

Abstract
The CACNA1C gene encodes the pore-forming subunit of the CaV1.2 L-type Ca2+ channel, a critical component of membrane physiology in multiple tissues, including the heart, brain and immune system. As such, mutations altering the function of these channels have the potential to impact a wide array of cellular functions. The first mutations identified within CACNA1C were shown to cause a severe, multisystem disorder known as Timothy syndrome (TS), which is characterized by neurodevelopmental deficits, long QT syndrome, life-threatening cardiac arrhythmias, craniofacial abnormalities and immune deficits. Since this initial description, the number and variety of disease-associated mutations identified in CACNA1C has grown tremendously, expanding the range of phenotypes observed in affected patients. CACNA1C channelopathies are now known to encompass multisystem phenotypes as described in TS, as well as more selective phenotypes where patients may exhibit predominantly cardiac or neurological symptoms. Here, we review the impact of genetic mutations on CaV1.2 function and the resultant physiological consequences.
Introduction
L-type calcium channels (LTCCs) are expressed throughout the body, where they play critical roles in the physiological function of numerous systems, including the heart, brain, smooth muscle, and the immune system(1, 2). Activation of these channels controls the entry of Ca2+ into the cell, which both shapes the morphology of the action potential and initiates numerous downstream signaling processes. Of the four L-type channel subtypes, CaV1.2 exhibits the widest expression across multiple tissues including the heart, brain, smooth muscle, endocrine and immune systems (3). Thus, disruption of these channels has the potential to impact a myriad of cellular functions across multiple organ systems. Advancements in genomic technologies have resulted in strong associations between CaV1.2 variants and clinical phenotypes, and have identified an increasing number and variety of genetic mutations affecting the functionality of these channels.
CaV1.2 in Cellular Physiology
LTCCs play a major role in excitation-contraction coupling in all muscle tissues. CaV1.2 is renowned for its role in the heart, where Ca2+ entry through the channel activates the ryanodine receptor (RyR2), thereby triggering the release of calcium from the sarcoplasmic reticulum (SR) in a process known as Ca2+-induced Ca2+ release (CICR)(4, 5). In addition, CaV1.2 plays a major role in shaping the cardiac action potential (AP)(6). Acting largely during the plateau phase, CaV1.2 channels are a major determinant of the duration of the duration of the cardiac AP (2, 7). In smooth muscle, the CaV1.2 channel triggers contraction of the muscle, making it a primary element in blood-pressure control.
In the brain, CaV1.2 comprise nearly 90% of all LTCCs(8). They are highly expressed within the hippocampus, cerebral cortex, and cerebellum, and are often detected in postsynaptic dendrites. CaV1.2 channels play a role in excitation-transcription coupling (9) as well as neuronal plasticity, and have been shown to play a critical role in long term potentiation and memory(10, 11) In addition, mouse models have described a role for CaV1.2 in anxiety(12).
LTCCs, including CaV1.2, have also been found to play a role in the immune system, although their role is less well understood. CaV1.2 has been shown to be highly expressed in T lymphocytes, and has been shown to play a role in B cells and dendritic cells(13, 14). While the precise role of CaV1.2 in these cells remains ambiguous, genetic mutations which disrupt the function of the channel result in a clear immune phenotype, demonstrating the importance of the channel in immune function(15). Moreover, CaV1.2 is expressed in chondrocytes and osteoblasts, and has been shown to be a requirement for normal mandibular development. Interestingly, this role for CaV1.2 in non-excitable cell types represents a relatively new avenue of research, which was preceded by identification of CaV1.2 mutations which caused marked phenotypes in non-excitable tissues(15, 16). As such, the function of these voltage-gated channels in non-excitable cells remains under investigation and promises new mechanisms for CaV1.2 function(16).
Concurrent with this idea of CaV1.2 function in non-excitable tissues, it has been increasingly recognized that the channel is capable of multiple cellular functions beyond directly controlling Ca2+ entry. In neurons, it has been shown that a voltage-dependent conformational change is required to initiate excitation-transcription coupling, and is required for normal synaptic function(17). Moreover, a cryptic promotor has been identified in the CACNA1C gene which results in expression of a C-terminal portion of the channel, which acts as a transcription regulator(18, 19). Thus, the role of CaV1.2 in normal and patho-physiology continues to expand.
CaV1.2 Regulation
In order to function across a broad distribution of different cell types, CaV1.2 channels can be tuned by multiple processes, including feedback regulation, alternative splicing, and subunit composition. To precisely control Ca2+ influx in various tissues and in response to a diverse array of stimuli, CaV1.2 channels utilize two major forms of feedback regulation: voltage-dependent inactivation (VDI) and Ca2+-dependent inactivation (CDI). VDI is thought to proceed through a hinged-lid mechanism(20), where the I-II linker docks with residues within the distal S6 regions, thus occluding the channel. CDI, on the other hand, represents as allosteric process, where channels open with a reduced open probability in response a rise in cytosolic Ca2+. The Ca2+ sensor for CaV1.2 CDI is calmodulin (CaM), a modulatory protein which is bound to the C-tail of the channel in its Ca2+ free state (apoCaM). Upon channel opening, Ca2+ binds to the resident CaM, resulting in a conformational rearrangement leading to reduced opening and CDI.
CaV1.2 channels can further be modified by alternative splicing. Sequencing has revealed 90 distinct CACNA1C transcript variants within the brain, with variable splicing profiles across different regions and tissues (21). Biophysical evaluation of many of the major splice variants has revealed substantial effects on channel gating and regulation, illustrating unique channel properties tailored to tissue expression or developmental state (22–25).
Finally, subunit composition can impact channel function. Like other voltage-gated calcium channels, CaV1.2 is generally expressed as a multi-subunit protein(26). The CACNA1C gene encodes the pore-forming alpha subunit, which typically associates with an auxiliary β subunit and an α2δ subunit. These subunits are known to modulate multiple channel properties, including trafficking, gating and second messenger regulation(27). These channels are also associated with a γ subunit in select tissues, which is capable of modulating channel function(28, 29). Disruption of any of these auxiliary proteins can result in significant disease(30–34). Here, we focus on the role of the pore-forming alpha subunit in the pathogenesis of CaV1.2 channelopathies.
Association with Neuropsychiatric Disorders
Multiple genome wide association studies (GWAS) studies have found that CACNA1C is highly associated with several psychiatric disorders(35). CACNA1C is commonly identified in GWAS studies of schizophrenia (SCZ) and bipolar disorder (BP) (36–38) (39). Further, polymorphisms in CACNA1C have been associated with additional psychiatric diseases, such as major depression(40, 41) and autism spectrum disorder (ASD)(42, 43). Cross-association of CACNA1C with multiple psychiatric diseases has identified commonalities across BP, major depression, attention deficit hyperactivity disorder, SCZ, and autism spectrum disorder (ASD)(1, 44), with CACNA1C identified as having a high association with BP, SCZ and major depression. Moreover, pathway analysis identified numerous Ca2+ channel activity genes within association for five major psychiatric diseases(44).
The most common neuropsychiatric-associated single nucleotide polymorphism (SNP) within CACNA1C has been identified as rs1006737 within intron 3 of the channel. This risk allele has been associated with BP (45–47), SCZ(48) and major depression(39), making it one to the most consistent associations in neuropsychiatric GWAS studies(49). This risk allele has been evaluated for impact on brain structure, where healthy individuals harboring the allele demonstrated an increase in multiple traits including depression, anxiety, startle reactivity and decreased verbal fluency (50–52). Moreover, rs1006737 has been associated with structural changes in the amygdala and prefrontal cortical regions(53–55), as well as changes in brain connectivity and grey matter structure and function(56). Interestingly, evidence exists for both gain(57, 58) or loss(59, 60) of function effects on CACNA1C in psychiatric disorders. Recent analysis of postmortem brain from schizophrenia patients identified a decrease in gene expression of CaV1.2(61). Thus, the pathogenic mechanism for CaV1.2 in neuropsychiatric disorders remains to be elucidated. Interestingly, the rs1006737 risk allele is also associated with changes in other systems where CaV1.2 is known to play a critical role, including blood pressure(62), demonstrating a potential wider impact of the allele outside the brain.
Timothy Syndrome
Timothy syndrome (TS) was initially identified in the early 1990s as a severe form of long-QT syndrome, comorbid with syndactyly(63, 64, 65). This rare clinical disorder would eventually be the first documented CaV1.2 channelopathy, and has been designated as long QT type 8 (LQT8). TS is a debilitating disease characterized by pronounced cardiac deficits, syndactyly, immune deficiencies, craniofacial abnormalities, and neurological symptoms(15, 66). Of particular clinical concern are the cardiac symptoms, which include early afterdepolarizations, AV block, torsades de pointes, ventricular tachycardia, ventricular fibrillation, and long-QT syndrome (LQTS)(66, 67). The prolongation of the QT interval (615-690 ms) makes LQT8 amongst the most severe type of LQTS yet described. This severe prolongation is largely responsible for the highly lethal nature of TS, which has an average age of mortality of approximately two and half years.
The genetic mechanisms underlying TS are informed by splice variations of the CACNA1C gene. Mature CaV1.2 channels can take the form of a multitude of CACNA1C spice isoforms, and of particular relevance to TS are the mutually exclusive exons 8 and 8A. Some ambiguity has arisen in the literature regarding the identity of these exons, as different groups have used different terminology(15, 23, 68). For the sake of clarity, the convention of using “exon 8” to refer to the upstream exon, and “exon 8a” to refer to the downstream one, will be used. The first causative mutation for TS was identified as G406R within exon 8a of CaV1.2, which places the mutation near the intracellular end of the domain I S6 transmembrane domain(15) (Fig. 1). In cardiac tissue, the ratio of exon 8 to exon 8a is approximately 4:1, and exon 8a is also expressed throughout other tissues that contain CaV1.2, a fact that contributes to the multisystem nature of TS.
Figure 1. CACNA1C mutations.
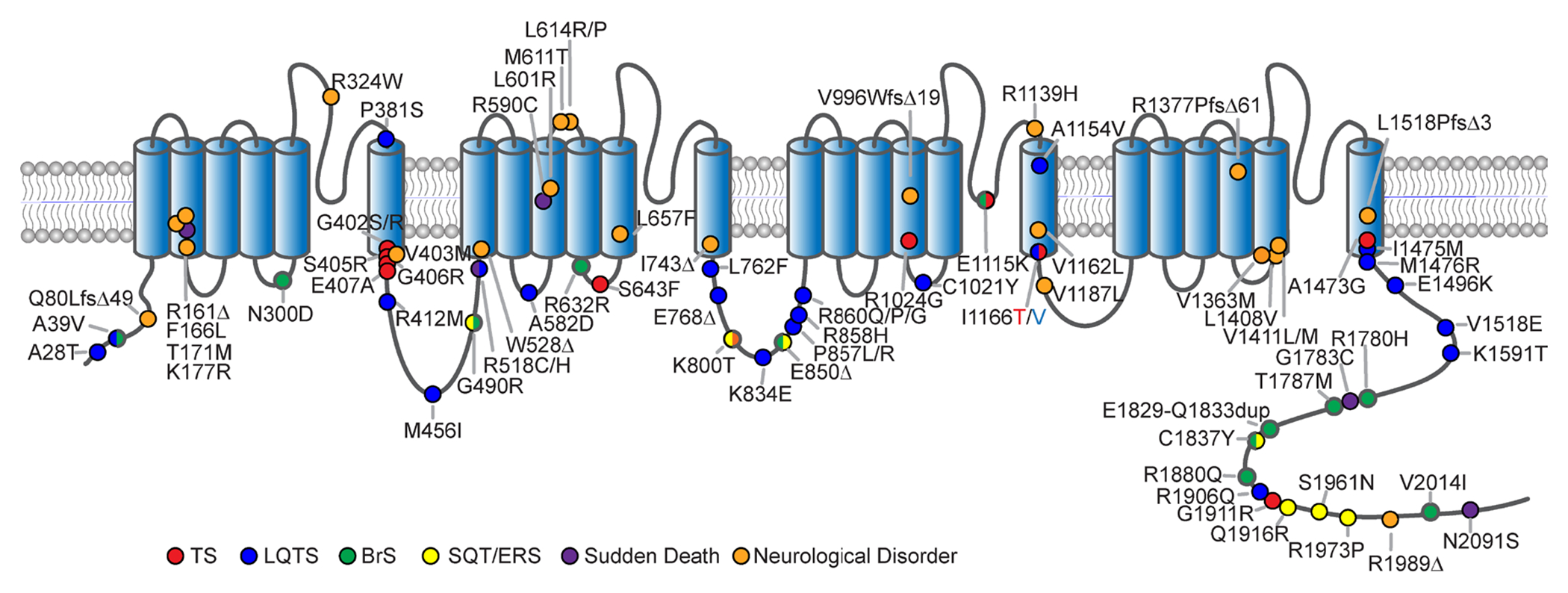
Cartoon depicting the membrane topology of the pore forming subunit of CaV1.2 indicating the locus of CACNA1C mutations across the channel. Phenotypes associated with each mutation are colored accordingly.
Biophysical studies revealed that channels containing the G406R mutation show a loss of VDI (Fig. 2a), decreased CDI(Fig. 2b) and a hyperpolarizing shift in the voltage dependance of channel activation (Fig. 2c) (15, 66, 69–71), all relatively clear components of a gain-of-function (GOF) phenotype. This channel behavior helps explain the effect of G406R on cardiac tissues, as the plateau phase of the cardiac action potential is enhanced by the excess entry of Ca2+ during depolarization. This, in turn, results in the characteristic prolongation of the QT interval observed in TS patients. Further, a mouse overexpressing rabbit CaV1.2 channels harboring the mutation indicated that even proportionally small amounts of the mutated channel were sufficient to cause large alterations to intracellular calcium concentrations, with increased SR Ca2+ leak and SR Ca2+ load, higher diastolic calcium, and increased Ca2+ spark activity in ventricular myocytes(72). The vast majority of patients harboring a G406R mutation in exon 8a (sometimes referred to as TS type 1 or TS1) suffer from the full range of characteristic symptoms of TS, but this is not universally true(73).
Figure 2. TS associated CACANA1C mutations alter the gating of CaV1.2 channels.
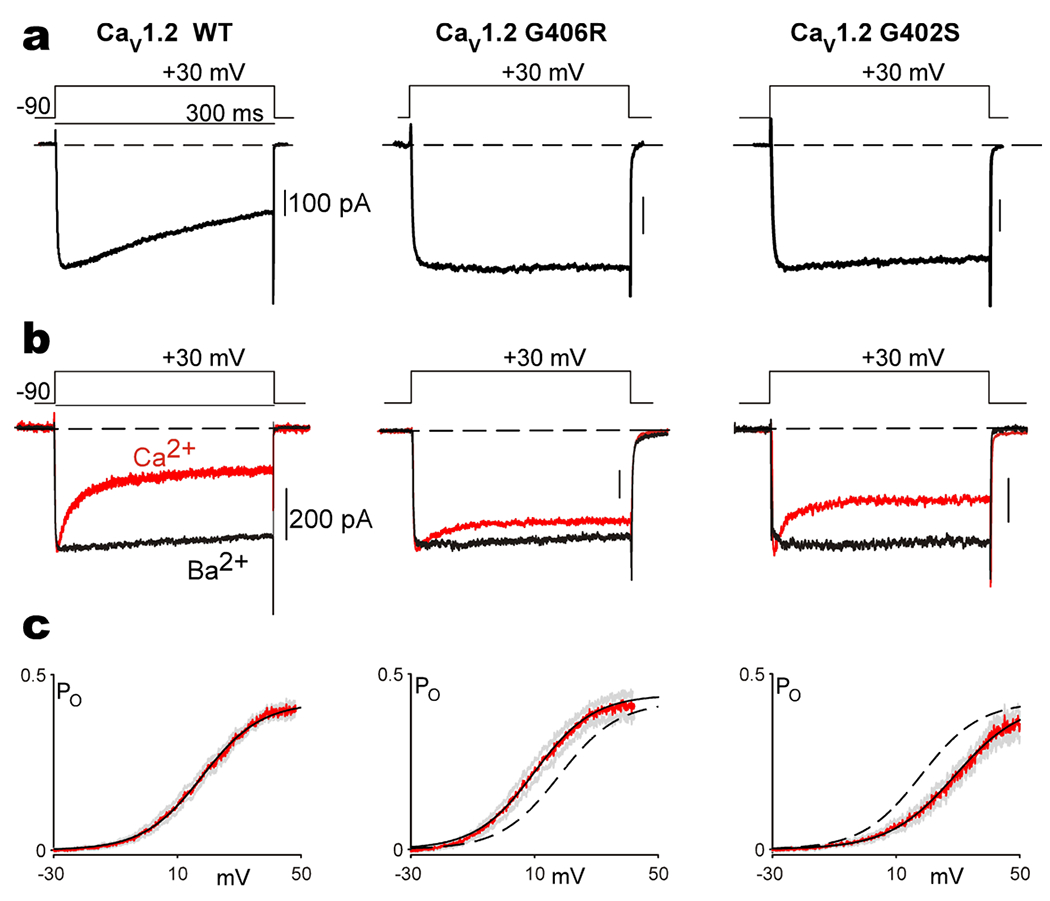
(a) Exemplar Ba2+ currents measured from CaV1.2 channels display typical VDI, seen as the decay in current over the time course of a 300 ms depolarization. Channels are expressed with the β1B auxiliary subunit, which is permissive of robust VDI. Both the G406R and G402S mutation nearly eliminate VDI in these channels. (b) Exemplar Ba2+ (black) and Ca2+ (red) currents measured from CaV1.2 channels enabling visualization of CDI, seen as the stronger decay in the Ca2+ current as compared to the Ba2+ current. CaV1.2 is expressed with the β2A -auxiliary subunit which decreases the amount of VDI, thus enabling measurement of CDI in relative isolation. Both the G406R and G402S mutations significantly blunt CDI. (c) Open-probability curves measured through single channel recordings of CaV1.2 opening as a function of voltage. Data is displayed in red, with SEM in gray, and a Boltzmann fit to the data shown as the solid black curve. The G406R mutation left shifts the activation of the channel as compared to WT, which is reproduced as the black dashed line (middle panel). The G402S mutation causes a right shift in channel activation (right panel) as compared to the WT data reproduced as the dashed black line. Reproduced with permission from (Dick et. al., 2016, Nat. Commun)(71).
Shortly following the initial characterization of the G406R mutation in exon 8a of CaV1.2, two more mutations in CACNA1C were observed in patients suffering similar symptoms(66). In 2005, a patient was observed to harbor a G406R mutation not in exon 8a, but in the more highly expressed alternative exon 8(66). Concurrently, a second mutation, G402S, was also identified within exon 8. Both mutations produced similar cardiac deficits, with QT intervals reaching as high as 730 ms, however they lacked the syndactyly characteristic of the original TS patients. This form of LQT8 therefore was designated as TS type 2 (TS2). TS2 patients harboring the G406R mutations generally present with a more severe cardiac pathology, with an earlier age of lethality as compared to TS1. This is likely due to the greater abundance of exon 8 in cardiac as compared to exon 8a.
In the case of the patient harboring the G402S mutation in exon 8, a similar but somewhat less severe LQT phenotype was observed, and similar neurological symptoms were described(66). However, the patient suffered from a cardiac arrest, which could somewhat complicate the understanding of the underlying effects of the mutation on neuronal function and development. Subsequent patients harboring the G402S mutation in exon 8 have been shown to lack the neurological phenotypes typically associated with TS. In one case, a child harboring the G402S mutation exhibited neurological impairments, however the patient’s affected sibling was neurologically healthy despite harboring the same mutation(74). This scenario points out two important features of TS. First, while most TS mutations have been described as de-novo, mosaicism can complicate the phenotype. In this case, a mosaic parent exhibited minimal symptoms, but passed the mutation onto two children. Second, the severe cardiac defects of TS can complicate identification of a causative link to neurological phenotypes, as the first sibling in this study likely displayed neurological deficits as a result of hypoxic injury rather than direct impact of the mutation. Further complicating the understanding of the pathology of the G402S mutation in exon 8 is a case report showing a patient with LQT, but no other syndromic features of TS(75), fitting with this mutation causing a less severe form of the disease. When examined for its biophysical properties, G402S, much like G406R, causes a loss of VDI (Fig. 2a) and a disruption of CDI (Fig. 2b)(66, 71). However, the voltage-dependance of activation exhibited a shift towards depolarized potentials, opposite of the hyperpolarizing shift seen in G406R (Fig. 2c)(71). The G402S mutation therefore combines a GOF phenotype shown in the effects on channel inactivation with an apparent loss-of-function (LOF) phenotype in channel activation, illustrating the limitations of characterizing mutations as solely enhancing or diminishing channel activity.
In terms of the neurological symptoms observed in patients, TS presents one of the most penetrant forms of autism spectrum disorder (ASD) yet observed, thus making CaV1.2 channels containing TS mutations the subject of much interest for researchers seeking to uncover molecular mechanisms that contribute to this often-debilitating neurological disorder(15). The first mice generated harboring the G406R mutation in exon 8a were engineered to contain a neomycin cassette to ensure survival to maturity(76). Behavioral evaluation revealed traits analogous to those seen to be at the core of ASD, including social and learning impairments, repetitive behavior patterns, and altered vocalizations(76). Brain tissue from this TS mouse model demonstrated increased development and myelination of cortical oligodendrocyte progenitor cells (OPCs) from these mice(77). Notably, altered myelination patterns and increased white matter volume have been noted in human cases of ASD(78, 79). Additionally, cortical neurons isolated from these mice showed activity-dependent dendritic retraction, a result which was replicated in neurons differentiated from induced pluripotent stem cell (iPSCs) derived from a TS patient(80). The G406R mutation was also shown to result in abnormal coupling of AKAP150 to the channel(81), and AKAP150’s association with the channel is thought to play a role in mediating the ability of CaV1.2 channels to couple cellular excitation to gene transcription(82).
Following the initial description of TS1 and TS2 mutations, an ever-growing number of mutations have been identified in CaV1.2, often within constitutively expressed exons. While a number of these mutations have been shown to alter channel properties and are observed in patients suffering from symptoms observed in TS, many of these mutations do not recapitulate the full multisystem TS disease phenotype. Mutations such as A1473G(83), I1166T(84), I1166V(85), and E407A(86) all occupy positions in or adjacent to S6 domains (Fig. 1), much like the originally described G406R and G402S mutations, but notably, I1166V and A407A appear to have primarily or exclusively cardiac symptoms (see below). Other mutations, such as S643F, appear to cause both cardiac and neurological symptoms but without syndactyly(87). Moreover, R1024G appears to cause neurological symptoms and syndactyly, but not LQTS(88). The expanding array of disease-associated mutations observed in CaV1.2 and the heterogeneity in clinical outcomes presents a challenge to researchers attempting to fully characterize the scope and pattern of these channelopathic impairments.
The multisystem nature of TS symptoms suggests that CaV1.2 functionality extends beyond its well-described role in excitable tissues, and indeed, RNA-seq data shows that the channel is widely expressed throughout the body(89). Prior to the implementation of molecular techniques that could identify the genetic identity of particular calcium channels, calcium currents and calcium dynamics were shown to influence the activity of T lymphocytes(90). Additionally, functional CaV1.2 channels were identified in oseteoblasts(91, 92), and exposure of these cells to the LTCC agonist BAY K 8644 stimulated the secretion of the bone matrix protein, osteocalcin, with evidence suggesting a role of the channels in bone remodeling(93). One of the symptoms of TS is mandibular malformation, and mice expressing CaV1.2 channels harboring TS mutations show enlarged mandibles as compared to those expressing wildtype channels(94). Evidence from mice and zebrafish indicates that CaV1.2 acts on mandibular development through calcineurin signaling. Thus the phenotype observed in TS patients has increased our understanding of the role of CaV1.2 in physiology, demonstrating a clear importance for the channel in non-excitable tissues(16).
Cardiac-selective CACNA1C Mutations
As the variety of known mutations in CACNA1C has continued to expand, so too does the spectrum of phenotypes associated with these mutations. While TS was first described as a multisystem disorder, it was not long before CACNA1C mutations were identified as causative of cardiac-selective phenotypes(32, 95). Mutations in CACNA1C (Fig. 1) have been identified among patients with Brugada syndrome (BrS), idiopathic ventricular fibrillation (IVF), short-QT syndrome (SQT) and early repolarization syndrome (ERS)(32, 96, 97). Unlike the full TS phenotype, these patients do not exhibit extra-cardiac symptoms. Moreover, these syndromes appear to result from a loss of function of the CaV1.2 channel. Lack of Ca2+ entry during the cardiac AP readily explains the decreased AP duration identified in ERS. In addition, when BrS is the result of a CACNA1C mutation, it often includes overlapping features of SQT(32, 96, 97), consistent with a loss of CaV1.2 current. Thus, a loss of CaV1.2 current can impact the morphology of the cardiac AP, resulting in arrhythmia via similar mechanisms as the gain-of-function mutations, but with opposing direction. While these loss-of-function mutations can manifest as several overlapping syndromes, the mechanism of pathogenesis does not appear to significantly affect other tissues where CaV1.2 is highly expressed.
The cardiac-selectivity of CACNA1C mutations, however, is not specific only to loss-of-function mutations. Even for gain-of-function mutations in CACNA1C, the multisystem effects described for canonical TS are not always present. Mutations in CACNA1C have increasingly been described in patients exhibiting cardiac-selective phenotype, sometimes called cardiac-only TS(95) or atypical TS(74, 98). Initially these patients were described to exhibit severe LQTS (LQT8), in complex with additional cardiac deficits including hypertrophic cardiomyopathy (HCM) or congenital heart defects(95, 99). Later, atrial fibrillation and sick sinus syndrome have also been described as occurring in cardiac-only TS patients, further expanding the spectrum of cardiac disorders in the absence of the full multisystem phenotype(100). In fact, since the first description of TS, more mutations in CACNA1C have been identified with cardiac-selective LQTS as compared to the full multisystem TS phenotype (Fig. 1). The pathogenesis of these LQT8 mutations is in line with other forms of LQTS, where a genetic mutation altering the function of an ion channel results in an abnormal prolongation of the action potential due to an excess entry of a depolarizing ion. However, like in the full multisystem manifestation, the QT prolongation of LQT8 patients often exceeds that of other LQTS phenotypes, making these patients highly susceptible to cardiac arrhythmia and sudden death.
CACNA1C mutations have been increasingly associated with sudden cardiac death (SCD) (96, 101, 102). This finding is often explained by the severe LQTS phenotype produced by gain-of-function mutations(15, 66, 95, 103), or as a result of BrS/SQT resulting from loss-of-function mutations(32, 96). Multiple postmortem studies of sudden cardiac death have identified mutations in the CACNA1C gene(104, 105). These studies have identified CACNA1C as associated with sudden cardiac death both in young and adult patients (101, 104), making it a strong risk factor for SCD. Thus, while CACNA1C mutations remain rare, their potential for severe pathological consequences is very high.
Neurological Phenotypes
The role of the CaV1.2 channel in the heart has long been known to be critical. It is therefore not surprising that the majority of CACNA1C mutations described thus-far have been associated with significant cardiac effects, sometimes with concurrent neurological features. However, as more pathogenic mutations in CACNA1C are being identified, this cardiac-inclusive phenotype has begun to change, and mutations have been identified as causative of primarily neurological phenotypes. While it is surprising that significant changes in CaV1.2 channel gating may spare cardiac function in some cases, this finding is consistent with the numerous studies which have identified a strong association of CACNA1C with schizophrenia, major depressive disorder, bipolar disorder and autism(39), generally in the absence of a described cardiac phenotype.
CACNA1C variants (V1363M and splice site variant c.3717+1_3717+2insA) have been reported in individuals with epilepsy in the form of Neonatal onset epileptic encephalopathy and late-onset epilepsy (Fig. 1) ((106). The individual with the V1363M had a normal ECG, but displayed hypotonia, syndactyly, and dysmorphic facial features. The patients with the splice variant in the described study exhibited learning disabilities, congenital cardiac anomalies, and dysmorphic facial features in addition to epilepsy, also without overt cardiac symptoms. Furthermore, a mutation in a CACNA1C intron (rs779393130) has been described as being associated with autosomal dominant cerebellar ataxia(107). In 2018, the mutation R1024G (Fig. 1) was described as associated with developmental and speech delay but no QT prolongation(88). This mutation results in the neutralization of the one of the gating charges in the IIIS4 voltage sensor. Though it has not been functionally characterized, it has been suggested that it may destabilize the VSD leading to a gating pore current(108). Thus, the pathogenic mechanism leading to a neuro-specific effect may be distinct from those underlying LQT8.
This class of CACNA1C mutations with predominantly neurological phenotypes is continuing to grow, with identification of patients with both neurodevelopmental phenotypes as well as epilepsy and/or ataxia. A recent study describes a number novel CACNA1C variants in which individuals display a predominantly neurological phenotype which includes developmental delay, intellectual disability, ASD, hypotonia, epilepsy and ataxia (109). Interestingly, the phenotypic differences among this cohort of patients could be broadly separated into those stemming from non-truncating vs. truncating mutations (Fig. 1). The non-truncating mutations in the study all occurred de novo and included both missense variants and in-frame deletions. All individuals with such mutations displayed severe global developmental delay/intellectual disability and impaired motor function. The majority of these patients also had a spectrum of epileptic phenotypes; from medically refractory infantile epileptic encephalopathy, to later onset focal or generalized seizures. Furthermore, most exhibited ataxia, orthopedic abnormalities and hypotonia. Only a single individual in showed mild QT prolongation.
Four missense mutations identified in this study (L614P, L614R, L657F, L1408V) were functionally characterized using whole-cell patch clamp electrophysiology. The L1408V mutation is located within the IVS5 region, near the cytosolic side of the channel (Fig. 1), and causes a decrease in current density and no change in activation kinetics. L657F is located in IIS5 (Fig. 1) and causes an increase in current density and a hyperpolarizing shift in the voltage dependent activation. In addition, L657F was associated with higher protein expression levels as seen via western blot, which could explain the increase in current density. Residue L614 is located in the IIS4 VSD (Fig. 1) and mutations L614R and L614P did not result in any detectable differences in the patch-clamp experiments, though L614P appeared to show a non-statistically significant increase in current.
Patients within this study identified as harboring truncation mutations exhibited distinct features. While the majority of these mutations were also de novo, a small fraction were paternally inherited. In addition, these patients were reported to have more selective developmental abnormalities involving expressive language. Except for a single individual, none of these patients showed aberrant early motor development, and one-third were described as having poor balance or coordination. ASD was also found to be more common in this subset of patients, and only a single individual had a history of seizures. None of the individuals who were tested with an ECG showed any cardiac abnormalities.
Finally, the mutation K800T appears to contradict the concurrence of LQTS and ASD described in TS(110). This mutation was associated with ASD and severe psychiatric symptoms including depressive and hypomanic episodes in a single patient. However, unlike in typical TS, a short QT phenotype accompanied the neurological symptoms. Thus, as more mutations are identified within CACNA1C, the spectrum of phenotypes continues to expand.
Beyond Loss-of-function/Gain-of-function
Numerous studies have evaluated the impact of a variety of CaV1.2 mutations on channel function, identifying changes in current amplitude, activation, and inactivation(15, 66, 69, 71, 84, 85, 95, 105, 111). In the heart, the impact of these gating changes correlates well with disease(15, 66, 71). For loss-of-function mutations, most studies identified an overall decrease in current amplitude(32, 96, 105, 112, 113), which can either be attributed to a loss of channels at the membrane surface, or a decrease in current though the channel. This decrease in Ca2+ influx through CaV1.2 is sufficient to explain the shortening of the AP and resulting arrhythmia within these patients. For gain-of-function mutations, it has been shown that mutations can increase CaV1.2 current either through a shift in the voltage-dependence of activation of the channel, a decrease in VDI or a decrease in CDI(71). Notably, the canonical G406R TS mutation exhibited all three of these gating changes (Fig. 2), resulting in a marked excess entry of Ca2+ through the channel during the cardiac AP, despite the fact that the mutant channel is expected to only account for ~10% of CaV1.2 channels in a typical human ventricle due to alternative splicing(15). Importantly, a loss of either form of inactivation, or a depolarizing shift in channel activation is predicted to be sufficient to explain the AP prolongation caused by these LQT8 mutations(114, 115). Even the G402S mutation, which exhibits a gain-of-function effect on CDI and VDI but a loss-of-function effect on channel activation (Fig. 2), is predicted to result in an overall increase in Ca2+ entry during the cardiac AP, explaining the LQTS phenotype of the mutation(71). However, while the cardiac features of CACANA1C mutations are well explained by the gating changes in the channel, the same cannot be said for the neurological phenotypes exhibited by these patients.
TS represents one of the most penetrant forms of ASD(116), and various mutations in CaV1.2 have been strongly associated with neurodevelopmental delay and epilepsy. Further, GWAS studies have identified a strong association with the CACNA1C gene, yet associating these neurological phenotypes with gain or loss of function effects has not been successful. One explanation includes the likelihood that neurological deficits can occur in response to either a decrease or increase in CaV1.2 current(109). However, if we consider just the strong neurodevelopmental phenotype associated with the gain-of-function TS mutations, it appears that the ASD identified in these patients does, indeed, associate with mutations shown to cause excess Ca2+ entry(111). Unfortunately, this observation does not explain why many CaV1.2 mutations which have been shown to produce similar gain-of-function effects on the channel result in a cardiac, but not neuronal, phenotypes. It seems likely that consideration of the distinct mechanisms of channel disruption are required, and simple gain-of-function descriptions are not sufficient to explain the unique impact of CaV1.2 mutations on neuronal function. This lack of clear correlation between excess Ca2+ entry through CaV1.2 and ASD has led to the consideration that some neurological deficits may result from mechanisms that are independent from Ca2+ influx through the channel(17, 117). However, the strong impact of these mutations on channel gating and the known importance of CaV1.2 channel function in the normal function of neurons makes it difficult to dismiss the likely impact of significant CaV1.2 gating changes in the brain.
The canonical G406R TS mutation caused a hyperpolarizing shift in the voltage dependence of CaV1.2 activation and deficits in both VDI and CDI (Fig. 2), and is causative of ASD in all patients. The G402S mutation caused a similar deficient in both CDI and VDI, however the effect of this mutation on channel activation was in the opposite (depolarizing) direction (Fig. 2). Interestingly, the neurodevelopmental phenotype first described for these patients is not consistent across the patient population and evidence suggests that it may be secondary to hypoxic injury(74). It is therefore tantalizing to speculate that this difference in activation shift direction may underly the lack of a neurological phenotype in these patients.
In fact, the left shift in channel activation has been hypothesized as being a requirement for the CaV1.2 related ASD phenotype(116, 118). Detailed study of the biophysical properties of multiple CaV1.2 mutations has demonstrated a correlation between a marked left shift in channel activation and neurodevelopmental disease, and it has been shown that the left shift in channel activation correlates with an altered response of the channels to a neuronal AP stimulus(111). Finally, this hypothesis is further strengthened by similar observations of the related CaV1.3 LTCC – where ASD occurs in patients harboring mutations which also produce a left shift in channel activation(118). Thus, it appears that the neurodevelopmental deficits of TS patients best track with a significant left shift in channel activation(111). While this result does not preclude additional mechanisms capable of modulating neuronal function, it is interesting that it is consistent with the reported importance of a voltage-dependent conformational change in the pathogenesis of TS(17).
Functional Channel Hotspots and the Emergence of S6-opathies
While mutations in CACNA1C occur throughout the length of the channel, select regions of known channel function have been noted to correlate with patient phenotypes, hinting at hotspots for pathogenesis. One of these is located on the cytosolic II-III loop of the channel, containing 6 mutations (Fig. 1) clustered near a known STAC (SH3 and cysteine rich domain) binding domain of the channel(119). Each of these mutations correlated with a significant increase in the QT interval of the patient. Interestingly, while STAC proteins have been shown to modulate CaV1.2 channel gating(120, 121), it is not clear that that this mechanism can fully explain the cardiac phenotype seen in these patients due to the locus of a functionally relevant STAC binding site on the C-tail of the channel, and unclear expression of STAC protein in the heart. Nonetheless, the overlapping patient phenotypes of mutations located within a known binding region point towards a common pathogenic mechanism of these mutations.
Yet another locus that appears to be emerging as a hotspot for severe disease is the distal S6 regions of the channel. Interestingly, out of the 11 mutations currently reported as producing a full, multisystem TS phenotype including both severe LQTS and neurological symptoms, 7 fall on a distal S6 region (Fig. 1, red). In addition, multiple non-syndromic LQTS and neurodevelopmental associated mutations fall within the same regions (Fig. 1, blue and orange). Interestingly, this pattern has emerged not only for mutations within CaV1.2 but for channelopathic mutations within other Ca2+ channels including CaV3.1, CaV3.2, CaV1.3, CaV2.1 and CaV2.3,(122, 123 , 124), and within other ion channels including NALCN(125) and HCN4(126). Thus, S6-opathies may represent a growing class of ion channelopathies(111, 122). As the distal S6 region is known to be a critical element controlling channel activation, it is not surprising that many of these CaV1.2 S6 mutations have demonstrated shifts in channel activation (Fig. 2)(71, 84, 102, 111, 116). Moreover, the distal S6 region contains residues critical for the docking of a the I-II linker region in a ‘hinged-lid’ mechanism underlying VDI(20, 127, 128), consistent with the impact of many CaV1.2 distal S6 mutations on VDI. Additionally, deficits in CDI have been shown to result from modulation of residues within this same S6 locus, often though a mechanism secondary to changes in channel activation(71, 111, 128). Thus, it is not unexpected that mutations within this region are likely to result in significant and physiologically impactful changes to channel gating, making these mutations highly deleterious. Finally, it must be considered that many disease-causing mutations identified within the S5; S4-5 linker may affect interactions with the S6 region, providing an expanded impact for altered S6 function. As the number of known mutations continues to grow, no doubt additional regions of pathogenic significance are likely to emerge.
Therapeutic Implications
Treatment for TS and LQT8 has remained challenging. As the cardiac symptoms are generally the most life-threatening component of CACNA1C channelopathies, they constitute the main focus for treatment for these patients. However, treatments that have been effective for other forms of LQTS have had limited benefit in the context of LQT8. Beta blockers have been used with little effect, and the sodium channel blocker mexiletine has shown partial efficacy in treating the QT prolongation of TS(129–131). Given that the LQTS phenotype due to CACNA1C mutations is the result of excess Ca2+ entry through the channel, L-type Ca2+ channel blockers (CCBs) would seem well positioned for the treatment of LQT8. Multiple CCBs are available and commonly used to treat cardiac arrhythmia, hypertension and angina pectoris. However, their efficacy in TS has remained limited, resulting in continued reliance on internal cardiac defibrillators in many patients(67, 75). Verapamil, in particular has been tried in the treatment of TS, however, while initial reports indicated that the drug was partially effective in reducing ventricular fibrillation, it did not reduce the QT interval(67), and over time resulted in increased atrial fibrillation and reduced atrial contraction(132). As a result, ranolazine, a multi-channel blocker, was added to the verapamil regiment, resulting in a decrease in atrial and ventricular fibrillation(133). Thus, it appears that targeting the CaV1.2 channel itself has not provided adequate results for the treatment of TS.
It appears that this lack of efficacy likely stems from the mechanism of CCB block of CaV1.2 channels. Three classes of CCBs have been described, phenylalkylamines, benzothiazepines, and dihydropyridines (DHPs). Each of these drugs are known to be state dependent blockers, such that channel opening during repeated depolarizations enhances drug binding(134, 135). Such use-dependent block by phenylalkylamines and benzothiazepines is thought to result from open and inactivated state block, where opening of the channel provides access of the drug to the receptor site. (136). Block by DHPs is proposed to result from preferential binding to the inactivated state of the channel. However, many TS mutations reduce channel inactivation, resulting in a loss of use-dependent block and an overall decrease in efficacy of the CCBs. Verapamil has been shown to exhibit a decrease in use-dependent block in the context of multiple pathogenic CaV1.2 mutations(137). This can be readily seen as a decrease in the activity-dependent shift of the concentration-response curve measured in HEK cells expressing WT versus G406R CaV1.2 channels (Fig. 3a vs. b), and has been shown to persist in the context of iPSC-derived cardiomyocytes(137). Importantly, this result translated to a decrease in AP shortening in iPSC derived cardiomyocytes derived from a TS (LQT8) patient harboring the G406R mutation (Fig. 3c). Likewise, DHPs have been shown to exhibit decreased efficacy in the context of pathogenic CaV1.2 mutations in HEK cells(15, 137, 138). Thus, the loss of inactivation due to the deleterious mutation causes a major challenge for treatment of these channelopathies, limiting the usefulness of currently available CCBs.
Figure 3. CACANA1C mutations reduce the efficacy of verapamil.
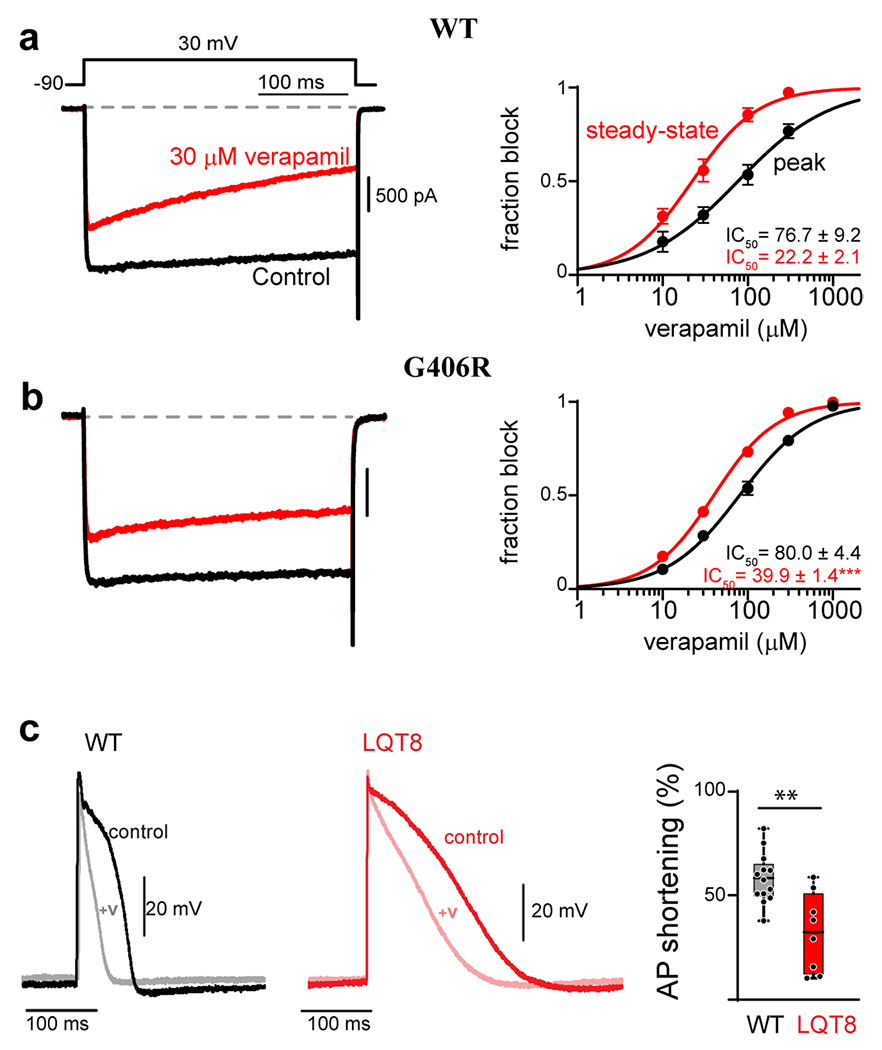
(a) Verapamil concentration-response curves measured for WT CaV1.2 in HEK 293 cells. Left: Exemplar Ba2+ current before (black) and after (red) the addition of 30 μM verapamil. Accumulation of block can be seen during the 300 ms depolarization in the presence of verapamil (red). Right: concentration-response curves can be generated by measuring the effect of the drug on the peak current amplitude immediately following depolarization (black), or at the end of the voltage step after 300 ms depolarization (red). The increased block measured from the steady-state value (red) is indicative of use-dependence of the drug. (b) The G406R mutation significantly reduces the use-dependent block of verapamil, seen as an increase in steady-state IC50. (c) Left: Exemplar AP recordings of WT iPSC derived cardiomyocytes before (black) and after (gray) the addition of 10 μM verapamil. Middle: Exemplar AP recording from an iPSC derived cardiomyocyte from a patient harboring the G406R mutation (red, LQT8) shows significant prolongation of the action potential as compared to WT (left, black). Addition of 10 μM verapamil (pink) reduces the AP duration to a lesser extent as compared to WT iPSC derived cardiomyocytes. Right: Quantification of the percent of AP shortening in WT vs. LQT8 (G406R) containing iPSC derived cardiomyocytes demonstrates a significant deficit in verapamil’s ability to shorten the AP in the context of patient derived cells harboring the G406R mutation (LQT8). Reproduced with permission from (Dick et. al., 2022, JMCC)(137).
Due to this lack of efficacy, research continues to explore alternative mechanisms for the treatment of these patients. One approach is the use of roscovitine, a cyclin-dependent kinase inhibitor and atypical LTCC blocker that has been reported to reverse the phenotype associated with TS in heterologous systems(139–141). Roscovitine has been shown to enhance VDI of LTCCs (139–143) and suppress early afterdepolarizations, which can lead to ventricular arrhythmias (143). It has also been reported that roscovitine can rescue the TS phenotype in both iPSC-derived cardiomyocytes and neurons differentiated from a TS patient(80, 142, 144, 145). In iPSC-derived CMs, roscovitine was able to reduce the irregular timing and amplitude of Ca2+ transients and significantly increased VDI(142, 144). Efforts continue to evaluate novel mechanisms for the treatment of TS, including the use of genetic approaches and identification of novel small molecule therapies.
References
- 1.Berger SM, Bartsch D, The role of L-type voltage-gated calcium channels Cav1.2 and Cav1.3 in normal and pathological brain function. Cell Tissue Res 357, 463–476 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Harvey RD, Hell JW, CaV1.2 signaling complexes in the heart. J Mol Cell Cardiol 58, 143–152 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zamponi GW, Striessnig J, Koschak A, Dolphin AC, The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol Rev 67, 821–870 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bers DM, Ca regulation in cardiac muscle. Med Sci Sports Exerc 23, 1157–1162 (1991). [PubMed] [Google Scholar]
- 5.Lederer WJ et al. , Excitation-contraction coupling in heart cells. Roles of the sodium-calcium exchange, the calcium current, and the sarcoplasmic reticulum. Annals of the New York Academy of Sciences 588, 190–206 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Nerbonne JM, Kass RS, Molecular physiology of cardiac repolarization. Physiol Rev 85, 1205–1253 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Fabiato A, Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. The American journal of physiology 245, C1–14 (1983). [DOI] [PubMed] [Google Scholar]
- 8.Sinnegger-Brauns MJ et al. , Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol 75, 407–414 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Ma H, Cohen S, Li B, Tsien RW, Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci Rep 33, 97–101 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moosmang S et al. , Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci 25, 9883–9892 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Striessnig J, Pinggera A, Kaur G, Bock G, Tuluc P, L-type Ca(2+) channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 3, 15–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee AS et al. , Forebrain elimination of cacna1c mediates anxiety-like behavior in mice. Molecular psychiatry 17, 1054–1055 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matza D, Flavell RA, Roles of Ca(v) channels and AHNAK1 in T cells: the beauty and the beast. Immunol Rev 231, 257–264 (2009). [DOI] [PubMed] [Google Scholar]
- 14.Davenport B, Li Y, Heizer JW, Schmitz C, Perraud AL, Signature Channels of Excitability no More: L-Type Channels in Immune Cells. Front Immunol 6, 375 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Splawski I et al. , Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Pitt GS, Matsui M, Cao C, Voltage-Gated Calcium Channels in Nonexcitable Tissues. Annual review of physiology 83, 183–203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li B, Tadross MR, Tsien RW, Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351, 863–867 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gomez-Ospina N et al. , A promoter in the coding region of the calcium channel gene CACNA1C generates the transcription factor CCAT. PLoS One 8, e60526 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R, The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell 127, 591–606 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stotz SC, Hamid J, Spaetgens RL, Jarvis SE, Zamponi GW, Fast inactivation of voltage-dependent calcium channels. A hinged-lid mechanism? J Biol Chem 275, 24575–24582 (2000). [DOI] [PubMed] [Google Scholar]
- 21.Clark MB et al. , Long-read sequencing reveals the complex splicing profile of the psychiatric risk gene CACNA1C in human brain. Molecular psychiatry 25, 37–47 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang ZZ et al. , Transcript scanning reveals novel and extensive splice variations in human l-type voltage-gated calcium channel, Cav1.2 alpha1 subunit. J Biol Chem 279, 44335–44343 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Liao P, Yong TF, Liang MC, Yue DT, Soong TW, Splicing for alternative structures of Cav1.2 Ca2+ channels in cardiac and smooth muscles. Cardiovascular research 68, 197–203 (2005). [DOI] [PubMed] [Google Scholar]
- 24.Liao P et al. , A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state-dependent inhibition by nifedipine. J Biol Chem 282, 35133–35142 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Liao P et al. , Alternative splicing generates a novel truncated Cav1.2 channel in neonatal rat heart. J Biol Chem 290, 9262–9272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J, International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev 57, 411–425 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Buraei Z, Yang J, The ss subunit of voltage-gated Ca2+ channels. Physiol Rev 90, 1461–1506 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei XY et al. , Heterologous regulation of the cardiac Ca2+ channel alpha 1 subunit by skeletal muscle beta and gamma subunits. Implications for the structure of cardiac L-type Ca2+ channels. J.Biol.Chem 266, 21943–21947 (1991). [PubMed] [Google Scholar]
- 29.Klugbauer N, Welling A, Specht V, Seisenberger C, Hofmann F, L-type Ca2+ channels of the embryonic mouse heart. Eur J Pharmacol 447, 279–284 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Ablinger C, Geisler SM, Stanika RI, Klein CT, Obermair GJ, Neuronal alpha2delta proteins and brain disorders. Pflugers Arch 472, 845–863 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolphin AC, Voltage-gated calcium channel alpha 2delta subunits: an assessment of proposed novel roles. F1000Res 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burashnikov E et al. , Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Heart Rhythm 7, 1872–1882 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Breitenkamp AF et al. , Rare mutations of CACNB2 found in autism spectrum disease-affected families alter calcium channel function. PLoS One 9, e95579 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cross-Disorder C Group of the Psychiatric Genomics, Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bhat S et al. , CACNA1C (Cav1.2) in the pathophysiology of psychiatric disease. Prog Neurobiol 99, 1–14 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Craddock N, Sklar P, Genetics of bipolar disorder. Lancet 381, 1654–1662 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Green EK et al. , Replication of bipolar disorder susceptibility alleles and identification of two novel genome-wide significant associations in a new bipolar disorder case-control sample. Molecular psychiatry 18, 1302–1307 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.G. C. B. D. W. G. Psychiatric, Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat Genet 43, 977–983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green EK et al. , The bipolar disorder risk allele at CACNA1C also confers risk of recurrent major depression and of schizophrenia. Molecular psychiatry 15, 1016–1022 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casamassima F et al. , L-type calcium channels and psychiatric disorders: A brief review. Am J Med Genet B Neuropsychiatr Genet 153B, 1373–1390 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Sullivan PF et al. , Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Molecular psychiatry 14, 359–375 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li J et al. , Schizophrenia Related Variants in CACNA1C also Confer Risk of Autism. PLoS One 10, e0133247 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sayad A et al. , Association Study of Sequence Variants in Voltage-gated Ca2+ Channel Subunit Alpha-1C and Autism Spectrum Disorders. Rep Biochem Mol Biol 8, 56–62 (2019). [PMC free article] [PubMed] [Google Scholar]
- 44.Pouget JG et al. , Cross-disorder analysis of schizophrenia and 19 immune-mediated diseases identifies shared genetic risk. Hum Mol Genet 28, 3498–3513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferreira MA et al. , Collaborative genome-wide association analysis supports a role for ANK3 and CACNA1C in bipolar disorder. Nat Genet 40, 1056–1058 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keers R, Farmer AE, Aitchison KJ, Extracting a needle from a haystack: reanalysis of whole genome data reveals a readily translatable finding. Psychol Med 39, 1231–1235 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Sklar P et al. , Whole-genome association study of bipolar disorder. Molecular psychiatry 13, 558–569 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu D et al. , CACNA1C (rs1006737) may be a susceptibility gene for schizophrenia: An updated meta-analysis. Brain Behav 9, e01292 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshimizu T et al. , Functional implications of a psychiatric risk variant within CACNA1C in induced human neurons. Molecular psychiatry 20, 162–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Erk S et al. , Hippocampal and frontolimbic function as intermediate phenotype for psychosis: evidence from healthy relatives and a common risk variant in CACNA1C. Biological psychiatry 76, 466–475 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Roussos P, Giakoumaki SG, Georgakopoulos A, Robakis NK, Bitsios P, The CACNA1C and ANK3 risk alleles impact on affective personality traits and startle reactivity but not on cognition or gating in healthy males. Bipolar Disord 13, 250–259 (2011). [DOI] [PubMed] [Google Scholar]
- 52.Krug A et al. , Effect of CACNA1C rs1006737 on neural correlates of verbal fluency in healthy individuals. Neuroimage 49, 1831–1836 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Koch K et al. , CACNA1C risk variant affects microstructural connectivity of the amygdala. Neuroimage Clin 22, 101774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lancaster TM, Foley S, Tansey KE, Linden DE, Caseras X, CACNA1C risk variant is associated with increased amygdala volume. Eur Arch Psychiatry Clin Neurosci 266, 269–275 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Wolf C et al. , CACNA1C genotype explains interindividual differences in amygdala volume among patients with schizophrenia. Eur Arch Psychiatry Clin Neurosci 264, 93–102 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Janiri D et al. , Genetic neuroimaging of bipolar disorder: a systematic 2017-2020 update. Psychiatr Genet 31, 50–64 (2021). [DOI] [PubMed] [Google Scholar]
- 57.Bigos KL et al. , Genetic variation in CACNA1C affects brain circuitries related to mental illness. Arch Gen Psychiatry 67, 939–945 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gershon ES et al. , A rare mutation of CACNA1C in a patient with bipolar disorder, and decreased gene expression associated with a bipolar-associated common SNP of CACNA1C in brain. Molecular psychiatry 19, 890–894 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dao DT et al. , Mood disorder susceptibility gene CACNA1C modifies mood-related behaviors in mice and interacts with sex to influence behavior in mice and diagnosis in humans. Biological psychiatry 68, 801–810 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Terrillion CE et al. , Reduced levels of Cacna1c attenuate mesolimbic dopamine system function. Genes Brain Behav 16, 495–505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schmitt A et al. , Post-mortem gene expression of calcium channels Cav1.2 and Cav1.3 in schizophrenia. Eur Arch Psychiatry Clin Neurosci 272, 1135–1137 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang H et al. , CACNA1C rs1006737 SNP increases the risk of essential hypertension in both Chinese Han and ethnic Russian people of Northeast Asia. Medicine (Baltimore) 100, e24825 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Napolitano C, Splawski I, Timothy KW, Bloise R, Priori SG, in GeneReviews(R), Pagon RA et al. , Eds. (Seattle (WA), 1993). [Google Scholar]
- 64.Reichenbach H, Meister EM, Theile H, [The heart-hand syndrome. A new variant of disorders of heart conduction and syndactylia including osseous changes in hands and feet]. Kinderarztl Prax 60, 54–56 (1992). [PubMed] [Google Scholar]
- 65.Marks ML, Whisler SL, Clericuzio C, Keating M, A new form of long QT syndrome associated with syndactyly. Journal of the American College of Cardiology 25, 59–64 (1995). [DOI] [PubMed] [Google Scholar]
- 66.Splawski I et al. , Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc Natl Acad Sci U S A 102, 8089–8096; discussion 8086-8088 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jacobs A, Knight BP, McDonald KT, Burke MC, Verapamil decreases ventricular tachyarrhythmias in a patient with Timothy syndrome (LQT8). Heart Rhythm 3, 967–970 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Soldatov NM, Molecular diversity of L-type Ca2+ channel transcripts in human fibroblasts. Proc Natl Acad Sci U S A 89, 4628–4632 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Barrett CF, Tsien RW, The Timothy syndrome mutation differentially affects voltage- and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc Natl Acad Sci U S A 105, 2157–2162 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Raybaud A et al. , The role of the GX9GX3G motif in the gating of high voltage-activated Ca2+ channels. J Biol Chem 281, 39424–39436 (2006). [DOI] [PubMed] [Google Scholar]
- 71.Dick IE, Joshi-Mukherjee R, Yang W, Yue DT, Arrhythmogenesis in Timothy Syndrome is associated with defects in Ca(2+)-dependent inactivation. Nat Commun 7, 10370 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Drum BM, Dixon RE, Yuan C, Cheng EP, Santana LF, Cellular mechanisms of ventricular arrhythmias in a mouse model of Timothy syndrome (long QT syndrome 8). J Mol Cell Cardiol 66, 63–71 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sepp R et al. , Timothy syndrome 1 genotype without syndactyly and major extracardiac manifestations. American journal of medical genetics. Part A 173, 784–789 (2017). [DOI] [PubMed] [Google Scholar]
- 74.Frohler S et al. , Exome sequencing helped the fine diagnosis of two siblings afflicted with atypical Timothy syndrome (TS2). BMC Med Genet 15, 48 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hiippala A, Tallila J, Myllykangas S, Koskenvuo JW, Alastalo TP, Expanding the phenotype of Timothy syndrome type 2: an adolescent with ventricular fibrillation but normal development. American journal of medical genetics. Part A 167A, 629–634 (2015). [DOI] [PubMed] [Google Scholar]
- 76.Bader PL et al. , Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci U S A 108, 15432–15437 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cheli VT et al. , Enhanced oligodendrocyte maturation and myelination in a mouse model of Timothy syndrome. Glia 66, 2324–2339 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Courchesne E et al. , Unusual brain growth patterns in early life in patients with autistic disorder: an MRI study. Neurology 57, 245–254 (2001). [DOI] [PubMed] [Google Scholar]
- 79.Carmody DP, Lewis M, Regional white matter development in children with autism spectrum disorders. Dev Psychobiol 52, 755–763 (2010). [DOI] [PubMed] [Google Scholar]
- 80.Pasca SP et al. , Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nature medicine 17, 1657–1662 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cheng EP et al. , Restoration of normal L-type Ca2+ channel function during Timothy syndrome by ablation of an anchoring protein. Circ Res 109, 255–261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Murphy JG et al. , AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7, 1577–1588 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gillis J et al. , Long QT, syndactyly, joint contractures, stroke and novel CACNA1C mutation: expanding the spectrum of Timothy syndrome. American journal of medical genetics. Part A 158A, 182–187 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boczek NJ et al. , Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Heart Rhythm 12, 211–219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wemhoner K et al. , Gain-of-function mutations in the calcium channel CACNA1C (Cav1.2) cause non-syndromic long-QT but not Timothy syndrome. J Mol Cell Cardiol 80, 186–195 (2015). [DOI] [PubMed] [Google Scholar]
- 86.Colson C et al. , Unusual clinical description of adult with Timothy syndrome, carrier of a new heterozygote mutation of CACNA1C. European journal of medical genetics 62, 103648 (2019). [DOI] [PubMed] [Google Scholar]
- 87.Ozawa J et al. , A novel CACNA1C mutation identified in a patient with Timothy syndrome without syndactyly exerts both marked loss- and gain-of-function effects. HeartRhythm Case Rep 4, 273–277 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kosaki R, Ono H, Terashima H, Kosaki K, Timothy syndrome-like condition with syndactyly but without prolongation of the QT interval. American journal of medical genetics. Part A 176, 1657–1661 (2018). [DOI] [PubMed] [Google Scholar]
- 89.Lachmann A et al. , Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun 9, 1366 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alcover A, Weiss MJ, Daley JF, Reinherz EL, The T11 glycoprotein is functionally linked to a calcium channel in precursor and mature T-lineage cells. Proc Natl Acad Sci U S A 83, 2614–2618 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chesnoy-Marchais D, Fritsch J, Voltage-gated sodium and calcium currents in rat osteoblasts. J Physiol 398, 291–311 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meszaros JG, Karin NJ, Akanbi K, Farach-Carson MC, Down-regulation of L-type Ca2+ channel transcript levels by 1,25-dihyroxyvitamin D3. Osteoblastic cells express L-type alpha1C Ca2+ channel isoforms. J Biol Chem 271, 32981–32985 (1996). [DOI] [PubMed] [Google Scholar]
- 93.Guggino SE, Lajeunesse D, Wagner JA, Snyder SH, Bone remodeling signaled by a dihydropyridine- and phenylalkylamine-sensitive calcium channel. Proc Natl Acad Sci U S A 86, 2957–2960 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ramachandran KV et al. , Calcium influx through L-type CaV1.2 Ca2+ channels regulates mandibular development. J Clin Invest 123, 1638–1646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Boczek NJ et al. , Identification and Functional Characterization of a Novel CACNA1C-Mediated Cardiac Disorder Characterized by Prolonged QT Intervals With Hypertrophic Cardiomyopathy, Congenital Heart Defects, and Sudden Cardiac Death. Circulation. Arrhythmia and electrophysiology 8, 1122–1132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Antzelevitch C et al. , Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fukuyama M et al. , L-type calcium channel mutations in Japanese patients with inherited arrhythmias. Circ J 77, 1799–1806 (2013). [DOI] [PubMed] [Google Scholar]
- 98.Bauer R, Timothy KW, Golden A, Update on the Molecular Genetics of Timothy Syndrome. Front Pediatr 9, 668546 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Boczek NJ et al. , Exome sequencing and systems biology converge to identify novel mutations in the L-type calcium channel, CACNA1C, linked to autosomal dominant long QT syndrome. Circulation. Cardiovascular genetics 6, 279–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gakenheimer-Smith L et al. , Expanding the phenotype of CACNA1C mutation disorders. Mol Genet Genomic Med 9, e1673 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Halvorsen M et al. , De novo mutations in childhood cases of sudden unexplained death that disrupt intracellular Ca(2+) regulation. Proc Natl Acad Sci U S A 118, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hennessey JA et al. , A CACNA1C variant associated with reduced voltage-dependent inactivation, increased CaV1.2 channel window current, and arrhythmogenesis. PLoS One 9, e106982 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dufendach KA et al. , Clinical Outcomes and Modes of Death in Timothy Syndrome: A Multicenter International Study of a Rare Disorder. JACC Clin Electrophysiol 4, 459–466 (2018). [DOI] [PubMed] [Google Scholar]
- 104.Narula N, Tester DJ, Paulmichl A, Maleszewski JJ, Ackerman MJ, Post-mortem Whole exome sequencing with gene-specific analysis for autopsy-negative sudden unexplained death in the young: a case series. Pediatr Cardiol 36, 768–778 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sutphin BS et al. , Molecular and Functional Characterization of Rare CACNA1C Variants in Sudden Unexplained Death in the Young. Congenit Heart Dis 11, 683–692 (2016). [DOI] [PubMed] [Google Scholar]
- 106.Bozarth X. et al. , Expanding clinical phenotype in CACNA1C related disorders: From neonatal onset severe epileptic encephalopathy to late-onset epilepsy. American journal of medical genetics. Part A 176, 2733–2739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen J, Sun Y, Liu X, Li J, Identification of a novel mutation in the CACNA1C gene in a Chinese family with autosomal dominant cerebellar ataxia. BMC Neurol 19, 157 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Striessnig J, Voltage-Gated Ca(2+)-Channel alpha1-Subunit de novo Missense Mutations: Gain or Loss of Function - Implications for Potential Therapies. Front Synaptic Neurosci 13, 634760 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rodan LH et al. , Phenotypic expansion of CACNA1C-associated disorders to include isolated neurological manifestations. Genet Med 23, 1922–1932 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Endres D et al. , New Cav1.2 Channelopathy with High-Functioning Autism, Affective Disorder, Severe Dental Enamel Defects, a Short QT Interval, and a Novel CACNA1C Loss-Of-Function Mutation. Int J Mol Sci 21, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bamgboye MA et al. , CaV1.2 channelopathic mutations evoke diverse pathophysiological mechanisms. J Gen Physiol 154, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Blancard M et al. , An African loss-of-function CACNA1C variant p.T1787M associated with a risk of ventricular fibrillation. Sci Rep 8, 14619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Beziau DM et al. , Complex Brugada syndrome inheritance in a family harbouring compound SCN5A and CACNA1C mutations. Basic research in cardiology 109, 446 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Morotti S, Grandi E, Summa A, Ginsburg KS, Bers DM, Theoretical study of L-type Ca(2+) current inactivation kinetics during action potential repolarization and early afterdepolarizations. J Physiol 590, 4465–4481 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Faber GM, Silva J, Livshitz L, Rudy Y, Kinetic properties of the cardiac L-type Ca2+ channel and its role in myocyte electrophysiology: a theoretical investigation. Biophys J 92, 1522–1543 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marcantoni A, Calorio C, Hidisoglu E, Chiantia G, Carbone E, Cav1.2 channelopathies causing autism: new hallmarks on Timothy syndrome. Pflugers Arch 472, 775–789 (2020). [DOI] [PubMed] [Google Scholar]
- 117.Krey JF et al. , Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nature neuroscience 16, 201–209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Calorio C et al. , Impaired chromaffin cell excitability and exocytosis in autistic Timothy syndrome TS2-neo mouse rescued by L-type calcium channel blockers. J Physiol 597, 1705–1733 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mellor GJ et al. , Type 8 long QT syndrome: pathogenic variants in CACNA1C-encoded Cav1.2 cluster in STAC protein binding site. Europace 21, 1725–1732 (2019). [DOI] [PubMed] [Google Scholar]
- 120.Niu J et al. , Allosteric regulators selectively prevent Ca(2+)-feedback of CaV and NaV channels. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Polster A et al. , Stac Proteins Suppress Ca(2+)-Dependent Inactivation of Neuronal l-type Ca(2+) Channels. J Neurosci 38, 9215–9227 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lory P, Nicole S, Monteil A, Neuronal Cav3 channelopathies: recent progress and perspectives. Pflugers Arch 472, 831–844 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shelley C, Whitt JP, Montgomery JR, Meredith AL, Phosphorylation of a constitutive serine inhibits BK channel variants containing the alternate exon “SRKR”. J Gen Physiol 142, 585–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Scholl UI et al. , Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat Genet 45, 1050–1054 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kschonsak M et al. , Structure of the human sodium leak channel NALCN. Nature 587, 313–318 (2020). [DOI] [PubMed] [Google Scholar]
- 126.Biel S et al. , Mutation in S6 domain of HCN4 channel in patient with suspected Brugada syndrome modifies channel function. Pflugers Arch 468, 1663–1671 (2016). [DOI] [PubMed] [Google Scholar]
- 127.Stotz SC, Zamponi GW, Identification of inactivation determinants in the domain IIS6 region of high voltage-activated calcium channels. J Biol Chem 276, 33001–33010 (2001). [DOI] [PubMed] [Google Scholar]
- 128.Tadross MR, Ben Johny M, Yue DT, Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J Gen Physiol 135, 197–215 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gao Y et al. , Inhibition of late sodium current by mexiletine: a novel pharmotherapeutical approach in timothy syndrome. Circulation. Arrhythmia and electrophysiology 6, 614–622 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Tunca Sahin G, Ergul Y, A case report: Is mexiletine usage effective in the shortening of QTC interval and improving the T-wave alternans in Timothy syndrome? Ann Noninvasive Electrocardiol 23, e12522 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hermida A et al. , Long-term follow-up of a patient with type 2 Timothy syndrome and the partial efficacy of mexiletine. Gene 777, 145465 (2021). [DOI] [PubMed] [Google Scholar]
- 132.Shah DP, Baez-Escudero JL, Weisberg IL, Beshai JF, Burke MC, Ranolazine Safely Decreases Ventricular and Atrial Fibrillation in Timothy Syndrome (LQT8). Pacing Clin Electrophysiol, (2010). [DOI] [PubMed] [Google Scholar]
- 133.Shah DP, Baez-Escudero JL, Weisberg IL, Beshai JF, Burke MC, Ranolazine safely decreases ventricular and atrial fibrillation in Timothy syndrome (LQT8). Pacing Clin Electrophysiol 35, e62–64 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Lee KS, Tsien RW, Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature 302, 790–794 (1983). [DOI] [PubMed] [Google Scholar]
- 135.Hering S et al. , Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: role of inactivation. Proc Natl Acad Sci U S A 94, 13323–13328 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Catterall WA, Lenaeus MJ, Gamal El-Din TM, Structure and Pharmacology of Voltage-Gated Sodium and Calcium Channels. Annual review of pharmacology and toxicology 60, 133–154 (2020). [DOI] [PubMed] [Google Scholar]
- 137.Bamgboye MA et al. , Impaired CaV1.2 inactivation reduces the efficacy of calcium channel blockers in the treatment of LQT8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Sheng X et al. , Two mechanistically distinct effects of dihydropyridine nifedipine on CaV1.2 L-type Ca(2)(+) channels revealed by Timothy syndrome mutation. Eur J Pharmacol 685, 15–23 (2012). [DOI] [PubMed] [Google Scholar]
- 139.Yarotskyy V, Elmslie KS, Roscovitine, a cyclin-dependent kinase inhibitor, affects several gating mechanisms to inhibit cardiac L-type (Ca(V)1.2) calcium channels. Br J Pharmacol 152, 386–395 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Yarotskyy V, Gao G, Peterson BZ, Elmslie KS, The Timothy syndrome mutation of cardiac CaV1.2 (L-type) channels: multiple altered gating mechanisms and pharmacological restoration of inactivation. J Physiol 587, 551–565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yarotskyy V et al. , Roscovitine binds to novel L-channel (CaV1.2) sites that separately affect activation and inactivation. J Biol Chem 285, 43–53 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yazawa M et al. , Using induced pluripotent stem cells to investigate cardiac phenotypes in Timothy syndrome. Nature 471, 230–234 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Angelini M et al. , Suppression of ventricular arrhythmias by targeting late L-type Ca2+ current. J Gen Physiol 153, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Song L et al. , Dual optical recordings for action potentials and calcium handling in induced pluripotent stem cell models of cardiac arrhythmias using genetically encoded fluorescent indicators. Stem Cells Transl Med 4, 468–475 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Song L, Park SE, Isseroff Y, Morikawa K, Yazawa M, Inhibition of CDK5 Alleviates the Cardiac Phenotypes in Timothy Syndrome. Stem Cell Reports 9, 50–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]