

Summary
Whole-exome sequencing of autism spectrum disorder (ASD) probands and unaffected family members has identified many genes harboring de novo variants suspected to play a causal role in the disorder. Of these, chromodomain helicase DNA-binding protein 8 (CHD8) is the most recurrently mutated. Despite the prevalence of CHD8 mutations, we have little insight into how CHD8 loss affects genome organization or the functional consequences of these molecular alterations in neurons. Here, we engineered two isogenic human embryonic stem cell lines with CHD8 loss-of-function mutations and characterized differences in differentiated human cortical neurons. We identified hundreds of genes with altered expression, including many involved in neural development and excitatory synaptic transmission. Field recordings and single-cell electrophysiology revealed a 3-fold decrease in firing rates and synaptic activity in CHD8+/− neurons, as well as a similar firing-rate deficit in primary cortical neurons from Chd8+/− mice. These alterations in neuron and synapse function can be reversed by CHD8 overexpression. Moreover, CHD8+/− neurons displayed a large increase in open chromatin across the genome, where the greatest change in compaction was near autism susceptibility candidate 2 (AUTS2), which encodes a transcriptional regulator implicated in ASD. Genes with changes in chromatin accessibility and expression in CHD8+/− neurons have significant overlap with genes mutated in probands for ASD, intellectual disability, and schizophrenia but not with genes mutated in healthy controls or other disease cohorts. Overall, this study characterizes key molecular alterations in genome structure and expression in CHD8+/− neurons and links these changes to impaired neuronal and synaptic function.
Keywords: autism, ASD, CHD8, chromodomain, helicase, neurodevelopment, CRISPR, isogenic, chromatin, synaptic transmission
Graphical abstract
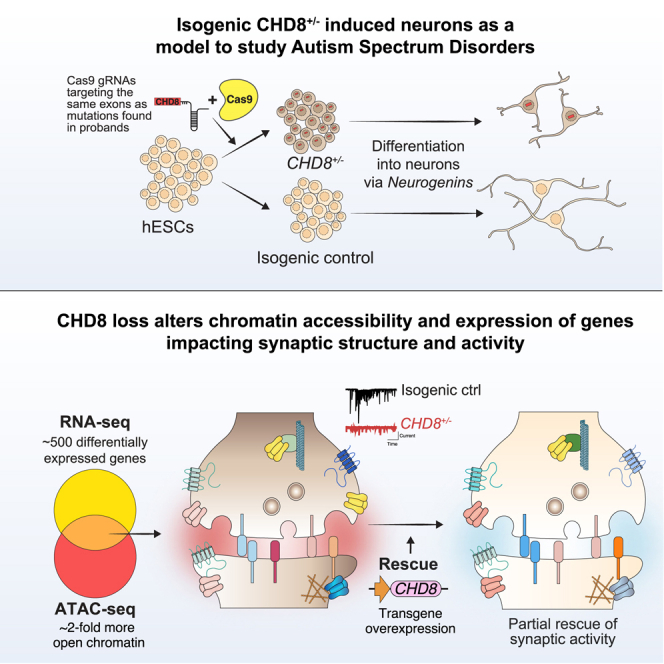
This study investigates the impact of CHD8 mutations on autism spectrum disorder (ASD). Using engineered heterozygous CHD8 human stem cells, we found alterations in gene expression, chromatin accessibility, and neuronal function. These findings provide insights into the molecular mechanisms underlying ASD and highlight potential therapeutic targets for the disorder.
Introduction
Autism spectrum disorder (ASD) is a common neurodevelopmental disorder—around 1 in 59 births in the United States—with significant phenotypic heterogeneity.1 ASD is characterized by a wide range of symptoms that include, but are not restricted to, impairments in social interaction, communication and language, repetitive patterns of behavior, anxiety, and sleeping and eating disorders. Despite this heterogeneity, there is a strong genetic basis to ASD incidence: recent studies estimate heritability at ∼80%.2 Whole-exome and whole-genome sequencing of ASD probands have identified ∼100 ASD risk genes carrying single de novo alterations.3,4 Together, rare de novo coding and copy-number variants contribute to 20%–30% of individuals with autism, and the most recurrently affected gene is chromodomain helicase DNA-binding protein 8 (CHD8).5
CHD8 was initially suggested as a likely causal factor in ASD when three unrelated probands with similar phenotypes were found to have large genomic deletions that always included the CHD8 locus.6 Recent targeted, whole-exome, and whole-genome sequencing studies have shown that de novo likely gene-disrupting mutations of CHD8 account for ∼0.4% of all ASD cases.3,7,8,9,10,11,12 Even though the loss of CHD8 function accounts for more cases of ASD than any other single de novo risk gene, our mechanistic understanding of how this gene drives ASD phenotypes is limited.
CHD8 is a broadly expressed ATP-dependent chromatin remodeler that regulates the expression of many genes.13 Individuals with CHD8 mutations have distinct phenotypes such as macrocephaly, hypertelorism, intellectual disability, sleep problems, and gastrointestinal issues.7,14 As a chromatin-remodeling enzyme, CHD8 most likely regulates multiple developmental pathways, which several groups have tried to dissect by using mouse models. Complete loss of Chd8 causes embryonic lethality in mice as a result of p53-mediated apoptosis.15 However, heterozygotes are fertile and have been reported by several groups.16,17,18,19 In all of these studies, Chd8+/− mice have increased brain volume, which is consistent with human CHD8 probands.14 However, few typical ASD-like behaviors are seen in Chd8+/− mice,16,20,21 making it challenging to design mouse models to capture the human disease phenotype. Recent studies profiled the transcriptional and some epigenetic changes driven by CHD8 loss in human neurons,22,23,24,25,26 yet only a few studies have reported neuronal phenotypes.27
In this study, we developed two isogenic CHD8+/− human embryonic stem cells (hESCs) that we differentiated into neurons (iNs) where we profiled CHD8-loss-driven changes in synaptic development and activity, chromatin accessibility, and gene expression. These iNs provide an in vitro cell model for studying ASD because they recapitulate many features of cortical excitatory neurons,28,29,30 a key cell type implicated in ASD by prior studies.31,32 Our work connects changes in chromatin accessibility in CHD8+/− neurons to differences in expression of genes that encode synaptic structural proteins and ion channels that regulate neuronal excitability. Furthermore, we find that this altered transcriptional state is associated with defects in synapse and network activity.
Many of the genes with changes in chromatin accessibility and/or expression overlap genes often mutated in ASD probands but not with genes mutated in three control cohorts (epilepsy, congenital heart disease, and unaffected individuals). With transgene rescue, we also show that these defects in neuronal function can be reversed if CHD8 levels are restored, highlighting a potential gene-therapy strategy for probands with CHD8 mutations. Using a combination of genome engineering, human neuron differentiation, multielectrode and high-resolution single-cell electrophysiology, high-throughput synaptic imaging, chromatin accessibility, gene expression, and population genetics, we present an integrative, deep characterization of this important de novo mutation in ASD.
Material and methods
hESC culture
HUES66 hESCs were maintained according to the feeder-free enhanced culture platform, which is more robust and less prone to spontaneous differentiation than the feeder-dependent standard culture platform and aids in culturing hESCs as single cells for selection after genome engineering.33,34,35 In brief, hESCs were cultured on LDEV-free Geltrex-coated plates (Thermo Fisher Scientific, A1413201) in mTeSR media (STEMCELL, 85850). Media were supplemented with 10 μM Y-27632 Rho-kinase inhibitor (Tocris, 1254) for 24 h after passaging and then removed.
Design of guide RNA and hESC transfection
We cloned a U6-driven single-guide RNA (sgRNA) cassette into the multicloning site of the pUC19 plasmid to yield the pU6 vector. We designed guide RNAs targeting CHD8 by using the Massachusetts Institute of Technology (MIT) CRISPR design tool and then cloned them individually into the pU6 vector.36 We co-transfected guide-RNA plasmids with EFS-Cas9-P2A-EGFP (Addgene, 63592) by using FuGENE HD (Promega, E2311). HUES66 cells were plated the day before transfection into 6-well dishes and transfected with 6 μg plasmid (5 μg Cas9 and 1 μg pU6, which is an approximately equal molar ratio) and 18 μL FuGENE HD. We diluted the plasmids into 300 μL OptiMEM (Thermo Fisher Scientific, 31985088) before adding the FugeneHD reagent. After vortexing and waiting 10 min, we added the mixture directly to the well.
Generation of iNs from hESCs
Constitutive rtTA3 expression driven by a human EF1α promoter and doxycycline-inducible human NEUROG2/1 expression driven by a TRE promoter were introduced into hESCs by lentiviral delivery as previously reported.33,37 A puromycin resistance gene was linked with NEUROG2/1 by a P2A linker (Figure 1A) for selecting cells expressing NEUROG2/1.
Figure 1.

Generation of two isogenic CHD8+/− hESCs via CRISPR editing
(A) ASD-associated mutations have been identified throughout the CHD8 coding sequence.
(B) Fluorescence-assisted cell-sorting strategy for isolating clonal isogenic hESCs with distinct heterozygous loss-of-function CHD8 mutations, termed CHD8-A and CHD8-B (resulting in premature stop codons at amino acid positions 1248 and 1247, respectively). These mutations are in CHD8 positions similar to the stop-gain mutations found in (A).
(C) CHD8+/− and an isogenic control hESCs retain pluripotency markers NANOG and OCT4 after genome engineering.
(D) qPCR in hESCs for CHD8+/− and isogenic control lines using primers that detect the indicated isoform or all isoforms of CHD8.
(E and F) CHD8 detection via western blot in CHD8+/− and isogenic control hESCs (E) and quantification (F).
All experiments were performed with three biological replicates. Statistical analyses were done with one-way ANOVA tests: ∗∗∗∗p < 10−4. Error bars indicate SEM.
Lentivirally transduced hESCs were plated on a Geltrex-coated 12-well Axion multielectrode array (MEA) plate (Axion Biosystems, Atlanta, GA, USA) at 1 × 104/well for MEA recording or on a 6-well cell-culture plate at 1 × 105/well for qPCR, immunocytochemistry (ICC), and patch-clamp recording. Geltrex-coated glass coverslips (Corning, 354087) were included in the 6-well cell-culture plate for ICC and patch-clamp recording. Doxycycline (2 μg/mL; Sigma-Aldrich, D3447) was applied for induction at day 0 of neuron differentiation. Puromycin (1 μg/mL) was applied from day 1 to day 4 for selection.
On days 1–3, we used a blended medium of mTeSR and neuron media (Neurobasal; Thermo Fisher Scientific, 21103049) supplemented with 1× B27 (Thermo Fisher Scientific, 17504044) to gently transition the induced neurons. Media were changed daily as follows: 75% mTeSR and 25% neuron media on day 1, 50% mTeSR and 50% neuron media on day 2, and 25% mTeSR and 75% neuron media on day 3. On day 4, we removed puromycin by exchanging the day 3 culture medium with 100% neuron media. Mouse glial cells were added on day 5 at 4 × 104 cells/well in one well of a 12-well MEA plate or at 4 × 105/well in one well of a 6-well cell-culture plate for ICC and patch-clamp recording. Primary mouse glial cells were cultured from the cortex of wild-type (WT) B6/C57 postnatal day 0 (P0) mice (Charles River, 027) and were subcultured for at least three passages before being used for co-culture experiments with iNs. On day 7, we added Ara-C (1 μM; Sigma-Aldrich, C1768) to control the growth of glia. From day 5 onward, we replaced 25% of the medium every 2–3 days to maintain the iN culture.
Chd8+/− mouse cortical neuron cell culture
Primary dissociated cortical neurons were prepared from P0 WT and Chd8+/−18 littermates according to a protocol similar to that described elsewhere.38 All of the animal work was conducted under the guidelines of the MIT Division of Comparative Medicine, and protocols (0414-024-17, 0414-027-17, and 0513-044-16) were approved by the MIT Committee for Animal Care, consistent with the 1996 Guide for Care and Use of Laboratory Animals from the National Research Council (institutional animal welfare assurance no. A-3125-01). In brief, cortical tissue was proteolyzed for 40 min at 37°C, triturated, and washed. The digestion solution consisted of dissection solution plus 20 U/mL papain (Worthington, 3126), 0.5 mM EDTA, 1.5 mM CaCl2, 1 mM l-cysteine, and 1 μg/mL DNase. For MEA recordings, dissociated neurons were plated onto substrate-embedded electrodes that had been pre-coated with 0.1% polyethyleneimine (Sigma) plus 20 μg/mL laminin (Thermo Fisher Scientific) at a density of 3 × 103 cells per mm2. For biochemical experiments, 3 × 105 cells were plated into one well of a 24-well plate that had been pre-coated with 20 μg/mL poly-d-lysine (Sigma) and 4 μg/mL laminin (Thermo Fisher Scientific). The cultures were treated with 1 μg/mL AraC (Sigma) on day 5 in vitro.
qPCR
RNA was extracted with the RNAeasy Mini Kit (Qiagen) and reverse transcribed with qScript cDNA SuperMix (Quanta). Transcript-specific primer pairs were designed with PrimerBLAST software (NCBI) using the following parameters: PCR product size = 90–200, exon junction span = “primer must span an exon-exon junction,” intron inclusion = “primer pair must be separated by at least one intron on the corresponding genomic DNA,” allow splice variants = “allow primer to amplify mRNA splice variants,” and max primer pairs to screen = 1,000. A version of PrimerBlast preconfigured with these design parameters is available at http://bit.ly/ns_qpcr. Primers were synthesized as standard desalted oligonucleotides (Integrated DNA Technologies), and qPCR was performed with Fast SYBR Green Master Mix (Thermo Fisher Scientific) according to the manufacturer’s protocol on a StepOnePlus qPCR thermocycler (Applied Biosystems).
Western blots
Samples for western blot were washed with ice-cold PBS and then lysed on ice in ice-cold cell-lysis buffer (Cell Signaling, 9803) with freshly added protease inhibitor cocktail (Sigma P8340). After scraping plates, we sonicated the samples for 5 min in a Bioruptor waterbath sonicator (Diagenode) with a 30 s on-off duty cycle. The soluble fraction of the lysate was isolated by centrifugation for 20 min at 13,000 rcf. We used the bicinchoninic acid assay to quantify protein concentrations (Thermo Fisher Scientific, 23225). For each blot, 20–100 μg protein was loaded onto a Bis-Tris precast gel (Thermo Fisher Scientific) in reducing conditions and run in MES buffer at 200 V. Proteins were transferred onto a nitrocellulose membrane at 60 V overnight at 4°C in a 20% methanol transfer buffer. The antibodies used included anti-CHD8 (1:1,000; Bethyl, A301-2214A), anti-FOXG1 (1:1,000; Abcam, ab18259), SV2 (1:1,000; DSHB), synaptophysin (SYP) (1:1,000; Invitrogen, 18-0130), PSD95 (1:1,000; Invitrogen, MA1-045), GRIA1 (GluR1) (1:1,000; Sigma, AB1504), and anti-GAPDH (1:5,000; Cell Signaling, 3683S). Membranes were developed by SuperSignal West Femto ECL (Thermo Fisher Scientific) and imaged with a BioRad ChemiDoc MP imaging system.
RNA sequencing and differential gene expression
RNA was extracted from iNs after 9 days of differentiation with the Quick-RNA Mini Kit (Zymo) and prepared for 3′ digital gene expression (3′-DGE) RNA sequencing (RNA-seq) with unique molecular identifiers as previously described.39 Sequencing was performed with 37-bp paired-end reads on a NextSeq (Illumina). Each sample was prepared with three independently differentiated biological replicates and analyzed with the DEseq2 package in RStudio.40
MEA electrophysiology
For multielectrode recordings from iNs, we used an Axion Maestro MEA system with 12-well MEA dishes (Axion Biosystems, M768-GLx). Each MEA well contained 64 gold microelectrodes (30 μm diameter) arranged over an 8 × 8 square grid with 200 μm center-to-center spacing in each well. The Maestro recording chamber was set to 35°C and supplied with a continuous perfusion of 5% carbon dioxide (with the balance air) (Airgas) throughout recording. All recordings were performed in neuron media and were started 5 min after the MEA plates had been placed on the recording chamber for a duration of 15 min.
Electrophysiology raw signals were acquired at 10 kHz and analyzed offline with Axion’s Integrated Studio software. Raw MEA data were filtered, and spikes were detected with a threshold-based detector as described previously.33 No spike sorting was performed. Timestamps of the spikes were collected and used for data analysis. In our analysis of the mean spike rate, we excluded electrodes that detected no spikes over the entire recording period. Spike raster plots were created in Neuroexplorer (Nex Technologies). We performed cross-correlation analyses in MATLAB (MathWorks) by comparing time-series data collected from different electrodes in the same MEA well.
Patch-clamp electrophysiology
For patch-clamp recordings, we used an inverted microscope (Zeiss Axiovert.A1) equipped with phase-contrast optics and a MultiClamp 700B Amplifier (Molecular Devices). We used a horizontal P-1000 pipette puller (Sutter Instrument) to pull borosilicate glass micropipettes. The extracellular solution (ECS) for patch-clamp recordings contained 145 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2 mM CaCl2, 5 mM HEPES, and 5 mM glucose, and the pH was adjusted to 7.4 with NaOH (all from Sigma). To isolate miniature synaptic events, we added 0.5 μM tetrodotoxin (Tocris, 1078) to the ECS.
We used current-clamp whole-cell configuration to record membrane potentials with pipettes filled with K-gluconate recording solution containing 131 mM K-gluconate, 17.5 mM KCl, 1 mM MgCl2, 10 mM HEPES, 1 mM EGTA, 2 mM MgATP, and 0.2 mM Na3GTP, and the pH was adjusted to 7.4 with KOH (all from Sigma). The same internal recording solution was also used for whole-cell recording of sodium-potassium currents in the voltage-clamp configuration. The ECS for recording Ca2+ current contained 130 mM NaCl, 10 mM BaCl2, 10 mM HEPES, and 0.5 mM μM tetrodotoxin (TTX) (for blocking Na+ currents), and the pH was adjusted to 7.3 with NaOH. The internal solution for recording Ca2+ current contained 126 mM CsCl, 10 mM EGTA, 1 mM EDTA, 10 mM HEPES, and 4 mM Mg-ATP, and the pH was adjusted to 7.3 with CsOH. Junction potentials (4–6 mV) were corrected for CsCl/NaCl solutions. Excitatory synaptic transmissions were recorded in the whole-cell patch-clamp mode with pipettes filled with K-gluconate recording solution as described above. Whole-cell voltage-clamp recordings were made at −70 mV in gap-free mode for at least 5 min for each cell, and access resistance was monitored throughout the recordings. All recordings were performed at room temperature (24°C–26°C). Data were digitized at 10 kHz with a 2 kHz low-pass filter (Molecular Devices). Data were analyzed offline with Clampfit 10.02 (Molecular Devices) and Mini Analysis Program (Synaptosoft).
ICC and image analysis
Cells were fixed by incubation with 4% paraformaldehyde and 4% sucrose in phosphate-buffered saline (PBS). Fixed cells were permeabilized with 0.1% Triton X-100 in PBS and stained with the following primary antibodies at 4°C: NANOG (1:500; Abcam, ab80892), OCT-3/4 (1:500; Santa Cruz, sc-5279), FOXG1 (1:1,000; Abcam, ab18259), MAP2 (1:500; Sigma, M1406; Synaptic Systems, 188004), NeuN (1:1,000; Millipore, MAB377), GAD2/65 (1:500; Sigma, AMAb91048), SV2 (1:1,000; DSHB), and SYP (1:1,000; Invitrogen, 18-0130). After the cells were washed three times with PBS, they were incubated with secondary antibody of goat anti-mouse, Alexa Fluor 555 (1:1,000; Thermo Fisher Scientific, A28180); goat anti-rabbit, Alexa Fluor 555 (1:1,000; Thermo Fisher Scientific, A21428); goat anti-mouse, Alexa Fluor 488 (1:1,000; Thermo Fisher Scientific, A32723); goat anti-rabbit, Alexa Fluor 488 (1:1,000; Thermo Fisher Scientific, A11034); or donkey anti-guinea pig, Alexa Fluor 647 (1:1,000; Jackson ImmunoResearch, 706-605-148). Nuclei were stained with 1 μg/mL DAPI (Sigma, D9542) for 15 min in the dark. The images were taken with an Axio Cam MRm (Zeiss) and a BZ-X800 automated microscope (Keyence).
We analyzed images with CellProfiler41 (https://cellprofiler.org/) by using custom-made pipelines and machine-learning-based classification with CellProfiler Analyst42 (https://cellprofileranalyst.org/). All image processing and analysis measurements were normalized to the WT controls. In brief, to count cells expressing the markers NeuN and GAD65, the image analysis pipeline identified the nuclei on the basis of DAPI signal and identified the neuronal body on the basis of GAD65 signal, allowing for classification of cells as positive or negative for the expression of these markers when using the machine-learning-based classifier function of CellProfiler Analyst. These analyses were done from images from three wells from three independent experiments. To measure changes in the cell area and neurite extension, the analysis pipeline uses the nucleus of the cell as a seed, which expands a defined number of pixels to the cytoplasm on the basis of the intensity of MAP2 signal. The analysis of the neurite length was done for >1,000 cells per condition.
To quantify changes in the density of pre- and post-synaptic markers in iNs, the pipeline identified puncta on the basis of their shape and higher intensity than background staining. These synaptic puncta were defined within a neurite territory determined by MAP2 staining. Next, the pipeline measured changes in the area and integrated intensity of puncta. For synaptic puncta in mouse neurons, we used an alternative quantification approach that uses changes in the integrated intensity. For each condition, 20 images containing a median of 30 cells each were analyzed per triplicate experiment.
CHD8 rescue and GFP mock lentiviruses
To create the CHD8 lentiviral transfer plasmid, we PCR amplified CHD8 from a WT HUES66 cDNA library by using primers specific to CHD8 isoform GenBank: NM_001170629 (transcript variant 1) and performed a Gibson ligation into lentiCas9-Blast (Addgene 52962) cut with AfeI and BamHI. The EGFP control plasmid was identical but included EGFP instead of CHD8. HUES66 WT and CHD8+/− cells were transduced with either a CHD8 or EGFP blasticidin-selectable lentivirus (pLenti-EFS-CHD8-P2A-Bla or pLenti-EFS-EGFP-P2A-Bla, respectively) as previously described.43 We performed two independent rescue experiments by using two independent batches of lentivirus (rescues 1 and 2, each with three biological replicate transductions).
ATAC-seq and analysis of differential open chromatin
We performed an assay of transposable and accessible chromatin sequencing (ATAC-seq) as previously described44 with an optimized lysis buffer to lower mitochondrial contamination.45 In brief, cell membranes were lysed in resuspension buffer (RSB) (10 mM Tris-HCL [pH 7.4], 3 mM MgCl2, and 10 mM NaCl) with freshly added 0.1% Tween-20. After pipetting up and down, we isolated nuclei by centrifuging at 500 rcf for 10 min at 4°C. After discarding the supernatant, we resuspended the nuclei in tagmentation DNA (TD) buffer with 2.5 μL Nextera enzyme (Illumina) and incubated them at 37°C for 30 min. After purification on a MinElute column (Qiagen), the purified, tagmented DNA was PCR amplified with Q5 enzyme (NEB) and barcoded primers for ten cycles (with an initial 72°C, 5 min step for Nextera end filling). The PCR product was purified via a 1.5× SPRI cleanup (Agencourt) and checked for a characteristic nucleosome banding pattern with a TapeStation (Agilent). Samples were sequenced with paired-end 37 bp reads on a NextSeq (Illumina). All three samples (WT, CHD8-A, and CHD8-B) were processed with two biological replicates that were separated cultured, tagmented, and analyzed.
The mitochondrial read rate was consistently low for all samples (<5%), as expected given our optimized lysis buffer.45 We used bowtie to align reads to the genome (hg19) and allowed for up to one mismatch. We removed duplicates (Picard MarkDuplicates) and mitochondrial reads (bowtie). All samples were downsampled to the same number of aligned reads per sample (14 M). We performed peak detection by using MACS2 software with the parameters “--nomodel --shift -100 --extsize 200.” After peak detection, we combined biological replicates by using bedtools merge. Unique peaks to each CHD8+/− cell line were identified with bedtools subtract. Peak-to-gene assignment was done with the bedtools closest function with parameter “-t first.”
To infer the overlap between ATAC-seq peaks and ENCODE3 candidate cis-regulatory elements (cCREs)46 (https://screen.encodeproject.org/), we used bedtools to intersect the concordant open-chromatin peaks with ENCODE cCREs (all human CREs [hg38], Registry V3, 2021). Then, using bedtools and bioMart, we mapped the peaks to their closest protein-coding genes from the UCSC Genome Browser (knowngene, hg38).47
Gene-set enrichment and analyses with whole-exome sequencing cohorts
We performed enrichment analyses by using WebGestalt (Web-Based Gene Set Analysis Toolkit).48 Specifically, we analyzed the set of 153 genes (RNA and ATAC) by using over-representation analysis against the Gene Ontology (GO)49,50 and Reactome databases.51 The background set for these analyses included all genes in the genome with false-discovery rate (FDR) correction. For the genes with changes in open chromatin and expression, we tested for enrichment of missense and likely gene-disrupting mutations in various disease (and healthy) patient cohorts.8 We performed all enrichment analyses by using the Genotypes and Phenotypes in Families (GPF) software: https://gpf.sfari.org/. This database includes de novo variants in unaffected children and children diagnosed with autism, epilepsy, intellectual disability, schizophrenia, developmental disorders, and congenital heart disease.
Results
Derivation of isogenic CHD8 heterozygous hESC lines by CRISPR mutagenesis
Over a decade ago, Zahir et al. found that a submicroscopic deletion in 14q11.2 led to serious neurodevelopmental symptoms and suggested that CHD8 was most likely the causal gene.6 Using whole-exome sequencing of probands in the Simons Simplex Collection, several groups have shown that CHD8 is the most recurrently mutated gene in de novo ASD.3,8,9,11,12 These studies found individuals with likely gene-disrupting mutations throughout CHD8 (Figure 1A). To characterize the effects of CHD8 haploinsufficiency in human excitatory cortical neurons, we designed a CRISPR-Cas9 sgRNA that targets near residue 1210. This sgRNA targets the same exon that harbors multiple de novo loss-of-function mutations in probands and is estimated to create loss-of-function alleles for 88% of repair outcomes.10,11,52 We co-transfected an EFS-Cas9-2A-EGFP plasmid and a U6-driven guide-RNA plasmid into HUES66 hESCs.53,54 Cells expressing GFP were purified by fluorescence-activated cell sorting and collected for single-cell-derived colony isolation (Figure 1B). Using Sanger sequencing, we isolated two colonies with premature stop codons. We termed these colonies CHD8-A, which had a 13-nt heterozygous deletion (resulting in p.Arg1210Leufs∗39 [GenBank: NP_001164100] on the modified allele), and CHD8-B, which had a 16-nt heterozygous deletion (resulting in p.Cys1208Aspfs∗40 [GenBank: NP_001164100] on the modified allele). The chosen guide RNA was optimized to have a minimal chance of off-target modification such that the closest off-target site had three mismatches, including one in the critical seed region for Cas9 binding, which makes any off-target activity highly unlikely (Figures S1A and S1B). To verify the lack of off-target genome modification, we sequenced the top predicted off-targets in both of the engineered lines and found no editing at these loci (Figure S1C). Consistent with the lethality reported for Chd8 biallelic knockout in mice,15 we were able to recover only cells with monoallelic premature stop codons and no clones with biallelic premature stop codons (n = 216 colonies sequenced).
The pluripotency of CHD8-A and CHD8-B hESCs after genome editing was confirmed by ICC staining for the pluripotency markers NANOG and OCT3/4 (Figure 1C). We performed qRT-PCR for CHD8 mRNA and found that both isoforms of CHD8 (GenBank: NM_001170629.2 and NM_020920.4) were reduced to 40%–45% of WT levels (Figure 1D). Using western blotting, we found a similar decrease in CHD8 protein in CHD8-A and CHD8-B, as expected given the genotype and mRNA expression (Figure 1E). After confirming the successful engineering of CHD8 heterozygous isogenic hESCs, we sought to derive mature neurons for further molecular and functional studies.
Loss of CHD8 leads to widespread changes in neuronal gene expression
Previously, we showed that overexpression of NEUROG2 and NEUROG1 (NEUROG2/1) in hESCs rapidly produces functional induced neurons, and we used these neurons to measure electrophysiological changes after the loss of a voltage-gated sodium channel.33,37 To investigate the effects of CHD8 loss on neuronal function, we introduced doxycycline-inducible expression of NEUROG2/1 into CHD8-A, CHD8-B, and WT control HUES66 hESCs and used the same differentiation method to convert them to NEUROG2/1-induced neurons (Figure 2A). 4 days after doxycycline induction, all cells from both CHD8+/− lines and from the isogenic control line expressed the pan-neuronal marker MAP2 and FOXG1, a key driver of forebrain development55 (Figure 2B), and were identified as neurons on the basis of the expression of the neuron-specific nuclear protein NeuN (Figures S2A and S2B). Across all genotypes, inhibitory neurons (GAD65 positive) constituted between 10% and 15% of the cell population (Figure S2C). This proportion of inhibitory cells remained unchanged in the CHD8+/− mutant cell lines. We also observed that CHD8 loss resulted in smaller cells, indicated by a reduction of the cellular area (MAP2) and nuclear area (DAPI) (Figures S2D and S2E). The nuclear area and cell area tend to be correlated in eukaryotic cells independently of growth conditions or exposure to physical environments.56,57,58 In addition, CHD8 mutants presented shorter neurites, defined by MAP2 extension, than did WT cells; this phenotype was apparent after 2 days of differentiation and became more significant with time (Figures S2F and S2G). After 1 week of differentiation, western blot analysis showed that CHD8-A and CHD8-B lines continued to express lower levels of CHD8 than did the isogenic control (Figure 2C).
Figure 2.

Differentiation of isogenic CHD8+/− neurons and RNA-seq of neurons at post-differentiation day 9
(A) Cortical neuron differentiation via overexpression of NEUROG2 and NEUROG1.
(B) ICC of FOXG1 and MAP2 after 4 days of differentiation.
(C) Western blot of CHD8 and FOXG1 and quantification of CHD8 after 4 days of differentiation.
(D) Quantification of gene expression in CHD8-A and CHD8-B isogenic cell lines after 9 days of differentiation.
(E) Volcano plot of differential gene expression between CHD8+/− neurons and isogenic control neurons.
(F) qPCR of several differentially expressed genes between CHD8+/− and isogenic control neurons (and also NEUROG2).
(G) Top five enriched GO processes in CHD8+/− neurons according to differential gene expression after 9 days of differentiation.
All experiments were performed with three biological replicates. Statistical analyses were done with one-way ANOVA tests: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 10−4. Error bars indicates SEM.
Given that CHD8 is a chromatin-remodeling protein, we hypothesized that global changes in CHD8 expression might lead to altered gene expression in NEUROG2/1 neurons. We used a 3′-DGE RNA-seq protocol with unique molecular identifiers (UMIs) to prepare sequencing libraries from CHD8-A, CHD8-B, and the isogenic control after 9 days of neuronal differentiation.39 These UMIs served to minimize any PCR bias during library amplification given that duplicate reads bearing the same UMI were eliminated from our analysis. To avoid amplifying genes from mouse glial cells, we differentiated NEUROG2/1 neurons in the absence of astrocyte co-culture for RNA-seq. Overall, both isogenic CHD8+/− cell lines produced highly correlated transcriptomes (Figure 2D). By modeling the count data by using a negative binomial distribution, we identified 475 genes with increased expression and 694 genes with decreased expression in CHD8+/− neurons (FDR q value < 0.1 for 15,574 genes detected) (Figure 2E; Table S1). For several genes with significant changes in gene expression, we performed an independent differentiation and validated the changes in their gene expression relative to that of the isogenic control by qRT-PCR (Figure 2F). We selected a handful of genes with the highest differential expression compared with that of isogenic control neurons and found increased expression in CHD8+/− neurons for the transcription initiation factor TAF9B and the long noncoding RNA PAX8-AS1. We found decreased expression in Rho GTPase-activating protein 29 (ARHGAP29) and alpha-2-macroglobulin pseudogene 1 (A2MP1). To further reinforce that these differences are due to CHD8 loss and not differential integration of Neurogenin lentivirus, we note that we found no change in NEUROG2 expression between CHD8+/− neurons and isogenic control neurons.
Given the hundreds of genes with significant changes in expression, we performed a GO49,50 analysis on differentially expressed genes and found that the top two enriched molecular processes involved synapse function: chemical synapse transduction and trans-synaptic signaling (Figure 2G). Enriched GO components included both presynaptic (axonal) and postsynaptic (dendritic) proteins and categories, suggesting possible widespread changes in synapse structure or function. Other significantly enriched GO terms for differentially expressed genes included focal adhesion, the cell cycle, and ubiquitin proteasome pathways, in agreement with other reports of CHD8 knockdown or heterozygous neurons.24,26 We next sought to understand how these alterations in expression of synapse-related genes might affect synaptic transmission between CHD8 heterozygous neurons.
CHD8 heterozygous neurons have lower spontaneous firing rates as a result of fewer functional synapses
Despite the strong enrichment of differential expression of synapse-associated genes, it is difficult to predict changes in neuronal spiking and synapse function on the basis of gene expression alone. Toward this end, we also differentiated CHD8-A, CHD8-B, and the parental (isogenic) control neurons on 64-channel MEAs and examined their spontaneous firing activities with repeated measurements over a post-induction period of 60 days. Consistent with our previous reports of WT stem cells,33,37 these directly differentiated neurons formed synapses and exhibited action potentials starting at approximately 2 weeks. We monitored network activity every 4–6 days from day 15 onward. Both CHD8-A and CHD8-B neurons had lower firing activity than the controls at all timepoints (Figures 3A and 3B). To examine changes in synchronized firing, we computed the correlation coefficient between all 64 electrodes in each dish. Between day 20 and day 25, the increase in synchronized firing activity seen in isogenic controls was not found in CHD8+/− neurons (Figure 3C). Although CHD8+/− neurons achieved synchronized firing similar to that of isogenic controls by day 60, there was a significant delay in synchronized activity in CHD8+/− neurons (days 20–25: isogenic control, p < 0.0001, t = 20.82, df = 1,178; CHD8-A, p = 0.19; CHD8-B, p = 0.17). The delayed development of synchronized firing suggests that CHD8 loss could result in a synaptic deficit.
Figure 3.

Characterization of electrophysiological and synaptic changes in human isogenic CHD8+/− neurons
(A) Example of field potential recording using MEAs.
(B) Average spike rate of human cortical neurons recorded every 5 days from post-differentiation day 15 to day 60.
(C) Average correlation coefficient between all pairs of electrodes. ∗Significant differences at a single time point. †Significant differences for a single cell line between neighboring time points.
(D) Voltage-clamp recordings of sEPSCs and quantification of sEPSC amplitude and frequency.
(E) Voltage-clamp recordings of mEPSCs using TTX and quantification of mEPSC amplitude and frequency.
(F) ICC for MAP2, SYP, and SV2 and quantification of the fold change over the isogenic control cell line in SYP and SV2 puncta area per unit dendrite (MAP2) area.
(G) Protein expression via western blot for both presynaptic proteins SYP and SV2 and postsynaptic proteins PSD95 and GRIA1 (GluR1) (top) and quantification relative to GAPDH (bottom) after 32 days of differentiation.
All experiments were performed with three biological replicates. Statistical analyses were done with one-way ANOVA tests: ∗∗p < 0.01, ∗∗∗p < 10−3, ∗∗∗∗p < 10−4. Error bars indicate SEM.
To test this directly, we performed high-resolution, whole-cell patch-clamp electrophysiology on post-differentiation day 30 and measured spontaneous excitatory postsynaptic currents (sEPSCs). We found that the amplitude of sEPSCs remained unchanged between CHD8+/− neurons and isogenic controls (Figure 3D). However, the frequency of spontaneous EPSCs recorded from CHD8+/− neurons was significantly lower than that recorded from isogenic controls (2.4 ± 0.71 hz in CHD8-A, 2.1 ± 0.40 hz in CHD8-B, 5.9 ± 0.86 hz in controls; CHD8-A: p = 0.004, t = 3.03, df = 51; CHD8-B: p < 0.0002, t = 3.99, df = 56). To identify the cause of the reduced sEPSCs, we measured the membrane properties of CHD8+/− induced neurons, such as resting membrane potential, membrane capacitance, neuronal excitability, and voltage-dependent Na+ and K+ currents (Figures S3A–S3I). Other than reduced membrane capacitance, which is in agreement with our earlier finding of smaller cells, no other significant differences were found between CHD8 mutant and control human neurons (Figure S3D). Given that most membrane properties were unaltered, we hypothesized that the change in sEPSCs might be due to a change in functional synapses.
To test this, we measured miniature excitatory postsynaptic currents (mEPSCs) by blocking voltage-gated sodium channels with TTX. We found that both lines of CHD8+/− neurons (CHD8-A and CHD8-B) had a more than 2-fold decrease in the frequency of mESPCs without altering the mEPSC amplitude (Figure 3E). Because the change in the frequency of mESPCs suggests an alteration in the number of functional synapses, we performed ICC to detect two presynaptic vesicle proteins, SV2 and SYP (synaptophysin), in dendrites delimited by the somato-dendritic cytoskeletal marker MAP2 (Figure 3F). High-content microscopy images showed an apparent reduction in the overall intensity of both presynaptic markers, particularly at focal points (synapses). Using automated image-analysis pipelines, we measured the area occupied by the focal accumulation of SV2 and SYP as a surrogate readout for functional synapses. The analysis revealed a 60%–80% decrease in SV2 area and a 40%–50% decrease in SYP area, supporting the reduction in functional synapses in CHD8+/− neurons. To understand whether the reduction in SV2 and SYP dendritic area resulted from changes in their expression levels, we performed western blotting (Figure 3G). We found significant (30%–40%) reductions in SV2 area in CHD8+/− neurons and smaller, non-significant (8%–19%) reductions in SYP area. To explore whether these changes were specific to the presynaptic compartment, we examined the expression levels of two postsynaptic proteins (GRIA1 [GluR1] and DLG4 [PSD95]), which were not affected by CHD8 loss.
Cortical neurons cultured from Chd8 heterozygous mice have similar alterations in firing rates and synapse density
To test whether the synaptic deficits and impaired synchronization of the neuronal network are specific to human neurons or whether they might be general cross-species features triggered by haploinsufficiency of CHD8, we also cultured postnatal cortical neurons from Chd8+/− mice and WT littermate controls.18 After culturing these murine neurons on MEAs, we found a similar reduction in the overall firing rate at day in vitro (DIV) 21 (2.9 ± 0.45 Hz in Chd8+/− mice; 4.6 ± 0.69 Hz in WT littermate controls) (Figures 4A and 4B). We performed ICC on these mouse neuron cultures at DIV 14 and 21 for SYP, PSD95, and MAP2 (Figure 4C). Given that changes in the dendritic area occupied by synaptic markers were already apparent from their intensity in human induced neurons, we used a simplified pipeline to measure changes in the intensity of synaptic markers in dendritic areas defined by MAP2. At DIV 21, we observed a 54% decrease in SYP and a 33% decrease in PSD95 in CHD8+/− neurons (Figures 4D and 4E). To confirm that the reduced spike rate and synaptic density were not due to a difference in the number of neurons, we measured neuronal density by using NeuN staining and found no change in neuronal density between Chd8+/− neuron cultures and WT controls (Figure 4F). Similar to hESC-derived human cortical neurons, we found large changes in neuron firing rates and in synaptic puncta in Chd8+/− mouse neurons. Altogether, our analysis of hESC-derived human cortical neurons and culture mouse neurons identified common deficits in synaptic transmission caused by CHD8 haploinsufficiency.
Figure 4.

Multielectrode array recordings and synaptic puncta in cortical neurons from Chd8 heterozygous mice
(A) Spike raster plot from mouse cortical neurons at DIV 14 and 21.
(B) Spike rate for cortical neurons recorded at DIV 14 and 21.
(C) ICC for MAP2, SYP, and PSD95 at DIV 21.
(D) Integrated PSD95 intensity per unit dendrite (MAP2) area.
(E) Integrated SYP intensity per unit dendrite (MAP2) area.
(F) Neuron density per unit area (mm2) by NeuN identification of neuronal nuclei.
All experiments were performed with three biological replicates. Statistical analyses were done with unpaired t tests: ∗p < 0.05, ∗∗p < 0.01. Error bars indicate SEM.
CHD8 overexpression in CHD8 heterozygous neurons rescues firing rate and synaptic defects
Given the observed defects in neuronal firing rates in both human and mouse CHD8+/− neurons, we further tested whether restoration of CHD8 expression via a transgene could rescue these defects. Using a blasticidin-selectable lentiviral vector, we introduced CHD8 under the control of the elongation factor 1a-short (EFS) promoter into one of the CHD8+/− hESCs (Figure 5A). To control for effects of lentiviral integration, we transduced the same lentivirus but with EGFP substituted for the CHD8 open reading frame into the same CHD8+/− hESC line and the isogenic control line (Figure 5A). After blasticidin selection, we isolated two clones from the polyclonal population and differentiated these into neurons as before via Neurogenin overexpression.
Figure 5.

Lentiviral rescue of CHD8 in human isogenic cells
(A) Lentiviral constructs for rescue experiments delivering CHD8 or EGFP coupled to a blasticidin selection gene. The transgenes are ubiquitously expressed by the EFS promoter.
(B) Transduction of CHD8-B cells with the CHD8 rescue lentivirus restores WT CHD8 levels.
(C) Spike rates from multielectrode array recording of CHD8-B cells with CHD8 rescue lentivirus in comparison with those of WT or CHD8-B cells with mock transduction (mean ± SD).
(D) ICC for MAP2, SYP, and SV2 at day 53 in WT cells with mock transduction, CHD8-B cells with mock transduction, and CHD8-B cells with CHD8 rescue lentivirus.
(E and F) Integrated SYP (E) and SV2 (F) intensity per unit dendrite (MAP2) area normalized to WT cells with mock transduction (mean + SD).
All experiments were performed with three biological replicates. Statistical analyses were done with one-way ANOVA tests: ∗∗p < 0.01, ∗∗∗p < 10−3, ∗∗∗∗p < 10−4. Error bars indicate SEM unless otherwise stated.
By western blot, we found that both CHD8 rescue lines displayed higher levels of CHD8 than did neurons from the mock (GFP-transduced) parental CHD8-B line and the isogenic control line (Figure 5B). On post-differentiation day 60, we found that both CHD8 rescue lines had nearly the same firing rate as the mock-transduced isogenic control (Figure 5C). In contrast, the mock-transduced CHD8 heterozygous line displayed an approximately 80% reduction in firing, similar to what we had observed before in CHD8+/− human and mouse neurons (Figures 3B, 4B, and 5C). We also performed ICC on CHD8 rescue and mock-transduced lines to examine the expression levels of SYP, SV2, and MAP2 (Figure 5D). Overexpression of CHD8 was able to rescue SYP expression in both lines (Figure 5E) and SV2 expression in one of the lines (Figure 5F). Supporting these findings, the work from Durak et al.26 overexpressed a shRNA-resistant human CHD8 construct, which reduced cell-cycle exit and restored patterns of neuronal differentiation and cortical layering in mouse brains. These results hold promise for the genetic rescue of synaptic deficits and altered firing caused by CHD8 loss but also highlight the need for further development of strategies for optimal control of CHD8 expression. Specifically, our approach relies on constitutive CHD8 expression; however, we know from in vivo studies that CHD8 is dynamically regulated across development and in different brain regions,16,17,18 and thus it is likely that precise control over CHD8 expression will be important for future therapeutic strategies.
CHD8 loss increases open chromatin in introns and intergenic regions
Given the prominent role in chromatin remodeling of CHD8 and its previously established interactions with the linker histone H1,59 we tested whether CHD8+/− hESCs and neurons have alterations in open and closed regions of chromatin with ATAC-seq.44 Using paired-end sequencing, we mapped transposon insertions, which tend to accumulate in genomic regions with more open chromatin and greater accessibility to transcription factor binding. In all samples, we found that the ATAC-seq fragments and sequenced libraries captured histone nucleosome spacing (∼146 bp) with clear mono-, di-, and multinucleosome bands (Figures S4A and S4B).
To compare open-chromatin regions, we initially binned all reads into 100-kb windows spanning the entire genome. We found a high degree of overlap between ATAC-seq biological replicates (Figure 6A). Specifically, in day 9 neurons, biological replicates of each cell line (CHD8-A, CHD8-B, and control) clustered together, and both CHD8+/− lines had greater similarity with each other than with the isogenic control line. One striking difference was that the CHD8+/− neurons had approximately 2-fold more open-chromatin peaks than isogenic controls (Figure 6B). Because peak calling can be influenced by read depth, we were careful to perform peak calling on the same number of aligned reads for each sample. To better understand where this increase in open chromatin was occurring across the genome, we mapped each ATAC-seq peak in all three lines to the nearest gene and ranked genes by differential ATAC-seq peaks. There was a good correlation between the genes that gained ATAC-seq peaks in CHD8-A and CHD8-B heterozygous neurons (Figure 6C; Table S2) (r = 0.70, p = 2 × 10−16). Autism susceptibility candidate 2 (AUTS2) had the largest change in ATAC-seq peaks after CHD8 loss: AUTS2 had 27 more peaks in CHD8-A neurons and 26 more peaks in CHD8-B neurons than in the isogenic controls. AUTS2 is a key subunit of the Polycomb PRC1 complex and is itself a chromatin modifier that is involved in regulating the expression of genes important for neural development (Figure 6D).60 Within AUTS2, we found large changes in open chromatin within the introns of the gene (Figure 6E). Previous work has shown that open-chromatin regions associate with more transcriptionally active genes.44 Concordant with the change in open chromatin, we found a nearly 2-fold increase in expression of AUTS2 in CHD8+/− hESCs (Table S1). Although we identified AUTS2 via its large gain in ATAC-seq peaks, other studies have shown that AUTS2 has an important neurodevelopmental function and acts as a master regulator of genes implicated in ASD-related pathways.61
Figure 6.

Chromatin accessibility of isogenic CHD8+/− neurons and comparison with genetic data from whole-exome sequencing of probands
(A) Clustering of aligned reads from ATAC-seq using 100-kb genomic windows. The analysis was done with two biological replicates per sample.
(B) MACS2-detected ATAC-seq peaks in human neurons at post-differentiation day 9 in CHD8+/− and isogenic controls.
(C) Mapping of novel peaks in CHD8+/− neurons to nearest gene for CHD8-A and CHD8-B lines.
(D) Schematic of PRC-AUTS2 complex.
(E) Examples of regions within AUTS2 with additional open-chromatin peaks.
(F) Fold change in ATAC-seq peaks across different types of genomic regions in CHD8+/− and isogenic control neurons.
(G) Changes in gene expression (upregulation or downregulation) for genes with gains in ATAC-seq peaks in CHD8+/− neurons.
(H) Venn diagram showcasing the common differentially expressed genes (padj < 0.05) and mapped novel ATAC-seq peaks in CHD8-A and CHD8-B cell lines.
(I) Top five enriched GO processes in CHD8+/− neurons of common genes identified in (H).
(J) Cohort analysis of whole-exome data shows that genes with significant changes in gene expression (RNA-seq) or chromatin compaction (ATAC-seq) are enriched with nonsense and/or missense mutations in ASD (n = 3,963 exomes), intellectual disability (n = 151), or schizophrenia (n = 964) cohorts but not in heart disease (n = 1,229), epilepsy (n = 264), or unaffected control (n = 2,303) cohorts.
Error bars indicate SEM.
Given these increases in intronic open chromatin at this locus and several other genes (Figure 6C), we wanted to more precisely map altered chromatin accessibility genome-wide. For this purpose, we examined increases in ATAC-seq peaks in different genomic regions, including the coding sequences (CDSs), 5′ untranslated regions (UTRs), 3′ UTRs, transcription start sites (TSSs), introns, and intergenic regions. In all categories, we found significantly more open chromatin in CHD8+/− neurons than in isogenic control neurons (Figure 6F). Of these genomic regions, we saw the greatest changes in open chromatin in CHD8+/− neurons in intronic and intergenic regions (2.7-fold ± 0.04 for introns and 3.4-fold ± 0.05 for intergenic regions in CHD8-A). Our analysis extends the regulatory landscape defined by CHD8 beyond TSSs, as previously identified by chromatin immunoprecipitation and sequencing (ChIP-seq).62 As with AUTS2, we compared changes in open chromatin with our earlier measurements of differential gene expression. We found that genes that are significantly upregulated in CHD8+/− neurons have, on average, 1.8-fold more ATAC-seq peaks than downregulated genes (n = 603 genes downregulated with ATAC-seq peaks; m = 424 genes upregulated with ATAC-seq peaks) (Figure 6G). This asymmetry was present when we restricted the ATAC-seq data to novel peaks (1.5-fold more peaks in upregulated genes) and restricted the ATAC-seq data to novel peaks found in gene bodies or promoters (1.1-fold more peaks in upregulated genes).
Then, we compared differentially expressed genes (padj < 0.05) with the genes closest to differentially accessible genomic regions from the ATAC-seq in both CHD8+/− mutant lines and isogenic controls. We identified 153 genes with both differential expression and changes in chromatin accessibility in CHD8+/− mutant lines (Figure 6H). Using pathway and GO analyses, we found an enrichment in genes that regulate neurite growth and axon development, synapse organization, vesicle-mediated transport at the synapse, axon guidance, and cell-cell adhesion pathways (Figure S4C).
Given the large increase in open chromatin in intron and intergenic regions, we hypothesized that changes in open chromatin might affect not only promoter accessibility but also the accessibility of cis-regulatory elements (CREs). We tested this hypothesis by quantifying the overlap between ATAC-seq peaks in CHD8 mutant neurons and ENCODE CREs.46 We found that 39% of ATAC-seq peaks gained in CHD8-A neurons and 35% of ATAC-seq peaks gained in CHD8-B neurons overlapped ENCODE CREs (Figure S4D). These ATAC-seq peaks overlapped both distal enhancer-like elements (dELS) and proximal enhancer-like elements (pELS). We mapped these ATAC-seq peaks overlapping dELS and pELS to their closest protein-coding gene and examined whether any of the mapped genes were differentially expressed in CHD8 mutants according to our RNA-seq data. We found that these dELS and pELS peaks were often nearby differentially expressed genes in CHD8+/− neurons, such as DNM3, and were enriched in biological processes related to glutamatergic synaptic transmission and the cell cycle (Figure S4E; Table S3). We found cell-cycle-related enriched GO categories; a recent CHD8 study from Ding et al. found small effects on the cell cycle and proliferation upon CHD8 loss63 and a similar enrichment in related gene expression.
Changes in chromatin compaction and gene expression specifically target genes that are often mutated in ASD and intellectual disability
Given that CHD8 was first identified through human sequencing studies of ASD probands, we tested whether the genes with significant changes in either expression or open chromatin in CHD8+/− neurons were also enriched with germline mutations associated with ASD and similar disorders. To quantify this, we analyzed whole-exome sequencing data from unaffected individuals and those with ASD, intellectual disability, schizophrenia, epilepsy, and heart disease. In total, we analyzed whole-exome data from 8,874 individuals, including 3,963 ASD probands. Genes with significant changes in expression (either an increase or decrease, q < 0.01) were over-represented in likely gene-disrupting mutations in ASD, intellectual disability, and schizophrenia probands (ASD: observed [O] = 18, expected [E] = 11.08, p = 0.05; intellectual disability: O = 1, E = 0.05, p = 0.04; schizophrenia: O = 6, E = 2.25, p = 0.03, χ² test) (Figure 6J). In contrast, we found no enrichment for mutations in these genes among three control cohorts: two with diseases of fundamentally different etiology (epilepsy and congenital heart disease) and the unaffected individuals.
We also sought to understand whether ASD probands were enriched with mutations in genes with altered chromatin accessibility in CHD8+/− neurons. When we examined genes with significant increases in the number of nearby ATAC-seq peaks (p < 0.05), we found that this gene set was over-represented in ASD and intellectual disability cohorts in both likely gene-disrupting and missense mutations but not in any of the other cohorts tested (ASD: O = 31, E = 13.47, p = 0.0008; intellectual disability: O = 2, E = 0.13, p = 0.006, χ² test) (Figure 6J). Together, these data suggest that heterozygous CHD8 loss causes widespread alterations in open chromatin and gene expression and that the genes affected by these changes are often disrupted in neurodevelopmental disorders but not in other disorders, such as heart disease or epilepsy, or in unaffected controls.
Discussion
Although CHD8 loss accounts for more cases of ASD than any other single de novo risk gene, there are few well-characterized models of how CHD8 is involved in neuronal function. Previous studies have focused on ASD-like behaviors in mouse models but have not examined the molecular function of CHD8 as a chromatin-remodeling enzyme.16,17,18 In this work, we engineered isogenic CHD8+/− human pluripotent stem cells by using Cas9 modification of normal embryonic stem cells, and using a rapid neuron-differentiation protocol, we derived human neurons carrying this mutation. The two isoforms of CHD8 (isoforms GenBank: NM_001170629 and NM_020920) are produced by alternative splicing and differ in length. GenBank: NM_020920 is shorter because it includes part of an intron containing an in-frame stop codon.15 Importantly, mutations in both isoforms correlate with increased risk of ASD.7,17,64,65 The CHD8+/− cell lines generated in our study show equal depletion of both isoforms, yielding accurate models of the monoallelic loss of both isoforms observed in ASD probands. Loss of CHD8 expression did not affect the differentiation of hESCs into neurons. However, the CHD8+/− mutants showed reduced cellular size and neurite length, suggesting that these neurons are less mature than the isogenic controls. Supporting this observation, using field recordings and single-cell patch-clamp electrophysiology, we found that CHD8+/− cortical neurons exhibited lower firing rates and a lower frequency of both miniature and spontaneous EPSCs. We also observed a decrease in the local accumulation of synaptic markers in dendrites for presynaptic SV2 and SYP, which could be associated with, but not exclusive of, a significant downregulation of their protein levels (i.e., SV2) in CHD8+/− neurons. These changes in firing rates and synaptic puncta were also seen in a heterozygous Chd8 mouse model.
One limitation of this study is that cultured neurons do not replicate the intricate three-dimensional organization and cellular complexity found in the developing brain.66 Other model systems, such as human brain organoids and nonhuman primates, grant access to unique cell types and developmental processes,67 as highlighted in several recent studies of CHD868,69 and our own work with brain organoids.70 Despite these limitations, this study provides a set of molecular and functional approaches for the robust characterization of ASD phenotypes in neuronal cells grown in monolayer culture, as well as a suitable framework for high-throughput characterization of ASD mutations.
Given CHD8’s role in chromatin remodeling,71 we measured changes in chromatin accessibility genome-wide. Gene regulation is highly dependent on both accessible chromatin, which allows for transcription factor binding and gene expression, and compacted chromatin, which is less accessible for transcription. Compared with matched isogenic controls, CHD8+/− neurons have sweeping large increases in open chromatin across the entire genome, including in intronic and intergenic regions. CHD8 has been previously found to bind these regions in studies using ChIP in neuronal and non-neuronal cell lines.72,73 In agreement, we found that more than a third of newly gained open-chromatin peaks in CHD8+/− neurons coincide with dELS situated in intergenic regions, as well as pELS located within introns. Notably, these regions with increased chromatin accessibility are located near differentially expressed genes in the CHD8 lines, suggesting that these changes in chromatin accessibility might drive altered gene expression (although further mechanistic work will need to demonstrate a causal relation). In total, we identified 153 genes with altered expression and chromatin accessibility across the CHD8 mutant lines. These genes were enriched in neurite and axon growth and development and synaptic organization pathways.
One of the enriched pathways (L1CAM) regulates the interactions between adhesion molecules and transmembrane receptors with the actin and microtubule cytoskeleton.74 L1CAM genes are essential for the control of neurite outgrowth and are associated with neurodevelopmental disorders, including autism.74,75 Several genes related to the L1CAM pathway, including DNM3, display increases in chromatin accessibility and expression in CHD8+/− cells. DNM3 is a pivotal microtubule-associated GTPase responsible for endocytosis and crucial for sustained neurotransmission.76 Previous studies have reported DNM3 upregulation in neuronal models of various psychiatric disorders, such as schizophrenia,77 but its exploration in autism has been lacking.
Another gene with significant changes in both open-chromatin peaks and gene expression in CHD8 mutants is KCNQ2, which encodes a voltage-dependent potassium channel with a key role in neuronal excitability.78 These channels are predominantly expressed at the axon initial segment and nodes of Ranvier and at lower densities at the soma, dendrites, and synaptic terminals of neurons. KCNQ2 has been identified as an ASD risk gene in whole-exome sequencing studies,79 and mutations in KCNQ2 cause neonatal epilepsy, characterized by severe early-onset seizures and impaired neurodevelopment.80 Depletion of KCNQ2 results in abnormal neuronal activity and reduced sEPSC frequency in induced pluripotent stem cells (iPSCs) differentiated to neurons.27 We found that KCNQ2 is significantly downregulated in CHD8 mutant neurons (among the top 20 most downregulated genes). Moreover, disruptive mutations in the highly conserved coiled-coil domain of KCNQ2, essential for protein interactions, lead to decreased neurite length (according to MAP2 staining) and reduced presynaptic puncta in neurons derived from the iPSCs of epileptic neonates, which were compared with an isogenic mutation-corrected control.81 Given the observed changes in neuronal excitability, our findings could nominate KCNQ2 as a primary mediator of the electrophysiological phenotypes that we observed upon CHD8 loss.
Of the 153 genes with changes in RNA expression and chromatin accessibility, CNTNAP2 had a substantial increase in open chromatin in both CHD8+/− mutant lines. CNTNAP2, which encodes the cell-adhesion glycoprotein Caspr2, is itself an autism risk gene and governs crucial aspects of neuronal development, such as differentiation, neurite growth, branching, and synapse formation.82,83 Loss-of-function mutations in CNTNAP2 lead to synaptic defects.83,84,85 Contactins, such as Caspr2, interact with ankyrins, which are also dysregulated in ASD86 and are required for the proper localization and clustering of voltage-gated channels (e.g., KCNQ2) and receptors at synapses.87 Together, our multiomic molecular datasets (RNA-seq and ATAC-seq) identify changes in both the structural proteins required for proper synapse formation and the channels that regulate neuronal excitability.
Remarkably, many of these differentially regulated genes with mapped CREs are associated with synaptic structure or activity, providing a mechanistic basis for the altered synaptic activity that we observed via electrophysiology and imaging. In addition to CRE-driven changes, differential splicing due to accessibility changes in introns might also play a role, as shown in a recent study.72
Using large-scale exome sequencing datasets, we found that these genes with altered expression are enriched with mutations found in probands with ASD, intellectual disability, and schizophrenia but not in unaffected controls or probands with cardiovascular disease or epilepsy. Furthermore, enrichment in ASD-risk genes regulated by CHD8 has been reported in brains from developing mice24 and in human neuronal progenitor cells.25
In order to assess whether CHD8-loss-driven changes in genome structure and gene expression are reversible, we delivered a CHD8 expression construct into CHD8+/− human pluripotent stem cells. Upon neuron differentiation, we found that decreases in firing activity and presynaptic proteins could be partially rescued by the CHD8 transgene. Similarly, ectopic overexpression of human CHD8 in Chd8 knockdown mice restored neuronal differentiation defects caused by CHD8 loss, including replenishing the depleted progenitor pool.26 There are several important caveats of our gene-overexpression approach, such as uncontrolled dosage of the EFS-driven CHD8 transcript, which could lead to supraphysiological levels of CHD8 and off-target binding. However, despite these caveats, our study suggests that restoring CHD8 by using gene-therapy approaches, including gene augmentation, has the potential to rescue WT neuronal function and might offer a viable treatment for CHD8 loss. An important next step will be to see whether restoring CHD8 after differentiation and/or in mature neurons can also provide a similar level of phenotypic rescue in human neurons.
Our approach also demonstrates the utility of isogenic models using genome engineering in human pluripotent stem cells and differentiated neurons. Compared with patient-derived iPSCs, the isogenic model can be produced efficiently in any lab without the need for patient access. For rare monogenic disorders, this kind of genetic model provides a more controlled (isogenic) comparison, which is important in phenotyping assays where there is greater biological noise, such as organoid cultures. These studies provide an initial view of the molecular, electrophysiological, and morphological consequences of loss-of-function mutations in CHD8 in human and mouse neurons and implicate CHD8 target genes in ASD and other neurological disorders. Our demonstration that CHD8+/− neurons display widespread changes in genome organization and synapse function opens new avenues for future work and therapeutic interventions.
Data and code availability
RNA-seq and ATAC-seq are available in the NCBI Gene Expression Omnibus under accession number GEO: GSE236994.
Acknowledgments
We thank the entire Sanjana, Pan, and Zhang laboratories for support and advice. We also thank Andrew Allen, Kaitlin Samocha, Ben Neale, Steve Hyman, and Ed Scolnick for helpful discussions. We thank Derek Peters and Chad Cowan for assistance with stem cell culture; C. Jimmie Ye, Rachel Gate, and Christine Cheng for assistance with ATAC-seq; Tarjei Mikkelson and Magali Soumillon for assistance with RNA-seq; and John Morris and Zoran Gajic for assistance with CRE analysis. SV2 monoclonal antibodies developed by K. Buckley were obtained from the Developmental Studies Hybridoma Bank, created by the National Institutes of Health (NIH) National Institute of Child Health and Human Development and maintained at the University of Iowa. A.C. is supported by the Swedish Research Council (VR). I.I. is supported by the Simons Foundation Autism Research Initiative (SFARI 529232). N.E.S. is supported by New York University and New York Genome Center startup funds, the Simons Foundation Autism Research Initiative (Genomics of ASD 896724), NIH National Human Genome Research Institute (R00HG008171 and DP2HG010099), NIH National Cancer Institute (R01CA279135), NIH National Institute of Allergy and Infectious Diseases (R01AI176601), and Brain & Behavior Foundation.
Author contributions
F.Z. and N.E.S. conceived of the study. F.Z., J.Q.P., and N.E.S. supervised the work. X.S., C.L., and N.E.S. designed the experiments. X.S., C.L., A.C., A.N., Y.Z., R.J.P., and N.E.S. performed the experiments and analyzed results. X.S., C.L., A.C., and N.E.S. wrote the manuscript with input from all authors.
Declaration of interests
F.Z. is a scientific advisor to and cofounder of Editas Medicine, Beam Therapeutics, Pairwise Plants, Arbor Biotechnologies, Proof Diagnostics, and Aera Therapeutics. F.Z. is a scientific advisor to Octant. N.E.S. is a scientific advisor to Qiagen and is a scientific advisor to and cofounder of OverT Bio.
Published: October 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2023.09.004.
Contributor Information
Jen Q. Pan, Email: jpan@broadinstitute.org.
Neville E. Sanjana, Email: neville@sanjanalab.org.
Supplemental information
References
- 1.Baio J., Wiggins L., Christensen D.L., Maenner M.J., Daniels J., Warren Z., Kurzius-Spencer M., Zahorodny W., Robinson Rosenberg C., White T., et al. Prevalence of autism spectrum disorder among children aged 8 years - autism and developmental disabilities monitoring network, 11 sites, United States, 2014. MMWR. Surveill. Summ. 2018;67:1–23. doi: 10.15585/mmwr.ss6706a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sandin S., Lichtenstein P., Kuja-Halkola R., Hultman C., Larsson H., Reichenberg A. The heritability of autism spectrum disorder. JAMA. 2017;318:1182–1184. doi: 10.1001/jama.2017.12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iossifov I., O’Roak B.J., Sanders S.J., Ronemus M., Krumm N., Levy D., Stessman H.A., Witherspoon K.T., Vives L., Patterson K.E., et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders S.J., He X., Willsey A.J., Ercan-Sencicek A.G., Samocha K.E., Cicek A.E., Murtha M.T., Bal V.H., Bishop S.L., Dong S., et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Turner T.N., Coe B.P., Dickel D.E., Hoekzema K., Nelson B.J., Zody M.C., Kronenberg Z.N., Hormozdiari F., Raja A., Pennacchio L.A., et al. Genomic patterns of de novo mutation in simplex autism. Cell. 2017;171:710–722.e12. doi: 10.1016/j.cell.2017.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zahir F., Firth H.V., Baross A., Delaney A.D., Eydoux P., Gibson W.T., Langlois S., Martin H., Willatt L., Marra M.A., Friedman J.M. Novel deletions of 14q11.2 associated with developmental delay, cognitive impairment and similar minor anomalies in three children. J. Med. Genet. 2007;44:556–561. doi: 10.1136/jmg.2007.050823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bernier R., Golzio C., Xiong B., Stessman H.A., Coe B.P., Penn O., Witherspoon K., Gerdts J., Baker C., Vulto-van Silfhout A.T., et al. Disruptive CHD8 mutations define a subtype of autism early in development. Cell. 2014;158:263–276. doi: 10.1016/j.cell.2014.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iossifov I., Ronemus M., Levy D., Wang Z., Hakker I., Rosenbaum J., Yamrom B., Lee Y.-H., Narzisi G., Leotta A., et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neale B.M., Kou Y., Liu L., Ma’ayan A., Samocha K.E., Sabo A., Lin C.-F., Stevens C., Wang L.-S., Makarov V., et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485:242–245. doi: 10.1038/nature11011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Roak B.J., Vives L., Fu W., Egertson J.D., Stanaway I.B., Phelps I.G., Carvill G., Kumar A., Lee C., Ankenman K., et al. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338:1619–1622. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Roak B.J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B.P., Levy R., Ko A., Lee C., Smith J.D., et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485:246–250. doi: 10.1038/nature10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanders S.J., Murtha M.T., Gupta A.R., Murdoch J.D., Raubeson M.J., Willsey A.J., Ercan-Sencicek A.G., DiLullo N.M., Parikshak N.N., Stein J.L., et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012;485:237–241. doi: 10.1038/nature10945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson B.A., Tremblay V., Lin G., Bochar D.A. CHD8 is an ATP-dependent chromatin remodeling factor that regulates beta-catenin target genes. Mol. Cell Biol. 2008;28:3894–3904. doi: 10.1128/MCB.00322-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yasin H., Gibson W.T., Langlois S., Stowe R.M., Tsang E.S., Lee L., Poon J., Tran G., Tyson C., Wong C.K., et al. A distinct neurodevelopmental syndrome with intellectual disability, autism spectrum disorder, characteristic facies, and macrocephaly is caused by defects in CHD8. J. Hum. Genet. 2019;64:271–280. doi: 10.1038/s10038-019-0561-0. [DOI] [PubMed] [Google Scholar]
- 15.Nishiyama M., Oshikawa K., Tsukada Y.i., Nakagawa T., Iemura S.i., Natsume T., Fan Y., Kikuchi A., Skoultchi A.I., Nakayama K.I. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 2009;11:172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gompers A.L., Su-Feher L., Ellegood J., Copping N.A., Riyadh M.A., Stradleigh T.W., Pride M.C., Schaffler M.D., Wade A.A., Catta-Preta R., et al. Germline Chd8 haploinsufficiency alters brain development in mouse. Nat. Neurosci. 2017;20:1062–1073. doi: 10.1038/nn.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katayama Y., Nishiyama M., Shoji H., Ohkawa Y., Kawamura A., Sato T., Suyama M., Takumi T., Miyakawa T., Nakayama K.I. CHD8 haploinsufficiency results in autistic-like phenotypes in mice. Nature. 2016;537:675–679. doi: 10.1038/nature19357. [DOI] [PubMed] [Google Scholar]
- 18.Platt R.J., Zhou Y., Slaymaker I.M., Shetty A.S., Weisbach N.R., Kim J.-A., Sharma J., Desai M., Sood S., Kempton H.R., et al. Chd8 mutation leads to autistic-like behaviors and impaired striatal circuits. Cell Rep. 2017;19:335–350. doi: 10.1016/j.celrep.2017.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Suetterlin P., Hurley S., Mohan C., Riegman K.L.H., Pagani M., Caruso A., Ellegood J., Galbusera A., Crespo-Enriquez I., Michetti C., et al. Altered neocortical gene expression, brain overgrowth and functional over-connectivity in Chd8 haploinsufficient mice. Cereb. Cortex. 2018;28:2192–2206. doi: 10.1093/cercor/bhy058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu Q., Liu Y.Y., Wang X., Tan G.H., Li H.P., Hulbert S.W., Li C.Y., Hu C.C., Xiong Z.Q., Xu X., Jiang Y.H. Autism-associated CHD8 deficiency impairs axon development and migration of cortical neurons. Mol. Autism. 2018;9:65. doi: 10.1186/s13229-018-0244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jung H., Park H., Choi Y., Kang H., Lee E., Kweon H., Roh J.D., Ellegood J., Choi W., Kang J., et al. Sexually dimorphic behavior, neuronal activity, and gene expression in Chd8-mutant mice. Nat. Neurosci. 2018;21:1218–1228. doi: 10.1038/s41593-018-0208-z. [DOI] [PubMed] [Google Scholar]
- 22.Sugathan A., Biagioli M., Golzio C., Erdin S., Blumenthal I., Manavalan P., Ragavendran A., Brand H., Lucente D., Miles J., et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. USA. 2014;111:E4468–E4477. doi: 10.1073/pnas.1405266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang P., Lin M., Pedrosa E., Hrabovsky A., Zhang Z., Guo W., Lachman H.M., Zheng D. CRISPR/Cas9-mediated heterozygous knockout of the autism gene CHD8 and characterization of its transcriptional networks in neurodevelopment. Mol. Autism. 2015;6:55. doi: 10.1186/s13229-015-0048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cotney J., Muhle R.A., Sanders S.J., Liu L., Willsey A.J., Niu W., Liu W., Klei L., Lei J., Yin J., et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nat. Commun. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilkinson B., Grepo N., Thompson B.L., Kim J., Wang K., Evgrafov O.V., Lu W., Knowles J.A., Campbell D.B. The autism-associated gene chromodomain helicase DNA-binding protein 8 (CHD8) regulates noncoding RNAs and autism-related genes. Transl. Psychiatry. 2015;5:e568. doi: 10.1038/tp.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durak O., Gao F., Kaeser-Woo Y.J., Rueda R., Martorell A.J., Nott A., Liu C.Y., Watson L.A., Tsai L.-H. Chd8 mediates cortical neurogenesis via transcriptional regulation of cell cycle and Wnt signaling. Nat. Neurosci. 2016;19:1477–1488. doi: 10.1038/nn.4400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deneault E., White S.H., Rodrigues D.C., Ross P.J., Faheem M., Zaslavsky K., Wang Z., Alexandrova R., Pellecchia G., Wei W., et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Rep. 2018;11:1211–1225. doi: 10.1016/j.stemcr.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu C., Shi X., Allen A., Baez-Nieto D., Nikish A., Sanjana N.E., Pan J.Q. Overexpression of NEUROG2 and NEUROG1 in human embryonic stem cells produces a network of excitatory and inhibitory neurons. FASEB J. 2019;33:5287–5299. doi: 10.1096/fj.201801110rr. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nehme R., Zuccaro E., Ghosh S.D., Li C., Sherwood J.L., Pietilainen O., Barrett L.E., Limone F., Worringer K.A., Kommineni S., et al. Combining NGN2 programming with developmental patterning generates human excitatory neurons with NMDAR-mediated synaptic transmission. Cell Rep. 2018;23:2509–2523. doi: 10.1016/j.celrep.2018.04.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y., Pak C., Han Y., Ahlenius H., Zhang Z., Chanda S., Marro S., Patzke C., Acuna C., Covy J., et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron. 2013;78:785–798. doi: 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Voineagu I., Wang X., Johnston P., Lowe J.K., Tian Y., Horvath S., Mill J., Cantor R.M., Blencowe B.J., Geschwind D.H. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Velmeshev D., Schirmer L., Jung D., Haeussler M., Perez Y., Mayer S., Bhaduri A., Goyal N., Rowitch D.H., Kriegstein A.R. Single-cell genomics identifies cell type–specific molecular changes in autism. Science. 2019;364:685–689. doi: 10.1126/science.aav8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu C., Shi X., Allen A., Baez-Nieto D., Nikish A., Sanjana N.E., Pan J.Q. Overexpression of NEUROG2 and NEUROG1 in human embryonic stem cells produces a network of excitatory and inhibitory neurons. FASEB J. 2019;33:5287–5299. doi: 10.1096/fj.201801110RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peters D.T., Cowan C.A., Musunuru K. StemBook. Harvard Stem Cell Institute; 2013. Genome editing in human pluripotent stem cells. [PubMed] [Google Scholar]
- 35.Schinzel R.T., Ahfeldt T., Lau F.H., Lee Y.-K., Cowley A., Shen T., Peters D., Lum D.H., Cowan C.A. Efficient culturing and genetic manipulation of human pluripotent stem cells. PLoS One. 2011;6 doi: 10.1371/journal.pone.0027495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O., et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Busskamp V., Lewis N.E., Guye P., Ng A.H.M., Shipman S.L., Byrne S.M., Sanjana N.E., Murn J., Li Y., Li S., et al. Rapid neurogenesis through transcriptional activation in human stem cells. Mol. Syst. Biol. 2014;10:760. doi: 10.15252/msb.20145508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu C., Chen Q., Zhou T., Bozic D., Fu Z., Pan J.Q., Feng G. Micro-electrode array recordings reveal reductions in both excitation and inhibition in cultured cortical neuron networks lacking Shank3. Mol. Psychiatry. 2016;21:159–168. doi: 10.1038/mp.2015.173. [DOI] [PubMed] [Google Scholar]
- 39.Soumillon M., Cacchiarelli D., Semrau S., Oudenaarden A. van, Mikkelsen T.S. Characterization of directed differentiation by high-throughput single-cell RNA-seq. bioRxiv. 2014 doi: 10.1101/003236. Preprint at. [DOI] [Google Scholar]
- 40.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpenter A.E., Jones T.R., Lamprecht M.R., Clarke C., Kang I.H., Friman O., Guertin D.A., Chang J.H., Lindquist R.A., Moffat J., et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol. 2006;7:R100. doi: 10.1186/gb-2006-7-10-r100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stirling D.R., Carpenter A.E., Cimini B.A. CellProfiler Analyst 3.0: accessible data exploration and machine learning for image analysis. Bioinformatics. 2021;37:3992–3994. doi: 10.1093/bioinformatics/btab634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanjana N.E., Wright J., Zheng K., Shalem O., Fontanillas P., Joung J., Cheng C., Regev A., Zhang F. High-resolution interrogation of functional elements in the noncoding genome. Science. 2016;353:1545–1549. doi: 10.1126/science.aaf7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rickner H.D., Niu S.-Y., Cheng C.S. ATAC-seq assay with low mitochondrial DNA contamination from primary human CD4+ T lymphocytes. J. Vis. Exp. 2019 doi: 10.3791/59120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.ENCODE Project Consortium. Moore J.E., Purcaro M.J., Pratt H.E., Epstein C.B., Shoresh N., Adrian J., Kawli T., Davis C.A., Dobin A., et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature. 2020;583:699–710. doi: 10.1038/s41586-020-2493-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S., et al. GENCODE: the reference human genome annotation for the ENCODE Project. Genome Res. 2012;22:1760–1774. doi: 10.1101/gr.135350.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J., Vasaikar S., Shi Z., Greer M., Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. doi: 10.1093/nar/gkx356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gene Ontology Consortium. Aleksander S.A., Balhoff J., Carbon S., Cherry J.M., Drabkin H.J., Ebert D., Feuermann M., Gaudet P., Harris N.L., et al. The Gene Ontology knowledgebase in 2023. Genetics. 2023;224 doi: 10.1093/genetics/iyad031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fabregat A., Sidiropoulos K., Viteri G., Forner O., Marin-Garcia P., Arnau V., D’Eustachio P., Stein L., Hermjakob H. Reactome pathway analysis: a high-performance in-memory approach. BMC Bioinf. 2017;18:142. doi: 10.1186/s12859-017-1559-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M., et al. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018;37:64–72. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bock C., Kiskinis E., Verstappen G., Gu H., Boulting G., Smith Z.D., Ziller M., Croft G.F., Amoroso M.W., Oakley D.H., et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cowan C.A., Klimanskaya I., McMahon J., Atienza J., Witmyer J., Zucker J.P., Wang S., Morton C.C., McMahon A.P., Powers D., Melton D.A. Derivation of embryonic stem-cell lines from human blastocysts. N. Engl. J. Med. 2004;350:1353–1356. doi: 10.1056/NEJMsr040330. [DOI] [PubMed] [Google Scholar]
- 55.Xuan S., Baptista C.A., Balas G., Tao W., Soares V.C., Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]
- 56.Neumann F.R., Nurse P. Nuclear size control in fission yeast. J. Cell Biol. 2007;179:593–600. doi: 10.1083/jcb.200708054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huber M.D., Gerace L. The size-wise nucleus: nuclear volume control in eukaryotes. J. Cell Biol. 2007;179:583–584. doi: 10.1083/jcb.200710156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim D.-H., Li B., Si F., Phillip J.M., Wirtz D., Sun S.X. Volume regulation and shape bifurcation in the cell nucleus. J. Cell Sci. 2015;128:3375–3385. doi: 10.1242/jcs.166330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nishiyama M., Skoultchi A.I., Nakayama K.I. Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol. Cell Biol. 2012;32:501–512. doi: 10.1128/MCB.06409-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gao Z., Lee P., Stafford J.M., von Schimmelmann M., Schaefer A., Reinberg D. An AUTS2-Polycomb complex activates gene expression in the CNS. Nature. 2014;516:349–354. doi: 10.1038/nature13921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oksenberg N., Haliburton G.D.E., Eckalbar W.L., Oren I., Nishizaki S., Murphy K., Pollard K.S., Birnbaum R.Y., Ahituv N. Genome-wide distribution of Auts2 binding localizes with active neurodevelopmental genes. Transl. Psychiatry. 2014;4:e431. doi: 10.1038/tp.2014.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sugathan A., Biagioli M., Golzio C., Erdin S., Blumenthal I., Manavalan P., Ragavendran A., Brand H., Lucente D., Miles J., et al. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc. Natl. Acad. Sci. 2014;111:E4468–E4477. doi: 10.1073/pnas.1405266111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding S., Lan X., Meng Y., Yan C., Li M., Li X., Chen J., Jiang W. CHD8 safeguards early neuroectoderm differentiation in human ESCs and protects from apoptosis during neurogenesis. Cell Death Dis. 2021;12:981. doi: 10.1038/s41419-021-04292-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Talkowski M.E., Rosenfeld J.A., Blumenthal I., Pillalamarri V., Chiang C., Heilbut A., Ernst C., Hanscom C., Rossin E., Lindgren A.M., et al. Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell. 2012;149:525–537. doi: 10.1016/j.cell.2012.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blumenthal I., Ragavendran A., Erdin S., Klei L., Sugathan A., Guide J.R., Manavalan P., Zhou J.Q., Wheeler V.C., Levin J.Z., et al. Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am. J. Hum. Genet. 2014;94:870–883. doi: 10.1016/j.ajhg.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lancaster M.A., Renner M., Martin C.-A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim J., Koo B.-K., Knoblich J.A. Human organoids: model systems for human biology and medicine. Nat. Rev. Mol. Cell Biol. 2020;21:571–584. doi: 10.1038/s41580-020-0259-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li C., Fleck J.S., Martins-Costa C., Burkard T.R., Stuempflen M., Vertesy Á., Peer A.M., Esk C., Elling U., Kasprian G., et al. Single-cell brain organoid screening identifies developmental defects in autism. bioRxiv. 2022 doi: 10.1101/2022.09.15.508118. Preprint at. [DOI] [Google Scholar]
- 69.Li B., Zhao H., Tu Z., Yang W., Han R., Wang L., Luo X., Pan M., Chen X., Zhang J., et al. CHD8 mutations increase gliogenesis to enlarge brain size in the nonhuman primate. Cell Discov. 2023;9:27. doi: 10.1038/s41421-023-00525-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Paulsen B., Velasco S., Kedaigle A.J., Pigoni M., Quadrato G., Deo A.J., Adiconis X., Uzquiano A., Sartore R., Yang S.M., et al. Autism genes converge on asynchronous development of shared neuron classes. Nature. 2022;602:268–273. doi: 10.1038/s41586-021-04358-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nishiyama M., Oshikawa K., Tsukada Y.i., Nakagawa T., Iemura S.i., Natsume T., Fan Y., Kikuchi A., Skoultchi A.I., Nakayama K.I. CHD8 suppresses p53-mediated apoptosis through histone H1 recruitment during early embryogenesis. Nat. Cell Biol. 2009;11:172–182. doi: 10.1038/ncb1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kerschbamer E., Arnoldi M., Tripathi T., Pellegrini M., Maturi S., Erdin S., Salviato E., Di Leva F., Sebestyén E., Dassi E., et al. CHD8 suppression impacts on histone H3 lysine 36 trimethylation and alters RNA alternative splicing. Nucleic Acids Res. 2022;50:12809–12828. doi: 10.1093/nar/gkac1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kita Y., Katayama Y., Shiraishi T., Oka T., Sato T., Suyama M., Ohkawa Y., Miyata K., Oike Y., Shirane M., et al. The autism-related protein CHD8 cooperates with C/EBPβ to regulate adipogenesis. Cell Rep. 2018;23:1988–2000. doi: 10.1016/j.celrep.2018.04.050. [DOI] [PubMed] [Google Scholar]
- 74.Maness P.F., Schachner M. Neural recognition molecules of the immunoglobulin superfamily: signaling transducers of axon guidance and neuronal migration. Nat. Neurosci. 2007;10:19–26. doi: 10.1038/nn1827. [DOI] [PubMed] [Google Scholar]
- 75.An J.Y., Cristino A.S., Zhao Q., Edson J., Williams S.M., Ravine D., Wray J., Marshall V.M., Hunt A., Whitehouse A.J.O., Claudianos C. Towards a molecular characterization of autism spectrum disorders: an exome sequencing and systems approach. Transl. Psychiatry. 2014;4:e394. doi: 10.1038/tp.2014.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fan F., Funk L., Lou X. Dynamin 1- and 3-mediated endocytosis is essential for the development of a large central synapse in vivo. J. Neurosci. 2016;36:6097–6115. doi: 10.1523/JNEUROSCI.3804-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Costas J., Carrera N., Alonso P., Gurriarán X., Segalàs C., Real E., López-Solà C., Mas S., Gassó P., Domènech L., et al. Exon-focused genome-wide association study of obsessive-compulsive disorder and shared polygenic risk with schizophrenia. Transl. Psychiatry. 2016;6:e768. doi: 10.1038/tp.2016.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Devaux J.J., Kleopa K.A., Cooper E.C., Scherer S.S. KCNQ2 is a nodal K+ channel. J. Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.C Yuen R.K., Merico D., Bookman M., L Howe J., Thiruvahindrapuram B., Patel R.V., Whitney J., Deflaux N., Bingham J., Wang Z., et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 2017;20:602–611. doi: 10.1038/nn.4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh N.A., Charlier C., Stauffer D., DuPont B.R., Leach R.J., Melis R., Ronen G.M., Bjerre I., Quattlebaum T., Murphy J.V., et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 81.Simkin D., Marshall K.A., Vanoye C.G., Desai R.R., Bustos B.I., Piyevsky B.N., Ortega J.A., Forrest M., Robertson G.L., Penzes P., et al. Dyshomeostatic modulation of Ca2+-activated K+ channels in a human neuronal model of KCNQ2 encephalopathy. Elife. 2021;10 doi: 10.7554/eLife.64434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rodenas-Cuadrado P., Ho J., Vernes S.C. Shining a light on CNTNAP2: complex functions to complex disorders. Eur. J. Hum. Genet. 2014;22:171–178. doi: 10.1038/ejhg.2013.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jang W.E., Park J.H., Park G., Bang G., Na C.H., Kim J.Y., Kim K.-Y., Kim K.P., Shin C.Y., An J.-Y., et al. Cntnap2-dependent molecular networks in autism spectrum disorder revealed through an integrative multi-omics analysis. Mol. Psychiatry. 2023;28:810–821. doi: 10.1038/s41380-022-01822-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peñagarikano O., Abrahams B.S., Herman E.I., Winden K.D., Gdalyahu A., Dong H., Sonnenblick L.I., Gruver R., Almajano J., Bragin A., et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Canali G., Garcia M., Hivert B., Pinatel D., Goullancourt A., Oguievetskaia K., Saint-Martin M., Girault J.-A., Faivre-Sarrailh C., Goutebroze L. Genetic variants in autism-related CNTNAP2 impair axonal growth of cortical neurons. Hum. Mol. Genet. 2018;27:1941–1954. doi: 10.1093/hmg/ddy102. [DOI] [PubMed] [Google Scholar]
- 86.Yoon S., Piguel N.H., Penzes P. Roles and mechanisms of ankyrin-G in neuropsychiatric disorders. Exp. Mol. Med. 2022;54:867–877. doi: 10.1038/s12276-022-00798-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lai H.C., Jan L.Y. The distribution and targeting of neuronal voltage-gated ion channels. Nat. Rev. Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ATAC-seq are available in the NCBI Gene Expression Omnibus under accession number GEO: GSE236994.