

Abstract
Background.
The basolateral amygdala (BLA) regulates mood and associative learning and has been linked to the development and persistence of alcohol use disorder (AUD). The GABAB receptor (GABABR) is a promising therapeutic target for AUD, and previous work suggests that exposure to ethanol and other drugs can alter neuronal GABABR-dependent signaling. The effect of ethanol on GABABR-dependent signaling in the BLA is unknown.
Methods.
GABABR-dependent signaling in the mouse BLA was examined using slice electrophysiology following repeated ethanol exposure. Neuron-specific viral genetic manipulations were then used to understand the relevance of ethanol-induced neuroadaptations in the BA to mood-related behavior.
Results.
The somatodendritic inhibitory effect of GABABR activation on principal neurons in the basal (BA) but not lateral (LA) sub-region of the BLA was diminished following ethanol exposure. This adaptation was attributable to the suppression of G protein-gated inwardly rectifying K+ (GIRK) channel activity and was mirrored by a re-distribution of GABABR and GIRK channels from the surface membrane to internal sites. While GIRK1 and GIRK2 subunits are critical for GIRK channel formation in BA principal neurons, GIRK3 is necessary for the ethanol-induced neuroadaptation. Viral suppression of GIRK channel activity in BA principal neurons from ethanol-naïve mice recapitulated some mood-related behaviors observed in C57BL/6J mice during ethanol withdrawal.
Conclusions.
The ethanol-induced suppression of GIRK-dependent signaling in BA principal neurons contributes to some of the mood-related behaviors associated with ethanol withdrawal in mice. Approaches designed to prevent this neuroadaptation and/or strengthen GIRK-dependent signaling may prove useful for treatment of AUD.
Keywords: Alcohol, GABAB receptor, Kir3, neuroadaptation, behavior
INTRODUCTION
Repeated cycles of exposure to and withdrawal from drugs of abuse such as alcohol foster neuroadaptations in brain regions that regulate mood, learning, and goal-directed behavior (1, 2). These adaptations likely drive hallmarks of alcohol use disorder (AUD) including heightened anxiety, cognitive deficits, craving, and compulsive drug-seeking (3). Collectively, these changes increase relapse susceptibility, while impeding the development of adaptive behaviors that could support abstinence (4). Thus, identifying neuroadaptations linked to ethanol exposure and withdrawal may suggest novel interventions for AUD.
The amygdala has been implicated in key aspects of AUD, including craving and relapse (2). A smaller amygdala volume is positively correlated with risk for developing AUD (5) and relapse (6). Studies in rodents suggest that the basolateral amygdala (BLA) is a key substrate for the development and persistence of AUD. For example, BLA neurons increase firing in response to drug-associated cues, and this activity has been linked to cue-induced relapse/reinstatement (7, 8). Furthermore, pharmacological inhibition of the BLA suppresses ethanol self-administration and cue-induced reinstatement of ethanol-seeking behavior (9–13). Moreover, acute and chronic ethanol exposure impacts BLA neurotransmission and neurophysiology (e.g., (14–19)).
The GABAB receptor (GABABR) is an intriguing target for treatment of AUD (20, 21). GABABR mediates the G protein-dependent presynaptic and somatodendritic inhibitory influence of GABA throughout the central nervous system (22). While acute ethanol exposure can enhance GABA release from presynaptic terminals and potentiate somatodendritic GABABR-dependent signaling (23, 24), the GABABR-selective agonist baclofen can suppress binge-like drinking, alcohol withdrawal sign severity, cue-induced reinstatement of alcohol-seeking behavior, and the reinforcing and motivational properties of alcohol in rodent models (25). In individuals with AUD, baclofen can suppress alcohol intake and craving, and it is used off-label for AUD treatment (21, 26).
Prior work from our lab and others has shown that psychostimulant exposure in mice weakens somatodendritic GABABR-dependent signaling in the mesocorticolimbic circuitry, including the ventral tegmental area (VTA) (27–30) and medial prefrontal cortex (31, 32). The goal of this study was to test whether ethanol impacts somatodendritic GABABR-dependent signaling in the BLA and, if it does, to understand the relevance of this ethanol-induced neuroadaptation to mood-related behaviors observed during ethanol withdrawal.
METHODS AND MATERIALS
Expanded methods and details are included in Supplemental Material.
Animals.
Experiments were approved by the University of Minnesota Institutional Animal Care and Use Committee. Generation of Girk1−/− (33), Girk2−/− (34), and Girk3−/− (35) mice, as well as conditional Girk1fl/fl mice (36), was described previously. Lines were backcrossed for at least 10 generations against the C57BL/6J strain before initiating this study. C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) for some studies. Unless otherwise noted, males and females were used in all experiments and groups were balanced by sex. Mice were maintained on a 14:10 h light/dark cycle and were provided ad libitum access to food and water.
Reagents.
Baclofen and barium chloride were purchased from Sigma (St. Louis, MO), and CGP54626 was purchased from Tocris (Bristol, UK).
Ethanol exposure.
For ethanol injection studies, C57BL/6J mice (8–10 wk) were given 4 daily injections (1000, 1200, 1400, and 1600 h) of saline or ethanol (2 g/Kg IP) over a 5-d period. Vapor chambers for chronic intermittent exposure (CIE) studies were constructed as described (37). Volatilized ethanol was produced by submerging an aeration stone in 100% ethanol and mixed with room air. Mice in the ethanol treatment group were injected with a priming dose of ethanol (1.5 g/Kg; 20% v/v) and the alcohol dehydrogenase inhibitor pyrazole (68.1 mg/Kg IP) prior to placement in the chamber. Delivery rate was titrated to yield BECs of 150–200 mg/dL, as assessed using trunk blood samples from age- and strain-matched sentinels and the EnzyChrom™ ethanol assay kit (BioAssay Systems, USA; Hayward, CA). Controls were similarly handled but administered saline and pyrazole before placement in a chamber where they were exposed to air. Mice were exposed to ethanol vapor or air for 16 h/d (in at 1700 h, out at 0900 h), followed by an 8-h withdrawal period for 4 d. At the end of day 4, mice began a 72-h period of abstinence. This protocol was conducted for 1 (CIE/1) or 4 (CIE/4) weeks.
Electrophysiology.
Somatodendritic currents, rheobase, and resting membrane potentials (RMP) were measured in principal neurons from acutely isolated coronal slices of the mouse BLA, as described (38).
In situ hybridization.
Multiplex fluorescent in situ hybridization for CaMKIIα, GIRK1, GIRK2, and GIRK3 was performed using RNAscope (39), using sections (16 μm) of the BLA from adult mice (8 wk), according to manufacturer specifications (Advanced Cell Diagnostics; Newark, CA). Fluorescence images of the BLA were acquired using a BZ-X810 Keyence fluorescence microscope with 10x/40x objectives (Keyence: Itasca, IL) and overlaid using ImageJ software (National Institute of Health; Bethesda, MD).
Behavioral testing.
Two testing batteries were used to evaluate the impact of ethanol or viral genetic manipulations on behavioral performance. The first battery began with an assessment of physical dependence by evaluating handling-induced convulsions (HIC). For studies involving mice subjected to the CIE/4 treatment protocol, HIC assessments were made 7-h after the final ethanol (or air) exposure session (Day 1). Subsequently, subjects were assessed in the light-dark box (Day 2), marble burying test (Day 4), and bottle brush test (Days 6 and 7). The second test battery involved elevated plus maze and delay fear conditioning tests, conducted on Day 2 and Days 3–5 after the ethanol (or air) exposure session (Day 1).
Intracranial viral manipulations.
Viral titers and volumes, as well as stereotaxic coordinates, were optimized previously to achieve maximal targeting of the BA along the rostro-caudal axis, while minimizing infection of neurons in adjacent structures (38).
Immunoelectron microscopy.
The subcellular distribution of GIRK2 and GABABR1 was assessed using pre-embedding immunoelectron microscopy, as described (40).
Statistical analysis.
Data are presented as mean ± SEM. Analyses were performed using Prism v. 8 (GraphPad Software, Inc.; La Jolla, CA). Unless otherwise noted, studies included male and female subjects. The impact of sex on experimental outcomes was assessed first. If no impact of sex was detected, then data from male and female subjects were pooled. For all comparisons, differences were considered significant if P<0.05.
RESULTS
Ethanol suppresses GABABR-dependent signaling in BA principal neurons
We used a 5-d injection protocol – with 4 ethanol injections (2 g/Kg IP) given daily – to examine the impact of ethanol exposure on somatodendritic GABABR-dependent signaling in the BLA of C57BL/6J mice (Fig. 1A). As a single injection of 2 g/Kg ethanol yields blood ethanol concentrations (BECs) of ~180 mg/dL (40 mM, 0.18%) in C57BL/6J mice (41), this protocol produces high intoxicating levels of ethanol for at least 7–8 h/d for 5 consecutive days.
Figure 1. Repeated ethanol injection suppresses somatodendritic GABABR-dependent signaling in BA principal neurons.
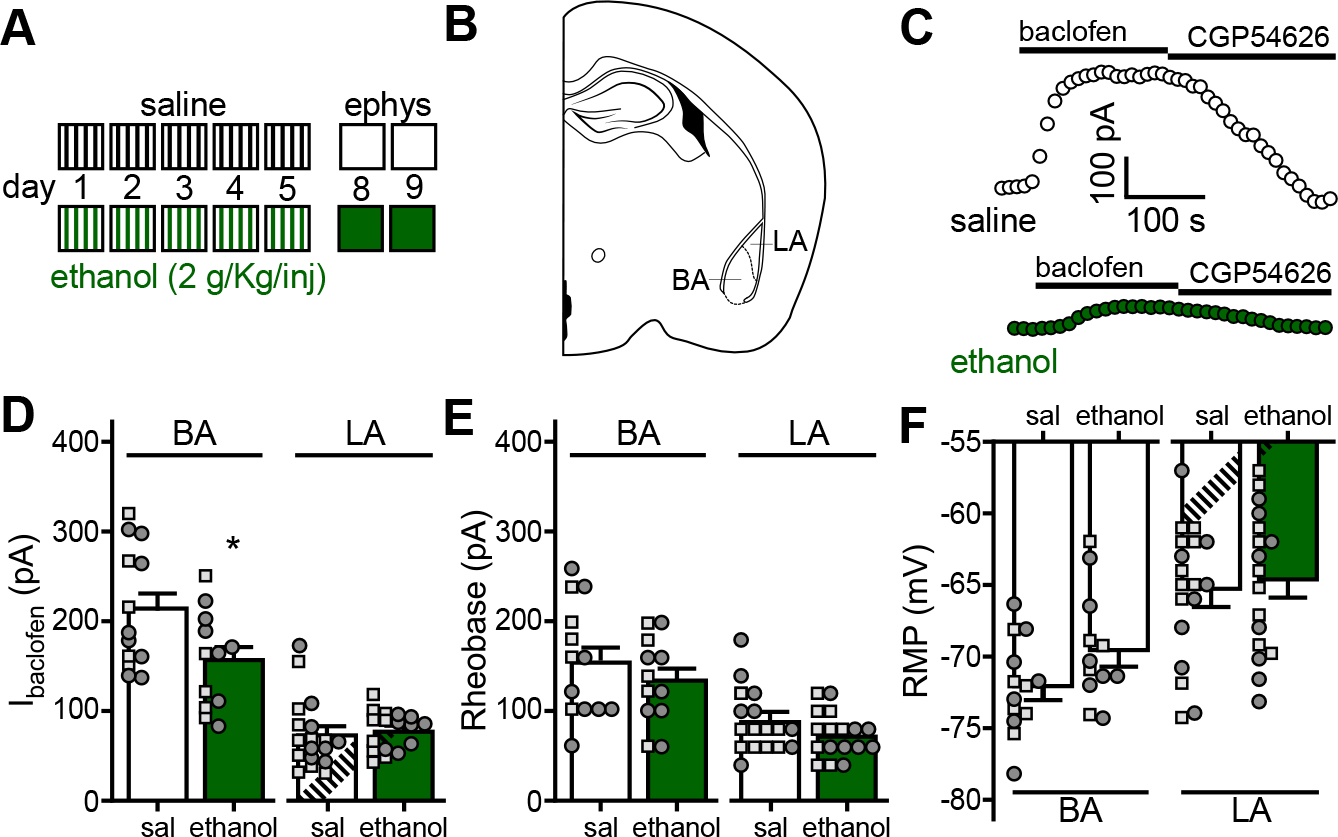
A. Depiction of the repeated ethanol injection protocol. C57BL/6J mice (34–41 d) were given 4 daily injections (1000, 1200, 1400, and 1600 h) of saline or ethanol (2 g/kg IP) over a consecutive 5-d period; injections are denoted by vertical lines. Electrophysiological analysis occurred on days 8 and 9.
B. Depiction of a coronal section containing the BLA, with BA and LA sub-regions highlighted.
C. Somatodendritic currents (Vhold = −60 mV) evoked by baclofen (200 μM) and reversed by the GABABR-selective antagonist CGP54626 (2 μM) in BA principal neurons, recorded 3 d after the last saline or ethanol injection.
D. Ibaclofen in principal neurons of the BA (t23=2.356, *P=0.0274; N=12–13/group; unpaired Student’s t-test) and LA (t26.81=0.375, P=0.711; N=17–18/group; unpaired Student’s t-test with Welch’s correction) from C57BL/6J mice treated with repeated saline (sal) or ethanol. Small squares and circles represent individual data points from male and female subjects, respectively.
E. Rheobase of principal neurons of the BA (t23=0.956, P=0.349; N=12–13/group; unpaired Student’s t-test) and LA (t33=1.527, P=0.136; N=17–18/group; unpaired Student’s t-test) from repeated saline- or ethanol-treated mice.
F. Resting membrane potential (RMP) of principal neurons of the BA (t23=1.823, P=0.0814; N=12–13/group unpaired Student’s t-test) and LA (t33=0.436, P=0.666; N=17–18/group; unpaired Student’s t-test) from repeated saline- or ethanol-treated mice.
Whole-cell recordings of BLA neurons were obtained 3–4 d after the final ethanol or saline injection. Most (85%) BLA neurons are glutamatergic principal neurons distinguishable from GABA neurons based on morphology, larger apparent capacitance, and longer action potential half-width (38). As the basal (BA) and lateral (LA) sub-regions of the BLA exhibit distinct connectivities and differentially contribute to behavior (42, 43), we evaluated BA and LA neurons separately (Fig. 1B). The GABABR agonist baclofen evoked outward somatodendritic currents (Ibaclofen) in BA (Fig. 1C) and LA (not shown) principal neurons; Ibaclofen in LA neurons was smaller than Ibaclofen in BA neurons (Fig. 1D). Ibaclofen was also smaller in BA principal neurons from ethanol-treated mice relative to saline-treated controls (Fig. 1C,D). In contrast, ethanol had no impact on Ibaclofen in LA principal neurons (Fig. 1D). Neither rheobase (Fig. 1E) nor resting membrane potential (RMP; Fig. 1F), measures of baseline neuronal excitability, were significantly impacted by repeated ethanol injection in BA or LA neurons.
We also used chronic intermittent exposure (CIE) to ethanol vapor, employing 1 wk (CIE/1) and 4 wk (CIE/4) protocols (Fig. 2A). Each week consisted of 4 d of 16-h vapor exposure sessions, followed by 3 d of abstinence. BECs reached 150–200 mg/dL during the sessions. Electrophysiological recordings were made 3–4 d after the final ethanol (or air control) exposure session. No impact of ethanol was observed on Ibaclofen in BA principal neurons from subjects in the CIE/1 group (Fig. 2C). In the CIE/4 cohort, however, Ibaclofen from ethanol-treated subjects was smaller than in air-exposed controls (Fig. 2B,C). Apparent capacitance values for BA principal neurons in the ethanol (192 ± 5 pF) and air (208 ± 9 pF) treatment groups were not different (t29.94=1.61; P=0.12), suggesting that Ibaclofen suppression is not linked to reduced cell size. Thus, repeated injection and CIE/4 vapor exposure protocols evoked comparable suppression of Ibaclofen in BA principal neurons. Consistent with a recent report (44), rheobase was also lower in the CIE/4 ethanol treatment group (Fig. 2D), whereas RMP was not affected by either treatment (Fig. 2E).
Figure 2. Chronic intermittent exposure to ethanol vapor suppresses somatodendritic GABABR-dependent signaling in BA principal neurons.
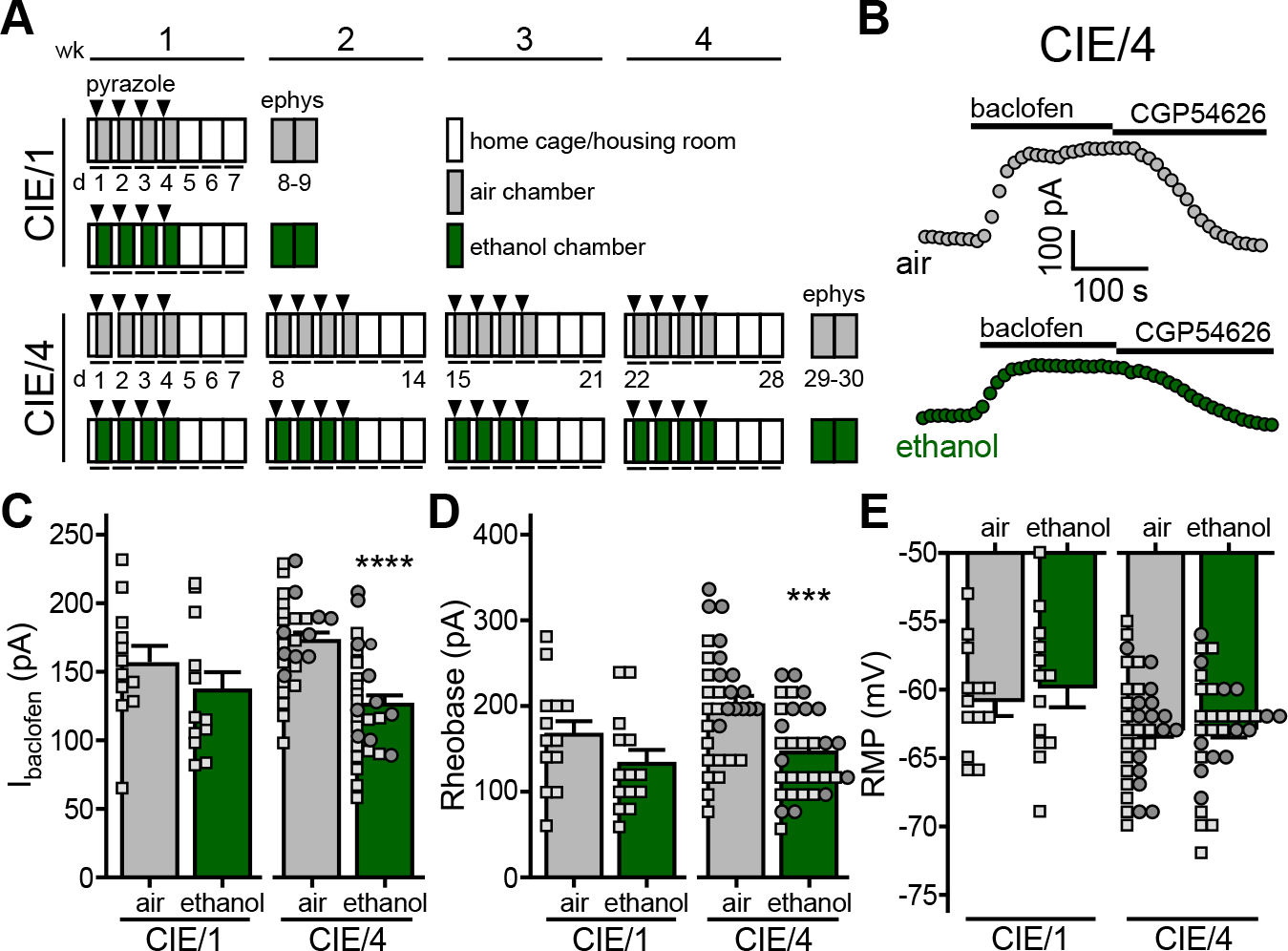
A. Depiction of chronic intermittent ethanol vapor exposure protocols. C57BL/6J mice (8 wk) were placed in ethanol vapor or air control chambers for 16 h sessions over 4 consecutive days, followed by 3 d of home cage housing. Prior to each session, mice were administered sodium pyrazole (68.1 mg/Kg IP; arrowheads) and either a priming dose of ethanol (1.5 g/Kg; 20% v/v) or saline (for air-treated controls). Separate cohorts of mice were run through 1-wk (CIE/1) and 4-wk (CIE/4) protocols, with electrophysiological assessments made 3–4 d after the final air or ethanol vapor exposure session.
B. Somatodendritic currents (Vhold = −60 mV) evoked by baclofen (200 μM) and reversed by CGP54626 (2 μM) in BA principal neurons from air- or ethanol-treated subjects following the CIE/4 protocol.
C. Ibaclofen in BA principal neurons from air- and ethanol-treated mice, 3–4 d after completing CIE/1 (t22=1.102, P=0.282; N=12/group; unpaired Student’s t-test) or CIE/4 (t58=5.072, ****P<0.001; N=30/group; unpaired Student’s t-test) protocols. Small squares and circles represent individual data points from male and female subjects, respectively.
D. Rheobase of BA principal neurons from air- and ethanol-treated mice, 3–4 d after completing CIE/1 (t25=1.523, P=0.140; N=13–14/group; unpaired Student’s t-test) or CIE/4 (t62=3.849, ***P=0.0003; N=32/group; unpaired Student’s t-test) protocols.
E. RMP of BA principal neurons from air- and ethanol-treated mice, 3–4 d after completing CIE/1 (t25=0.552, P=0.586; N=13–14/group; unpaired Student’s t-test) or CIE/4 (t65=0.0113, P=0.991; N=32–35/group; unpaired Student’s t-test) protocols.
GIRK channel activation contributes to Ibaclofen in BA principal neurons
Ibaclofen is mediated primarily by G protein-gated inwardly rectifying K+ (GIRK) channel activation in neurons evaluated to date (32, 35, 45–47). Neuronal GIRK channels are homo- and heterotetrameric complexes formed primarily by 3 subunits (GIRK1, GIRK2, GIRK3) expressed in overlapping patterns in the CNS (48). We probed for GIRK subunit expression in the mouse BLA using multiplex fluorescence in situ hybridization. GIRK1, GIRK2, and GIRK3 were detected throughout the BLA, including the BA and LA, of adult C57BL/6J mice (Fig. 3A). All 3 subunits overlapped completely with the glutamatergic marker CaMKIIα in the BA (Fig. 3B).
Figure 3. GIRK channel activation mediates GABABR-dependent somatodendritic currents in BA principal neurons.
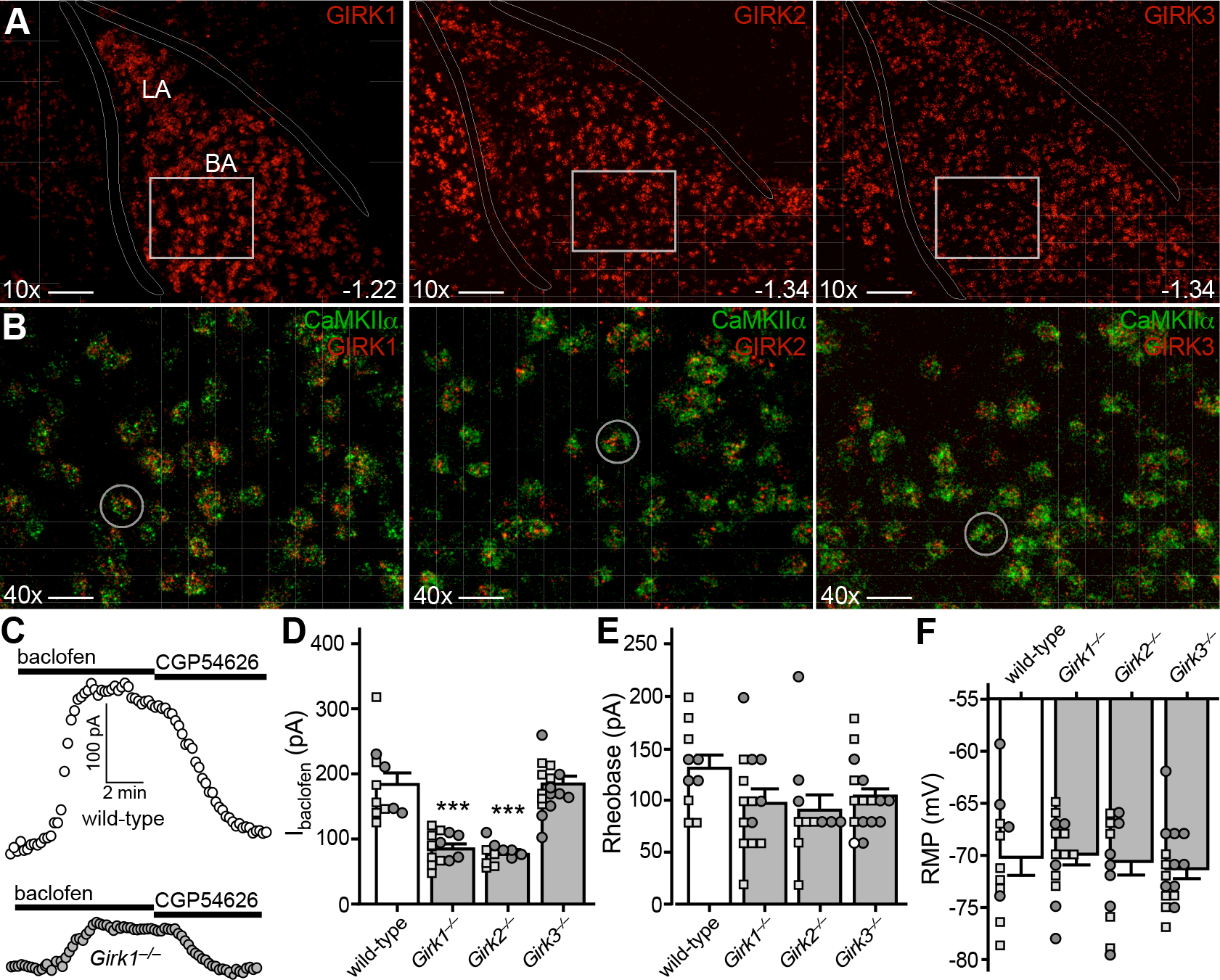
A. Expression of GIRK1, GIRK2, and GIRK3 subunit mRNAs (red) in the adult mouse BLA. Anterior/posterior coordinates are provided in the lower right-hand corner of the images. White rectangles in the upper panels are expanded in panel B. Scale bar = 100 μm.
B. Co-localization of GIRK subunit (red) and CaMKIIα (green) expression in the BA. White circles highlight representative CaMKIIα-positive cell bodies containing GIRK subunit mRNAs. Scale bar = 25 μm.
C. Somatodendritic currents (Vhold = −60 mV) evoked by baclofen (200 μM) and reversed by CGP54626 (2 μM) in BA principal neurons from wild-type (white, upper trace) and Girk1−/− (grey, lower trace) mice.
D. Currents evoked by baclofen (200 μM) in BA principal neurons from wild-type and Girk−/− mice (F3,48=33.63, P<0.0001; one-way ANOVA) (N=11–16/group). Symbols: ***P<0.001 vs. wild-type. Small squares and circles represent individual data points from male and female subjects, respectively.
E. Rheobase (F3,47=1.856, P=0.150; one-way ANOVA) of BA principal neurons from wild-type and Girk−/− mice (N=11–16/group).
F. RMP (F3,48=0.298, P=0.833; one-way ANOVA) of BA principal neurons from wild-type, and Girk−/− mice (N=11–16/group).
We next measured Ibaclofen in BA principal neurons from wild-type and Girk−/− mice. Ibaclofen was ~50% smaller in neurons from Girk1−/− or Girk2−/− mice as compared to controls (Fig. 3C,D). There was no impact of genotype on apparent capacitance values (F3,25=0.53, P=0.16; one-way ANOVA), suggesting that genotype differences in Ibaclofen do not reflect differences in neuron size. GIRK channel ablation did not significantly impact the rheobase of BA principal neurons (Fig. 3E), nor did it impact RMP (Fig. 3F). Despite the presence of GIRK3 mRNA in BA principal neurons, Girk3 ablation had no impact on Ibaclofen or excitability measures (Fig. 3D–F). Thus, Ibaclofen in BA principal neurons is mediated largely by activation of GIRK channels containing GIRK1 and GIRK2.
Ethanol suppresses the GIRK component of Ibaclofen
Extracellular Ba2+ (0.1–0.3 mM) blocks outward current mediated by some inwardly rectifying K+ channels, including GIRK channels. Ibaclofen in BA principal neurons from C57BL/6J mice was ~50% lower when measured in the presence of 0.3 mM extracellular Ba2+ (Fig. 4A), consistent with results from Girk1−/− and Girk2−/− mice (Fig. 3). To determine whether ethanol suppresses the GIRK and/or non-GIRK component of the GABABR-dependent response, we measured Ibaclofen in BA principal neurons from ethanol-treated and control subjects in the presence of extracellular Ba2+. The Ba2+-insensitive component of Ibaclofen was not impacted by the CIE/4 protocol (Fig. 4B,C), suggesting that the ethanol-induced suppression of somatodendritic GABABR-dependent current involves the selective suppression of GIRK channel activity.
Figure 4. Ethanol induces a GIRK3-dependent suppression of Ibaclofen in BA principal neurons.
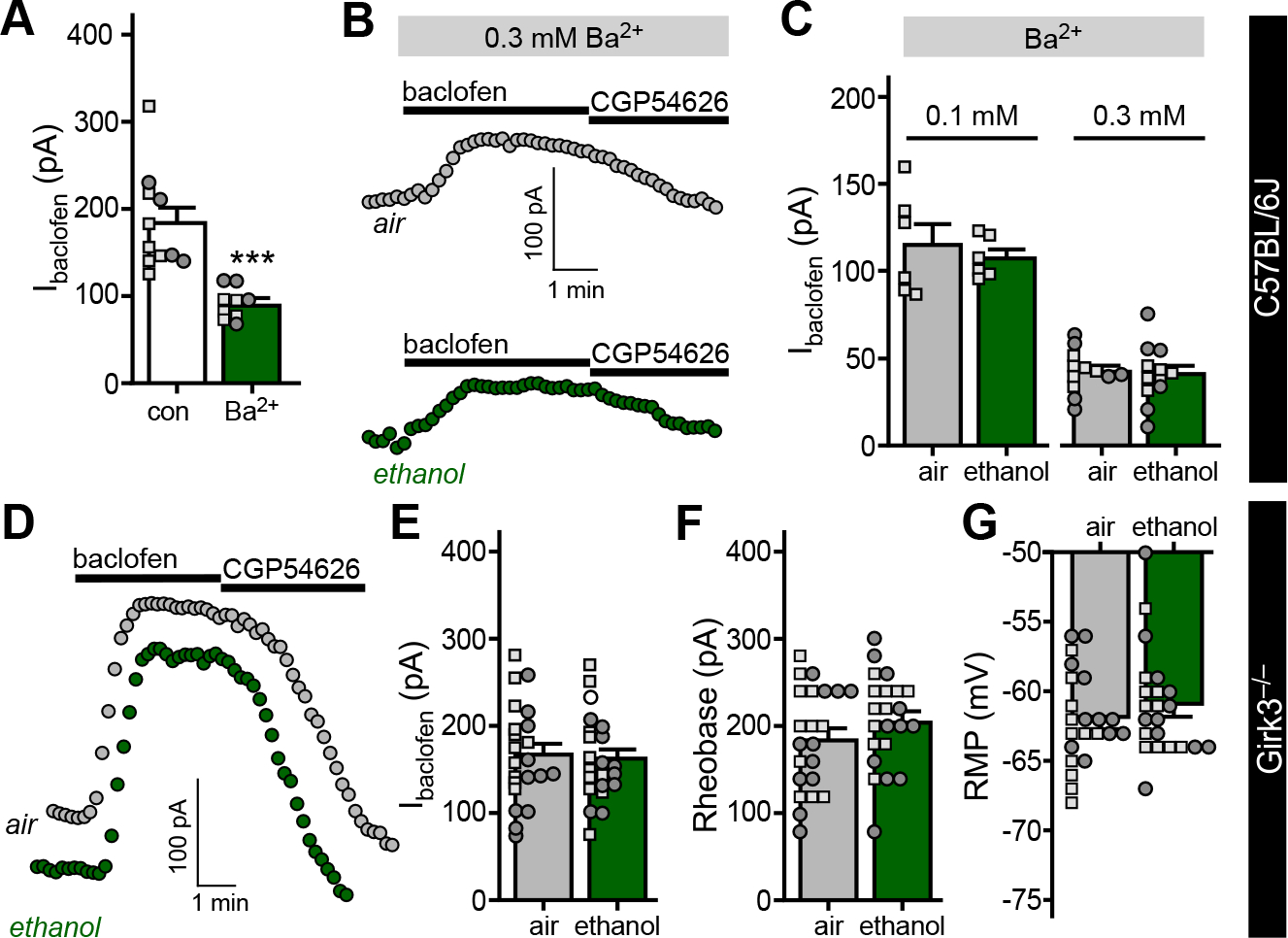
A. Ibaclofen in BA principal neurons from C57BL/6J mice, measured in the absence or presence of 0.3 mM extracellular Ba2+ (t12.43=5.218, ***P=0.0002, Student’s t-test with Welch’s correction; N=9–11/group). Small squares and circles represent individual data points from male and female subjects, respectively.
B. Somatodendritic currents (Vhold = −60 mV) evoked by baclofen (200 μM) and reversed by CGP54626 (2 μM) in BA principal neurons from C57BL/6J mice, measured in the presence of 0.3 mM extracellular Ba2+, 3 d after completion of the CIE/4 protocol.
C. Ibaclofen in BA principal neurons from C57BL/6J mice, 3–4 d after completing the CIE/4 protocol, measured in the presence of 0.1 mM (t10=0.617, P=0.551; N=6/group; unpaired Student’s t-test) or 0.3 mM (t22=0.236, P=0.816; N=12/group; unpaired Student’s t-test) extracellular Ba2+.
D. Somatodendritic currents (Vhold = −60 mV) evoked by baclofen (200 μM) and reversed by CGP54626 (2 μM) in BA principal neurons, measured in slices from air- and ethanol vapor-treated Girk3−/− mice (70–90 d), 3 d after completing the CIE/4 protocol.
E. Ibaclofen in BA principal neurons from Girk3−/− mice, 3–4 d after completing the CIE/vapor exposure (t42=0.317, P=0.753, N=22/group; unpaired Student’s t test).
F. Rheobase of BA principal neurons from Girk3−/− mice 3–4 d after completing the CIE/vapor exposure (t42=1.191, P=0.240, N=22/group; unpaired Student’s t test).
G. RMP of BA principal neurons from Girk3−/− mice 3–4 d after completing the CIE/vapor exposure (t42=0.923, P=0.361, N=22/group; unpaired Student’s t test).
While GIRK3 ablation has little-or-no impact on GIRK current amplitude in neurons (32, 47), GIRK3 has been implicated in neuroadaptations involving GIRK channels (30, 49, 50). Thus, we asked whether the ethanol-induced suppression of Ibaclofen in BA principal neurons required GIRK3. Consistent with a key role for GIRK3 in this neuroadaptation, the CIE/4 treatment protocol did not impact Ibaclofen in BA principal neurons from Girk3−/− mice (Fig. 4D,E). Rheobase (Fig. 4F) and RMP (Fig. 4G) were similarly unaffected.
Ethanol triggers the subcellular redistribution of GABABR and GIRK channels
Psychostimulants can alter the subcellular distribution of GIRK channels and GABABR (27, 28, 32). As such, we performed quantitative immunoelectron microscopy on BLA sections from mice that underwent the CIE/4 protocol. Consistent with our electrophysiological data, the number of GIRK2 (Fig. 5A,E1–4) and GABABR1 (Fig. 5B,E5–8) immunoparticles in the plasma membrane was lower in BA principal neurons from ethanol-exposed mice relative to air-exposed controls. Immunoparticle density changes were observed in both dendrites and spines (Fig. 5C,D). The reduction in surface membrane-associated GIRK2 and GABABR1 in ethanol-exposed mice correlated with an increase in the number of intracellular immunoparticles. Notably, total and surface membrane GIRK2 and GABABR1 immunoparticle counts were lower in principal neurons of the LA relative to BA (Fig. S1A,B) in air-exposed controls, which aligns with the smaller Ibaclofen in LA neurons (Fig. 1D). Furthermore, ethanol did not provoke a subcellular redistribution of GIRK2 or GABABR1 immunoparticles in LA principal neurons (Fig. S1C–G), consistent with the lack of impact of ethanol on Ibaclofen in these neurons (Fig. 1D). Given that ethanol had no impact on Ibaclofen in LA principal neurons and it selectively suppressed the GIRK-dependent component of Ibaclofen in BA principal neurons (Fig. 4B,C), these ultrastructural data suggest that the ethanol-induced suppression of Ibaclofen in BA principal neurons is mediated by internalization of GABABR and/or GIRK channels.
Figure 5. Ethanol triggers internalization of GIRK channels and GABABR.
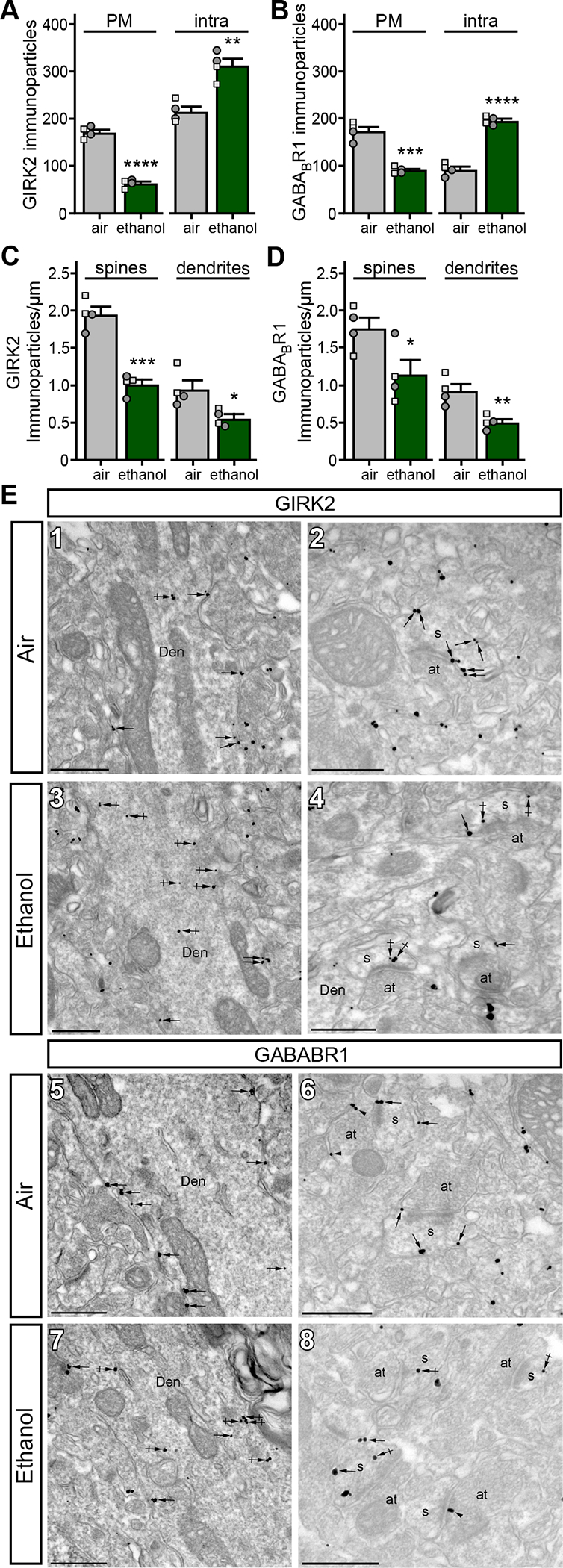
A. Distribution of GIRK2 immunoparticles at the plasma membrane (PM; t6=14.19, ****P<0.0001; N=4/group; unpaired Student’s t-test) and intracellular sites (intra; t6=5.221, **P=0.002; N=4/group; unpaired Student’s t-test) in BA principal neurons from air- and ethanol-exposed mice. Small squares and circles represent individual data points from male and female subjects, respectively.
B. Distribution of GABABR1 immunoparticles at the plasma membrane (PM; t6=8.078, ***P=0.0002; N=4/group; unpaired Student’s t-test) and intracellular sites (intra; t6=12.74, ****P<0.0001; N=4/group; unpaired Student’s t-test) in BA principal neurons from air- and ethanol-exposed mice.
C. Plasma membrane-associated immunoparticle density for GIRK2 in spines (t6=7.566, ***P=0.0003; N=4/group; unpaired Student’s t-test) and dendrites (t6=2.892, *P=0.0276; N=4/group; unpaired Student’s t-test) from BA principal neurons from air- and ethanol-exposed mice.
D. Plasma membrane-associated immunoparticle density for GABABR1 in spines (t6=2.529, *P=0.0447; N=4/group; unpaired Student’s t-test) and dendrites (t6=3.916, **P=0.0078; N=4/group; unpaired Student’s t-test) from BA principal neurons from air- and ethanol-exposed mice.
E. Electron micrographs showing immunoparticles for GIRK2 (panels 1&2) and GABABR1 (panels 5&6) in the BA of air-exposed mice, and for GIRK2 (panels 3&4) and GABABR1 (panels 7&8) in ethanol-exposed mice. In sections from air-exposed mice, most immunoparticles for GIRK2 and GABABR1 were located along the extrasynaptic plasma membrane (arrows) of dendritic shafts (Den) and spines (s) of BA principal neurons. GIRK2 and GABABR1 immunoparticles were also detected at intracellular sites (crossed arrows). In sections from ethanol-exposed mice, GIRK2 and GABABR1 immunoparticles were commonly located at intracellular sites (crossed arrows), and less frequently along the extrasynaptic plasma membrane (arrows) of dendritic shafts (Den) and spines (s) of BA principal neurons. at, axon terminal. Scale bars: 500 nm.
Ethanol alters mood-related behavior in C57BL6/J mice
To understand the impact of ethanol exposure and withdrawal on behaviors linked to the BLA, we evaluated C57BL/6J mice in light-dark box, marble burying, and bottle brush tests following completion of the CIE/4 treatment protocol. Subjects were first assessed for handling-induced convulsions (HIC) 7 h after the last ethanol (or air) session. Ethanol-treated mice exhibited elevated HIC scores (Fig. 6A), indicating that the CIE/4 protocol induced physical dependence. In the light-dark box test (Day 2), ethanol-treated mice spent less time in the light side of the box (Fig. 6B). In the marble burying test (Day 4), ethanol exposure correlated with increased number of marbles buried (Fig. 6C). Ethanol-treated mice also exhibited elevated irritability, as assessed by the number of aggressive and defensive responses in the bottle-brush test (Fig. 6D).
Figure 6. Ethanol triggers a GIRK3-dependent disruption of mood-related behavior.
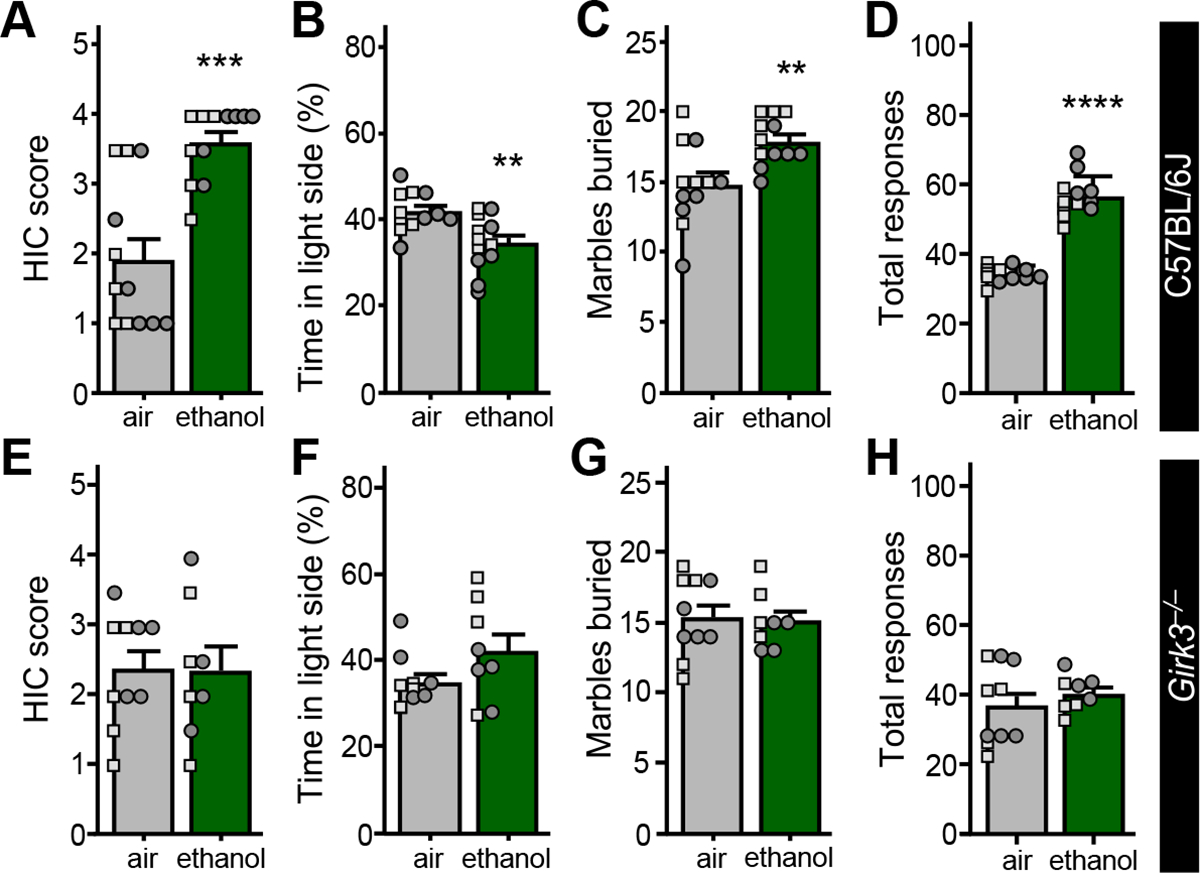
A. Handling-induced convulsion (HIC) scores of C57BL/6J mice, measured 7 h after the final ethanol (or air control) exposure session (t16.12=4.989, ***P=0.0001, N=12/group; unpaired Student’s t-test with Welch’s correction). Small squares and circles represent individual data points from male and female subjects, respectively.
B. Percentage of time spent in the light side of the light-dark box by air-treated and ethanol-treated C57BL/6J mice (t22=3.208, **P=0.0041, N=12/group; unpaired Student’s t-test), measured on Day 2.
C. Number of marbles buried by air-treated and ethanol-treated C57BL/6J mice (t22=3.174, **P=0.0044, N=12/group; unpaired Student’s t-test), measured on Day 4.
D. Total irritability-like responses of C57BL/6J mice (t22=12.19, ****P=<0.0001, N=12/group; unpaired Student’s t test), measured on Days 6 and 7.
E. HIC scores of air-treated and ethanol-treated Girk3−/− mice, measured 7 h after completing the CIE/4 ethanol vapor (or air control) treatment protocol (t16=0.0590, P=0.954, N=8–10/group; unpaired Student’s t test).
F. Percentage of time spent in the light side of the light-dark box by air-treated and ethanol-treated Girk3−/− mice (t16=1.719, P=0.105, N=8–10/group; unpaired Student’s t test), measured on Day 2.
G. Number of marbles buried by air-treated and ethanol-treated Girk3−/− mice (t16=0.233, P=0.819, N=8–10/group; unpaired Student’s t test), measured on Day 4.
H. Total irritability-like responses observed by air-treated and ethanol-treated Girk3−/− mice, (t16=0.822, P=0.423, N=8–10/group; unpaired Student’s t test), measured on Days 6 and 7.
A separate cohort of ethanol- and air-exposed control mice was evaluated in elevated plus maze (EPM) and fear conditioning tests. Ethanol treatment did not impact time spent in open (Fig. S2A) or closed (Fig. S2B) arms of the EPM, or on total distance traveled (Fig. S2C). Similarly, no differences between ethanol and control groups were found in context or cue fear learning (Fig. S2D). Thus, CIE/4 treatment provoked aberrant behavior in a subset of mood-related behavioral tests.
Ethanol-induced alterations in mood-related behavior require GIRK3
Given that GIRK3 is required for the ethanol-induced suppression of GABABR-GIRK signaling in BA principal neurons (Fig. 4), we evaluated behavioral performance in Girk3−/− mice following CIE/4 treatment. Consistent with published observations (51), CIE/4 treatment failed to produce signs of physical dependence in Girk3−/− mice as assessed by HIC severity (Fig. 6E). Similarly, ethanol treatment had no impact on the performance of Girk3−/− mice in light-dark box (Fig. 6F), marble burying (Fig. 6G), or bottle brush (Fig. 6H) tests. Thus, GIRK3 is required for the change in mood-related behaviors seen during withdrawal following CIE/4 treatment.
Suppression of GIRK channel activity recapitulates some withdrawal-related behaviors
To probe the behavioral implications of the ethanol-induced suppression of GABABR-GIRK signaling in BA principal neurons, we used conditional Girk1 knockout (Girk1fl/fl) mice and a viral Cre approach to suppress GIRK channel activity in BA principal neurons in ethanol-naïve mice. CaMKIIα promoter-based AAV vectors have been used extensively to drive transgene expression in principal neurons of the BLA, including BA principal neurons (38). We delivered AAV8-CaMKIIα-Cre(mCherry) or control vector to the BA of Girk1fl/fl mice (Fig. 7A). After 4–6 wks, viral Cre-treated BA principal neurons from Girk1fl/fl mice exhibited smaller Ibaclofen relative to controls (Fig. 7B,C). Viral Cre suppression of GIRK channel activity also decreased rheobase (Fig. 7D) but had no impact on RMP (Fig. 7E).
Figure 7. GIRK channel ablation in BA principal neurons recapitulates some ethanol-induced mood-related behavioral deficits.
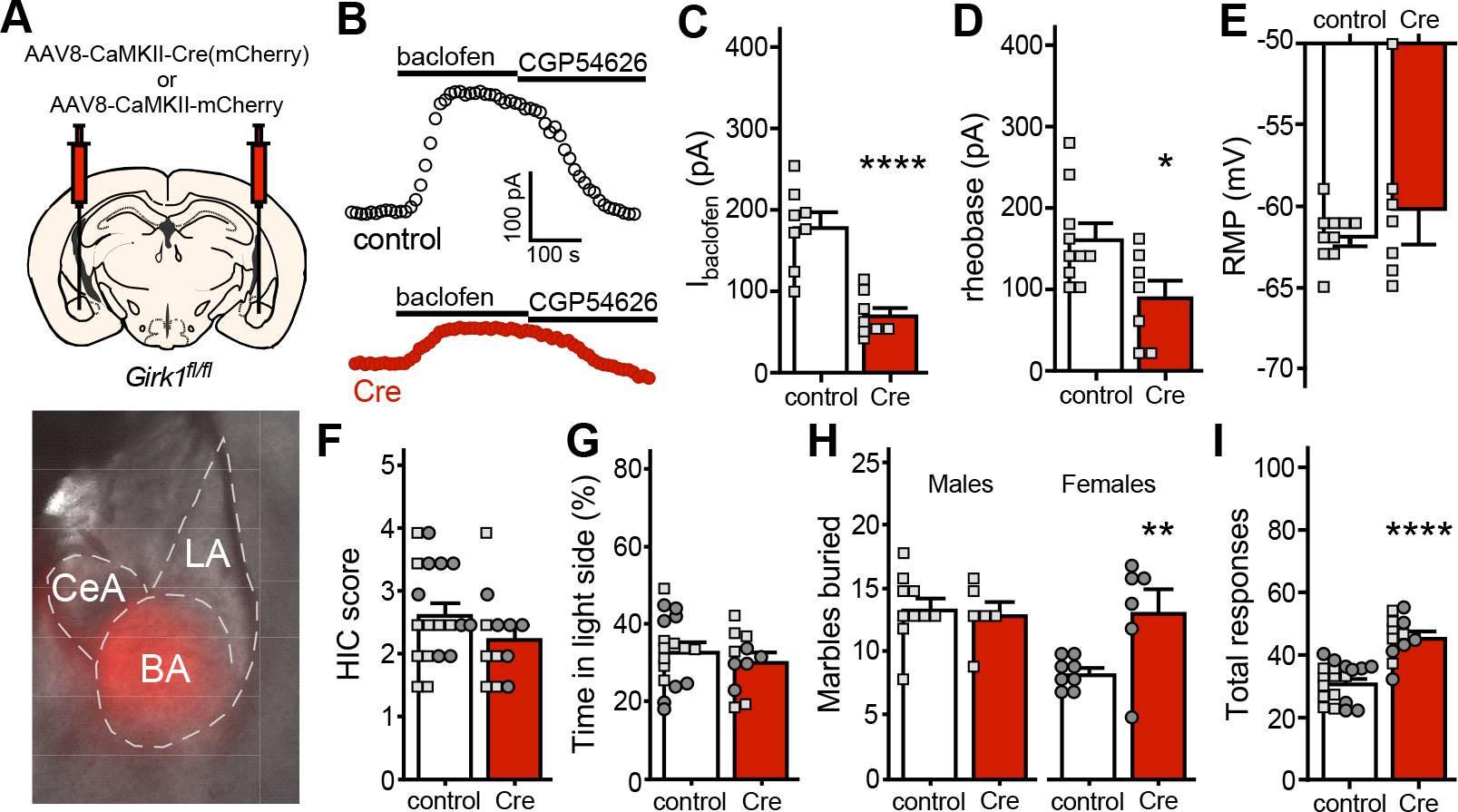
A. Depiction of viral targeting of the BA of Girk1fl/fl mice using AAV8-CaMKIIα-Cre(mCherry) or control (AAV8-CaMKIIα-mCherry) vector; the image below shows selective targeting of the BA in a mouse treated with the control vector.
B. Somatodendritic currents (Vhold=−60 mV) evoked by baclofen (200 μM) and reversed by CGP54626 (2 μM) in BA principal neurons from Girk1fl/fl mice, measured 5 wk after treatment with intra-BA AAV8-CaMKIIα-Cre(mCherry) or control (AAV8-CaMKIIα-mCherry) vector.
C. Ibaclofen in BA principal neurons from male Girk1fl/fl mice, measured 4–6 wk after intra-BA treatment with AAV8-CaMKIIα-Cre(mCherry) or AAV8-CaMKIIα-mCherry vector (t14=5.551, ****P<0.0001; N=8/group; unpaired Student’s t test).
D. Rheobase in BA principal neurons from male Girk1fl/fl mice treated with AAV8-CaMKIIα-Cre(mCherry) or AAV8-CaMKIIα-mCherry vector (t15=2.503, *P=0.0243; N=7–10/group; unpaired Student’s t test).
E. RMP in BA principal neurons from male Girk1fl/fl mice treated with AAV8-CaMKIIα-Cre(mCherry) or AAV8-CaMKIIα-mCherry vector (t6.771=0.792, P=0.455; N=7–10/group; unpaired Student’s t-test with Welch’s correction).
F. Handling-induced convulsion (HIC) scores of ethanol-naïve Girkfl/fl mice, assessed 5–6 wk after intra-BA treatment with AAV8-CaMKIIα-Cre(mCherry) or AAV8-CaMKIIα-mCherry vector (t29=1.397, P=0.173, N=12–19/group; unpaired Student’s t test), measured on Day 1.
G. Percentage of time spent in the light side of the light-dark box by Girkfl/fl mice treated with AAV8-CaMKIIα-Cre(mCherry) or control (AAV8-CaMKIIα-mCherry) vector (t26=0.841, P=0.408, N=11–17mice/group; unpaired Student’s t test), measured on Day 2.
H. Number of marbles buried by Girkfl/fl mice treated with AAV8-CaMKIIα-Cre(mCherry) or control (AAV8-CaMKIIα-mCherry) vector, measured on Day 4. A significant interaction was observed between sex and viral treatment (F(1,26)=6.377, P=0.0180; 2-way ANOVA); Sidak’s post hoc test revealed a viral treatment effect in females (**P=0.0071, N=6–8 mice/group) but not males (P=0.9453, N=6–10 mice/group).
I. Total irritability-like responses of Girkfl/fl mice treated with AAV8-CaMKIIα-Cre(mCherry) or control (AAV8-CaMKIIα-mCherry) vector (t29=6.319, ****P<0.0001, N=12–19 mice/group; unpaired Student’s t test), measured on Days 6 and 7.
Cre- and control-treated Girk1fl/fl mice were next run through behavioral testing. As expected, ethanol-naïve viral Cre-treated Girk1fl/fl mice exhibited no evidence of ethanol dependence as measured by HIC (Fig. 7F). Similarly, suppression of GIRK channel activity in BA principal neurons did not impact performance in the light-dark box test (Fig. 7G). In the marble burying test, suppression of GIRK channel activity in BA principal neurons correlated with increased marbles buried by female but not male mice (Fig. 7H). Irritability-like behavior was also higher in viral Cre-treated mice (Fig. 7I). Thus, suppression of GIRK channel activity in BA principal neurons from ethanol-naive mice recapitulates some of the mood-related behaviors observed in C57BL/6J mice during ethanol withdrawal.
DISCUSSION
Of the many ion channels and receptors implicated in the cellular and behavioral effects of ethanol (52, 53), GIRK channels have emerged as an intriguing target. While the canonical pathway for GIRK channel activation involves the direct binding of the G protein Gβγ subunit (48), GIRK channels are also activated by ethanol in a Gβγ-independent manner (54). Several ethanol-related behaviors, including consumption, reward, preference, and acute withdrawal are altered or absent in mice lacking GIRK subunits (51, 55–57). Girk2−/− mice, for example, consumed more ethanol than wild-type counterparts but failed to develop ethanol conditioned place preference (55). Moreover, genetic variation in the GIRK2/KCNJ6 gene is associated with vulnerability to alcohol dependence in humans (58).
GIRK-dependent signaling is also subject to plasticity evoked by drugs of abuse, experience, and neuronal activity (48). For example, psychostimulant exposure suppressed GIRK-dependent signaling in the VTA (27–30) and prelimbic cortex (32), and aversive experience suppressed GIRK-dependent signaling in the lateral habenula (59). A repeated ethanol injection protocol comparable to that used in this study enhanced the D2 dopamine receptor-dependent activation of GIRK channels in mouse VTA dopamine neurons (60). In addition, withdrawal following ethanol vapor exposure, as well as voluntary ethanol consumption, correlated with reduced inhibitory influence of dopamine, norepinephrine, and serotonin on neuronal excitability in the lateral orbitofrontal cortex; suppression of GIRK channel activity was implicated in this effect, at least in female subjects (61, 62). Here, we show that GIRK-dependent signaling in mouse BA principal neurons was weakened by repeated ethanol exposure. Using either a repeated injection or CIE ethanol vapor exposure protocol, we found that GABABR-GIRK currents in BA (but not LA) principal neurons were suppressed during early withdrawal. Thus, ethanol exposure and/or withdrawal provoke neuron-specific adaptations (enhancement or suppression) of GIRK-dependent signaling in addiction-relevant neuron populations.
GIRK1 and GIRK2 subunits play an essential role in the formation of neuronal GIRK channels (48), including the GIRK channel in BA principal neurons. Ethanol treatment correlated with decreased surface labeling (and increased intracellular labeling) of GIRK2 in BA principal neurons. This, together with the lack of impact of ethanol on the GIRK-independent (Ba2+-insensitive) component of Ibaclofen, suggests that the ethanol-induced suppression of GIRK-dependent signaling involves a reduction in GIRK channel number on the cell surface. Ethanol could impact other Ibaclofen signaling pathway elements, however, including inhibitory G proteins or GABABR itself. Indeed, ethanol also led to a reduction in the surface level of GABABR in BA principal neurons. Given the lack of impact of ethanol on the GIRK-independent component of Ibaclofen, this suggests the intriguing possibility that ethanol selectively impacts discrete intracellular branches of GABABR-dependent signaling in BA principal neurons. To address this issue, it will be important to complement the assessment of ethanol effects on Ibaclofen reported herein with investigations of synaptic GABABR-dependent inhibitory postsynaptic currents and presynaptic GABABR-dependent inhibition. Relatedly, it will be important to determine whether signaling pathways involving other inhibitory receptors that may couple to GIRK channels in BA principal neurons are similarly impacted by ethanol.
Despite the widespread distribution of GIRK3 in the rodent brain (63), functional roles for this subunit are less apparent. GIRK3 ablation, for example, has little impact on the amplitude of GIRK-dependent currents in most neurons (32, 47, 64, 65), as we show here in BA principal neurons. Nevertheless, the ethanol-induced suppression of GABABR-GIRK signaling in BA principal neurons requires GIRK3. While GIRK3 may function within BA principal neurons to mediate this neuroadaptation, it is also possible that the adaptation is driven by ethanol withdrawal, which is diminished in Girk3−/− mice (51). Thus, GIRK3 may exert its influence on GABABR-GIRK signaling in BA principal neurons and related behaviors indirectly, from outside the BA.
GIRK3 is also required for the psychostimulant-induced suppression of GIRK-dependent signaling (30) and the bi-directional influence of firing rate on GIRK channel activity (50) in VTA dopamine neurons. The influence of GIRK3 in these settings likely relates to its ability to negatively regulate the surface trafficking of neuronal GIRK channels (66). This is an intriguing mechanism given that ethanol exposure triggers the subcellular re-distribution of GIRK channels and GABABR in BA principal neurons. Increased internalization of GIRK channels and GABABR has been implicated in other neuroadaptations involving Ibaclofen (28, 32, 59). The influence of GIRK3 on the subcellular trafficking of GIRK channels appears to involve an interaction between GIRK3 and sorting nexin 27, which promotes the endosomal trafficking of GIRK channels (67).
Genetic studies in mice have identified GIRK3 as a key regulator of behavioral sensitivity to several classes of drugs. Differential expression of GIRK3 has been implicated in mouse strain differences in analgesic sensitivity to morphine, clonidine, and WIN55,212, and Girk3−/− mice are less sensitive to the analgesic effects of these drugs (68). GIRK3 was identified as a candidate gene exerting influence on withdrawal severity for ethanol, pentobarbital, and zolpidem (51), and Girk3−/− mice exhibit little-or-no withdrawal symptoms in following exposure to these drugs. While these findings suggest that GIRK3 confers increased sensitivity to the behavioral effects of ethanol and other drugs, Girk3−/− mice also exhibit enhanced ethanol conditioned place preference (57) and increased ethanol consumption under binge conditions (69). Moreover, the acute ethanol-induced increase in firing rate of VTA dopamine neurons is lower in Girk3−/− mice than in control (69). Collectively, evidence suggests that GIRK3 exerts neuron-specific influence on ethanol sensitivity and related behaviors.
The relationships between ethanol exposure, Ibaclofen, neuronal excitability (rheobase and RMP), and behavioral outcomes in this study warrant discussion. For example, the repeated injection approach suppressed Ibaclofen in BA principal neurons (Fig. 1D), while the similar duration CIE/1 exposure protocol did not (Fig. 2C). This may relate to differences in routes or timing of exposure, and/or cumulative ethanol exposure levels. It could also reflect significant within-group variability in Ibaclofen and related electrophysiological measures, combined with moderate effect sizes. Indeed, CIE/1 and CIE/4 treatments exerted qualitatively similar influence on Ibaclofen and rheobase in BA principal neurons (Fig. 2C,D), but only the larger CIE/4 study showed significant differences in these measures between ethanol- and air-treated subjects.
Decreased GIRK-dependent signaling in neurons generally correlates with increased excitability (32, 45, 47). However, we show here that ablation of Girk1 or Girk2 eliminated most or all of the GIRK component of Ibaclofen in BA principal neurons (Fig. 3C,D & 4A), yet neither rheobase (Fig. 4E) nor RMP (Fig. 4F) were significantly impacted. In contrast, viral Cre ablation of GIRK1 in BA principal neurons suppressed Ibaclofen and decreased rheobase (Fig. 7B–D). This discrepancy might reflect the contribution of another factor(s) that regulates BA principal neuron excitability and compensates for the loss of GIRK channel activity throughout development. Relatedly, the ethanol-induced suppression of Ibaclofen in BA principal neurons correlated with decreased rheobase in the CIE/4 model (Fig. 2D), but not the repeated injection approach (Fig. 1E). This suggests that other influences on BA principal neuron excitability may be altered by ethanol in manners that depend on route, duration, timing, and/or cumulative level of exposure.
Suppression of GIRK-dependent signaling in BA principal neurons was sufficient to recapitulate some but not all behaviors observed in C57BL/6J mice during withdrawal from ethanol exposure. The lack of perfect alignment in this case suggests that other adaptations and neuron populations contribute to withdrawal-related behaviors. The selective impact of viral Cre suppression of GIRK channel activity in BA principal neurons on marble burying behavior in female subjects is particularly intriguing. This finding is reminiscent of sex differences in the behavioral impact of viral Cre ablation of GIRK-dependent signaling in prelimbic pyramidal neurons (70), and is consistent with evidence that addiction-related behaviors can be regulated by different molecular and cellular mechanisms in males and females (71).
In sum, this study highlights a neuroadaptation in the BA associated with repeated ethanol exposure in mice. Interventions targeting GABABR-GIRK signaling, particularly those designed to enhance GIRK channel activity and selectively tailored toward GIRK3, warrant further investigation as therapeutic approaches in AUD. Identification and elucidation of alcohol-induced neuroadaptations may reveal opportunities to disrupt the cycle of intoxication, withdrawal, and craving that complicate the treatment of AUD.
Supplementary Material
KEY RESOURCES TABLE.
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Antibody | Rabbit anti-GIRK2 | Frontier Institute Co. Japan | Rb-Af290 | aa. 390–421 of mouse GIRK2A-1; RRID: AB_2571712 |
| Mouse anti-GABABR1 | Neuromab, CA, USA | clone N93A/49 | ||
| Goat anti-rabbit IgG | Nanoprobes Inc | #2003 | coupled to 1.4 nm gold | |
| Goat anti-mouse IgG | Nanoprobes Inc | #2001 | coupled to 1.4 nm gold | |
| Bacterial or Viral Strain | AAV8-CaMKIIa-mCherry | University of Minnesota Viral Vector and Cloning Core | Custom made from pAAV-CaMKIIα-hChR2(C128S/D156A)-mCherry (Addgene plasmid #35502, a gift from Dr. Karl Deisseroth) | |
| AAV8-CaMKIIa-Cre-mCherry | University of North Carolina Vector Core | |||
| Chemical Compound or Drug | Baclofen | Sigma | B5399 | |
| Barium Chloride | Sigma | B0750 | ||
| CGP54626 | Tocris | 1088 | ||
| Pyrazole | Sigma | P56607 | ||
| Ethanol | Pharmco | 111000200 | ||
| Commercial Assay Or Kit | EnzyChromeTM ethanol assay kit | BioAssay Systems, USA | ECET-100 | |
| Organism/Strain | C57BL/6J | The Jackson Laboratory | Strain #:000664 | |
| Girk1−/− | PMID: 12374786 | |||
| Girk2−/− | PMID: 9023358 | |||
| Girk3−/− | PMID: 12040038 | |||
| Girk1flox/flox | PMID: 28131769 | |||
| Sequence-Based Reagent | CaMKIIa RNAScope probe | Advanced Cell Diagnostics | 445231-C1 | NM_009792.3; target nt region, 896–1986 |
| GIRK1 RNAScope probe | Advanced Cell Diagnostics | 523951-C2 | NM_008426.2; target nt region, 658–1679 | |
| GIRK2 RNAScope probe | Advanced Cell Diagnostics | 472321-C2 | NM_001025584.2; target nt region, 282–1456 | |
| GIRK3 RNAScope probe | Advanced Cell Diagnostics | 508871-C2 | NM_008429.2; target nt region, 84–1276 | |
| Software; Algorithm | Prism v.8 | GraphPad Software, Inc |
ACKNOWLEDGEMENTS
This project was supported by NIH grants to KW (DA034696, AA027544), MET (AA025978), MCD (AA028726), TRR (DA007234), and the University of Minnesota Viral Vector and Cloning Core (DA048742), as well as a Wallin Neuroscience Discovery Fund award (KW) and Doctoral Dissertation Fellowships to BV and TRR from the University of Minnesota. RL is supported by grants from the Spanish Ministerio de Economica y Competitividad (PID2021-125875OB-I00), Junta de Comunidades de Castilla-La Mancha (SBPLY/21/180501/000064), and MCIN/AEI/10.13039/501100011033/“ERDF A way of making Europe”. The authors would like to thank Zhilian Xia, Hannah Oberle, Mehrsa Zahiremami, and Courtney Wright for exceptional care of the mouse colony.
Footnotes
DISCLOSURES
The authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Feltenstein MW, See RE (2013): Systems level neuroplasticity in drug addiction. Cold Spring Harb Perspect Med 3:a011916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koob GF, Volkow ND (2016): Neurobiology of addiction: a neurocircuitry analysis. Lancet Psychiatry 3:760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grant BF, Goldstein RB, Saha TD, Chou SP, Jung J, Zhang H, et al. (2015): Epidemiology of DSM-5 Alcohol Use Disorder: Results From the National Epidemiologic Survey on Alcohol and Related Conditions III. JAMA Psychiatry 72:757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le Berre AP, Fama R, Sullivan EV (2017): Executive Functions, Memory, and Social Cognitive Deficits and Recovery in Chronic Alcoholism: A Critical Review to Inform Future Research. Alcohol Clin Exp Res 41:1432–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dager AD, McKay DR, Kent JW Jr., Curran JE, Knowles E, Sprooten E, et al. (2015): Shared genetic factors influence amygdala volumes and risk for alcoholism. Neuropsychopharmacology 40:412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wrase J, Makris N, Braus DF, Mann K, Smolka MN, Kennedy DN, et al. (2008): Amygdala volume associated with alcohol abuse relapse and craving. Am J Psychiatry 165:1179–1184. [DOI] [PubMed] [Google Scholar]
- 7.Stalnaker TA, Roesch MR, Franz TM, Calu DJ, Singh T, Schoenbaum G (2007): Cocaine-induced decision-making deficits are mediated by miscoding in basolateral amygdala. Nat Neurosci 10:949–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ambroggi F, Ishikawa A, Fields HL, Nicola SM (2008): Basolateral amygdala neurons facilitate reward-seeking behavior by exciting nucleus accumbens neurons. Neuron 59:648–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marinelli PW, Funk D, Juzytsch W, Le AD (2010): Opioid receptors in the basolateral amygdala but not dorsal hippocampus mediate context-induced alcohol seeking. Behav Brain Res 211:58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sinclair CM, Cleva RM, Hood LE, Olive MF, Gass JT (2012): mGluR5 receptors in the basolateral amygdala and nucleus accumbens regulate cue-induced reinstatement of ethanol-seeking behavior. Pharmacol Biochem Behav 101:329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chaudhri N, Woods CA, Sahuque LL, Gill TM, Janak PH (2013): Unilateral inactivation of the basolateral amygdala attenuates context-induced renewal of Pavlovian-conditioned alcohol-seeking. Eur J Neurosci 38:2751–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCool BA, Christian DT, Fetzer JA, Chappell AM (2014): Lateral/basolateral amygdala serotonin type-2 receptors modulate operant self-administration of a sweetened ethanol solution via inhibition of principal neuron activity. Front Integr Neurosci 8:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Butler TR, Chappell AM, Weiner JL (2014): Effect of beta3 adrenoceptor activation in the basolateral amygdala on ethanol seeking behaviors. Psychopharmacology (Berl) 231:293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu PJ, Lovinger DM (2006): Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J Neurophysiol 96:433–441. [DOI] [PubMed] [Google Scholar]
- 15.Perra S, Pillolla G, Luchicchi A, Pistis M (2008): Alcohol inhibits spontaneous activity of basolateral amygdala projection neurons in the rat: involvement of the endocannabinoid system. Alcohol Clin Exp Res 32:443–449. [DOI] [PubMed] [Google Scholar]
- 16.Lack AK, Ariwodola OJ, Chappell AM, Weiner JL, McCool BA (2008): Ethanol inhibition of kainate receptor-mediated excitatory neurotransmission in the rat basolateral nucleus of the amygdala. Neuropharmacology 55:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Silberman Y, Ariwodola OJ, Weiner JL (2009): Differential effects of GABAB autoreceptor activation on ethanol potentiation of local and lateral paracapsular GABAergic synapses in the rat basolateral amygdala. Neuropharmacology 56:886–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Christian DT, Alexander NJ, Diaz MR, Robinson S, McCool BA (2012): Chronic intermittent ethanol and withdrawal differentially modulate basolateral amygdala AMPA-type glutamate receptor function and trafficking. Neuropharmacology 62:2430–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindemeyer AK, Liang J, Marty VN, Meyer EM, Suryanarayanan A, Olsen RW, et al. (2014): Ethanol-induced plasticity of GABAA receptors in the basolateral amygdala. Neurochem Res 39:1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soyka M, Muller CA (2017): Pharmacotherapy of alcoholism - an update on approved and off-label medications. Expert Opin Pharmacother 18:1187–1199. [DOI] [PubMed] [Google Scholar]
- 21.Logge WB, Morley KC, Haber PS (2022): GABAB Receptors and Alcohol Use Disorders: Clinical Studies. Curr Top Behav Neurosci 52:195–212. [DOI] [PubMed] [Google Scholar]
- 22.Rose TR, Wickman K (2022): Mechanisms and Regulation of Neuronal GABAB Receptor-Dependent Signaling. Curr Top Behav Neurosci 52:39–79. [DOI] [PubMed] [Google Scholar]
- 23.Federici M, Nistico R, Giustizieri M, Bernardi G, Mercuri NB (2009): Ethanol enhances GABAB-mediated inhibitory postsynaptic transmission on rat midbrain dopaminergic neurons by facilitating GIRK currents. Eur J Neurosci 29:1369–1377. [DOI] [PubMed] [Google Scholar]
- 24.Kelm MK, Criswell HE, Breese GR (2011): Ethanol-enhanced GABA release: a focus on G protein-coupled receptors. Brain Res Rev 65:113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colombo G, Gessa GL (2018): Suppressing Effect of Baclofen on Multiple Alcohol-Related Behaviors in Laboratory Animals. Front Psychiatry 9:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Beaurepaire R, Sinclair JMA, Heydtmann M, Addolorato G, Aubin HJ, Beraha EM, et al. (2018): The Use of Baclofen as a Treatment for Alcohol Use Disorder: A Clinical Practice Perspective. Front Psychiatry 9:708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arora D, Hearing M, Haluk DM, Mirkovic K, Fajardo-Serrano A, Wessendorf MW, et al. (2011): Acute cocaine exposure weakens GABA(B) receptor-dependent G-protein-gated inwardly rectifying K+ signaling in dopamine neurons of the ventral tegmental area. J Neurosci 31:12251–12257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Padgett CL, Lalive AL, Tan KR, Terunuma M, Munoz MB, Pangalos MN, et al. (2012): Methamphetamine-evoked depression of GABA(B) receptor signaling in GABA neurons of the VTA. Neuron 73:978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharpe AL, Varela E, Bettinger L, Beckstead MJ (2014): Methamphetamine self-administration in mice decreases GIRK channel-mediated currents in midbrain dopamine neurons. Int J Neuropsychopharmacol 18:pyu073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munoz MB, Padgett CL, Rifkin R, Terunuma M, Wickman K, Contet C, et al. (2016): A Role for the GIRK3 Subunit in Methamphetamine-Induced Attenuation of GABAB Receptor-Activated GIRK Currents in VTA Dopamine Neurons. J Neurosci 36:3106–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jayaram P, Steketee JD (2004): Effects of repeated cocaine on medial prefrontal cortical GABAB receptor modulation of neurotransmission in the mesocorticolimbic dopamine system. J Neurochem 90:839–847. [DOI] [PubMed] [Google Scholar]
- 32.Hearing M, Kotecki L, Marron Fernandez de Velasco E, Fajardo-Serrano A, Chung HJ, Lujan R, et al. (2013): Repeated cocaine weakens GABA(B)-Girk signaling in layer 5/6 pyramidal neurons in the prelimbic cortex. Neuron 80:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bettahi I, Marker CL, Roman MI, Wickman K (2002): Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKACh. J Biol Chem 277:48282–48288. [DOI] [PubMed] [Google Scholar]
- 34.Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M (1997): Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci U S A 94:923–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K (2002): G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J Neurosci 22:4328–4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marron Fernandez de Velasco E, Carlblom N, Xia Z, Wickman K (2017): Suppression of inhibitory G protein signaling in forebrain pyramidal neurons triggers plasticity of glutamatergic neurotransmission in the nucleus accumbens core. Neuropharmacology 117:33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morton RA, Diaz MR, Topper LA, Valenzuela CF (2014): Construction of vapor chambers used to expose mice to alcohol during the equivalent of all three trimesters of human development. J Vis Exp 89:51839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tipps M, Marron Fernandez de Velasco E, Schaeffer A, Wickman K (2018): Inhibition of Pyramidal Neurons in the Basal Amygdala Promotes Fear Learning. eNeuro 5:ENEURO.0272–18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, et al. (2012): RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn 14:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marron Fernandez de Velasco E, Hearing M, Xia Z, Victoria NC, Lujan R, Wickman K (2015): Sex differences in GABA(B)R-GIRK signaling in layer 5/6 pyramidal neurons of the mouse prelimbic cortex. Neuropharmacology 95:353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crabbe JC, Cotnam CJ, Cameron AJ, Schlumbohm JP, Rhodes JS, Metten P, et al. (2003): Strain differences in three measures of ethanol intoxication in mice: the screen, dowel and grip strength tests. Genes Brain Behav 2:201–213. [DOI] [PubMed] [Google Scholar]
- 42.Calhoon GG, Tye KM (2015): Resolving the neural circuits of anxiety. Nat Neurosci 18:1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tovote P, Fadok JP, Luthi A (2015): Neuronal circuits for fear and anxiety. Nat Rev Neurosci 16:317–331. [DOI] [PubMed] [Google Scholar]
- 44.Wang N, Liu X, Li XT, Li XX, Ma W, Xu YM, et al. (2021): 7,8-Dihydroxyflavone Alleviates Anxiety-Like Behavior Induced by Chronic Alcohol Exposure in Mice Involving Tropomyosin-Related Kinase B in the Amygdala. Mol Neurobiol 58:92–105. [DOI] [PubMed] [Google Scholar]
- 45.Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA (1997): G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19:687–695. [DOI] [PubMed] [Google Scholar]
- 46.Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Lüscher C (2004): Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci 7:153–159. [DOI] [PubMed] [Google Scholar]
- 47.Koyrakh L, Lujan R, Colon J, Karschin C, Kurachi Y, Karschin A, et al. (2005): Molecular and cellular diversity of neuronal G-protein-gated potassium channels. J Neurosci 25:11468–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luo H, Marron Fernandez de Velasco E, Wickman K (2022): Neuronal G protein-gated K+ channels. Am J Physiol Cell Physiol 323:C439–C460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Labouebe G, Lomazzi M, Cruz HG, Creton C, Lujan R, Li M, et al. (2007): RGS2 modulates coupling between GABAB receptors and GIRK channels in dopamine neurons of the ventral tegmental area. Nat Neurosci 10:1559–1568. [DOI] [PubMed] [Google Scholar]
- 50.Lalive AL, Munoz MB, Bellone C, Slesinger PA, Luscher C, Tan KR (2014): Firing modes of dopamine neurons drive bidirectional GIRK channel plasticity. J Neurosci 34:5107–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kozell LB, Walter NA, Milner LC, Wickman K, Buck KJ (2009): Mapping a barbiturate withdrawal locus to a 0.44 Mb interval and analysis of a novel null mutant identify a role for Kcnj9 (GIRK3) in withdrawal from pentobarbital, zolpidem, and ethanol. J Neurosci 29:11662–11673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McCool BA (2011): Ethanol modulation of synaptic plasticity. Neuropharmacology 61:1097–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zorumski CF, Mennerick S, Izumi Y (2014): Acute and chronic effects of ethanol on learning-related synaptic plasticity. Alcohol 48:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bodhinathan K, Slesinger PA (2014): Alcohol modulation of G-protein-gated inwardly rectifying potassium channels: from binding to therapeutics. Front Physiol 5:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Blednov YA, Stoffel M, Chang SR, Harris RA (2001): Potassium channels as targets for ethanol: studies of G-protein-coupled inwardly rectifying potassium channel 2 (GIRK2) null mutant mice. J Pharmacol Exp Ther 298:521–530. [PubMed] [Google Scholar]
- 56.Hill KG, Alva H, Blednov YA, Cunningham CL (2003): Reduced ethanol-induced conditioned taste aversion and conditioned place preference in GIRK2 null mutant mice. Psychopharmacology (Berl) 169:108–114. [DOI] [PubMed] [Google Scholar]
- 57.Tipps ME, Raybuck JD, Kozell LB, Lattal KM, Buck KJ (2016): G Protein-Gated Inwardly Rectifying Potassium Channel Subunit 3 Knock-Out Mice Show Enhanced Ethanol Reward. Alcohol Clin Exp Res 40:857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nishizawa D, Ikeda K (2013): [Genetic polymorphisms commonly associated with sensitivity to various addictive substances]. Nihon Shinkei Seishin Yakurigaku Zasshi 33:205–209. [PubMed] [Google Scholar]
- 59.Lecca S, Pelosi A, Tchenio A, Moutkine I, Lujan R, Herve D, et al. (2016): Rescue of GABA and GIRK function in the lateral habenula by protein phosphatase 2A inhibition ameliorates depression-like phenotypes in mice. Nat Med 22:254–261. [DOI] [PubMed] [Google Scholar]
- 60.Perra S, Clements MA, Bernier BE, Morikawa H (2011): In Vivo Ethanol Experience Increases D(2) Autoinhibition in the Ventral Tegmental Area. Neuropsychopharmacology 36:993–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nimitvilai S, Lopez MF, Mulholland PJ, Woodward JJ (2017): Ethanol Dependence Abolishes Monoamine and GIRK (Kir3) Channel Inhibition of Orbitofrontal Cortex Excitability. Neuropsychopharmacology 42:1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nimitvilai S, Lopez MF, Woodward JJ (2018): Effects of monoamines on the intrinsic excitability of lateral orbitofrontal cortex neurons in alcohol-dependent and non-dependent female mice. Neuropharmacology 137:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karschin C, Dissmann E, Stuhmer W, Karschin A (1996): IRK(1–3) and GIRK(1–4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J Neurosci 16:3559–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Marker CL, Lujan R, Colon J, Wickman K (2006): Distinct populations of spinal cord lamina II interneurons expressing G-protein-gated potassium channels. J Neurosci 26:12251–12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arora D, Haluk DM, Kourrich S, Pravetoni M, Fernandez-Alacid L, Nicolau JC, et al. (2010): Altered neurotransmission in the mesolimbic reward system of Girk−/− mice. J Neurochem 114:1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ma D, Zerangue N, Raab-Graham K, Fried SR, Jan YN, Jan LY (2002): Diverse trafficking patterns due to multiple traffic motifs in G protein-activated inwardly rectifying potassium channels from brain and heart. Neuron 33:715–729. [DOI] [PubMed] [Google Scholar]
- 67.Lunn ML, Nassirpour R, Arrabit C, Tan J, McLeod I, Arias CM, et al. (2007): A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci 10:1249–1259. [DOI] [PubMed] [Google Scholar]
- 68.Smith SB, Marker CL, Perry C, Liao GC, Sotocinal SG, Austin JS, et al. (2008): Quantitative trait locus and computational mapping identifies Kcnj9 (GIRK3) as a candidate gene affecting analgesia from multiple drug classes. Pharmacogenet Genomics 18:231–241. [DOI] [PubMed] [Google Scholar]
- 69.Herman MA, Sidhu H, Stouffer DG, Kreifeldt M, Le D, Cates-Gatto C, et al. (2015): GIRK3 gates activation of the mesolimbic dopaminergic pathway by ethanol. Proc Natl Acad Sci U S A 112:7091–7096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson EM, Loke S, Wrucke B, Engelhardt A, Demis S, O’Reilly K, et al. (2021): Suppression of pyramidal neuron G protein-gated inwardly rectifying K+ channel signaling impairs prelimbic cortical function and underlies stress-induced deficits in cognitive flexibility in male, but not female, mice. Neuropsychopharmacology 46:2158–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Orsini CA, Brown TE, Hodges TE, Alonso-Caraballo Y, Winstanley CA, Becker JB (2022): Neural Mechanisms Mediating Sex Differences in Motivation for Reward: Cognitive Bias, Food, Gambling, and Drugs of Abuse. J Neurosci 42:8477–8487. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.