

Abstract
Human dedifferentiated liposarcoma (DDLPS) is a rare but lethal cancer with no driver mutations being identified, hampering the development of targeted therapies. We and others recently reported that constitutive activation of Notch signaling through overexpression of the Notch1 intracellular domain (NICDOE) in murine adipocytes leads to tumors resembling human DDLPS. However, the mechanisms underlying the oncogenic functions of Notch activation in DDLPS remains unclear. Here, we show that Notch signaling is activated in a subset of human DDLPS and correlates with poor prognosis and expression of MDM2, a defining marker of DDLPS. Metabolic analyses reveal that murine NICDOE DDLPS cells exhibit markedly reduced mitochondrial respiration and increased glycolysis, mimicking the Warburg effect. This metabolic switch is associated with diminished expression of peroxisome proliferator-activated receptor gamma coactivator 1α (Ppargc1a, encoding PGC-1α protein), a master regulator of mitochondrial biogenesis. Genetic ablation of the NICDOE cassette rescues the expression of PGC-1α and mitochondrial respiration. Similarly, overexpression of PGC-1α is sufficient to rescue mitochondria biogenesis, inhibit the growth and promote adipogenic differentiation of DDLPS cells. Together, these data demonstrate that Notch activation inhibits PGC-1α to suppress mitochondrial biogenesis and drive a metabolic switch in DDLPS.
INTRODUCTION
Human liposarcomas (LPS) arise from adipocytes and account for ~20% of sarcomas in adults [1]. Based on histological features, LPS are classified into four subtypes, well-differentiated liposarcoma (WDLPS), dedifferentiated liposarcoma (DDLPS), myxoid/round cell liposarcoma, and pleomorphic liposarcoma [2, 3]. Among these, DDLPS constitute 15–20% of LPS and has a potential metastasis rate of 15–20% [4]. Whole genome sequencing analysis of human LPS identifies amplifications of chromosome 12q13–15 as a hallmark of DDLPS. This region includes MDM2, CDK4, and HMGA2 genes, leading to MDM2 overexpression [5]. The MDM2-encoded protein is an E3 ubiquitin ligase that promotes ubiquitination and degradation of p53 tumor suppressor [6]. However, no study has indicated MDM2 as a driver mutation of LPS [7]. Additionally, targeted therapy using MDM2 inhibitors alone has an unsatisfactory objective response rate of 5% in patients with DDLPS [8–10]. Therefore, oncogenic driver genes of LPS have yet to be identified, but heterogeneity of tumor cells that often contain many different genetic alterations even within the same subtype hampers identification of critical drivers of LPS [11].
Notch signaling plays a key role in the initiation, proliferation, and stemness of cancer cells [12–15]. We have previously identified Notch signaling as a driver mutation of DDLPS tumorigenesis in a transgenic mouse model and developed a Notch-driven murine DDLPS cell line named mLPS1 cells [16, 17]. The mLPS1 cells express a constitutively active Notch1 intracellular domain (NICD) under the control of Rosa26 locus [17]. Consistent with our findings in mice, analysis of whole genomic sequences of human DDLPS reveals that 7% of the patients had Notch1 DNA copy number amplification [18]. Although this is a low frequency as a common driver mutation, many other tumor-driving mutations have been shown to converge on the Notch signaling pathway, making Notch signaling the top pathway implicated in head and neck squamous cell carcinomas [19]. Consistent with this notion, the DDLPS marker MDM2 have been shown to positively regulate Notch signaling through directly modulate Notch receptor activity or indirectly modulate degradation of Numb, an inhibitor of Notch signaling [20–22]. However, the mechanism by which Notch signaling regulates tumorigenesis of DDLPS cells remains unclear.
Notch signaling has been reported to rewire cellular metabolism towards glycolysis in Drosophila wing discs [23]. Cellular metabolic switching is a hallmark of tumorigenesis that facilitates the adaptation of cancer cells to various environmental stresses and stemness. Depending on the context, cancer cells utilize anaerobic glycolysis to support fast proliferation, known as the Warburg effect [24] or oxidative phosphorylation to generate energy more efficiently. In addition to regulating cancer cell proliferation, several studies have indicated that glycolytic metabolism plays a functional role in regulating human pluripotent stem cell states [25]. Specifically, high rates of glycolysis diminish the differentiation of human embryonic stem cells [26]. In accordance with the function of glycolysis in human pluripotent stem cells, changes in cell metabolism may similarly regulate cell differentiation and switch the status of cancer stem cells.
In this study, we tested the hypothesis that Notch signaling reprograms cellular metabolism in liposarcomas to maintain cancer cell stemness. We first examined expression of Notch pathway genes in human DDLPS tissues and cells. We found that the levels of Notch-related genes were positively correlated with MDM2 expression. Furthermore, we took advantage of our newly established mLPS1 cells to show that Notch signaling regulates cancer stem cell states by inhibiting mitochondrial biogenesis to facilitate glycolysis. In addition, RNA-seq analysis indicated that mitochondrial function and oxidative phosphorylation (OXPHOS) genes were significantly repressed in Notch-driven DDLPS. We further showed that Notch activation suppresses the expression of PGC-1α, a master regulator of mitochondrial biogenesis [27]. Overexpression of PGC-1α in mLPS1 cells increased mitochondrial biogenesis, altered cancer stem cell metabolism, and stimulated cell differentiation. These observations point to a previously unrecognized role of Notch signaling in regulating metabolism of DDLPS.
RESULTS
NOTCH1 is frequently overexpressed in human DDLPS
To assess the role and clinical significance of NOTCH signaling in DDLPS, we measured mRNA levels of MDM2 (a defining marker of DDLPS) and NOTCH pathway genes in human LPS samples. MDM2 oncogene encodes an E3 ubiquitin-protein ligase that promotes p53 ubiquitination and proteasomal degradation. In human DDLPS, more than 95% of the patients showed MDM2 amplification and high MDM2 expression was correlated to cancer proliferation and poor prognosis [1, 6]. Pearson’s correlation coefficient analysis revealed that NOTCH1 mRNA expression was positively correlated with MDM2 levels in WDLPS and DDLPS tissues (Fig. 1A). We further focused on DDLPS, a more malignant LPS subtype with a lower patient survival rate compared to WDLPS. Both NOTCH1 and its downstream target HES1 mRNA levels were strongly correlated with MDM2 levels in DDLPS (Fig. 1B, C).
Fig. 1. Notch activation is correlated with MDM2 expression and poor prognosis in human liposarcomas.
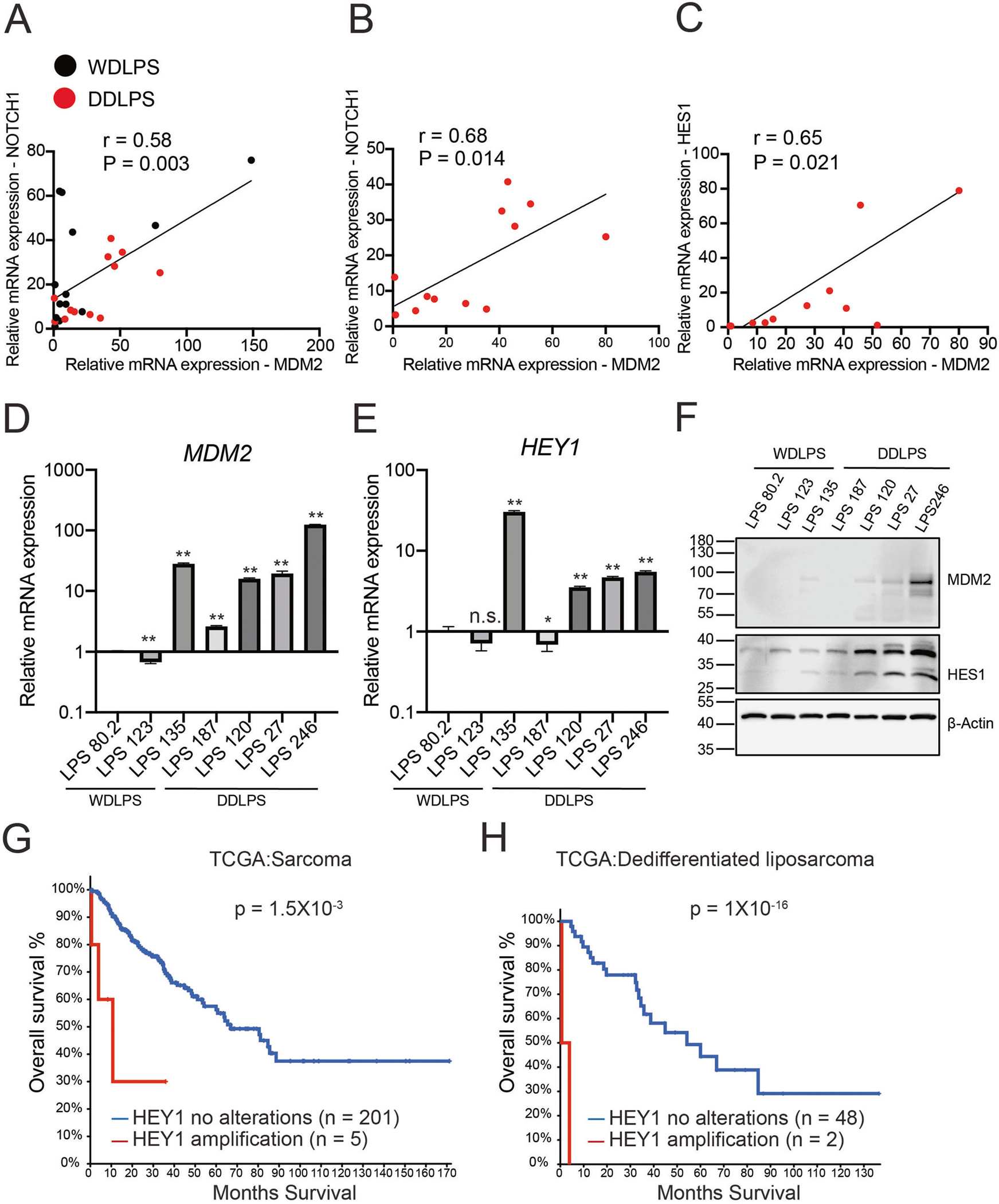
A–C Positive correlation of Notch signaling related genes (NOTCH1, HES1) with MDM2 in human well-differentiated liposarcoma (WDLS, n = 13) and dedifferentiated liposarcoma (DDLPS, n = 12). Black dots, WDLPS. Red dots, DDLPS. R, correlation coefficient, and P values were generated using Pearson’s correlation. D, E MDM2 and HEY1 mRNA levels in 7 human liposarcoma cell lines (n = 3). Data are presented as mean ± SD, *P < 0.05, **P < 0.001. F Immunoblots of MDM2 and HES1 proteins in various human liposarcoma cells. G, H Kaplan–Meier curve of overall survival in sarcoma and dedifferentiated liposarcoma patients from the TCGA dataset using cBioPortal platform analysis. The sarcoma samples were segregated into HEY1 amplification (2.4%, n = 5), HEY1 no alterations (n = 201) groups. The dedifferentiated liposarcoma patients were segregated into HEY1 amplification (4%, n = 2), HEY1 no alterations (n = 48). The P value was calculated using a log-rank (Mantel–Cox) test.
Next, we examined the mRNA levels of MDM2 and NOTCH target HEY1 in human primary liposarcoma cells. Consistent with our finding in human DDLPS tissues, primary DDLPS cells more frequently had elevated mRNA levels of MDM2 and HEY1 compared with WDLPS cells (Fig. 1D, E). Moreover, analysis of various human primary liposarcoma cells confirmed that HES1 protein expression was elevated in human DDLPS and positively correlated with MDM2 expression (Fig. 1F). In addition, analysis of the liposarcoma dataset in The Cancer Genome Atlas (TCGA) [28] revealed that patients with HEY1 DNA amplification had worse overall survival than patients without HEY1 amplification in both sarcomas (Fig. 1G) and DDLPS (Fig. 1H) cohorts. These results together suggest that NOTCH signaling is activated in human DDLPS and associated with tumor malignancy and poor prognosis.
Inhibition of NICD transcriptional activity suppressed growth of human and mouse DDLPS cells
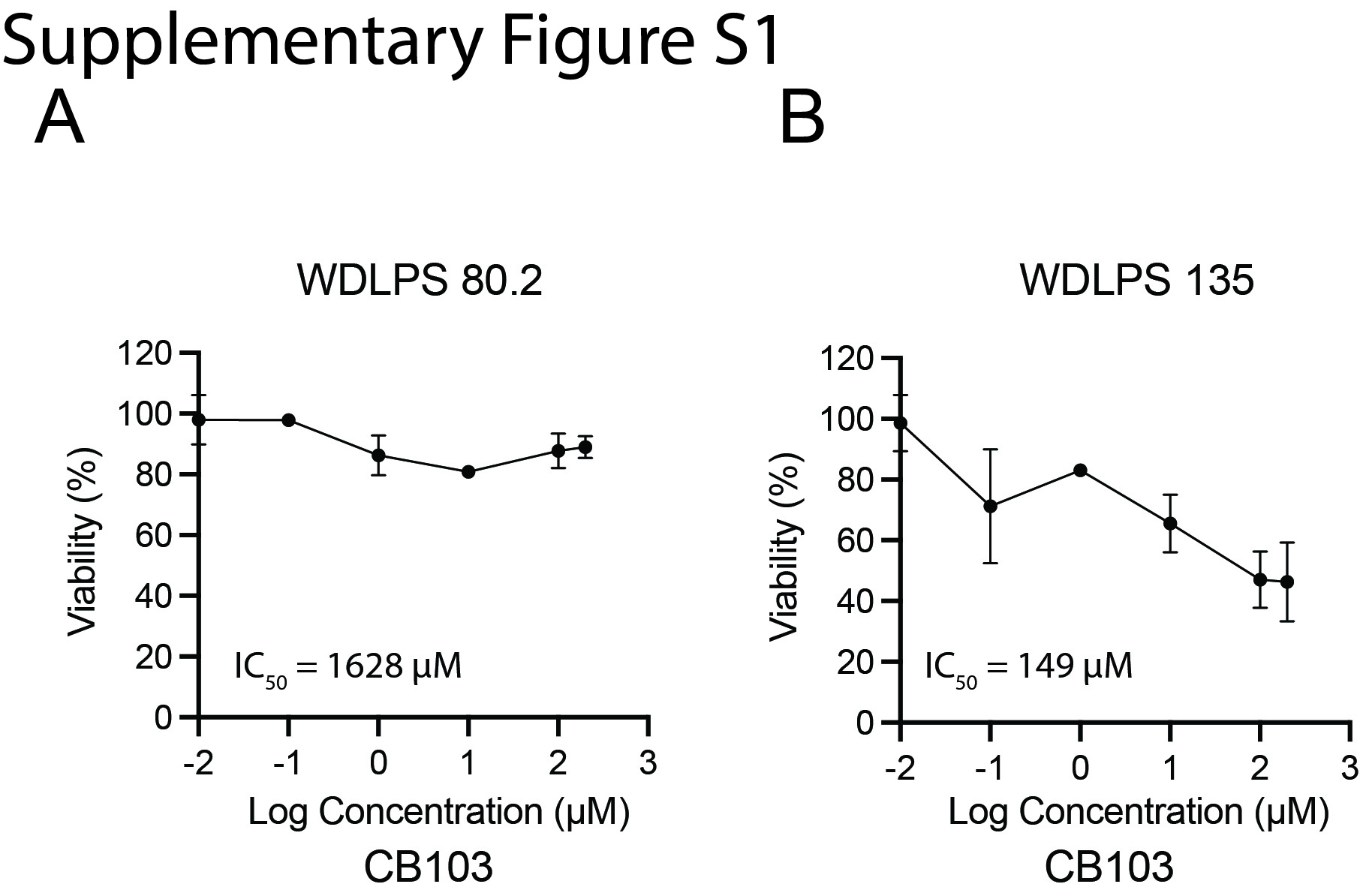
We next sought to directly examine the role of NOTCH signaling in DDLPS with inhibitors. Gamma secretase inhibitors (GSIs) have been widely used to inhibit the cleavage and generation of NICD [29], but these inhibitors carry two major limitations. The first is the off-target effects that cause gastrointestinal toxicity [30, 31] and the second is the lack of efficiency in tumors featuring NICD amplification independent of cleavage of full-length Notch receptors. To overcome the limitations, we took advantage of two small-molecule compounds, IMR1 and CB103, that target the NICD transcriptional complex to inhibit the expression of downstream target genes [32, 33]. RT-qPCR analysis showed that treatment of human dedifferentiated LPS246 cells with 1 μM IMR1 or 1 μM CB103 significantly reduced NOTCH1 and HES1 expression (Fig. 2A, B). The reduced levels of Notch-related genes were associated with lower cell viabilities, with the half maximal inhibitory concentration (IC50) of 61 μM and 119 μM for IMR1 and CB103, respectively (Fig. 2C, D). In addition, human WDLPS cells with a higher HEY1 level (WDLPS135) were more sensitive to the Notch inhibitors than WDLPS80.2 cells that have a lower HEY1 level (see Fig. 1E) with a much lower IC50 of 149 μM (Supplementary Fig. S1). We also evaluated the effects of IMR1 and CB103 on mLPS1 cells, a murine NICDOE DDLPS cell line that we established from the AdipoqCre-RosaNICD mouse DDLPS tumor tissue [17]. In agreement with the human cell line results, IMR1 or CB103 inhibited the expression of Notch1 and its downstream target genes at 20 μM (Fig. 2E, F). Furthermore, IMR1 and CB103 suppressed mLPS1 cell viability, with IC50 values of 98 and 83 μM, respectively (Fig. 2G, H). The treatment of Notch inhibitor had no effects on cell apoptosis in mLPS1 and LPS246 cells (Supplementary Fig. S2A), suggesting the reduced viability upon Notch inhibitor treatment is mainly due to a reduction in proliferation. This result is consistent with the current finding that abrogating oncogene in liposarcoma diminishes cancer stem cell phenotypes and inhibits cancer growth but does not stimulate cell apoptosis [34].
Fig. 2. Effect of Notch transcriptional inhibitors (IMR1 and CB103) on human LPS246 and murine mLPS1 cells.
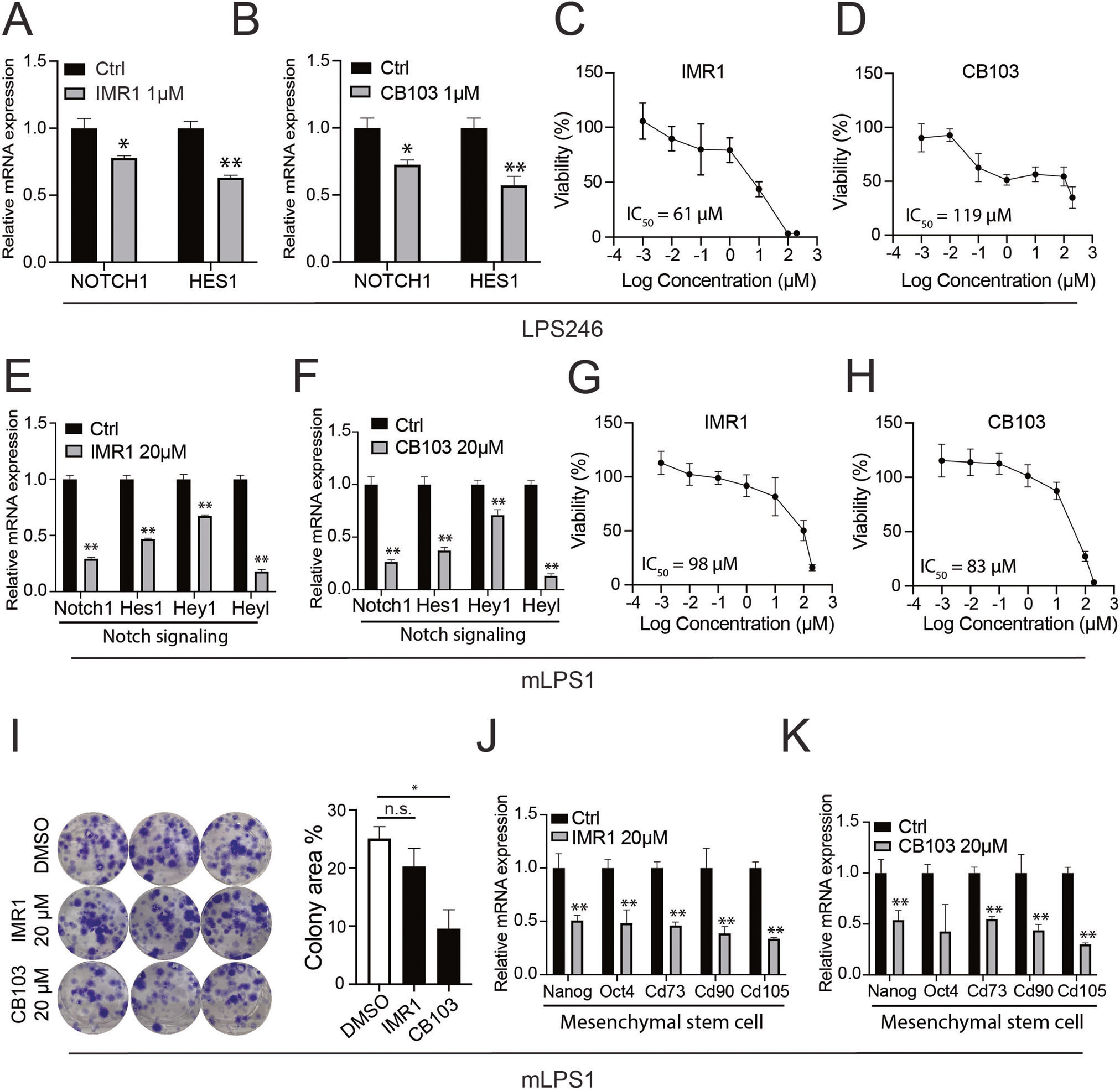
A, B RT-qPCR showing that IMR1 and CB103 inhibits NOTCH1 and HES1 gene expression in LPS246 cells (n = 3). C, D Representative dose-response curves of LPS246 to varying concentrations of IMR1 and CB103, and the corresponding and IC50 values (n = 6). E, F Relative levels of Notch related genes in mLPS1 cells treated with IMR1 or CB103 (n = 3). G, H Representative dose-response curves of mLPS1 to varying concentrations of IMR1 and CB103 and the corresponding IC50 values (n = 6). I Clone formation assay of mLPS1 cells treated with vehicle control (DMSO), IMR1, or CB103 (n = 3) (left), and the quantification of colony area (right). J, K Effect of IMR1 and CB103 on mRNA levels of cancer stem cell markers in mLPS1 cells (n = 3). Data are presented as mean ± SD, *P < 0.05, **P < 0.001.
The cellular response to the NICD inhibitors in mLPS1 cells was further determined using a colony formation assay. We observed that IMR1 and CB103 (20 μM) significantly reduced the colony area (determined by colony density and size) of mLPS1 cells (Fig. 2I). We next examined the tumor-suppressing ability of CB103 by treating mice bearing mLPS1-tumors after subcutaneous cell transplantation (Supplementary Fig. S3A). The daily treatment with 40 mg/kg CB103 tended to reduce tumor growth and obviously reduced tumor-associated skin necrosis (Supplementary Fig. S3B, C). At the end of the treatment, all except one of the CB103 treated LPS were smaller than vehicle treated LPS, leading to a lower average tumor weight (Supplementary Fig. S3D, E), though the difference was not statistically significant. These results demonstrate a key role of Notch signaling in supporting mouse and human DDLPS cell growth.
Notch signaling is essential for maintaining cancer stem cell (CSC) stemness [35]. To explore the effect of NICD inhibition in CSC marker gene expression of DDLPS, we performed RT-qPCR analysis in mLPS1 cells after treatment with IMR1 or CB103. The results indicated that the mRNA levels of the CSC markers Nanog, Oct4, Cd73, Cd90, and Cd105 were all downregulated by IMR1 or CB103 (Fig. 2J, K). These results highlight a role of Notch signaling in CSC marker gene expression in DDLPS.
Reversal of NICDOE in mLPS1 cells suppresses their growth and tumorigenesis
Tumor cells often exhibit “oncogene addiction”, where their malignancy is dependent on the expression of an oncogene, such as Myc [36]. To determine if the NICDOE mLPS1 cells are addicted to Notch signaling, we used CRISPR-CAS9 genome editing to remove the NICDOE cassette in the mLPS1 cells. We created the NICDOE-cassette knockout (mLPS1ΔNICD) cell line using two guide RNAs targeting exons 29–30 and 31–32 in the NICDOE cDNA, respectively (Fig. 3A). This targeting strategy will leave the endogenous Notch1 gene intact as both gRNAs spanned exons. The fidelity of the targeting was validated by genomic DNA sequencing, showing a 2-bp deletion in both gRNA-targeting regions (Fig. 3A). The mRNA levels of Notch signaling pathway genes Notch1, Hes1, Hey1, and Heyl, were all significantly downregulated in the mLPS1ΔNICD cells by more than 90%, compared to mLPS1 cells (Fig. 3B). Furthermore, western blot analysis confirmed that NICD protein was decreased to an undetectable level in the mLPS1ΔNICD cells, the protein levels of GFP that is independently translated via an internal ribosomal entry sequence (IRES) was only moderately reduced (Fig. 3C).
Fig. 3. NICD is responsible for the tumorigenic potential of mLPS1 cells.
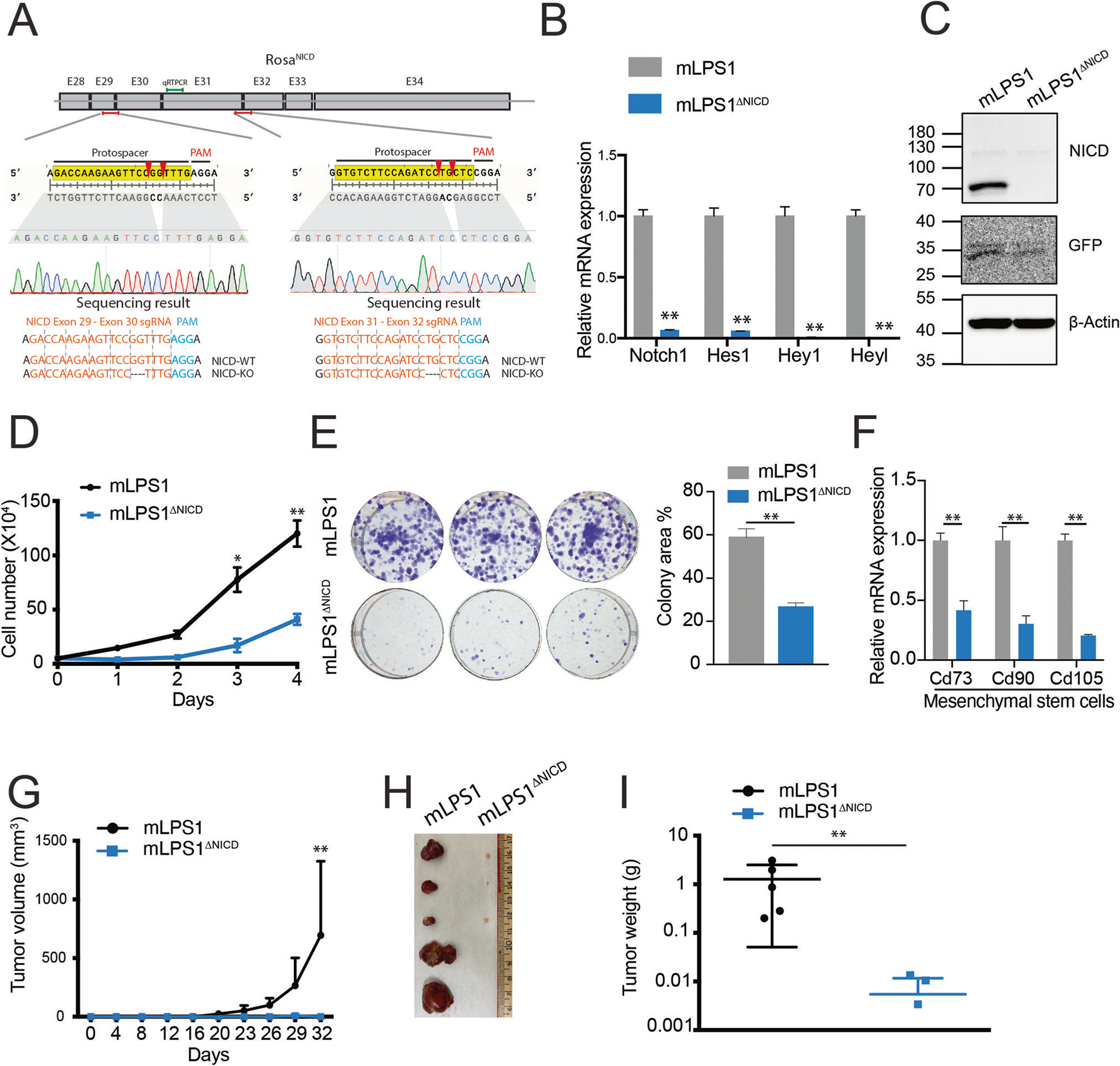
A CRISPR targeting strategy to ablate the Rosa-NICDOE cassette in mLPS1 cells. The NICD cDNA containing exon 28 to 34 of Notch1 gene was inserted into the Rosa26 locus. Two guide RNAs (gRNAs) each spanning two exons were designed to target the NICDOE transgene without affecting the endogenous Notch1 gene. The sequencing results validated the correct targeting. B Relative mRNA levels of Notch related genes in mLPS1 and mLPS1 NICD knockout (mLPS1ΔNICD) cells (n = 3). C NICD and GFP protein levels in mLPS1 and mLPS1ΔNICD cells. D Cell proliferation of mLPS1 and mLPS1ΔNICD cells (n = 5). E Colony formation assay of mLPS1 and mLPS1ΔNICD cells (n = 3). Representative pictures of colony size (left) and quantification of colony area (right) were shown. F RT-qPCR analysis of mesenchymal stem cell markers Cd73, Cd90, and Cd105 in mLPS1 and mLPS1ΔNICD cells (n = 3). G Tumorigenicity of mLPS1 and mLPS1ΔNICD cells after subcutaneous transplantation into the left and right flanks of NRG mice, respectively (n = 5). H Images of the grafted tumors after surgical removal from the NRG recipient mice. I The average weights of the tumors (n = 5). Data are presented as mean ± SD, *P < 0.05, **P < 0.001.
The mLPS1ΔNICD cells exhibited much reduced growth kinetics (Fig. 3D) and clonogenicity (Fig. 3E) when compared to their parental mLPS1 cells. This is most likely due to reduced cell proliferation as the inhibitors had no effects on cell apoptosis (Supplementary Fig. S2B). Consistently, the expression of mesenchymal and cancer stem cell markers, Cd73, Cd90, and Cd105, was also significantly lower in the mLPS1ΔNICD cells than in mLPS1 cells (Fig. 3F). We further investigated the tumorigenicity of mLPS1 and mLPS1ΔNICD cells after subcutaneous transplantation into immune deficient NRG mice. Tumorigenicity and propagation of the mLPS1ΔNICD cells was completely abolished based on tumor volume measurements (Fig. 3G, H). By contrast, all transplanted mLPS1 cells grew into tumors of various sizes (Fig. 3G, H). At the end point, the weight of transplanted mLPS1ΔNICD cells was 100-fold lower than the tumors grew from the mLPS1 cells (Fig. 3I). These results demonstrate that Notch signaling is essential for mLPS1 proliferation and tumorigenesis; and further suggest that Notch signaling addiction may underly pathogenesis of mLPS1 cells.
Notch signaling restricts re-differentiation of liposarcoma cells
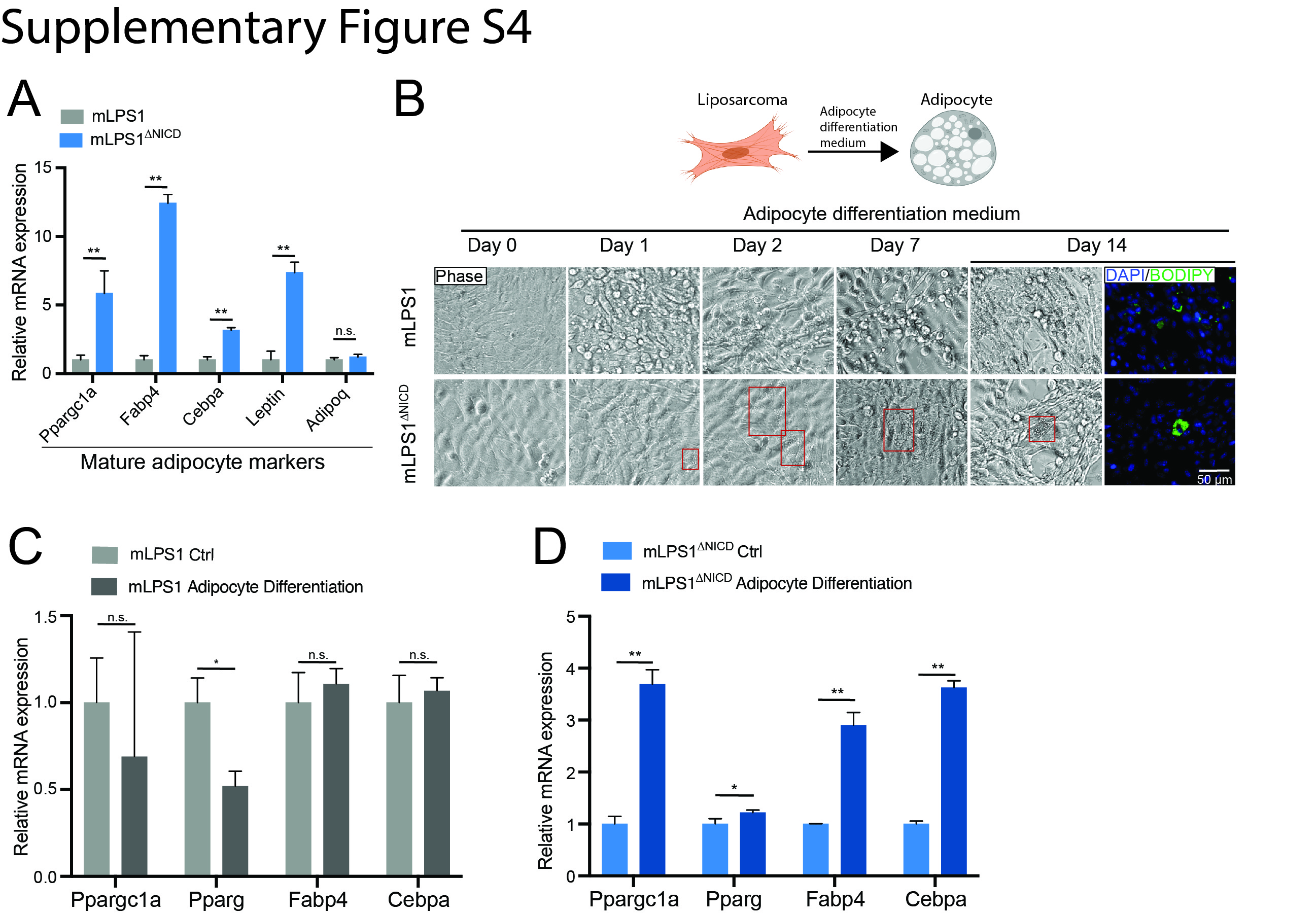
Notch signaling is a well-established master regulator of organ development that determines cell fate and controls cell differentiation [37, 38]. Notch signaling suppresses differentiation of human adipose-derived mesenchymal stem cells towards mature adipocytes [39]. Furthermore, pathological profiling revealed that 10% of the WDLPS cases transform into DDLPS [1], a poorly differentiated subtype, even though the two subtypes had similar genomic mutations [5]. Thus, we examined whether Notch signaling restricts adipogenic differentiation to promote DDLPS. We evaluated the expression of mature adipocyte markers in mLPS1 and mLPS1ΔNICD cells. The mRNA levels of adipogenic differentiation-related markers were all up-regulated in mLPS1ΔNICD cells (Fig. 4A), suggesting that Notch activation suppresses adipogenic differentiation of liposarcoma cells. We treated mLPS1 and mLPS1ΔNICD cells with adipogenic medium and analyzed differentiation by immunofluorescent staining with BODIPY, a lipid probe (Fig. 4B). Lipid droplets were only found in mLPS1ΔNICD but not mLPS1 cells, although the rate of re-differentiation is very low and the differentiated cells did not resemble adipocytes. Furthermore, gene expression analysis revealed that the mLPS1 cells were resistant to adipogenic induction and showed no changes (or even reductions) in the expression of mature adipocyte markers (Fig. 4C). In contrast, the mLPS1ΔNICD cells expressed significantly higher levels of mature adipocyte markers after treatment with adipogenic medium (Fig. 4D). Together, these results demonstrate that the constitutive activated Notch signaling in mLPS1 cells restricts their re-differentiation into adipocytes.
Fig. 4. Notch activation suppresses mLPS1 cell differentiation into adipocytes.
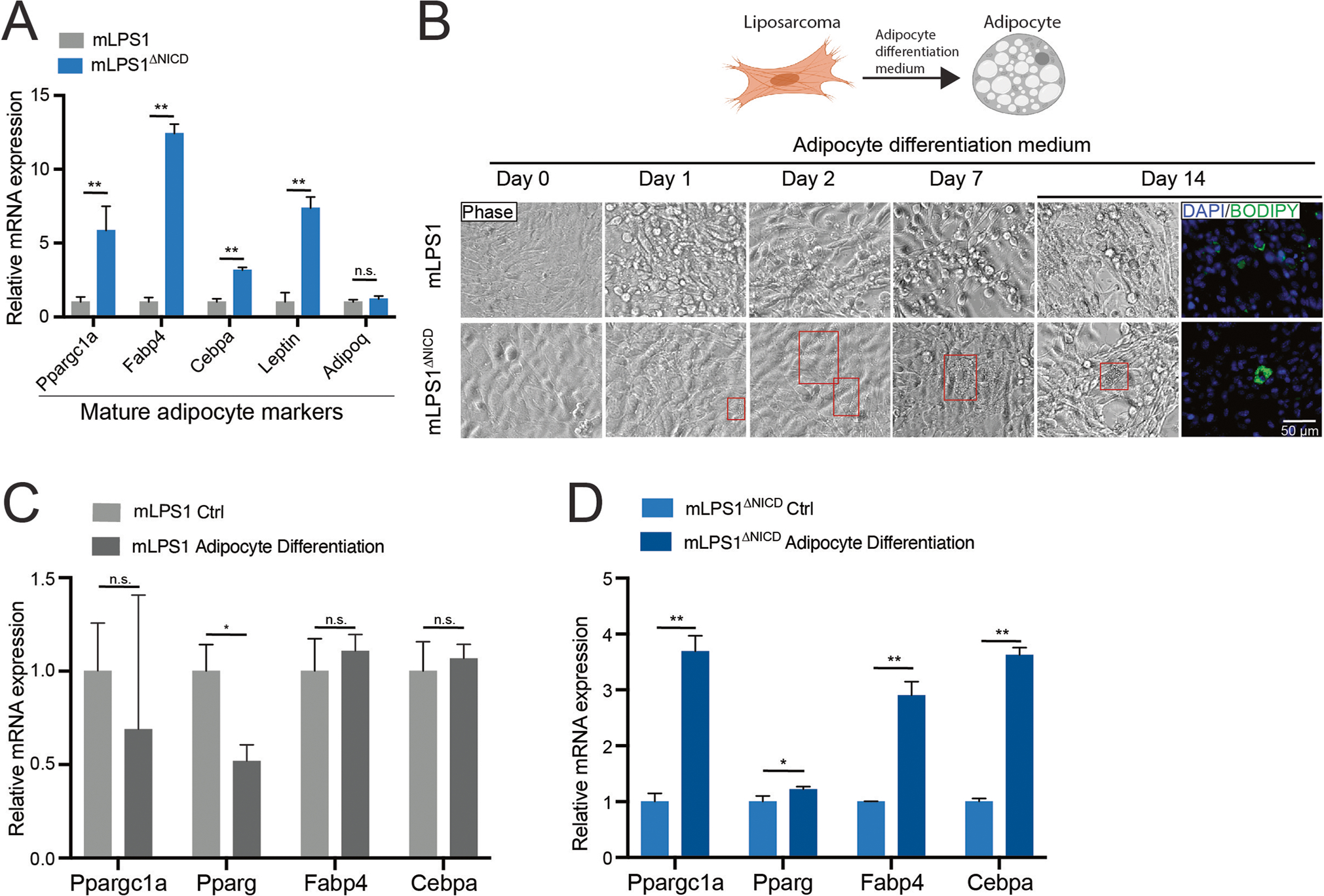
A RT-qPCR analysis showing expression of mature adipocyte marker genes in mLPS1 and mLPS1ΔNICD cells. B Fluorescent staining images of lipid droplets (labeled with BODIPY in green as indicator of adipogenic differentiation) in mLPS1 and mLPS1ΔNICD cells after treated with adipocyte differentiation medium, visualized by immunofluorescence microscopy. Scale bars, 50 μm. Nuclei were counterstained with DAPI (blue). C RT-qPCR analysis showing relative levels of mature adipocyte markers in mLPS1 cells at 24 h after expose to adipocyte differentiation medium. D RT-qPCR analysis of mature adipocyte marker expression levels in mLPS1ΔNICD cells at 24 h after expose to adipocyte differentiation medium. Data are represented as mean ± SD. *P < 0.05, **P < 0.001. n.s., not significant.
Notch signaling regulates mitochondria biogenesis and oxidative phosphorylation in mLPS1 cells
Tumorigenesis of cancer cells is preceded by metabolic reprogramming [40]. Notch signaling is a well-established driver of many types of cancers [41]. Notch mutations have been reported to cause infrequent long-tail mutations [19], promote chromosomal instability [42], and regulate cellular metabolism [43]. We hypothesized that Notch signaling underlies cancer cell stemness through regulating cellular metabolism. To test this hypothesis, we first measured the mitochondrial DNA content of mLPS1 and mLPS1ΔNICD cells. Mitochondrial to nuclear DNA ratios were significantly lower in mLPS1 cells than in mLPS1ΔNICD cells (Fig. 5A), suggesting that NICDOE inhibits mitochondrial biogenesis. Indeed, flow cytometric analysis of cells stained with MitoTracker further revealed that mLPS1 cells had significantly lower mitochondrial intensity than did mLPS1ΔNICD cells (Fig. 5B). Since mitochondria play an essential role in energy generation by regulating glycolysis and oxidative phosphorylation (OXPHOS), we further evaluated how NICDOE affects energy metabolism. The mLPS1ΔNICD cells secreted less lactate despite higher glucose uptake (Fig. 5C, D), and produced more ATP (Fig. 5E) than the mLPS1 cells. We further examined energy metabolism by measuring oxygen consumption rate (OCR) and extracellular acidification rate (ECAR). We observed higher OCR associated with both basal and maximal respiration in mLPS1ΔNICD cells than in mLPS1 cells (Fig. 5F). Conversely, ECAR associated with basal glycolysis and glycolytic capacity were lower in mLPS1ΔNICD cells than in mLPS1 cells (Fig. 5G). These results indicate that Notch activation promotes Warburg glycolysis and Notch inhibition increases mitochondrial biogenesis and stimulates oxidative phosphorylation, thereby acting as an anti-Warburg agent.
Fig. 5. Notch signaling regulates mitochondrial biogenesis and respiration in mLPS1 cells.
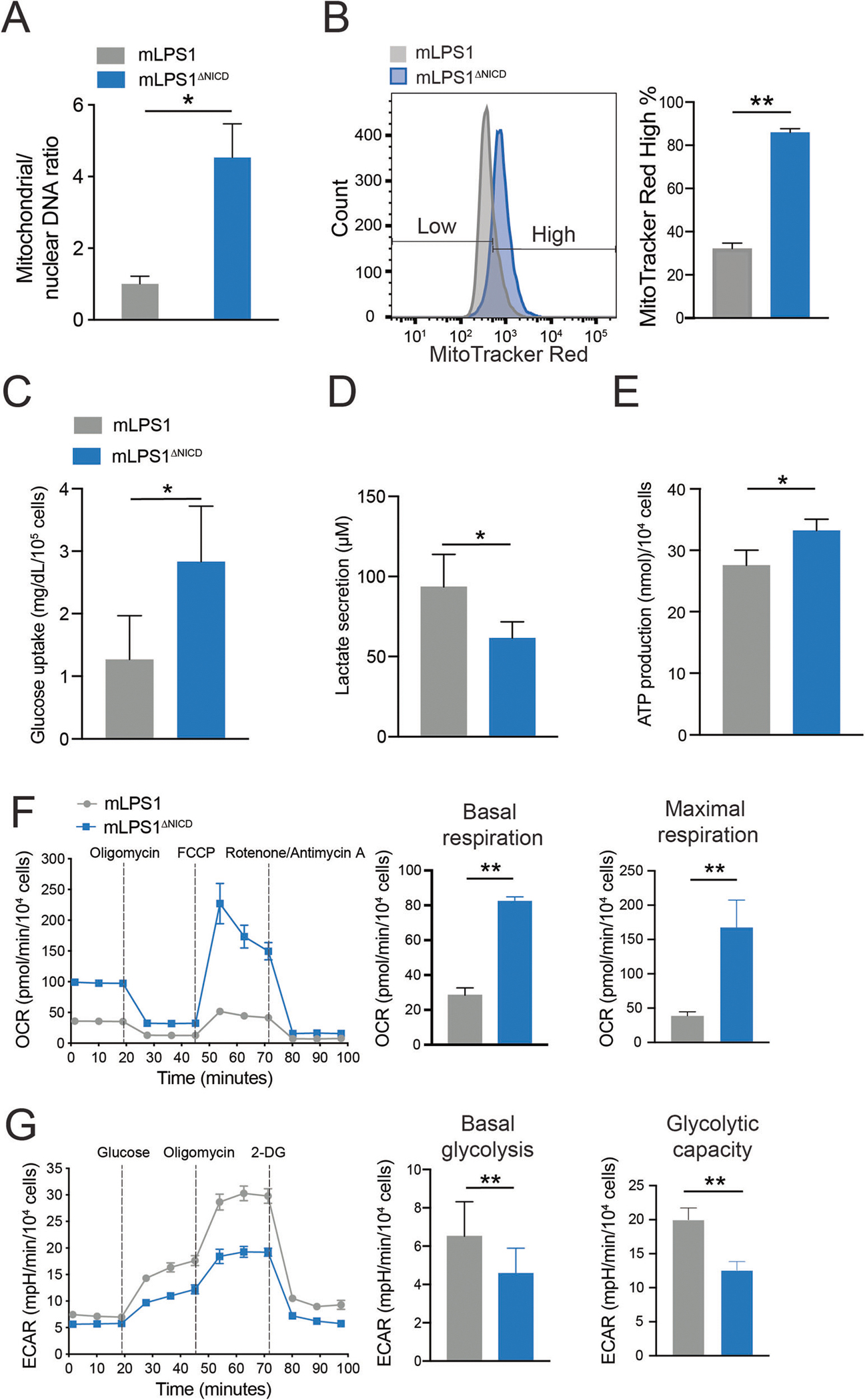
A RT-qPCR analysis of mitochondrial DNA (MT-ND1) versus nuclear DNA (HK2) content in mLPS1 and mLPS1ΔNICD cells (n = 3). B mLPS1 and mLPS1ΔNICD cells were stained with the MitoTracker Red FM probe and analyzed by Flow cytometry (n = 3). Histograms show the fluorescence intensity corresponding to mitochondrial mass (left) and quantification of high MitoTracker signal cells (right). C Glucose uptake of mLPS1 and mLPS1ΔNICD cells based on measuring reduction of glucose in culture media over 6 h (n = 4). D lactate production (n = 6) of mLPS1 and mLPS1ΔNICD cells based on measuring lactate concentration in culture media over 16 h. E ATP production (n = 6) of mLPS1 and mLPS1ΔNICD cells based on measuring chemical luminescence signaling in cell after 20 min incubation with reaction buffer. F Cellular respiration was monitored using the Seahorse bioscience extracellular flux analyzer (Left). The oxygen consumption rate (OCR) was normalized to protein abundance. The OCR corresponding to basal respiration (middle) and maximal respiratory capacity (right) in mLPS1 and mLPS1ΔNICD cells were shown (n = 5). G Seahorse bioscience extracellular flux analysis of extracellular acidification rate (ECAR) in mLPS1 and mLPS1ΔNICD cells (left). The basal glycolysis (middle) and maximal glycolytic capacity (right) were quantified in bar graphs (n = 5). Data are presented as mean ± SD of three reading cycles, for each cycle n = 5, *P < 0.05, **P < 0.001.
Notch signaling inhibits mitochondrial biogenesis by downregulating PGC-1α
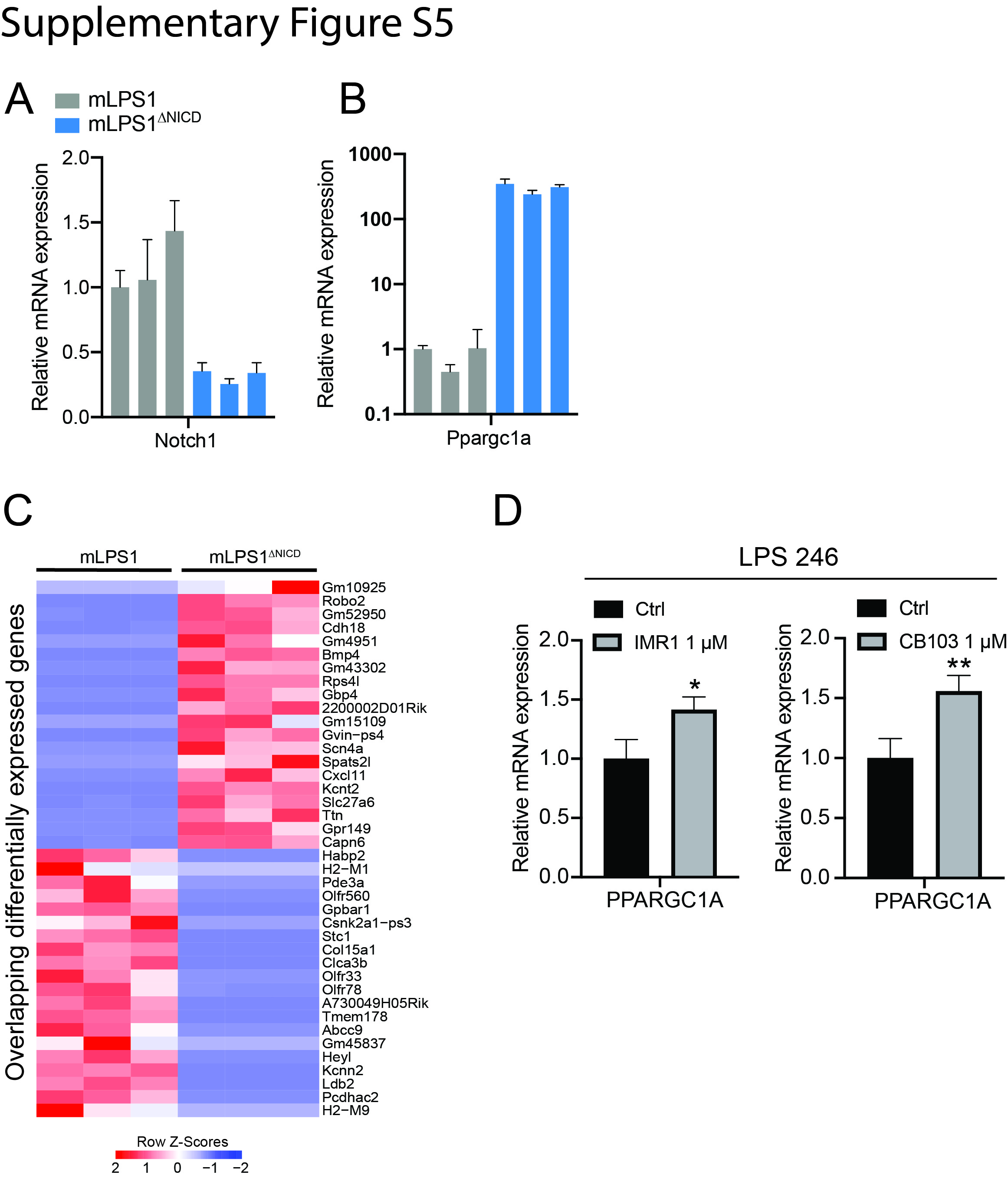
To elucidate how Notch signaling induces metabolic reprogramming and oncogenic transformation of adipocytes, we performed RNA-seq to compared gene expression of mLPS1 and mLPS1ΔNICD cells. This analysis revealed 6,876 genes whose expression were downregulated or upregulated by more than 1-fold between the two cell lines. By overlapping this RNA-seq with our previous RNA-seq comparing NICDOE liposarcoma with NICDOE adipose tissues (GSE80433), we identified 584 co-upregulated and 703 co-downregulated genes (Fig. 6A). The RNA-seq results were confirmed by RT-qPCR analysis of Notch1 and Ppargc1a expression in mLPS1 and mLPS1ΔNICD cells (Supplementary Fig. S4A, B). Selective co-upregulated and co-downregulated genes were illustrated in (Fig. 6B, Supplementary Fig. S4C). Among these are Ppargc1a, a master regulator of mitochondrial biogenesis that is upregulated in the mLPS1ΔNICD cells relative to mLPS1 cells (Fig. 6C). This observation is consistent with the report that Notch signaling effector HES1 binds to the Ppargc1a promoter region to inhibit its transcription [44]. We also treated LPS246 with Notch inhibitor IMR1 and CB103, and found that the treatment elevated PPARGC1A mRNA levels (Supplementary Fig. S4D). Consistent with our earlier finding that mLPS1ΔNICD cells have higher levels of mitochondrial respiration, we found that mitochondrial complex-associated genes were upregulated in mLPS1ΔNICD cells compared to mLPS1 cells (Fig. 6D). Furthermore, KEGG and GO analyses revealed that the “Metabolic pathways” and “Lipid metabolic process” were the most upregulated pathways/processes in the mLPS1ΔNICD cells (Fig. 6E). These results support our finding that Notch signaling regulates cellular metabolism in DDLPS.
Fig. 6. Notch signaling regulates mLPS1 cell metabolism and PGC-1α, whose reduced expression is correlated with poor prognosis of sarcomas, and with dedifferentiated status of liposarcomas.
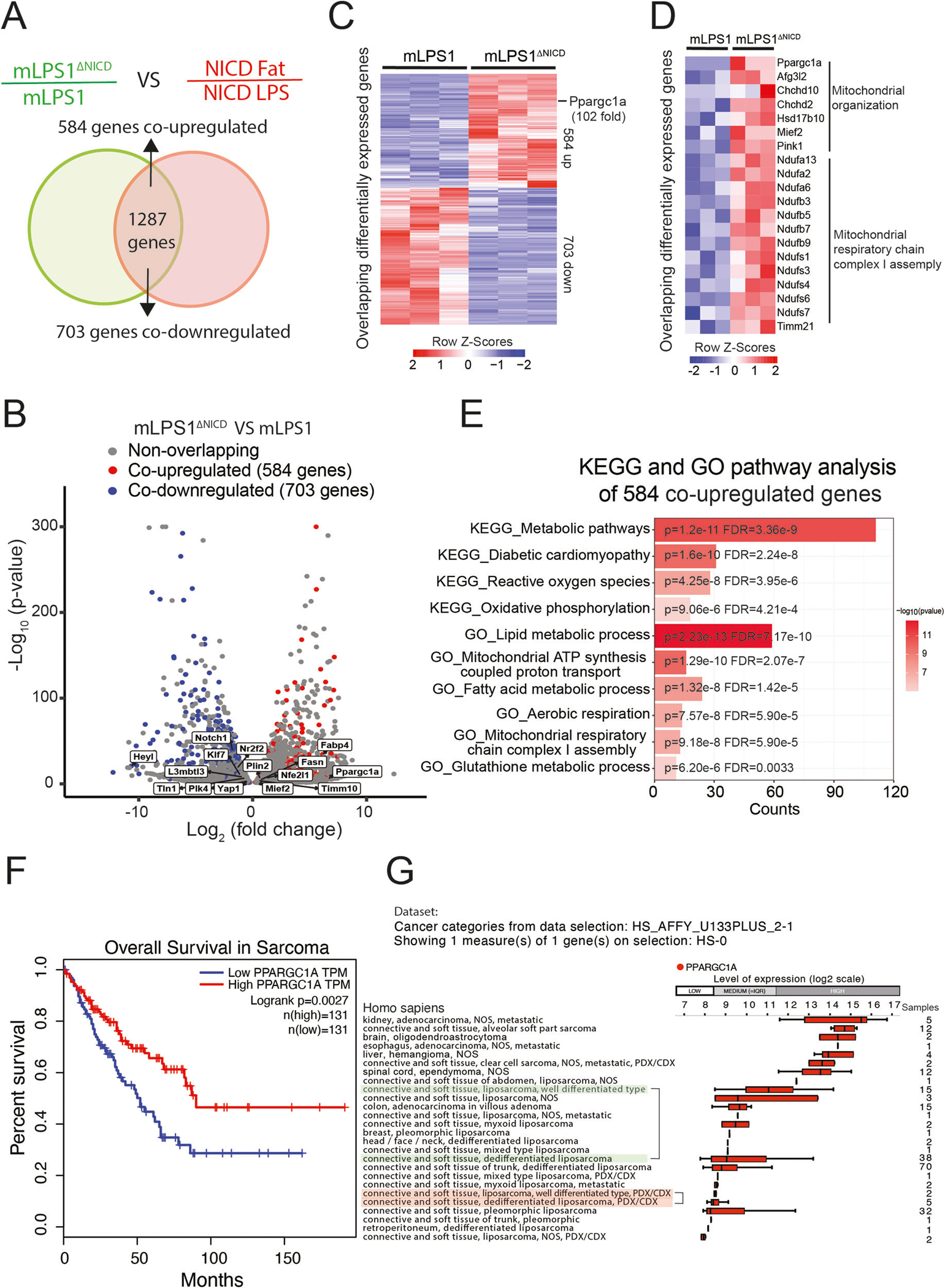
A Venn diagram showing the overlap of differentially expressed genes in mLPS1 vs mLPS1ΔNICD cells (green) and NICDOE LPS vs adipose tissues (red). Co-upregulated genes represent those suppressed by NICD highly expressed in adipocytes, and the co-downregulated genes represent those activated by NICD highly expressed in LPS cells. B Volcano plot of differentially expressed genes in mLPS1 and mLPS1ΔNICD cells and those co-upregulated or co-downregulated in A. C Heatmap visualization of the co-upregulated and co-downregulated genes. D Heatmap showing mitochondrial metabolic genes regulated by NICD. E Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses of the top 10 activated pathways in mLPS1ΔNICD cells compared to mLPS1 cells. Results of KEGG and GO analysis were determined to be significant by using a criterion of FDR.Q < 0.05 and a P < 0.05 is shown. F Kaplan–Meier curve of overall survival of sarcoma patients based on PPARGC1A transcripts per million (TPM), data from TCGA database (BMC Cancer, 2014) analyzed with the PROGgeneV2 platform. G Bar chart showing PPARGC1A expression in human liposarcoma tissues (Genevestigator) ranked by expression level from high (Top) to low (Bottom). Different types of human liposarcomas and neoplasms are shown on the left-hand side. Tumor tissue from the same region and source were grouped in the same color marker for comparison. The numbers of samples are listed on the right. Error bars represent standard deviation.
We next analyzed PPARGC1A mRNA levels in human sarcoma patients available in the TCGA database. The results showed that patients with high PPARGC1A expression had significantly higher survival rates and better prognosis (Fig. 6F). We further explored PPARGC1A expression in different human tumors using genome array analysis. The results revealed that patients with DDLPS had relatively lower PPARGC1A expression levels than those with WDLPS (Fig. 6G). These data support a model in which NICDOE regulates cellular metabolism by inhibiting PGC-1α mediated mitochondrial biogenesis.
Overexpression of PGC-1α restores mitochondrial biogenesis and respiration in mLPS1 cells
To investigate the role of PGC-1α in DDLPS, we overexpressed PGC-1α in mLPS1 cells using a doxycycline (Dox) inducible system (mLPS1 TetO-PGC1αOE). As high concentrations of Dox inhibit cell proliferation and metabolism [45], we tested Dox dose response and found that 1μg/mL of Dox induced PGC-1α expression without affecting mLPS1 cell growth (data not shown). mLPS1 TetO-PGC1αOE cells were treated with 1μg/mL Dox to induce PGC-1α expression, at both mRNA (Fig. 7A) and protein (Fig. 7B) levels. Control cells that were transfected with empty vector were also treated with Dox identically. To further explore the contribution of PGC-1α to the metabolic phenotype of mLPS1 cells, we examined mitochondrial abundance in mLPS1 TetO-PGC1αOE cells after Dox induction. We observed that PGC-1α significantly increased mitochondrial density based on mitochondrial DNA content (Fig. 7C). Next, we examined the mitochondrial content using MitoTracker staining in mLPS1 TetO-PGC1αOE and mLPS1 TetO-empty vector cells after Dox treatment. Consistently, PGC-1α expression in mLPS1 cells increased the mitochondrial density (Fig. 7D). We also used the Seahorse bioanalyzer to measure OCR in mLPS1 TetO-PGC1αOE cells and mLPS1 TetO-empty vector control cells. Expression of PGC-1α in mLPS1 cells led to higher basal and maximal respirations (Fig. 7E) and reduced anaerobic glycolysis with lower ECAR production (Fig. 7F). Together, these results indicate that PGC-1α expression facilitates mitochondrial biogenesis, which promotes oxidative phosphorylation and reduces anaerobic glycolysis in mLPS1 cells.
Fig. 7. PGC-1α expression promotes mitochondrial biogenesis and respiration in mLPS1 cells.
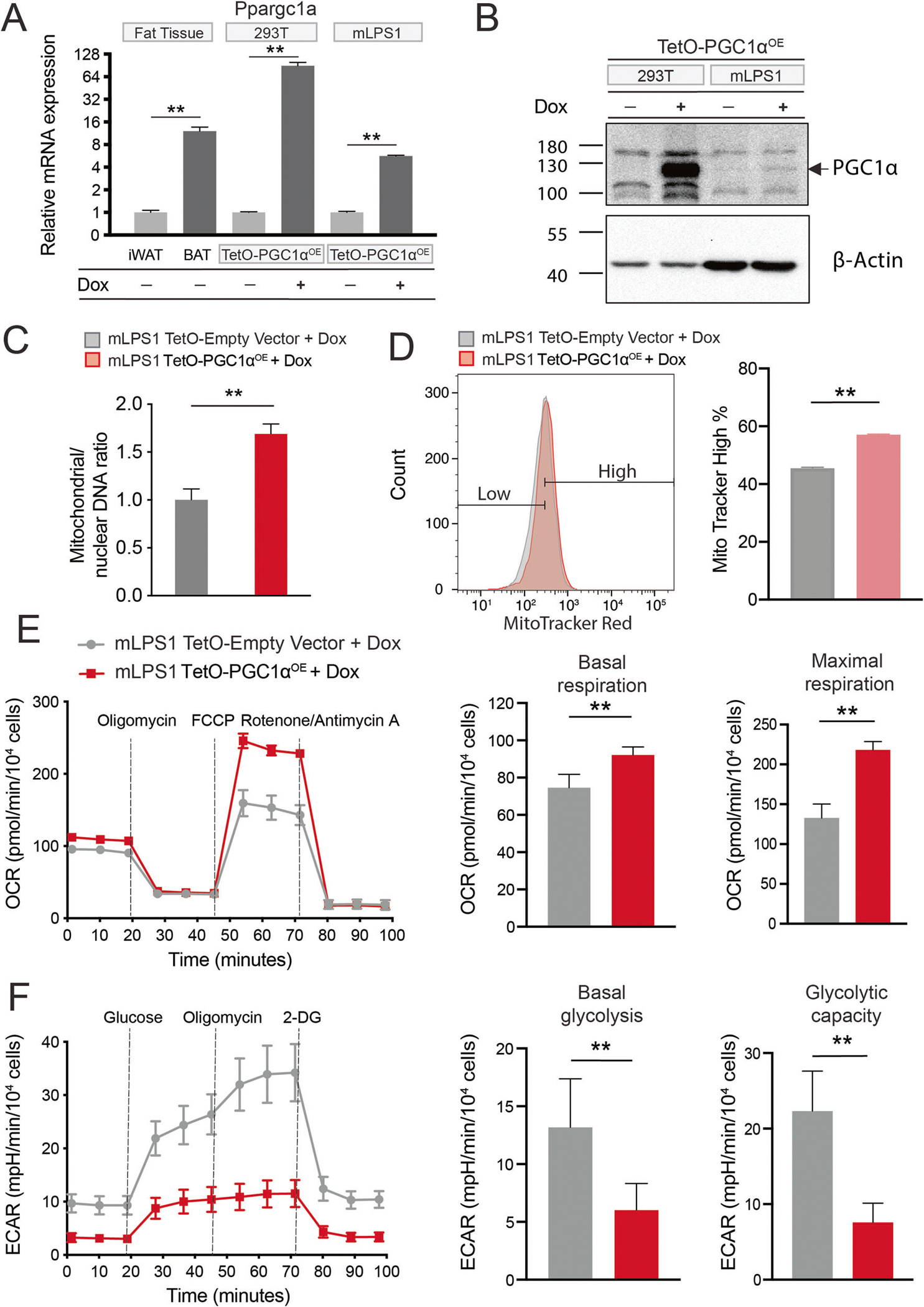
A Ppargc1a mRNA levels in mouse inguinal white adipose tissue, brown adipose tissue, TetO-mPGC1αOE stably transfected 293 T cells, and TetO-mPGC1αOE stably transfected mLPS1 cells with or without doxycycline (Dox) induction (n = 3). B PGC-1α protein levels in TetO-mPGC1αOE 293 T and mLPS1 cells treated with or without Dox. C RT-qPCR analysis of mitochondrial DNA (MT-ND1) and nuclear DNA (HK2) in TetO-Empty vector and TetO-mPGC1αOE mLPS1 cells with Dox treatment (n = 3). D TetO-Empty vector and TetO-mPGC1αOE mLPS1 cells were stained with the MitoTracker Red FM probe and analyzed by flow cytometry (n = 3). E The oxygen consumption rate (OCR) was measured with Seahorse in TetO-Empty vector and TetO-mPGC1αOE mLPS1 cells treated with Dox. OCR corresponding to basal mitochondrial respiration (middle) and maximal mitochondrial respiratory capacity (right) were shown (n = 5). F The extracellular acidification rate (ECAR) of TetO-Empty vector and TetO-mPGC1αOE mLPS1 cells treated with Dox. ECAR corresponding to basal glycolysis (middle) and glycolytic capacity (right) were shown (n = 5). Data are presented as mean ± SD of three reading cycles for each cycle, n = 5, *P < 0.05, **P < 0.001.
PGC-1α suppress mLPS1 cell growth in vitro and in vivo
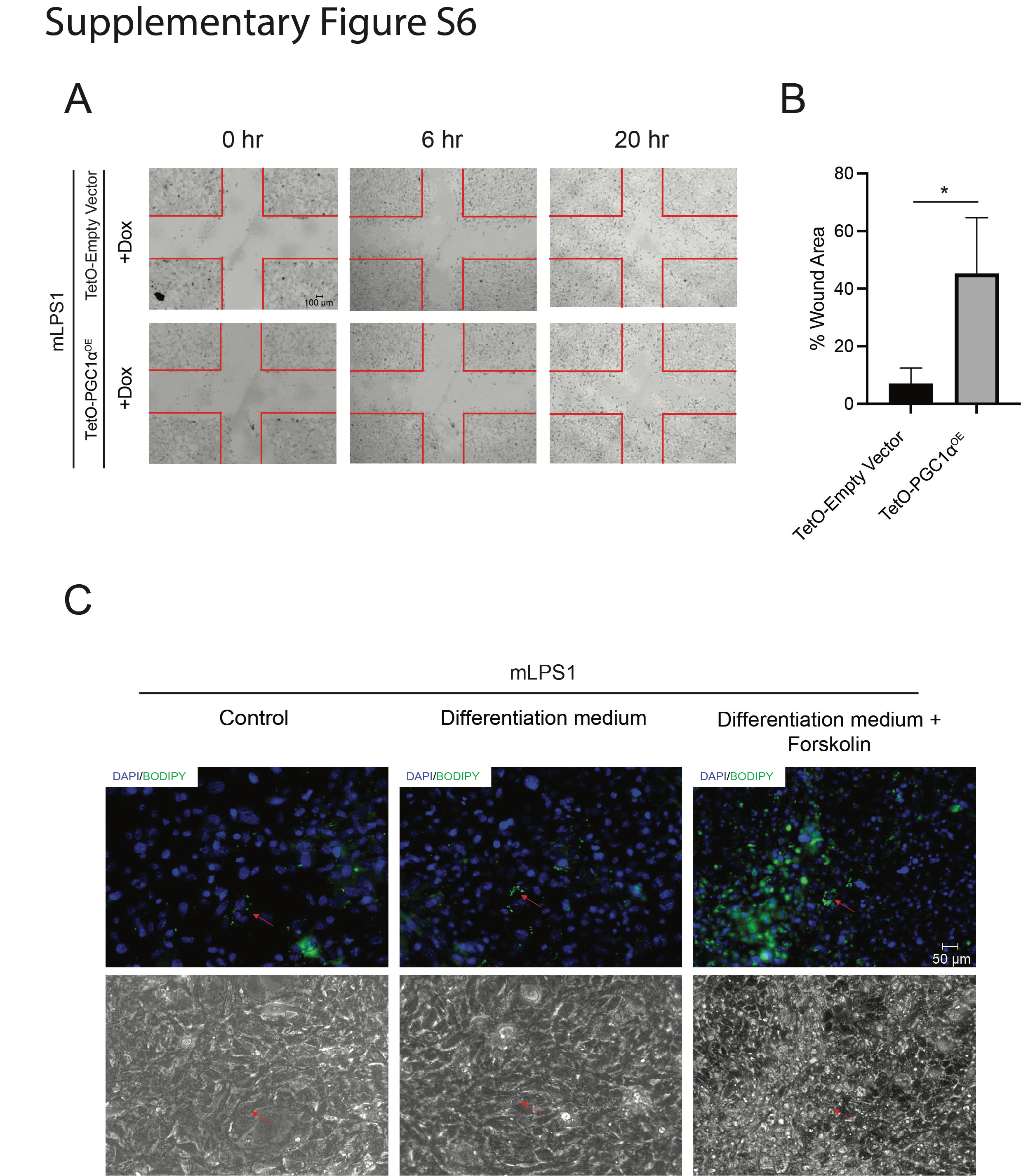
We next investigated how PGC-1α overexpression affects growth of mLPS1 cells. The mLPS1 TetO-PGC1αOE cells and empty vector control cells were treated with Dox or vehicle control. The results showed that only the Dox treated TetO-PGC1αOE cells exhibited reduced cell growth in vitro (Fig. 8A). In addition, Dox treatment in TetO-PGC1αOE cells expressed significantly lower levels of the cancer stem cell markers Nanog, Oct4, and Sox2 (Fig. 8B). In consistence with the result, PGC-1α overexpression decrease cell migration ability (Supplementary Fig. S5A, B). To test the function of PGC-1α in stimulating cancer cell differentiation, we treat mLPS1 cell with adipocyte differentiation medium combine with PGC-1α agonist Forskolin. As expected, Forskolin stimulation of PGC-1α enhanced adipogenic differentiation of mLPS1 cells (Supplementary Fig. S5C). To further evaluate the role of PGC-1α in tumor growth and maintenance in vivo, NRG mice were injected subcutaneously with mLPS1 TetO-empty vector cells in the left flank and mLPS1 TetO-PGC1αOE cells in the right flank so that both control and PGC1αOE cells are grown in the identical host environments. Dox-induced PGC1αOE effectively prevented skin necrosis induced by mLPS1 cells (Fig. 8C), and significantly decreased tumor growth in terms of volume (Fig. 8D), but only marginally decreased the average tumor weight (Fig. 8E, F). RT-qPCR analysis confirmed that PGC-1α expressing tumor cohorts exhibited high Ppargc1a mRNA levels (Fig. 8G). Moreover, we observed that the percentages of Ki67-positive cells were significantly decreased in allograft tumors derived from mLPS1 TetO-PGC1αOE cells (Fig. 8H, I). These results demonstrate that even partial PGC-1α restoration inhibits in vitro proliferation and in vivo tumor growth in DDLPS.
Fig. 8. PGC-1α expression attenuates mLPS1 cell proliferation and tumorigenesis.
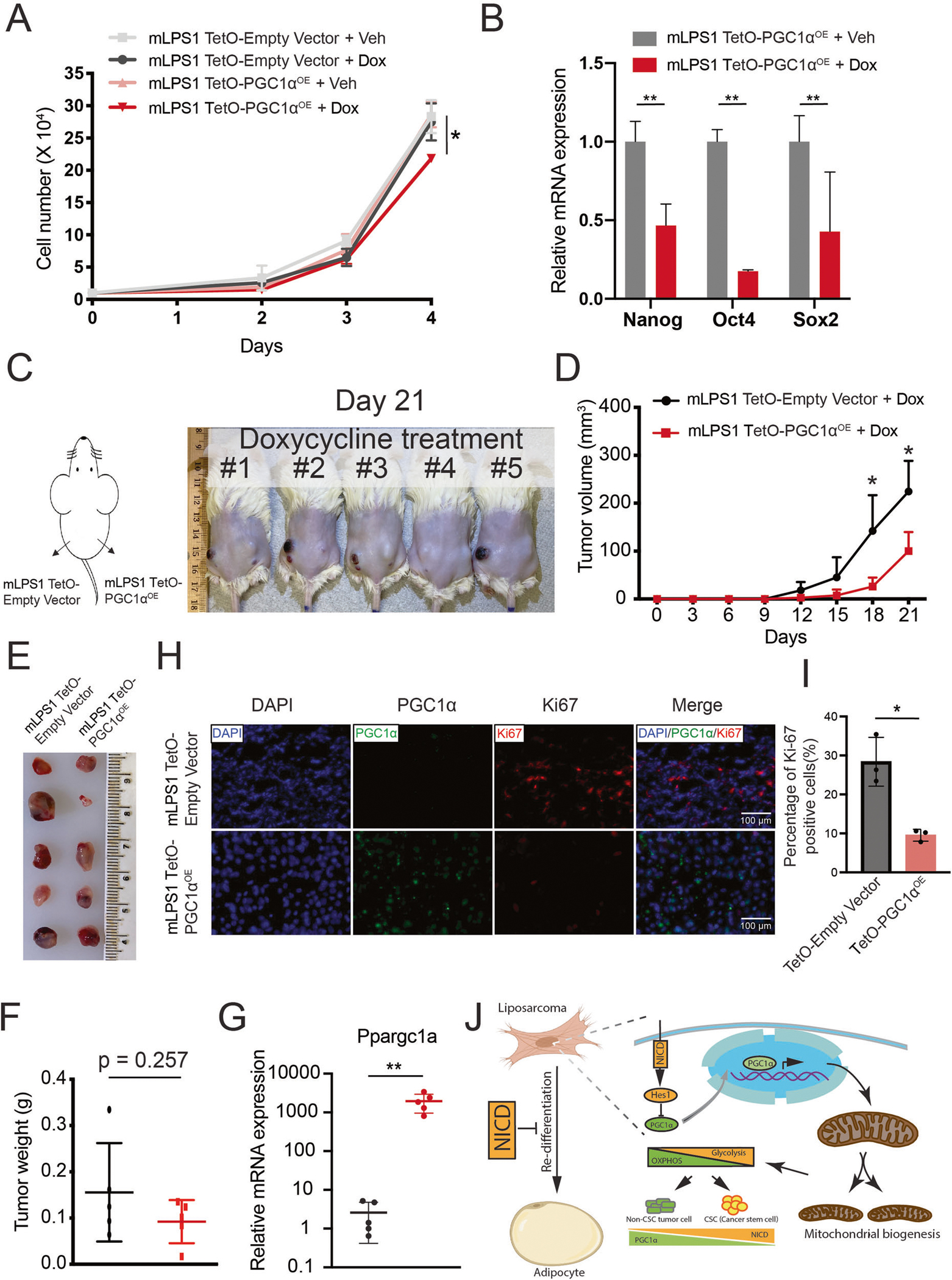
A Cell proliferation of TetO-Empty vector and TetO-mPGC1αOE stable transfected mLPS1 cells treated with doxycycline (dox) or vehicle control (veh) (n = 3). B RT-qPCR analysis of the relative mRNA levels of cancer stem cell markers in Veh or Dox treated TetO-mPGC1αOE mLPS1 cells (n = 3). C The mLPS1 TetO-mPGC1αOE cells and mLPS1 TetO-Empty vector cells were subcutaneously transplanted into NRG mice and treated with Dox via drinking water (n = 5) for 21 days. D Tumor growth curves based on tumor volume calculation obtained from caliper measurement. E Morphology and (F) average weight of the transplanted tumors. G RT-qPCR analysis of PGC-1α expression in the transplanted tumors. H Immunofluorescent staining shows the relative expression of PGC-1α and Ki67 in the mLPS1 TetO-Empty vector and the mLPS1 TetO-mPGC1αOE allograft tumors. Nuclei were stained by DAPI. I Quantitation of Ki67 positive cells in H. Data are represented as mean ± SD. *P < 0.05, **P < 0.001. J Graphic illustration of how Notch signaling regulates cancer cell differentiation and mitochondrial function in liposarcoma.
Notch inhibitors enhance liposarcoma response to chemotherapeutic drugs
To investigate the clinical potential of Notch inhibition in human LPS treatment, we treat liposarcoma cells with the first-line chemotherapy drug Doxorubicin combined with Notch inhibitor CB103. In vitro treatment of Notch inhibitor enhanced Doxorubicin efficacy in inhibiting LPS246 cell growth (Supplementary Fig. S6A). Further, combining Notch inhibitor with adipocyte differentiation medium stimulated expression of mature adipocyte markers Pparg, Adipoq, and Leptin (Supplementary Fig. S6B). These results establish a foundation for future development of Notch inhibitor-based treatments against human liposarcomas.
DISCUSSION
DDLPS is the most malignant subtype of liposarcomas and has a poor patient survival rate. Although rapid advances in the molecular analysis of DDLPS have identified many oncogenic mutations in human DDLPS [5], the specific driver mutation of DDLPS remains unclear and has not been recapitulated in animal models [46]. Here, we implicate Notch signaling as a driver mutation in murine DDLPS by demonstrating that reversal of Notch overexpression abrogates tumorigenicity of mLPS1 cells. We also provide evidence that Notch signaling supports LPS proliferation and regulates LPS metabolism. We further extend murine data to show that Notch activation is associated with MDM2 expression, a defining marker of human LPS tissues and cells, and show that inhibition of Notch signaling reduces proliferation of human LPS246 cells.
Notch signaling is critical for homeostasis of cancer stem cells (CSCs) through regulating cell fate, differentiation, and metabolism. However, the underlying mechanism of Notch signaling in DDLPS is unclear. In this study, we establish a model in which Notch signaling regulates LPS cellular metabolism by inhibiting mitochondrial biogenesis and facilitates anaerobic glycolysis to maintain CSCs function (Fig. 8J). To acquire cell stemness, cancer cells leverage the cellular metabolism to better utilize available nutrients and oxygen to support cell survival and proliferation. However, the mechanism of metabolic reprogramming in CSCs in DDLPS is unclear. In DDLPS, cancer cells prefer glycolysis to maintain cell proliferation, and a recent study reports that overexpression of miR-133a causes a metabolic shift from glycolysis to mitochondria oxidative phosphorylation to suppress cell proliferation [47]. Here, we show that Notch signaling also plays a role in reprogramming cell metabolism by suppressing PGC-1α expression and mitochondrial respiration. Linking these two independent studies, miR-133 has recently been reported to inhibit Notch signaling to suppress tumorigenesis of oral squamous cell carcinoma [48]. Our results further suggest that activation of PGC-1α may represent a potential therapy for re-differentiating LPS cells into well-differentiated lipid-accumulating cells.
PGC-1α is a master regulator of mitochondrial biogenesis. Overexpression of PGC-1α promoted mitochondrial respiration in the Notch overexpressing mLPS1 cells. Consistently, mLPS1 proliferation in vitro and tumor growth in vivo were both reduced by overexpression of PGC-1α. However, the overall tumor size and weight were only marginally reduced when tumors were isolated from mice. This may be attributed to the dosage- and context-dependent effects of PGC-1α on cancer cells and niche supporting cells that both contribute to tumorigenesis. In this scenario, PGC-1α overexpression in the cancer cells significantly reduced lesion to the tumor microenvironment, manifested by remarkably less skin necrosis (indicated by purple) at the site of tumor burden. The skin lesion may have contributed to the significantly larger tumor size, when tumors were measured in situ with a caliper without removing the skin. In addition, the PGC-1α level is very high in our dox-induced overexpression model, such high levels of PGC-1α expression may bring about non-specific effects that counteract its tumor-suppressor function. In other studies, PGC-1α stimulates breast cancer metastasis without inhibiting cell proliferation [49], but it suppresses the proliferation and metastatic capacity of prostate cancer cells [50]. The extremely low levels of PGC-1α in Notch overexpressing mLPS1 cells suggest that an inhibition of mitochondrial biogenesis leads to the Warburg effect. Nevertheless, other Notch targets may also play a role in regulating LPS cell metabolism. Future CRISPR screening will facilitate identification of such targets.
Notch signaling plays multiple roles in DDLPS. Besides stimulating glycolysis, Notch activation inhibits cancer cell differentiation. RNA-seq analysis revealed that expression of Nr2f2 (nuclear receptor subfamily 2 group F member 2) was elevated in Notch-overexpressing cells. Nr2f2 has been reported to repress adipocyte differentiation [51]. Thus, the high level of Nr2f2 in Notch-driven liposarcoma cells may have put a brake on the differentiation program to maintain the cancer cells in a dedifferentiated state. Consistent with this prediction, CRISPR/CAS9 deletion of the NICD-overexpressing cassette only had little effects on the differentiation of mLPS1 cells when induced in an adipogenic medium. Although Notch inhibition significantly upregulated the expression of adipogenic genes in mLPS1 cells, the vast majority NICD KO cells cannot fully mature into lipid-laden cells resembling mature adipocytes. Therefore, Notch inhibition appears to mainly function to initiate re-differentiation of liposarcoma cells but not sufficient to promote maturation of the cells into large lipid droplet containing adipocytes. Future studies should explore if co-targeting Notch signaling and its downstream effectors such as Nr2f2 will promote more efficient re-differentiation of mLPS1 cells.
We examined two newly developed Notch inhibitors (IMR1 and CB103) and target NICD transcriptional complex independent of NICD cleavage from the Notch receptors [33, 52]. These new inhibitors are advantageous in treatment of cancers such as leukemia and a rare population of LPS driven by Notch amplification mutations that produce NICD independent of ligand binding. While these inhibitors robustly inhibited cell proliferation, they had limited effects in suppressing tumor growth in vivo. Future studies should focus on optimizing the dosage, treatment duration and route of drug delivery to see if the inhibitor has better effects. Considering that both Notch inhibitor and PGC-1α activation reduces skin lesions, combinatorial treatment of tumors with Notch inhibitor and PGC-1α activator may improve the in vivo efficacy. Notably, CB-103 has been tested in phase I/II study to investigate the safety, tolerability and efficacy in solid tumors and blood caners (NCT03422679). Studies in mouse models of LPS will further establish its potential in human LPS treatment.
In summary, our findings demonstrate a new role of Notch signaling in supporting DDLPS progression through regulating cell metabolism. Specifically, activation of Notch favors glycolytic metabolism and inhibition of Notch promotes mitochondrial oxidative respiration through upregulation of PGC-1α. Despite the promising data demonstrating in vitro efficacy in inhibiting LPS cell proliferation, pharmacological inhibition of NICD or genetic activation of PGC-1α has limited effects on LPS suppression. Given that genetic abrogation of ectopic Notch expression abolishes the tumorigenicity of mLPS1 cells in vivo, the lack of efficacy of the pharmacological inhibitors suggests that they either does not fully block Notch signaling or presents side effects that support tumor growth. In light of these limitations, future study should aim to identify more potent Notch inhibitors with less side effects, or to optimize the regime of drug delivery and dosage to accomplish more effective Notch inhibition. Another future direction is to explore if combining Notch inhibitor with other targeting strategies would produce better efficacy in LPS treatment. For example, our data show that activation of PGC-1α promotes cancer cell differentiation in vitro and partially reduce tumor growth in vivo, it is plausible to predict that combining Notch inhibitor and PGC-1α activator will lead to better targeting efficacy. Notch inhibitors can also be combined with inhibitors of glycolytic metabolism [53], or with conventional chemotherapies to achieve better therapeutic effects.
METHODS
Patients and clinical samples
De-identified frozen human liposarcoma tissues were obtained from the CHTN tissue bank or collected by co-author Raphael Pollock under the approved institutional review board protocol. Liposarcoma subtypes were classified according to the surgical pathology reports.
Mice
Tumor allograft experiments were approved by the Animal Care and Use Committee of Purdue University. NRG (NOD-RAG1−/−IL2Rγ−/−) mice (2–4 months old) provided by Purdue University Biological Evaluation Shared Resource were used as recipients. Cells (1 × 106) were resuspended in 50–100 μL PBS and injected into the left or right flank. After cell injection, the mice were monitored daily, and tumor length and width were measured using a digital caliper once palpable tumors were detected and randomized assign to each group. The grafted mice were treated with vehicle, CB103 (40 mg/kg/day), or 1 mg/mL doxycycline (Dox) in water (Tokyo Chemical Industry, D4116) supplemented with 1% sucrose (Fisher Scientific, S5–500), to inhibit Notch or to activate PGC-1α. The allograft tumors were dissected for downstream analysis at the end of the experiment or if the length reaches 3 cm, whichever comes first. Mice were housed under standard laboratory conditions, including a 12 hr light/dark cycle, with free access to food and water.
Cell culture
Human primary WDLPS cells LPS80.2, LPS123, and LPS135 and primary DDLPS cells LPS187, LPS120, and LPS27 were established by Raphael Pollock. Human LPS246 (DDLPS) cells were provided by L. Chin (MD Anderson Cancer Center, Houston, TX, USA) [54]. Human primary liposarcoma cells and cell line were cultured in DMEM (Sigma, D5796) containing 10% FBS (Corning, 35-010-CV) and penicillin (100U/ mL)/streptomycin (100 μg/mL) (Hyclone #SV30010). The mouse liposarcoma cell lines mLPS1, mLPS1ΔNICD, mLPS1 stably transfected with TetO-Empty or TetO-PGC1αOE vectors were cultured in RPMI1640 (Corning, 10-040-CV) containing 10% FBS and penicillin (100 U/mL)/streptomycin (100 μg/mL). Cells were certified as mycoplasma-free by RT-qPCR analysis of the mycoplasma-specific 16 s rRNA gene region.
Vector construction and cell transfection
The doxycycline-inducible TetO-PGC1αOE mLPS1 and empty vector control cells were generated using the Sleeping Beauty transposon system (Addgene #60497). The PGC-1α cDNA sequence was subcloned from the TRIPZ-haPGC1A plasmid (kindly provided by Arkaitz Carracedo) [50]. mLPS1ΔNICD cells were generated by clustered regularly interspaced short palindromic repeats (CRISPR) targeting, catalyzed by CRISPR-associated protein 9 (CAS9) and directed by a guide RNA (gRNA) that recognizes specific sequences of NICD (Addgene #62988). The efficiency of gene silencing or overexpression was verified using qPCR and western blotting.
Genotyping analysis of CRISPR editing cells
PCR was performed using genomic DNA extracted from the mLPS1ΔNICD cells. For Sanger sequencing, PCR amplification was performed using the forward primer 5′-AGCTCTGGTTCCCTGAGGGTTT-3′ and reverse primer 5′-CAGTCTCATAGCTGCCCTCAC GG-3′ to generate a 910 bp product.
Cell growth assay and adipogenic differentiation assay
Cell proliferation was evaluated by counting the number of cells using a hemocytometer. Stock Doxorubicin (Cayman 15007) was dissolved in DMSO at 10 mM and diluted in culture medium to a final concentration of 300 nM. For combination treatment, CB103 concentration was 20 μM. LPS246 cells were seeded onto 24-well plates (1 × 105 cells/well) and incubated at 37 °C with 5% CO2. Doxorubicin and/or CB103 were added to cells next day and cells were continually cultured for up to 5 days. Cell numbers were examined daily. For adipogenic differentiation of LPS cells, the cells were seeded into 24-well culture plates until they reached 90% confluency. To induce adipogenic differentiation, culture medium was replaced with the adipocyte differentiation medium containing 33 μM biotin, 0.5 μM human insulin, 17 μM pantothenate, 0.1 μM dexamethasone, 2 nM triiodothyronine, 500 μM IBMX (3-isobutyl-1-methylxanthine), 30 μM indomethacin, and 2% FBS for 8 days and medium were refreshed every 3 days. After 8 days in differentiation medium, culture medium was changed to adipocyte maturation medium containing 0.5 μM human insulin, 2 nM triiodothyronine, and 2% FBS for 6 days. Adipogenic differentiation was confirmed by staining with BODIPY (Thermo Scientific, #D6003) and Hoechst 33342 (#H3570; Invitrogen). Images were acquired using a fluorescence microscope (Leica DMI6000B; Wetzlar, Germany).
Methylthiazolyldiphenyl-tetrazolium bromide (MTT) assay
Cells were seeded in 96-well plates at a density of 3 × 103 cells/well. Cells were treated with vehicle control, CB-103 (HY-135145, Medchemexpress) or IMR-1 (HY-100431, Medchemexpress) at various concentrations in 200 μL complete medium for 48 h.
Colony formation
Liposarcoma cancer cells at a density of 5 × 102 cells/well were placed in six-well plates and cells were treated with 20 μM CB-103 or 20 μM IMR1 for 14 days. The cells were then washed with PBS and fixed with methanol/acetic acid 3:1 (vol/vol) for 5 min at room temperature. The colonies were stained with 0.1% crystal violet (Coleman & Bell Company). Stained colonies were photographed and analyzed using the ImageJ software.
Cell death assay by TUNEL (Terminal dUTP nick end labeling)
The mLPS1 cells were treated with IMR1, CB103 treatment for 24 hr and deoxyribonuclease (Invitrogen, AM1907) were used as the positive control. Cell apoptosis were detected with an apoptosis detection kit (Biotium) and a fluorescence microscope (Leica DMI6000B; Wetzlar, Germany).
Wound-healing assay
The mLPS1 TetO-Vector and mLPS1 TetO-PGC1αOE cells were seeded into six-well plates in 80% confluence. Upon cell attachment (~6 h), 1 ug/mL doxycycline was added into the medium. After cells reached 100% confluence as a monolayer, a scratch in the monolayer of cells was generated using a 100 μl plastic pipette tip. Images were captured with a light Leica microscope at 0, 6, and 20 h. ImageJ software was used to determine the relative migration in each group.
Western blot
Membranes were blocked with 5% non-fat milk for 1 h at room temperature, followed by overnight incubation at 4 °C with the following specific primary antibodies: actin (A5441, 1:5000; Sigma-Aldrich), NICD (MAB5352, 1:1000; Sigma-Aldrich), HES1 (AB5702, 1:1000; Sigma-Aldrich), MDM2 (OP46, 1:1000; Calbiochem), and PGC-1α (ST1202, 1:1000; Sigma-Aldrich). HRP-conjugated secondary antibodies were incubated for 1 h at room temperature. Membranes were developed using the western blot luminol reagent (Santa Cruz Biotechnology) and imaged using the FluorChem R image system (ProteinSimple).
Real-time quantitative PCR and mitochondrial DNA (mtDNA) content determination
Total RNA was extracted using TriReagent (Sigma-Aldrich #T9424) and converted into cDNA using M-MLV reverse transcriptase (Invitrogen #28025–021). All reactions were run in triplicate and normalized using 18 s as the internal control gene. To determine mtDNA content, total DNA was extracted from the cell samples using phenol/chloroform. qPCR was run to quantify the mtDNA copy number. MT-ND1 was used as the standard for mitochondrial DNA (mtDNA), and HK2 was used as the nuclear DNA (nDNA) normalizer to calculate the mtDNA/nDNA ratio. Quantitative PCR was performed on a LightCycler 96 real-time PCR system (Roche). Fold changes in gene expression were calculated using the comparative Ct method (ΔΔCt). The mouse-specific primer sequences are listed in Supplementary Table 1.
MitoTracker Staining
Cells were incubated with MitoTracker Deep Red FM reagent (Invitrogen, M46753) 1:10000 in PBS at 37 °C for 30 min. After staining was complete, cells were gently washed once with PBS. The samples were analyzed by flow cytometry using a BD FACS Aria III flow cytometer at the Purdue Flow Cytometry and Cell Separation Facility. The data were analyzed using FlowJo version 10.4.
Glucose uptake, lactate production, and ATP assay
For glucose uptake, mLPS1 and mLPS1ΔNICD cells were seeded in 24-well plates at a density of 1 × 105 cells/well and co-cultured in RPMI 1640 medium supplemented with 10% FBS for 16 h. The cells were then washed with PBS and cultured for 6 h in fresh RPMI 1640 medium. The culture medium was collected, and glucose uptake was analyzed using a OneTouch Ultra 2 Blood Glucose Meter (Lifescan). For the lactate production and ATP assays, the lactate production was analyzed using the L-Lactate Assay Kit II (Eton Bioscience) according to the manufacturer’s instructions. Simultaneously, cell lysates were collected to measure ATP production using an ATP Detection Assay Kit (Cayman, 700410), following the manufacturer’s instructions.
Seahorse XF cell mitochondria stress and glycolysis stress analysis
The mitochondrial respiratory capacity was determined using the XF Cell Mito Stress Test Kit (Agilent Technologies, 103015–100). The cells were seeded in an XFe24 cell culture microplate at a density of 4 × 104 cells/well. Five replicates were set up for each of the following groups: (1) mLPS1, (2) mLPS1ΔNICD, (3) mLPS1 TetO-Vector + Dox and (4) mLPS1 TetO-PGC1αOE + Dox. Briefly, cells were seeded in a microplate for 12 h at 37 °C. The mLPS1 TetO-Vector and mLPS1 TetO-PGC1αOE cells were pretreated with 1 mM doxycycline for 48 h prior to seeding into an XFe24 cell culture microplate. The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured following the manufacturer’s instructions. OCR and ECAR values were normalized to the number of cells in each well [55].
RNA-seq analysis
The mLPS1 and mLPS1ΔNICD cells were collected from three batches of different passage numbers. Total RNA was extracted using TriReagent (Sigma-Aldrich #T9424) and prepared for RNA-seq analysis at BGI America (Cambridge, MA, USA). RNA-seq libraries were constructed and sequenced using the DNBSEQ platform (DNBSEQ Technology). The clean reads were quality-checked using FastQC and subsequently aligned to the mouse reference genome (version mm10) using Salmon with default parameters [56]. Differential expression was determined using a cutoff significance level of FDR (padj) < 0.05, using the DESeq2 [57]. RStudio software was used for data visualization, and pathway analysis was performed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) platforms. Transcriptome data were deposited into the GSE210457 dataset.
Kaplan–Meier curve of overall survival of liposarcoma patients
Kaplan–Meier curves of disease-specific survival of patients with liposarcoma and dedifferentiated liposarcoma, stratified by tumor expression of HEY1, were generated using cBioPortal (http://www.https://cbioportal.org) from TCGA Cell, 2017 dataset. Overall survival of sarcoma patients stratified by tumor expression of PPARGC1A from TCGA database (BMC Cancer, 2014) using PROGgeneV2 platform analysis (http://gepia.cancer-pku.cn/index.html). PPARGC1A expression levels in human liposarcoma tissues were determined using Genevestigator (https://genevestigator.com) in the HS_AFFY_U133PLUS_2–1 dataset.
Statistical analysis
All experiments were independently repeated at least thrice, and the number of replicates is provided in the figure legends. Statistical analyses and graphs were generated using Microsoft Excel and GraphPad Prism, version 9. Data are presented as the mean ± SD. Statistical analysis was performed using a two-tailed unpaired t-test with the following significance indicators: *P < 0.05, **P < 0.001.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGEMENTS
This work was supported by grants from the National Cancer Institute of the US National Institutes of Health (R01CA212609) and Purdue University Center for Cancer. Research (P30CA023168). We thank the Cooperative Human Tissue Network for providing human liposarcoma samples, and the Ohio State University Comprehensive. Cancer Center for supporting human liposarcoma samples and cell lines. We also thank the Purdue University Flow cytometry core for FACS data collection, DR. Jason Hanna (Purdue Biological Sciences) for providing stock Doxorubicin solutions, and Jun Wu for technical support.
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41388-023-02768-6.
Reprints and permission information is available at http://www.nature.com/reprints
DATA AVAILABILITY
The data supporting this publication are available in the GSE210457 repository at https://www.ncbi.nlm.nih.gov/gds/.
REFERENCES
- 1.Thway K Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin Diagn Pathol. 2019;36:112–21. [DOI] [PubMed] [Google Scholar]
- 2.Jones RL, Lee ATJ, Thway K, Huang PH. Clinical and molecular spectrum of liposarcoma. J Clin Oncol. 2018;36:151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang L, Chen S, Luo P, Yan W, Wang C. Liposarcoma: Advances in cellular and molecular genetics alterations and corresponding clinical treatment. J Cancer. 2020;11:100–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koseła-Paterczyk H, Wągrodzki M. Liposarcoma — spectrum of disease. Oncol Clin Pract. 2019;14:341–7. [Google Scholar]
- 5.Keung EZ, Somaiah N. Overview of liposarcomas and their genomic landscape. J Transl Genet Genom. 2019. [Google Scholar]
- 6.Casadei L, Calore F, Braggio DA, Zewdu A, Deshmukh AA, Fadda P, et al. MDM2 derived from dedifferentiated liposarcoma extracellular vesicles induces MMP2 production from preadipocytes. Cancer Res. 2019;79:4911–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sciot R Mdm2 amplified sarcomas: A literature review. Diagnostics 2021;11:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Livingston JA, Bugano D, Barbo A, Lin H, Madewell JE, Wang WL, et al. Role of chemotherapy in dedifferentiated liposarcoma of the retroperitoneum: Defining the benefit and challenges of the standard. Sci Rep. 2017;7:11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gahvari Z, Parkes A. Dedifferentiated liposarcoma: systemic therapy options. Curr Treat Options Oncol. 2020;21:15. [DOI] [PubMed] [Google Scholar]
- 10.Italiano A, Toulmonde M, Cioffi A, Penel N, Isambert N, Bompas E, et al. Advanced well-differentiated/dedifferentiated liposarcomas: Role of chemotherapy and survival. Ann Oncol. 2012;23:1601–7. [DOI] [PubMed] [Google Scholar]
- 11.Taylor BS, DeCarolis PL, Angeles CV, Brenet F, Schultz N, Antonescu CR, et al. Frequent alterations and epigenetic silencing of differentiation pathway genes in structurally rearranged liposarcomas. Cancer Discov. 2011;1:587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiao W, Gao Z, Duan Y, Yuan W, Ke Y. Notch signaling plays a crucial role in cancer stem-like cells maintaining stemness and mediating chemotaxis in renal cell carcinoma. J Exp Clin Cancer Res. 2017;36:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.de Almeida Magalhães T, Cruzeiro GAV, de Sousa GR, da Silva KR, Lira RCP, Scrideli CA, et al. Notch pathway in ependymoma RELA-fused subgroup: upregulation and association with cancer stem cells markers expression. Cancer Gene Ther. 2020;27:509–12. [DOI] [PubMed] [Google Scholar]
- 14.Fang S, Liu M, Li L, Zhang FF, Li Y, Yan Q, et al. Lymphoid enhancer-binding factor-1 promotes stemness and poor differentiation of hepatocellular carcinoma by directly activating the NOTCH pathway. Oncogene 2019;38:4061–74. [DOI] [PubMed] [Google Scholar]
- 15.Jiang H, Zhou C, Zhang Z, Wang Q, Wei H, Shi W, et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat Commun. 2020;11:5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bi P, Yue F, Karki A, Castro B, Wirbisky SE, Wang C, et al. Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J Exp Med. 2016;213:2019–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tien PC, Quan M, Kuang S. Sustained activation of notch signaling maintains tumor-initiating cells in a murine model of liposarcoma. Cancer Lett. 2020;494:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Somaiah N, Beird HC, Barbo A, Song J, Mills Shaw KR, Wang WL, et al. Targeted next generation sequencing of well-differentiated/ dedifferentiated liposarcoma reveals novel gene amplifications and mutations. Oncotarget 2018;9:19891–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loganathan SK, Schleicher K, Malik A, Quevedo R, Langille E, Teng K, et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020;367:1264–9. [DOI] [PubMed] [Google Scholar]
- 20.Pettersson S, Sczaniecka M, McLaren L, Russell F, Gladstone K, Hupp T, et al. Non-degradative ubiquitination of the Notch1 receptor by the E3 ligase MDM2 activates the Notch signalling pathway. Biochem J. 2013;450:523–36. [DOI] [PubMed] [Google Scholar]
- 21.Sczaniecka M, Gladstone K, Pettersson S, McLaren L, Huart AS, Wallace M. MDM2 protein-mediated ubiquitination of numb protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. J Biol Chem. 2012;287:14052–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yogosawa S, Miyauchi Y, Honda R, Tanaka H, Yasuda H. Mammalian Numb is a target protein of Mdm2, ubiquitin ligase. Biochem Biophys Res Commun. 2003;302:869–72. [DOI] [PubMed] [Google Scholar]
- 23.Slaninova V, Krafcikova M, Perez-Gomez R, Steffal P, Trantirek L, Bray SJ, et al. Notch stimulates growth by direct regulation of genes involved in the control of glycolysis and the tricarboxylic acid cycle. Open Biol. 2016;6:150155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu X, Chen HH, Gao CY, Zhang XX, Jiang JX, Zhang Y, et al. Energy metabolism in cancer stem cells. World J Stem Cells. 2020;12:448–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gu W, Gaeta X, Sahakyan A, Chan AB, Hong CS, Kim R, et al. Glycolytic metabolism plays a functional role in regulating human pluripotent stem cell state. Cell Stem Cell. 2016;19:476–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huppertz I, Perez-Perri JI, Mantas P, Sekaran T, Schwarzl T, Russo F, et al. Riboregulation of Enolase 1 activity controls glycolysis and embryonic stem cell differentiation. Mol Cell. 2022;82:2666–80.e11. [DOI] [PubMed] [Google Scholar]
- 27.Luo C, Widlund HR, Puigserver P. PGC-1 coactivators: shepherding the mitochondrial biogenesis of tumors. Trends Cancer. 2016;2:619–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abeshouse A, Adebamowo C, Adebamowo SN, Akbani R, Akeredolu T, Ally A, et al. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell 2017;171:950–65.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ran Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, et al. γ-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med. 2017;9:950–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haapasalo A, Kovacs DM. The many substrates of presenilin/γ-secretase. J Alzheimer’s Dis. 2011;25:3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gounder MM, Rosenbaum E, Wu N, Dickson MA, Sheikh TN, D’Angelo SP, et al. A Phase Ib/II Randomized Study of RO4929097, a Gamma-Secretase or Notch Inhibitor with or without Vismodegib, a Hedgehog Inhibitor, in Advanced Sarcoma. Clin Cancer Res. 2022;28:1586–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Astudillo L, da Silva TG, Wang Z, Han X, Jin K, VanWye J, et al. The small molecule IMR-1 inhibits the notch transcriptional activation complex to suppress tumorigenesis. Cancer Res. 2016;76:3593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehal R, Zaric J, Vigolo M, Urech C, Frismantas V, Zangger N, et al. Pharmacological disruption of the Notch transcription factor complex. Proc Natl Acad Sci USA. 2020;117:16292–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Zhang H, La Ferlita A, Sp N, Goryunova M, Sarchet P, et al. Phosphorylation of IWS1 by AKT maintains liposarcoma tumor heterogeneity through preservation of cancer stem cell phenotypes and mesenchymal-epithelial plasticity. Oncogenesis 2023;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Venkatesh V, Nataraj R, Thangaraj GS, Karthikeyan M, Gnanasekaran A, Kaginelli SB, et al. Targeting notch signalling pathway of cancer stem cells. Stem Cell Investig. 2018;5:5–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinstein IB. Cancer: Addiction to oncogenes - The Achilles heal of cancer. Science. 2002;297:63–4. [DOI] [PubMed] [Google Scholar]
- 37.Siebel C, Lendahl U. Notch signaling in development, tissue homeostasis, and disease. Physiol Rev. 2017;97:1235–94. [DOI] [PubMed] [Google Scholar]
- 38.Mašek J, Andersson ER. The developmental biology of genetic notch disorders. Dev (Camb). 2017;144:1743–63. [DOI] [PubMed] [Google Scholar]
- 39.Osathanon T, Subbalekha K, Sastravaha P, Pavasant P. Notch signalling inhibits the adipogenic differentiation of single-cell-derived mesenchymal stem cell clones isolated from human adipose tissue. Cell Biol Int. 2012;36:1161–70. [DOI] [PubMed] [Google Scholar]
- 40.Hanahan D Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12:31–46. [DOI] [PubMed] [Google Scholar]
- 41.Rota R, Ciarapica R, Miele L, Locatelli F. Notch signaling in pediatric soft tissue sarcomas. BMC Med. 2012;10:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang XF, Yang SA, Gong S, Chang CH, Portilla JM, Chatterjee D, et al. Polyploid mitosis and depolyploidization promote chromosomal instability and tumor progression in a Notch-induced tumor model. Dev Cell. 2021;56:1976–88.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SY, Long F. Notch signaling suppresses glucose metabolism in mesenchymal progenitors to restrict osteoblast differentiation. J Clin Investig. 2018;128:5573–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bi P, Shan T, Liu W, Yue F, Yang X, Liang XR, et al. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med. 2014;20:911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahler E, Sullivan WJ, Cass A, Braas D, York AG, Bensinger SJ, et al. Doxycycline alters metabolism and proliferation of human cell lines. PLoS One. 2013;8:e64561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Codenotti S, Mansoury W, Pinardi L, Monti E, Marampon F, Fanzani A. Animal models of well-differentiated/dedifferentiated liposarcoma: utility and limitations. Onco Targets Ther. 2019;12:5257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu PY, Lopez G, Braggio D, Koller D, Bill KLJ, Prudner BC, et al. miR-133a function in the pathogenesis of dedifferentiated liposarcoma. Cancer Cell Int. 2018;18:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W, Shi X, Wang B. microRNA-133a exerts tumor suppressive role in oral squamous cell carcinoma through the Notch signaling pathway via downregulation of CTBP2. Cancer Gene Ther. 2022;29:62–72. [DOI] [PubMed] [Google Scholar]
- 49.Lebleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. 2014;16:992–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat Cell Biol. 2016;18:645–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xu Z, Yu S, Hsu CH, Eguchi J, Rosen ED. The orphan nuclear receptor chicken ovalbumin upstream promoter- transcription factor II is a critical regulator of adipogenesis. Proc Natl Acad Sci USA. 2008;105:2421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alvarez-Trotta A, Guerrant W, Astudillo L, Lahiry M, Diluvio G, Shersher E, et al. Pharmacological disruption of the notch1 transcriptional complex inhibits tumor growth by selectively targeting cancer stem cells. Cancer Res. 2021;81:3347–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pelicano H, Martin DS, Xu RH, Huang P. Glycolysis inhibition for anticancer treatment. Oncogene 2006;25:4633–46. [DOI] [PubMed] [Google Scholar]
- 54.Peng T, Zhang P, Liu J, Nguyen T, Bolshakov S, Belousov R, et al. An experimental model for the study of well-differentiated and dedifferentiated liposarcoma; Deregulation of targetable tyrosine kinase receptors. Lab Investig. 2011;91:392–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Orellana E, Kasinski A. Sulforhodamine B (SRB) assay in cell culture to investigate cell proliferation. Bio Protoc. 2016;6:e1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. 2017;14:417–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting this publication are available in the GSE210457 repository at https://www.ncbi.nlm.nih.gov/gds/.