

Abstract
Disturbances in the brain’s capacity to meet its energy demand increase the risk of synaptic loss, neurodegeneration, and cognitive decline. Nutritional and metabolic interventions that target metabolic pathways combined with diagnostics to identify deficits in cerebral bioenergetics may therefore offer novel therapeutic potential for Alzheimer’s disease (AD) prevention and management. Many diet-derived natural bioactive components can govern cellular energy metabolism but their effects on brain aging are not clear. This review examines how nutritional metabolism can regulate brain bioenergetics and mitigate AD risk. We focus on leading mechanisms of cerebral bioenergetic breakdown in the aging brain at the cellular level, as well as the putative causes and consequences of disturbed bioenergetics, particularly at the blood-brain barrier with implications for nutrient brain delivery and nutritional interventions. Novel therapeutic nutrition approaches including diet patterns are provided, integrating studies of the gut microbiome, neuroimaging, and other biomarkers to guide future personalized nutritional interventions.
1 |. INTRODUCTION
The human adult brain utilizes ~20% of the whole body’s energy resources, yet it only represents ~2% of the body mass. This underscores an enormous metabolic workload, which is mainly fueled by glucose and several nutrients derived from the circulation.1 Interplay between disrupted intracellular bioenergetic pathways is likely to be relevant to degenerative brain diseases. It is already suspected that relationships between changing efficiency of glycolysis, the pentose phosphate shunt, the Krebs cycle, and oxidative phosphorylation fluxes affect signaling and transcription changes observed in age-related neurodegenerative diseases. These metabolic relationships are currently being addressed on a mechanistic level, but extending such investigations to clinical interventions in diseases like Alzheimer’s disease (AD) could provide valuable insights for prevention and therapeutics.2 Specifically, these investigations should aim to substantiate the role of cerebral bioenergetics as a causal factor for the onset of AD rather than a consequence of brain cell degeneration. This may include demonstrations that AD genetic risk loci impact bioenergetics, bioenergetic deficits precede and contribute to the classical hallmarks of AD pathology (i.e., plaque and tangles), and, ultimately, restoration of cerebral bioenergetic disturbances to promote cognitive health and/or prevention of neurodegeneration in formal randomized-controlled interventions.
This review focuses on key mechanisms involved in cerebral bioenergetics, and how deficits in these pathways may contribute to cognitive decline in AD. Cell, animal, and clinical studies are presented that enhance our understanding of how genetic, environmental risk factors and emerging factors such as the gut microbiome affect brain uptake and utilization of nutrients. We then discuss current mechanistic understandings of mitochondrial energetic changes in aging and AD, which suggest that insufficient nutritional support of cerebral bioenergetics may occur as early as mid-life prior to significant neuropathology and compounds with disturbed nutrient delivery to the brain. Finally, we present examples of promising single nutrients or combinations paired together with diet patterns and identify new areas of basic research, blood, and imaging biomarkers to guide future clinical interventions.
2 |. NUTRITIONAL REGULATION OF BRAIN ENERGY METABOLISM IN AD PATHOGENESIS
It is estimated that the brain consumes 120 g of glucose per day or about 20% of the body’s glucose utilization during a resting state. Glucose undergoes glycolysis to pyruvate, which is metabolized to acetyl CoA as the key carbon source for respiration or oxidative phosphorylation to generate adenosine triphosphate (ATP) in mitochondria (Figure 1A). In resting conditions, the favored metabolic route is mitochondrial oxidative phosphorylation, while highly demanding activity states such as synaptic plasticity, learning, and memory require the additional contribution from glycolysis or lactate metabolism.3–5 When the brain is starved for glucose during hypoglycemia or as a result of defective glucose transporters (GLUT) function,6 cells utilize liver-derived ketone bodies such as acetoacetate and 3-hydroxybutyrate to form acetyl CoA. These ketone bodies temporarily replace glucose as the energy source in the brain, but the ketogenic brain must revert to a precise regulation of glucose oxidation to maintain prolonged CNS function.7
FIGURE 1.
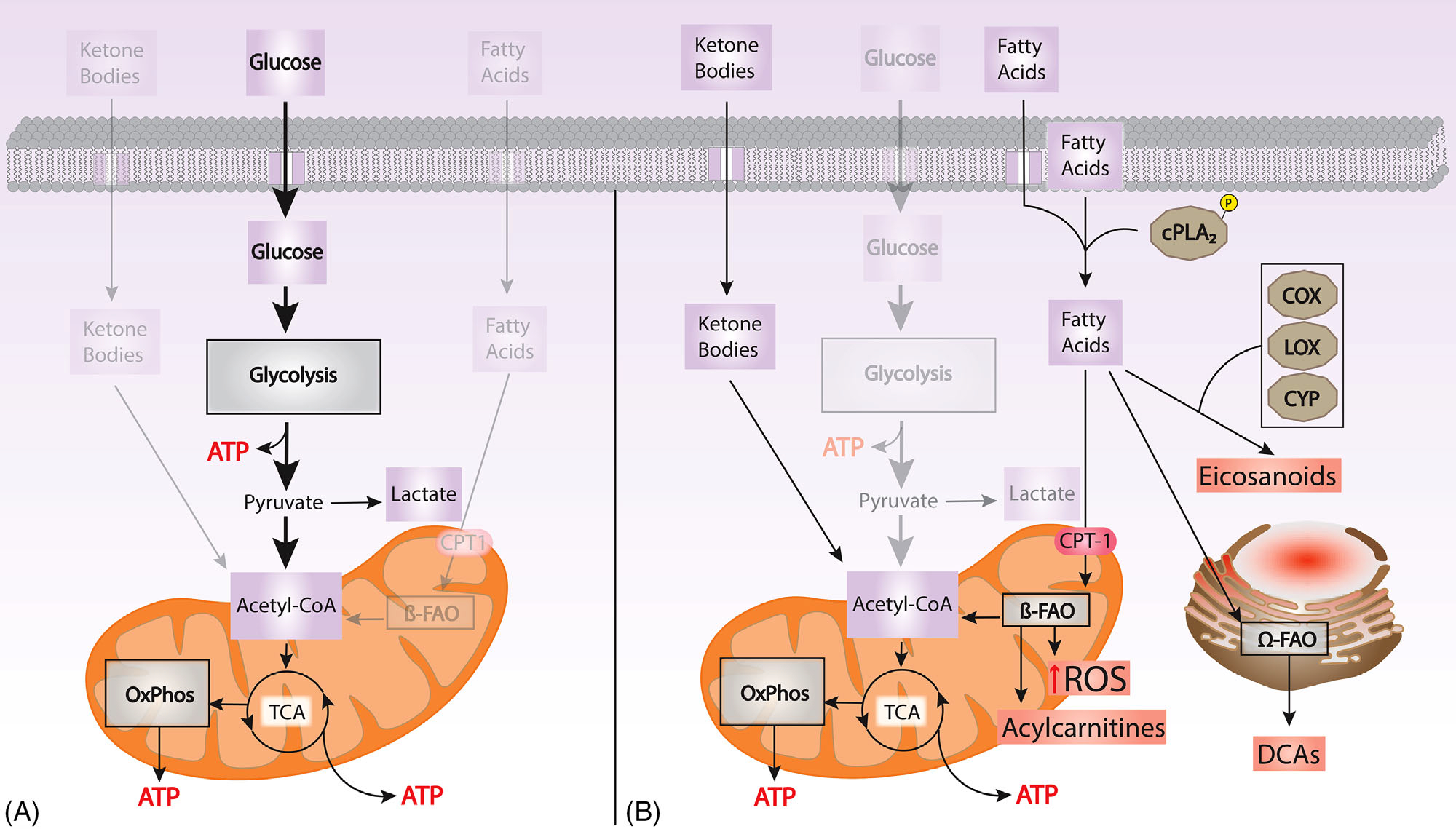
(A) Glucose is the main energy substrate in the brain. Glucose undergoes glycolysis to pyruvate, which is metabolized to acetyl CoA, as the key source of carbon for oxidative phosphorylation (Oxphos) to generate adenosine triphosphate (ATP). In resting conditions, the favored metabolic route is Oxphos, while highly demanding activity require contributions from glycolysis or lactate metabolism. When the brain is starved for glucose, ketone bodies are metabolized to form acetyl CoA. (B) Failure to utilize glucose and energetic stress shifts metabolism into oxidation of fatty acids, which leads to ROS production. The activation of cPLA2 promotes the formation of inflammatory lipid mediators (eicosanoids). Incomplete fatty acid oxidation (FAO) in the mitochondria is reflected by levels of acylcarnitines in the blood and the brain. The oxidation of n-3 fatty acids in the ER results in DCA accumulation, a urinary biomarker of bioenergetic stress. COX, cyclooxygenase; cPLA2, calcium dependent phospholipase A2; OxPhos, oxidative phosphorylation; CPT-1, carnitine palmitoyltransferase 1; CYP, cytochrome P450; DCA, dicarboxylic acids; ER, endoplasmic reticulum; FAO, fatty acid oxidation; LOX, lipoxygenase; ROS, reactive oxygen species; TCA, tricarboxylic acid
Several nutrients, including vitamins, amino acids, trace elements (e.g., iron and magnesium) and fatty acids derived from dietary intake of carbohydrates, fats, and proteins are concentrated in the brain and may have a role in meeting brain energy demand and their brain delivery is affected by AD (Table 1). Several nutrients serve as cellular bioenergetic catalysts to produce key energetic substrates for sustained neuronal function (i.e., ATP). Some micronutrients serve as coenzymes and cofactors for enzymatic reactions while others play a structural role within the enzyme and the mitochondrial cytochromes. Others serve as electron and proton carriers in the ATP-generating respiratory chain, including B vitamins and ascorbic acid.8,9 For example, thiamin pyrophosphate (TPP; vitamin B1), pantothenic acid (vitamin B5; precursor for co-enzyme A synthesis), flavin mononucleotide (FMN; derived from vitamin B2), flavin adenine dinucleotide (FAD; derived from vitamin B2) and nicotinamide adenine dinucleotide (NAD; derived from nicotinamide) are involved in the Krebs cycle and I and II complexes of the respiratory chain. Biotin, CoA, and FAD are involved in heme biosynthesis, which is an essential part of the cytochromes and important for mitochondrial respiratory chain reactions. Succinyl-CoA can nourish the respiratory chain or the Krebs cycle depending upon specific cellular needs.10,11 Iron has important roles in maintaining the high metabolic and energetic requirements of neuronal tissues and is also involved in myelin synthesis, neurotransmitter synthesis, and metabolism.12
TABLE 1.
Nutrients with a putative role in cerebral bioenergetics: function, concentration gradient, and transport mechanisms
| Nutrient and reference | Function, concentration gradient | Transporters and location |
|---|---|---|
| B1 thiamine238,239 | CSF/serum ratio 2.1:1 for thiamine, 8.3:1 for thiamin monophosphate | SLC25A19 mitochondria SLC19A3 brain unknown SLC19A2 ubiquitous |
| B6 pyridoxine, pyridoxal, PLP46 | CSF/plasma ratio 1.3:1 PL = pyridoxal is most abundant in CSF; requires phosphorylation of PL to PLP centrally; |
BBB Unknown bidirectional transport; facilitated diffusion BCSFB |
| B7 biotin240 | CSF/serum ratio 1:3.2 CSF/serum ratio lower in AD |
SMVT BCSFB |
| B9 folate241,242 | CSF-blood ratio is 4:1 CSF folate levels are lower in AD Folate in enriched in mitochondrial fractions |
FOLR1 BCSFB BBB folate receptor at the BCSFB; FR alpha blood to CSF; receptor mediated endocytosis |
| B12 cobalamin243–245 | CSF/serum ratio 1:20 CSF/serum ratio is lower in AD Required for the oxidation of odd-chain fatty acids and catabolism of ketogenic amino acids Homocysteine metabolism |
Cubam receptors at the BCSFB |
| Vitamin C, ascorbic acid246,247 | CSF/Serum ratio 3:1 Essential for carnitine synthesis, involved in transport of long chain fatty acids into the mitochondria and fatty acid oxidation. Facilitates transport and uptake of non-heme iron at the mucosa, and reduction of folic acid intermediates. |
SVCT-2 basolateral BCSFB GLUT1 (SLC2A1) as dehydroascorbic acid at BBB GLUT4 as dehydroascorbic acid within Astrocytes |
| DHA, docosahexaenoic acid108,248 | CSF to plasma ratio 1:75 Component of structural phospholipids in the CNS CSF to plasma ratio lower in AD |
MFSD2A109 Fatty acid binding proteins249 |
| Vitamin D, cholecalciferol, 25 hydroxyvitamin D250–252 |
CSF to serum ratio 1.4:1 Lower CSF/serum ratio in AD 1alpha-hydroxylase the enzyme responsible for the formation of the active form in the human brain |
VDR VDBP |
Abbreviations: PLP, Pyridoxal 5'-phosphate; PL, pyridoxal; SMVT, sodium dependent multivitamin transporter; BCSFB, blood cerebrospinal fluid barrier; SVCT, sodium dependent vitamin C transporter; BBB, blood brain barrier; CSF, cerebrospinal fluid; FOLR1, Folate receptor 1; FR alpha, folate receptor alpha; GLUT, glucose transporter; VDR, Vitamin D Receptor; VDBP, Vitamin D Binding Protein, MFSD2A, major facilitator superfamily domain-containing protein 2a
Fatty acids are a minor source to fuel ATP production in the brain.13 First, the high oxygen consumption associated with fatty acid β-oxidation (FAO) increases the risk of neurons becoming hypoxic because oxygen pressure is nonuniform and low.14 Second, the utilization of fatty acids as fuel increases susceptibility to oxidative damage. The mitochondria are a major source of reactive oxygen species (ROS) generation.15 Superoxide is formed by a one-electron transfer from certain sites within the mitochondrial electron transfer chain (ETC) to molecular oxygen.15 FAO in both the mitochondria and peroxisomes is a source of superoxide production. Neuronal membranes are rich in polyunsaturated fatty acids (PUFAs), namely the n-6 arachidonic acid (AA) and n-3 docosahexaenoic acid (DHA), which constitutes a significant proportion of gray matter fatty acids.16,17 The double bonds in PUFAs are particularly vulnerable to oxidative stress, so neurons are susceptible to oxidative damage and require constant antioxidant protection. In activated brain areas, the local increase in oxygen consumption is lower than that of the glucose consumption,18 supporting the need to uncouple glucose use from oxygen consumption for ATP production. Indeed, elevated lactate observed during high neuronal activity supports this uncoupling of glucose use from oxygen consumption and indicates that selective activation of glycolysis is a major driver of brain ATP production,19 when oxidative ATP supply is at maximal capacity. In a resting, awake brain, most of the glucose is completely oxidized to CO2 and water.
2.1 |. Cell-specific bioenergetics
Brain cell types approach their energy needs and bioenergetic fluxes differently, and these routes align specific parts of the brain with cell-specific functions to determine nutrient needs. This impact is seen at the molecular level and involves glycolysis, the Krebs cycle, mitochondrial function, and overall strategies for making ATP. These signaling-related energy demands also involve metabolic plasticity, which is achieved by epigenetic and transcriptional activities.
2.1.1 |. Neuron-astrocyte energy shuttles
Astrocytes are the most numerous glial cells and have a major role in supporting neuronal energy demands. Interestingly, despite their need to respire, neuronal mitochondria are not structured to perform FAO, which could reflect the need of neurons to maintain an extensive membrane surface area. Conversely, respiration appears less critical for obtaining the overall bioenergetic needs of astrocytes. Astrocytes appear to maintain high rates of glycolysis, and to some extent manage the gateway through which glucose enters the brain parenchyma. Unlike neuronal mitochondria, astrocytic mitochondria can perform FAO, but this function is limited by low rates of long-chain fatty acid transport.20 At face value, this might seem to contradict the astrocyte’s reduced dependence on respiration for generating ATP. The logical explanation for this relates to the fact that astrocytes can provide carbon intermediates that fuel neuron mitochondrial respiration.21 In the brain, lactate produced via glycolysis or stored glycogen (glycogenolysis) is released from astrocytes via the monocarboxylate transporter 4 (MCT4). It is then taken up by monocarboxylate transporter 2 (MCT2) present on neuronal membranes.22 This lactate, upon conversion to pyruvate, undergoes oxidation by neuronal mitochondria. The described “astrocyte-neuron lactate shuttle hypothesis” is thus activated under aerobic conditions in which activity at neuronal glutamatergic synapses promotes the production of lactate by astrocytes that supplements the basal production of neuronal ATP normally coming from the oxidation of glucose carbon derived directly from neuronal glycolysis.23 In low glucose conditions, lactate glycogenolysis in astrocytes may be upregulated to fulfill demand by neurons. Lactate shuttling and the supply of carbon to neurons can be disrupted by various processes, including: a generalized decrease in glucose uptake via down-regulation of glucose transporter at the BBB, reduced expression of hexokinase, and alterations in the expression of astrocytic MCT4 and neuronal MCT2 during aging.24,25 A similar “ketone body shuttle” is also postulated where beta-hydroxybutyrate and acetoacetate are generated from FAO in astrocytes that additionally support neuron mitochondrial respiration.26 This reduced reliance on respiration further enables astrocytic mitochondria to recover carbon for macromolecule synthesis.
2.1.2 |. Oligodendrocytes
Oligodendrocytes are the glial cells that produce myelin. Recently findings have increased appreciation for the roles of myelin in the optimal functioning of the nervous system besides axonal integrity. Myelin provides trophic and metabolic support to axons by secretion of growth factors and by transfer of energy metabolites.27,28 Oligodendrocytes also provide direct metabolic support to neurons as a source of lactate to the axon.29,30 In the CNS, monocarboxylate transporter 1 (MCT1) is mainly expressed by oligodendrocytes.31 Lactate is released to the periaxonal space, the space between the axonal membrane and the myelin sheath, through the MCT1 and is transported into the axonal cytoplasm via MCT2.29,30 Oligodendrocytes can also supply glucose to the axons, as seen in the corpus callosum32 and thalamus.32 Oligodendrocytes also deliver glucose and lactate to the axons together with astrocytes.33 Moreover, oligodendrocytes can directly modulate axonal energy metabolism by transferring sirtuin 2 (SIRT2) in exosomes, which in turn promote axonal mitochondrial ATP production by deacetylation of mitochondrial proteins.34
2.1.3 |. Microglia
Microglia are mononuclear phagocytes in the CNS that are derived from myeloid progenitors outside of the brain during embryonic development.35 Microglia predominantly rely on ATP as an energy substrate via glucose metabolism through glycolysis, the TCA cycle, and oxidative phosphorylation to remove apoptotic neurons,36 prune non-functional synapses,37 and produce trophic factors that increase neuronal survival38 under homeostatic conditions. Lactate may also be an energy substrate for microglial metabolism, as recent reports demonstrated that microglia express MCTs and lactate dehydrogenase B which is responsible for catabolizing lactate into pyruvate.39 Microglia show remarkable metabolic flexibility depending on the environmental conditions of the brain parenchyma in vivo, as recent studies using time-lapse two-photon imaging of the mouse brain demonstrate that microglia can adapt to use glutamine as a metabolic fuel in the absence of glucose.40 This flexibility suggests that the microglia phenotypes and functions to maintain homeostasis in the central nervous system are directly tied to energy metabolism.
2.1.4 |. Endothelial cells
Endothelial cells (EC) are key components of the blood-brain barrier (BBB). The layer of ECs represents a tightly sealed cell that that results in high trans-endothelial electrical resistance and low paracellular and transcellular permeability,41 separating the brain from the blood-stream. The endothelium of the BBB has multiple specific proteins acting as transporters and receptors (Figure 2A), which are responsible for the passage of metabolites, nutrients, and junction proteins. Glucose transport is regulated by GLUTs, and solute carrier family proteins (SLCs) Among them, the most highly expressed transporter is GLUT1.42 SLCs, SLC carriers, organic cation transporters (OCTs), and facilitated diffusion carriers are additional transporters to facilitate the uptake of creatine,43 choline,44 amino acids (glutamate, aspartate, GABA, and glycine),45 nucleosides, nucleoside triphosphate, nucleobases,42 water-soluble vitamins, that is, thiamine, biotin, folate, and ascorbic acid46 as well as essential metals acting as enzyme cofactors.47 Interestingly, the number of mitochondria in ECs is five or six times greater than other tissues of the human body,48 reflecting the high energy needs of ECs to maintain brain homeostasis. The major micronutrients with a putative role in cerebral bioenergetics, their function, concentration gradient, and transport mechanisms through ECs are summarized in Table 1.
FIGURE 2.
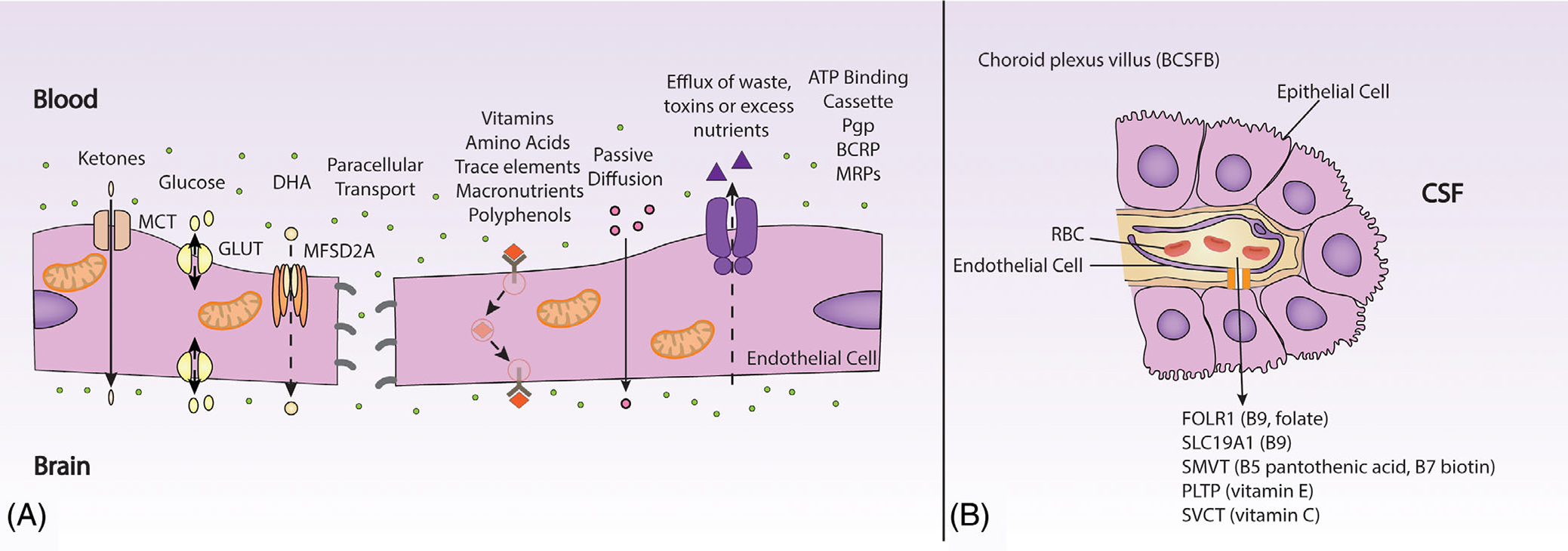
(A) Key transport proteins at the BBB include MCT for ketones, GLUTs for glucose, MFSD2A for DHA. Efflux of waste, drugs or excess nutrients is mediated by the Pgp, ATP binding cassette, BCRP and MRPs. Vitamin and nutrients are taken up via solute carriers and receptor mediated endocytosis pathways. An energy dependent mechanism regulates nutrient brain uptake transport. Endothelial cells of the BBB have a high density of mitochondria. Loss of BBB function affects gapping of tight junctions, cell surface transporters all of which are linked to mitochondrial dysfunction. (B) Key nutrient transporters at the choroid villus. ATP binding cassette, adenosine triphosphate binding cassette; BBB, blood-brain barrier; BCRP, breast cancer resistance protein; BCSFB, blood cerebrospinal fluid barrier; CSF, cerebrospinal fluid; FOLR1, Folate receptor 1; GLUTs, glucose transporters; MCT, monocarboxylate transporters; MFSD2A, major facilitator superfamily domain-containing protein 2a; MRPs, multidrug resistance proteins; Pgp, P-glycoprotein; PLTP, phospholipid transfer protein; RBC, Red blood cell; SLC19A1: Solute Carrier Family 19 Member 1; SMVT, sodium dependent multivitamin transporter; SVCT, sodium dependent vitamin C transporter
2.1.5 |. Epithelial cells
The blood CSF barrier (BCSFB) found at the lateral, third and fourth ventricle of the brain is composed of CSF facing cuboidal epithelial cells of the choroid plexus. These mitochondria-abundant epithelia have a Golgi apparatus, smooth endoplasmic reticulum, and lysosome-like vesicles to synthesize and aid in sustaining CSF and also facilitate CNS nutriture, including known transporters for vitamin C, B vitamins (folate, pantothenic acid, biotin) and vitamin E (PLTP)49 (Figure 2B).
2.1.6 |. Pericytes
Pericytes play an important role in the development of cerebral microcirculation and maintaining BBB integrity. Within the brain, pericytes actively relax or contract to change cerebral blood flow in response to localized changes in neuronal activity.50
2.2 |. Cerebral bioenergetics supply and dysfunction in AD
A myriad of cerebral bioenergetics failures in AD have been described that occur with changes in the immune system, protein and lipid homeostatic systems and intersect with brain nutrient delivery (Figures 1B and 3). These observations have led to the generation of hypotheses that impairment in the supply and utilization of energy sources to the brain can be a cause of synaptic dysfunction, neurodegeneration, and cognitive decline in AD, rather than just being a consequence of AD neuropathology and cell death characterized by the formation of extracellular amyloid-beta plaques and intracellular, insoluble tau tangles. In support of this rationale, amyloid plaques first appear in brain regions that feature high levels of aerobic glycolysis in patients with AD,51 which suggests that brain regions that are most dependent on astrocyte assistance for meeting their bioenergetic needs are those that are most likely to acquire plaques. Moreover, mitochondrial bioenergetic stress associates with measures of chronic inflammation and oxidative stress.52
FIGURE 3.
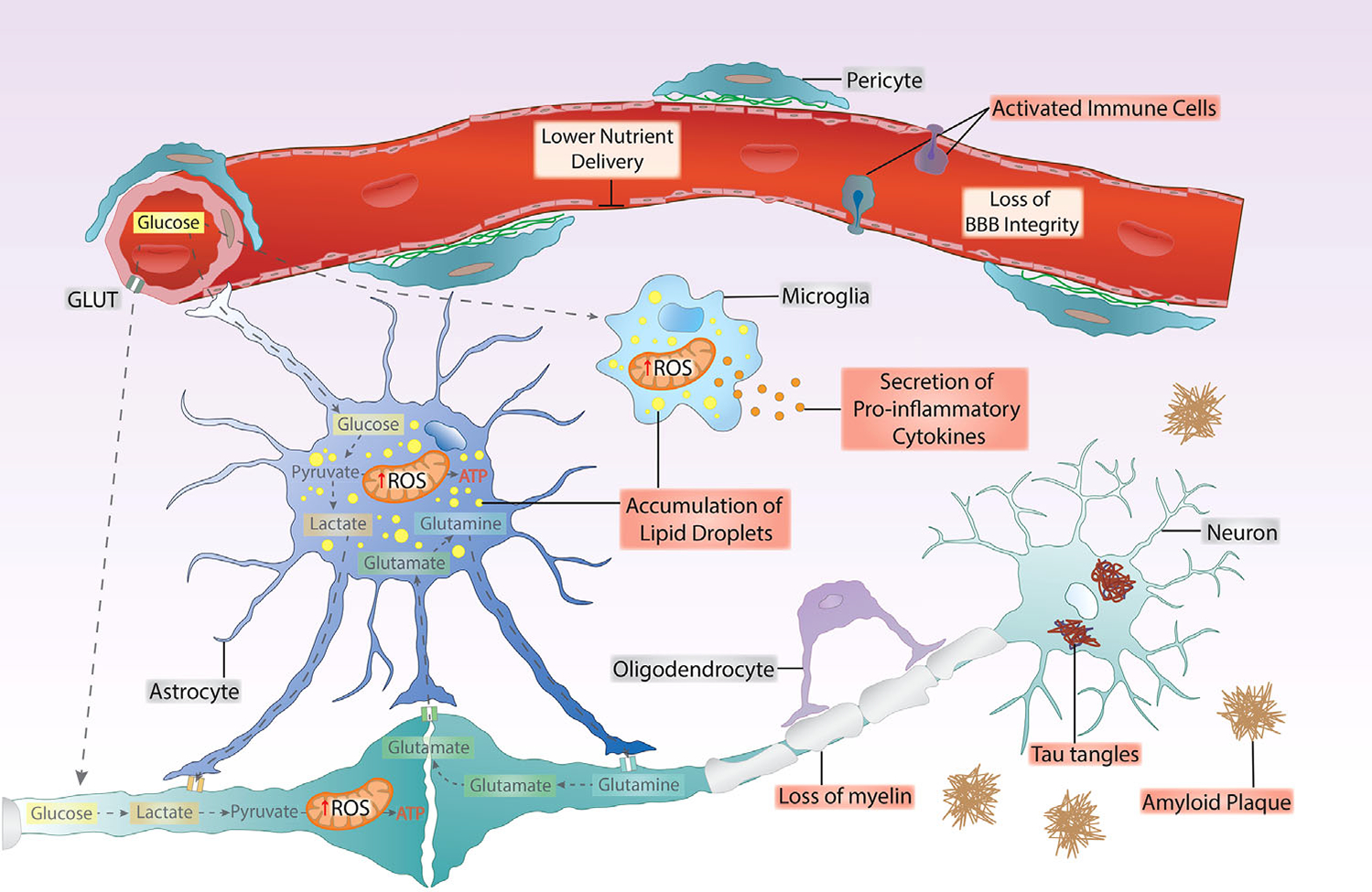
Cell-specific brain changes in Alzheimer’s disease, showing interactions of brain nutrient uptake and utilization with blood-brain barrier (BBB) function. Bioenergetic failure and loss of BBB function is associated with chronic inflammation and oxidative stress (ROS) in several brain cell types, associating with an increase in lipid droplets in glial cells, loss of myelin, and the appearance of classic AD pathology markers of amyloid plaques and neurofibrillary tangles. Bioenergetic stress is reflected initially by an increase in glycolytic flux and dysfunctional mitochondrial oxidative phosphorylation.
2.2.1 |. Dysregulated glucose metabolism
Imaging biomarkers of brain glucose uptake, oxygen utilization, and blood flow in cognitively normal adults from 20 to 82 years revealed that aging associates with lower brain glucose uptake that exceeds the decrease in oxygen use, resulting in declining brain glycolysis.53 In patients with mild cognitive impairment, lower brain glucose uptake using 18F-fluorodeoxyglucose (FDG)-positron emission tomography (PET) scans predicts progression to AD dementia54. Consistent with lower brain glucose uptake and neurovascular coupling, microvascular glucose transporters GLUT1 were shown to be decreased by nearly 50% in the cerebral endothelium of patients with AD dementia.55 GLUT3 concentrations are also decreased in AD compared with age-matched controls.56,57 Preclinical evidence suggests that changes in brain glycolysis is associated with defects in mitochondrial oxidative phosphorylation, reflecting an energy crisis.58 The need for more energy substrates drives greater mobilization of fatty acids from neuronal membranes, increasing susceptibility to oxidative damage. For example, the metabolism of arachidonic acid (AA) leads to an increase in neuroinflammatory responses from astrocytes and microglia and oxidative stress byproducts.59,60 Prostaglandins, leukotrienes, and pro-resolvins derived from liberated PUFAs appear to promote a state of unresolved chronic inflammation in AD that can ultimately lead to neuronal loss.61,62
2.2.2 |. Impaired mitochondrial FAO
There is good evidence of impaired FAO in AD. Disturbance in respiration-associated components are observed in AD. These include: pyruvate dehydrogenase complex, which catalyzes the entry of carbons derived from glucose into the citrate cycle; the α-ketoglutarate dehydrogenase complex comprises a key catalytic step in the citrate cycle and is an enzyme of glutamate metabolism; and cytochrome oxidase or complex IV, the component of the electron transport chain which uses molecular oxygen as one of its substrates.63,64 When glucose supply is reduced to the brain during metabolic challenges, FAO in mitochondria provides ketone bodies that can be a supplemental energy source for brain bioenergetics.65 Compared to neurons and microglia, astrocytes preferentially express carnitine palmitoyltransferase-1 (CPT-1), which combines L-carnitine with long chain fatty acid (LCFA) to generate acylcarnitines that then enter mitochondria for FAO.20 As such, carnitine/acylcarnitines metabolism may play an important role in brain bioenergetic failure states where FAO is detrimental for normal neuronal function. Medium chain acylcarnitines (MCA) are key substrates for FAO.66 MCA can be converted to ketone bodies in the periphery and transported to the brain to support bioenergetics.67 The contributions of MCA and LCA to FAO in AD warrant further investigation, as reports have shown that MCA levels are decreased68–70 or elevated71,72 in plasma and cerebrospinal fluid (CSF) of AD patients. Given that LCFA is enriched in polyunsaturated fatty acids that have numerous double bonds, they are susceptible to lipid peroxidation, thereby contributing to ROS generation. On the other hand, medium chain fatty acids (MCFA) likely contain fewer double-bonds, and this may contribute to lower levels of ROS generation.
2.2.3 |. Oxidative damage
There is evidence of oxidative damage to proteins and lipids in AD,71 and we anticipate effects from ω-oxidation will be manifest in AD. Ω-oxidation of lipids resulting in short- and medium-chain dicarboxylic acid (DCA) production is normally a minor pathway for normal fatty acid metabolism72; it is considered a rescue pathway that compensates for defects in FAO, and urinary excretion of DCAs increase in persons who have defects in fatty acid metabolism.73 In addition, increased ω-oxidation may provide succinyl-CoA for the Krebs cycle and gluconeogenesis.73,74 Ω-oxidation is also upregulated with increased cytochrome P450 enzyme activity.75 C4-DCA modifies (succinylates) several mitochondrial proteins,76 while other DCAs such as azelaic acid (C9-DCA) may have anti-inflammatory, antioxidative, and bactericidal activity.77 Thus, changes in DCA levels can directly dysregulate energy pathways and influence AD-related mechanisms.
2.2.4 |. Cell-specific bioenergetic failure in AD
All brain cell types all show evidence of inflammation, oxidative and bioenergetic stress contributing to neuronal energy failure (Figure 3). Oligodendrocyte dysfunction and white matter loss are often among the earliest brain changes in AD,78–82 suggesting that the supply of energy substrates from oligodendrocytes to the axons such as lactate will be diminished, resulting in aggravation of the neuronal dysfunction. Aβ accumulation and tau hyperphosphorylation may lead to further disruption of myelin integrity and oligodendrocyte maturation and metabolism through oxidative stress, neuroinflammation, and/or excitotoxicity.80,83–87 Like oligodendrocytes, astrocytic morphology and functions are abnormal in AD, resulting in deleterious effects that include reduced carbon delivery to neurons for oxidative phosphorylation, and dysregulated linkages between neuronal energy demand and regional blood supply.88 Similarly, microglial metabolism shifts from oxidative phosphorylation to aerobic glycolysis as observed upon acute exposure to stressors such as fatty acids or Aβ.89,90 This metabolic shift to aerobic glycolysis in microglia can be a driver of AD pathology, as the increased production of lactate in microglia results in epigenetic modifications that activate glycolytic genes in a positive impact loop in the 5XFAD mouse model.91 Endothelial cells and pericytes regulate the energy sensing pathways in response to changes in nutrient availability, allowing them to move between quiescent and proliferative states.92,93 Dysfunctional FAO in pericytes can cause them to undergo apoptosis and detach from the endothelial cell, contributing to capillary pruning.93 Pericyte loss plays an important role in neurovascular dysfunction. It leads to brain vascular damage by a reduction in brain microcirculation causing diminished brain capillary perfusion, cerebral blood flow, and cerebral blood flow responses to brain activation, which ultimately mediates chronic perfusion stress and hypoxia.94 Taken together, these data suggest that changes to cellular bioenergetics are key factors that influences the onset AD pathophysiology. However, many questions remain about the causal role of individual cell type bioenergetics and AD, and future studies should aim to define these metabolic changes more clearly over the entire AD time course.
2.2.5 |. Blood-brain barrier dysfunction, nutrient transport, and maintenance in the CNS
BBB breakdown as assessed with dynamic contrast enhanced MRI or through lumbar puncture derived CSF albumin index is associated with aging,95,96 neuroinflammation,97 AD,98 and accelerated cognitive decline.97,98 Compromise of BBB integrity is associated with brain accumulation of toxic molecules involved in neuronal degenerative changes.99 Several types of neurotoxic blood-derived molecules that can enter brain after BBB breakdown leading to synaptic and neuronal dysfunction.100 These include iron species derived from hemoglobin; fibrinogen that activates microglia promoting neuroinflammatory response, and also causing retraction of neurites and white matter injury to oligodendrocytes and pericytes; thrombin that is directly neurotoxic and reviewed in more detail by Sweeney et al.100 A pro-inflammatory response may also cause BBB disruption via upregulating various factors, and this can cause leukocytes to enter perivascular spaces and may further cause an inflammatory cascade. Dysregulation in BBB function occurs with known risk factors for AD such as aging,95 APOE genotype,101 and conditions such as metabolic syndrome.102
The functional changes at the BBB and BCSFB can alter brain perfusion and delivery of glucose, oxygen, and other nutrients to the brain from the blood, which may have implications for nutritional interventions across disease stages and age strata, even in the absence of whole-body tissue deficiencies. This may occur through elevated rates of catabolism due to accumulating neurovascular pathology (i.e., cerebral amyloid angiopathy and arteriosclerosis) or inflammation or a combination of both. Aging and conditions leading to BBB breakdown may disturb the brain’s capacity to meet its nutritional requirements through several mechanisms, including gapping of tight junctions of BBB (endothelia) or BCSFB (cuboidal epithelia), cell surface transporter dysfunction (i.e., solute carriers and receptor mediated transcytosis damage), and mitochondrial dysfunction leading to loss of energy potentials.103 BBB breakdown may cripple the brain’s capacity to maintain nutrient concentrations to battle reactive oxygen species (i.e., leaking of vitamin C from the CNS into periphery or damage to SVCT at the choroid plexus)104 and support DNA methylation reactions (one carbon metabolism).105,106 Defects in BBB function affect notonlybrainglucosemetabolism107 and some micronutrients but also important fatty acids such as the transport of DHA to the brain108 via MFSD2A.109
2.3 |. APOE4 and cerebral bioenergetics
Apolipoprotein E ε4 polymorphism (APOE4) is by far the strongest genetic risk factor for late-onset AD.110 As its name suggests, apolipoprotein E (ApoE) is a surface protein component of lipoprotein particles, and thus it is perhaps unsurprising that it affects several metabolic functions. This encompasses ApoE isoform-specific alterations ranging from cholesterol trafficking and efflux, intracellular lipid storage, glucose uptake and utilization patterns, brain insulin signaling, and mitochondrial function.110 These various APOE-associated changes in cerebral (and peripheral) metabolism have been extensively reviewed.111–115 APOE4 may affect BBB integrity before the onset of cognitive decline through chronic unresolved inflammation,101 is associated with presymptomatic brain glucose hypometabolism,116 and may lower delivery of n-3 PUFAs to the brain prior to the onset of dementia.108 Whether APOE4 affects the transport of ketones into the brain is still not clear.
More recent studies have highlighted emerging areas of interest regarding the effects of APOE4 on cerebral metabolism, particularly in glial cells. Glial carbohydrate metabolism was shown to be a primary pathway associated with cognitive impairment and AD pathology by a comprehensive proteomics analysis.117 Although the effect of APOE was muted in the study, several other publications highlight important roles for APOE in modulating glucose metabolism. For example, APOE4-expressing astrocytes increase glycolytic flux relative to APOE3 and APOE2 astrocytes.118 A follow-up translational study suggests that this APOE4-associated increase in glycolysis in astrocytes appears to be mirrored in whole-body metabolic measures of APOE4 mice as well as in the plasma metabolome and breath-based calorimetry of young women carrying APOE4.119 Effects of APOE isoforms on glial metabolism appear to also include microglia, as a recent study showed that APOE4-expressing human iPSC-derived microglia have lower rates of both oxygen consumption and glycolysis.120 Younger APOE4 carriers have higher brain uptake of DHA,121 suggesting preference for PUFAs, possibly due to increased DHA consumption.
Multiple studies also highlight a role for APOE in modulating the accumulation and storage of intracellular lipids in astrocytes. For example, the expression of APOE4 by astrocytes led to an increased accumulation of lipid droplets in multiple model systems.122–124 Qi et al. recently expanded upon these findings by similarly showing that APOE4 leads to lipid accumulation in astrocytes and further suggested that a decreased capacity of these astrocytes to buffer neuronal lipids and degrade fatty acids via oxidation or other mechanisms may compromise metabolic support to neurons.125 Similarly, a recent study by Rawat et al. highlighted the susceptibility of apoE4 protein aggregates to decrease recycling and lipidating functions of ATP binding cassette A1 (ABCA1).126 While there is strong evidence that FAO is impaired in AD, particularly among APOE4 carriers, the mechanism by which APOE genotype contributes to bioenergetic deficits in AD remains to be fully investigated. Studies show that APOE4 carriers affect the metabolism of PUFAs,121,127 which could be due to their increased breakdown or reduced transport into the brain.128 Deficits in maintaining adequate brain bioenergetics in APOE4 carriers can also be attributed to FAO of intracellular lipid pools.122 APOE4 carriers with dementia are unable to adequately compensate energy generation through FAO.129 Mouse models of human APOE show that acylcarnitine levels decline with age in APOE4 mice, whereas APOE3 mice maintain adequate supplies and APOE2 mice have elevated acylcarnitine levels in blood with age.130 In sum, these studies highlight APOE4-associated increases in intracellular lipid accumulation, decreased cholesterol efflux, and incomplete FAO, with some of thes changes appearing early in life and impacting brain energy metabolism.
2.4 |. Gut microbiome dysbiosis and cerebral bioenergetics
A key factor that determines nutrient absorption and metabolism from dietary components are the trillions of microorganisms that reside in the gut and the vast array of enzymes encoded in their metagenomes, colloquially known as the gut microbiome. Diet and nutritional status play a major role in the symbiotic relationship between the gut microbial diversity and host metabolism. Nutritional imbalance impacts both microbial communities in the gut and host physiological functions including brain homeostasis that is heavily dependent on gut-derived secondary metabolites, which include neurotransmitters.131 The gut microbiota-derived secondary metabolites exert their effects either locally or via systemic circulation as stored energy reserves in external organs.132 Gut-brain signaling has been widely studied, and one of the areas of interest linking the digestive system with the brain is satiety and appetite regulation. However, in the last decade, the focus has dramatically shifted towards a better understanding of the microbiota-gut-brain relationship, which is frequently referred to as the MGB axis.133 MGB represents a bidirectional communication pathway mediated by microbiota and their secondary derivatives. Gut microbes release a range of neuromodulatory metabolites, including γ-aminobutyric acid (GABA), dopamine, serotonin, acetylcholine, trimethylamine oxide (TMAO) and short chain fatty acids (SCFAs).134–136 Bacteria-derived neuromodulators enter the brain either via afferent vagal nerve fibers that directly connect the enteric nervous system (ENS) and central nervous system (CNS) or cross the BBB from portal circulation.137,138
Age is a key factor that influences gut microbiome composition and function. While normal health in adulthood is characterized by a stable microbiome,139 late life changes in gut microbiome composition shifts toward less stable states. A persistent imbalance of gut microbial communities, termed dysbiosis, has been reported in a variety of neuropsychiatric and neurodegenerative disorders, including AD, prompting the question of whether age-related microbiome changes combined with differential gut metabolites could contribute to mitochondrial dysfunction linked to neuropsychiatric disorders.140–146 Mounting evidence also links an abnormal increase in specific enteric microbes to a range of chronic late life inflammatory diseases with primary pathologies outside the GI, including CNS disorders.132,137 Many neurological disorders are shown to be associated with microbiota-associated GI symptoms and possibly stemming from altered gut microbial diversity.
Microbiome-derived secondary metabolites may contribute to brain bioenergetic dysfunction during aging. Microbiota-derived SCFAs including butyrate, acetate and propionate, are essential for normal brain health.147 Several studies have highlighted the importance of SCFAs in understanding pathogenesis associated with multiple neurodegenerative disorders, including AD.142,143 Although the precise mechanism of action of SCFAs on brain is still being explored, mounting data suggest SCFAs are essential factors for the effective functioning of mitochondria. SCFAs are associated with the mitochondrial function specifically with the ETC involved in ATP production, oxygen consumption and membrane potential in the brain. A recent study in germ-free mice demonstrated that gut microbiota and the acetate they produce are important contributors to brain mitochondrial bioenergetics, particularly microglia function via regulation of mitochondrial oxidative phosphorylation.148 SCFA concentrations are lower in CSF of AD participants, providing further support that changes in microbiota metabolism may contribute to brain energetic failure in AD.149 Conversely, TMAO, a gut metabolite derived from microbial metabolism of dietary carnitine and choline, has been shown to influence brain function. A combination of gut dysbiosis and increased levels of TMAO is observed in anxiety and cognitive deficits,150 and TMAO is reported to be elevated in the CSF of dementia patients.151 TMAO also promotes vascular aging as well as impairment of mitochondrial function and increased oxidative stress.150 This evidence strongly suggests that gut microbiota and their metabolites may play a role in aging and mitochondrial dysfunction observed in AD.
3 |. EVIDENCE FOR TARGETING CEREBRAL BIOENERGETICS DEFICITS IN AD WITH NUTRITION
The effect of diet on the brain is complex, modulated by several factors that may affect the transport and metabolism of nutrients making them readily available to the brain. Novel modeling approaches such as network analysis is needed in a much more diverse sample.152 This network may vary by region, social, and cultural practices and includes genetics (e.g., APOE4), vascular risk factors (hypertension, exposure to air pollution), nutritional intake, diet patterns, or other lifestyle factors such as social, cognitive, and physical activities. The complex interaction of these factors on brain energy homeostasis and neuronal survival has implications toward personalized recommendations, and is illustrated in Figure 4. Given this complexity, it is not surprising that most dietary supplement interventions using a single or a few nutrients in a single “average population” have been disappointing in their ability to slow cognitive decline or AD progression152 compared to the effect of the major diet patterns on dementia risk seen in observational cohorts (Table 2). These issues notwithstanding, there remain open questions of whether certain targeted nutrient interventions can restore cerebral bioenergetics in populations selected with signs of disturbed nutritional and cerebral bioenergetic dysfunction when guided by selected biomarkers. Efforts to advance our understanding of the role of cerebral bioenergetics in AD etiology will need to account for the appearance and characteristic biology of its classic histopathology including beta amyloid or tau perturbations the field associates with AD.
FIGURE 4.
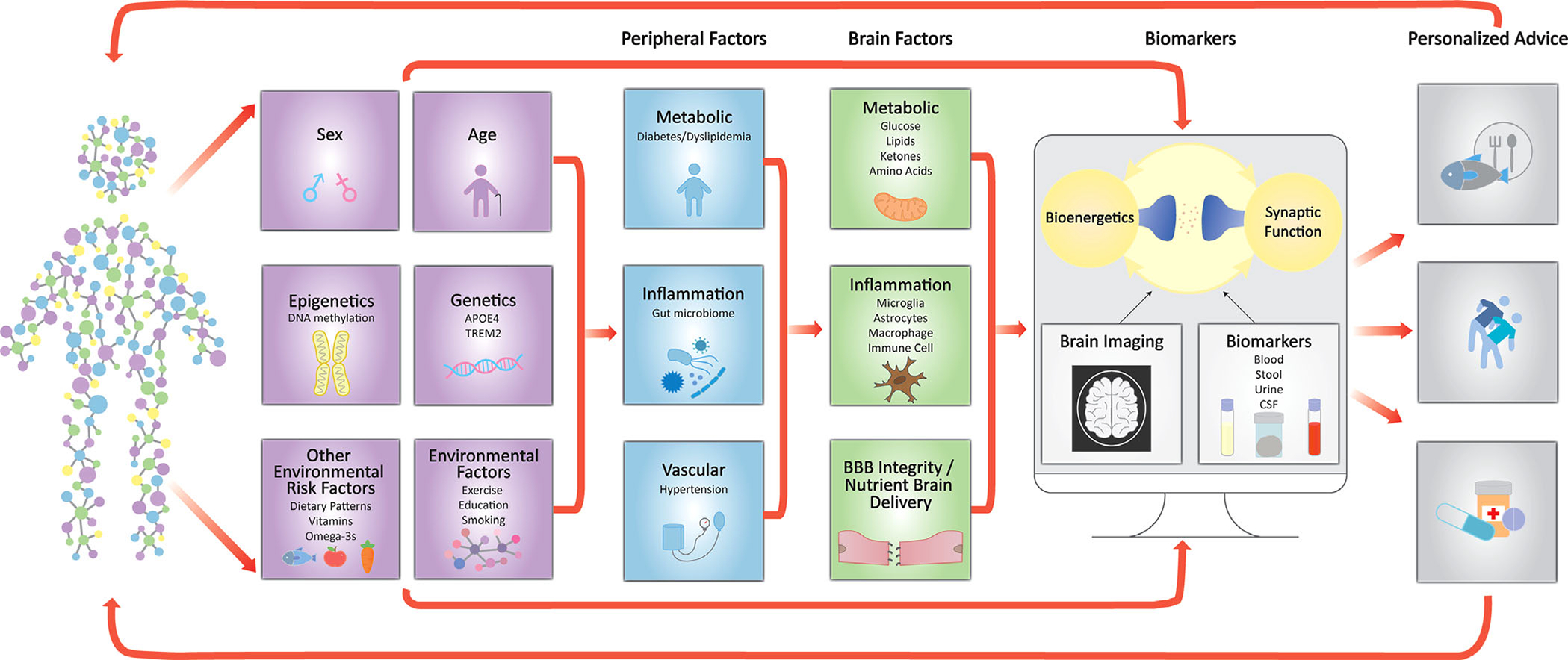
An illustration of how capturing information on environmental, genetic, systemic and brain risk factors that affect neuronal survival and synaptic function can lead to personalized dietary and lifestyle recommendations. BBB, blood-brain barrier; CSF, cerebrospinal fluid
TABLE 2.
Dietary and nutrient biomarker patterns associated with cognitive function and dementia risk
| Diet or biomarker pattern | Micro/macronutrient emphasis | Dementia risk, cognitive outcomes and biomarkers |
|---|---|---|
| MIND diet173,253 | Higher intake of vitamin E, folate, flavonoids, carotenoids, dietary fiber, monounsaturated fats, and lower intake of saturated and trans fatty acids | Lower incident AD and cognitive decline |
| DASH diet254 | Higher intake in potassium, magnesium, fiber, calcium, monounsaturated fats, and protein; low in saturated fats, cholesterol, and sodium | Less cognitive decline and lower incident AD255 |
| MeDi256 | High intake of folate, vitamin E, carotenoids, flavonoids, and other antioxidants, dietary fiber, omega-3 fatty acids; lower intake of saturated fatty acids | Lower incident AD257 |
| MMKD174 | Low carbohydrate (5%-10%), high fat (60%-65%) and 30% protein. Like MeDi guidelines that emphasizes protein sources low in saturated fat (fish, lean meats), healthy fats, fruits and vegetables, wholegrains, and 1 glass of wine per day | Lower indication of AD pathology (higher CSF abet42 and lower CSF tau); Increased cerebral perfusion and increased cerebral ketone body uptake (11C-acetoacetate PET) following MMKD |
| Nutrient biomarker pattern Oregon Brain Aging Study258 | Higher blood levels of B1, B2, B6, folate, B12, C, D, E, omega-3 fatty acids, carotenoids (lutein and zeaxanthin) and lower levels of trans fatty acids | Superior cognitive function, less white matter lesions and higher total brain volume |
| Nutrient biomarker pattern Three-City study259 | Higher blood levels of vitamin D, carotenoids, and polyunsaturated fatty acids and lower levels of saturated fats associated with less risk of dementia | Lower incident dementia |
| Nutritional Risk Index Multi-domain Alzheimer prevention trial260 | Higher blood levels of vitamin Dand omega-3 fatty acids and lower levels of homocysteine | Participants with optimum nutritional status had superior cognitive performance |
Abbreviations: DASH, dietary approaches to stop hypertension; MeDi, mediterranean style diet; MIND, mediterranean-DASH diet intervention for neurodegenerative delay; MMKD, modified mediterranean ketogenic diet.
3.1 |. Single or few nutrient trials
Some dietary supplements, B vitamins, n-3 PUFAs, and ketones have shown preliminary promising effects on brain bioenergetics in the aging brain. Closer examination of these nutrient trials identifies the need for more targeted or personalized approaches and to define neuroprotective ranges of nutrients to enroll individuals outside this beneficial range for intervention.153 We discuss here some encouraging single or few nutrient interventions:
3.1.1 |. B1 vitamins
There is evidence linking abnormalities in thiamine (vitamin B1) availability and metabolism to the pathophysiology of AD through brain bioenergetics. Thiamine-deficient humans with Wernicke Korsakoff syndrome have significant neurofibrillary tangles.154,155 In mice, thiamine deficiency increases the phosphorylation of tau,156 and treatment with benfotiamine diminishes phosphorylation of tau in at least three different animal models of AD.157,158 These results stimulated a single site blinded Phase 2a randomized placebo-controlled pilot trial of benfotiamine to provide preliminary evidence of feasibility, safety, and efficacy. The trial tested whether a 12-month treatment with benfotiamine would delay clinical decline in amyloid positive patients (ascertained with amyloid PET) with amnestic mild cognitive impairment (MCI) (MMSE > = 26) or mild AD (26 > MMSE > 21) compared to placebo.159 The primary clinical outcome was AD Assessment Scale-Cognitive Subscale (ADAS-Cog), and secondary outcomes were the clinical dementia rating (CDR) score and brain glucose uptake measured by FDG-PET. The trial showed that benfotiamine at a dose of 600 mg per day is safe in patients with early AD. The treatment delivery was efficacious as shown by a 161-fold mean increase in blood thiamine. The change in the primary outcome did not reach statistical significance, and there were signals that secondary outcomes were improving. These encouraging preliminary findings support validation in targeted/personalized interventions with responsive clinical outcomes.
3.1.2 |. Vitamin B12, vitamin B6, and folate
A systematic review of 21 trials explored the preventive efficacy of vitamin B12, vitamin B6, or folic acid alone or in combination in any form on cognitive decline of patients with mild cognitive impairment or elderly adults without cognitive impairment.160 This analysis demonstrated that vitamin B supplements significantly lowered the levels of serum homocysteine levels and prevented the decline of global cognitive function, but did not affect other cognitive domains. Nevertheless, the effect sizes for those treated with vitamin B supplements compared to placebo were modest. Like B1, targeted and well-designed randomized controlled trial (RCT) in participants with high baseline homocysteine levels and responsive cognitive outcomes can clarify whether they have preventive efficacy.
3.1.3 |. PUFAs
Brain PUFAs have important roles in brain bioenergetics and synaptic functions, but PUFA supplementation clinical trials have not succeeded in showing benefit on cognition, despite several observational cohorts linking higher n-3 PUFA intake or blood levels with better cognition or lower AD incidence.153,161 Brain autopsy studies in APOE4 carriers with dementia demonstrate the activation of pathways that increase the breakdown of brain PUFAs (e.g., via calcium-dependent phospholipase A2 [cPLA2] activation162) and PUFAs with numerous double-bonds are more susceptible to lipid peroxidation, thereby contributing to ROS generation. However, there might be a role for PUFA supplementation in AD prevention for vulnerable populations. Using 11-C DHA PET imaging, the APOE4 brain appears to extract more DHA from blood than the non-APOE4 brain several decades before any evidence of cognitive decline.121 This greater dependence on systemic DHA as a brain substrate in APOE4 poses vulnerability to lower DHA intake, since blood DHA levels are largely determined by dietary consumption. As the APOE4 brain ages with a lower BBB function and the accompanied lower ability to extract DHA from the circulation,108 the ability of dietary DHA intake to reverse AD pathology appears to wane. The field awaits more personalized interventions (e.g., younger APOE4 carriers without high n-3 PUFA intake and prior to the onset of clinical disease) with high dose and long term PUFA supplementation to address this question.163
3.1.4 |. Ketones
Given the difficulty in restoring disrupted brain glucose metabolism, another therapeutic strategy involves improving brain energy metabolism with ketones in MCI or AD. This question was addressed with a ketogenic medium chain triglyceride (kMCT) supplement. The largest investigation to date included 152 older participants with mild to moderate AD, followed-up over 90 days with 20 g/day of kMCT (tricaprylin) supplementation. A significant improvement in the ADAS-Cog score was observed only in APOE4 non-carriers.164 A higher dose of kMCT (42 g/day of tricaprylin) improved cognitive stability in mild-moderate AD in 20 participants who underwent a 6-month double-blinded, randomized, placebo-controlled cross-over study with a 6-month open label phase.165 In older adults with MCI, Fortier et al. showed that 30 g of kMCT supplementation per day over a 6-month period significantly improved cognitive function.166 At baseline, the brain glucose deficit was about 10%, which was reduced to about 7% by ketones from the kMCT.166 Post-intervention, scores for episodic memory, executive function, and language were all significantly improved in the kMCT group only.167 These cognitive improvements were directly associated with increased brain ketone uptake and/or increased plasma ketones in the kMCT group. However, there were no changes in brain FDG uptake, cortical thickness, volume of brain regions, or brain blood flow. Analysis of diffusion and PET imaging before and at the end of the intervention period showed that ketones were actively utilized by multiple white matter tracts in the kMCT group and that the uptake of ketones in these tracts was directly related to improvement in processing speed168 and attention.169 A double-blind, randomized, placebo-controlled crossover study of kMCT was undertaken in 53 mild to moderate AD patients and showed cognitive improvements in the ADAS-Cog-C among APOE4 non-carriers.170 In a recent systematic review of RCTs with ketogenic therapy in AD, an APOE4 genotype treatment effect was noted with APOE4 negative participants having a better treatment response than those who were APOE4 positive.171 In addition to AD, impaired brain glucose metabolism is a feature of other neurodegenerative disorders including frontotemporal dementia.172 Large randomized controlled trials with long-term follow-up and appropriate clinical and biological outcomes (including plasma, CSF, or brain metabolism biomarkers) are needed to validate the outcomes and long-term acceptability of ketone interventions in MCI or AD. The effect of APOE4 on ketone brain uptake requires further studies.
3.2 |. Diet patterns and whole diet interventions
While specific nutrients uniquely impact bioenergetics pathways potentially leading to AD, these nutrients are contained in foods and consumed together within a diet. Moreover, they may have synergistic (or at least additive) effects on metabolic pathways, so that their co-consumption within a diet may potentiate benefits (or harms) in relation to brain health. Thus, dietary patterns are tremendously important to consider when studying nutrition and the brain. There is emerging evidence from human neuroimaging studies that diet patterns overall may influence cerebral bioenergetics, possibly as early as middle adulthood, prior to any effect on other AD biomarkers and brain structural changes.
The Mediterranean diet (MeDi), DASH (Dietary Approaches to Stop Hypertension), and the MIND (Mind-DASH Intervention for Neurodegenerative Delay) are each associated with less cognitive decline and dementia incidence (Table 2). These diets are characterized by high intake of vegetables, fruits, fish, legumes, and less processed foods, red meat, and confectionary foods. Although there are similarities among these dietary patterns (fish and vegetable intake), there are also differences such as berries and emphasis of dark leafy greens in the MIND and olive oil (monounsaturated fatty acids) as the main source of fat in both the MeDi and MIND.173 A modified Mediterranean Ketogenic Diet that requires low carbohydrate and high mono and polyunsaturated fat intake and lower saturated fat intake has been associated with lower signs of AD pathology and increases in cerebral blood flow in early interventions.174
Other studies have identified dietary patterns or nutrient biomarker patterns associated with less brain atrophy and white matter lesions,175 but the estimated effects on outcomes related to cerebral bioenergetics such as arterial spin labeling derived cerebral blood flow, FDG PET and BOLD imaging have been limited. Measuring brain glucose metabolism with FDG PET as a proxy of neuronal activity, a few longitudinal neuroimaging studies of cognitively intake adults (aged 30–60 years) with other risk factors for AD suggested that healthy diets, such as the Mediterranean diet, or healthy nutrient patterns, influence brain glucose metabolism in mid-to-late adulthood. For example, an analysis of 70 participants from the cohort found that lower adherence to a Mediterranean diet was associated with reduced FDG-PET and a faster rate of FDG decline over 3 years in AD-affected regions (3.3% per year decline in those with lower adherence to a Mediterranean diet [i.e., scored 0–4 over a total of 9 possible points], versus 0.3% per year in those with higher adherence [score 5–9]).176 This reported decrease in glucose metabolism in persons with lower adherence to Mediterranean diet paralleled significant 11C-Pittsburgh compound B (PIB) increases, while volumetric MRI results were unaffected by treatment. In 52 participants from the same cohort study, a healthy nutrient pattern combining unsaturated fats; vitamins A, B12, C, D, and E; zinc; carotenoids; and fiber, was associated with increased FDG-PET glucose metabolism177 providing some evidence of potential key nutrients influencing brain glucose metabolism. In fact, there are emerging studies of the impact of antioxidant-rich fruits and vegetables, n-3 PUFA and low-glycemic index carbohydrates,178 zinc,179 fish oil supplementation rich in n-3 PUFA,180 vitamins B12, E, D, PUFA, antioxidants and fibers,177 and multi-nutrient supplements181,182on FDG PET derived cerebral glucose metabolism. There are also diet patterns such a Mediterranean-type diet that have been associated with beneficial cerebral metabolism profiles in both cross-sectional183 and longitudinal176 studies.
3.3 |. Emerging targets for cerebral bioenergetic restoration
There are emerging bioenergetic targets that have yet to be fully explored in human trials, but preliminary evidence is promising. These mechanisms include targeting FAO, the gut microbiome, and epigenetic modifications to restore cerebral bioenergetics in AD.
3.3.1 |. Acetyl-L-carnitine
Acetyl-L-carnitine is a mitochondrial bioenergetic substrate that may improve brain functions through FAO, but clinical trial findings are inconsistent. A small-scale study placebo-controlled randomized trial (RCT) of acetyl-l-carnitine in 36 AD participants showed some trends in improvement in short-term memory.184 Another RCT of acetyl-L-carnitine in 130 clinically diagnosed AD patients showed that participants treated with acetyl-L-carnitine had significantly better scores on tests that assessed verbal abilities and verbal memory as well as attention.185 However, another RCT of acetyl-L-carnitine among early onset AD cases45–60,62–66 did not achieve its primary outcome of cognitive decline.186 Others have explored acetyl-L-carnitine as part of nutritional cocktail showing some efficacy on cognitive outcomes,187 but these studies lack efficacy biomarkers other than supporting overall bioenergetic processes in the brain. Given the uncertainty with acetyl-L-carnitine doses, brain delivery and with the short duration of these preliminary trials, the potential therapeutic effects of targeting FAO via L-carnitine remain unknown. Whether acetyl-L-carnitine is serving as a direct supplement for FAO or is being converted into ketone bodies in the periphery and then supplied to the brain also remains unknown.
3.3.2 |. Nicotinamide adenine dinucleotide (NAD+) precursors
NAD is a coenzyme for multiple metabolic reactions, including glycolysis, the electron transport chain and sirtuin activity.188 The ratio of reduced (NADH) to oxidized NAD (NAD+) increases with aging.188 It has been postulated that NAD+ boosters may improve mitochondrial metabolism during aging. Two major NAD+ precursors are nicotinamide riboside (NR) and nicotinic mononucleotide (NMN) that within cells can be converted to NAD+ and may raise NAD+. Some of the animal studies with NAD+ supplements have shown promise with aging188 and reducing microglial inflammation and cellular senescence,189 although in the largest mouse span extension study, Intervention Testing Program (ITP), NAD+ precursors did not extend lifespan.190 The challenge with these supplements is the lack of efficacy biomarkers at the cellular level, their unclear brain delivery, and very limited data for cognitive outcomes, although several smaller trials are in progress.191
3.3.3 |. Microbial metabolites and probiotics
Direct supplementation of secondary metabolites or stimulation of their production by providing either a specific medication, appropriate bacterial strains (i.e., probiotics), or microbiota-enhancing diet features (prebiotics), are an emerging opportunity for a wide range of disorders, including AD. Increases in SCFA production can be achieved by direct supplementation, or via high fiber supplementation.192 In general, in vitro experiments have shown SCFAs inhibits AD-related Aβ aggregation.143 However, SCFA supplementation in preclinical animal models of neurodegenerative diseases has yielded conflicting results. Butyrate supplementation inhibits neuroinflammation and helps maintain BBB integrity in mice,193 possibly through improvement of mitochondrial function by reducing oxidative stress and increasing oxidative phosphorylation.194 However, in a pre-clinical model of Parkinson’s disease, microbiomes from PD patients and propionate led to enhanced motor dysfunction.195 Synbiotics, defined as combinations of probiotics and prebiotics mixed in appropriate proportions have been shown to ameliorate a host of CNS disorder pathologies. Lactobacillus salivarius, a probiotic when used in combination with prebiotics has engendered enhanced mitochondrial function in 6-hydroxydopamine (6-OHDA)-induced PD rats.196 Similarly, Lactobacillus paracasei, another probiotic belonging to the same genus and a prebiotic XOS, when used as synbiotics, is shown to attenuate mitochondrial dysfunction and prevent hippocampal oxidative stress and apoptosis.197 However, Akkermansia muciniphila as a standalone probiotic supplement has been shown to produce increased CSF nicotinamide levels in mouse model of amyotrophic lateral sclerosis.198
3.3.4 |. Mitochondrial complex IV
Complex IV is part of the electron transport chain and this complex becomes negatively affected in AD. Importantly, Complex IV is the site of molecular oxygen consumption. Interestingly, studies with dietary creatine,199 and also other compounds,200 have shown promise to reverse Complex IV deficits in transgenic mouse models. These findings become important for therapeutically targeting aging and age-related diseases, such as AD and obesity with potential nutritional interventions that may slow or reverse these processes. In fact, key roles have been found for Complex IV dysfunction during aging that have been linked to age-dependent obesity201 and associated inflammatory processes. Surprisingly, there are currently no FDA-approved mitochondrial medicines and only one European Medicines Agency approved drug Idebenone (Raxone) is available for treating mitochondrial dysfunction in Leber’s hereditary optic neuropathy (LHON). However, so-called mito cocktails containing vitamins and minerals, mitochondrial transfusions, and druggable mitochondrial targets are now being aggressively explored.
4 |. FUTURE DIRECTIONS TO GUIDE NUTRITION INTERVENTIONS TARGETING CEREBRAL BIOENERGETICS
As the aging brain and AD risk factors such as APOE4 adversely affect the delivery of nutrients to the brain even before the onset of dementia,108,121 certain nutritional interventions during mid-life may prove more effective if started before the onset of clinical symptoms, while others may work later in the disease process. Ultimately, improved insulin sensitivity seems to have an important role in reducing the risk of AD. A better understanding of brain energy utilization pathways throughout the lifespan and in both healthy aging and disease states can inform personalized nutrition trials aimed at testing the hypothesis that providing adequate energy to the dementia-at-risk brain limits the progression to neurodegenerative diseases such as AD. Here, brain bioenergetics biomarkers can have key roles in guiding the efficiency of supplements and nutrients. We provide here examples of promising molecular pathways, modeling of dietary patterns, imaging and blood biomarkers, and target populations that can guide nutritional interventions. There are major research and funding gaps that exist and therefore the following recommendations are made.
4.1 |. Preclinical studies of chronic inflammation and cerebral bioenergetics
Given the coupling of disrupted brain energy utilization with chronic inflammation, future studies targeting brain inflammation pathways promise to restore brain energy utilization and limit oxidative damage that leads to synaptic loss and neurodegeneration. For example, repairing the BBB can enhance brain glucose and nutrient uptake, but this remains to be determined. One such target involves cyclophilin A—matrix metalloproteinase 9 (CypA–MMP9) pathway that is activated in APOE4 carriers and associated with BBB integrity loss and cognitive decline.101,99 Another example involves the increase activation of cPLA2 that promotes greater breakdown of neuronal fatty acids, leading to neuroinflammation and oxidative stress. Inhibiting cPLA2 activity may limit the oxidation of brain PUFAs, and shift brain energy utilization toward alternative sources of energy production and repair the BBB.202 Moreover, targeting systemic inflammation in the periphery may hold promise in restoring cerebral bioenergetics in the brain, as a recent study demonstrated that blockade of prostaglandin E2 signaling in peripheral myeloid cells was sufficient to restore cognition in aged mice.203 Another aspect implicates targeting enzymes or other indirect processes involved in cerebral lipid storage, particularly in glia, to support neuronal energetics. For example, depletion of lipoprotein lipase in microglia increased the accumulation of lipid droplets, decreased cholesterol efflux, decreased PPAR signaling, and increased expression of inflammatory markers suggesting that microglial LPL deficiency could play a role in dysregulated lipid metabolism,204 and conversely facilitating cholesterol and lipid efflux via ABCA1126 or ABCA7222 ameliorates AD pathology and inflammation. Several other pathways involving oxidative stress, inflammatory signaling, insulin sensitivity, and lipid metabolism influence brain bioenergetics and still need to be explored. It will be crucial to elucidate how these pathways complement nutrition-based interventions in AD.
4.2 |. Epigenetic and translational network malfunctions
Network malfunctions during AD or cognitive aging such as the destabilization of firing rates, synaptic dysfunction, or learning and memory deficits205–207 are accompanied by metabolic malfunctions such as decreased oxygen and glucose consumption, dysregulated histone acetylation, as well as altered gene expression profiles.208–212 Conversely, interventions aimed at restoring glucose metabolism, like lifestyle paradigms to enhance exercise and mental stimulation, not only significantly improve cognitive dysfunctions,213 but also reinstate chromatin and transcriptional plasticity.214,215 What is more, several bioactive dietary components are known to influence cognitive performance through the regulation of cellular physiology and function, via the intermediate of different epigenetic states.216,217 For instance, the reduction in vitamin A observed in AD patients has been linked to aberrantHAT-dependent histone acetylation patterns, subsequent learning and memory deficits, as well as accumulations of Aβ aggregates.218,219 In addition, many other nutrients such as folate, B, C, D, and E vitamins, methionine, turmeric, catechins, and betaine range among potent dietary epigenetic mediators affecting DNA methylation and histone acetylation.220,221 Another example of how nutrients can affect brain energy utilization through epigenetic modifications is provided with the B1 vitamin thiamin. The complexity of how thiamine maintains brain function has been studied for decades, but new technologies suggest that the field exponentially underestimated its roles. Future research needs to address how thiamine dependent enzymes regulate cell function through hundreds of post-translational modifications under conditions that do not alter ATP. Experiments must address the role of thiamine dependent enzymes in the nucleus and their role in epigenetic modifications.
4.3 |. Biomarkers
One of the major challenges in nutrition and cognition research is patient heterogeneity that associates with a large variability in the risk of cognitive decline and dementia incidence. Focusing on individuals with dementia risk factors223 but before the onset of clinical symptoms is of great interest. Cost-effective biomarkers to easily identify dementia risk factors in diverse populations and enrich future studies with these participants are urgently needed. While amyloid and tau-based biomarkers have been useful in identifying evolving AD pathology with increases in amyloid deposition and tau hyperphosphorylation in the brain, to date, there are no validated biomarkers available that can provide information about cerebral bioenergetic changes that precede or follow amyloid and tau changes in the brain. As such, there is a need for validated diagnostic and prognostic biomarkers of cerebral bioenergetics failure that predict future cognitive decline in AD and indicate cerebral bioenergetic changes that correspond with disease progression in AD. We summarize promising nutritional and metabolic biomarkers in Table 3. The pathogenesis in AD begins decades before the onset of clinical symptoms and, among ε4-carriers, bioenergetic changes can be detected prior to amyloid deposition in the brain. As such, biomarkers that can detect APOE genotype effects on brain bioenergetics, particularly in conjunction to nutritional status will be extremely valuable.
TABLE 3.
Promising bioenergetic and nutritional biomarkers and their implications for brain functions
| Biomarker | Implications for nutritional and metabolic interventions |
|---|---|
| Fluid biomarkers | |
| Omega-3 index | Lower blood omega-3 index is associated with increased dementia risk.161,261,262 Screen vulnerable populations (with low blood omega-3 index) for LC n-3 PUFA supplementation before the onset of AD dementia. |
| DHA/AA ratio | A lower plasma or CSF DHA/AA ratio in AD and APOE4127,263 may reflect activation of lipid catabolism pathways and serve as a biomarker for therapies (such as cPLA2 inhibitors).162 |
| Homocysteine | An increase in plasma homocysteine is associated with increased risk of AD.264–266 Reducing homocysteine using vitamin B complexes maybe beneficial to reducing AD risk before the onset of clinical dementia. |
| 25-OH vitamin D | Screen vulnerable populations (with 25-OH-D deficiency [<25 nmol/L]) or insufficiency [25–50 nmol/L]) for vitamin D supplementation.267,268 |
| Acylcarnitines | Lower levels ACC found in AD serum,269 as a biomarker of dysfunctional FAO. |
| sPDGFRβ | CSF sPDGFRβ levels is a marker of pericyte function, lower in APOE4 and those with lower cognitive function, independently of CSF amyloid beta42.101 Interventions that restore pericyte integrity may improve brain nutrient delivery. |
| SCFA | SCFA concentrations are lower in CSF of AD participants and are markers of brain energetic failure in AD.149 SCFA levels can guide screening and validation of multitudes of pre- and probiotic co-culture combinations. |
| Urinary DCA | Biomarker of unsaturated fatty acids oxidative damage.270 Urine DCAs can help select individuals with oxidative damage and guide antioxidant therapeutics. |
| Imaging biomarkers | |
| DHA PET scans | APOE4 carriers may have a greater vulnerability to omega-3 deficiency before onset of clinical disease.121 DHA PET scan can help identify vulnerable groups and guide supplementation interventions and selection of cognitive outcomes. |
| Ketone PET scans | Ketone PET guides the uptake of ketones in brain regions directly related to improvements in cognitive outcomes166 including processing speed168 and attention.169 Ketone PET scans can help identify individuals responsive to a ketogenic intervention and selection of cognitive outcomes. |
| FDG-H215O PET scans | FDG-PET is a presymptomatic marker of brain hypometabolismand AD risk. Dietary behaviors may affect regional cerebral blood flow which maybe used as an efficacy endpoint for appropriate initial trial fine tuning.271 |
| Infra-red spectroscopy | The effects of different interventions (caffeine, wine and tea polyphenols such as resveratrol orepigallocatechin gallate, creatine, DHA-rich fish oil) in infra-red spectroscopy markers may provide surrogate end points in proof-of-concept nutritional trials.272–277 |
| MR proton or phosphorus spectroscopy | The effect of potential interventions with precursors of brain neurotransmitters important for brain metabolism such as choline or citicholine that are derived from diet may be directly measured in the brain using MR spectroscopy approaches.278,279 |
| MRI CBV | Cerebral blood volume has been enhanced in concert with improved cognitive performance in the hippocampal dentate gyrus afteraflavanol intervention. CBV can be used as preliminary efficacy target.280 |
| Structural connectivity MRI DTI | The effect of potential interventions with a variety of nutrients (ω3 and ω6, ω6, vitamin E, different lipids236,281) and dietary patterns such as the MeDi282 may be estimated with structural connectivity measures. |
| Functional connectivity fMRI | The effects of potential interventions236 including ketones,169 MUFA, SFA, n-3 and n-6 PUFAs,283 B vitamins, vitamin E, carotenoid, carotene and lycopene,283 resveratrol,284,285 cholesterol related lipids,286 beetroot juice287 can be captured with functional connectivity neuroimaging techniques. |
| MRI DCE | DCE MRI can define BBB integrity and is lower with APOE4 and AD.95 Interventions that restore BBB integrity may improve brain nutrient uptake and metabolism. |
Abbreviations: AA, Arachidonic Acid, ACC, Acylcarnitines; DHA, Docosahexaenoic acid; CBV, cerebral blood volume; CSF, cerebrospinal fluid; cPLA2, Calcium-dependent phospholipase A2; DCA, Dicarboxylic Acid; DCE, Dynamic contrast enhancement; DTI, diffusor tensor imaging; FAO, fatty acid oxidations; FDG, Fluorodeoxyglucose; PDGFRβ, soluble platelet-derived growth factor receptor β; SCFA, short chain fatty acids.BBB, blood-brain barrier; MUFA, monounsaturated fatty acids; SFA, saturated fatty acids; PUFAs, polyunsaturated fatty acids; MeDi, mediterranean style diet;
4.3.1 |. Blood, urinary, and CSF based biomarkers
Lipidomics and metabolomics provide an unprecedented opportunity to map the multiple biological system failures occurring in dementia/AD. However, these methods are limited by intrinsically large heterogeneity (i.e., varying over time, across sex, according to exposures and disease status), which hampers reliability and reproducibility of measurements and replicability of findings. Most studies have used targeted metabolomics leveraging a specific platform, and comparability with findings obtained with a different platform is limited. Moreover, gains in sensitivity and reproducibility through using targeted metabolomic approaches must ignore some parts of the metabolome and, therefore, do not capture the system globally.224 Once validated, candidates resulting from lipidomic and metabolomic screens can help model brain mitochondrial functions in relation to cognitive decline and neurodegeneration. Careful longitudinal studies with cognitive and imaging outcomes will help validate the information from these biomarkers. They may also assist as surrogate biomarkers of dietary or other interventions aimed at targeting lipid metabolism and bioenergetics in aging and in AD. There are several biomarkers that can provide insight direct or indirect evidence of brain energy metabolism. For example, brain and blood acylcarnitines could help identify the role of FAO deficiencies in AD pathogenesis, particularly among ε4 carriers before the onset of dementia. Lipidomics and metabolomics have further identified lower Krebs cycle components (succinate and glutamate) and higher levels of oxidized dicarboxylic acids as evidence of abnormal mitochondrial stress in early and established AD.225 Isoprostanes formed by free-radical-mediated peroxidation of PUFAs in response to inflammation are AD biomarkers,226 though studies associate them with other dementias.227 Novel metabolomic studies can reveal several potential AD biomarkers in urine, including DCAs and lysophospholipids.228 The extensive loss of brain tissue in AD may alter urinary lipid-derived metabolites, tested against cognitive performance, CSF, and imaging biomarkers.229 Another promising biomarker includes the platelet-derived growth factor receptor-β (PDGFRβ), that is expressed in the brain by vascular mural cells-brain capillary pericytes and arterial vascular smooth muscle cells. BBB disruption and increased permeability positively correlate with elevated levels of soluble PDGFRβ in CSF of patients with mild dementia.230
4.3.2 |. Neuroimaging biomarkers
The use of neuroimaging tools in nutritional research has increased substantially over the past 2 decades. However, this is the case mostly for structural imaging studies,175,231–234 while investigations in nutrition research using functional imaging approaches such as the above that bring us closer to cerebral metabolism and bioenergetics has been relatively more limited. Additionally, not many functional imaging studies have been performed in elderly at risk for cognitive decline individuals. Furthermore, most such studies have been of relatively limited power and mainly cross-sectional while multiple other methodological limitations are not uncommon.235 Addressing such limitations235,236 in future larger and prospective studies in appropriate middle aged or elderly populations may considerably enhance our understanding of the effects of nutrition in brain energy metabolism. While imaging brain glucose uptake provided insights on the reduced glucose uptake that associate with neurodegeneration, imaging ketone, and fatty acid uptake121,237 may provide insights into early change in brain lipid remodeling occurring decades before the onset of cognitive decline and likely influenced by the diet. As improved MR spectroscopy techniques are being developed nutritional neuroimaging may help us better comprehend the effects of diet in a variety of additional neurotransmitters. As new PET radiotracers are developed, nutritional effects in additional mechanisms affecting brain bioenergetics including inflammation, oxidative stress, etc., will be better understood. At the same time, different imaging modalities may have a unique explanatory power relative to each other and the prospect of the simultaneous inclusion of multiple imaging outcome measures may provide us with deeper understanding of nutritional effects in brain metabolism. We are in an era of impressive expansion of the list of biological specimens derived nutrient biomarkers and of impressive technological advances of all neuroimaging techniques. A parallel progress of analytical methods could bring us closer to a more precision medicine, personalized-type understanding153 of the nutrition-cerebral metabolism relation.
4.4 |. Microbiota sampling in human studies
The need for novel alternatives for early diagnosis, prevention, and treatment aimed to restore cerebral bioenergetic dysfunction in aging and AD cannot be overstated. Microbiome-based treatments to reverse mitochondrial dysfunction are likely to be validated for use in the near foreseeable future. Although it is important to recognize the need for further investigation of microbiome-based interventions, current evidence-based assessments demonstrate an efficacious and promising outcome post-treatment in other disease conditions in addition to AD. However, longitudinal clinical studies on human blood and stool metabolites and their link to neuropsychiatric disorders and cognitive function would further establish the benefits of this novel mode of treatment regime. Stool sampling, which is a non-invasive, cost-effective biomarker assessment method, when combined with in depth target-based, bioenergetic pathway screening is likely to provide data on selective biomarkers to consider for future clinical trials. For example, delineating homo- and heterofermentative Lactobacillus spp and their synergistic association with other probiotics like Bifidobacterium spp may offer a better understanding of the probiotic mutualism for effective drug design. This is particularly important in deciphering and identifying the right prebiotics, probiotics and postbiotics that positively influence cerebral bioenergetics. Preclinical studies have established SCFAs as important contributors to cerebral bioenergetics. Therefore, future studies involving screening and validation of multitudes of pre- and probiotic co-culture combinations that produce postbiotics with the greatest potential to restore bioenergetic homeostasis are likely to yield best-in-class microbiome-based treatments.
5 |. CONCLUSIONS
AD represents a complex disease with cell-specific energy failure affected by nutrient availability early in the preclinical disease phases and impacting nutrient brain delivery and metabolism later as the disease progresses. Many questions remain on the individual cell-specific metabolic changes that contribute to neuronal bioenergetics failures. Such mechanistic studies are important to design targeted therapeutics. One can speculate that drugs or supplements that can better support astroglial mitochondria FAO and spare neuronal mitochondria might be advantageous. Basic research into mechanisms of impaired FAO could lead to novel drugs that shift metabolism away for oxidative or glycolytic stress. The integrated bioenergetic system of astrocytes, pericytes and endothelial cells has direct effects on vascular blood flow and BBB functions. Maintaining BBB integrity is a highly regulated and metabolically demanding task that appears to fail early in AD, particularly among APOE4 carriers. Since BBB function has a major impact on nutrient brain delivery, understanding the mechanisms that restore cerebral bioenergetics promise to enhance the role of nutritional interventions on brain functions. As the nutrition field moves further toward personalized medicine, single or a combination of a few nutrients may still have a role in AD prevention when targeted to vulnerable populations and guided by validated biomarkers including the microbiome. Current efforts are ongoing to explore potential effects of dietary pattern interventions such as the MIND trial,173 the BEAT-AD trial of a modified ketogenic diet (NCT03472664), the Multicultural Health Diet (MHD) an anti-inflammatory diet (NCT03240406) and multiple World Wide FINGER initiatives (where dietary patterns are considered within the framework of multidomain lifestyle interventions). Such efforts need to be expanded and supplemented by parallel biomarker investigations toward aiding in better understanding of dietary pattern effects in brain bioenergetics. It will also be important to design and implement these nutritional interventions in ethno-racially diverse populations to fully explore cerebral bioenergetics as a viable target for the prevention of cognitive decline and dementia.
Supplementary Material
RESEARCH IN CONTEXT.
1. Systematic review:
Does providing adequate energy to the dementia-at-risk brain limit the progression to neurodegenerative diseases such as AD? Systematic reviews of randomized trials of dietary and nutritional supplements have reported largely null effects on cognitive outcomes.
2. Interpretation:
Because the aging brain and AD risk factors affect the delivery of nutrients to the brain before the onset of dementia, preventive nutritional interventions during mid-life may prove more effective when started early, while other nutritional interventions (such as the ketogenic diet) may work later in the disease process.
3. Future directions:
A better understanding of brainenergy utilization pathways throughout the lifespan and in both healthy aging and disease states guided by nutrition-brain-specific biomarkers can inform personalized nutrition clinical trials.
ACKNOWLEDGMENTS
We thank Brianna Liu for helping with the figures and Thomas Urich for proofreading the manuscript and assisting with the references. H.N.Y. is supported by R13AG069386, R21AG056518, R01AG055770, R01AG054434, R01AG067063, RF1AG076124 and RF1AG078362 from the National Institute on Aging as well as Alzheimer’ Association and receives consulting fees from Huron Consulting Firm. H.N.Y. was the NMD PIA co-chair 2019–2021, and sits on the ISTAART Advisory board 2022–2024. G.G. is supported by 2P01AG014930–15A1, R01AG043679 from Alzheimer’s Drug Discovery Foundation as well as Method for treating neurodegenerating diseases with benfotiamine or derivatives thereof for Patents planned, issued or pending. M.G.H. is supported by 1R01AG054434, 2P01AG052350–06, RO1AG063857, RO1 AG055770 from L.K. Whittier Foundation and W.M. Keck Foundation as well as R01NS072497 for grants or contracts. M.G.H. is also a member of NINDS Study Section ZRG1 BDCN-W, NINDS Headache CDE Working Group, Co-Chair of Biomarkers Committee. R.S. is supported by P30072973 from the National Institute on Aging as well as honoraria for lectures from Nestle Health. S.C. receives funding from Nestlé Health Science SC: Payment to institution—Abitec SC: Research materials. S.C. has License for BrainXpert product. S.C.: Paid to me and institution. S.C. receives consulting fees from Nestlé Health Science, Cargill and Abbott. S.C. receives payment or honoraria from Nestlé Health Science SC: Paid to me. S.C. receives support for attending meetings and/or travel from Nestlé Health Science. S.C. has Patents planned, issued or pending from MCT formulation, co-inventor with Bernard Cuenoud, Nestlé Health Science. S.C. has participation on a Data Safety Monitoring Board or Advisory Board from AB—Nestlé Health Science, AB—Abbott, AB—Cargill SC: Paid to me, and receives equipment, materials, drugs, medical writing, gifts or other services from Abitec, research materials SC: Institution. N.S. receives funding from EISAI—Local Pi of recruiting site for multinational, multicenter industry sponsored phase III treatment trial for Alzheimer’s disease NS: Payments to Institution, and EPAD—Local PI of recruiting site for multinational, multicenter Innovative Medicines Initiative (IMI) sponsored observational study of prodromal stages of dementia NS: Payments to Institution, as well as NovoNordisc—Local PI of recruiting site for multinational, multicenter industry sponsored phase III treatment trial for Alzheimer’s disease NS: Payments to Institution. N.S. chairs and receives funding from the Albert Einstein College of Medicine Data Safety Monitoring Board- NIH funded study NS. J.G. receives funding from ERC Starting Grant, StG 678832 as well as patents planned, issued or pending on Inventor for European Patent No. 16180434.9 “Methods of Diagnosis Alzheimer’s Disease.” Co-Inventor for U.S. patent 12/917,402 “The Use of CI-994 and Dinaline for the Treatment of Memory/Cognition and Anxiety Disorders.” L.A.J. is supported by R01 AG062550, R01 AG060056 from the National Institute on Aging as well as Cure Alzheimer’s Fund. J.L. is supported by Springer for co-editor of the book Diabetes and the Brain. J.L. also receives consulting fees from vTv therapeutics. H.F. receives funding from Annovis (QR Pharma), Vivoryon (Probiodrug), AC immune, and LuMind; service agreements for consulting activities with Genetech (DSMB), Roche/Banner (DMC), Tau Consortium (SAB), Samus Theraputics, Biosplice Therapeutics, Axon Neurosciences, Novo Nordisk Inc., Janssen Research & Development LLC; and travel funding from World Events Forum (ADDF) with no personal funds received and all payments to UCSD. HF also reports a philanthropic donation from the Epstein Family Alzheimer’s Disease Collaboration for therapeutic research in AD. G.L.B. is supported by R01 AG043398 from NIH/NIA as well as R90AT008924 from NIH/NCCIH. G.L.B. is a founder 2015, served as co-chair 2015–2019 and immediate past co-chair 2019–2021. H.Y., G.L.B., B.A., C.S., and W.S. are all members of the NMD PIA, ISTAART.
Funding information
National Institute on Aging, Grant/Award Numbers: R13AG069386, R21AG056518, R01AG055770, R01AG054434, R01AG067063, RF1AG076124, RF1AG078362, 2P01AG014930-15A1, R01AG043679; L.K. Whittier Foundation; W.M. Keck Foundation, Grant/Award Numbers: R01NS072497, P30072973; ERC Starting, Grant/Award Numbers: StG 678832, R01 AG062550, R01 AG060056, R01AG043398; NIH/NCCIH, Grant/Award Number: NCCIHR90AT008924
Footnotes
CONFLICT OF INTEREST
All other authors report no relevant disclosures. Author disclosures are available in the supporting information.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Mink JW, Blumenschine RJ, Adams DB. Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis. Am J Physiol. 1981;241(3):R203–12. doi: 10.1152/ajpregu.1981.241.3.R203 [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow RH. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxid Redox Signaling. 2012;16(12):1434–1455. doi: 10.1089/ars.2011.4149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goyal MS, Hawrylycz M, Miller JA, Snyder AZ, Raichle ME. Aerobic glycolysis in the human brain is associated with development and neotenous gene expression. Cell Metab. 2014;19(1):49–57. doi: 10.1016/j.cmet.2013.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris JJ, Jolivet R, Attwell D. Synaptic energy use and supply. Neuron. 2012;75(5):762–777. doi: 10.1016/j.neuron.2012.08.019 [DOI] [PubMed] [Google Scholar]
- 5.Magistretti PJ, Allaman I. Lactate in the brain: from metabolic end-product to signalling molecule. Nat Rev Neurosci. 2018;19(4):235–249. doi: 10.1038/nrn.2018.19 [DOI] [PubMed] [Google Scholar]
- 6.Benarroch EE. Brain glucose transporters. Implications for neurologic disease. Neurology. 2014;82(15):1374–1379. doi: 10.1212/wnl.0000000000000328 [DOI] [PubMed] [Google Scholar]
- 7.Kapogiannis D, Avgerinos KI. Brain glucose and ketone utilization in brain aging and neurodegenerative diseases. Int Rev Neurobiol. 2020;154:79–110. doi: 10.1016/bs.irn.2020.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ames BN. Prolonging healthy aging: longevity vitamins and proteins. Proc Natl Acad Sci U S A. 2018;115(43):10836–10844. doi: 10.1073/pnas.1809045115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross C, Caballero B, Cousins R, Tucker K, Zeigler T. Modern Nutrition In Health and Disease. Wolters Kluwer Business, Lippincott Williams & Wilkins; 2014. [Google Scholar]
- 10.Dong Y, Brewer GJ. Global metabolic shifts in age and Alzheimer’s disease mouse brains pivot at NAD+/NADH redox sites. J Alzheimers Dis. 2019;71(1):119–140. doi: 10.3233/JAD-190408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Tapias V, Acosta D, Xu H, Chen H, Bhawal R, et al. Altered succinylation of mitochondrial proteins, APP and tau in Alzheimer’s disease. Nat Commun. 2022;13(1):159. doi: 10.1038/s41467-021-27572-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerlach M, Ben-Shachar D, Riederer P, Youdim MB. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J Neurochem. 1994;63(3):793–807. doi: 10.1046/j.1471-4159.1994.63030793.x [DOI] [PubMed] [Google Scholar]
- 13.Schönfeld P, Reiser G. Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain. J Cereb Blood Flow Metab. 2013;33(10):1493–1499. doi: 10.1038/jcbfm.2013.128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erecińska M, Silver IA. Tissue oxygen tension and brain sensitivity to hypoxia. Respir Physiol. 2001;128(3):263–276. doi: 10.1016/s0034-5687(01)00306-1 [DOI] [PubMed] [Google Scholar]
- 15.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1–13. doi: 10.1042/BJ20081386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brenna JT, Diau GY. The influence of dietary docosahexaenoic acid and arachidonic acid on central nervous system polyunsaturated fatty acid composition. Prostaglandins Leukot Essent Fatty Acids. 2007;77(5–6):247–250. doi: 10.1016/j.plefa.2007.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Umhau JC, Zhou W, Carson RE, et al. Imaging incorporation of circulating docosahexaenoic acid into the human brain using positron emission tomography. J Lipid Res. 2009;50(7):1259–1268. doi: 10.1194/jlr.M800530-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fox PT, Raichle ME, Mintun MA, Dence C. Nonoxidative glucose consumption during focal physiologic neural activity. Science. 1988;241(4864):462–464. doi: 10.1126/science.3260686 [DOI] [PubMed] [Google Scholar]
- 19.Sappey-Marinier D, Calabrese G, Fein G, Hugg JW, Biggins C, Weiner MW. Effect of photic stimulation on human visual cortex lactate and phosphates using 1H and 31P magnetic resonance spectroscopy. J Cereb Blood Flow Metab. 1992;12(4):584–592. doi: 10.1038/jcbfm.1992.82 [DOI] [PubMed] [Google Scholar]
- 20.Jernberg JN, Bowman CE, Wolfgang MJ, Scafidi S. Developmental regulation and localization of carnitine palmitoyltransferases (CPTs) in rat brain. J Neurochem. 2017;142(3):407–419. doi: 10.1111/jnc.14072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pellerin L, Magistretti PJ. Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. The neuroscientist: a review journal bringing neurobiology. Neurol Psychiatry. 2004;10(1):53–62. doi: 10.1177/1073858403260159 [DOI] [PubMed] [Google Scholar]
- 22.Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94(1):1–14 [DOI] [PubMed] [Google Scholar]
- 23.Pellerin L, Magistretti PJ. Food for thought: challenging the dogmas. J Cereb Blood Flow Metab. 2003;23(11):1282–1286. doi: 10.1097/01.WCB.0000096064.12129.3D [DOI] [PubMed] [Google Scholar]
- 24.Beard E, Lengacher S, Dias S, Magistretti PJ, Finsterwald C. Astrocytes as key regulators of brain energy metabolism: new therapeutic perspectives. Front Physiol. 2021;12:825816. doi: 10.3389/fphys.2021.825816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Powell CL, Davidson AR, Brown AM. Universal glia to neurone lactate transfer in the nervous system: physiological functions and pathological consequences. Biosensors (Basel). 2020;10(11):183. doi: 10.3390/bios10110183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guzman M, Blazquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12(4):169–173 [DOI] [PubMed] [Google Scholar]
- 27.Ettle B, Schlachetzki JCM, Winkler J. Oligodendroglia and myelin in neurodegenerative diseases: more than just bystanders? Mol Neurobiol. 2016;53(5):3046–3062. doi: 10.1007/s12035-015-9205-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010;11(4):275–283. doi: 10.1038/nrn2797 [DOI] [PubMed] [Google Scholar]
- 29.Mot AI, Depp C, Nave KA. An emerging role of dysfunctional axonoligodendrocyte coupling in neurodegenerative diseases. Dialogues Clin Neurosci. 2018;20(4):283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jha MK, Morrison BM. Glia-neuron energy metabolism in health and diseases: new insights into the role of nervous system metabolic transporters. Exp Neurol. 2018;309:23–31. doi: 10.1016/j.expneurol.2018.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee Y, Morrison BM, Li Y, et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487(7408):443–448. doi: 10.1038/nature11314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer N, Richter N, Fan Z, et al. Oligodendrocytes in the mouse corpus callosum maintain axonal function by delivery of glucose. Cell Rep. 2018;22(9):2383–2394. doi: 10.1016/j.celrep.2018.02.022 [DOI] [PubMed] [Google Scholar]
- 33.Philippot C, Griemsmann S, Jabs R, Seifert G, Kettenmann H, Steinhauser C. Astrocytes and oligodendrocytes in the thalamus jointly maintain synaptic activity by supplying metabolites. Cell Rep. 2021;34(3):108642. doi: 10.1016/j.celrep.2020.108642 [DOI] [PubMed] [Google Scholar]
- 34.Chamberlain KA, Huang N, Xie Y, et al. Oligodendrocytes enhance axonal energy metabolism by deacetylation of mitochondrial proteins through transcellular delivery of SIRT2. Neuron. 2021;109(21):3456–3472 e8. doi: 10.1016/j.neuron.2021.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. doi: 10.1126/science.1194637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sierra A, Encinas JM, Deudero JJ, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529 [DOI] [PubMed] [Google Scholar]
- 38.Parkhurst CN, Yang G, Ninan I, et al. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monsorno K, Buckinx A, Paolicelli RC. Microglial metabolic flexibility: emerging roles for lactate. Trends Endocrinol Metab. 2022;33(3):186–195. doi: 10.1016/j.tem.2021.12.001 [DOI] [PubMed] [Google Scholar]
- 40.Bernier L-P, York EM, Kamyabi A, Choi HB, Weilinger NL, MacVicar BA. Microglial metabolic flexibility supports immune surveillance of the brain parenchyma. Nat Commun. 2020;11(1):1559. doi: 10.1038/s41467-020-15267-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–150. doi: 10.1038/nrneurol.2017.188. Epub 2018/01/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchida Y, Zhang Z, Tachikawa M, Terasaki T. Quantitative targeted absolute proteomics of rat blood–cerebrospinal fluid barrier transporters: comparison with a human specimen. J Neurochem. 2015;134(6):1104–1115. doi: 10.1111/jnc.13147 [DOI] [PubMed] [Google Scholar]
- 43.Mak C, Waldvogel H, Dodd J, et al. Immunohistochemical localisation of the creatine transporter in the rat brain. Neuroscience. 2009;163(2):571–585. [DOI] [PubMed] [Google Scholar]
- 44.Kang YS, Lee KE, Lee NY, Terasaki T. Donepezil, tacrine and alpha-phenyl-n-tert-butyl nitrone (PBN) inhibit choline transport by conditionally immortalized rat brain capillary endothelial cell lines (TR-BBB). Arch Pharm Res. 2005;28(4):443–450. doi: 10.1007/bf02977674 [DOI] [PubMed] [Google Scholar]
- 45.Shawahna R, Uchida Y, Declèves X, et al. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol Pharm. 2011;8(4):1332–1341. doi: 10.1021/mp200129p [DOI] [PubMed] [Google Scholar]
- 46.Spector R, Johanson CE. Vitamin transport and homeostasis in mammalian brain: focus on Vitamins B and E. J Neurochem. 2007;103(2):425–438. doi: 10.1111/j.1471-4159.2007.04773.x [DOI] [PubMed] [Google Scholar]
- 47.Montalbetti N, Simonin A, Kovacs G, Hediger MA. Mammalian iron transporters: families SLC11 and SLC40. Mol Aspects Med. 2013;34(2–3):270–287. doi: 10.1016/j.mam.2013.01.002 [DOI] [PubMed] [Google Scholar]
- 48.Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann Neurol. 1977;1(5):409–417. [DOI] [PubMed] [Google Scholar]
- 49.Campos-Bedolla P, Walter FR, Veszelka S, Deli MA. Role of the blood-brain barrier in the nutrition of the central nervous system. Arch Med Res. 2014;45(8):610–638. doi: 10.1016/j.arcmed.2014.11.018 [DOI] [PubMed] [Google Scholar]
- 50.Hall CN, Reynell C, Gesslein B, et al. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508(7494):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vlassenko AG, Vaishnavi SN, Couture L, et al. Spatial correlation between brain aerobic glycolysis and amyloid-beta (Abeta) deposition. Proc Natl Acad Sci U S A. 2010;107(41):17763–17767. 10.1073/pnas.1010461107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin F, Sancheti H, Patil I, Cadenas E. Energy metabolism and inflammation in brain aging and Alzheimer’s disease. Free Radical Biol Med. 2016;100:108–122. doi: 10.1016/j.freeradbiomed.2016.04.200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goyal MS, Vlassenko AG, Blazey TM, et al. Loss of brain aerobic glycolysis in normal human aging. Cell Metab. 2017;26(2):353–360.e3. doi: 10.1016/j.cmet.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6(8):734–746. doi: 10.1016/s1474-4422(07)70178-3 [DOI] [PubMed] [Google Scholar]
- 55.Kalaria RN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem. 1989;53(4):1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x [DOI] [PubMed] [Google Scholar]
- 56.Simpson IA, Davies P. Reduced glucose transporter concentrations in brains of patients with Alzheimer’s disease. Ann Neurol. 1994;36(5):800–801. doi: 10.1002/ana.410360522 [DOI] [PubMed] [Google Scholar]
- 57.Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Brain glucose transporters, O-GlcNAcylation and phosphorylation of tau in diabetes and Alzheimer’s disease. J Neurochem. 2009;111(1):242–249. doi: 10.1111/j.1471-4159.2009.06320.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim Y, Zheng X, Ansari Z, et al. Mitochondrial aging defects emerge in directly reprogrammed human neurons due to their metabolic profile. Cell Rep. 2018;23(9):2550–2558. doi: 10.1016/j.celrep.2018.04.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chao CC, Gutierrez-Vazquez C, Rothhammer V, et al. Metabolic control of astrocyte pathogenic activity via cPLA2-MAVS. Cell. 2019;179(7):1483–1498 e22. doi: 10.1016/j.cell.2019.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chuang DY, Simonyi A, Kotzbauer PT, Gu Z, Sun GY. Cytosolic phospholipase A 2 plays a crucial role in ROS/NO signaling during microglial activation through the lipoxygenase pathway. J Neuroinflammation. 2015;12(1):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ebright B, Assante I, Poblete RA, Wang S, Duro MV, Bennett DA, et al. Eicosanoid lipidome activation in post-mortem brain tissues of individuals with APOE4 and Alzheimer’s dementia. Alzheimer’s Research & Therapy. 2022;14:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Duro MV, Ebright B, Yassine HN. Lipids and brain inflammation in APOE4-associated dementia. Curr Opin Lipidol. 2022;33(1):16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Blass JP, Sheu KF, Piacentini S, Sorbi S. Inherent abnormalities in oxidative metabolism in Alzheimer’s disease: interaction with vascular abnormalities. Ann N Y Acad Sci. 1997;826:382–385. doi: 10.1111/j.1749-6632.1997.tb48488.x [DOI] [PubMed] [Google Scholar]
- 64.Sharma C, Kim S, Nam Y, Jung UJ, Kim SR. Mitochondrial dysfunction as a driver of cognitive impairment in Alzheimer’s disease. Int J Mol Sci. 2021;22(9):4850. doi: 10.3390/ijms22094850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Knottnerus SJG, Bleeker JC, Wust RCI, et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord. 2018;19:93–106. doi: 10.1007/s11154-018-9448-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lehmann R, Zhao X, Weigert C, et al. Medium chain acylcarnitines dominate the metabolite pattern in humans under moderate intensity exercise and support lipid oxidation. PLoS One. 2010;5(7):e11519. doi: 10.1371/journal.pone.0011519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005;28(2):109–121. doi: 10.1007/s10545-005-5518-0 [DOI] [PubMed] [Google Scholar]
- 68.Cristofano A, Sapere N, La Marca G, et al. Serum levels of acylcarnitines along the continuum from normal to Alzheimer’s Dementia. PLOS ONE. 2016;11(5):e0155694. doi: 10.1371/journal.pone.0155694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Horgusluoglu E, Neff R, Song W-M, et al. Integrative metabolomics-genomics approach reveals key metabolic pathways and regulators of Alzheimer’s disease. Alzheimers Dement. 2022;18(6):1260–1278. doi: 10.1002/alz.12468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huguenard CJC, Cseresznye A, Evans JE, et al. APOE ε4 and Alzheimer’s disease diagnosis associated differences in L-carnitine, GBB, TMAO and acylcarnitines in blood and brain. Curr Res Transl Med. 2022:103362. doi: 10.1016/j.retram.2022.103362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signaling. 2006;8(11–12):2021–2037. doi: 10.1089/ars.2006.8.2021 [DOI] [PubMed] [Google Scholar]
- 72.Gregersen N, Mortensen PB, Kolvraa S. On the biologic origin of C6-C10-dicarboxylic and C6-C10-omega-1-hydroxy monocarboxylic acids in human and rat with acyl-CoA dehydrogenation deficiencies: in vitro studies on the omega- and omega-1-oxidation of medium-chain (C6-C12) fatty acids in human and rat liver. Pediatr Res. 1983;17(10):828–834. doi: 10.1203/00006450-198310000-00013 [DOI] [PubMed] [Google Scholar]
- 73.Gregersen N, Ingerslev J. The excretion of C6-C10-dicarboxylic acids in the urine of newborn infants during starvation. Evidence for omega-oxidation of fatty acids in the newborn. Acta Paediatr Scand. 1979;68(5):677–681. doi: 10.1111/j.1651-2227.1979.tb18437.x [DOI] [PubMed] [Google Scholar]
- 74.Johnson DC, Brunsvold RA, Ebert KA, Ray PD. Gluconeogenesis in rabbit liver. I. Pyruvate-derived dicarboxylic acids and phosphoenolpyruvate formation in rabbit liver. J Biol Chem. 1973;248(3):763–770 [PubMed] [Google Scholar]
- 75.Orrenius S Cytochrome P-450 in the omega-oxidation of fatty acids. Biochem J. 1969;115(5):25P–6P. doi: 10.1042/bj1150025p [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y, Gibson GE. Succinylation links metabolism to protein functions. Neurochem Res. 2019;44(10):2346–2359. doi: 10.1007/s11064-019-02780-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sieber MA, Hegel JK. Azelaic acid: properties and mode of action. Skin Pharmacol Physiol. 2014;27(Suppl 1):9–17. doi: 10.1159/000354888 [DOI] [PubMed] [Google Scholar]
- 78.Bartzokis G Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25(1):5–18. doi: 10.1016/j.neurobiolaging.2003.03.001 [DOI] [PubMed] [Google Scholar]
- 79.Nasrabady SE, Rizvi B, Goldman JE, Brickman AM. White matter changes in Alzheimer’s disease: a focus on myelin and oligodendrocytes. Acta Neuropathol Commun. 2018;6(1):22. doi: 10.1186/s40478-018-0515-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rivera AD, Chacon-De-La-Rocha I, Pieropan F, Papanikolau M, Azim K, Butt AM. Keeping the ageing brain wired: a role for purine signalling in regulating cellular metabolism in oligodendrocyte progenitors. Pflugers Arch. 2021;473(5):775–783. doi: 10.1007/s00424-021-02544-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bartzokis G Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32(8):1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Teipel SJ, Bayer W, Alexander GE, et al. Progression of corpus callosum atrophy in Alzheimer disease. Arch Neurol. 2002;59(2):243–248. doi: 10.1001/archneur.59.2.243 [DOI] [PubMed] [Google Scholar]
- 83.Wu Y, Ma Y, Liu Z, Geng Q, Chen Z, Zhang Y. Alterations of myelin morphology and oligodendrocyte development in early stage of Alzheimer’s disease mouse model. Neurosci Lett. 2017;642:102–106. doi: 10.1016/j.neulet.2017.02.007 [DOI] [PubMed] [Google Scholar]
- 84.Vanzulli I, Papanikolaou M, De-La-Rocha IC, et al. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol Aging. 2020;94:130–139. doi: 10.1016/j.neurobiolaging.2020.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Butt AM, De La, Rocha IC, Rivera A. Oligodendroglial cells in Alzheimer’s disease. Adv Exp Med Biol. 2019;1175:325–333. doi: 10.1007/978-981-13-9913-8_12 [DOI] [PubMed] [Google Scholar]
- 86.Zhang X, Wang R, Hu D, et al. Oligodendroglial glycolytic stress triggers inflammasome activation and neuropathology in Alzheimer’s disease. Sci Adv. 2020;6(49):eabb8680. doi: 10.1126/sciadv.abb8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC, Bowers WJ. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am J Pathol. 2010;177(3):1422–1435. doi: 10.2353/ajpath.2010.100087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Acosta C, Anderson HD, Anderson CM. Astrocyte dysfunction in Alzheimer disease. J Neurosci Res. 2017;95(12):2430–2447. doi: 10.1002/jnr.24075 [DOI] [PubMed] [Google Scholar]
- 89.Baik SH, Kang S, Lee W, et al. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019;30(3):493–507.e6. doi: 10.1016/j.cmet.2019.06.005 [DOI] [PubMed] [Google Scholar]
- 90.Rubio-Araiz A, Finucane OM, Keogh S, Lynch MA. Anti-TLR2 antibody triggers oxidative phosphorylation in microglia and increases phagocytosis of β-amyloid. J Neuroinflammation. 2018;15(1):247. doi: 10.1186/s12974-018-1281-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pan R-Y, He L, Zhang J, et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab. 2022;34(4):634–648.e6. doi: 10.1016/j.cmet.2022.02.013 [DOI] [PubMed] [Google Scholar]
- 92.Liu Y, Zhang H, Wang S, et al. Reduced pericyte and tight junction coverage in old diabetic rats are associated with hyperglycemia-induced cerebrovascular pericyte dysfunction. Am J Physiol Heart Circ Physiol. 2021;320(2):H549–H62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fisslthaler B, Fleming I. Activation and signaling by the AMP-activated protein kinase in endothelial cells. Circ Res. 2009;105(2):114–127. [DOI] [PubMed] [Google Scholar]
- 94.Nwadozi E, Rudnicki M, Haas TL. Metabolic coordination of pericyte phenotypes: therapeutic implications. Front Cell Dev Biol. 2020;8:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Montagne A, Barnes SR, Sweeney MD, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. doi: 10.1016/j.neuron.2014.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mooradian AD. Effect of aging on the blood-brain barrier. Neurobiol Aging. 1988;9(1):31–39. doi: 10.1016/s0197-4580(88)80013-7 [DOI] [PubMed] [Google Scholar]
- 97.Bowman GL, Dayon L, Kirkland R, et al. Blood-brain barrier breakdown, neuroinflammation, and cognitive decline in older adults. Alzheimers Dement. 2018;14(12):1640–1650. doi: 10.1016/j.jalz.2018.06.2857 [DOI] [PubMed] [Google Scholar]
- 98.Bowman GL, Kaye JA, Moore M, Waichunas D, Carlson NE, Quinn JF. Blood-brain barrier impairment in Alzheimer disease: stability and functional significance. Neurology. 2007;68(21):1809–1814. doi: 10.1212/01.wnl.0000262031.18018.1a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bell RD, Winkler EA, Singh I, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012;485(7399):512–516. doi: 10.1038/nature11087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21–78. doi: 10.1152/physrev.00050.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature. 2020;581(7806):71–76. doi: 10.1038/s41586-020-2247-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Van Dyken P, Lacoste B. Impact of metabolic syndrome on neuroinflammation and the blood-brain barrier. Front Neurosci. 2018;12:930. doi: 10.3389/fnins.2018.00930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Deane R, Sagare A, Hamm K, et al. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118(12):4002–4013. doi: 10.1172/jci36663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bowman GL, Dodge H, Frei B, et al. Ascorbic acid and rates of cognitive decline in Alzheimer’s disease. J Alzheimers Dis. 2009;16(1):93–98. doi: 10.3233/JAD-2009-0923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dayon L, Guiraud SP, Corthesy J, et al. One-carbon metabolism, cognitive impairment and CSF measures of Alzheimer pathology: homocysteine and beyond. Alzheimers Res Ther. 2017;9(1):43. doi: 10.1186/s13195-017-0270-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stover PJ, Durga J, Field MS. Folate nutrition and blood-brain barrier dysfunction. Curr Opin Biotechnol. 2017;44:146–152. doi: 10.1016/j.copbio.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV. Establishment and dysfunction of the blood-brain barrier. Cell. 2015;163(5):1064–1078. doi: 10.1016/j.cell.2015.10.067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Arellanes IC, Choe N, Solomon V, et al. Brain delivery of supplemental docosahexaenoic acid (DHA): a randomized placebo-controlled clinical trial. EBioMedicine. 2020;59:102883. doi: 10.1016/j.ebiom.2020.102883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nguyen LN, Ma D, Shui G, et al. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature (London). 2014;509(7501):503–506. doi: 10.1038/nature13241 [DOI] [PubMed] [Google Scholar]
- 110.Yassine HN, Finch CE. APOE alleles and diet in brain aging and Alzheimer’s disease. Front Aging Neurosci. 2020;12(150):1–18. doi: 10.3389/fnagi.2020.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Brandon JA, Farmer BC, Williams HC, Johnson LA. APOE and Alzheimer’s disease: neuroimaging of metabolic and cerebrovascular dysfunction. Front Aging Neurosci. 2018;10:180. doi: 10.3389/fnagi.2018.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Farmer BC, Johnson LA, Hanson AJ. Effects of apolipoprotein E on nutritional metabolism in dementia. Curr Opin Lipidol. 2019;30(1):10–15. doi: 10.1097/mol.0000000000000566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Martínez-Martínez AB, Torres-Perez E, Devanney N, Del Moral R, Johnson LA, Arbones-Mainar JM. Beyond the CNS: the many peripheral roles of APOE. Neurobiol Dis. 2020;138:104809. doi: 10.1016/j.nbd.2020.104809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fernandez CG, Hamby ME, McReynolds ML, Ray WJ. The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer’s disease. Front Aging Neurosci 2019;11:14. doi: 10.3389/fnagi.2019.00014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wolf AB, Caselli RJ, Reiman EM, Valla J. APOE and neuroenergetics: an emerging paradigm in Alzheimer’s disease. Neurobiol Aging. 2013;34(4):1007–1017. doi: 10.1016/j.neurobiolaging.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Reiman EM, Chen K, Alexander GE, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc Natl Acad Sci. 2004;101(1):284–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Johnson ECB, Dammer EB, Duong DM, et al. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med. 2020;26(5):769–780. doi: 10.1038/s41591-020-0815-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Williams HC, Farmer BC, Piron MA, et al. APOE alters glucose flux through central carbon pathways in astrocytes. Neurobiol Dis. 2020;136:104742. doi: 10.1016/j.nbd.2020.104742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Farmer BC, Williams HC, Devanney NA, et al. APOΕ4 lowers energy expenditure in females and impairs glucose oxidation by increasing flux through aerobic glycolysis. Mol Neurodegener. 2021;16(1):62. doi: 10.1186/s13024-021-00483-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Konttinen H, Cabral-da-Silva MEC, Ohtonen S, et al. PSEN1ΔE9, APPswe, and APOE4 confer disparate phenotypes in human iPSC-Derived Microglia. Stem Cell Reports. 2019;13(4):669–683. doi: 10.1016/j.stemcr.2019.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Yassine HN, Croteau E, Rawat V, et al. DHA brain uptake and APOE4 status: a PET study with [1–11 C]-DHA. Alzheimers Res Ther. 2017;9(1):23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Farmer BC, Kluemper J, Johnson LA. Apolipoprotein E4 alters astrocyte fatty acid metabolism and lipid droplet formation. Cells. 2019;8(2):182. doi: 10.3390/cells8020182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sienski G, Narayan P, Bonner JM, et al. APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med. 2021;13(583):eaaz4564. doi: 10.1126/scitranslmed.aaz4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Lin YT, Seo J, Gao F, et al. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron. 2018;98(6):1141–1154.e7. doi: 10.1016/j.neuron.2018.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Qi G, Mi Y, Shi X, Gu H, Brinton RD, Yin F. ApoE4 impairs neuron-astrocyte coupling of fatty acid metabolism. Cell Rep. 2021;34(1):108572. doi: 10.1016/j.celrep.2020.108572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rawat V, Wang S, Sima J, et al. ApoE4 alters ABCA1 membrane trafficking in astrocytes. J Neurosci. 2019;39(48):9611–9622. doi: 10.1523/jneurosci.1400-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tomaszewski N, He X, Solomon V, et al. Effect of APOE genotype on plasma docosahexaenoic acid (DHA), eicosapentaenoic acid, arachidonic acid, and hippocampal volume in the Alzheimer’s disease cooperative study-sponsored DHA clinical trial. J Alzheimers Dis. 2020;74(3):975–990. doi: 10.3233/JAD-191017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Chouinard-Watkins R, Plourde M. Fatty acid metabolism in carriers of apolipoprotein E epsilon 4 allele: is it contributing to higher risk of cognitive decline and coronary heart disease? Nutrients. 2014;6(10):4452–4471. doi: 10.3390/nu6104452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zilberter Y, Zilberter M. The vicious circle of hypometabolism in neurodegenerative diseases: ways and mechanisms of metabolic correction. J Neurosci Res. 2017;95(11):2217–2235. doi: 10.1002/jnr.24064 [DOI] [PubMed] [Google Scholar]
- 130.Zhao N, Ren Y, Yamazaki Y, et al. Alzheimer’s risk factors age, APOE genotype, and sex drive distinct molecular pathways. Neuron. 2020;106(5):727–742 e6. doi: 10.1016/j.neuron.2020.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ezra-Nevo G, Henriques SF, Ribeiro C. The diet-microbiome tango: how nutrients lead the gut brain axis. Curr Opin Neurobiol. 2020;62:122–132. doi: 10.1016/j.conb.2020.02.005 [DOI] [PubMed] [Google Scholar]
- 132.Lal S, Kirkup AJ, Brunsden AM, Thompson DG, Grundy D. Vagal afferent responses to fatty acids of different chain length in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;281(4):G907–G915. doi: 10.1152/ajpgi.2001.281.4.G907 [DOI] [PubMed] [Google Scholar]
- 133.Baj A, Moro E, Bistoletti M, Orlandi V, Crema F, Giaroni C. Glutamatergic signaling along the microbiota-gut-brain axis. Int J Mol Sci. 2019;20(6):1482. doi: 10.3390/ijms20061482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lyte M Probiotics function mechanistically as delivery vehicles for neuroactive compounds: microbial endocrinology in the design and use of probiotics. BioEssays. 2011;33(8):574–581. doi: 10.1002/bies.201100024 [DOI] [PubMed] [Google Scholar]
- 135.Sarkar A, Lehto SM, Harty S, Dinan TG, Cryan JF, Burnet PWJ. Psychobiotics and the manipulation of bacteria-gut-brain signals. Trends Neurosci. 2016;39(11):763–781. doi: 10.1016/j.tins.2016.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vogt NM, Romano KA, Darst BF, et al. The gut microbiota-derived metabolite trimethylamine N-oxide is elevated in Alzheimer’s disease. Alzheimers Res Ther. 2018;10(1):124. doi: 10.1186/s13195-018-0451-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. 2016;167(4):915–932. doi: 10.1016/j.cell.2016.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci. 2018;12:49. doi: 10.3389/fnins.2018.00049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nagpal R, Mainali R, Ahmadi S, et al. Gut microbiome and aging: physiological and mechanistic insights. Nutr Healthy Aging. 2018;4(4):267–285. doi: 10.3233/NHA-170030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gevezova M, Sarafian V, Anderson G, Maes M. Inflammation and mitochondrial dysfunction in autism spectrum disorder. CNS Neurol Disord Drug Targets. 2020;19(5):320–333. doi: 10.2174/1871527319666200628015039 [DOI] [PubMed] [Google Scholar]
- 141.Wu SC, Cao ZS, Chang KM, Juang JL. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat Commun. 2017;8(1):24. doi: 10.1038/s41467-017-00040-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liu J, Wang F, Liu S, et al. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J Neurol Sci. 2017;381:176–181. doi: 10.1016/j.jns.2017.08.3235 [DOI] [PubMed] [Google Scholar]
- 143.Ho L, Ono K, Tsuji M, Mazzola P, Singh R, Pasinetti GM. Protective roles of intestinal microbiota derived short chain fatty acids in Alzheimer’s disease-type beta-amyloid neuropathological mechanisms. Expert Rev Neurother. 2018;18(1):83–90. doi: 10.1080/14737175.2018.1400909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Liu S, Li E, Sun Z, et al. Altered gut microbiota and short chain fatty acids in Chinese children with autism spectrum disorder. Sci Rep. 2019;9(1):287. doi: 10.1038/s41598-018-36430-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vogt NM, Kerby RL, Dill-McFarland KA, et al. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537. doi: 10.1038/s41598-017-13601-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Zeng Q, Shen J, Chen K, et al. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci Rep. 2020;10(1):12998. doi: 10.1038/s41598-020-69845-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramakrishna BS, Roediger WE. Bacterial short chain fatty acids: their role in gastrointestinal disease. Dig Dis. 1990;8(6):337–345. doi: 10.1159/000171266 [DOI] [PubMed] [Google Scholar]
- 148.Erny D, Dokalis N, Mezo C, et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021;33(11):2260–2276 e7. doi: 10.1016/j.cmet.2021.10.010 [DOI] [PubMed] [Google Scholar]
- 149.Fonteh AN, Cipolla M, Chiang J, Arakaki X, Harrington MG. Human cerebrospinal fluid fatty acid levels differ between supernatant fluid and brain-derived nanoparticle fractions, and are altered in Alzheimer’s disease. PLoS One. 2014;9(6):e100519. doi: 10.1371/journal.pone.0100519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li D, Ke Y, Zhan R, et al. Trimethylamine-N-oxide promotes brain aging and cognitive impairment in mice. Aging Cell. 2018;17(4):e12768. doi: 10.1111/acel.12768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Del Rio D, Zimetti F, Caffarra P, et al. The gut microbial metabolite trimethylamine-N-oxide is present in human cerebrospinal fluid. Nutrients. 2017;9(10):1053. doi: 10.3390/nu9101053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yassine HN, Samieri C, Livingston G, et al. Nutrition state of science and dementia prevention: recommendations of the nutrition for dementia prevention working group. Lancet Healthy Longev. 2022;3(7):e501–e12. doi: 10.1016/s2666-7568(22)00120-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Samieri C, Yassine HN, Melo van Lent D, et al. Personalized nutrition for dementia prevention. Alzheimers Dement. 2021;18(7):1424–1437. doi: 10.1002/alz.12486 [DOI] [PubMed] [Google Scholar]
- 154.Cullen KM, Halliday GM. Neurof ibrillary tangles in chronic alcoholics. Neuropathol Appl Neurobiol. 1995;21(4):312–318. doi: 10.1111/j.1365-2990.1995.tb01065.x [DOI] [PubMed] [Google Scholar]
- 155.Cullen KM, Halliday GM, Caine D, Kril JJ. The nucleus basalis (Ch4) in the alcoholic Wernicke-Korsakoff syndrome: reduced cell number in both amnesic and non-amnesic patients. J Neurol Neurosurg Psychiatr. 1997;63(3):315. doi: 10.1136/jnnp.63.3.315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Karuppagounder SS, Xu H, Shi Q, et al. Thiamine deficiency induces oxidative stress and exacerbates the plaque pathology in Alzheimer’s mouse model. Neurobiol Aging. 2009;30(10):1587–1600. doi: 10.1016/j.neurobiolaging.2007.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Pan X, Gong N, Zhao J, et al. Powerful beneficial effects of benfotiamine on cognitive impairment and beta-amyloid deposition in amyloid precursor protein/presenilin-1 transgenic mice. Brain. 2010;133(Pt 5):1342–1351 [DOI] [PubMed] [Google Scholar]
- 158.Tapias V, Jainuddin S, Ahuja M, et al. Benfotiamine treatment activates the Nrf2/ARE pathway and is neuroprotective in a transgenic mouse model of tauopathy. Hum Mol Genet. 2018;27(16):2874–2892. doi: 10.1093/hmg/ddy201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Gibson GE, Luchsinger JA, Cirio R, et al. Benfotiamine and cognitive decline in Alzheimer’s disease: results of a randomized placebo-controlled phase IIa clinical trial. J Alzheimers Dis. 2020;78(3):989–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Li S, Guo Y, Men J, Fu H, Xu T. The preventive efficacy of vitamin B supplements on the cognitive decline of elderly adults: a systematic review and meta-analysis. BMC Geriatrics. 2021;21(1):367. doi: 10.1186/s12877-021-02253-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Thomas A, Baillet M, Proust-Lima C, et al. Blood polyunsaturated omega-3 fatty acids, brain atrophy, cognitive decline, and dementia risk. Alzheimers Dement. 2020;17(3):407–416. doi: 10.1002/alz.12195 [DOI] [PubMed] [Google Scholar]
- 162.Wang S, Li B, Solomon V, et al. Calcium-dependent cytosolic phospholipase A2 activation is implicated in neuroinflammation and oxidative stress associated with ApoE4. Mol Neurodegener. 2022;17(1):42. doi: 10.1186/s13024-022-00549-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bowman GL, Scarmeas N. Dietary patterns in early life pay dividends for midlife cognitive performance. Neurology. 2019;92(14):645–646. doi: 10.1212/WNL.0000000000007229 [DOI] [PubMed] [Google Scholar]
- 164.Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond). 2009;6:31. doi: 10.1186/1743-7075-6-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Juby AG, Blackburn TE, Mager DR. Use of medium chain triglyceride (MCT) oil in subjects with Alzheimer’s disease: a randomized, double-blind, placebo-controlled, crossover study, with an open-label extension. Alzheimers Dement (N Y). 2022;8(1):e12259–e. doi: 10.1002/trc2.12259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Fortier M, Castellano CA, Croteau E, et al. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019;15(5):625–634. doi: 10.1016/j.jalz.2018.12.017 [DOI] [PubMed] [Google Scholar]
- 167.Fortier M, Castellano CA, St-Pierre V, et al. A ketogenic drink improves cognition in mild cognitive impairment: results of a 6-month RCT. Alzheimers Dement. 2021;17(3):543–552. doi: 10.1002/alz.12206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Roy M, Fortier M, Rheault F, et al. A ketogenic supplement improves white matter energy supply and processing speed in mild cognitive impairment. medRxiv. 2021;7(1):e12217. doi: 10.1101/2021.03.18.21253884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Roy M, Edde M, Fortier M, et al. A ketogenic intervention improves dorsal attention network functional and structural connectivity in mild cognitive impairment. Neurobiol Aging. 2022;115:77–87. doi: 10.1016/j.neurobiolaging.2022.04.005 [DOI] [PubMed] [Google Scholar]
- 170.Xu Q, Zhang Y, Zhang X, et al. Medium-chain triglycerides improved cognition and lipid metabolomics in mild to moderate Alzheimer’s disease patients with APOE4(−/−): a double-blind, randomized, placebo-controlled crossover trial. Clin Nutr. 2020;39(7):2092–2105 [DOI] [PubMed] [Google Scholar]
- 171.Grammatikopoulou MG, Goulis DG, Gkiouras K, et al. To keto or not to keto? A systematic review of randomized controlled trials assessing the effects of ketogenic therapy on Alzheimer Disease. Adv Nutr. 2020;11(6):1583–1602. doi: 10.1093/advances/nmaa073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Cunnane SC, Trushina E, Morland C, et al. Brain energy rescue: an emerging therapeutic concept for neurodegenerative disorders of ageing. Nat Rev Drug Discovery. 2020;19(9):609–633. doi: 10.1038/s41573-020-0072-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Morris MC, Tangney CC, Wang Y, Sacks FM, Bennett DA, Aggarwal NT. MIND diet associated with reduced incidence of Alzheimer’s disease. Alzheimers Dement. 2015;11(9):1007–1014. doi: 10.1016/j.jalz.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Neth BJ, Mintz A, Whitlow C, et al. Modified ketogenic diet is associated with improved cerebrospinal fluid biomarker profile, cerebral perfusion, and cerebral ketone body uptake in older adults at risk for Alzheimer’s disease: a pilot study. Neurobiol Aging. 2020;86:54–63. doi: 10.1016/j.neurobiolaging.2019.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Jensen DEA, Leoni V, Klein-Flügge MC, Ebmeier KP, Suri S. Associations of dietary markers with brain volume and connectivity: a systematic review of MRI studies. Ageing Res Rev. 2021;70:101360. doi: 10.1016/j.arr.2021.101360 [DOI] [PubMed] [Google Scholar]
- 176.Berti V, Walters M, Sterling J, et al. Mediterranean diet and 3-year Alzheimer brain biomarker changes in middle-aged adults. Neurology. 2018;90(20):e1789–e98. doi: 10.1212/wnl.0000000000005527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Berti V, Murray J, Davies M, et al. Nutrient patterns and brain biomarkers of Alzheimer’s disease in cognitively normal individuals. J Nutr Health Aging. 2015;19(4):413–423. doi: 10.1007/s12603-014-0534-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Small GW, Silverman DH, Siddarth P, et al. Effects of a 14-day healthy longevity lifestyle program on cognition and brain function. Am J Geriatr Psychiatry. 2006;14(6):538–545. doi: 10.1097/01.JGP.0000219279.72210.ca [DOI] [PubMed] [Google Scholar]
- 179.Kim JW, Byun MS, Yi D, et al. Serum zinc levels and in vivo beta-amyloid deposition in the human brain. Alzheimers Res Ther. 2021;13(1):190. doi: 10.1186/s13195-021-00931-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Nugent S, Croteau E, Pifferi F, et al. Brain and systemic glucose metabolism in the healthy elderly following fish oil supplementation. Prostaglandins Leukot Essent Fatty Acids. 2011;85(5):287–291. doi: 10.1016/j.plefa.2011.04.008 [DOI] [PubMed] [Google Scholar]
- 181.Scheltens NME, Briels CT, Yaqub M, et al. Exploring effects of Souvenaid on cerebral glucose metabolism in Alzheimer’s disease. Alzheimers Dement (N Y). 2019;5:492–500. doi: 10.1016/j.trci.2019.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Manzano Palomo MS, Anaya Caravaca B, Balsa Bretón MA, et al. Mild cognitive impairment with a high risk of progression to Alzheimer’s Disease Dementia (MCI-HR-AD): effect of Souvenaid(®) treatment on cognition and (18)F-FDG PET scans. J Alzheimers Dis Rep. 2019;3(1):95–102. doi: 10.3233/adr-190109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Matthews DC, Davies M, Murray J, et al. Physical activity, mediterranean diet and biomarkers-assessed risk of Alzheimer’s: a multi-modality brain imaging study. Adv J Mol Imaging. 2014;4(4):43–57. doi: 10.4236/ami.2014.44006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Rai G, Wright G, Scott L, Beston B, Rest J, Exton-Smith AN. Double-blind, placebo controlled study of acetyl-l-carnitine in patients with Alzheimer’s dementia. Curr Med Res Opin. 1990;11(10):638–647. doi: 10.1185/03007999009112690 [DOI] [PubMed] [Google Scholar]
- 185.Spagnoli A, Lucca U, Menasce G, et al. Long-term acetyl-L-carnitine treatment in Alzheimer’s disease. Neurology. 1991;41(11):1726–1732. doi: 10.1212/wnl.41.11.1726 [DOI] [PubMed] [Google Scholar]
- 186.Thal LJ, Calvani M, Amato A, Carta A. A 1-year controlled trial of acetyl-l-carnitine in early-onset AD. Neurology. 2000;55(6):805–810. doi: 10.1212/wnl.55.6.805 [DOI] [PubMed] [Google Scholar]
- 187.Remington R, Bechtel C, Larsen D, et al. Maintenance of cognitive performance and mood for individuals with Alzheimer’s disease following consumption of a nutraceutical formulation: a one-year, open-label study. J Alzheimers Dis. 2016;51(4):991–995. doi: 10.3233/jad-151098 [DOI] [PubMed] [Google Scholar]
- 188.Fang EF, Lautrup S, Hou Y, et al. NAD+ in aging: molecular mechanisms and translational implications. Trends Mol Med. 2017;23(10):899–916. doi: 10.1016/j.molmed.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Hou Y, Wei Y, Lautrup S, et al. NAD(+) supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc Natl Acad Sci U S A. 2021;118(37):e2011226118. doi: 10.1073/pnas.2011226118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Harrison DE, Strong R, Reifsnyder P, et al. 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell. 2021;20(5):e13328. doi: 10.1111/acel.13328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Reiten OK, Wilvang MA, Mitchell SJ, Hu Z, Fang EF. Preclinical and clinical evidence of NAD+ precursors in health, disease, and ageing. Mech Ageing Dev. 2021;199:111567. doi: 10.1016/j.mad.2021.111567 [DOI] [PubMed] [Google Scholar]
- 192.de Vadder F, Mithieux G. Gut-brain signaling in energy homeostasis: the unexpected role of microbiota-derived succinate. J Endocrinol. 2018;236(2):R105–R8. doi: 10.1530/JOE-17-0542 [DOI] [PubMed] [Google Scholar]
- 193.Braniste V, Al-Asmakh M, Kowal C, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. 2014;6(263):263ra158. doi: 10.1126/scitranslmed.3009759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Valvassori SS, Resende WR, Budni J, et al. Sodium butyrate, a histone deacetylase inhibitor, reverses behavioral and mitochondrial alterations in animal models of depression induced by early- or late-life stress. Curr Neurovasc Res. 2015;12(4):312–320. doi: 10.2174/1567202612666150728121121 [DOI] [PubMed] [Google Scholar]
- 195.Sampson TR, Debelius JW, Thron T, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s Disease. Cell. 2016;167(6):1469–1480 e12. doi: 10.1016/j.cell.2016.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nurrahma BA, Tsao SP, Wu CH, et al. Probiotic supplementation facilitates recovery of 6-OHDA-induced motor deficit via improving mitochondrial function and energy metabolism. Front Aging Neurosci. 2021;13:668775. doi: 10.3389/fnagi.2021.668775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Chunchai T, Thunapong W, Yasom S, et al. Decreased microglial activation through gut-brain axis by prebiotics, probiotics, or synbiotics effectively restored cognitive function in obese-insulin resistant rats. J Neuroinflammation. 2018;15(1):11. doi: 10.1186/s12974-018-1055-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Blacher E, Bashiardes S, Shapiro H, et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature. 2019;572(7770):474–480. doi: 10.1038/s41586-019-1443-5 [DOI] [PubMed] [Google Scholar]
- 199.Snow WM, Cadonic C, Cortes-Perez C, et al. Sex-specific effects of chronic creatine supplementation on hippocampal-mediated spatial cognition in the 3xTg mouse model of Alzheimer’s disease. Nutrients. 2020;12(11):3589. doi: 10.3390/nu12113589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Adlimoghaddam A, Odero GG, Glazner G, Turner RS, Albensi BC. Nilotinib improves bioenergetic profiling in brain astroglia in the 3xTg mouse model of Alzheimer’s disease. Aging Dis. 2021;12(2):441–465. doi: 10.14336/ad.2020.0910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Soro-Arnaiz I, Li QOY, Torres-Capelli M, et al. Role of mitochondrial complex IV in age-dependent obesity. Cell Rep. 2016;16(11):2991–3002. doi: 10.1016/j.celrep.2016.08.041 [DOI] [PubMed] [Google Scholar]
- 202.Hartz AMS, Rempe RG, Soldner ELB, et al. Cytosolic phospholipase A2 is a key regulator of blood-brain barrier function in epilepsy. FASEB J. 2019;33(12):14281–14295. doi: 10.1096/fj.201901369RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Minhas PS, Latif-Hernandez A, McReynolds MR, et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature. 2021;590(7844):122–128. doi: 10.1038/s41586-020-03160-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Loving BA, Tang M, Neal MC, et al. Lipoprotein lipase regulates microglial lipid droplet accumulation. Cells. 2021;10(2):198. doi: 10.3390/cells10020198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Buckner RL, Andrews-Hanna JR, Schacter DL. The brain’s default network: anatomy, function, and relevance to disease. Ann N Y Acad Sci. 2008;1124:1–38. doi: 10.1196/annals.1440.011 [DOI] [PubMed] [Google Scholar]
- 206.Frere S, Slutsky I. Alzheimer’s disease: from firing instability to homeostasis network collapse. Neuron. 2018;97(1):32–58. doi: 10.1016/j.neuron.2017.11.028 [DOI] [PubMed] [Google Scholar]
- 207.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer’s disease. Lancet Neurol. 2011;10(2):187–198. doi: 10.1016/s1474-4422(10)70277-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Kyrtata N, Emsley HCA, Sparasci O, Parkes LM, Dickie BR. A systematic review of glucose transport alterations in Alzheimer’s disease. Front Neurosci. 2021;15:626636. doi: 10.3389/fnins.2021.626636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bradshaw PC. Acetyl-CoA metabolism and histone acetylation in the regulation of aging and lifespan. Antioxidants (Basel). 2021;10(4):572. doi: 10.3390/antiox10040572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Gräff J, Rei D, Guan JS, et al. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature. 2012;483(7388):222–226. doi: 10.1038/nature10849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Nativio R, Donahue G, Berson A, et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat Neurosci. 2018;21(4):497–505. doi: 10.1038/s41593-018-0101-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Nativio R, Lan Y, Donahue G, et al. An integrated multi-omics approach identifies epigenetic alterations associated with Alzheimer’s disease. Nat Genet. 2020;52(10):1024–1035. doi: 10.1038/s41588-020-0696-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. Embo j. 2017;36(11):1474–1492. doi: 10.15252/embj.201695810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Wang X, Meng ZX, Chen YZ, et al. Enriched environment enhances histone acetylation of NMDA receptor in the hippocampus and improves cognitive dysfunction in aged mice. Neural Regen Res. 2020;15(12):2327–2334. doi: 10.4103/1673-5374.285005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fischer A, Sananbenesi F, Wang X, Dobbin M, Tsai LH. Recovery of learning and memory is associated with chromatin remodelling. Nature. 2007;447(7141):178–182. doi: 10.1038/nature05772 [DOI] [PubMed] [Google Scholar]
- 216.Gomez-Pinilla F, Tyagi E. Diet and cognition: interplay between cell metabolism and neuronal plasticity. Curr Opin Clin Nutr Metab Care. 2013;16(6):726–733. doi: 10.1097/MCO.0b013e328365aae3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Davinelli S, Calabrese V, Zella D, Scapagnini G. Epigenetic nutraceutical diets in Alzheimer’s disease. J Nutr Health Aging. 2014;18(9):800–805. doi: 10.1007/s12603-014-0552-y [DOI] [PubMed] [Google Scholar]
- 218.Hou N, Ren L, Gong M, et al. Vitamin A deficiency impairs spatial learning and memory: the mechanism of abnormal CBP-dependent histone acetylation regulated by retinoic acid receptor alpha. Mol Neurobiol. 2015;51(2):633–647. doi: 10.1007/s12035-014-8741-6 [DOI] [PubMed] [Google Scholar]
- 219.Ono K, Yamada M. Vitamin A and Alzheimer’s disease. Geriatr Gerontol Int. 2012;12(2):180–188. doi: 10.1111/j.1447-0594.2011.00786.x [DOI] [PubMed] [Google Scholar]
- 220.Chiu S, Woodbury-Fariña MA, Shad MU, et al. The role of nutrient-based epigenetic changes in buffering against stress, aging, and Alzheimer’s disease. Psychiatr Clin North Am. 2014;37(4):591–623. doi: 10.1016/j.psc.2014.09.001 [DOI] [PubMed] [Google Scholar]
- 221.Athanasopoulos D, Karagiannis G, Tsolaki M. Recent findings in Alzheimer disease and nutrition focusing on epigenetics. Adv Nutr. 2016;7(5):917–927. doi: 10.3945/an.116.012229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tomioka M, Toda Y, Mañucat NB, et al. Lysophosphatidylcholine export by human ABCA7. Biochim Biophys Acta Mol Cell Biol Lipids. 2017;1862(7):658–665. doi: 10.1016/j.bbalip.2017.03.012 [DOI] [PubMed] [Google Scholar]
- 223.Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet commission. Lancet. 2020;396(10248):413–446. doi: 10.1016/s0140-6736(20)30367-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Toledo JB, Arnold M, Kastenmüller G, et al. Metabolic network failures in Alzheimer’s disease: a biochemical road map. Alzheimers Dement. 2017;13(9):965–984. doi: 10.1016/j.jalz.2017.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tokuoka SM, Kita Y, Shimizu T, Oda Y. Isobaric mass tagging and triple quadrupole mass spectrometry to determine lipid biomarker candidates for Alzheimer’s disease. PLoS One. 2019;14(12):e0226073. doi: 10.1371/journal.pone.0226073. 358 Shimadzu Co., and Eisai Co. Ltd. This does not alter our adherence to PLOS ONE policies on sharing data and materials. There are no patents, products in development or marketed products to declare [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Montine TJ, Markesbery WR, Morrow JD, Roberts LJ 2nd. Cerebrospinal fluid F2-isoprostane levels are increased in Alzheimer’s disease. Ann Neurol. 1998;44(3):410–413. doi: 10.1002/ana.410440322 [DOI] [PubMed] [Google Scholar]
- 227.Montine TJ, Kaye JA, Montine KS, McFarland L, Morrow JD, Quinn JF. Cerebrospinal fluid abeta42, tau, and f2-isoprostane concentrations in patients with Alzheimer disease, other dementias, and in age-matched controls. Arch Pathol Lab Med. 2001;125(4):510–512. doi: 10.5858/2001-125-0510-CFATAF [DOI] [PubMed] [Google Scholar]
- 228.Cui Y, Liu X, Wang M, et al. Lysophosphatidylcholine and amide as metabolites for detecting Alzheimer disease using ultrahigh-performance liquid chromatography-quadrupole time-of-flight mass spectrometry-based metabonomics. J Neuropathol Exp Neurol. 2014;73(10):954–963. doi: 10.1097/NEN.0000000000000116 [DOI] [PubMed] [Google Scholar]
- 229.Fonteh AN, Harrington RJ, Huhmer AF, Biringer RG, Riggins JN, Harrington MG. Identification of disease markers in human cerebrospinal fluid using lipidomic and proteomic methods. Dis Markers. 2006;22(1–2):39–64. doi: 10.1155/2006/202938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Sagare AP, Sweeney MD, Makshanoff J, Zlokovic BV. Shedding of soluble platelet-derived growth factor receptor-β from human brain pericytes. Neurosci Lett. 2015;607:97–101. doi: 10.1016/j.neulet.2015.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.de Wilde MC, Kamphuis PJ, Sijben JW, Scheltens P. Utility of imaging for nutritional intervention studies in Alzheimer’s disease. Eur J Pharmacol. 2011;668(Suppl 1):S59–S69. doi: 10.1016/j.ejphar.2011.07.011 [DOI] [PubMed] [Google Scholar]
- 232.Scarmeas N, Luchsinger JA, Stern Y, et al. Mediterranean diet and magnetic resonance imaging-assessed cerebrovascular disease. Ann Neurol. 2011;69(2):257–268. doi: 10.1002/ana.22317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Gardener H, Scarmeas N, Gu Y, et al. Mediterranean diet and white matter hyperintensity volume in the Northern Manhattan Study. Arch Neurol. 2012;69(2):251–256. doi: 10.1001/archneurol.2011.548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gu Y, Brickman AM, Stern Y, et al. Mediterranean diet and brain structure in a multiethnic elderly cohort. Neurology. 2015;85(20):1744–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Smeets PAM, Dagher A, Hare TA, et al. Good practice in food-related neuroimaging. Am J Clin Nutr. 2019;109(3):491–503. doi: 10.1093/ajcn/nqy344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Jensen DEA, Leoni V, Klein-Flügge MC, Ebmeier KP, Suri S. Associations of dietary markers with brain volume and connectivity: a systematic review of MRI studies. Ageing Res Rev. 2021;70:101360. doi: 10.1016/j.arr.2021.101360 [DOI] [PubMed] [Google Scholar]
- 237.Esposito G, Giovacchini G, Liow JS, et al. Imaging neuroinflammation in Alzheimer’s disease with radiolabeled arachidonic acid and PET. J Nucl Med. 2008;49(9):1414–1421. doi: 10.2967/jnumed.107.049619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Tallaksen CM, Bohmer T, Bell H. Concentrations of the water-soluble vitamins thiamin, ascorbic acid, and folic acid in serum and cerebrospinal fluid of healthy individuals. Am J Clin Nutr. 1992;56(3):559–564. doi: 10.1093/ajcn/56.3.559 [DOI] [PubMed] [Google Scholar]
- 239.Abdou E, Hazell AS. Thiamine deficiency: an update of pathophysiologic mechanisms and future therapeutic considerations. Neurochem Res. 2015;40(2):353–361. doi: 10.1007/s11064-014-1430-z [DOI] [PubMed] [Google Scholar]
- 240.Anagnostouli M, Livaniou E, Nyalala JO, et al. Cerebrospinal fluid levels of biotin in various neurological disorders. Acta Neurol Scand. 1999;99(6):387–392. doi: 10.1111/j.1600-0404.1999.tb07369.x [DOI] [PubMed] [Google Scholar]
- 241.Balashova OA, Visina O, Borodinsky LN. Folate action in nervous system development and disease. Dev Neurobiol. 2018;78(4):391–402. doi: 10.1002/dneu.22579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Serot JM, Christmann D, Dubost T, Béne MC, Faure GC. CSF-folate levels are decreased in late-onset AD patients. J Neural Transm (Vienna). 2001;108(1):93–99. doi: 10.1007/s007020170100 [DOI] [PubMed] [Google Scholar]
- 243.Nielsen MJ, Rasmussen MR, Andersen CBF, Nexø E, Moestrup SK. Vitamin B12 transport from food to the body’s cells—a sophisticated, multistep pathway. Nat Rev Gastroenterol Hepatol. 2012;9(6):345–354. doi: 10.1038/nrgastro.2012.76 [DOI] [PubMed] [Google Scholar]
- 244.Larsen C, Etzerodt A, Madsen M, Skjødt K, Moestrup SK, Andersen CBF. Structural assembly of the megadalton-sized receptor for intestinal vitamin B12 uptake and kidney protein reabsorption. Nat Commun. 2018;9(1):5204. doi: 10.1038/s41467-018-07468-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Ikeda T, Furukawa Y, Mashimoto S, Takahashi K, Yamada M. Vitamin B12 levels in serum and cerebrospinal fluid of people with Alzheimer’s disease. Acta Psychiatr Scand. 1990;82(4):327–329. doi: 10.1111/j.1600-0447.1990.tb01395.x [DOI] [PubMed] [Google Scholar]
- 246.Quinn J, Suh J, Moore MM, Kaye J, Frei B. Antioxidants in Alzheimer’s disease-vitamin C delivery to a demanding brain. J Alzheimers Dis. 2003;5(4):309–313. doi: 10.3233/jad-2003-5406 [DOI] [PubMed] [Google Scholar]
- 247.Harrison FE. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease. J Alzheimers Dis. 2012;29(4):711–726. doi: 10.3233/jad-2012-111853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Cole GM, Frautschy SA. DHA may prevent age-related dementia. J Nutr. 2010;140(4):869–874. doi: 10.3945/jn.109.113910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Pan Y, Scanlon MJ, Owada Y, Yamamoto Y, Porter CJH, Nicolazzo JA. Fatty acid-binding protein 5 facilitates the blood–brain barrier transport of docosahexaenoic acid. Mol Pharmaceutics. 2015;12(12):4375–4385. doi: 10.1021/acs.molpharmaceut.5b00580 [DOI] [PubMed] [Google Scholar]
- 250.Eyles DW, Liu PY, Josh P, Cui X. Intracellular distribution of the vitamin D receptor in the brain: comparison with classic target tissues and redistribution with development. Neuroscience. 2014;268:1–9. doi: 10.1016/j.neuroscience.2014.02.042 [DOI] [PubMed] [Google Scholar]
- 251.Lee D-H, Kim JH, Jung MH, Cho M-C. Total 25-hydroxy vitamin D level in cerebrospinal fluid correlates with serum total, bioavailable, and free 25-hydroxy vitamin D levels in Korean population. PLoS One. 2019;14(3):e0213389–e. doi: 10.1371/journal.pone.0213389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Soares JZ, Valeur J, Šaltyte Benth J, et al. Vitamin D in Alzheimer’s˙ disease: low levels in cerebrospinal fluid despite normal amounts in serum. J Alzheimers Dis. 2022;86(3):1301–1314. doi: 10.3233/jad215536 [DOI] [PubMed] [Google Scholar]
- 253.Morris MC, Tangney CC, Wang Y, et al. MIND diet slows cognitive decline with aging. Alzheimers Dement. 2015;11(9):1015–1022. doi: 10.1016/j.jalz.2015.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Appel LJ, Moore TJ, Obarzanek E, et al. A clinical trial of the effects of dietary patterns on blood pressure. DASH collaborative research group. N Engl J Med. 1997;336(16):1117–1124. doi: 10.1056/NEJM199704173361601 [DOI] [PubMed] [Google Scholar]
- 255.van den Brink AC, Brouwer-Brolsma EM, Berendsen AAM, van de Rest O. The Mediterranean, dietary approaches to stop hypertension (DASH), and Mediterranean-DASH intervention for neurodegenerative delay (MIND) diets are associated with less cognitive decline and a lower risk of Alzheimer’s disease-a review. Adv Nutr. 2019;10(6):1040–1065. doi: 10.1093/advances/nmz054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Panagiotakos DB, Tzima N, Pitsavos C, et al. The association between adherence to the Mediterranean diet and fasting indices of glucose homoeostasis: the ATTICA Study. J Am Coll Nutr. 2007;26(1):32–38. doi: 10.1080/07315724.2007.10719583 [DOI] [PubMed] [Google Scholar]
- 257.Scarmeas N, Stern Y, Tang MX, Mayeux R, Luchsinger JA. Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. 2006;59(6):912–921. doi: 10.1002/ana.20854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Bowman GL, Silbert LC, Howieson D, et al. Nutrient biomarker patterns, cognitive function, and MRI measures of brain aging. Neurology. 2012;78(4):241–249. doi: 10.1212/WNL.0b013e3182436598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Amadieu C, Lefevre-Arbogast S, Delcourt C, et al. Nutrient biomarker patterns and long-term risk of dementia in older adults. Alzheimers Dement. 2017;13(10):1125–1132. doi: 10.1016/j.jalz.2017.01.025 [DOI] [PubMed] [Google Scholar]
- 260.Bowman GL, Dodge HH, Guyonnet S, et al. A blood-based nutritional risk index explains cognitive enhancement and decline in the multidomain Alzheimer prevention trial. Alzheimers Dement (N Y). 2019;5:953–963. doi: 10.1016/j.trci.2019.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Pottala JV, Yaffe K, Robinson JG, Espeland MA, Wallace R, Harris WS. Higher RBC EPA + DHA corresponds with larger total brain and hippocampal volumes: wHIMS-MRI study. Neurology. 2014;82(5):435–442. doi: 10.1212/wnl.0000000000000080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Ammann EM, Pottala JV, Robinson JG, Espeland MA, Harris WS. Erythrocyte omega-3 fatty acids are inversely associated with incident dementia: secondary analyses of longitudinal data from the Women’s Health Initiative Memory Study (WHIMS). Prostaglandins Leukot Essent Fatty Acids. 2017;121:68–75. doi: 10.1016/j.plefa.2017.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Abdullah L, Evans JE, Emmerich T, et al. APOE epsilon4 specific imbalance of arachidonic acid and docosahexaenoic acid in serum phospholipids identifies individuals with preclinical mild cognitive impairment/Alzheimer’s disease. Aging. 2017;9(3):964–985. doi: 10.18632/aging.101203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Luzzi S, Papiri G, Viticchi G, et al. Association between homocysteine levels and cognitive profile in Alzheimer’s Disease. J Clin Neurosci. 2021;94:250–256. doi: 10.1016/j.jocn.2021.09.033 [DOI] [PubMed] [Google Scholar]
- 265.Wang Q, Zhao J, Chang H, Liu X, Zhu R. Homocysteine and folic acid: risk factors for Alzheimer’s disease-an updated meta-analysis. Front Aging Neurosci. 2021;13:665114. doi: 10.3389/fnagi.2021.665114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Zuin M, Cervellati C, Brombo G, Trentini A, Roncon L, Zuliani G. Elevated blood homocysteine and risk of Alzheimer’s dementia: an updated systematic review and meta-analysis based on prospective studies. J Prev Alzheimers Dis. 2021;8(3):329–334. doi: 10.14283/jpad.2021.7 [DOI] [PubMed] [Google Scholar]
- 267.Kalra A, Teixeira AL, Diniz BS. Association of vitamin D levels with incident all-cause dementia in longitudinal observational studies: a systematic review and meta-analysis. J Prev Alzheimers Dis. 2020;7(1):14–20. doi: 10.14283/jpad.2019.44 [DOI] [PubMed] [Google Scholar]
- 268.Goodwill AM, Szoeke C. A systematic review and meta-analysis of the effect of low vitamin D on cognition. J Am Geriatr Soc. 2017;65(10):2161–2168. doi: 10.1111/jgs.15012 [DOI] [PubMed] [Google Scholar]
- 269.Ciavardelli D, Piras F, Consalvo A, et al. Medium-chain plasma acylcarnitines, ketone levels, cognition, and gray matter volumes in healthy elderly, mildly cognitively impaired, or Alzheimer’s disease subjects. Neurobiol Aging. 2016;43:1–12. doi: 10.1016/j.neurobiolaging.2016.03.005 [DOI] [PubMed] [Google Scholar]
- 270.Castor KJ, Shenoi S, Edminster SP, et al. Urine dicarboxylic acids change in pre-symptomatic Alzheimer’s disease and reflect loss of energy capacity and hippocampal volume. PLoS One. 2020;15(4):e0231765. doi: 10.1371/journal.pone.0231765. analysis by our consultant (JMP) from Cipher Biostatistics & Reporting does not alter our adherence to PLOS ONE policies on sharing data and materials. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.DelParigi A, Chen K, Salbe AD, et al. Successful dieters have increased neural activity in cortical areas involved in the control of behavior. Int J Obes (Lond). 2007;31(3):440–448. doi: 10.1038/sj.ijo.0803431 [DOI] [PubMed] [Google Scholar]
- 272.Kennedy DO, Haskell CF. Cerebral blood flow and behavioural effects of caffeine in habitual and non-habitual consumers of caffeine: a near infrared spectroscopy study. Biol Psychol. 2011;86(3):298–306. doi: 10.1016/j.biopsycho.2010.12.010 [DOI] [PubMed] [Google Scholar]
- 273.Dodd FL, Kennedy DO, Riby LM, Haskell-Ramsay CF. A double-blind, placebo-controlled study evaluating the effects of caffeine and L-theanine both alone and in combination on cerebral blood flow, cognition and mood. Psychopharmacology (Berl). 2015;232(14):2563–2576. doi: 10.1007/s00213-015-3895-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Kennedy DO, Wightman EL, Reay JL, et al. Effects of resveratrol on cerebral blood flow variables and cognitive performance in humans: a double-blind, placebo-controlled, crossover investigation. Am J Clin Nutr. 2010;91(6):1590–1597. doi: 10.3945/ajcn.2009.28641 [DOI] [PubMed] [Google Scholar]
- 275.Wightman EL, Haskell CF, Forster JS, Veasey RC, Kennedy DO. Epigallocatechin gallate, cerebral blood flow parameters, cognitive performance and mood in healthy humans: a double-blind, placebo-controlled, crossover investigation. Hum Psychopharmacol. 2012;27(2):177–186. doi: 10.1002/hup.1263 [DOI] [PubMed] [Google Scholar]
- 276.Watanabe A, Kato N, Kato T. Effects of creatine on mental fatigue and cerebral hemoglobin oxygenation. Neurosci Res. 2002;42(4):279–285. doi: 10.1016/s0168-0102(02)00007-x [DOI] [PubMed] [Google Scholar]
- 277.Jackson PA, Reay JL, Scholey AB, Kennedy DO. DHA-rich oil modulates the cerebral haemodynamic response to cognitive tasks in healthy young adults: a near IR spectroscopy pilot study. Br J Nutr. 2012;107(8):1093–1098. doi: 10.1017/s0007114511004041 [DOI] [PubMed] [Google Scholar]
- 278.Cohen BM, Renshaw PF, Stoll AL, Wurtman RJ, Yurgelun-Todd D, Babb SM. Decreased brain choline uptake in older adults. An in vivo proton magnetic resonance spectroscopy study. JAMA. 1995;274(11):902–907 [PubMed] [Google Scholar]
- 279.Silveri MM, Dikan J, Ross AJ, et al. Citicoline enhances frontal lobe bioenergetics as measured by phosphorus magnetic resonance spectroscopy. NMR Biomed. 2008;21(10):1066–1075. doi: 10.1002/nbm.1281 [DOI] [PubMed] [Google Scholar]
- 280.Brickman AM, Khan UA, Provenzano FA, et al. Enhancing dentate gyrus function with dietary flavanols improves cognition in older adults. Nat Neurosci. 2014;17(12):1798–1803. doi: 10.1038/nn.3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Gu Y, Vorburger RS, Gazes Y, et al. White matter integrity as a mediator in the relationship between dietary nutrients and cognition in the elderly. Ann Neurol. 2016;79(6):1014–1025. doi: 10.1002/ana.24674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Pelletier A, Barul C, Feart C, et al. Mediterranean diet and preserved brain structural connectivity in older subjects. Alzheimers Dement. 2015;11(9):1023–1031. doi: 10.1016/j.jalz.2015.06.1888 [DOI] [PubMed] [Google Scholar]
- 283.Zwilling CE, Talukdar T, Zamroziewicz MK, Barbey AK. Nutrient biomarker patterns, cognitive function, and fMRI measures of network efficiency in the aging brain. Neuroimage. 2019;188:239–251. doi: 10.1016/j.neuroimage.2018.12.007 [DOI] [PubMed] [Google Scholar]
- 284.Huhn S, Beyer F, Zhang R, et al. Effects of resveratrol on memory performance, hippocampus connectivity and microstructure in older adults - a randomized controlled trial. Neuroimage. 2018;174:177–190. doi: 10.1016/j.neuroimage.2018.03.023 [DOI] [PubMed] [Google Scholar]
- 285.Witte AV, Kerti L, Margulies DS, Flöel A. Effects of resveratrol on memory performance, hippocampal functional connectivity, and glucose metabolism in healthy older adults. J Neurosci. 2014;34(23):7862–7870. doi: 10.1523/jneurosci.0385-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Meusel LC, Anderson ND, Parrott MD, et al. Brain function is linked to LDL cholesterol in older adults with cardiovascular risk. J Am Geriatr Soc. 2017;65(2):e51–e5. doi: 10.1111/jgs.14663 [DOI] [PubMed] [Google Scholar]
- 287.Petrie M, Rejeski WJ, Basu S, et al. Beet root juice: an ergogenic aid for exercise and the aging brain. J Gerontol A Biol Sci Med Sci. 2017;72(9):1284–1289. doi: 10.1093/gerona/glw219 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.