

Significance
In this study, we create a three-dimensional (3D) organotypic model of lymphatic vessels that emulates lymphatic junctional structure and drainage function. Using the lymphatic drainage-on-chip model, we reveal a previously unappreciated ROCK2/JAM-A protein complex that regulates lymphatic endothelial cell–cell junctions and the drainage of interstitial fluid into the lymphatic circulation and propose ROCK2 as a potential therapeutic target for lymphedema. This work also emphasizes how recapitulating appropriate architectures can enable the emergence of multicellular functions, and the advancement of such biomimetic cultures is providing a unique platform for bridging traditional in vitro and in vivo models.
Keywords: lymphatics-on-chip, cell–cell adhesion, lymphatic drainage, lymphedema, ROCK2
Abstract
Impaired lymphatic drainage and lymphedema are major morbidities whose mechanisms have remained obscure. To study lymphatic drainage and its impairment, we engineered a microfluidic culture model of lymphatic vessels draining interstitial fluid. This lymphatic drainage-on-chip revealed that inflammatory cytokines that are known to disrupt blood vessel junctions instead tightened lymphatic cell–cell junctions and impeded lymphatic drainage. This opposing response was further demonstrated when inhibition of rho-associated protein kinase (ROCK) was found to normalize fluid drainage under cytokine challenge by simultaneously loosening lymphatic junctions and tightening blood vessel junctions. Studies also revealed a previously undescribed shift in ROCK isoforms in lymphatic endothelial cells, wherein a ROCK2/junctional adhesion molecule-A (JAM-A) complex emerges that is responsible for the cytokine-induced lymphatic junction zippering. To validate these in vitro findings, we further demonstrated in a genetic mouse model that lymphatic-specific knockout of ROCK2 reversed lymphedema in vivo. These studies provide a unique platform to generate interstitial fluid pressure and measure the drainage of interstitial fluid into lymphatics and reveal a previously unappreciated ROCK2-mediated mechanism in regulating lymphatic drainage.
Lymphatic vessels maintain fluid homeostasis by draining excess interstitial fluid through openings at lymphatic endothelial cell (LEC)–cell junctions and via active transcytosis (1–3). Lymphedema, the excess accumulation of interstitial fluid resulting from impaired fluid drainage into the lymphatic vasculature (4), affects more than 150 million individuals worldwide, and yet, with no effective treatments, the standard of care is largely palliative (5). A lack of experimental models of normal and impaired lymphatic drainage has been a major obstacle to understanding and treating lymphedema. Animal models of lymphedema are often difficult to use to establish the pathophysiologic mechanisms underlying this multifactorial disease, owing to the inability to distinguish the relative contributions of biological and biophysical factors. In contrast, two-dimensional (2D) cell culture models are highly controllable but do not recapitulate the 3D organization and function of lymphatics in vivo (6). Therefore, there is a clear, unmet need for 3D in vitro models that reconstitute human lymphatic vasculature to study lymphatic biology. Based on this need, 3D lymphatic cultures have been developed to investigate lymphatic sprouting, permeability, and interactions in the context of lymph nodes, tumor cells, and immune cell trafficking (7–15). However, a platform to generate interstitial fluid pressure and measure the drainage of interstitial fluid into lymphatic capillaries is still lacking.
To address this challenge, we developed a microfluidic 3D culture model featuring an engineered initial lymphatic vessel that can drain interstitial fluid. This lymphatic drainage-on-chip revealed that inflammatory cytokines that disrupt blood endothelial junctions instead tightened lymphatic endothelial junctions and impaired lymphatic drainage. Under cytokine challenge, inhibition of rho-associated protein kinase (ROCK) normalized fluid drainage by loosening lymphatic junctions and tightening blood vessel junctions, as demonstrated in our platform and further validated in a mouse model of surgically induced secondary lymphedema. Mechanistically, we further observed a cytokine-induced shift in the expression of the ROCK2 isoform in LECs and identified a unique ROCK2/junctional adhesion molecule-A (JAM-A) complex that was responsible for the cytokine-induced lymphatic junction zippering. Knockdown of ROCK2 in the human LECs abrogated cytokine-induced drainage dysfunction, and lymphatic-specific knockout of ROCK2 reversed lymphedema in vivo.
Results
Lymphatic Drainage-on-Chip Recapitulates Lymphatic Structure, Drainage, and Dysfunction.
We created a microfluidic 3D culture model of lymphatic vessels that can drain interstitial fluid. The device consists of a polydimethylsiloxane (PDMS) housing and two parallel cylindrical channels within a 3D collagen hydrogel (Fig. 1A and SI Appendix, Fig. S1). Human dermal LECs seeded in one channel generated an engineered lymphatic vessel that expressed lymphatic endothelial markers such as lymphatic vessel endothelial hyaluronan receptor 1 (LYVE-1) and podoplanin (PDPN) (Fig. 1 A and B). LECs were polarized in the device, reflected by the appropriate apical orientation of PDPN, a mucin-related glycoprotein receptor (Fig. 1B) (16). Furthermore, stimulation with the lymphangiogenic vascular endothelial growth factor C (VEGFC) triggered lymphatic sprouting (Fig. 1C). To characterize the integrity of the lymphatic endothelium within the device, we examined their junctional organization by immunofluorescence. Staining for JAM-A revealed weak tight junctions (TJs) in the LEC-generated lymphatic vessels, in contrast to the strongly established TJs in the blood microvascular endothelial cell (BEC)-generated blood vessels as controls (Fig. 1D). Moreover, VE-cadherin and CD31 staining showed intermittent, discontinuous adherens junctions (AJs) in the lymphatic vessels versus continuous AJs in the blood vessels (Fig. 1E; red arrows: VE-cadherin positive areas, yellow arrows: CD31 positive areas), reminiscent of the “button-like junctions” in the initial lymphatic vessels observed in vivo (1). Together, these data suggested the structural characteristics of a rudimentary lymphatic vessel.
Fig. 1.

Lymphatic drainage-on-chip recapitulates lymphatic structure, drainage, and dysfunction. (A) A schematic of the lymphatic drainage-on-chip platform. (B) Apical podoplanin (PDPN) expression on the luminal side of the vessel. (C) Lymphatic sprouting in response to VEGFC stimulation. (D) Immunostaining of LEC-generated lymphatic vessels and BEC-generated blood vessels with a tight junction marker, JAM-A. (E) Immunostaining of lymphatic vessels and blood vessels with an adherens junction marker, VE-cadherin (VE-cad), and CD31. Red and yellow arrows indicate exclusive expression of VE-cad and CD31, respectively, showing interdigitated, discontinuous expression of VE-cad in LECs. (F) A schematic of a biomimetic lymphatic drainage-on-chip model system. The engineered lymphatic vessel (LV) in the right-side channel functions as an initial LV to drain interstitial lymph fluid that is introduced through the left-side channel. (G) Transport of lymph fluid. The left-side channel pressured with lymph fluid induces fluid transport. The pressure gradient between two channels results in fluid convection from the left channel to the engineered LV. The lymph fluid is drained by the engineered LV and accumulated in two right-side reservoirs. Total drained fluid is analyzed to obtain the number of drained lymph molecules. (H) Percent drainage of lymph molecules through LEC and BEC-line channels as compared to acellular channels (No cell), (N = 4). (I) Representative images of BODIPY-C16 fatty acid drainage by LEC-generated lymphatic vessels and BEC-generated blood vessels. (J) Percent lymphatic drainage after cytokine exposure (BODIPY-C16 fatty acid drainage), (N = 5). (K) VE-cadherin staining of engineered lymphatics after cytokine exposure. (L) Percent zipper junction in engineered lymphatics after cytokine exposure, (n = 8, N = 3 experiments). [Scale bars (A–E and I), 100 μm; Scale bars (K), 50 μm.] *P < 0.05 and **P < 0.01 indicate statistical significance.
To assess the functionality of this rudimentary lymphatic vessel required measurements of lymphatic drainage (Fig. 1F). To accomplish this, we pressurized the second, cell-free channel with a surrogate lymph fluid composed of fluorescently labeled lymph molecules, including fatty acids, phospholipids, albumin, and insoluble particles. The differential pressure between the two channels was adjusted to induce interstitial fluid transport with an initial flow rate of 1 µm/s into the engineered lymphatic vessel, given the physiological range of interstitial flow rate in healthy tissues (0.0 to 1.0 µm/s) (17, 18). Lymph fluid drained by the lymphatic vessel accumulated in the connected reservoirs and was collected for analysis (Fig. 1G). Human dermal BEC generated blood vessels or cell-free channels were used as controls. For all tested lymph molecules, engineered lymphatic vessels showed superior draining ability, compared to engineered blood vessels (Fig. 1H). Time-lapse microscopy of drainage further confirmed that fluid containing fatty acids or albumin entered the luminal space of lymphatic vessels faster than that of blood vessels (Fig. 1I and Movie S1). Surprisingly, whereas blood vessels appeared to be compressed by the increased interstitial fluid pressure, lymphatic vessel lumens widened under the same conditions (Movie S1), consistent with in vivo observations that lymphatics dilate with elevated interstitial fluid pressure (3). Together, these findings suggested that our 3D culture model of lymphatic drainage recapitulated the structural and functional characteristics of native lymphatic vessels.
Inflammation is a primary driver of lymphedema (19), so we sought to test whether inflammatory cytokines could impair drainage in our system. We tested nine inflammatory cytokines that have been reported to be involved in lymphedema progression in patients or in animal models, including IL-2, thrombin, histamine, IL-10, IL-4, IL-6, TNF-a, GM-CSF, and G-CSF (20). Among them, IL-2, GM-CSF, and G-CSF each markedly decreased lymphatic drainage (Fig. 1J and SI Appendix, Fig. S2) and tightened lymphatic AJs (Fig. 1K), forming more zipper junctions (Fig. 1L).
ROCK Inhibition Normalizes LEC and BEC Junctions and Reverses Lymphedema on-chip and In Vivo.
To begin to characterize what downstream signaling pathways might be involved in tightening the lymphatic junctions, we screened inhibitors that might impact IL-2-induced junction assembly (SI Appendix, Fig. S3): PP2, an Src family of protein tyrosine kinase inhibitor; Wortmannin, a PI3K inhibitor; Y27632, a ROCK inhibitor; GGTI298, a Rap1 inhibitor; NSC23766, a Rac1 inhibitor; Blebbistatin, a Myosin II inhibitor; and Cpd22, an integrin-linked kinase inhibitor. Only the ROCK inhibitor, Y27632, completely rescued the impaired drainage in IL-2 treated lymphatic vessels by normalizing zippered LEC junctions (SI Appendix, Fig. S3 and Fig. 2 A and B). Y27632 also normalized GM-CSF and G-CSF-treated conditions (Fig. 2C and SI Appendix, Fig. S4), and clinically used ROCK inhibitors Fasudil and Ripasudil (21) similarly improved lymphatic drainage (Fig. 2D). Taken together, these data suggest a role for ROCK in inflammatory modulation of lymphatic junctions and drainage.
Fig. 2.

ROCK inhibition normalizes LEC and BEC junctions and reverses lymphedema. (A) VE-cadherin staining of engineered lymphatics exposed to IL-2 with or without Y27632. (B) Percent zipper junction in engineered lymphatics exposed to IL-2 with or without Y27632 (n = 8, N = 3). (C) Percent lymphatic drainage with Y27632 (BODIPY-C16 fatty acid drainage) (N = 4). (D) Percent lymphatic drainage with Y27632, Fasudil, and Ripasudil (BODIPY-C16 fatty acid drainage) (N = 5). (E) Dye-leakage-based permeability assays and VE-cadherin images of engineered blood vessels. (F) The permeability coefficient of the engineered blood vessels in inflammation with or without Y27632 (N = 3). (G) Effects of ROCK inhibition on LEC and BEC junctions. (H) Tail volume change with Y27632 in a murine lymphedema model (N = 5). (I) Tail volume change with Y27632 in the lymphedema model when swelling is peaked in the control group (N = 5). (J) Representative images of tails with Y27632 treatment. [Scale bars (A and E, Lower), 50 μm; Scale bars (E, Upper), 200 μm; Scale bars (J), 5 mm.] *P < 0.05 indicates statistical significance.
Interestingly, inflammatory cytokines have the opposite effects on capillary blood vessels, increasing vessel permeability by loosening cell–cell junctions (22), which can also contribute to lymphedema. Human dermal BECs were seeded in our one-channel devices and cultured under luminal shear flow for 2 d as described previously (23). Fluorescent dextran was introduced into the reservoirs. Diffusion of the dextran to the ECM was imaged, and the resulting diffusion profile was fitted to a dynamic mass conservation equation with the diffusive permeability coefficient (Pd) defined by the following equation: J = Pd (Cvessel – CECM), where J is the mass flux of dextran, Cvessel is the concentration of dextran in the vessel, and CECM is the concentration of dextran in the perivascular extracellular matrix (ECM). Indeed, in our engineered BEC-lined vessels, IL-2, GM-CSF, and G-CSF all disrupted vascular barrier function (Fig. 2 E and F). Here, treatment with Y27632 rescued barrier function (Fig. 2F), suggesting that ROCK inhibition might ameliorate lymphedema by simultaneously reducing fluid leakage from blood vessels and increasing lymphatic drainage (Fig. 2G).
Previous studies have reported ROCK to modulate lymphatic function, though they have shown different effects in different experimental models (24–26). In the small intestine, pan-ROCK inhibitor treatment tightened LEC junctions and prevented chylomicron uptake in wild-type mice (24). In the eyes, however, pan-ROCK inhibitor treatment loosened LEC junctions and increased aqueous humor clearance through the Schlemm’s canal, the ocular lymphatic endothelium in a glaucoma condition (25, 26). In this study, we found that pan-ROCK inhibition loosened dermal LEC junctions but tightened dermal BEC junctions in a condition of inflammation and lymphedema (Fig. 2G). To investigate the potential efficacy of ROCK inhibition in secondary lymphedema, we tested Y27632 in an established mouse model of a surgically induced, secondary lymphedema (27, 28). Lymphedema was induced by surgical excision of peripheral lymphatics in mouse tails. Swelling in tails was observed in both control and Y27632 treated groups after the surgical procedure for several days. However, while control animals continued to increase swelling for nearly 3 wk, Y27632-treated mice by day 10 began to show significantly reduced swelling (Fig. 2H). At the peak of lymphedema in the control group (day 19), the Y27632-treated group showed a 55% reduced tail volume compared to the control group (Fig. 2 I and J).
ROCK2 Forms a Unique Tight Junction Complex in Inflamed LECs, but Not in BECs.
While these results suggested a promising avenue for treating lymphedema, it remained unclear how ROCK activity could assemble LEC junctions while having the opposite effect on blood endothelial cell (BEC) junctions, or how pan-ROCK inhibition would have different effects in different settings of lymphatic dysfunction. We postulated that this difference might arise from different functions of the two isoforms of ROCK, ROCK1, and ROCK2 (29). Under basal conditions, ROCK1 was detected at comparable levels in LECs and BECs, whereas ROCK2 was expressed in LECs but was nearly undetectable in BECs (Fig. 3A). Consistent with our findings in human cells, studies of isolated murine LECs and BECs showed that ROCK2 was highly expressed in LECs compared to BECs (30). Interestingly, IL-2 in LECs substantially down-regulated ROCK1, leaving ROCK2 as the dominant isoform (Fig. 3B). To test whether changes in ROCK1/2 levels might alter LEC junctions and drainage, we examined the effects of siRNA-mediated knock-down of ROCK1/2 (Fig. 3C). Indeed, knocking down ROCK1 decreased drainage in LECs, phenocopying the effects of IL-2 exposure, while loss of ROCK2 phenocopied Y27632, maintaining drainage regardless of IL-2 (Fig. 3D). These striking differences between ROCK1 and ROCK2 were also reflected by the phenotype of adherens junctions (Fig. 3 E and F). Loss of ROCK1 tightened junctions while loss of ROCK2 loosened junctions in LECs. Given the prominent expression of ROCK1 in BECs, we examined the effects of silencing ROCK1 also in these cells. As before, control BECs showed high barrier function (low permeability, smaller Pd values) in basal conditions that is lost with exposure to IL-2 (Fig. 3 G, Left). Consistent with the effects in LECs, knocking down ROCK1 allowed BECs to maintain high barrier function (Fig. 3 G, Right) and intact junctions (SI Appendix, Fig. S5). Together, these data suggest that ROCK1/2 isoforms differentially regulate junctional stability in LECs and BECs. Whereas ROCK1 disrupts or loosens junctions, ROCK2 is required for cytokine-induced junction tightening in LECs, and inhibition of ROCK2 normalizes these junctions to support lymphatic drainage.
Fig. 3.
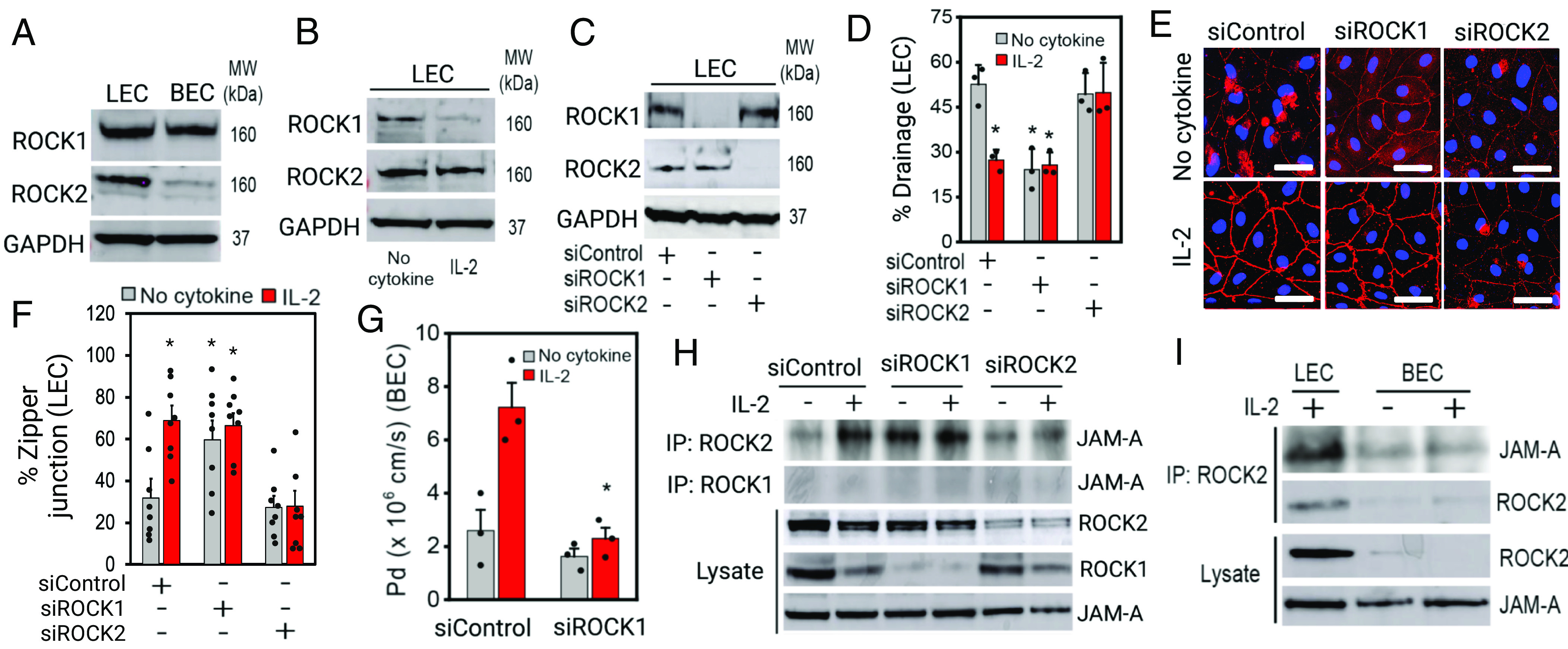
ROCK2 forms a unique tight junction complex in inflamed LECs, but not in BECs. (A) Western blot showing basal levels of ROCK1/2 expression in LECs and BECs. (B) Western blot showing ROCK1 downregulation in inflamed LECs. (C) siRNA-mediated knock-down of ROCK1 and ROCK2 in LECs. (D) Percent lymphatic drainage by LEC knocked-down ROCK1 or ROCK2 in normal or IL-2 condition (BODIPY-C16 fatty acid drainage) (N = 3). (E) VE-cadherin images of the engineered lymphatics with or without ROCK1/2 knock-down in normal or IL-2 condition. (F) Percent zipper junction in engineered lymphatics with or without ROCK1/2 knock-down in normal or IL-2 condition (n = 8, N = 3). (G) Permeability coefficient (Pd) of the engineered blood vessels with or without ROCK1 knock-down in normal or IL-2 condition (N = 3). (H) Immunoprecipitation data showing ROCK2 interactions with junctional adhesion molecule-A (JAM-A) in inflamed LECs or in ROCK1 kd LECs. (I) Immunoprecipitation data showing that ROCK2-down-regulating BEC does not form ROCK2/JAM-A protein complex in normal or IL-2 condition. [Scale bars (E), 50 μm.] *P < 0.05 indicates statistical significance.
Although the role of ROCK1 in disrupting BEC junctions has been investigated (31), how ROCK2 might support junction assembly in LECs remains unknown. By isolating endogenous ROCK2 via immunoprecipitation, we identified an interaction with JAM-A that emerges in cytokine-exposed LECs (Fig. 3H). ROCK1 was not in the junction complex in any conditions, but knockdown of ROCK1 induced ROCK2/JAM-A complex assembly, phenocopying the effects of IL-2. In contrast, ROCK2-silenced LECs interfered with the formation of the ROCK2/JAM-A complex (Fig. 3H). However, these ROCK2/JAM-A complexes were not observed in BECs (Fig. 3I). Taken together, ROCK2 associates with JAM-A and stabilizes lymphatic junctions when inflammatory signals are present.
Inhibition of ROCK2 Reverses Lymphedema In Vivo.
To test whether ROCK2 alone might be critical for the development of lymphedema in vivo, LEC-specific inducible ROCK2 knockout (LEC-iROCK2KO) mice were generated by crossing ROCK2 flox/flox mouse with Cre deleter mouse strain Prox 1 CreERT2 and injecting 4-hydroxytamoxifen into those mice to induce ROCK2 depletion prior to utilization, wherein 4-hydroxytamoxifen injection into littermate mice lacking Cre or into WT mice with Cre served as controls. Lymphedema was induced by surgical ligation of peripheral lymphatics in tails of control and LEC-iROCK2KO mice. Tail edema was observed in both WT control and LEC-iROCK2KO groups in the first 1 to 2 wk following injury, after which edema began to resolve in the LEC-iROCK2KO group while the control group continued to worsen (Fig. 4A). At peak edema in control animals (day 28), the LEC-iROCK2KO group showed a 75% reduction in tail swelling (Fig. 4 B and C). Taken together, LEC-specific ROCK2 knockout reverses lymphedema in vivo.
Fig. 4.
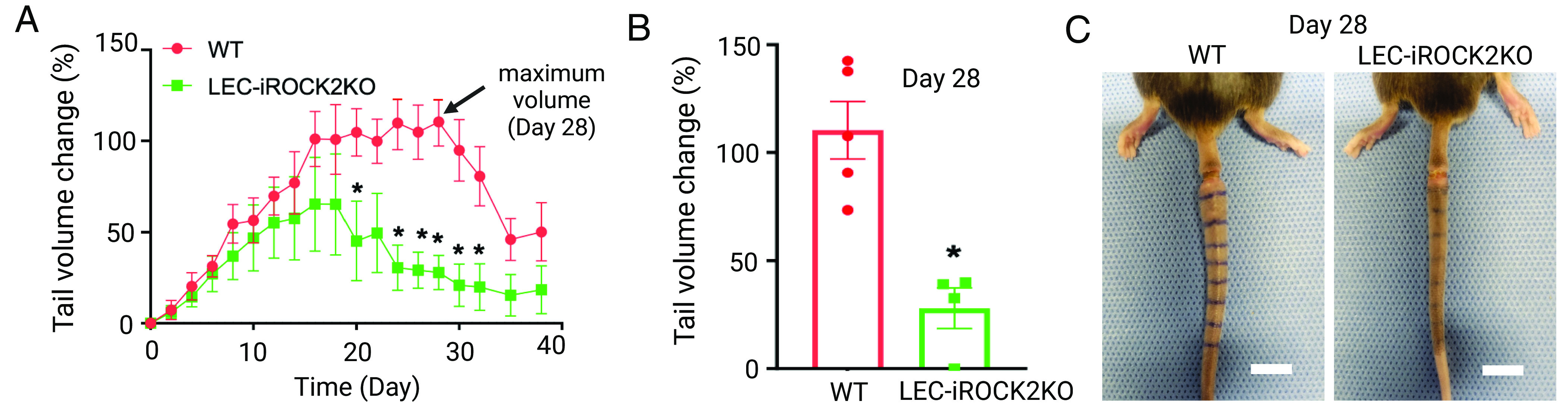
Inhibition of lymphatic endothelial ROCK2 reverses lymphedema in vivo. (A) Tail volume change in WT or LEC-iROCK2KO mice after having tail lymphedema procedure (N = 4 to 5). (B) Tail volume changes when swelling is peaked in the control group (day 28) (N = 4 to 5). (C) Representative images of tails after lymphedema surgery in WT and LEC-iROCK2KO mice at day 28. [Scale bars (C), 5 mm.] *P < 0.05 indicates statistical significance.
Discussion
Rho-associated protein kinase 1 (ROCK1) and ROCK2 are two isoforms of ROCK that are ubiquitously expressed in mammals and are downstream targets of small GTPases RhoA, RhoB, and RhoC (22, 29). ROCK1 has been extensively studied, as a regulator of cell contractility through the formation of stress fibers and focal adhesions by phosphorylating myosin light chain 2 (MLC2) (29, 31). In BECs, VEGFR2 activates this ROCK1-MLC2 pathway to destabilize BEC junctions (32, 33). In contrast, less is known about ROCK2, although it has largely been assumed to play a redundant role to ROCK1. In this study, using a 3D organotypic model of lymphatic vessels that emulates lymphatic junctional structure and drainage function, we revealed that ROCK1 and ROCK2 appear to have opposing effects: In normal LECs, the presence of ROCK1 appears to allow lymphatic drainage by preventing the formation of a ROCK2/JAM-A protein complex, while inflammatory cytokine-mediated ROCK1 downregulation disinhibits the formation of this complex leading to junction tightening and reduced lymphatic drainage. Indeed, direct knockdown of ROCK1 in LECs induces junction zippering and reduced drainage directly, phenocopying cytokine exposure (Fig. 3). Indeed, one study has shown that the CCM complex in BECs can selectively scaffold ROCK2 at junctions and that ROCK1 and ROCK2 acted differentially on endothelial barrier function (34). Given the opposing roles of ROCK1 and ROCK2 in junctional regulation, it is not surprising that nonspecific, pan-ROCK inhibitors might have differential effects on lymphatics in different contexts, perhaps as a result of the relative importance of ROCK1 versus ROCK2 in different organs and experimental models (24–26). Regardless, pan-ROCK inhibitors are of limited clinical utility due to severe hypotension resulting from loss of ROCK1-mediated vascular smooth muscle tone (21). Here, our studies suggest that specific targeting of ROCK2 could be a more viable path to treat lymphedema with reduced safety concerns. One ROCK2 inhibitor has recently been approved for human use in graft-vs-host disease (belumosudil) (35), suggesting a potential avenue forward.
This study also highlights a previously unappreciated ROCK2/ JAM-A protein complex that regulates lymphatic cell–cell junctions and the passage of fluid back into the lymphatic circulation. Further characterizing the tissue-specific mechanisms that LECs use to dynamically regulate these adhesions will be essential to normalizing perfusion. Last, the work underscores how recapitulating appropriate architectures can enable the emergence of multicellular functions previously only observed in vivo, and the advancement of such biomimetic cultures is proving to play a key role in bridging traditional in vitro and in vivo models of physiology and disease.
Materials and Methods
Study Design.
All in vitro experiments were performed using chemicals and materials from reputable manufacturers and according to published protocols. For the in vivo experiments, power analyses were performed to estimate the minimum animal group size based on the results observed in preliminary studies (36–38) (α = 0.05, P = 0.8). We used both male and female animals considering sex as a potential biological variable. Animals were randomly assigned into control and treatment groups and experimenters were double-blinded. In the murine lymphedema model, we stopped our data collection once after the control group showed the maximum tail volume. All experimental sample numbers (N) were indicated in the figure legends.
Cell Culture.
Primary human dermal LECs were gifted from Young Kwon Hong (USC) (9). LECs were isolated from dermal tissues of neonatal donors. Primary human dermal BECs were purchased from Lonza (CC-2813, HMVEC-dBlNeo, Lonza). BECs were derived from dermal tissues of neonatal donors. Both endothelial cell types were originated from the same tissue type (“skin”) of “neonatal” donors. LECs were cultured in EBM media (Lonza, CC-3121), containing 15% fetal bovine serum (Sigma), 2 mM L-glutamine (Sigma, G7513), 10 μg/mL hydrocortisone acetate (Sigma, H0396), 2.5 × 10−2 mg/mL N-6,2′-O-dibutyryl-adenosine 3′,5′-cyclic monophosphate (Sigma, D0627), and 50 μg/mL Gentamycin (Life Technologies). BECs were cultured in EGM-2MV media (Lonza, CC-3156 & CC-3202). All the cells were used at passages 3-6 and maintained in standard tissue culture incubators at 37 °C, 95% humidity, and 5% CO2.
Microfluidics.
Microfluidic devices (SI Appendix, Fig. S1) were fabricated using soft lithography. The lymphatic drainage-on-chip device consists of a PDMS housing and two parallel cylindrical microchannels within the 3D collagen hydrogel. PDMS (Sylgard 184, Dow-Corning, DC2065622) was mixed with a curing agent, provided in the Sylgard 184 PDMS kit, at 10:1 weight ratio (base:curing agent) and cured overnight at 60 °C on a silicon master. The PDMS was removed from the silicon master, trimmed, and surface activated by plasma etching for PDMS bonding to a cover glass (22 × 40 mm, Fisher, 12-545-C). Four reservoir walls were embedded in the PDMS (not in collagen 1) by making four holes through the PDMS device using a biopsy punch (with a 6-mm diameter). Bonding of the PDMS to the glass was followed by curing at 60 °C overnight for permanent bonding. The devices were plasma etched, treated with 0.01% poly-L-lysine (Sigma, P8920) for 1 h, followed by 1% glutaraldehyde (Electron Microscopy Sciences, 16310) for 30 min and then rinsed three times with sterile water, and incubated overnight in sterile water at room temperature. The next day, the devices were sterilized with 70% ethanol for 30 min. Steel acupuncture needles (0.25 × 50 mm, Hawato, HS 25 × 50) were sterilized with 70% ethanol and BSA-coated (1 mg/mL in PBS), then introduced into the devices as a scaffold of a “casting method.” The needle-inserted devices were air-gun dried and UV sterilized for 30 min. Collagen type I (Corning, 356236) was buffered with PBS, titrated to a pH of 8.0 with NaOH (Sigma), yielding a final concentration of 2.5 mg/mL, that was pipetted into the microfluidic devices and polymerized for 50 min at 37 °C. Cell growth medium was then added to the devices overnight, and needles were carefully removed to create 250 μm diameter channels in the collagen gel. After overnight incubation with cell growth medium, the devices were seeded with human dermal LECs. Briefly, enzymatically dislodged (0.05% Trypsin/EDTA) LECs were resuspended at 2 × 106 cells/mL in LEC media, and 100 μL of cell suspension was introduced into the right-side channel of the device via the media reservoirs that are connected to the right-side channel, allowing cells to adhere to the collagen matrix for 10 min before washing with growth medium. The devices were incubated for 2 d on a rocking platform in a humidified tissue culture incubator (5% CO2, 37 °C), replenishing culture media daily.
Lymphatic Drainage Assay.
To measure lymphatic drainage in the microfluidic devices, fluorescently labeled lymph molecules were introduced into the devices via the media reservoirs that are connected to the empty (left-sided) channel. The lymph molecules include BODIPY® FL C16 (Thermo Fisher, D3821), 2 μg/mL in LEC media; BODIPY® FL C12 (Thermo Fisher, D3822), 2 μg/mL; β-BODIPY® FL C5-HPC (Thermo Fisher, D3803), 2 μg/mL; FluoSpheres® Carboxylate-Modified Microspheres, 0.2 µm, red fluorescent (580/605), 2% solids (Thermo Fisher, F-8810), 0.004% solid; Bovine Serum Albumin (BSA)-FITC conjugate (Thermo Fisher, A23015), 250 μg/mL, and fluid only (LEC media). Having 50 μL of lymph fluid in both channels with equal volumes (25 μL: left, 25 μL: right), we additionally loaded excess lymph fluid in the left-side of channel, making 7 mm fluid height difference between two channels triggered interstitial fluid transport with an initial flow rate of 1 µm/s. To have the 1 μm/s flow rate, we had two channels 7 mm apart and made the height difference of lymph fluid between two channels 7 mm, so the initial flow rate was calculated to be 1 μm/s. This initial flow rate was continuously reduced during the experiment as the height difference of fluid diminished. During the drainage, the devices were put on the rocking platform to maintain luminal shear flow in the channels. After 15 h of drainage, fluid in the left-side reservoirs and the right-side reservoirs was gathered, and the fluid volumes and the concentrations of fluorescently labeled lymph molecules in the fluid were measured to quantify the number of remained and drained lymph molecules. As control, we kept the right-side channel empty without seeding cells and quantified the drainage as 100% drainage of lymph molecules through the “No cell” barrier. All the LEC or BEC drainage data were normalized by this 100% drainage. To emulate inflammation, 50 ng/mL of IL-2 (Peprotech, 200-02), GM-CSF (Peprotech, 300-03), and G-CSF (Peprotech, 300-23) were included in the lymph fluid. For drug testing, PP2 (Tocris, 1407, 10 μM), Cpd22 (EMD Millipore, 407331, 1 μM), Wortmannin (Tocris, 1232, 30 μM), Y27632 (Sigma, 688000, 10 μM), human recombinant VEGFC (R&D systems, 100 ng/mL), GGTI298 (Sigma, G5169, 10 μM), NSC23766 (Sigma, SML0952, 100 μM), Ripasudil (Selleckchem, S7995, 10 μM), Fasudil (Sigma, CDS021620, 10 μM), and Blebbistatin (Abcam, ab120425, 10 μM) were included in the lymph fluid. The concentration of the inhibitors was optimized for cell survival and assay based on previous literature.
Blood Vascular Permeability Assay.
Blood vascular permeability in microfluidic devices was measured as described previously (23). Briefly, one-channel devices were fabricated using a soft lithography method. Primary human dermal BECs were seeded in the devices and cultured under luminal shear flow of 4.5 dyne/cm2 for 2 d. Fluorescent dextran (500 kDa, FITC, Life Technologies, D7136) was introduced into perfusion media at a concentration of 25 μg/mL, and diffusion of the dextran was imaged in real time with a Yokogawa CSU-21/Zeiss Axiovert 200M inverted spinning disk microscope with a 10× objective and an Evolve EMCCD camera (Photometrics). Time-lapse microscopy was used to measure the flux of dextran into the collagen gel, and the resulting diffusion profile was fitted to a dynamic mass conservation equation as previously described (23) with the diffusive permeability coefficient (PD) defined by the following equation: J = PD (Cvessel − CECM), where J is the mass flux of dextran, Cvessel is the concentration of dextran in the vessel, and CECM is the concentration of dextran in the perivascular ECM (23).
Cell Imaging in 3D On-chip Models.
For live imaging, a lymphatic drainage-on-chip device was transferred from the tissue culture incubator to a temperature and CO2 equilibrated environmental microscope chamber, and BODIPY® FL C16 (Thermo Fisher, D3821) lymph fluid was introduced to the left channel of the device. The right channel decorated with LECs or BECs was imaged every minute for 2 h on the Nikon Ti Eclipse epifluorescence microscope with a Yokogawa CSU-21/Zeiss Axiovert 200M inverted spinning disk microscope with a Zeiss LD C-Apochromat 40×/1.1 NA water-immersion objective and an Evolve EMC CCD camera (Photometrics). With the obtained images, we performed the maximum z-projections and adjusted for brightness and contrast using ImageJ (39). The finalized time-lapse images were consecutively opened and saved as one video file using ImageJ (39). For immunofluorescent staining and imaging, LECs or BECs embedded in the 3D collagen bulk of the device were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, 15710) in EBM2 (Lonza, CC-3156) for 1 h at room temperature. Fixed devices were permeated with PBST (0.3% Triton-X in PBS) for 45 min at room temperature and then blocked with 3% BSA in PBS overnight at 4 °C. Primary antibodies detecting VE-cadherin (Abcam, ab33168, 1:400), CD31 (DAKO, JC70A, 1:200), JAM-A (Santa Cruz, 1H2A9, sc-53624, 1:100), LYVE-1 (Abcam, ab36993, 1:100), Podoplanin (Abcam, ab10288, 1:100), and ROCK2 (EMD Millipore, ABS436, 1:100) were incubated in blocking buffer overnight at 4 °C. After incubation with primary antibodies, the samples were washed for 24 h in PBS at 4 °C. Secondary antibodies (all from Life Technologies, 1:500), Phalloidin (Life Technologies, A22287, 1:200), and DAPI (Sigma, 1:500) were subsequently incubated in blocking buffer overnight at 4 °C in dark. The secondary antibodies, phalloidin, and DAPI were washed for 24 h in PBS at 4 °C in the dark to remove the fluorescent background before confocal microscopy. Confocal images were acquired with either an Axiovert 200M inverted microscope (Zeiss) equipped with a CSU10 spinning disk confocal scan head (Yokogawa Electric Corporation) and an Evolve EMCCD camera (Photometrics); or a Leica HC FLUOTAR L 40×/0.95 water-immersion objective on an upright TCS SP8 multiphoton microscope (Leica); or an inverted Leica SP8 confocal microscope. With the obtained images, we performed the maximum z-projections and adjusted for brightness and contrast using ImageJ (39).
Quantification of VE-Cadherin Junction Type.
We quantified % of zipper junctions by using the “Segmented line” tool in ImageJ as described previously (40, 41). Briefly Segmented line tool draws lines over VE-Cadherin+ junctions and measures their length using the “Measure” function. Eight different fields of view of three different devices were analyzed per group (n = 8, N = 3 experiments). Types of junctions (buttons vs. zippers) have been defined by the measured length of junctions: the length of zipper junction >7.85 μm; buttons and others (e.g., intermediate) <7.85 μm in length (40). We normalized zipper junctions as a percentage of the total length, and the % of zipper junctions was determined.
Mouse Line Generation.
ROCK2 floxed mouse was purchased from Taconic (model # 12905, C57BL/6-ROCK2 tm1.1 Mrl). LEC-specific inducible ROCK2 knockout (LEC-iROCK2KO) mice were generated by crossing ROCK2 fl/fl mouse with Cre Deleter mouse strain Prox 1 CreERT2 (37, 38). Inducible LEC-specific ROCK2 KO mice were obtained by injection of 4-hydroxytamoxifen (10 mg/kg, i.p.) seven times every other day as previously described (37, 38), along with injection into littermate mice lacking Cre or WT mice with Cre to serve as a control. Both age, sex, and genetic background-matched male and female mice were used for the analyses.
Mouse Tail Lymphedema Model.
For the murine tail lymphedema model, C57/BL6 WT or LEC-iROCK2KO mice were anesthetized with 3% isoflurane. To sever the dermal lymphatic vessels of the tail, a circumferential incision was made through the dermis at 1 cm down from the tail base using a stainless-steel surgical blade (Feather, Japan, #5005093). The edges of this incision were pushed apart with a cauterizing iron to the deeper lymphatic vessels while preventing superficial bleeding and creating a 3 mm gap to delay wound closure (36). A thin layer of triple antibiotic ointment (Actavis, #1851583), containing Bacitracin zinc 400 units; Neomycin 3.5 mg; Polymyxin B sulfate 5,000 units, was administrated to cover the circumferential incision using a sterile swab to prevent infections. After the surgical procedure, Loxicom (meloxicam, 5 mg/kg) was administered subcutaneously to relieve pain. Volume measurements of the tails were taken every 2 d by measuring the circumference of the tail from the tip to the distal edge of the wound using a digital caliper. For tail measurement, the animal was anesthetized in 3% isoflurane without restraining the animals. For pharmacological inhibition of ROCK signaling, pan-ROCK inhibitor Y27632 (Sigma, 688000, 10 mg/kg/day, via i.p. injection; 100 μL per injection, N = 5) was administered at day 3 after incision, as it has been previously established that after lymphatic ablation, lymphangiogenesis occurs during the first 3 d after surgery; the degree to which this occurs may serve to limit the extent of lymphedema development (36). 0.9% sodium chloride injection was done as a vehicle treatment (N = 5). All animal procedures were approved by IACUC and performed in a sterile environment of a laminar flow cabinet housed in the animal facility at Boston Children’s Hospital, Harvard Medical School, and Cornell University.
siRNA-Based Knock-Down.
Knock-down of ROCK1, ROCK2, and JAM-A in LECs and BECs was performed with reagents purchased from Dharmacon. The reagents include SMARTpool: ON-TARGETplus ROCK1 siRNA (L-003536-00-0005); SMARTpool: ON-TARGETplus ROCK2 siRNA (L-004610-00-0005); SMARTpool: ON-TARGETplus F11R (JAM-A) siRNA (L-005053-00-0005); ON-TARGETplus Nontargeting siRNA #2 (D-001810-02-05); DharmaFECT 1 Transfection Reagent (T-2001); and 5× siRNA buffer (B-002000-UB-100). Knock-down was achieved using the manufacturer’s procedures. Briefly, we prepared 1× siRNA buffer via 1:5 dilution of 5× siRNA buffer in RNAse-free water (Qiagen, 129112). Then, we made 5 μM siRNA stock solution in 1× siRNA buffer for storage at −20 °C. The 5 μM siRNA stock solution was further diluted (1:20 dilution ratio) with the siRNA Opti-MEM™ I Reduced Serum Medium (Gibco, 31985062) to result in 250 nM siRNA solution, followed by 5 min incubation at room temperature. DharmaFECT 1 Transfection Reagent was diluted (1:25 dilution ratio) in the siRNA Opti-MEM™ I Reduced Serum Medium and incubated for 5 min at room temperature. 250 nM siRNA solution and the diluted DharmaFECT 1 solution were mixed together and incubated for 15 min at room temperature. The siRNA/DharmaFECT mixture was finally diluted in complete LEC media or BEC media to make a final concentration of 25 nM siRNA solution. Approximately 40 to 50% confluent LECs or BECs plated on 50 μg/mL collagen I (Corning, 356236) coated six-well plates were treated with 2 mL of 25 μM siRNA solution per well. After incubation in standard tissue culture incubators for 24 h, the siRNA solution was replaced with fresh LEC or BEC media. After 3 d after transfection, we used the knock-down or scrambled control LECs or BECs for cell-based experiments. Knock-down of ROCK1, ROCK2, and JAM-A was confirmed with western blotting.
Western Blot Analysis.
Confluent LECs or BECs plated on 50 μg/mL collagen I (Corning, 356236) were rinsed with cold EBM2 (Lonza, CC-3156) three times, then lysed in cold Pierce™ IP Lysis Buffer (Thermo Fisher, 87788) with 2× protease and phosphatase inhibitors: PhosSTOP™ (Sigma, 4906845001), cOmplete™ ULTRA Tablets, Mini, EASYpack Protease Inhibitor Cocktail (Sigma, 5892970001). Lysates were drawn through a 21G syringe 15 times and incubated on ice for 20 min before clarification at 14,000 × g for 30 min at 4 °C. Clarified lysates were equalized for protein content using Quick StartTM Bradford Protein Assay (BioRad, 5000201). The lysate was mixed with 1× NuPAGE™ LDS Sample Buffer (Life Technologies, NP0007) and 3% 2-Mercaptoethanol (Sigma, M6250) and then boiled at 95 °C for 8 min. The lysate proteins were fractionated by SDS-PAGE using NUPAGE 4-12% Bis-tris gels (Life Technologies, NP0335), 1× NUPAGE MOPS SDS running buffer (Life Technologies, NP0001) in the Mini Gel Tank (Life Technologies, A25977) at 150 V. The fractionated proteins in the Bis-tris gel were transferred to a nitrocellulose membrane using Trans-Blot® Turbo™ Mini Nitrocellulose Transfer Packs (BioRad, 1704158EDU) and the Trans-Blot® Turbo™ Transfer Starter System (BioRad, 170-4155). After protein transfer, the nitrocellulose membrane was rinsed with 1× TBST (Cell Signaling, 9997) and blocked with 5% Nonfat dry milk (Cell Signaling, 9999S) and 2% BSA (Sigma, A4503) in 1× TBST for 1 h at room temperature. Primary antibodies (diluted in 5% Nonfat dry milk and 2% BSA) and secondary antibodies (diluted in 5% Nonfat dry milk) include anti-ROCK2 antibody (R&D systems, AF4790, 1:200), anti-ROCK1 antibody (R&D systems, MAB4590, 1:500), anti-GAPDH antibody (Cell Signaling, 5174s, 1:2,000), anti-JAM-A antibody (Santa Cruz, 1H2A9, sc-53624, 1:200), HRP-linked anti-rabbit IgG antibody (Cell Signaling, 7074, 1:2,000), and HRP-link anti-goat IgG antibody (Santa Cruz, sc-2354, 1:2,000), and HRP-linked anti-mouse IgG antibody (Cell Signaling, 7076, 1:2,000). The protein bands were visualized by 4 min membrane exposure to Super Signal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher, 34580), and the chemiluminescent HRP was detected by the Amersham Imager 600 (CCD-based, GE Healthcare, #29083461). The western blot images were adjusted for brightness and contrast using ImageJ (39).
Immunoprecipitation.
Confluent LECs or BECs cultured on 50 μg/mL collagen I (Corning, 356236) were rinsed with cold EBM2 (Lonza, CC-3156) three times and then lysed with cold Pierce™ IP Lysis Buffer (Thermo Fisher, 87788) with 2× protease and phosphatase inhibitors: PhosSTOP™ (Sigma, 4906845001) and complete™ ULTRA Tablets, Mini, EASYpack Protease Inhibitor Cocktail (Sigma, 5892970001). Lysates were drawn through a 21G syringe 15 times and incubated on ice for 20 min before clarification at 14,000 × g for 30 min at 4 °C. Clarified lysates were equalized for protein content and volume and incubated with 3 μg of anti-ROCK2 antibody (Santa Cruz, D-11, sc-398519), 3 μg of anti-ROCK1 antibody (Santa Cruz, G-6, sc-17794), and 3 μg of anti-JAM-A antibody (Santa Cruz, 1H2A9, sc-53624) for 2 h at 4 °C with end-over-end rotation. Antibody complexes were purified with 50 μL Protein A/G beads (Santa Cruz, sc-2003) per microcentrifuge tube for 2 h at 4 °C with end-over-end rotation. Bead pellets were washed three times with 800 μL cold lysis buffer including 2× protease inhibitors by gentle pipetting with a 1000T pipette and spinning for 60 s at 2,000 × g at 4 °C. Protein complexes were extracted in 2× NUPAGE® LDS Sample Buffer (Life Technologies, NP0007) containing 3% 2-Mercaptoethanol (Sigma, M6250) and analyzed by SDS-PAGE and immunoblotting with chemiluminescent HRP detection. Details of the running and probing western blots are described in the western blot section.
Statistics.
Independent two-sample populations were compared using unpaired, two-sample t tests with a normal distribution assumption. For group analyses, one-way ANOVAs with Tukey’s HSD (Honestly Significant Difference) tests were used to compare the mean values. All P values were two-sided, and *P < 0.05 was considered statistically significant. Statistical analyses were performed using Microsoft Excel. All data points on the graphs represent average values, and error bars depict the SEM.
Supplementary Material
Appendix 01 (PDF)
A time-lapse movie of active response of an engineered LV and BV under interstitial flow. A 2-hour time-lapse movie shows active responses of the engineered LV and BV to interstitial flow of BODIPY-C16 (pseudo red-colored). BODIPY-C16 entered the luminal space of the engineered LV faster than that of the engineered BV, indicating faster fluid drainage in LVs than BVs. The engineered LV was relaxed under interstitial flow, displaying an enlarging lumen (bright field). By contrast, the engineered BV was squeezed by the interstitial flow.
Acknowledgments
This work was supported in part by funding from the NIH (EB025765; EB000262; HL133216; and HL141858), the NSF (CMMI-1548571; EEC-1647837), and the Wellcome Leap HOPE program (Human Organs Physiology and Engineering). E.L. was supported by the Lymphatic Education and Research Network and the National Center for Advancing Translational Sciences at the NIH (TL1TR001410). E.L., A.M.K., and T.J.K. were supported by the NIH (HL165135; CA279560; and AI166772). W.J.P. was supported by the NIH (Ruth L. Kirchstein National Research Service Award F32 HL129733; and Organ Design and Engineering Training program T32 EB16652). M.L.K. was supported by the NIH (K99-CA226366-01A1). We thank Young Kwon Hong for providing human dermal lymphatic endothelial cells (LECs). We thank Scott Wong for generating and phenotyping LEC-specific ROCK2 knock-out mice. We thank Anne Eichmann and Peter Baluk for their scientific discussions. This work was performed in part at the Cornell NanoScale Facility, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the NSF (Grant NNCI-2025233), and Cornell Center for Materials Research (grant number: DMR-1719875).
Author contributions
E.L. and C.S.C. designed research; E.L., S.-L.C., Y.L., W.J.P., S.K., A.W., D.-H.T.N., M.L.K., S.A., A.M.K., T.J.K., J.E., D.R.B., H.C., and C.S.C. performed research; E.L., S.-L.C., Y.L., W.J.P., S.K., A.W., D.-H.T.N., M.L.K., S.A., A.M.K., T.J.K., J.E., D.R.B., H.C., and C.S.C. analyzed data; and E.L. and C.S.C. wrote the paper.
Competing interests
C.S.C. is a founder and owns shares of Innolign Biomedical, a company that is developing engineered organ models for pharmaceutical research and development, and Satellite Biosciences, a company that is developing cell-based therapies.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Esak Lee, Email: el767@cornell.edu.
Christopher S. Chen, Email: chencs@bu.edu.
Data, Materials, and Software Availability
All study data are included in the article and/or supporting information.
Supporting Information
References
- 1.Baluk P., et al. , Functionally specialized junctions between endothelial cells of lymphatic vessels. J. Exp. Med. 204, 2349–2362 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Triacca V., Guc E., Kilarski W. W., Pisano M., Swartz M. A., Transcellular pathways in lymphatic endothelial cells regulate changes in solute transport by fluid stress. Circ. Res. 120, 1440–1452 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Tammela T., Alitalo K., Lymphangiogenesis: Molecular mechanisms and future promise. Cell 140, 460–476 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Oliver G., Kipnis J., Randolph G. J., Harvey N. L., The lymphatic vasculature in the 21(st) century: Novel functional roles in homeostasis and disease. Cell 182, 270–296 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rockson S. G., Update on the biology and treatment of lymphedema. Curr. Treat Options Cardiovasc. Med. 14, 184–192 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Schwartz M. A., Chen C. S., Cell biology. Deconstructing dimensionality. Science 339, 402–404 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Kim S., Chung M., Jeon N. L., Three-dimensional biomimetic model to reconstitute sprouting lymphangiogenesis in vitro. Biomaterials 78, 115–128 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Osaki T., Serrano J. C., Kamm R. D., Cooperative effects of vascular angiogenesis and lymphangiogenesis. Regen. Eng. Transl. Med. 4, 120–132 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choi D., et al. , Laminar flow downregulates Notch activity to promote lymphatic sprouting. J. Clin. Invest. 127, 1225–1240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bruyere F., et al. , Modeling lymphangiogenesis in a three-dimensional culture system. Nat. Methods 5, 431–437 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Gibot L., et al. , Cell-based approach for 3D reconstruction of lymphatic capillaries in vitro reveals distinct functions of HGF and VEGF-C in lymphangiogenesis. Biomaterials 78, 129–139 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Gong M. M., et al. , Human organotypic lymphatic vessel model elucidates microenvironment-dependent signaling and barrier function. Biomaterials 214, 119225 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henderson A. R., Ilan I. S., Lee E., A bioengineered lymphatic vessel model for studying lymphatic endothelial cell-cell junction and barrier function. Microcirculation 28, e12730 (2021), 10.1111/micc.12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bovay E., et al. , Multiple roles of lymphatic vessels in peripheral lymph node development. J. Exp. Med. 215, 2760–2777 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birmingham K. G., et al. , Lymph node subcapsular sinus microenvironment-on-a-chip modeling shear flow relevant to lymphatic metastasis and immune cell homing. iScience 23, 101751 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kashyap B., Kullaa A. M., Regulation of mucin 1 expression and its relationship with oral diseases. Arch. Oral Biol. 117, 104791 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Chary S. R., Jain R. K., Direct measurement of interstitial convection and diffusion of albumin in normal and neoplastic tissues by fluorescence photobleaching. Proc. Natl. Acad. Sci. U.S.A. 86, 5385–5389 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wasson E. M., Dubbin K., Moya M. L., Go with the flow: Modeling unique biological flows in engineered in vitro platforms. Lab Chip 21, 2095–2120 (2021). [DOI] [PubMed] [Google Scholar]
- 19.Rockson S. G., The lymphatics and the inflammatory response: Lessons learned from human lymphedema. Lymphat. Res. Biol. 11, 117–120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ly C. L., Kataru R. P., Mehrara B. J., Inflammatory manifestations of lymphedema. Int. J. Mol. Sci. 18, 171 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Y., LoGrasso P. V., Defert O., Li R., Rho Kinase (ROCK) inhibitors and their therapeutic potential. J. Med. Chem. 59, 2269–2300 (2016). [DOI] [PubMed] [Google Scholar]
- 22.de Almeida O. P., Bohm G. M., Vascular permeability in the rat gingiva. A model of vessel response in chronic inflammation. J. Pathol. 127, 27–34 (1979). [DOI] [PubMed] [Google Scholar]
- 23.Polacheck W. J., et al. , A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 552, 258–262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang F., et al. , Lacteal junction zippering protects against diet-induced obesity. Science 361, 599–603 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Overby D. R., et al. , Altered mechanobiology of Schlemm’s canal endothelial cells in glaucoma. Proc. Natl. Acad. Sci. U.S.A. 111, 13876–13881 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanihara H., et al. , Intra-ocular pressure-lowering effects of a Rho kinase inhibitor, ripasudil (K-115), over 24 hours in primary open-angle glaucoma and ocular hypertension: A randomized, open-label, crossover study. Acta Ophthalmol. 93, e254–e260 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Schneider M., Ny A., Ruiz de Almodovar C., Carmeliet P., A new mouse model to study acquired lymphedema. PLoS Med. 3, e264 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian W., et al. , Leukotriene B4 antagonism ameliorates experimental lymphedema. Sci. Transl. Med. 9, eaal3920 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa O., et al. , ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Lett. 392, 189–193 (1996). [DOI] [PubMed] [Google Scholar]
- 30.Jurisic G., et al. , An unexpected role of semaphorin3a-neuropilin-1 signaling in lymphatic vessel maturation and valve formation. Circ. Res. 111, 426–436 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimokawa H., Sunamura S., Satoh K., RhoA/Rho-kinase in the cardiovascular system. Circ. Res. 118, 352–366 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Bryan B. A., et al. , RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 24, 3186–3195 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Nieuw Amerongen G. P., Koolwijk P., Versteilen A., van Hinsbergh V. W., Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol. 23, 211–217 (2003). [DOI] [PubMed] [Google Scholar]
- 34.Lisowska J., et al. , The CCM1-CCM2 complex controls complementary functions of ROCK1 and ROCK2 that are required for endothelial integrity. J. Cell Sci. 131, jcs216093 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Cutler C., et al. , Belumosudil for chronic graft-versus-host disease after 2 or more prior lines of therapy: The ROCKstar Study. Blood 138, 2278–2289 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ongstad E. L., et al. , Lymphangiogenesis-independent resolution of experimental edema. Am. J. Physiol. Heart Circ. Physiol. 299, H46–54 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X., et al. , Temporal and spatial regulation of epsin abundance and VEGFR3 signaling are required for lymphatic valve formation and function. Sci. Signal 7, ra97 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H., et al. , Epsin deficiency promotes lymphangiogenesis through regulation of VEGFR3 degradation in diabetes. J. Clin. Invest. 128, 4025–4043 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schindelin J., et al. , Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jannaway M., et al. , VEGFR3 is required for button junction formation in lymphatic vessels. Cell Rep. 42, 112777 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng W., et al. , Angiopoietin 2 regulates the transformation and integrity of lymphatic endothelial cell junctions. Genes Dev. 28, 1592–1603 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
A time-lapse movie of active response of an engineered LV and BV under interstitial flow. A 2-hour time-lapse movie shows active responses of the engineered LV and BV to interstitial flow of BODIPY-C16 (pseudo red-colored). BODIPY-C16 entered the luminal space of the engineered LV faster than that of the engineered BV, indicating faster fluid drainage in LVs than BVs. The engineered LV was relaxed under interstitial flow, displaying an enlarging lumen (bright field). By contrast, the engineered BV was squeezed by the interstitial flow.
Data Availability Statement
All study data are included in the article and/or supporting information.