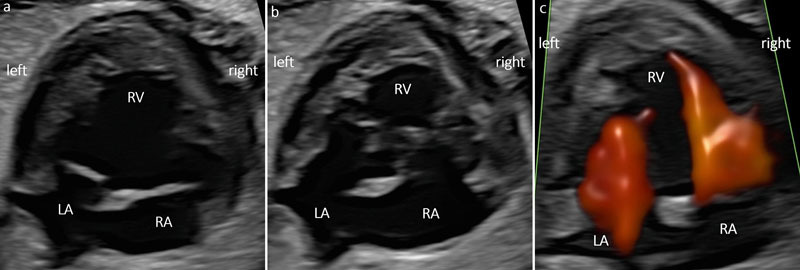
a DIRV (30 + 1 weeks). An interventricular septum (IVS) is missing. Note the coarse apical trabeculations. Grayscale a) systole. b diastole. c Color Doppler ventricular inflow during diastole.
Usually there is a malposition of the great vessels (MGA). In our collective this was true in 90% of the cases, with two thirds being D-MGA and one third L-MGA ( Fig. 2 c and Fig. 4 ). A small series of eight cases of DILV described L-MGA in all cases 10 . The frequency of a Holmes heart is given in the literature as 15% 11 . In our collective, this variant was seen in 16.6%.
Fig. 4.
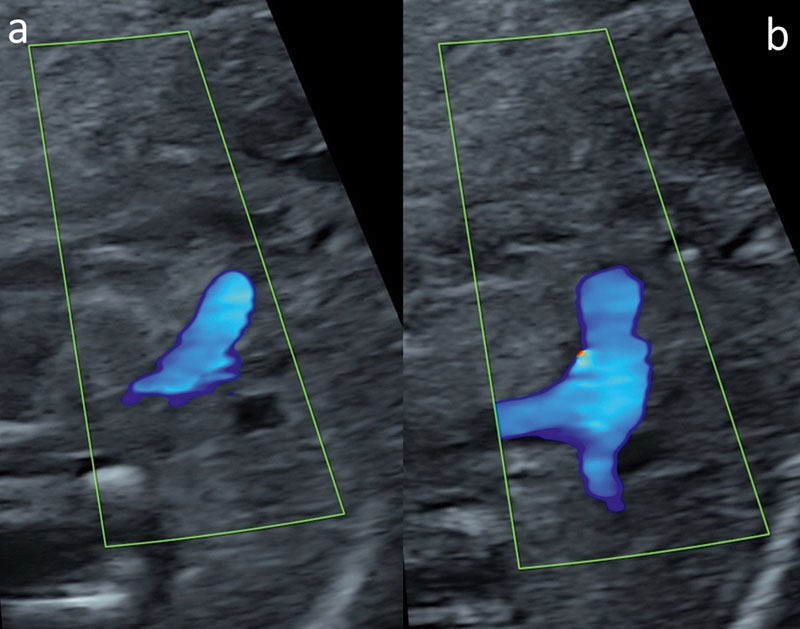
DILV with L-MGA (20 + 1 weeks). Color Doppler in systole with the narrow aorta coming from the rudimentary outlet chamber in levo-malposition ( a ) and the pulmonary trunk (TP) originating from the LV ( b ). Because of different levels, both outflow tracts cannot be visualized on the same plane.
Color Doppler provides information about the patency of the AV valves, restriction of flow across the interventricular communication, and outflow tract obstruction. If the aorta arises from the rudimentary outlet chamber, the size of the interventricular communication can be responsible for the degree of aortic arch stenosis. The smaller the interventricular communication, the smaller the aortic arch (CoA, interrupted aortic arch, rare).
Differential diagnoses
In the case of an absent AV connection, the prenatal distinction between imperforate and atretic AV connection is difficult since both have the same pathophysiological effect. Especially TA and DILV with atresia of the right AV valve present quite similarly in fetal echocardiography. Here, knowing the exact position of the atretic tricuspid valve is relevant. If it is above the rudimentary outlet chamber, there is TA. However, if the atretic tricuspid valve is in communication with the dominant LV, DILV with TA is present. Even though both entities require Fontan palliation, it is useful to differentiate between them, as they seem to differ in outcome. Therefore, Lan et al. reviewed the outcomes of 164 consecutive pediatric patients with DILV (n=105) or TA with transposed great arteries (n=35; lost to follow-up n=24) who underwent surgical palliation and showed patients with DILV having a significantly lower mortality rate than patients with TA with TGA 12 . Thus, while in the case of a right-sided imperforate or absent AV connection there is a TA, it is usually referred to as mitral atresia when this is the case on the left side. DIV can occur with any atrial arrangement and prenatal morphological differentiation between mitral and tricuspid valve is commonly not possible 9 . In our study, we had to exclude 2 cases because the prenatal suspicion of DILV was not confirmed. These patients had TA postnatally. In contrast to DIV, in association with TA, chromosomal anomalies may be present, in up to 10% of cases in one series 13 .
Another differential diagnosis is the unbalanced AVSD, where the common AV valve is skewed more to one side. It is called AVSD with DIV if more than two-thirds drain into the dominant ventricle. This, however, is often difficult to distinguish prenatally and the terminology is soft. In our collective, there were no such cases.
With regard to the position of the great vessels and the univentricular situation, ccTGA 14 15 and hypoplasia of the left ventricle 16 are also included as differential diagnostic considerations.
Outcome
For patients with a functionally univentricular heart, Fontan palliation has become established in the course. To date, only a few studies describe the outcome of patients with DIV. Most data in this regard come from subgroup analyses of larger collectives of univentricular hearts and there is hardly any data regarding the quality of life which can be crucial for prenatal counseling.
In 2015, Beroukhim et al. described the perinatal outcome after prenatal diagnosis of single-ventricle defects (hypoplastic left heart syndrome, TA, DILV, PA with intact ventricular septum). The prenatal outcome of cases with single-ventricle cardiac defects was similar between those with a dominant RV and those with a dominant LV. However, postnatal intermediate-term survival favored those with a dominant LV 17 . Consistent with these findings, in 2016, Wolter et al. published a retrospective analysis of 155 patients with a prenatally diagnosed functionally univentricular heart between 2008 and 2015, including 103 with a dominant RV and 52 with a dominant LV. Outcome after surgery reached 93.7% with higher survival rates for patients with dominant LV than patients with dominant RV 18 .
The outcome has so far been studied mainly in relation to the main group of univentricular hearts. However, in 2005, Earing et al. analyzed 225 patients with DILV who all had Fontan operations from 1974 to 2001. The median age at Fontan operation was 9 years. During a median follow-up period of 12 years, there were 22 deaths (9.8%), all within 30 days after the Fontan operation. The survival for the remaining 203 patients at 5, 10, 15, and 20 years was 91%, 80%, 73%, and 69%, respectively 19 . In 2008, Tham et al. examined the outcome of 171 patients with DILV from 1990 to 2004. Transplant-free survival was 88%, 82%, 79%, and 76% at 1 month, 1 year, 5 years, and 10 years, respectively. Prenatal diagnosis was made in 65 cases, with 43 being born alive. The proportion of prenatally diagnosed patients increased significantly during the study period. However, survival did not improve with prenatal diagnosis 20 . In 2017, Alsoufi et al. described the results of single ventricle palliation in 58 patients with DILV with an overall survival rate of 94% and 87% at 1 and 10 years, respectively 21 .
In 2011, Gidvani et al. described 8 cases of DILV focusing of prenatal course and outcome. The mean gestational age at diagnosis was 24.7 weeks. All had L-MGA. Four fetuses (50%) had PA. One fetus (12.5%) also had TA and CoA and died at 17 months of age. Complete heart block was present in one fetus (12.5%), who died shortly after birth. Six (75%) infants are alive, the oldest one at 41 months of age 10 . The results of this small case series agree in parts with ours. We had L-MGA in only 35.7% (10/28). Pulmonary outflow tract obstruction was present in 13 cases (39.4%). In our cohort, no deterioration could be recorded antenatally. There were no signs of cardiac insufficiency. Doppler sonographic examinations of the ductus venosus (DV) were unremarkable in all pregnancies. We too did not find chromosomal abnormalities.
The earliest diagnosis in our cohort was made at 14+1 weeks of gestation. As DIV is characterized by an abnormal four-chamber view, there is the potential for diagnosis in early pregnancy. Recently, a 14-year retrospective study of 26,805 transvaginal ultrasound screening examinations between 14 and 16 weeks of gestation was performed. 14 cases of DILV were diagnosed 22 . As with other cardiac heart defects that are readily diagnosable in the four-chamber view, the timing of diagnosis depends on the timing of the first detailed fetal anomaly scan in the respective population.
The question of whether it is morphologically a left or a right ventricle might be prognostically relevant. It seems logical that the right ventricle was not created to maintain the systemic circulation after Fontan procedure. However, there is not much robust data for this. Julsrud et al. showed in their retrospective study that right ventricular morphology is associated with a worse short-term outcome (up to 6 months) 20 . One study showed better survival in cases with a dominant left ventricle (DILV, TA, PA with intact ventricular septum) compared to HLHS 17 . Differences in myofiber architecture were analyzed through cardiac magnetic resonance imaging and could in part explain these results. The RV adapts suboptimally as a solitary pumping chamber in the Fontan circulation as compared with the LV. Therefore, it seems that single right ventricles have a higher end-systolic wall and fiber stress, a higher rate of ventricular dilation, lower circumferential fiber shortening, and similar longitudinal shortening than single left ventricles 23 . We, however, could not confirm these results in our cohort. Of the 7 cases of right ventricular morphology in our collective, 4 were terminated and 3 are alive.
The development of pulmonary hypertension remains a feared complication of circulatory conversion. PAB is designed to reduce pulmonary blood flow to prevent pulmonary hypertension after birth. As primary surgical correction is the goal nowadays, PAB is performed less frequently, but remains a useful tool, especially in cases where PS is absent 24 . In our collective PAB was performed in 22.7% (5 of 22 neonates).
Probably because of direct injury to the sinus node or its vascular supply, the development of arrhythmias after Fontan procedure has been reported in 4–16%. Sometimes, antiarrhythmic or pacemaker therapy is indicated 25 . In our cohort there were four children who required pacemaker therapy (18.2%).
Despite the undeniable improvement in survival after Fontan palliation, one of these patients, now almost 16 years old, developed a failing Fontan with progressive edema in relation to protein-losing enteropathy, a complication seen after Fontan operation in about 3–15% 25 .
Conclusion
The course of pregnancy is commonly uneventful for most affected fetuses. Chromosomal or associated extracardiac anomalies are usually absent. However, serial follow-up scans are required for detection of worsening of outflow tract obstructions or insufficiency of atrioventricular valves. In order to determine the type of DIV, the ventricular muscular structure should be inspected closely. The most common form is DILV, followed by DIRV and very rarely DIIV. When there is DIRV, DORV is usually present. Special attention should be paid to the interventricular communication, especially if the aorta arises from the incomplete ventricle, as the smaller its size the higher the chance of CoA or interrupted aortic arch. In addition, the presence or absence of pulmonary obstruction indicates the need for postnatal PAB. From an obstetric point of view, the affected fetuses can be delivered vaginally. Fontan palliation remains the common final course of action. The majority received a first stage of univentricular palliation within the first three weeks of life followed by PCPC on average at 6–7 months and lastly TCPC at the age of roughly three years with high survival rates. The assessment of quality of life, of course, is determined on an individual basis. However, it might be assessed as positive overall. All children have to undergo major surgical procedures and need to adhere to the prescribed regular, long-term check-ups as well as possible drug therapies. This can put a lot of strain on the individual and also the whole family. If all recommendations are implemented and there are no post-operative complications, these children can go to school regularly and are of normal intelligence. They tend to show physical growth differences and slight physical limitations. However, these interpretations are limited as it is a small group of cases (9/22 under 2 years).
The results of this study may contribute to better counseling for affected families in the future.
Footnotes
Conflict of Interest The authors declare that they have no conflict of interest.
References
- 1.Morris JK, Springett AL, Greenlees R et al. Trends in congenital anomalies in Europe from 1980 to 2012. PLoS One. 2018;13(04):e0194986. doi: 10.1371/journal.pone.0194986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Samánek M, Vorísková M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatr Cardiol. 1999;20(06):411–417. doi: 10.1007/s002469900502. [DOI] [PubMed] [Google Scholar]
- 3.Paige SL, Yang W, Priest JR et al. Risk factors associated with the development of double-inlet ventricle congenital heart disease. Birth Defects Res. 2019;111(11):640–648. doi: 10.1002/bdr2.1501. [DOI] [PubMed] [Google Scholar]
- 4.van Praagh R, Ongley PA, Swan HJ. Anatomic types of single or common ventricle in man. Am J Cardiol. 1964;13(03):367–386. doi: 10.1016/0002-9149(64)90453-9. [DOI] [PubMed] [Google Scholar]
- 5.Anderson RH, Franklin RCG, Spicer DE. Anatomy of the functionally univentricular heart. World J Pediatr Congenit Heart Surg. 2018;9(06):677–684. doi: 10.1177/2150135118800694. [DOI] [PubMed] [Google Scholar]
- 6.Cook AC, Anderson RH. The anatomy of hearts with double inlet ventricle. Cardiol Young. 2006;16:22–6. doi: 10.1017/S1047951105002283. [DOI] [PubMed] [Google Scholar]
- 7.Dobell AR, van Praagh R. The Holmes heart: historic associations and pathologic anatomy. Am Heart J. 1996;132(02):437–445. doi: 10.1016/s0002-8703(96)90443-3. [DOI] [PubMed] [Google Scholar]
- 8.Holmes WF. Case of malformation of the heart. Trans Med Chir Soc Edin. 1824(01):252–9. [PMC free article] [PubMed] [Google Scholar]
- 9.Penny DJ, Anderson RH. Philadelphia, PA: Churchill Livingstone; 2010. Other forms of functionally univentricular hearts; pp. 665–686. [Google Scholar]
- 10.Gidvani M, Ramin K, Gessford E et al. Prenatal diagnosis and outcome of fetuses with double-inlet left ventricle. Am J Perinatol Rep. 2011;1(02):123–128. doi: 10.1055/s-0031-1293515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weichert J, Axt-Fliedner R, Gembruch U et al. Holmes heart – a simple antenatal diagnosis of a complex cardiac anomaly? Fetal echocardiographic findings and review. Congenit Heart Dis. 2013;8(06):579–84. doi: 10.1111/j.1747-0803.2011.00621.x. [DOI] [PubMed] [Google Scholar]
- 12.Lan Y-T, Chang R-K, Laks H. Outcome of patients with double-inlet left ventricle or tricuspid atresia with transposed great arteries. J Am Coll Cardiol. 2004;43(01):113–119. doi: 10.1016/j.jacc.2003.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Berg C, Lachmann R, Kaiser C et al. Prenatal diagnosis of tricuspid atresia: intrauterine course and outcome. Ultrasound Obstet Gynecol. 2010;35(02):183–190. doi: 10.1002/uog.7499. [DOI] [PubMed] [Google Scholar]
- 14.Vorisek CN, Enzensberger C, Willomeit S et al. Prenatal diagnosis and outcome of congenital corrected transposition of the great arteries – a multicenter report of 69 cases. Ultraschall in Med. 2021;42(03):291–296. doi: 10.1055/a-1069-7698. [DOI] [PubMed] [Google Scholar]
- 15.Krummholz A, Gottschalk I, Geipel A et al. Prenatal diagnosis, associated findings and postnatal outcome in fetuses with congenitally corrected transposition of the great arteries. Arch Gynecol Obstet. 2021;303(06):1469–1481. doi: 10.1007/s00404-020-05886-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vorisek CN, Bischofsberger L, Kurkevych A et al. Fetal echocardiography in predicting postnatal outcome in borderline left ventricle. Ultraschall in Med. 2021 doi: 10.1055/a-1530-5240.. [DOI] [PubMed] [Google Scholar]
- 17.Beroukhim RS, Gauvreau K, Benavidez OJ et al. Perinatal outcome after prenatal diagnosis of single-ventricle cardiac defects. Ultrasound Obstet Gynecol. 2015;45(06):657–663. doi: 10.1002/uog.14634. [DOI] [PubMed] [Google Scholar]
- 18.Wolter A, Nosbüsch S, Kawecki A et al. Prenatal diagnosis of functionally univentricular heart, associations and perinatal outcomes. Prenat Diagn. 2016;36(06):545–554. doi: 10.1002/pd.4821. [DOI] [PubMed] [Google Scholar]
- 19.Earing MG, Cetta F, Driscoll DJ et al. Long-term results of the Fontan operation for double-inlet left ventricle. Am J Cardiol. 2005;96(02):291–298. doi: 10.1016/j.amjcard.2005.03.061. [DOI] [PubMed] [Google Scholar]
- 20.Tham EBC, Wald R, McElhinney DB et al. Outcome of fetuses and infants with double inlet single left ventricle. Am J Cardiol. 2008;101(11):1652–1656. doi: 10.1016/j.amjcard.2008.01.048. [DOI] [PubMed] [Google Scholar]
- 21.Alsoufi B, McCracken C, Kanter K et al. Current results of single ventricle palliation of patients with ouble inlet left ventricle. Ann Thorac Surg. 2017;104(06):2064–2071. doi: 10.1016/j.athoracsur.2017.04.031. [DOI] [PubMed] [Google Scholar]
- 22.Khatib N, Bronshtein M, Beloosesky R et al. Early prenatal diagnosis of double inlet left ventricle. J Matern Fetal Neonatal Med. 2021:1–5. doi: 10.1080/14767058.2021.1974385. [DOI] [PubMed] [Google Scholar]
- 23.Ghelani SJ, Colan SD, Azcue N et al. Impact of ventricular morphology on fiber stress and strain in Fontan patients. Circ Cardiovasc Imaging. 2018;11(07):e006738. doi: 10.1161/CIRCIMAGING.117.006738. [DOI] [PubMed] [Google Scholar]
- 24.Rao PS. Management of congenital heart disease: state of the art-part II-cyanotic heart defects. Children (Basel) 2019;6(04) doi: 10.3390/children6040054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rychik J, Cohen MI. Long-term outcome and complications of patients with single ventricle. Prog Pediatr Cardiol. 2002;16(01):89–103. [Google Scholar]