

Abstract
Haematopoietic development is regulated by nuclear protein complexes that coordinate lineage-specific patterns of gene expression. Targeted mutagenesis in embryonic stem cells and mice has revealed roles for the X-linked gene Gata1 in erythrocyte and megakaryocyte differentiation1–4. GATA-1 is the founding member of a family of DNA-binding proteins that recognize the motif WGATAR through a conserved multifunctional domain consisting of two C4-type zinc fingers5–8. Here we describe a family with X-linked dyserythropoietic anaemia and thrombocytopenia due to a substitution of methionine for valine at amino acid 205 of GATA-1. This highly conserved valine is necessary for interaction of the amino-terminal zinc finger of GATA-1 with its essential cofactor, FOG-1 (for friend of GATA-1; refs 9–12). We show that the V205M mutation abrogates the interaction between Gata-1 and Fog-1, inhibiting the ability of Gata-1 to rescue erythroid differentiation in an erythroid cell line deficient for Gata-1 (G1E). Our findings underscore the importance of FOG-1:Gata-1 associations in both megakaryocyte and erythroid development, and suggest that other X-linked anaemias or thrombocytopenias may be caused by defects in GATA1.
A woman with mild chronic thrombocytopenia had two pregnancies complicated by severe fetal anaemia requiring in utero red blood cell transfusions. The offspring were male half-siblings who were anaemic and severely thrombocytopenic at birth and thereafter. Abnormalities were present in the erythrocyte and platelet lineages. Their peripheral blood (Fig. 1a) showed a paucity of platelets, and erythrocytes were abnormal in size and shape (poikilocytosis and anisocytosis). The bone marrow was hypercellular and contained an abundance of large, multinucleated, erythroid precursors (dyserythropoiesis; Fig. 1c) and numerous small, dysplastic megakaryocytes (Fig. 1b,d). Electron microscopy13 also revealed dysplastic changes in megakaryocytes and platelets (Fig. 2). Both boys had cryptorchidism. There were three asymptomatic female siblings (Fig. 3a).
Fig. 1.
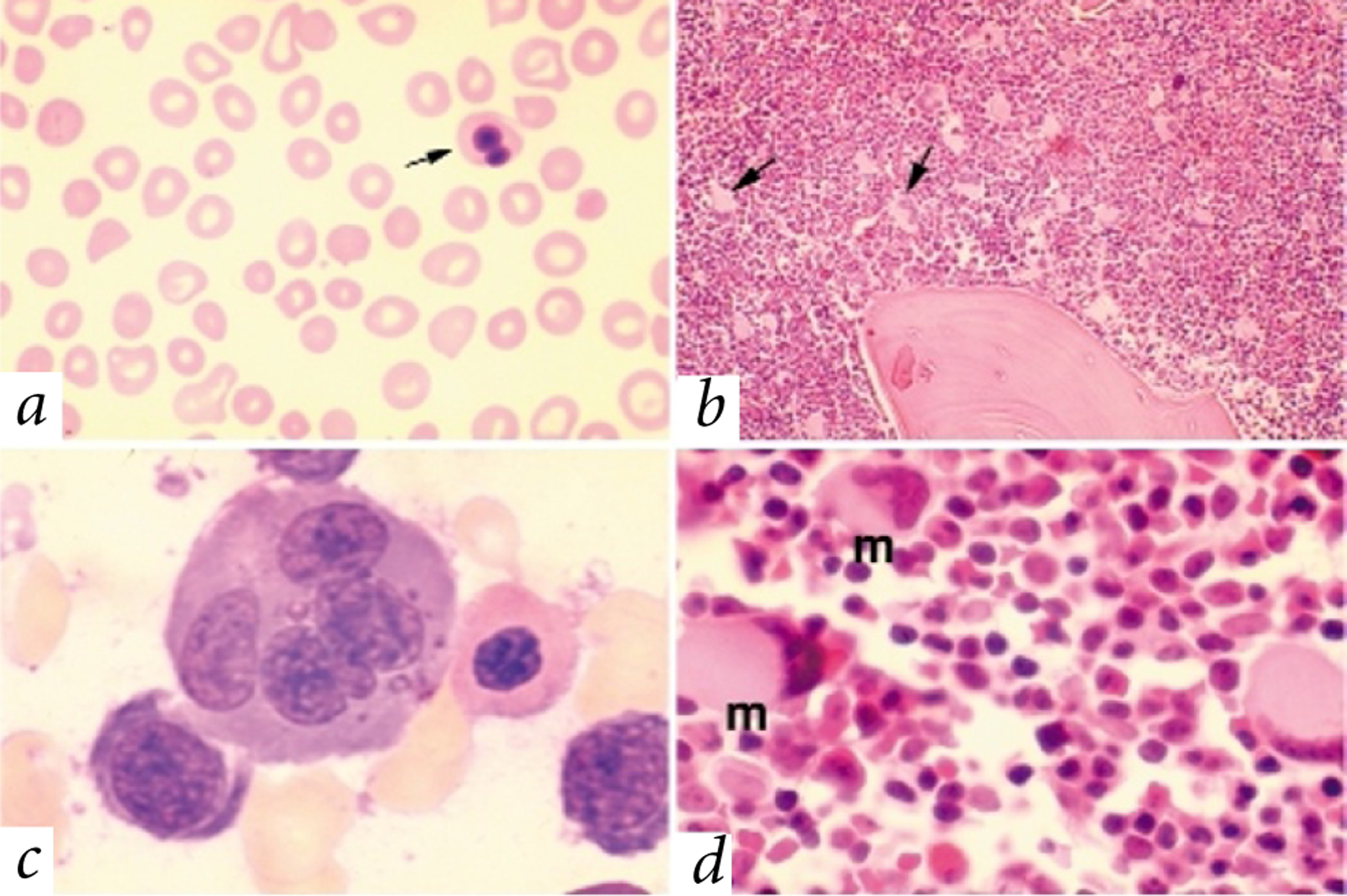
Peripheral blood and bone marrow abnormalities in patients II-1 and II-2. a, Peripheral blood smear. Note the severe deficiency of platelets and heterogeneity in red blood cell size (poikilocytosis) and shape (anisocytosis). An abnormal red blood cell with a bilobed nucleus is indicated (arrow). Original magnification ×50. b, Bone marrow core biopsy. Megakaryocytes, distinguished by their pale pink cytoplasm, are abundant and abnormally small. Representative megakaryocytes are indicated (arrows). Original magnification ×10. c, Bone marrow aspirate showing a cluster of developing erythroid precursors. Dyserythropoiesis is illustrated by the large, multinuclear erythroblast (centre). Original magnification ×100. d, Bone marrow core biopsy. Megakaryocytes (m) are dysplastic with small ecentric nuclei. Original magnification ×50.
Fig. 2.

Ultrastructural abnormalities in megakaryocytes and platelets revealed by electron microscopy. a, Low-power micrograph of a normal megakaryocyte. Note the centrally located nucleus (Nu) and the well-developed demarcation membrane system (DM). Original magnification ×2,880. b, Megakaryocyte from affected patient showing showing abnormal features that include an abundance of smooth endoplasmic reticulum (Ser), ecentric nucleus and a relative paucity of granules. Original magnification ×2,880. c, High-power micrograph of a normal megakaryocyte showing well-organized membrane demarcation system. Original magnification ×20,000. d, High-power micrograph of mutant megakaryocyte showing abnormal abundance of smooth endoplasmic reticulum (Ser). The membrane demarcation system is poorly developed. Original magnification ×20,000. e, Platelet from normal individual. Note the abundance of a-granules (G). Original magnification ×20,000. f,g,h, Circulating platelets from affected patients. Note the paucity of granules (G), abundance of smooth endoplasmic reticulum (Ser) and abnormal membranous complexes (MC). Original magnification ×20,000.
Fig. 3.

Mutational analysis of GATA1 in the affected pedigree. a, Partial pedigree of the affected kindred and DNA sequence analysis of GATA1. Patients with severe manifestations are represented by filled symbols, and patients with milder symptoms by half-filled symbols. Genotypes are shown below the pedigree members (other family members were not tested). b, Amino acid alignment of the N-terminal zinc finger of GATA-1 protein from various species and the Drosophila GATA-A protein. Amino acids known to interact with FOG-1 are labelled with black dots. The V205M mutation is indicated. c, Three-dimensional structure of the N-terminal zinc finger. Amino acids presumed to interact with DNA are shown in red. Valine 205 is indicated in yellow. Other FOG-1–interacting residues are shown in green.
The haematopoietic defects in combination with a pedigree structure that was consistent with an X-linked disorder suggested the possibility of GATA-1 deficiency3,4,14,15. Therefore we analysed GATA1 genomic DNA sequence from available family members at each of the five coding exons and approximately 800 bp of the promoter region (Fig. 3a). We identified a hemizygous G→A transition at nt 613 in both affected boys (II-1 and II-2). This mutation is predicted to convert valine to methionine at amino acid 205, a highly conserved residue within the GATA-1 protein. The boys inherited this mutation from their mother (I-2), who was heterozygous at nt 613. We did not detect the G613A mutation in peripheral blood leukocyte DNA from 50 normal females (data not shown).
The alteration of valine to methionine occurs in the N-terminal zinc finger, a region that is highly conserved between GATA-1 proteins of all species and in related proteins such as GATA-2–6 and the GATA-A (Pannier) protein of Drosophila melanogaster (Fig. 3b, and data not shown). In vitro studies have shown that the Gata-1 N-terminal zinc finger is essential for erythroid maturation, in large part due to its interaction with the zinc-finger protein Fog-1 (ref. 12). Valine 205 is one of several amino acids critical for direct association of Fog-1 with Gata-1 (Fig. 3c; refs 11,12,16). FOG-1 contains nine zinc fingers of multiple types, which presumably act as docking sites to link promoter-bound GATA-1 to other nuclear factors. Such multi-protein assemblies are likely to exert complex overall effects on gene expression, as FOG-1 can either activate or repress the activity of GATA-1, depending on cellular and promoter contexts9,17.
The V205M mutation was recently identified in a mutagenic screen designed to isolate N-terminal zinc finger mutants with impaired binding to Fog-1 (ref. 12). To assess whether V205M interferes with Fog-1 binding in the context of full-length Gata-1, we introduced this mutation into mouse cDNA. Wild-type or V205M Gata-1 were coexpressed in COS cells with a FLAG epitope-tagged, truncated version of Fog-1 consisting of zinc fingers 5 and 6. Following immunoprecipitation with anti-FLAG antibody, we analysed samples by western blot with anti-Gata-1 antibody (Fig. 4a). In contrast to wild-type Gata-1, interaction between the V205M mutant and Fog-1 was reduced. Thus, the mutation severely impairs the interaction between Gata-1 and Fog-1 in mammalian, as well as yeast12, cells.
Fig. 4.

Biochemical characterization of mutant V205M Gata-1. a, The V205M mutation impairs binding of Gata-1 to Fog-1. Wild-type or V205M Gata-1 cDNAs were transfected into COS cells along with a FLAG-tagged version of Fog-1 consisting of zinc fingers 5 and 6. Gata-1 bound to Fog-1 was detected by immunoprecipitation with anti-FLAG antibody (α-FLAG IP) followed by western-blot analysis with anti-Gata-1 antibody (top). Gata-1 is indicated (arrow). The upper band (asterisk) corresponds to the IgG heavy chain of the anti-FLAG antibody. Total Gata-1 was determined by western-blot analysis of nuclear extracts from the transfected cells (NE, bottom). Equivalent amounts of FLAG-tagged Fog-1 were immunoprecipitated in Zfpm1 cDNA-transfected samples as determined by western blot with anti-FLAG antibody (data not shown). b, V205M Gata-1 binds DNA normally. The stability of Gata-1–DNA interactions was measured by a dissociation gel shift assay. Labelled probe bound to Gata-1 (arrow) migrates more slowly than free probe seen in the lower portion of the panel. V205M and wild-type (WT) Gata-1 dissociate from the labelled probe at similar rates. In contrast, the C204R mutation, which disrupts the N-terminal zinc finger, increases the dissociation rate between Gata-1 and bound DNA probe.
In addition to its role in mediating protein interactions, the Gata-1 N-terminal zinc finger cooperates with the carboxyl finger to stabilize DNA binding at a subset of Gata-consensus motifs, in particular those that contain palindromic repeats18–20. Gel shift analysis of V205M Gata-1 showed that the mutation has no discernable effect on the stability of Gata-1 interaction with a palindromic DNA consensus motif, in contrast to the C204R mutation, which increases the rate of dissociation from bound DNA (Fig. 4b). Therefore, the deleterious consequences of this mutation most likely result from impaired interaction with Fog-1, rather than reduced affinity to critical GATA motifs in erythroid and megakaryocyte target genes.
To assess the biological significance of the V205M substitution, we examined the capacity of mutant protein to promote maturation of an erythroid cell line deficient for Gata-1 (G1E). We showed previously that stable expression of a conditional form of Gata-1 (Gata-1/ER, wild-type protein fused to the ligand-binding domain of the oestrogen receptor) renders G1E cells oestrogen-dependent for erythroid maturation9,21. We introduced wild-type or mutant V205M Gata-1/ER into G1E cells and selected stably expressing clones. Expression of wild-type Gata-1/ER imparted β-oestradiol-dependent maturation reproducibly, but the V205M mutant was inert in eight independent clones (Fig. 5a). For example, clone V205M-8 was unaffected by β-oestradiol, despite Gata-1/ER protein expression equivalent to that of clone WT-4, in which cells exhibited changes in morphology and gene expression characteristic of terminal erythroid maturation, and nearly 100% became positive for benzidine, indicating haemoglobin production (Fig. 5b,c). Thus, the V205M mutation impairs the ability of Gata-1 to promote erythroid differentiation in vitro.
Fig. 5.

Impaired function of V205M mutant Gata-1 in erythroid cells. a, Plasmids encoding oestrogen-inducible forms of wild-type (WT) or V205M Gata-1 were introduced into the Gata-1–null erythroid line G1E. Nuclear extracts from stably expressing clones were analysed by western blot for Gata-1 expression (arrow). Cells were treated with β-oestradiol for 72 h and erythroid maturation was determined by benzidine staining, which detects haemoglobin. Note that V205M/Gata-1 fails to induce differentiation in eight independent clones. b, Representative data from clones V205M-8 and WT-4. May Grunwald-Giemsa (MGG) and benzidine staining after 72 h of β-oestradiol treatment. c, Northern-blot analysis of clones V205M-8 and WT-4 after treatment with β-oestradiol for the indicated times. WT-4 exhibits mRNA changes that are characteristic of erythroid maturation, including induction of α-globin and band 3, and concommitant downregulation of Gata-2. No changes in gene expression are seen in V205M-8 cells after β-oestradiol treatment. Each lane represents 15 μg of total RNA.
Our data indicate that the V205M mutation in GATA1 causes human haematopoietic disease, and underscore the biological significance of the GATA-1:FOG-1 complex in erythroid and megakaryocytic development. The phenotypic effects of the V205M substitution seen here are of particular interest when compared with those of targeted mutations introduced into mouse Gata1 and Zfpm1 (encoding Fog-1). Homozygous disruption of Gata1 in mice is embryonic lethal due to extreme anaemia resulting from maturation arrest and apoptosis of embryonic (also termed yolk sac or primitive) and adult-type (also termed fetal liver/bone marrow or definitive) erythroid precursors1,3,22. The erythroid defect is slightly less severe in Zfpm1−/− mice, but is still embryonic lethal10. In comparison, a milder erythroid phenotype is seen in mice harbouring a mutation in a region upstream of Gata1 that reduces its expression in erythroid cells by four- to fivefold14. In the megakaryocyte lineage of these ‘knockdown’ mice, Gata-1 expression is absent, leading to severe thrombocytopenia with hyperproliferation of dysplastic megakaryocytes4,15.
The haematological findings in the boys with the V205M mutation most closely resemble those of mice with the ‘knockdown’ Gata1 mutation. The relatively milder consequences of the V205M substitution during erythrocyte development (compared with complete loss of Gata1) may point to FOG-1–independent actions of GATA-1 in the erythroid lineage, or reflect residual weak interactions between FOG-1 and the V205M mutant GATA-1 in vivo. The profound disturbance of megakaryopoiesis caused by the V205M substitution reveals for the first time a role for the FOG-1:GATA-1 interaction during megakaryocyte development and platelet formation. The absence of megakaryocytes in mice with deletion of Zfpm1 previously precluded an in vivo assessment of the requirement for this interaction during megakaryocyte differentiation10. The mild thrombocytopenia noted in patient I-2, who is heterozygous for the V205M substitution, may be due to skewed X-chromosome inactivation resulting in a relative abundance of megakaryocytic precursors expressing mutant GATA-1.
Although Gata-1 is expressed in Sertoli cells of the testis in young mice23, it is unclear whether the V205M mutation is responsible for cryptorchidism in the male patients described here. In mice, high expression of Gata-1 in Sertoli cells is not observed until several days post-natally, after the testes have descended23,24. Moreover cryptorchidism is not present in mice with selective loss of Gata-1 in testis (Y. Fujiwara and S.H.O., unpublished data). Interspecies differences may exist, or the V205M mutant may be more deleterious to testicular development than loss of GATA-1 alone. Alternatively, cryptorchidism, which is relatively common in humans25, may be unrelated to the GATA1 mutation in these patients.
Our data provide the first evidence for mutations of GATA1 in human disease and should encourage the search for other GATA1 mutations in families with haematologic disorders. Although reports of X-linked anaemia and thrombocytopenia are rare26, quantitative or qualitative defects in GATA-1 protein expression might lead to selective X-linked defects of either platelet or erythrocyte production. In this regard, the gene mutated in Wiskott-Aldrich syndrome (WAS), an inherited disorder characterized by immunodeficiency and thrombocytopenia, is closely linked to GATA1 at Xp11.22–23 (see Genemap99, http://www.ncbi.nlm.nih.zgov/genemap/map.cgi?MAP=GB4&BIN=610&MARK=stSG43166). Familial thrombocytopenias linked to this region, in which no mutations are detected at the WAS locus, might be caused by defects in GATA1.
Methods
Patients.
A family with hereditary anaemia and thrombocytopenia was identified at the Children’s Hospital of Philadelphia. Patient II-2 is the third child born to a mother (I-2) with chronic mild thrombocytopenia (platelet count 50,000–140,000/μl). At 16 weeks, the pregnancy was complicated by severe fetal hydrops requiring three intrauterine transfusions of packed red blood cells. After an uncomplicated delivery, II-2 remained anaemic (haemoglobin <8.4 g/dl) and thrombocytopenic (platelet count <20,000/μl). Although he required platelet transfusions during the first two years of life, after the perinatal period he no longer required red blood cell transfusions. White blood count and leukocyte differential were normal. At age 24 months he received an HLA-matched bone marrow transplant from his sister (II-3), whose haematologic parameters were normal. The clinical course of patient II-1 was similar to that of his half brother, with the exception that he underwent transplantation of HLA-matched unrelated donor bone marrow. Both boys had bilateral cryptorchidism. Speciments obtained during surgical correction showed normal testicular tissue with germ cells present.
Haematologic indices at the time of presentation to Children’s Hospital of Philadelphia are shown (Table 1). For hemizygous patients II-1 and II-2, low platelet numbers and clinically indicated platelet transfusions prevented aggregation studies.
Table 1 •.
Haematologic profiles of affected family members
| Patient | Age | Hgb (g/dl) |
Hct (%) | RBC (×106/μl) |
MCV (μm3) |
MCH (pg) |
Retic (%) (0.5–1.5) |
nRBC (%) 0 |
RDW (11.5–14.5) |
Plt (×103/μl) (150–400) |
|---|---|---|---|---|---|---|---|---|---|---|
| II-2 | 23 months | 8.5 (10.5–12.0) |
29.2 (33–36) |
3.3 (3.9–4.6) |
88.6 (70–78) |
26.1 (23–27) |
3 | 29 | 22.0 | 24 |
| II-1 | 8 months | 8.2 (10.5–12.0) |
27.4 (33–36) |
2.9 (3.9–4.6) |
96.2 (70–78) |
28.8 (23–27) |
3 | 25 | 19.1 | 11 |
| I-2 | 28 y | 11.3 (12–16) |
33.5 (36–46) |
3.6 (4.0–5.2) |
94.0 (80–100) |
31.6 (26–34) |
1 | 0 | 13.1 | 53 |
Haematologic indices at the time of presentation to Children’s Hospital of Philadelphia are shown for male hemizygous patients (II-1 and II-2) and their heterozygous mother (I-2). Age-adjusted normal values are shown in parentheses. Hgb, haemoglobin concentration; Hct, haematocrit; RBC, red blood cell number; MCV, mean corpuscular volume; MCH, mean corpuscular haemoglobin; Retic, reticulocytes as percentage of total red blood cells; nRBC, number of nucleated red blood cells per 100 white blood cells; RDW, red blood cell distribution width; Plt, platelet number.
Following informed consent, we obtained peripheral blood from patients I-2, II-1, II-2 and II-4 for genetic analysis. We isolated mononuclear cells by Ficoll Hypaque (Pharmacia) sedimentation and extracted genomic DNA using standard methods. We obtained peripheral blood from patients II-1 and II-2 before bone marrow transplantation.
DNA sequencing.
We obtained GATA1 intron sequences by automated sequence analysis of a human BAC clone (Research Genetics) containing the entire GATA1 gene. We screened patient-derived DNA samples for mutations using a nested PCR strategy. An initial long-range PCR reaction was used to amplify a ~3-kb genomic fragment containing the GATA1 coding sequence using Expand Taq polymerase (Roche) and the following primers: Hg1 forward, 5′–GTCCTCGCAGGTTAATCCCC–3′; and Hg1 reverse, 5′–GGCTACAAGAGGAGAAGGAC–3′. Conditions were 94 °C for 5 min; 94 °C, 30 s; 60 °C, 30 s; 68 °C, 3 min; for a total of 25 cycles. A second PCR was performed using primers that flanked each of the five coding GATA1 exons: 2S, 5′–GAGGAGCAGGTGAAAGGAGGTGG–3′; 2A, 5′–GCCAAGGATCTCCATGGCAACCC–3′; 3S, 5′–GGA ACTTGGCCACCATGTTGG–3′; 3A, 5′–GAGCTAGGCTCAGCTCAGC TTTAC–3′; 4S, 5´–GAGGTGGGAGGGGTGGCCCAAAG–3′; 4A, 5′–CTGTAATCATGAGAACAGCGTTCC–3′; 5S, 5′–GGCATCACCCTGTA AACAAAGCC–3′; 5A, 5′–GGGCAGGAGTTCTCATGCAGG–3′; 6S, 5′–GTGTCCCTGGTTGACACAGAG–3′. The Hg1 reverse primer was used as the antisense primer to amplify exon 6 of GATA1. To amplify the GATA1 promoter, we used the primers Hg1 promoter sense, 5´–GAG GAATCATCCCTGGCTCCC–3´, and Hg1 promoter antisense, 5´–GTG TCCTGGCTGGCCTTGGC–3´. Conditions for exon and promoter PCR reactions were 95 °C for 5 min; 95 °C, 30 s; 60 °C, 30 s; 72 °C, 45 s; for 20 cycles. PCR products were prepared using Quiaquick PCR purification columns (Qiagen) to remove unincorporated nucleotides, and subjected to automated nucleotide sequencing (ABI 373; Applied Biosystems). To verify results of heterozygote sequence analysis, we inserted PCR products into the TA-Topo vector (Invitrogen) and evaluated 6–10 independent clones by automated sequence analysis.
Cell culture.
Growth and retroviral transduction of G1E cells were performed as described27.
Site-directed mutagenesis, immunoprecipitations and northern-blot analysis.
We performed these analyses as described12.
Electophoretic mobility shifts.
Nuclear extracts of COS cells expressing wild-type or mutant GATA-1 were incubated with a 32P-labelled, double-stranded oligonucleotide probe containing a palindromic GATA site. Unlabelled competitor DNA was added and after various incubation times the samples were subjected to nondenaturing polyacrylamide gel electrophoresis.
Acknowledgements
We thank Y. Fujiwara, E. Neufeld and S. McKenzie for helpful insights and discussions; D. Haber for assistance with DNA sequencing and review of the manuscript; and J. Mackay and M. Crossley for insightful discussions and providing data on the three-dimensional structure of the GATA-1 N-terminal zinc finger. This work was supported in part by NIH grants AI K11AI01331-05 (K.E.N.), R01 CA78545 (J.M.M.) and HL40387 (M.P.).
References
- 1.Pevny L et al. Erythroid differentiation in chimaeric mice blocked by a targeted mutation in the gene for transcription factor GATA-1. Nature 349, 257–260 (1991). [DOI] [PubMed] [Google Scholar]
- 2.Weiss MJ, Keller G & Orkin SH Novel insights into erythroid development revealed through in vitro differentiation of GATA-1− embryonic stem cells. Genes Dev. 8, 1184–1197 (1994). [DOI] [PubMed] [Google Scholar]
- 3.Fujiwara Y, Browne CP, Cunniff K, Goff SC & Orkin SH Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc. Natl Acad. Sci. USA 93, 12355–12358 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shivdasani RA, Fujiwara Y, McDevitt MA & Orkin SH A lineage-selective knockout establishes the critical role of transcription factor GATA-1 in megakaryocyte growth and platelet development. EMBO J. 16, 3965–3973 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsai SF et al. Cloning of cDNA for the major DNA-binding protein of the erythroid lineage through expression in mammalian cells. Nature 339, 446–451 (1989). [DOI] [PubMed] [Google Scholar]
- 6.Evans T & Felsenfeld G The erythroid-specific transcription factor Eryf1: a new finger protein. Cell 58, 877–885 (1989). [DOI] [PubMed] [Google Scholar]
- 7.Orkin SH GATA-binding transcription factors in hematopoietic cells. Blood 80, 575–581 (1992). [PubMed] [Google Scholar]
- 8.Weiss MJ & Orkin SH GATA transcription factors: key regulators of hematopoiesis. Exp. Hematol 23, 99–107 (1995). [PubMed] [Google Scholar]
- 9.Tsang AP et al. FOG, a multitype zinc finger protein, acts as a cofactor for transcription factor GATA-1 in erythroid and megakaryocytic differentiation. Cell 90, 109–119 (1997). [DOI] [PubMed] [Google Scholar]
- 10.Tsang AP, Fujiwara Y, Hom DB & Orkin SH Failure of megakaryopoiesis and arrested erythropoiesis in mice lacking the GATA-1 transcriptional cofactor FOG. Genes Dev. 12, 1176–1188 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fox AH, Kowalski K, King GF, Mackay JP & Crossley M Key residues characteristic of GATA N-fingers are recognized by FOG. J. Biol. Chem 273, 33595–33603 (1998). [DOI] [PubMed] [Google Scholar]
- 12.Crispino JD, Lodish MB, MacKay JP & Orkin SH Use of altered specificity mutants to probe a specific protein-protein interaction in differentiation: the GATA-1:FOG complex. Mol. Cell 3, 219–228 (1999). [DOI] [PubMed] [Google Scholar]
- 13.White JG Use of the electron microscope for diagnosis of platelet disorders. Semin. Thromb. Hemost 24, 163–168 (1998). [DOI] [PubMed] [Google Scholar]
- 14.McDevitt MA, Shivdasani RA, Fujiwara Y, Yang H & Orkin SH A “knockdown” mutation created by cis-element gene targeting reveals the dependence of erythroid cell maturation on the level of transcription factor GATA-1. Proc. Natl Acad. Sci. USA 94, 6781–6785 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vyas P, Ault K, Jackson CW, Orkin SH & Shivdasani RA Consequences of GATA-1 deficiency in megakaryocytes and platelets. Blood 93, 2867–2875 (1999). [PubMed] [Google Scholar]
- 16.Kowalski K, Czolij R, King GF, Crossley M & Mackay JP The solution structure of the N-terminal zinc finger of GATA-1 reveals a specific binding face for the transcriptional co-factor FOG. J. Biomol. NMR 13, 249–262 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Fox AH et al. Transcriptional cofactors of the FOG family interact with GATA proteins by means of multiple zinc fingers. EMBO J. 18, 2812–2822 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin DIK & Orkin SH Transcriptional activation and DNA binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes Dev. 4, 1886–1898 (1990). [DOI] [PubMed] [Google Scholar]
- 19.Yang H-Y & Evans T Distinct roles for the two cGATA-1 fingers. Mol. Cell. Biol 12, 4562–4570 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trainor CD et al. A palindromic regulatory site within vertebrate GATA-1 promoters requires both zinc fingers of the GATA-1 DNA-binding domain for high-affinity interaction. Mol. Cell. Biol 16, 2238–2247 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregory T et al. GATA-1 and erythropoietin cooperate to promote erythroid cell survival by regulating bcl-xL expression. Blood 94, 87–96 (1999). [PubMed] [Google Scholar]
- 22.Weiss MJ & Orkin SH Transcription factor GATA-1 permits survival and maturation of erythroid precursors by preventing apoptosis. Proc. Natl Acad. Sci. USA 92, 9623–9627 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yomogida K et al. Developmental stage- and spermatogenic cycle-specific expression of the transcription factor GATA-1 in mouse Sertoli cells. Development 120, 1759–1766 (1994). [DOI] [PubMed] [Google Scholar]
- 24.Viger RS, Mertineit C, Trasler JM & Nemer M Transcription factor GATA-4 is expressed in a sexually dimorphic pattern during mouse gonadal development and is a potent activator of the Mullerian inhibiting substance promoter. Development 125, 2665–2675 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Hutson JM, Baker M, Terada M, Zhou B & Paxton G Hormonal control of testicular descent and the cause of cryptorchidism. Reprod. Fertil. Dev 6, 151–156 (1994). [DOI] [PubMed] [Google Scholar]
- 26.Thompson AR, Wood WG & Stamatoyannopoulos G X-linked syndrome of platelet dysfunction, thrombocytopenia, and imbalanced globin chain synthesis with hemolysis. Blood 50, 303–316 (1977). [PubMed] [Google Scholar]
- 27.Weiss MJ, Yu C & Orkin SH Erythroid-cell-specific properties of transcription factor GATA-1 revealed by phenotypic rescue of a gene-targeted cell line. Mol. Cell. Biol 17, 1642–1651 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]