

Abstract
Introduction:
Historically, the majority of childhood cancers, including acute lymphoblastic leukemia (ALL), were not thought to have a hereditary basis. However, recent germline genomic studies have revealed that at least 5 – 10% of children with cancer (and approximately 3 – 4% of children with ALL) develop the disease due to an underlying genetic predisposition.
Areas covered:
This review discusses several recently identified ALL predisposing conditions and provides updates on other more well-established syndromes. It also covers topics related to the evaluation and management of children and family members at increased ALL risk.
Expert opinion:
Germline predisposition is gaining recognition as an important risk factor underlying the development of pediatric ALL. The challenge now lies in how best to capitalize on germline genetic information to improve ALL diagnosis, treatment, and perhaps even prevention.
Keywords: Acute lymphoblastic leukemia (ALL), familial studies, genetic predisposition, genome wide association studies (GWAS), hematologic malignancy, transcription factors
1. Introduction
Acute lymphoblastic leukemia (ALL) serves as a paradigm for the study and successful treatment of childhood cancer [1]. Sixty years ago, little was known about the genetic factors underlying development of childhood ALL. Thanks to decades of study and the application of high-throughput sequencing approaches, it is now recognized that ALL consists of multiple subtypes based on the presence of defining somatic genetic lesions, some of which influence the response to therapy and overall prognosis [2]. Accordingly, recent efforts to incorporate somatic genetic information into clinical care has moved ALL treatment in the direction of ‘precision medicine’ by informing the use of targeted agents such as tyrosine kinase inhibitors for children whose ALL blasts express the BCR-ABL oncoprotein or harbor mutations affecting the JAK-STAT pathway [2].
Germline genomic investigations have also steadily increased current knowledge surrounding the host genetic factors that influence ALL risk, with sequencing of rare leukemia-prone kindreds and large ALL cohorts revealing an expanding array of leukemia predisposing genes and associated genetic conditions (Table 1). Notably, for several of these conditions, the causal genes are the same as those targeted by somatic mutations in ALL blasts (e.g. PAX5, IKZF1, ETV6). As with somatic genetic information, germline genetic data can significantly impact clinical care by identifying children who are at increased risk to develop therapy-associated toxicities, second malignant neoplasms, and non-oncologic manifestations. These factors must be carefully considered when developing a treatment plan for a child with an ALL predisposition. In this review, we discuss some of the recent advances surrounding genetic predisposition to pediatric ALL. As there have been excellent articles published recently on childhood cancer predisposition [3–5], we will focus on the newer discoveries and their implications on ALL pathogenesis and treatment.
Table 1.
Genetic syndromes that predispose to pediatric ALL.
| Genes | Locus | Mode of inheritance | ALL Subtype | Other associated neoplasms | Reference | |
|---|---|---|---|---|---|---|
| DNA Damage Response and Cell Cycle/Apoptosis | ||||||
| Ataxia Telangiectasia (AT) | ATM | 11q22.3 | AR | T-ALL | T-PLL, NHL, HL, B-CLL, breast, ovarian, gastric, melanoma, leiomyomas, and sarcomas | [6] |
| Bloom syndrome (BS) | BLM | 15q26.1 | AR | ALL | MDS, AML, NHL, GI (upper/lower), GU, oropharyngeal, skin, breast | [7,110] |
| Constitutional mismatch repair deficiency syndrome (CMMRD) | MLH1 | 3p22.2 2p21-p16 | AR | B-ALL or T-ALL | NHL, AML, CML, GI, brain | [113–115] |
| MSH2 | 2p16.3 7p22.1 | |||||
| MSH6 | 2p21 | |||||
| PMS2 | ||||||
| EPCAM | ||||||
| Fanconi anemia (FA) | FANCA | 16q24.3 | AR | ALL | MDS, AML, squamous cell carcinoma of the head, neck, esophagus, and vulva, cervical cancer, liver tumors | [8] |
| FANCC | 9q22.32 | |||||
| FANCD2 | 3p25.3 | |||||
| BRCA2 | 13q13.1 | |||||
| FANCE | 6p21.31 | |||||
| FANCI | 15q26.1 | |||||
| FANCL | 2p16.1 | |||||
| RAD51C | 17q22 | |||||
| SLX4 | 16p13.3 | |||||
| ERCC4 | 16p13.12 | |||||
| UBE2T | 1q32.1 | |||||
| XRCC2 | 7q36.1 | |||||
| MAD2L2 | 1p36.22 | |||||
| RFWD3 | 16q23.1 | |||||
| FANCB | Xp22.2 | XLR | ||||
| RAD51A | 15q15.1 | AD | ||||
| FANCF | 11p14.3 | Unknown | ||||
| XRCC9 | 9p13.3 | |||||
| BRIP1 | 17q23.2 | |||||
| FANCM | 14q21.2 | |||||
| PALB2 | 16p12.2 | |||||
| Li-Fraumeni syndrome (LFS) | TP53 | 17p13.1 | AD | B-ALL | MDS, AML (therapy-associated), NHL, brain, bone, soft tissue, breast, adrenocortical carcinoma | [101–105] |
| Nijmegen breakage syndrome (NBS) | NBN | 8q21.3 | AR | T-ALL | NHL, AML, APL, medulloblastoma, glioma, RMS | [9] |
| Lymphocyte Differentiation | ||||||
| Familial platelet disorder with predisposition to myeloid malignancy (FPDMM) | RUNX1 | 21q22.12 | AD | T-ALL | AML | [79,84,85] |
| IKZF1-associated leukemia predisposition | IKZF1 | 7p12.2 | AD | B-ALL | None | [46] |
| PAX5-associated leukemia predisposition | PAX5 | 9p13.2 | AD | B-ALL | None | [19,20] |
| ETV6-associated leukemia predisposition | ETV6 | 12p13.2 | AD | B-ALL | AML, CML, NHL, MDS, multiple myeloma | [30,32–36] |
| Signaling | ||||||
| Neurofibromatosis, type 1 (NF1) | NF1 | 17q11.2 | AD | ALL | JMML, AML, brain, PNST, RMS, GIST, breast, pheochromocytoma | [69] |
| Noonan syndrome (NS) | PTPN11 | 12q24.13 | AD | ALL | JMML, CMML, AML, RMS, neuroblastoma | [51,53,54] |
AML, acute myeloid leukemia; APL, acute prolymphocytic leukemia; AT, Ataxia Telangiectasia; B-CLL, B cell chronic lymphocytic leukemia; BS, Bloom syndrome; CML, chronic myeloid leukemia; CMML, chronic myelomonocytic leukemia; CMMRD, Constitutional mismatch repair deficiency; FA, Fanconi anemia; FPDMM, Familial platelet disorder with predisposition to myeloid malignancies; GI, gastrointestinal; GIST, gastrointestinal stromal tumor; GU, genitourinary; HL, Hodgkin lymphoma; JMML, juvenile myelomonocytic leukemia; LFS, Li-Fraumeni syndrome; MDS, myelodysplastic syndrome; NBS, Nijmegen breakage syndrome; NF1, Neurofibromatosis type 1; NHL, Non-Hodgkin lymphoma; NS, Noonan syndrome; PNST, peripheral nerve sheath tumor; RMS, rhabdomyosarcoma; T-PLL, T cell prolymphocytic leukemia.
2. Genetic predisposition to leukemia: Mendelian single gene disorders
Some of first reports discussing familial ALL appeared in the literature in the early 1950s-1960s with descriptions of kindreds in which multiple close relatives were affected by the disease [10–12]. Although it was initially proposed that these familial aggregations might be due to inheritance of specific HLA alleles [13], it was not until 1990 when germline TP53 variants were discovered as the cause for Li-Fraumeni syndrome (LFS) [14] that the genetic basis for familial ALL truly began to emerge. Since this initial finding, it is now known that there are several Mendelian genetic conditions that increase ALL risk, with the majority following an autosomal dominant pattern of inheritance. For the most part, ALL predisposing conditions are caused by pathogenic germline alterations affecting critical genes implicated in several cellular processes such as differentiation, proliferation, apoptosis, repair of DNA damage, and intracellular signaling (Figure 1). Not surprisingly, the clinical phenotypes of many ALL-predisposing conditions correlate with the expression and function of the affected genes, with variants in genes controlling B-lymphocyte differentiation predisposing primarily to B-ALL, while variants in genes with a broader expression and function predispose to a wider spectrum of cancers and other manifestations.
Figure 1.
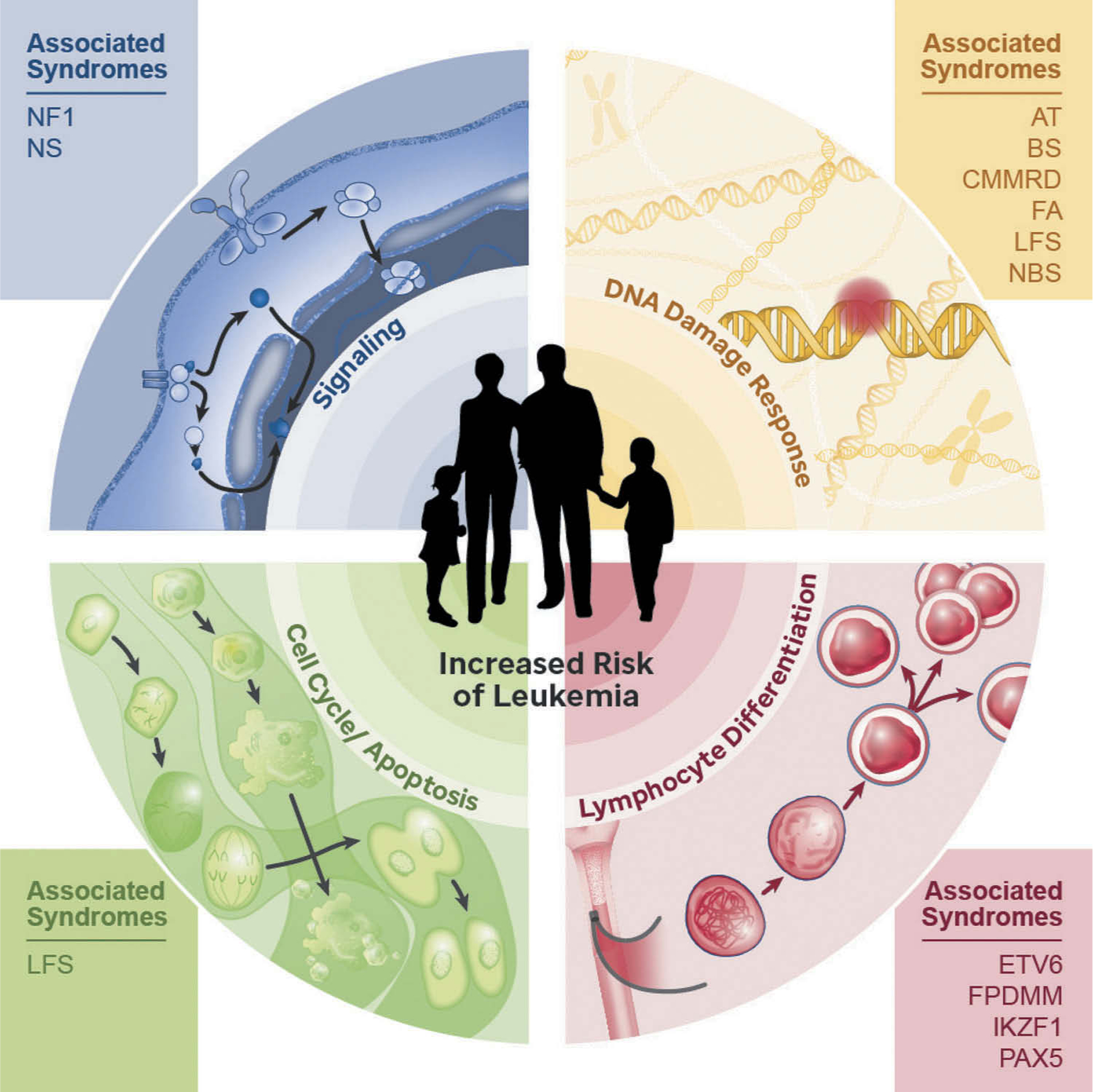
Predisposition to pediatric acute lymphoblastic leukemia results from germline variants affecting genes that encode proteins involved in critical pathways such as cellular signaling, DNA damage response, cell cycle, cell death, and lymphocyte differentiation. AT, Ataxia Telangiectasia; BS, Bloom syndrome; CMMRD, Constitutional mismatch repair deficiency; ETV6, ETV6-associated predisposition; FA, Fanconi anemia; FPDMM, Familial platelet disorder with predisposition to myeloid malignancy; IKZF1, IKZF1-associated predisposition to B-ALL; LFS, Li-Fraumeni syndrome; NBS, Nijmegen breakage syndrome; NF1, Neurofibromatosis type 1; NS, Noonan syndrome; PAX5, PAX5-associated leukemia predisposition.
2.1. Syndromes that primarily predispose to ALL
2.1.1. PAX5-associated leukemia predisposition (susceptibility to ALL 3; OMIM #167414)
PAX5-associated leukemia predisposition is an autosomal dominant ALL predisposition caused by germline variants in PAX5 (Paired Box Gene 5). PAX5 encodes a transcription factor with important roles in B cell development and B-leukemogenesis [15]. In mice, the genetic ablation of PAX5 leads to a profound block in B cell differentiation [16], while in humans, somatic mutations or translocations affecting PAX5 comprise the most common genetic lesions occurring in one-third of all B-ALL cases [17,18]. In 2013, three unrelated families were reported in which individuals affected by ALL harbored the same heterozygous germline PAX5 p.Gly183Ser missense variant [19,20]. Within these families, several PAX5 variant carriers did not develop ALL, suggesting this germline variant exhibits incomplete penetrance. To date, all germline PAX5 variant-associated ALL samples examined exhibit loss of the wildtype PAX5 allele, either through formation of an isochromosome or dicentric chromosome 9q [20]. Thus, PAX5 appears to function as a classic tumor suppressor with germline mutations acting as the first ‘hit’ and loss of the remaining wild-type (WT) allele serving as the second hit within leukemic blasts.
Studies are beginning to elucidate the mechanisms by which germline PAX5 variants promote B-leukemogenesis. PAX5 is most highly expressed in the B cell lineage (www.bloodspot.eu), where it regulates the expression of key downstream target genes involved in B cell ontogeny, including CD19, CD79A, and BLNK, as well as genes involved in metabolic programming, such as NR3C1 (which encodes the glucocorticoid receptor) and TXNIP (which encodes a glucose-feedback sensor). Notably, PAX5+/− mice do not spontaneously develop leukemia, even after long periods of observation [21,22]. However, they do exhibit alterations in B cell development with higher proportions of immature B-progenitors [21]. These findings suggest that PAX5 heterozygosity favors development of an abnormal B-precursor population that may be susceptible to oncogenic transformation. Consistent with this notion, delayed exposure of PAX5+/– animals to pathogens [21; see below Section 4.0 Germline Leukemia Predisposition and Delayed Infection Hypothesis] or to chemical or viral mutagens [23] promotes ALL development with leukemic blasts exhibiting somatic genetic lesions similar to those observed in human B-ALL samples. The results of these and other investigations support the concept that PAX5 protein normally functions to promote differentiation of B-lymphoid progenitors while restricting glucose and energy supply. In cells that are deficient in PAX5 function, development and metabolism are disrupted in a manner that favors malignant transformation upon acquisition of cooperating somatic genetic lesions [24].
2.1.2. ETV6-associated leukemia predisposition (thrombocytopenia V; OMIM #616216)
ETV6 (ETS Variant 6) encodes an ETS family transcriptional repressor [25,26] that is essential for establishing mouse bone marrow (BM) hematopoiesis and maintaining survival of hematopoietic stem cells (HSCs) [27–29]. The ETV6 protein contains two functional domains, including the pointed domain (enables homo- and hetero-dimerization) and the ETS domain (mediates DNA binding). ETV6 was first identified as an ALL predisposition gene in 2015 following the identification of eight unrelated families exhibiting autosomal dominant transmission of B-ALL and thrombocytopenia in which damaging germline ETV6 variants co-segregated with disease [30,32–34]. Subsequently, Moriyama and colleagues performed targeted ETV6 sequencing using remission blood samples from 4,405 pediatric ALL cases and identified germline variants in ~1% of cases [30]. In both studies, many ETV6 variants clustered in the DNA binding domain or were predicted to produce a truncated protein lacking all or part of this domain.
To date, 96 individuals from 23 families have been described [30–36]. Among these 96 individuals, 25–30% are reported to have developed leukemia [30,32–36], most commonly B-ALL with a hyperdiploid karyotype. Rarer cases of mixed-phenotype leukemia, acute or chronic myeloid leukemia, B cell non-Hodgkin lymphoma, and multiple myeloma, have also been reported [35]. In a study of 38 ALL patients with germline ETV6 variants, two developed secondary MDS/AML suggesting a possible association with onset of therapy-associated myeloid neoplasms [37]. Although some ETV6 variant carriers have developed solid tumors, such as colorectal, duodenal, breast and renal carcinoma, breast fibroadenoma, and skin cancer [32–34,38], the relationship of these cancers to germline ETV6 status remains unclear. Mild to moderate thrombocytopenia is present in almost every germline ETV6 variant carrier with some exhibiting a bleeding tendency and/or platelet functional defects [31,39]. Severe thrombocytopenia only appears when associated with myelodysplastic syndrome [31,33]. Analysis of the BM in ETV6 variant positive individuals without leukemia reveals megakaryocyte hyperplasia, hypolobulated megakaryocytes, mild dyserythropoiesis, and abnormal myeloid cells [33,34,39].
The mechanisms by which germline ETV6 variants contribute to B-ALL remain poorly understood. The fact that many variants cluster within, and truncate or remove, the DNA binding domain suggests that interference with transcriptional activity is central to the leukemogenic process. In line with this possibility, in vitro studies show that ETV6 variant proteins exhibit impaired repressor activity, reduced DNA binding, and aberrant subcellular localization [32–34]. Furthermore, when co-expressed with WT ETV6, ALL-associated variant proteins exert a dominant negative effect on transcriptional repression mediated by WT ETV6 [34]. Based on these studies, it appears likely that germline ETV6 variants alter gene expression within hematopoietic progenitors, thereby disrupting normal B-lymphoid and megakaryocyte development, as well as platelet function.
2.1.3. IKZF1-associated predisposition to B-ALL (OMIM #616873)
IKZF1 (IKAROS Family Zinc Finger 1) encodes the founding member of the IKAROS family of transcription factors [40]. IKAROS contains six zinc fingers, four N-terminal and two C-terminal, which mediate DNA binding and dimerization with self or other members of the IKAROS family [41,42]. IKAROS is widely expressed among cells of the hematopoietic system where it is required for specification of lymphoid lineages and involved in differentiation of pro-B cells into pre-B cells (reviewed in [43]). Somatic mutations affecting IKZF1, often resulting in loss of function or dominant negative effects, are found in high risk B-ALL, especially in cases expressing the BCR-ABL oncoprotein (also known as the Philadelphia chromosome [Ph+]) and those lacking the oncoprotein but exhibiting a similar transcriptional profile (Ph-like) [44,45]. In these cases, the presence of somatic IKZF1 mutations appears to confer an even poorer response to therapy [44,45].
In 2018, Churchman and colleagues [46] described a family with five individuals who harbored a heterozygous germline IKZF1 variant (p.Asp186fs) that is predicted to truncate the protein within the region of the N-terminal zinc fingers. Among these five IKZF1 mutation carriers, two developed B-ALL, lending credence to the possible role of this variant as a leukemia predisposing allele [46]. To further examine this possibility, these investigators performed targeted sequencing of IKZF1 in a cohort of 4,963 pediatric ALL cases and identified 27 unique non-silent IKZF1 coding variants among 43 individuals (0.9%), with all but one affected with B-ALL [46]. Genotype-phenotype studies revealed no obvious correlations; however, several of the IKZF1 mutation carriers exhibited B lymphopenia, suggesting the presence of an underlying immunodeficiency.
Prior to the report by Churchman, germline IKZF1 variants had been described in individuals with common variable immune deficiency, where variants primarily clustered in the N-terminal zinc fingers and resulted in impaired DNA binding [47,48]. In contrast, B-ALL-associated germline IKZF1 variants appear distributed across the gene with some clustering in the C-terminal zinc fingers. Although it is poorly understood why there is clustering in this region, in vitro and in vivo functional studies of B-ALL-associated germline IKZF1 variants, including dimerization, DNA binding, transcriptional repression, nuclear localization, cellular adhesion, and interference with drug responsiveness, revealed that 22 of 28 unique variants (79%) interfered with least one of these activities, regardless of localization within the gene [46]. It remains to be determined how these variants alter hematopoiesis and promote leukemogenesis.
2.2. Syndromes predisposing to a variety of hematologic malignancies, including ALL
2.2.1. Noonan syndrome (NS; OMIM #163950)
NS is an autosomal dominant condition resulting from germline variants affecting genes encoding components of the RAS/mitogen activated protein kinase (MAPK) pathway [49–51]. Approximately 50% of NS patients harbor heterozygous germline variants in PTPN11 [51], with fewer patients carrying changes in genes such as SOS1, KRAS, RAF1, BRAF, and MEK1, among others. NS occurs with a frequency of 1 in 1000 to 1 in 2500 live births [49] with common non-oncologic manifestations including distinctive facial features, congenital heart defects, short stature, chest deformity, abnormal pigmentation (café-au-lait macules [CALMs]), variable developmental impairment, thrombocytopenia, and platelet dysfunction [49,52].
Hematological malignancies, including juvenile myelomonocytic leukemia (JMML) and ALL represent the most frequently occurring cancers [53]. Two large studies encompassing over 1,800 RASopathy patients revealed that ALL is: 1) present in 0.3–0.5% of NS patients and characterized by a median age of onset of 6 years (range: 1.5–17 years); 2) exclusively of B cell phenotype; and 3) associated with a germline PTPN11 or SOS1 variant [51,54]. NS patients harboring germline PTPN11 variants are more likely to develop hyperdiploid ALL and their disease generally responds favorably to therapy [51]. Although late toxicities are not commonly documented, one report indicated that one-fifth of NS patients who were treated for ALL later presented with myelodysplasia (MDS) that either resolved spontaneously but exhibited persistent thrombocytopenia, or evolved into a JMML-like neoplasm or acute myeloid leukemia (AML) [51].
PTPN11 encodes Src homology region 2 containing protein tyrosine phosphatase 2 (SHP2), a phosphatase that positively regulates signaling downstream of growth factor receptors [55]. Consistent with this notion, human JMML cells, which commonly harbor somatic gain-of-function mutations in PTPN11 [56], exhibit hypersensitivity to granulocyte-macrophage colony stimulating factor (GM-CSF) [57]. In mice, SHP2 is a positive regulator of hematopoiesis, where the loss of expression or decrease in catalytic activity is associated with reduced stem and progenitor cell numbers and function [58], and altered expression of important hematopoietic transcription factors, such as GATA2 and CEBPA [59,60]. Conversely, expression of an activating PTPN11 mutant protein enhanced HSC proliferation and repopulating ability, leading to development of myeloproliferative neoplasms and AML [61,62]. Altogether, these human and murine studies suggest that the inability to attenuate RAS signaling is a critical step in myeloid leukemogenesis. Since somatic alterations in RAS pathway genes, including PTPN11, are observed in over 40% of pediatric B-ALL cases, including high hyperdiploid B-ALL, Ph-like, and non-Ph-like ‘B-other’ phenotypes [63], it is possible that similar mechanisms underlie development of lymphoid leukemia.
2.2.2. Neurofibromatosis type 1 (NF1; OMIM #162200)
NF1 is one of the more common genetic disorders, affecting approximately 1 in every 3000 births [64,65]. NF1 exhibits autosomal dominant inheritance and is caused by germline variants in NF1, the gene encoding neurofibromin, a guanosine triphosphatase (GTPase)-activating protein that normally binds to and negatively regulates RAS [66,67]. The clinical features of NF1 include dermatologic findings such as CALMs and axillary and inguinal freckling; bony abnormalities including osteopenia or osteoporosis, dysplasia of long bones and sphenoid wing, and scoliosis; cardiovascular problems including systemic and pulmonary hypertension, vasculopathy, and pulmonary valve stenosis; neurologic issues including learning disabilities, behavior problems, and less commonly seizures; development of benign and malignant tumors such as Lisch nodules (iris hamartomas), cutaneous and plexiform neurofibromas, central nervous system (CNS) tumors; and less commonly hematologic malignancies [68,69].
The tendency for NF1 patients to develop leukemia, including chronic myelomonocytic leukemia (CMML), JMML, AML, and to a lesser extent ALL, has been known for decades [70–72]. JMML is the most common leukemia, with children with NF1 exhibiting a 200–500-fold increase in risk compared to children without NF1 [68]. A large population-based study of leukemia and non-Hodgkin lymphoma associated with NF1 performed in the United Kingdom revealed only 12 cases of ALL (relative risk 5.4; 95% CI 2.8–9.4) [73]. Among the 12 ALL cases identified, three were of T-lineage and 9 of B-lineage origin. The average age of diagnosis was 5.1 years for B-ALL (range: 1.3–12.3 years) and 11.4 years for T-ALL (range: 9.8–14.5 years) [73]. In over 50% of NF1-associated myeloid malignancies, NF1 loss-ofheterozygosity (LOH) is detected [66]. It remains to be determined whether this is also the case for ALL occurring in the context of NF1.
Consistent with the low prevalence of ALL in individuals with NF1, somatic NF1 mutations are only rarely reported in ALL, with 3% of pediatric and young adult T-ALL cases harboring such mutations [2,70,74,75]. Curiously, somatic NF1 alterations are common in near-haploid B-ALL, where 30 of 68 (44%) samples examined exhibited sequence alterations or focal microdeletions [76]. Nf1-deficient mouse hematopoietic progenitors exhibit hypersensitivity to cytokines with abnormal proliferation of immature and lineage-restricted progenitor populations [77], suggesting that the mechanisms of tumorigenesis are similar to those observed for PTPN11 perturbations.
2.2.3. Familial platelet disorder with predisposition to myeloid malignancy (FPDMM; OMIM #601399)
FPDMM is an autosomal dominant condition caused by germline variants in RUNX1, the gene encoding RUNT-related transcription factor 1 (RUNX1; previously CBFA2 [Core-Binding Factor Subunit Alpha 2] or AML1 [Acute Myeloid Leukemia 1]). RUNX1 functions as a partner of Core-Binding Factor Subunit Beta (CBFB) to form a heterodimeric transcription factor that is one the most frequently altered genes in leukemias, including MDS, AML, ALL, and CMML [78]. RUNX1 is involved in the common t(12;21) ETV6-RUNX1 translocation in pediatric B-ALL, the t(8;21) RUNX1T1-RUNX1 in AML [79], and more than 50 other translocations [80]. In addition to translocations, somatic RUNX1 mutations have been identified in 4.5% (12/264) of pediatric and young adult T-ALL cases [75] and 30% (4/12) of early thymocyte precursor (ETP)-ALL cases [81].
Germline RUNX1 variants were first identified in 1999 in six unrelated families exhibiting functional platelet defects and a tendency to develop AML, with each of the identified variants predicted to result in RUNX1 haploinsufficiency [82]. To date, over 80 families harboring pathogenic germline RUNX1 variants have been described [78,83]. The presentation of FPDMM includes lifelong thrombocytopenia and functional platelet defects that may or may not be associated with bleeding [84]. Individuals with germline RUNX1 variants are at increased risk to develop MDS and AML, and to a lesser extent T-ALL [79], with 30–40% of mutation carriers developing a hematologic malignancy. It has been proposed that individuals with dominant negative RUNX1 variants have a higher incidence of MDS and acute leukemia than patients with loss-of-function variants [85,86]. To date, 11 RUNX1 carriers have been reported to develop T-ALL [84,85,87–90], with three of these individuals developing AML less than five years after the initial T-ALL diagnosis [84,85]. Although somatic RUNX1 mutations have been identified in 7% of B-ALL cases [78], we are aware of only rare individuals with FPDMM who has developed B-ALL (K. Nichols, unpublished).
Mice globally deficient for Runx1 expression lack definitive hematopoiesis [91–93], while animals deficient for Runx1 selectively in adult BM progenitors exhibit impaired B and T cell development, reduced platelet production, and development of abnormal myeloproliferation [94]. These manifestations are similar to humans with FPDMM, whose leukemia samples commonly harbor mutations within the remaining RUNX1 allele [95]. Together, these results suggest that germline RUNX1 variants impair hematopoietic development, leading to the emergence of abnormal populations that are then prone to transformation upon loss of WT RUNX1 [95].
2.3. Syndromes predisposing to solid tumors in addition to ALL
2.3.1. Li-Fraumeni syndrome (LFS; OMIM #151623)
LFS is caused by pathogenic germline variants in TP53, the gene encoding the critical tumor suppressor p53. Due to its central role in maintaining genomic stability and preventing the proliferation of cells with damaged DNA, p53 has been dubbed the ‘guardian of the genome’ [96]. Approximately 50% of all tumors contain somatic TP53 mutations, and many of the remaining cases acquire other genetic or epigenetic alterations that compromise p53 function [97,98]. Accordingly, p53 is the most commonly inactivated protein in all human cancers [99].
LFS is inherited in an autosomal dominant manner and confers a significantly increased lifetime risk for cancer [100]; nearly 100% of individuals with LFS develop cancer by age 70, and 50% of patients develop a second primary cancer within 10 years of the initial cancer diagnosis [101]. While individuals with LFS have a high tendency to develop bone and soft tissue sarcomas, brain tumors, early onset breast cancer, and adrenocortical carcinoma, they also develop leukemia, albeit at a lower frequency (leukemia comprises 2–4% of all LFS cancers) [101–105]. The leukemias associated with LFS include ALL, AML, and MDS, with most, but not all, ALL cases exhibiting a ‘low’ hypodiploid phenotype in which the leukemic blasts contain 32–39 chromosomes [76,106,107]. ALL tends to occur later in children with LFS, with a median age at onset of 15.5 years (versus 7.3 years for children without LFS), and presenting leukocyte counts tend to be lower [106]. Importantly, patients harboring germline TP53 variants appear to have an inferior event-free and overall survival compared to individuals without these variants due to a high-risk for second cancers [106]. Hypodiploid ALL occurring in the context of LFS almost universally shows TP53 LOH, together with alterations in IKZF2 (65%) and RAS pathway genes (9%) [76].
In a study of 47 breast cancer survivors with therapy-associated leukemia, three (6%) individuals harbored pathogenic germline TP53 variants [108]. Notably, two of these three individuals had secondary ALL, an unusual treatment-related hematopoietic malignancy. Compared to primary ALL developing in children with LFS, these two cases of therapy-related leukemia did not exhibit a low hypodiploid phenotype. Hence, for cancer patients who develop therapy-associated ALL, regardless of its cytogenetic make-up, consideration should be given to germline TP53 testing.
2.3.2. Constitutional mismatch repair deficiency (OMIM #276300)
Constitutional mismatch repair deficiency (CMMRD; also known as biallelic mismatch repair deficiency) is an autosomal recessive condition caused by homozygous or compound heterozygous mutations affecting the mismatch repair (MMR) genes, namely MSH2, MSH6, MLH1, PMS2, or rarely, by deletions of the 3ʹ region of EPCAM, a gene adjacent to the MSH2 promoter [109]. The MMR pathway operates to correct single strand DNA mutations that arise during replication [110]. Proteins encoded by mutated MMR genes can no longer correct point mutations, resulting in microsatellite instability, compromised DNA integrity, and an increase in the number of mutations [111].
Heterozygous pathogenic variants in the MMR genes cause Lynch syndrome, an autosomal dominant disorder associated with development of adult-onset cancers, commonly colon and endometrial carcinoma [112]. In contrast, CMMRD is associated with an extremely high incidence of cancer starting in early childhood [113–115], and many patients also have CALMs (a feature not observed in Lynch syndrome). Approximately one-third of CMMRD patients develop a hematologic malignancy, with a median age at diagnosis of 6 years (range: 0–21 years) [114–116]. Non-Hodgkin lymphoma (NHL), generally of T cell origin, is the most frequently reported hematologic malignancy, with fewer cases of ALL, also mostly of T cell origin [114–117]. Hematologic malignancies are more prevalent in individuals harboring MLH1 or MSH2 variants (38%), as compared to MSH6 (25%) or PMS2 (16%) carriers [117]. In addition, first malignancies occur at a younger age in MLH1 or MSH2 mutation carriers (mean age at diagnosis of 2.5 years), as opposed to 7 and 9 years for MSH6 and PMS2 carriers, respectively [117].
Patients with CMMRD generally do not experience undue toxicities following treatment with conventional ALL chemotherapy; however, they are prone to leukemia relapse. There are several possible explanations for this finding. First, ALL backbone therapy includes 6-mercaptopurine (6-MP), an agent that requires intact mismatch repair to exert it cytotoxic effects [118–120]. Thus, 6-MP might function with lower efficacy against ALL blasts from CMMRD patients. Second, there is mounting evidence that certain chemotherapeutic agents (e.g. temozolamide) can induce somatic mutations in CMMRD-associated tumor cells [121,122]. Furthermore, most CMMRD-associated tumors already exhibit a hypermutator phenotype with >100 mutations per megabase of DNA [123]. This striking accumulation of somatic mutations in CMMRD-associated tumors, whether treatment-associated or not, likely contributes to therapy resistance.
Most recently, it has been proposed that the high mutational burden in MMR-deficient tumors could result in generation of neoantigens capable of being recognized by the immune system [124,125]. Curiously, CMMRD-associated tumors often contain a high number of infiltrating lymphocytes [124,125]. Thus, immune-checkpoint inhibitors might serve as an attractive treatment for these patients. Toward this end, recent reports describe the beneficial effects of pembrolizumab in adults with MMR-deficient tumors [125] and nivolumab in three children [126,127] and an adolescent [128] with CMMRD-associated gliomas. Whether immune-checkpoint inhibitors will have roles in the treatment of ALL or other hematologic malignancies remains to be determined.
2.4. Genetic predisposition to leukemia due to constitutional chromosome alterations
2.4.1. Down syndrome (DS; OMIM #190685)
DS, a genetic condition associated with constitutional trisomy for chromosome 21, was one of the first genetic syndromes associated with increased risk for childhood leukemia with initial reports dating back to the 1930s [129,130]. DS is the most common chromosomal disorder and occurs in approximately 1 in every 700 live births (www.CDC.gov). Children with DS present with a variety of congenital abnormalities, most commonly affecting the heart and gastrointestinal tract [131]. Additional features include intellectual disabilities and characteristic facies. While the majority of DS cases are not familial, in rare cases DS can be caused by transmission of an abnormal chromosome harboring a Robertsonian translocation involving chromosome 21. In so-called ‘translocation’ DS (t-DS), the affected baby inherits a chromosome (usually from the mother) that contains two copies of the long arm of chromosome 21. Children with t-DS exhibit similar phenotypic features as children with DS caused by an extra copy of the entire chromosome 21.
Children with DS have a 20-fold increased risk of developing ALL compared to children without DS, and a 500-fold increased risk of developing acute megakaryoblastic leukemia (AMKL) [132]. Remarkably, children with the rare Robertsonian translocation, rob(15;21)(q10;q10)c have a 2,700-fold increased risk of developing ALL [133]. The age of leukemia onset in DS is bimodal, with the first peak in newborns and the second at 3–6 years of age; the increased risk may also extend to adulthood. The leukemias occurring in infants with DS are almost exclusively myeloid [129]. In contrast, ALL occurs in children with DS at ages similar to children without DS [129]. The vast majority of ALL cases seen in children with DS are of B cell precursor origin. In a study of 700 patients with DS-ALL, only five cases of T-ALL were observed [134]. DS-ALL has historically been considered to have a worse prognosis than non-DS ALL due to therapy resistance and/or treatment related mortality [134,135]. However, recent reports have suggested that DS patients with ALL fare as well as those without DS when given appropriate supportive care [136,137].
Common somatic genetic abnormalities observed in sporadic B-ALL are less frequent in DS B-ALL, such as ETV6-RUNX1 and hyperdiploidy [134,138]. In contrast, up to 60% of DS-ALL cases overexpress Cytokine Receptor Like Factor 2 (CRLF2), the a type I cytokine receptor that binds thymic stromal lymphopoietin (TSLP) and is often associated with JAK-STAT pathway activating mutations [139,140]. In one study, Bercovitch and colleagues showed that somatic mutations in JAK2 were observed in 16 of 88 DS patients with B-ALL and only 1 of 109 non-DS patients with B-ALL [141]. Notably, this one case also harbored an isochromosome 21q. The authors went on to express the observed JAK2 mutants in mouse hematopoietic progenitor cells in vitro and observed cytokine independent growth due to constitutive JAK-STAT pathway activation, consistent with gain-of-function effects [141]. In line with these findings, recent genetic and epigenetic investigations reveal that Ph-like ALL is the most common DS-ALL subtype [142].
3. Genetic predisposition to leukemia: common variants acting as risk factors for ALL
Over the last decade, several genome-wide association studies (GWAS) have sought to elucidate how common germline variants, including single nucleotide polymorphisms (SNPs) in coding and non-coding regions of the genome, contribute to ALL. While the SNPs identified through GWAS generally confer a lower risk for ALL when compared to rare germline variants revealed through familial studies, these SNPs can nevertheless provide important insights into the biology of ALL. Consistent with this notion, prior GWAS have revealed susceptibility loci associated with pediatric ALL and involving genes known to play essential roles in hematopoiesis, such as IKZF1, ARID5B, CEBPE, GATA3, BMI1, and CDKN2A, among others (Table 2) [143–146]. GWAS have also identified risk loci specifically associated with drug responsiveness [147], relapse [146], and development of ALL in specific ethnic groups (potentially explaining racial or ancestral discrepancies [146,148,149]), and most recently, in children with Down syndrome [150].
Table 2.
Risk loci associated with increased risk for pediatric ALL.
| Locus | Gene | SNP ID | Cohort | ALL Subtype | Reference |
|---|---|---|---|---|---|
| 2q22.3 | rs17481869 | Multi-ethnic | ETV6-RUNX1 B-ALL | [151] | |
| 3q28 | TP63 | rs17505102 | European | ETV6-RUNX1 B-ALL | [152] |
| 7p12.2 | IKZF1 | rs11978267 | European | B-ALL | [143–145,153,154] |
| 7p15.3 | SP4 | rs2390536 | Multi-ethnic | B-ALL | [155] |
| 8q24.21 | rs28665337 | Multi-ethnic | B-ALL or T-ALL | [151,155] | |
| 9p21.3 | CDKN2A/B | rs3731217, rs77728904 | European | B-ALL or T-ALL | [153,154,156] |
| 10p12.2 | PIP4K2A | rs7088318 | Multi-ethnic | B-ALL or T-ALL | [145,149] |
| 10p14 | GATA3 | rs3824662 | Multi-ethnic | Ph-like B-ALL | [145,146] |
| 10q21.2 | ARID5B | rs10821936 | European | Hyperdiploid ALL or T-ALL | [143–145,154] |
| 10q26.13 | LHPP | rs35837782 | European | B-ALL or T-ALL | [157] |
| 12q23.1 | ELK3 | rs4762284 | European | B-ALL | [157] |
| 14q11.2 | CEBPE | rs2239633 | European | B-ALL | [143,145,154] |
| 16p13.2 | USP7 | rs74010351 | Multi-ethnic | T-ALL | [158] |
| 17q12 | IKZF3 | rs2290400 | Multi-ethnic | B-ALL or T-ALL | [155] |
| 21q22.2 | ERG | rs2836365 | Multi-ethnic | ALL | [148] |
ALL, acute lymphoblastic leukemia; B-ALL, B cell precursor ALL; T-ALL, T cell ALL.
Earlier this year, Qian and colleagues performed a GWAS involving 1,191 children with T-ALL and identified the first germline risk locus associated exclusively with this leukemia subtype [158]. This locus resides at 16p13.2 and includes several SNPs involving USP7 (Ubiquitin specific peptidase-7), the gene encoding a ubiquitin specific protease that is somatically mutated in 12% of pediatric and young adult T-ALL cases [75]. Interestingly, the most common SNP in USP7 (rs74010351) is overrepresented in TAL1 mutation positive T-ALL, a subtype also commonly harboring somatic USP7 mutations. The USP7 risk allele was also over-represented in individuals of African descent, thus contributing to the observed higher incidence of T-ALL in this population [158]. This SNP is located proximal to the USP7 transcription start and resides in a cis-regulatory region marked by active histone modifications and open chromatin segments, suggesting that it might influence USP7 expression. Further supporting this notion, in vitro luciferase reporter assays reveal that the presence of this SNP leads to a 2.9-fold decrease in luciferase expression [158]. Curiously, germline SNPs involving USP7 co-occur only rarely with somatic USP7 mutations in T-ALL blasts. These data suggest that germline and somatic lesions play similar roles during the development of T-ALL, with somatic mutations likely exerting stronger effects. It remains to be determined how reduced USP7 expression contributes to T-leukemogenesis.
4. Germline leukemia predisposition and delayed infection hypothesis
It is well accepted that ALL develops due to the acquisition of multiple genetic lesions within a susceptible hematopoietic cell. The initiating genetic event may be somatic or germline, but by itself, it is unlikely to cause leukemia. The mechanisms that induce the additional genetic lesions needed to promote leukemia development are likely multifactorial, and include chance as well as environmental, infectious, and dietary factors, among others. In 1988, two hypotheses were put forth to implicate infection as a risk factor for ALL [159,160]. These hypotheses suggested that delayed exposure to infection, or exposure to a specific pathogen, might promote an abnormal immune response that then triggers the acquisition of additional genetic lesions at an age commensurate with increased lymphoid-cell proliferation. Consistent with the concept of delayed exposure to infection, a recent report by Marcotte and colleagues describes a decreased risk for ALL in children exposed to influenza or respiratory syncytial virus early in life (e.g. the first three months) as compared to children exposed later (e.g. 9–12 months of age) [161].
Two recent studies have used mouse models to further investigate the contribution of delayed exposure to infection in the development of ALL. In the first, Martin-Lorenzo and colleagues show that PAX5+/– mice born and raised in a specific pathogen free environment do not spontaneously develop B-ALL. However, when these mice were moved to a ‘common infectious environment’, 22% develop B-ALL [162]. Remarkably, this penetrance is comparable to that observed in the few human carriers of germline PAX5 variants [19,20]. In a second report, Rodriguez-Hernandez and colleagues use a similar experimental design to examine the influence of delayed infection in mice expressing the somatic ETV6-RUNX1 fusion protein within hematopoietic cells. The t (12;21) chromosomal translocation encoding this fusion is proposed to act as a ‘first hit’ in humans, much like germline leukemia-predisposing variants. Similar to the PAX5+/– mice, delayed transfer to a ‘common infectious environment’ led to emergence of B-ALL in 11% of ETV6-RUNX1-expressing animals [21]. This again resembles the low penetrance of B-ALL development in human children harboring an ETV6-RUNX1 translocation positive clone [163,164]. In the latter mouse model, leukemia initiation following infection exposure was accompanied by differential regulation of epigenetic regulator genes of the lysine demethylase (KDM) family. It is interesting to note that the authors did not find a high proportion of differentially regulated epigenetic regulator genes when PAX5+/− mice were exposed to late infection, indicating that this is a phenomenon specific to malignant transformation of cells expressing the ETV6-RUNX1 fusion protein.
Together, these epidemiological and animal modeling investigations suggest that a delayed exposure to infection might increase the risk for ALL while earlier exposure is protective. However, additional studies are needed to further examine and understand this phenomenon. For example, there still remains the major question of how exactly does delayed exposure to infection facilitate the acquisition of mutations that promote leukemogenesis? Similarly, how does early exposure provide protection? By testing the effects of exposure to various pathogens during different developmental windows using animal models, investigators are likely to gain novel and important insights into these poorly understood phenomena that can then be further examined in humans.
5. Evaluation and management of children with hereditary predisposition to ALL
As more and more children with germline predisposition are identified, there is increasing urgency to develop clinical management guidelines, not only for those already affected by cancer, but also for relatives who carry a predisposing germline variant but have not developed the disease. As limited data on the optimal methods and schedule of cancer screening exist, many of the current recommendations (summarized below and in Figure 2) are based on expert opinion.
Figure 2.
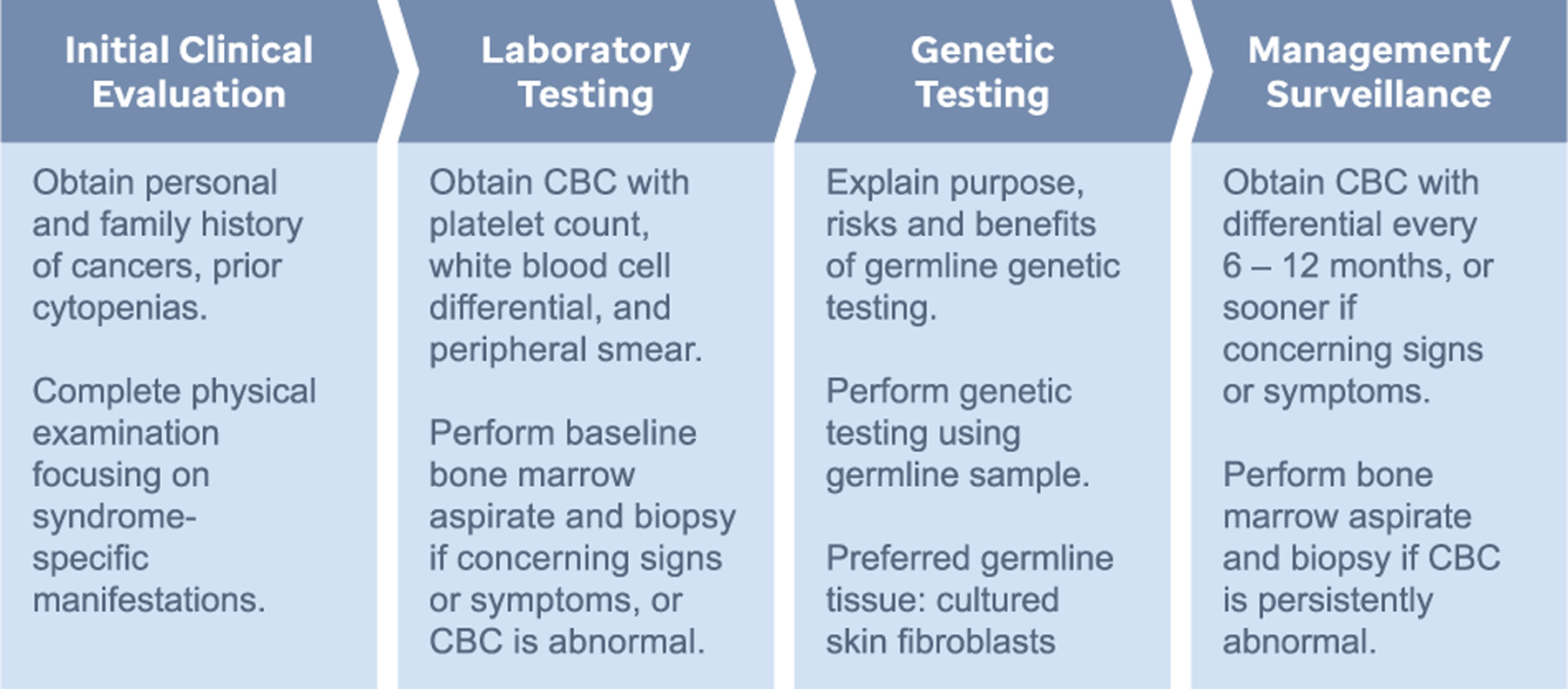
Schematic describing the steps involved in the clinical evaluation of patients with suspected germline predisposition to ALL. CBC, complete blood count.
5.1. Screening for hereditary predisposition to leukemia
Given the increasing number, as well as the phenotypic and genotypic heterogeneity of leukemia predisposing conditions, it is highly recommended that genetic evaluations be completed in consultation with providers who are familiar with these disorders. Generally, these providers include oncologists, geneticists, and genetic counselors who will: 1) collect a medical history of the patient to assess for features such as easy bruising or bleeding (an indicator of possible thrombocytopenia or platelet functional defects as in ETV6-related predisposition to ALL or FPDMM) and developmental delays/behavioral problems (as can be seen in NF1); 2) gather family history information about close blood relatives (first or second degree) who may have also developed leukemia or other cancers, or exhibited evidence of cytopenias; 3) complete a physical examination to look for syndrome-specific features such as CALMs (NF1/RASopathy, CMMRD), axillary/inguinal freckling (NF1), and typical facial and other physical features (RASopathy); and 4) review tumor data focusing on findings such as low hypodiploidy (LFS), isochromosome or dicentric chromosome 9q (PAX5), hypermutator phenotype (CMMRD), and presence of somatic variants within genes indicative of a possible predisposition [165]. Clinical germline testing should be considered for any individual with leukemia who exhibits one or more concerning features, such as: 1) a personal history of multiple cancers; 2) a strong family history of cancers, especially those developing at earlier than expected ages; 3) hematopoietic malignancies diagnosed in two individuals within a three-generation pedigree; 4) a personal history or close relative with one or more cytopenias; and 5) a personal history or close relative with physical findings associated with a known predisposition syndrome. Genetic testing of leukemic blasts can also provide clues to an underlying leukemia predisposition, such as presence of pathogenic variants affecting PAX5 (with isochromosome or dicentric chromosome 9q), ETV6, IKZF1, NF1, PTPN11, or TP53 at an allele frequency of ~30–50%. Such cases suggest the possibility of germline heterozygosity and should prompt genetic testing, as discussed below (Section 5.2 Genetic testing for germline predisposition to ALL).
5.2. Genetic testing for germline predisposition to ALL
Following appropriate counseling about the testing process, its associated risks and benefits, and the potential outcomes, the counselor and/or primary care team must determine which germline tissue to test. As leukemia cells circulate in the blood, blood and BM do not represent the optimal tissues for germline testing, especially during early phases of treatment. While some laboratories do accept a remission blood sample, it may be difficult to determine whether any identified mutations are germline or somatic in origin. Indeed, somatic mutations associated with clonal hematopoiesis of indeterminate potential (CHIP) can persist in the blood even during remission [166,167]. Similarly, saliva can be contaminated with circulating blood cells. Therefore, fibroblasts cultured from a skin biopsy are the optimal tissue to be used for germline testing in a patient with a hematopoietic malignancy [168]. One downside of using cultured skin fibroblasts is the long time required to generate a cell line, usually on the order of 3–6 weeks. Therefore, in cases where a result is needed urgently, a remission peripheral blood sample (preferably in a patient with negative minimum residual disease) can be tested. If there are no pathogenic variants identified, then there is less concern for a germline predisposition syndrome. If, on the other hand, certain mutations are identified, it may be necessary to also test cultured skin fibroblasts. Most recently, it has been reported that plucked hairs with attached follicles provide a rapid and accessible source of germline DNA that may be used as an alternative for genetic analysis [169,170].
5.3. Management of children with an underlying germline predisposition
Learning that a child has an underlying ALL predisposing condition can provide solace since it explains why the leukemia developed. Parents and children can be counseled about the risks for additional neoplasms as well as the risks for recurrence in offspring. Finally, blood relatives can be counseled and tested. Any who test positive can be offered screening for the early detection of cancer. This approach, while less helpful for individuals at risk for acute leukemias such as ALL, holds potential to improve long-term outcomes for those at risk for solid cancers, MDS, and/or BM failure. For children undergoing allogeneic hematopoietic stem cell transplantation (HSCT), germline genetic information is critical to avoid choosing a related donor who also harbors a predisposing germline variant.
It is generally recommended that a complete blood count (CBC) with manual differential be performed at the initial visit for all patients at increased risk for ALL [171]. This CBC provides a baseline for future comparison. In asymptomatic patients, follow-up CBCs could be performed every 6–12 months and more frequently if there are signs or symptoms concerning for leukemia. Any identified abnormalities should prompt a repeat CBC, with the timing dependent upon the severity of the finding. A baseline BM examination with cytogenetic analysis and molecular testing should be considered for all patients with an underlying predisposition syndrome, particularly individuals with significant abnormalities on the baseline CBC. Cytogenetic analysis and molecular testing may disclose early signs of clonal evolution prior to the onset of overt leukemia. Patients with worsening BM dysplasia, increasing numbers of blasts, or progressive BM clonal abnormalities should be followed closely and potentially transitioned to allogeneic HSCT. In children with inherited susceptibility to ALL and a normal initial BM evaluation, there is no consensus regarding the need or use of routine follow-up BM examinations [171,172].
For children with ALL due to an underlying predisposition, germline genetic information can inform the risk for therapy-associated toxicities and provide prognostic information. For individuals with genetic conditions associated with impaired responses to DNA damage, standard-dose therapies can result in serious complications. Patients with Ataxia Telangiectasia (AT) can experience accelerated neurological compromise following exposure to vinca alkaloids [173], pancytopenia and hemorrhagic cystitis following exposure to alkylating agents [174], and mucositis and hemorrhagic colitis following exposure to anthracyclines [174]. Similarly, patients with LFS are at an increased risk to develop chemotherapy-associated leukemias [108,175]. Finally, individuals with AT, Nijmegen breakage syndrome (NBS), and LFS are at significantly increased risk for radiation-induced toxicities, such as leukoencephalopathy (AT) and secondary malignant neoplasms (AT, NBS, LFS) [174,176,177]. While patients with CMMRD are not at risk for therapy-associated toxicities, they are prone to relapse due to therapy resistance [121,122]. Accordingly, information about germline predisposition can allow clinicians to tailor therapy to reduce the risk for these untoward side effects and intensify monitoring for possible disease recurrence or development of secondary neoplasms.
5.4. The advantages and challenges of surveillance for children with ALL predisposition
Many ALL predisposing conditions have only recently been identified and thus remain poorly defined in terms of the clinical manifestations and age-specific leukemia risks. Moreover, given the rapid onset of ALL and the excellent outcomes using current therapies, there is little evidence that surveillance provides any medically relevant benefits. That said, surveillance (as described in Section 5.3 Management of children with an underlying germline predisposition) can prove useful for the early detection of cancer in children predisposed to MDS and/or solid malignant neoplasms [171]. Regardless of the underlying genetic condition, patients, parents, and other affected relatives should be educated about the signs and symptoms of leukemia and encouraged to seek medical advice promptly should they experience any concerning signs or symptoms.
5.5. Allogeneic stem cell transplantation (HSCT) for children with ALL predispositions
While overall survival in pediatric ALL is now close to 90% in developed countries [1], high risk forms of ALL and relapsed disease remain major clinical challenges and comprise the primary indications for allogeneic HSCT. However, with expanding appreciation of germline predisposition, thought is being put toward the use of preemptive allogeneic HSCT to replace a genetically affected bone marrow with a healthy donor marrow before the overt onset of leukemia. Recently, Hamilton and colleagues commented on the factors to be considered when offering preemptive HSCT to patients predisposed to AML, such as the: 1) nature of the underlying syndrome (syndromes associated with solid tumor development will not be helped by allogeneic HSCT); 2) age of the patient (children and young adults tolerate HSCT better than older adults); 3) disease penetrance (preemptive HSCT is more likely to benefit individuals with highly penetrant conditions compared to those with conditions characterized by lower disease penetrance); 4) donor type and availability (fully HLA matched genetically unaffected donors are preferred); and of course, 5) patient and family preferences [178]. It remains to be determined whether preemptive HSCT will prove useful for children with an underlying genetic predisposition to ALL.
6. Expert opinion
Germline predisposition is gaining increasing recognition as an important factor contributing to the development of pediatric cancers, including ALL. Over the last 4 years, studies have shown that 5–10% of children with cancer (including 3–4% with ALL), carry a pathogenic germline variant in a cancer predisposing gene [107,179–181]. With continued examination of additional cohorts and deeper scrutiny of germline data, these percentages will undoubtedly increase as novel variants are uncovered. As the initiating events in tumorigenesis, germline variants perturb cell growth and differentiation and set the stage for malignant transformation. Accordingly, the study of germline variants, and their associated genetic syndromes, is very likely to provide new and important insights into the biology of childhood cancers, including ALL.
Despite the opportunities afforded, there remain several challenges that must be overcome to fully reap the benefits of germline genetic information. For example, it is important to distinguish whether any newly identified germline variants are truly related to cancer and not simply incidental findings. To strengthen association, investigators should examine germline data from cancer cohorts as well as control cohorts. In so doing, it is possible to evaluate for enrichment of predisposing variants in individuals with cancer as compared to controls. For these studies, cohorts should also be carefully examined for differences in ethnicity, which can complicate interpretation of findings.
A second challenge relates to determining the functional relevance of any identified germline variants. Although in silico approaches are commonly used to assist with this process, these approaches do not always accurately predict the true impact of germline variants on the function(s) of the encoded proteins. In this regard, Churchman and colleagues showed that only 65% of the germline IKZF1 variants characterized as damaging in their study were accurately classified using in silico methods [46]. To address this challenge, investigators must use alternative approaches, such as expression of variant proteins in cell lines or live animals, followed by evaluation for downstream effects. Unfortunately, these approaches can be costly as well as time and labor intensive, and they too may have limitations. This is especially apparent for hematopoietic stem cells and B cells, where mouse and human cells have unique cytokine dependencies and express different cell surface markers, suggesting that the mouse and human systems may not be entirely complementary [182]. The use of induced pluripotent stem cells (iPSCs) provides one additional approach to model inherited cancer syndromes and confirm any phenotypes arising through the use of cell lines and mouse models. Indeed, patient-derived iPSCs have already provided insights to how GATA2 deficiency affects human hematopoiesis [183].
A third challenge stems from poor understanding of the impacts of germline variants on clinical phenotypes, which severely limits our ability to optimize the management of children and adults with an underlying predisposition. Indeed, for many cancer predisposing conditions, the age-specific cancer risks have not yet been defined. As such, the degree of cancer risk might be higher or lower, and the spectrum of cancers wider or narrower, than originally proposed. In addition, it remains poorly understood how germline variants influence responses to therapy and overall outcomes. Historically, most individuals to undergo genetic testing for cancer were those who met clinical criteria for a specific hereditary syndrome. As might be expected, this process confirmed and further strengthened associations between specific phenotypes and underlying germline genotypes. However, recent non-biased sequencing studies are revealing individuals with cancer who harbor germline variants that never would have been expected based on their clinical phenotype. For many cases, it remains to be determined whether these novel germline variants are truly causal. To improve knowledge surrounding specific genotype-phenotype associations, investigators must uniformly collect and share clinical, family history, and germline genetic data.
In recognition of the emerging importance of germline predisposition in the diagnosis and management of patients with hematopoietic malignancies, the World Health Organization (WHO) provisionally recognized myeloid neoplasms with germline predisposition in the 2016 revision of the classification of tumors of hematopoietic and lymphoid tissues [184]. Given the continued expansion in knowledge surrounding genetic predisposition to ALL, it is very likely that lymphoid neoplasms will soon join these ranks. Over the next five years, genetic counseling and germline genetic testing will become increasingly employed in pediatric oncology clinics and the resulting information will be used to inform the care of children with cancers, including ALL. The challenge now remains in determining how best to capitalize on this information to further improve the overall cure rates and enhance long-term outcomes for at-risk children and their families.
Article highlights
Recent family-based and large scale high-throughput sequencing studies have uncovered several novel genetic conditions predisposing to acute lymphoblastic leukemia (ALL).
The genes associated with these ALL predisposing conditions are often the same as those targeted by somatic mutations in ALL blasts (e.g. PAX5, IKZF1, ETV6, PTPN11).
For some conditions, such as those caused by germline alterations affecting PAX5, IKZF1, and ETV6, affected individuals are primarily predisposed to develop ALL.
For others, such as Noonan syndrome (NS), Neurofibromatosis 1 (NF1), Li-Fraumeni syndrome (LFS), and Constitutional mismatch repair deficiency (CMMRD), affected individuals are at risk for ALL as well as other hematologic malignancies and/or solid tumors.
Examination of the genetic status of leukemic blasts can provide clues to the underlying ALL predisposing condition. For example, blasts in LFS commonly exhibit a low hypodiploid karyotype, while blasts in CMMRD have a hypermutator phenotype.
Knowledge of an underlying ALL predisposing condition informs leukemia treatment through avoidance of carcinogenic therapies. It also enables the initiation of screening programs for the early detection and treatment of second primary or therapy-associated cancers.
When considering genetic testing in a child with ALL, it is important to choose the appropriate germline tissue; cultured skin fibroblasts are the preferred source of germline DNA.
Pre-emptive allogeneic hematopoietic stem cell transplantation before the overt development of leukemia is a consideration for some individuals with ALL predisposing conditions.
Continued efforts are warranted to better define the spectrum and penetrance of known ALL predisposing conditions, discover new genes and germline variants associated with ALL risk, elucidate the interactions between germline genetic variation and environmental exposures in the development of ALL, and optimize clinical care based on germline genetic information.
Acknowledgments
The authors wish to thank Joshua Stokes and colleagues in Biomedical Communications at St. Jude for their assistance in preparing the figure.
Funding
This manuscript was funded in part by the American Lebanese Syrian Associated Charities and St. Jude Children’s Research Hospital.
Footnotes
Declaration of interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Pui CH, Yang JJ, Bhakta N, et al. Global efforts toward the cure of childhood acute lymphoblastic leukaemia. Lancet Child Adolesc Health. 2018. Jun;2(6):440–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iacobucci I, Mullighan CG. Genetic basis of acute lymphoblastic leukemia. J Clin Oncol. 2017. Mar 20;35(9):975–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pui CH, Nichols KE, Yang JJ. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat Rev Clin Oncol. 2019. Apr;16(4):227–240. [DOI] [PubMed] [Google Scholar]
- 4.Plon SE, Lupo PJ. Genetic predisposition to childhood cancer in the genomic era. Annu Rev Genomics Hum Genet. 2019;20:241–263. [DOI] [PubMed] [Google Scholar]
- 5.Scollon S, Anglin AK, Thomas M, et al. A comprehensive review of pediatric tumors and associated cancer predisposition syndromes. J Genet Couns. 2017;26(3):387–434. [DOI] [PubMed] [Google Scholar]
- 6.Adam M, Ardinger H, Pagon R, et al. Ataxia-Telangiectasia–GeneReviews®.
- 7.Flanagan M, Cunniff C. Bloom syndrome. Seattle: GeneReviews® [Internet]: University of Washington; 2019. [Google Scholar]
- 8.Mehta PA, Tolar J. Fanconi anemia. Seattle: GeneReviews®[Internet]: University of Washington; 2018. [Google Scholar]
- 9.Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, et al. Nijmegen breakage syndrome (NBS). Orphanet J Rare Dis. 2012;7 (1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson RC. Familial leukemia: a report of leukemia in five siblings, with a brief review of the genetic aspects of this disease. AMA Am J Dis Children. 1951;81(3):313–322. [DOI] [PubMed] [Google Scholar]
- 11.Jr CW H, Moloney WC. Familial leukemia: five cases of acute leukemia in three generations. N Engl J Med. 1965;272(17):882–887. [DOI] [PubMed] [Google Scholar]
- 12.Gunz F, Fitzgerald P, Crossen P, et al. Multiple cases of leukemia in a sibship. Blood. 1966;27(4):482–489. [PubMed] [Google Scholar]
- 13.Blattner WA, Naiman JL, Mann DL, et al. Immunogenetic determinants of familial acute lymphocytic leukemia. Ann Intern Med. 1978;89(2):173–176. [DOI] [PubMed] [Google Scholar]
- 14.Malkin D, Li FP, Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250(4985):1233–1238. [DOI] [PubMed] [Google Scholar]
- 15.Matthias P, Rolink AG. Transcriptional networks in developing and mature B cells. Nat Rev Immunol. 2005. Jun;5(6):497–508. [DOI] [PubMed] [Google Scholar]
- 16.Urbanek P, Wang ZQ, Fetka I, et al. Complete block of early B cell differentiation and altered patterning of the posterior midbrain in mice lacking PAX5/BSAP. Cell. 1994. Dec 2;79(5):901–912. [DOI] [PubMed] [Google Scholar]
- 17.Mullighan CG, Goorha S, Radtke I, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007. Apr 12;446(7137):758–764. [DOI] [PubMed] [Google Scholar]
- 18.Kuiper RP, Schoenmakers EF, van Reijmersdal SV, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007. Jun;21 (6):1258–1266. [DOI] [PubMed] [Google Scholar]
- 19.Auer F, Ruschendorf F, Gombert M, et al. Inherited susceptibility to pre B-ALL caused by germline transmission of PAX5 c.547G>A. Leukemia. 2014;28:1136–1138. [DOI] [PubMed] [Google Scholar]
- 20. •.Shah S, Schrader KA, Waanders E, et al. A recurrent germline PAX5 mutation confers susceptibility to pre-B cell acute lymphoblastic leukemia. Nat Genet. 2013;45(10):1226–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]; First report of families with ALL harboring pathogenic germline PAX5 variants.
- 21.Rodriguez-Hernandez G, Hauer J, Martin-Lorenzo A, et al. Infection exposure promotes ETV6-RUNX1 precursor B-cell leukemia viaim-paired H3K4 demethylases. Cancer Res. 2017. Aug 15;77 (16):4365–4377. [DOI] [PubMed] [Google Scholar]
- 22.Heltemes-Harris LM, Willette MJ, Ramsey LB, et al. Ebf1 or PAX5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med. 2011;208(6):1135–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dang J, Wei L, de Ridder J, et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. 2015;125(23):3609–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chan LN, Chen Z, Braas D, et al. Metabolic gatekeeper function of B-lymphoid transcription factors. Nature. 2017;542(7642):479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guidez F, Petrie K, Ford AM, et al. Recruitment of the nuclear receptor corepressor N-CoR by the TEL moiety of the childhood leukemia–associated TEL-AML1 oncoprotein. Blood. 2000;96 (7):2557–2561. [PubMed] [Google Scholar]
- 26.Chakrabarti SR, Nucifora G. The leukemia-associated gene TEL encodes a transcription repressor which associates with SMRT and mSin3A. Biochem Biophys Res Commun. 1999;264 (3):871–877. [DOI] [PubMed] [Google Scholar]
- 27.Wang LC, Kuo F, Fujiwara Y, et al. Yolk sac angiogenic defect and intra-embryonic apoptosis in mice lacking the Ets-related factor TEL. Embo J. 1997;16(14):4374–4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang LC, Swat W, Fujiwara Y, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12(15):2392–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hock H, Meade E, Medeiros S, et al. Tel/ETV6 is an essential and selective regulator of adult hematopoietic stem cell survival. Genes Dev. 2004;18(19):2336–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. •.Moriyama T, Metzger ML, Wu G, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015;16(16):1659–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]; Large study of 4,405 childhood ALL cases that identified damaging germline ETV6 variants in 1% of cases.
- 31.Melazzini F, Palombo F, Balduini A, et al. Clinical and pathogenic features of ETV6-related thrombocytopenia with predisposition to acute lymphoblastic leukemia. Haematologica. 2016. Nov;101 (11):1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topka S, Vijai J, Walsh MF, et al. Germline ETV6 mutations confer susceptibility to acute lymphoblastic leukemia and thrombocytopenia. PLoS Genet. 2015;11(6):e1005262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015. May;47 (5):535–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang MY, Churpek JE, Keel SB, et al. Germline ETV6 mutations in familial thrombocytopenia and hematologic malignancy. Nat Genet. 2015. Feb;47(2):180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Di Paola J, Porter CC. ETV6-related thrombocytopenia and leukemia predisposition. Blood. 2019. Aug 22;134(8):663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rampersaud E, Ziegler DS, Iacobucci I, et al. Germline deletion of ETV6 in familial acute lymphoblastic leukemia. Blood Adv. 2019. Apr 9;3(7):1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Junk SV, Klein N, Schreek S, et al. TP53, ETV6 and RUNX1 germline variants in a case series of patients developing secondary neoplasms after treatment for childhood acute lymphoblastic leukemia. Haematologica. 2019;104(9):e402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang M, Gu D, Du M, et al. Common genetic variation in ETV6 is associated with colorectal cancer susceptibility. Nat Commun. 2016. May;5(7):11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Poggi M, Canault M, Favier M, et al. Germline variants in ETV6 underlie reduced platelet formation, platelet dysfunction and increased levels of circulating CD34+ progenitors. Haematologica. 2017;102(2):282–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Merkenschlager M Ikaros in immune receptor signaling, lymphocyte differentiation, and function. FEBS Lett. 2010. Dec 15;584 (24):4910–4914. [DOI] [PubMed] [Google Scholar]
- 41.Georgopoulos K The making of a lymphocyte: the choice among disparate cell fates and the IKAROS enigma. Genes Dev. 2017. Mar 1;31(5):439–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olsson L, Johansson B. Ikaros and leukaemia. Br J Haematol. 2015;169(4):479–491. [DOI] [PubMed] [Google Scholar]
- 43.Nutt SL, Kee BL. The transcriptional regulation of B cell lineage commitment. Immunity. 2007;26(6):715–725. [DOI] [PubMed] [Google Scholar]
- 44.Mullighan CG, Phillips LA, Su X, et al. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008. Nov 28;322(5906):1377–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009. Jan 29;360 (5):470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. ••.Churchman ML, Qian M, Te Kronnie G, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. 2018. May 14;33(5):937–948.e938. [DOI] [PMC free article] [PubMed] [Google Scholar]; Report describing genetic and functional analyses of germline IKZF1 variants in children with ALL.
- 47.Boutboul D, Kuehn HS, Van de Wyngaert Z, et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. 2018. Jul 2;128(7):3071–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuehn HS, Boisson B, Cunningham-Rundles C, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. 2016. Mar 17;374(11):1032–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romano AA, Allanson JE, Dahlgren J, et al. Noonan syndrome: clinical features, diagnosis, and management guidelines. Pediatrics. 2010. Oct;126(4):746–759. [DOI] [PubMed] [Google Scholar]
- 50.Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001. Dec;29(4):465–468. [DOI] [PubMed] [Google Scholar]
- 51.Cave H, Caye A, Strullu M, et al. Acute lymphoblastic leukemia in the context of RASopathies. Eur J Med Genet. 2016. Mar;59 (3):173–178. [DOI] [PubMed] [Google Scholar]
- 52.Singer ST, Hurst D, Addiego JE Jr. Bleeding disorders in Noonan syndrome: three case reports and review of the literature. J Pediatr Hematol Oncol. 1997. Mar-Apr;19(2):130–134. [DOI] [PubMed] [Google Scholar]
- 53.Jongmans MC, van der Burgt I, Hoogerbrugge PM, et al. Cancer risk in patients with Noonan syndrome carrying a PTPN11 mutation. Eur J Hum Genet. 2011. Aug;19(8):870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kratz CP, Franke L, Peters H, et al. Cancer spectrum and frequency among children with Noonan, Costello, and cardio-facio-cutaneous syndromes. Br J Cancer. 2015. Apr 14;112(8):1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Neel BG, Gu H, Pao L. The ‘Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28(6):284–293. [DOI] [PubMed] [Google Scholar]
- 56.Tartaglia M, Niemeyer CM, Fragale A, et al. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet. 2003. Jun;34 (2):148–150. [DOI] [PubMed] [Google Scholar]
- 57.Emanuel PD, Bates LJ, Castleberry RP, et al. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77(5):925–929. [PubMed] [Google Scholar]
- 58.Pandey R, Saxena M, Kapur R. Role of SHP2 in hematopoiesis and leukemogenesis. Curr Opin Hematol. 2017. Jul;24(4):307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan G, Cheung LS, Yang W, et al. Essential role for PTPN11 in survival of hematopoietic stem and progenitor cells. Blood. 2011. Apr 21;117(16):4253–4261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu HH, Ji K, Alderson N, et al. Kit-Shp2-Kit signaling acts to maintain a functional hematopoietic stem and progenitor cell pool. Blood. 2011. May 19;117(20):5350–5361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong L, Yu W-M, Zheng H, et al. Leukaemogenic effects of PTPN11 activating mutations in the stem cell microenvironment. Nature. 2016;539(7628):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu D, Wang S, Yu W-M, et al. A germline gain-of-function mutation in PTPN11 (Shp-2) phosphatase induces myeloproliferative disease by aberrant activation of hematopoietic stem cells. Blood. 2010;116 (18):3611–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jerchel IS, Hoogkamer AQ, Aries IM, et al. RAS pathway mutations as a predictive biomarker for treatment adaptation in pediatric B-cell precursor acute lymphoblastic leukemia. Leukemia. 2018. Apr;32(4):931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riccardi VM. The prenatal diagnosis of NF-1 and NF-2. J Dermatol. 1992. Nov;19(11):885–891. [DOI] [PubMed] [Google Scholar]
- 65.Wimmer K, Rosenbaum T, Messiaen L. Connections between constitutional mismatch repair deficiency syndrome and neurofibromatosis type 1. Clin Genet. 2017. Apr;91(4):507–519. [DOI] [PubMed] [Google Scholar]
- 66.Shannon KM, O’Connell P, Martin GA, et al. Loss of the normal NF1 allele from the bone marrow of children with type 1 neurofibromatosis and malignant myeloid disorders. N Engl J Med. 1994. Mar 3;330(9):597–601. [DOI] [PubMed] [Google Scholar]
- 67.Le DT, Kong N, Zhu Y, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004. Jun 1;103(11):4243–4250. [DOI] [PubMed] [Google Scholar]
- 68.McClatchey AI. Neurofibromatosis. Annu Rev Pathol. 2007;2:191–216. [DOI] [PubMed] [Google Scholar]
- 69.Friedman J. Neurofibromatosis 1, GeneReviews. Seattle: Copyright University of Washington; 2013. 1997. [Google Scholar]
- 70.Balgobind BV, Van Vlierberghe P, van den Ouweland AM, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. 2008. Apr 15;111(8):4322–4328. [DOI] [PubMed] [Google Scholar]
- 71.Philpott C, Tovell H, Frayling IM, et al. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. 2017. Jun 21;11(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bader JL, Miller RW. Neurofibromatosis and childhood leukemia. J Pediatr. 1978. Jun;92(6):925–929. [DOI] [PubMed] [Google Scholar]
- 73.Stiller CA, Chessells JM, Fitchett M. Neurofibromatosis and childhood leukaemia/lymphoma: a population-based UKCCSG study. Br J Cancer. 1994. Nov;70(5):969–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mullighan CG. The genomic landscape of acute lymphoblastic leukemia in children and young adults. Hematology Am Soc Hematol Educ Program. 2014. Dec 5;2014(1):174–180. [DOI] [PubMed] [Google Scholar]
- 75.Liu Y, Easton J, Shao Y, et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat Genet. 2017. Aug;49(8):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013. Mar;45 (3):242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang YY, Vik TA, Ryder JW, et al. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J Exp Med. 1998. Jun 1;187(11):1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sood R, Kamikubo Y, Liu P. Role of RUNX1 in hematological malignancies. Blood. 2017. Apr 13;129(15):2070–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schlegelberger B, Heller PG. RUNX1 deficiency (familial platelet disorder with predisposition to myeloid leukemia, FPDMM). Semin Hematol. 2017. Apr;54(2):75–80. [DOI] [PubMed] [Google Scholar]
- 80.De Braekeleer E, Douet-Guilbert N, Morel F, et al. RUNX1 translocations and fusion genes in malignant hemopathies. Future Oncol. 2011. Jan;7(1):77–91. [DOI] [PubMed] [Google Scholar]
- 81.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012. Jan 11;481 (7380):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999. Oct;23 (2):166–175. [DOI] [PubMed] [Google Scholar]
- 83.Kanagal-Shamanna R, Loghavi S, DiNardo CD, et al. Bone marrow pathologic abnormalities in familial platelet disorder with propensity for myeloid malignancy and germline RUNX1 mutation. Haematologica. 2017. Oct;102(10):1661–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Preudhomme C, Renneville A, Bourdon V, et al. High frequency of RUNX1 biallelic alteration in acute myeloid leukemia secondary to familial platelet disorder. Blood. 2009. May 28;113(22):5583–5587. [DOI] [PubMed] [Google Scholar]
- 85.Latger-Cannard V, Philippe C, Bouquet A, et al. Haematological spectrum and genotype-phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet J Rare Dis. 2016. Apr 26;11:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Michaud J, Wu F, Osato M, et al. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood. 2002. Feb 15;99 (4):1364–1372. [DOI] [PubMed] [Google Scholar]
- 87.Nishimoto N, Imai Y, Ueda K, et al. T cell acute lymphoblastic leukemia arising from familial platelet disorder. Int J Hematol. 2010. Jul;92(1):194–197. [DOI] [PubMed] [Google Scholar]
- 88.Bluteau D, Gilles L, Hilpert M, et al. Down-regulation of the RUNX1-target gene NR4A3 contributes to hematopoiesis deregulation in familial platelet disorder/acute myelogenous leukemia. Blood. 2011. Dec 8;118(24):6310–6320. [DOI] [PubMed] [Google Scholar]
- 89.Antony-Debre I, Duployez N, Bucci M, et al. Somatic mutations associated with leukemic progression of familial platelet disorder with predisposition to acute myeloid leukemia. Leukemia. 2016;30:999–1002. [DOI] [PubMed] [Google Scholar]
- 90.Prebet T, Carbuccia N, Raslova H, et al. Concomitant germ-line RUNX1 and acquired ASXL1 mutations in a T-cell acute lymphoblastic leukemia. Eur J Haematol. 2013. Sep;91(3):277–279. [DOI] [PubMed] [Google Scholar]
- 91.Wang Q, Stacy T, Binder M, et al. Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci U S A. 1996;93(8):3444–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Okuda T, Van Deursen J, Hiebert SW, et al. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell. 1996;84(2):321–330. [DOI] [PubMed] [Google Scholar]
- 93.Sasaki K, Yagi H, Bronson RT, et al. Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci U S A. 1996;93(22):12359–12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Growney JD, Shigematsu H, Li Z, et al. Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood. 2005;106(2):494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bellissimo DC, Speck NA. RUNX1 mutations in inherited and sporadic leukemia. Front Cell Dev Biol. 2017;5:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. [DOI] [PubMed] [Google Scholar]
- 97.Pinto EM, Ribeiro RC, Figueiredo BC, et al. TP53-associated pediatric malignancies. Genes Cancer. 2011. Apr;2(4):485–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Weisz L, Oren M, Rotter V. Transcription regulation by mutant p53. Oncogene. 2007. Apr 2;26(15):2202–2211. [DOI] [PubMed] [Google Scholar]
- 99.Lane D p53 and human cancers. Br Med Bull. 1994;50(3):582–599. [DOI] [PubMed] [Google Scholar]
- 100.Correa H Li-Fraumeni syndrome. J Pediatr Genet. 2016. Jun;5 (2):84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mai PL, Best AF, Peters JA, et al. Risks of first and subsequent cancers among TP53 mutation carriers in the national cancer institute Li-Fraumeni syndrome cohort. Cancer. 2016. Dec 1;122 (23):3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gonzalez KD, Noltner KA, Buzin CH, et al. Beyond Li Fraumeni syndrome: clinical characteristics of families with p53 germline mutations. J Clin Oncol. 2009;27(8):1250–1256. [DOI] [PubMed] [Google Scholar]
- 103.Ruijs MW, Verhoef S, Rookus MA, et al. TP53 germline mutation testing in 180 families suspected of Li–Fraumeni syndrome: mutation detection rate and relative frequency of cancers in different familial phenotypes. J Med Genet. 2010;47(6):421–428. [DOI] [PubMed] [Google Scholar]
- 104.Bougeard G, Renaux-Petel M, Flaman JM, et al. Revisiting Li-Fraumeni syndrome from TP53 mutation carriers. J Clin Oncol. 2015. Jul 20;33(21):2345–2352. [DOI] [PubMed] [Google Scholar]
- 105.Nichols KE, Malkin D, Garber JE, et al. Germ-line p53 mutations predispose to a wide spectrum of early-onset cancers. Cancer Epidemiol Biomarkers Prev. 2001. Feb;10(2):83–87. [PubMed] [Google Scholar]
- 106. ••.Qian M, Cao X, Devidas M, et al. TP53 germline variations influence the predisposition and prognosis of B-cell acute lymphoblastic leukemia in children. J Clin Oncol. 2018. Feb 20;36(6):591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]; Large study showing that germline TP53 variants are associated with inferior event-free and overall survival for children with ALL due to a much higher risk of second cancers.
- 107.Zhang J, Walsh MF, Wu G, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med. 2015;373 (24):2336–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Churpek JE, Marquez R, Neistadt B, et al. Inherited mutations in cancer susceptibility genes are common among survivors of breast cancer who develop therapy-related leukemia. Cancer. 2016. Jan 15;122(2):304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wimmer K, Etzler J. Constitutional mismatch repair-deficiency syndrome: have we so far seen only the tip of an iceberg? Hum Genet. 2008;124(2):105–122. [DOI] [PubMed] [Google Scholar]
- 110.Quinn E, Nichols KE. Cancer predisposition syndromes associated with myeloid malignancy. Semin Hematol. 2017. Apr;54(2):115–122. [DOI] [PubMed] [Google Scholar]
- 111.Miyashita K, Fujii K, Taguchi K, et al. A specific mode of microsatellite instability is a crucial biomarker in adult T-cell leukaemia/lymphoma patients. J Cancer Res Clin Oncol. 2017. Mar;143 (3):399–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kohlmann W, Gruber S. Lynch syndrome, GeneReviews. Seattle: Copyright University of Washington; 1993. [Google Scholar]
- 113.Tabori U, Hansford JR, Achatz MI, et al. Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res. 2017. Jun 1;23(11):e32–e37. [DOI] [PubMed] [Google Scholar]
- 114.Lavoine N, Colas C, Muleris M, et al. Constitutional mismatch repair deficiency syndrome: clinical description in a French cohort. J Med Genet. 2015. Nov;52(11):770–778. [DOI] [PubMed] [Google Scholar]
- 115.Vasen HF, Ghorbanoghli Z, Bourdeaut F, et al. Guidelines for surveillance of individuals with constitutional mismatch repair-deficiency proposed by the European consortium “Care for CMMRD” (C4CMMR-D). J Med Genet. 2014. May;51(5):283–293. [DOI] [PubMed] [Google Scholar]
- 116.Ripperger T, Schlegelberger B. Acute lymphoblastic leukemia and lymphoma in the context of constitutional mismatch repair deficiency syndrome. Eur J Med Genet. 2016. Mar;59(3):133–142. [DOI] [PubMed] [Google Scholar]
- 117.Wimmer K, Kratz CP, Vasen HF, et al. Diagnostic criteria for constitutional mismatch repair deficiency syndrome: suggestions of the European consortium ‘care for CMMRD’ (C4CMMRD). J Med Genet. 2014. Jun;51(6):355–365. [DOI] [PubMed] [Google Scholar]
- 118.Lage H, Dietel M. Involvement of the DNA mismatch repair system in antineoplastic drug resistance. J Cancer Res Clin Oncol. 1999;125 (3–4):156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shinsato Y, Furukawa T, Yunoue S, et al. Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget. 2013. Dec;4 (12):2261–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Stritzelberger J, Distel L, Buslei R, et al. Acquired temozolomide resistance in human glioblastoma cell line U251 is caused by mismatch repair deficiency and can be overcome by lomustine. Clin Transl Oncol. 2018. Apr;20(4):508–516. [DOI] [PubMed] [Google Scholar]
- 121.Fink D, Aebi S, Howell SB. The role of DNA mismatch repair in drug resistance. Clin Cancer Res. 1998. Jan;4(1):1–6. [PubMed] [Google Scholar]
- 122.Bardelli A, Cahill DP, Lederer G, et al. Carcinogen-specific induction of genetic instability. Proc Natl Acad Sci U S A. 2001. May 8;98 (10):5770–5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Campbell BB, Light N, Fabrizio D, et al. Comprehensive analysis of hypermutation in human cancer. Cell. 2017. Nov 16;171 (5):1042–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Westdorp H, Kolders S, Hoogerbrugge N, et al. Immunotherapy holds the key to cancer treatment and prevention in constitutional mismatch repair deficiency (CMMRD) syndrome. Cancer Lett. 2017. Sep;10(403):159–164. [DOI] [PubMed] [Google Scholar]
- 125.Le DT, Uram JN, Wang H, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015. Jun 25;372 (26):2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bouffet E, Larouche V, Campbell BB, et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol. 2016. Jul 1;34(19):2206–2211. [DOI] [PubMed] [Google Scholar]
- 127.AlHarbi M, Ali Mobark N, AlMubarak L, et al. Durable response to nivolumab in a pediatric patient with refractory glioblastoma and constitutional biallelic mismatch repair deficiency. Oncologist. 2018. Dec;23(12):1401–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pavelka Z, Zitterbart K, Noskova H, et al. Effective immunotherapy of glioblastoma in an adolescent with constitutional mismatch repair-deficiency syndrome. Klin Onkol. 2019;32(1):70–74. [DOI] [PubMed] [Google Scholar]
- 129.Fong C-T, Brodeur GM. Down’s syndrome and leukemia: epidemiology, genetics, cytogenetics and mechanisms of leukemogenesis. Cancer Genet Cytogenet. 1987;28(1):55–76. [DOI] [PubMed] [Google Scholar]
- 130.Brewster H, Cannon H Acute lymphatic leukemia: report of case in eleventh month mongolian idiot. New Orleans Med Surg J. 1930;82:872–873. [Google Scholar]
- 131.Stoll C, Dott B, Alembik Y, et al. Associated congenital anomalies among cases with Down syndrome. Eur J Med Genet. 2015;58 (12):674–680. [DOI] [PubMed] [Google Scholar]
- 132.Mateos MK, Barbaric D, Byatt S-A, et al. Down syndrome and leukemia: insights into leukemogenesis and translational targets. Transl Pediatr. 2015;4(2):76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Li Y, Schwab C, Ryan SL, et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature. 2014;508(7494):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Buitenkamp TD, Izraeli S, Zimmermann M, et al. Acute lymphoblastic leukemia in children with Down syndrome: a retrospective analysis from the Ponte di Legno study group. Blood. 2014;123 (1):70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Roberts I, Izraeli S. Haematopoietic development and leukaemia in Down syndrome. Br J Haematol. 2014;167(5):587–599. [DOI] [PubMed] [Google Scholar]
- 136.Athale UH, Puligandla M, Stevenson KE, et al. Outcome of children and adolescents with Down syndrome treated on Dana-Farber cancer institute acute lymphoblastic leukemia consortium protocols 00–001 and 05–001. Pediatr Blood Cancer. 2018;65(10):e27256. [DOI] [PubMed] [Google Scholar]
- 137.Matloub Y, Rabin KR, Ji L, et al. Excellent long-term survival of children with Down syndrome and standard-risk ALL: a report from the Children’s Oncology Group. Blood Adv. 2019;3 (11):1647–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Forestier E, Izraeli S, Beverloo B, et al. Cytogenetic features of acute lymphoblastic and myeloid leukemias in pediatric patients with Down syndrome: an iBFM-SG study. Blood. 2008;111 (3):1575–1583. [DOI] [PubMed] [Google Scholar]
- 139.Mullighan CG, Collins-Underwood JR, Phillips LA, et al. Rearrangement of CRLF2 in B-progenitor–and Down syndrome–associated acute lymphoblastic leukemia. Nat Genet. 2009;41 (11):1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hertzberg L, Vendramini E, Ganmore I, et al. Down syndrome acute lymphoblastic leukemia, a highly heterogeneous disease in which aberrant expression of CRLF2 is associated with mutated JAK2: a report from the international BFM study group. Blood. 2010;115 (5):1006–1017. [DOI] [PubMed] [Google Scholar]
- 141.Bercovich D, Ganmore I, Scott LM, et al. Mutations of JAK2 in acute lymphoblastic leukaemias associated with Down’s syndrome. Lancet. 2008;372(9648):1484–1492. [DOI] [PubMed] [Google Scholar]
- 142.Kubota Y, Uryu K, Ito T, et al. Integrated genetic and epigenetic analysis revealed heterogeneity of acute lymphoblastic leukemia in Down syndrome. Cancer Sci. 2019;110(10):3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Papaemmanuil E, Hosking FJ, Vijayakrishnan J, et al. Loci on 7p12.2, 10q21.2 and 14q11.2 are associated with risk of childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1006–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Treviño LR, Yang W, French D, et al. Germline genomic variants associated with childhood acute lymphoblastic leukemia. Nat Genet. 2009;41(9):1001–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Migliorini G, Fiege B, Hosking FJ, et al. Variation at 10p12.2 and 10p14 influences risk of childhood B-cell acute lymphoblastic leukemia and phenotype. Blood. 2013;122(19):3298–3307. [DOI] [PubMed] [Google Scholar]
- 146.Perez-Andreu V, Roberts KG, Harvey RC, et al. Inherited GATA3 variants are associated with Ph-like childhood acute lymphoblastic leukemia and risk of relapse. Nat Genet. 2013;45(12):1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ramsey LB, Bruun GH, Yang W, et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012;22(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Qian M, Xu H, Perez-Andreu V, et al. Novel susceptibility variants at the ERG locus for childhood acute lymphoblastic leukemia in Hispanics. Blood. 2019;133(7):724–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Xu H, Yang W, Perez-Andreu V, et al. Novel susceptibility variants at 10p12.31–12.2 for childhood acute lymphoblastic leukemia in ethnically diverse populations. J Natl Cancer Inst. 2013;105 (10):733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Brown AL, de Smith AJ, Gant VU, et al. Inherited genetic susceptibility of acute lymphoblastic leukemia in Down syndrome. Blood. 2019. doi: 10.1182/blood.2018890764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vijayakrishnan J, Studd J, Broderick P, et al. Genome-wide association study identifies susceptibility loci for B-cell childhood acute lymphoblastic leukemia. Nat Commun. 2018;9(1):1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ellinghaus E, Stanulla M, Richter G, et al. Identification of germline susceptibility loci in ETV6-RUNX1-rearranged childhood acute lymphoblastic leukemia. Leukemia. 2012;26(5):902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu H, Zhang H, Yang W, et al. Inherited coding variants at the CDKN2A locus influence susceptibility to acute lymphoblastic leukaemia in children. Nature Comm. 2015;6:7553. doi: 10.1038/ncomms8553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sherborne AL, Hosking FJ, Prasad RB, et al. Variation in CDKN2A at 9p21.3 influences childhood acute lymphoblastic leukemia risk. Nat Genet. 2010;42(6):492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wiemels JL, Walsh KM, de Smith AJ, et al. GWAS in childhood acute lymphoblastic leukemia reveals novel genetic associations at chromosomes 17q12 and 8q24.21. Nat Commun. 2018;9(1):286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hungate EA, Vora SR, Gamazon ER, et al. A variant at 9p21.3 functionally implicates CDKN2B in paediatric B-cell precursor acute lymphoblastic leukaemia aetiology. Nat Commun. 2016;7:10635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vijayakrishnan J, Kumar R, Henrion MY, et al. A genome-wide association study identifies risk loci for childhood acute lymphoblastic leukemia at 10q26.13 and 12q23.1. Leukemia. 2017;31 (3):573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. ••.Qian M, Zhao X, Devidas M, et al. Genome-wide association study of susceptibility loci for T-cell acute lymphoblastic leukemia in children. J Natl Cancer Inst. 2019. Apr 2. DOI: 10.1093/jnci/djz043. [DOI] [PMC free article] [PubMed] [Google Scholar]; GWAS study that identified USP7 as the first germline risk locus associated with T-ALL.
- 159.Kinlen L Evidence for an infective cause of childhood leukaemia: comparison of a Scottish new town with nuclear reprocessing sites in Britain. Lancet. 1988. Dec 10;2(8624):1323–1327. [DOI] [PubMed] [Google Scholar]
- 160.Greaves MF. Speculations on the cause of childhood acute lymphoblastic leukemia. Leukemia. 1988. Feb;2(2):120–125. [PubMed] [Google Scholar]
- 161.Marcotte EL, Ritz B, Cockburn M, et al. Exposure to infections and risk of leukemia in young children. Cancer Epidemiol Biomarkers Prev. 2014;23(7):1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Martin-Lorenzo A, Hauer J, Vicente-Duenas C, et al. Infection exposure is a causal factor in B-cell precursor acute lymphoblastic leukemia as a result of PAX5-inherited susceptibility. Cancer Discov. 2015. Dec;5(12):1328–1343. [DOI] [PubMed] [Google Scholar]
- 163.Ford AM, Palmi C, Bueno C, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-β pathway in early B lineage progenitor cells. J Clin Invest. 2009;119(4):826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mori H, Colman SM, Xiao Z, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci U S A. 2002;99(12):8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Drazer MW, Kadri S, Sukhanova M, et al. Prognostic tumor sequencing panels frequently identify germ line variants associated with hereditary hematopoietic malignancies. Blood Adv. 2018;2 (2):146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014. Dec 25;371(26):2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014. Dec 25;371(26):2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Desai AV, Perpich M, Godley LA. Clinical assessment and diagnosis of germline predisposition to hematopoietic malignancies: the University of Chicago experience. Front Pediatr. 2017;5:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chaudhary G, Dogra T, Raina A. Evaluation of blood, buccal swabs, and hair follicles for DNA profiling technique using STR markers. Croat Med J. 2015;56(3):239–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Škerl P, Krajc M, Blatnik A, et al. Genetic testing and counseling of a recipient after bone marrow transplant from a sibling harboring a germline BRCA1 pathogenic mutation. Oncol Rep. 2017;38 (1):279–282. [DOI] [PubMed] [Google Scholar]
- 171.Porter CC, Druley TE, Erez A, et al. Recommendations for surveillance for children with leukemia-predisposing conditions. Clin Cancer Res. 2017. Jun 1;23(11):e14–e22. [DOI] [PubMed] [Google Scholar]
- 172.McReynolds LJ, Savage SA. Pediatric leukemia susceptibility disorders: manifestations and management. Hematology Am Soc Hematol Educ Program. 2017. Dec 8;2017(1):242–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Toledano SR, Lange BJ. Ataxia-telangiectasia and acute lymphoblastic leukemia. Cancer. 1980;45(7):1675–1678. [DOI] [PubMed] [Google Scholar]
- 174.Sandoval C, Swift M. Treatment of lymphoid malignancies in patients with ataxia-telangiectasia. Med Pediatr Oncol. 1998;31 (6):491–497. [DOI] [PubMed] [Google Scholar]
- 175.Schulz E, Valentin A, Ulz P, et al. Germline mutations in the DNA damage response genes BRCA1, BRCA2, BARD1 and TP53 in patients with therapy related myeloid neoplasms. J Med Genet. 2012. Jul;49 (7):422–428. [DOI] [PubMed] [Google Scholar]
- 176.Eyre J, Gardner-Medwin D, Summerfield G. Leukoencephalopathy after prophylactic radiation for leukaemia in ataxia telangiectasia. Arch Dis Child. 1988;63(9):1079–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Yanofsky RA, Seshia SS, Dawson AJ, et al. Ataxia-telangiectasia: atypical presentation and toxicity of cancer treatment. Can J Neurosci Nurs. 2009;36(4):462–467. [DOI] [PubMed] [Google Scholar]
- 178.Hamilton KV, Maese L, Marron JM, et al. Stopping leukemia in its tracks: should preemptive hematopoietic stem-cell transplantation be offered to patients at increased genetic risk for acute myeloid leukemia? J Clin Oncol. 2019. Jun 6:Jco1900181. [DOI] [PubMed] [Google Scholar]
- 179.Wang Z, Wilson CL, Easton J, et al. Genetic risk for subsequent neoplasms among long-term survivors of childhood cancer. J Clin Oncol. 2018;36(20):2078–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Parsons DW, Roy A, Yang Y, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016;2(5):616–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mody RJ, Wu Y-M, Lonigro RJ, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314(9):913–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Parekh C, Crooks GM. Critical differences in hematopoiesis and lymphoid development between humans and mice. J Clin Immunol. 2013;33(4):711–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Jung M, Cordes S, Zou J, et al. GATA2 deficiency and human hematopoietic development modeled using induced pluripotent stem cells. Blood Adv. 2018;2(23):3553–3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. [DOI] [PubMed] [Google Scholar]