

Abstract
Genome haploidization at meiosis depends on two consecutive nuclear divisions, which are controlled by an oscillatory system consisting of Cdk1‐cyclin B and the APC/C bound to the Cdc20 activator. How the oscillator generates exactly two divisions has been unclear. We have studied this question in yeast where exit from meiosis involves accumulation of the APC/C activator Ama1 at meiosis II. We show that inactivation of the meiosis I‐specific protein Spo13/MEIKIN results in a single‐division meiosis due to premature activation of APC/CAma1. In the wild type, Spo13 bound to the polo‐like kinase Cdc5 prevents Ama1 synthesis at meiosis I by stabilizing the translational repressor Rim4. In addition, Cdc5‐Spo13 inhibits the activity of Ama1 by converting the B‐type cyclin Clb1 from a substrate to an inhibitor of Ama1. Cdc20‐dependent degradation of Spo13 at anaphase I unleashes a feedback loop that increases Ama1's synthesis and activity, leading to irreversible exit from meiosis at the second division. Thus, by repressing the exit machinery at meiosis I, Cdc5‐Spo13 ensures that cells undergo two divisions to produce haploid gametes.
Keywords: cyclin phosphorylation, meiotic exit, polo‐like kinase, Spo13/MEIKIN, translational repression
Subject Categories: Cell Cycle
Polo‐like kinase Cdc5 and yeast Spo13 synergize in repressing the cell division exit machinery to prevent meiosis with only a single division.

Introduction
Meiosis consists of two consecutive nuclear divisions whereby chromosomes segregate reductionally at meiosis I and equationally at meiosis II. Reductional segregation depends on recombination, which forms bivalent chromosomes by generating crossovers between homologous, maternal, and paternal chromosomes (Petronczki et al, 2003). In addition, sister kinetochores fuse their microtubule‐binding interfaces so that maternal and paternal centromeres are pulled to opposite spindle poles. In yeast, this process, called sister kinetochore mono‐orientation, requires recruitment of the monopolin complex to kinetochores (Toth et al, 2000; Rabitsch et al, 2003; Petronczki et al, 2006). At meiosis I, dyad chromosomes segregate because the separase protease cleaves cohesin on chromosome arms (Klein et al, 1999; Buonomo et al, 2000), while centromeric cohesin is protected from cleavage by a complex consisting of shugoshin (Sgo1 in yeast) and the phosphatase PP2A (Riedel et al, 2006; Katis et al, 2010). Segregation of chromatids at meiosis II requires the removal of monopolin and Sgo1‐PP2A from centromeres, so that sister kinetochores orient towards opposite spindle poles and separase gains access to centromeric cohesin.
The meiosis I‐specific behaviour of centromeres depends on a group of weakly conserved proteins represented by Spo13 in budding yeast, Moa1 in fission yeast, and MEIKIN in mammals (Nasmyth, 2015). These proteins appear intrinsically disordered, are present only at meiosis I, and bind to polo‐like kinase (PLK1, Cdc5 in yeast). Indeed, they are thought to promote sister kinetochore mono‐orientation and centromeric cohesin protection by recruiting Cdc5/PLK1 to centromeres (Kim et al, 2015; Galander et al, 2019; Ma et al, 2021). However, centromere‐related functions cannot account for a striking aspect of the spo13Δ phenotype. While mutants lacking monopolin and the ability to protect centromeric cohesin undergo two divisions (Toth et al, 2000; Rabitsch et al, 2003), spo13Δ cells exit from meiosis after a single division (Klapholz & Esposito, 1980b; Shonn et al, 2002). Similarly, spermatocytes from Meikin −/− mice undergo only one division (Kim et al, 2015). Thus, a hitherto unexplained aspect of the spo13Δ and the Meikin −/− phenotype is premature exit from meiosis, indicating that Spo13/MEIKIN controls processes beyond the centromere. Indeed, the finding that spo13Δ cells undergo nuclear division in the absence of the APC/C activator Cdc20 implies a dramatic change in cell cycle regulation (Katis et al, 2004).
Meiosis is characterized by a period of low Cdk1 activity at prophase followed by two consecutive waves of Cdk1 activity during nuclear divisions. Unravelling how this activity pattern is generated and how it accommodates checkpoint‐dependent delays is crucial to our understanding of meiosis. Cdk1‐cyclin B and APC/CCdc20 constitute an oscillatory system capable of generating successive M phases (Sha et al, 2003; Tyson & Novak, 2008). Cdk1 bound to B‐type cyclins (Clbs in yeast) induces spindle formation and is required for the ubiquitin‐ligase activity of APC/CCdc20. Once activated, APC/CCdc20 induces chromosome segregation by mediating the degradation of the separase‐inhibitor Pds1/securin and cyclin B. As cyclin B levels decline, the activity of APC/CCdc20 decreases, which allows re‐accumulation of Pds1 and cyclin B, resulting in entry into meiosis II. The Cdk1‐APC/CCdc20 oscillator is modulated by checkpoint controls. For instance, in the absence of tension exerted by bipolar spindle forces, kinetochores inhibit APC/CCdc20 and thereby prolong metaphase through a signalling cascade, known as the spindle assembly checkpoint (SAC; Musacchio, 2015). To ensure a two‐division meiosis, SAC activity at metaphase I has to inhibit not only entry into anaphase I but also processes specific to meiosis II, although mRNAs encoding meiosis II‐specific proteins are already present at meiosis I (Brar et al, 2012).
In budding yeast, two meiosis‐specific proteins, the transcription factor Ndt80 and the APC/C activator Ama1, are crucial for starting and stopping the oscillator (Chu & Herskowitz, 1998; Cooper et al, 2000). During prophase, the recombination checkpoint represses Ndt80 and thereby the synthesis of M phase regulators (Chu & Herskowitz, 1998; Hepworth et al, 1998). In addition, low levels of APC/CAma1 activity render M phase regulators unstable (Okaz et al, 2012). Once recombination is complete, Ndt80 produces Clbs and Cdc20, the key components of the oscillator (Chu et al, 1998). As cells enter meiosis II, Ama1 accumulates to high levels, which suppresses the Cdk1‐APC/CCdc20 oscillator due to degradation of M phase regulators, including Ndt80. Similar mechanisms operate in fission yeast: The transcription factor Mei4 produces M phase regulators (Horie et al, 1998; Murakami‐Tonami et al, 2007), while high levels of the APC/C activator Fzr1/Mfr1 induce exit from meiosis (Asakawa et al, 2001; Blanco et al, 2001). Ama1 is related to the APC/C activator Cdh1, which is active during pre‐meiotic G1 but inhibited at other stages of meiosis (Oelschlaegel et al, 2005; Holt et al, 2007). Mammalian genomes encode only two APC/C activators, namely Cdc20 and Cdh1, whereby Cdh1's role resembles that of Ama1. Cdh1 prevents premature accumulation of cyclin B at prophase (Holt et al, 2011, 2014), and hyperactive Cdh1 causes spermatocytes to exit from meiosis after a single division (Tanno et al, 2020).
How exit from meiosis is confined to the second division is poorly understood. APC/CAma1 is inhibited by high levels of Cdk1 activity (Oelschlaegel et al, 2005; Okaz et al, 2012). Thus, by initiating Clb degradation, APC/CCdc20 might activate APC/CAma1 at anaphase II and thereby induce exit from meiosis. How APC/CAma1 is inhibited at meiosis I is unclear. Three properties of Ama1 might make this a challenging task: First, Ama1 accumulates at metaphase II of an unperturbed meiosis. However, it appears already at metaphase I, when the latter is prolonged due to the inhibition of Cdc20 (Oelschlaegel et al, 2005). Second, by destroying its inhibitor, Cdk1‐Clb, APC/CAma1 increases its own activity. Once unleashed, this feedback loop triggers an irreversible exit from M phase (Okaz et al, 2012). Third, since Ama1 is not affected by the SAC, APC/CAma1 activity threatens to curtail metaphase I. Confining APC/CAma1 activity to meiosis II might therefore be crucial for the ability of the SAC to prolong metaphase I and, indeed, for performing a second division.
We show here that the Cdc5‐Spo13 kinase ensures a two‐division meiosis by preventing premature activation of APC/CAma1. At metaphase I, Cdc5‐Spo13 inhibits the synthesis of Ama1 by stabilizing the translational repressor Rim4. Furthermore, by phosphorylating the cyclin Clb1, Cdc5‐Spo13 generates an inhibitor of APC/CAma1. At anaphase I, Cdc20‐dependent degradation of Spo13 unleashes synthesis and activation of Ama1, leading to exit from meiosis at the second division. In the spo13 mutant, premature accumulation and activation of Ama1 causes exit from meiosis after a single division, even in the absence of Cdc20.
Results
Properties of the single‐division meiosis in the spo13Δ mutant
The single division of the spo13Δ mutant shows at least three unusual properties: First, these cells perform only one round of spindle formation (metaphase) and release of the Cdc14 phosphatase from the nucleolus (anaphase), which implies a single wave of Cdk1 and APC/C activity (Fig 1A). However, the division is preceded by recombination, an aspect of meiosis I, but concluded by exit from meiosis and spore formation, which are features of meiosis II (Klapholz & Esposito, 1980b). Second, spo13Δ mutants undergo Cdc14 release, nuclear division, and sporulation even when Cdc20 is depleted, which causes otherwise normal cells to arrest at metaphase I (Katis et al, 2004; Appendix Fig S1A). Thus, the spo13Δ‐division might be promoted by Ama1 or Cdh1, although these APC/C activators are thought to be inhibited during M phase (Zachariae et al, 1998; Oelschlaegel et al, 2005). Third, the spo13Δ‐division depends on the duration of metaphase I. While a normal metaphase I lasts 26 min, spo13Δ cells show a metaphase of 66 min, which is reduced to 10 min upon deletion of the SAC protein Mad2 (Fig 1A and B; Shonn et al, 2002). Remarkably, spo13Δ mad2Δ cells undergo two rounds of spindle formation and Cdc14 release (Fig 1B). However, 40% of these cells do not undergo nuclear division until entry into meiosis II, probably because sister kinetochores biorient in the absence of Spo13 (Katis et al, 2004; Lee et al, 2004). Indeed, the meiosis I‐division is restored by rec8‐18D, a mutation that causes cleavage of centromeric cohesin at anaphase I (Appendix Fig S1B; Arguello‐Miranda et al, 2017). Thus, spo13Δ cells perform a single division when metaphase I is long but two divisions when metaphase I is short. We considered the possibility that spo13Δ cells undergo unrestrained sporulation, thereby cutting off the second division when the first is delayed. However, blocking spore formation does not restore a second division in spo13Δ cells (Appendix Fig S1C). We therefore investigated the idea that Spo13 prevents Ama1 or Cdh1 from mediating exit from meiosis when Cdc20 is inhibited.
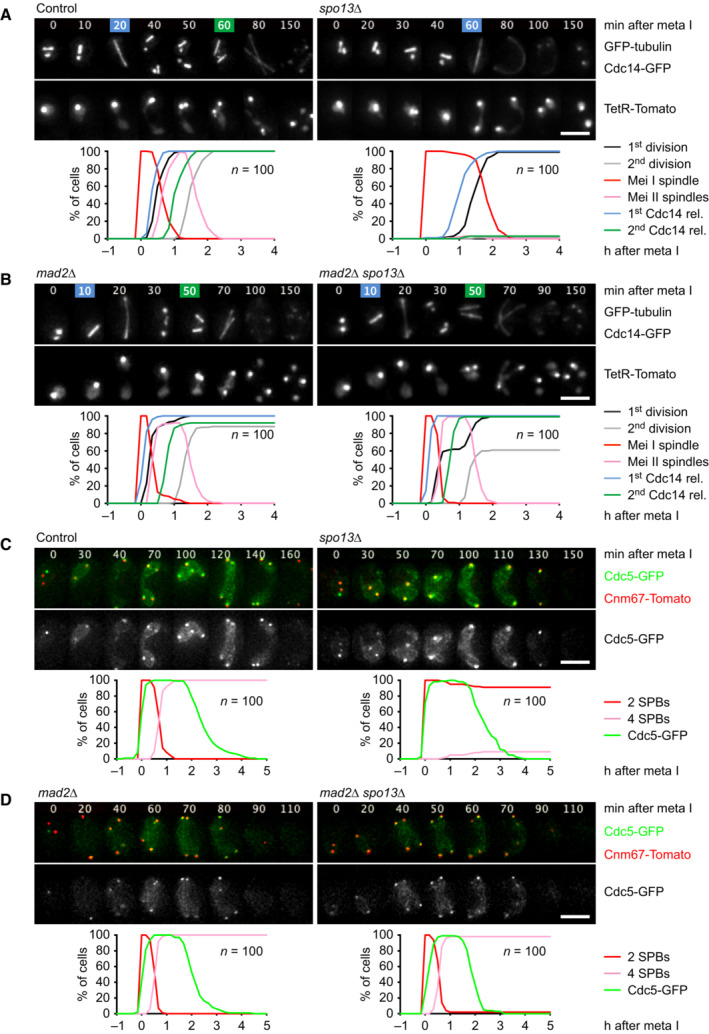
Figure 1. Analysis of meiotic progression in spo13Δ mutants.
-
A, BImaging of spindles (GFP‐tubulin), nucleolar release of Cdc14‐GFP, and TetR‐Tomato, which labels the nucleoplasm (diffuse signal) and the centromeres of one copy of chromosome V (dots). Top, time‐lapse series. First (blue) and second (green) Cdc14 release are marked. Bottom, meiotic events quantified in cells synchronized in silico to spindle formation at metaphase I (t = 0). (A) spo13Δ cells show a longer metaphase I (66 ± 21 min) than control cells (26 ± 9 min, P < 0.0001) and a single round of spindle formation and Cdc14 release. (B) mad2Δ spo13Δ cells show a short metaphase I (10 ± 5 min), similar to mad2Δ cells (10 ± 8 min, P = 0.68), and undergo two rounds of spindle formation and Cdc14 release. Cells in (A) and (B) were filmed together.
-
C, DImaging of Cdc5‐GFP and SPBs (Cnm67‐Tomato). Top, time‐lapse series. Bottom, the presence of Cdc5 and SPB reduplication quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0). (C) While control cells degrade Cdc5 at meiosis II (four SPBs), spo13Δ cells do so at meiosis I (two SPBs). (D) mad2Δ and mad2Δ spo13Δ cells degrade Cdc5 at meiosis II (four SPBs). Cells in (C) and (D) were filmed together.
Data information: Data are representative of three (A) or two (B and C) independent experiments. Means were compared using Welch's t‐test. Scale bar, 4 μm.
Source data are available online for this figure.
Spo13 prevents accumulation of the APC/C activator Ama1 at metaphase I
To monitor the activity of APC/CAma1, we imaged its substrate Cdc5‐GFP together with Cnm67‐tdTomato, a component of the spindle‐pole body (SPB, the yeast centrosome; Fig 1C and D). SPBs duplicate at S phase and separate at metaphase I to assemble the meiosis I‐spindle. SPBs then reduplicate at metaphase II to form a pair of meiosis II‐spindles (Moens & Rapport, 1971). As previously shown (Arguello‐Miranda et al, 2017), control cells degrade Cdc5 at anaphase II, after SPB reduplication. While spo13Δ cells prolong metaphase I and do not reduplicate SPBs, they degrade Cdc5 with similar kinetics as control cells (Fig 1C). Thus, spo13Δ cells degrade Cdc5 at meiosis I. By contrast, spo13Δ mad2Δ cells degrade Cdc5 after SPB reduplication, that is, at anaphase II (Fig 1D). One interpretation is that spo13Δ cells cannot restrain APC/CAma1 activity when Cdc20 is inhibited by the SAC. To investigate this idea, we used the mitosis‐specific SCC1 promoter to deplete cells of Cdc20. P SCC1 ‐CDC20 cells arrest at metaphase I, whereby APC/C substrates persist and nuclei do not divide. However, deletion of SPO13 results in the degradation of APC/C substrates and nuclear division, which depends on Ama1 (Fig 2A), but not on Cdh1 (Appendix Fig S2A). Our data suggest that Spo13 prevents the activation of APC/CAma1, and thereby premature exit from metaphase I, when Cdc20 is inhibited by the SAC or completely absent.
Figure 2. Spo13 prevents accumulation of Ama1 at metaphase I.
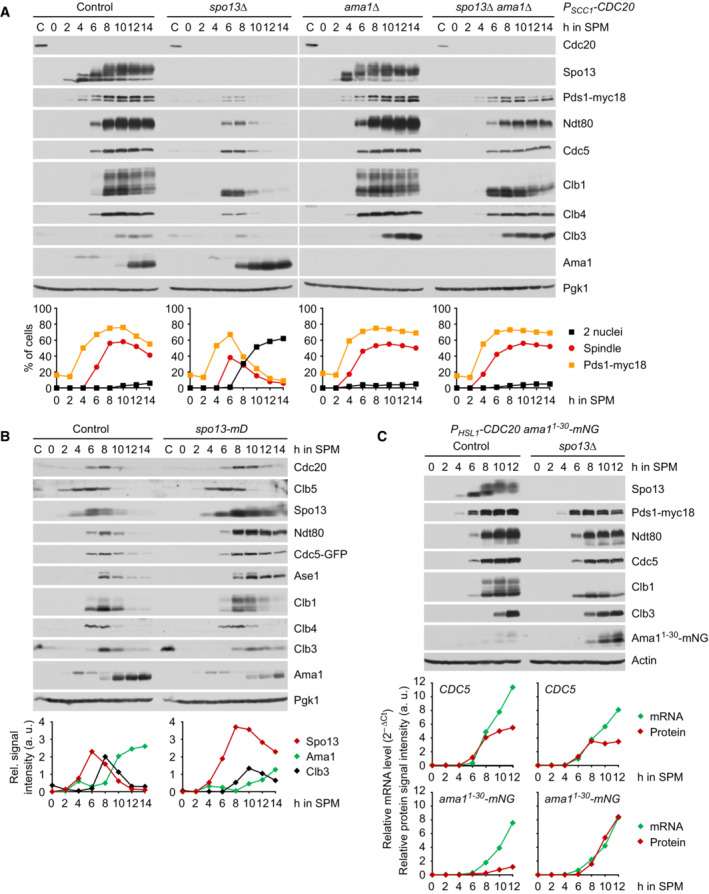
- Deletion of SPO13 causes Ama1‐dependent proteolysis and nuclear division in P SCC1 ‐CDC20 cells. Top, immunoblot detection of proteins. C, sample from proliferating cells. Bottom, progression of meiosis quantified in fixed cells.
- Spo13‐mD reduces Ama1 accumulation and Ama1‐dependent proteolysis at meiosis II. Top, immunoblot detection of proteins. Bottom, relative signal intensities of proteins.
- Analysis of mRNA and protein encoded by the ama1 1‐30 ‐mNG locus in P HSL1 ‐CDC20 control and spo13Δ cells. Top, immunoblot detection of proteins. Bottom, mRNA levels relative to the ACT1 transcript were quantified by RT‐qPCR and the 2−ΔCt method. Signal intensities of proteins were normalized to Act1/actin. While control and spo13Δ cells produce ama1 1‐30 ‐mNG mRNA, only spo13Δ cells synthesize mNG protein.
Data information: (B) is representative of two independent experiments.
Source data are available online for this figure.
How does Spo13 restrain the activity of APC/CAma1? In the wild‐type, Ama1 is present at low levels at prophase but accumulates to high levels as cells enter meiosis II at ~8 h after induction of meiosis (Fig 2B, left; Oelschlaegel et al, 2005). Indeed, degradation of Cdc5 and Ndt80 at anaphase II requires high levels of Ama1 and is blocked when Ama1 is expressed at prophase‐levels (Arguello‐Miranda et al, 2017). Depletion of Cdc20 strongly delays, but ultimately fails to suppress, the accumulation of Ama1. Thus, P SCC1 ‐CDC20 cells start to accumulate Ama1 at 12 h after induction of meiosis (Fig 2A). Remarkably, deletion of SPO13 advances accumulation and binding to the APC/C of Ama1 by 4 h (Fig 2A and Appendix Fig S2B). These data suggest that Ama1 accumulation is promoted by Cdc20 and repressed by Spo13, a substrate of APC/CCdc20 (Sullivan & Morgan, 2007). Deletion of SPO13 also advances the appearance of the Clb3 cyclin (Fig 2A). This is more obvious in strains in which Clb3 is stable due to the absence of both Cdc20 and Ama1. Similar to Ama1, the accumulation of Clb3 is usually confined to meiosis II (Carlile & Amon, 2008).
To test whether Ama1 accumulation requires Cdc20‐dependent degradation of Spo13, we mutated the D‐box of Spo13 (Sullivan & Morgan, 2007). Spo13‐mD accumulates to higher levels and persists longer than wild‐type Spo13 (Fig 2B; Oz et al, 2022). In spo13‐mD cells, Ama1 levels do not exceed 50% of those in the control. This does not affect the degradation of Clb4 or Clb5, which are targeted by Cdc20 as well as Ama1. However, Spo13‐mD increases the formation of slow‐migrating species of Clb1, which persist slightly longer than the unmodified form. We shall analyse this modification further below. Importantly, Ase1, Cdc5, and Ndt80 are stabilized in spo13‐mD cells. These proteins are normally degraded at anaphase II in a manner dependent on high levels of Ama1 (Okaz et al, 2012; Arguello‐Miranda et al, 2017). Live‐imaging confirms that spo13‐mD cells fail to properly degrade Cdc5 at meiosis II (Appendix Fig S2C). Our data suggest that Cdc20‐dependent degradation of Spo13 at anaphase I is required for Ama1 accumulation and Ama1‐dependent proteolysis at meiosis II. Interestingly, Spo13 degradation is also required for normal accumulation of Clb3 (Fig 2B).
Spo13 is required for repressing AMA1 translation at metaphase I
While CLB3 transcription is induced by Ndt80 at metaphase I, CLB3 translation is repressed until entry into meiosis II (Carlile & Amon, 2008; Berchowitz et al, 2013). To test whether AMA1 expression is controlled by a similar mechanism, we used cells expressing Cdc20 from the mitosis‐specific HSL1 promoter and replaced AMA1 sequences downstream of codon 30 with a fragment encoding the fluorescent protein mNeonGreen (mNG; Shaner et al, 2013). Since these cells lack Ama1 activity, they stably arrest at metaphase I, even when SPO13 is deleted. In control cells, ama1 1‐30 ‐mNG and CDC5 transcripts start to accumulate at 6 h after induction of meiosis (Fig 2C, green lines). While CDC5 mRNA is promptly translated, the mNG protein is barely detectable by immunoblotting (Fig 2C, red lines) or live‐imaging (Fig EV1A). Importantly, deletion of SPO13 has little effect on ama1 1‐30 ‐mNG transcription but causes accumulation of mNG. These data confirm that AMA1 transcription is induced at metaphase I (Chu et al, 1998; Primig et al, 2000), while translation is repressed by a Spo13‐dependent mechanism. Recent work has identified at least 20 genes involved in spore formation, which show Ndt80‐dependent transcription and delayed translation, either at entry into metaphase II (e.g., GAT4 and GIP1) or at late anaphase II (e.g., SPS4 and SSP2; Brar et al, 2012; Whinston et al, 2013; Jin et al, 2015). To monitor translation at metaphase I, we replaced coding sequences with the mNG fragment in P HSL1 ‐CDC20 ama1Δ cells (Fig EV1B). mNG integrated at any one of these genes is not translated at metaphase I unless SPO13 is deleted. This suggests that several sporulation genes are subject to Spo13‐dependent translational repression at meiosis I and explains how spo13Δ mutants can sporulate at meiosis I (Oz et al, 2022).
Figure EV1. Regulation of translation by Spo13.
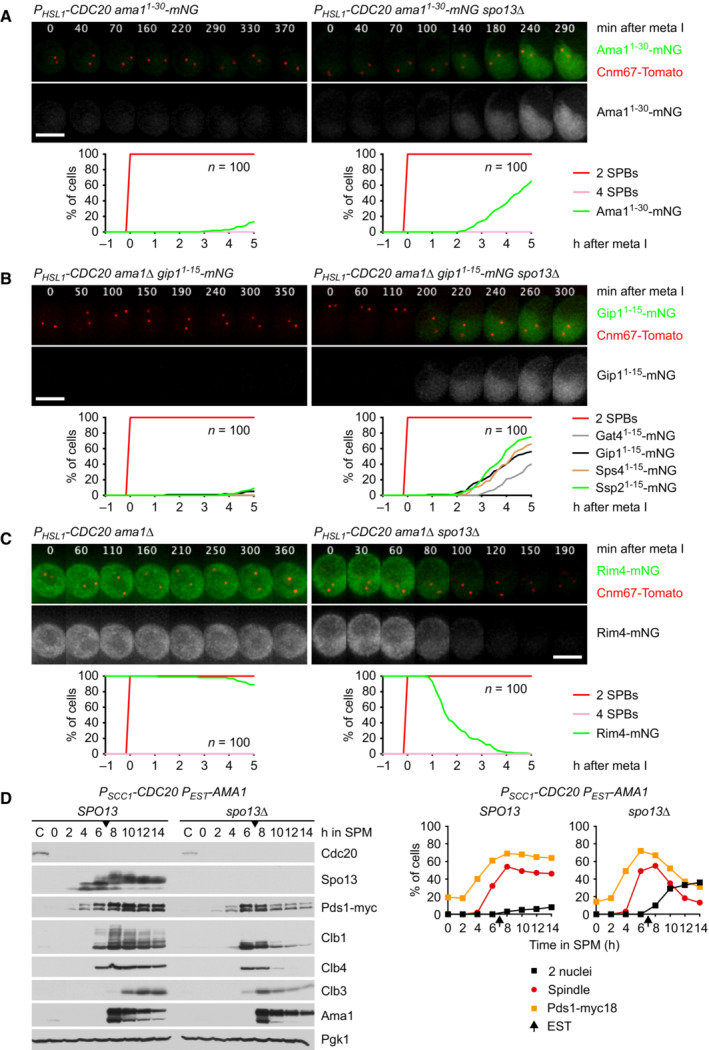
- Deletion of SPO13 causes AMA1 translation in Cdc20‐depleted cells. Top, time‐lapse series from the imaging of mNG and SPBs (Cnm67‐Tomato) in P HSL1 ‐CDC20 control and spo13Δ cells carrying ama1 1‐30 ‐mNG in place of AMA1. Bottom, the presence of mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
- Sporulation genes showing Spo13‐dependent translational repression in metaphase I‐arrested P HSL1 ‐CDC20 ama1Δ strains. Top, time‐lapse series from the imaging of mNG and SPBs (Cnm67‐Tomato) in control and spo13Δ cells carrying gip1 1‐15 ‐mNG in place of GIP1. Bottom, the presence of mNG quantified in cells in which mNG replaces coding sequences downstream of codon 15 of GAT4, GIP1, SPS4, or SSP2. Cells have been synchronized in silico to SPB separation at metaphase I (t = 0).
- Deletion of SPO13 causes Rim4 degradation in metaphase I‐arrested P HSL1 ‐CDC20 ama1Δ cells. Top, time‐lapse series from the imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato). Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
- Deletion of SPO13 causes degradation of APC/C substrates and nuclear division in Cdc20‐depleted (P SCC1 ‐CDC20) cells expressing AMA1 from an estradiol‐inducible GAL promoter (P EST ‐AMA1) at t = 7 h in SPM (arrows). Left, immunoblot detection of proteins. Right, progression of meiosis quantified in fixed cells.
Data information: Data are representative of three (A and C) or two (B) independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Translation of CLB3 and SPS4 is repressed by the RNA‐binding protein Rim4, which is degraded at entry into metaphase II. Furthermore, these mRNAs and those encoding Ama1 were found among mRNAs copurifying with Rim4 (Berchowitz et al, 2013; Jin et al, 2015; Carpenter et al, 2018). To test whether Rim4 represses AMA1 translation at metaphase I, we used an auxin‐inducible degradation (AID) system (Yesbolatova et al, 2020) to remove Rim4 from Cdc20‐depleted cells (Fig 3A). Indeed, removal of Rim4 advances the accumulation of Ama1 by ~4 h. As a result, cells degrade APC/C substrates and undergo nuclear division in an Ama1‐dependent manner. Consistent with previous work (Berchowitz et al, 2013), removal of Rim4 also advances the synthesis of Clb3, which is most evident in strains lacking both Cdc20 and Ama1 (Fig 3A). We conclude that Rim4 prevents accumulation of Ama1 as well as Clb3 when metaphase I is prolonged due to the inhibition of Cdc20.
Figure 3. Spo13 controls Ama1 accumulation via the translational repressor Rim4.
-
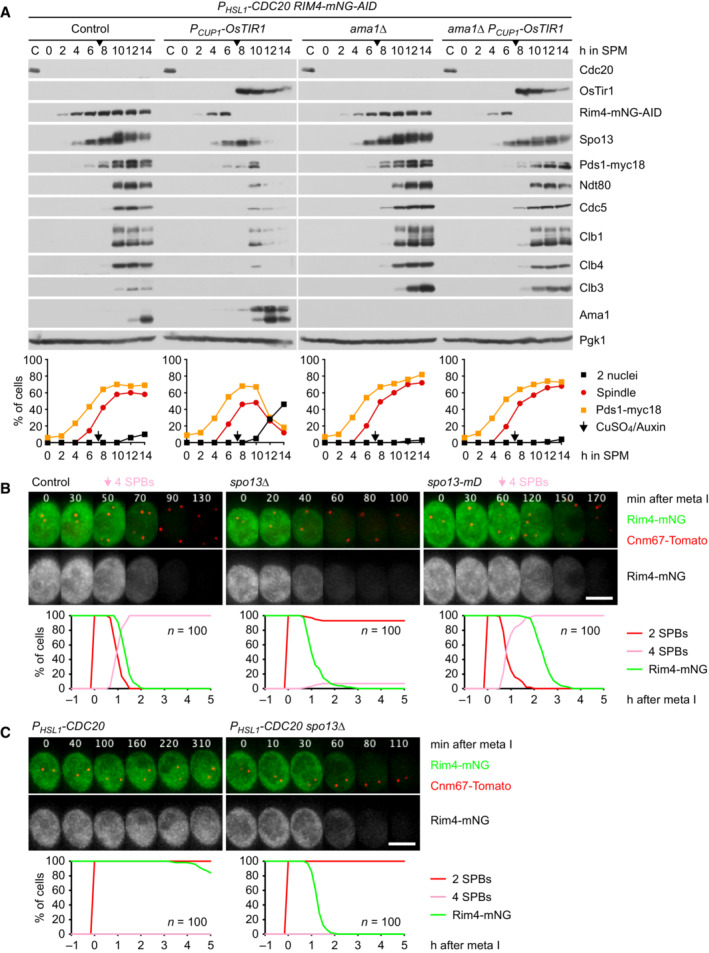
AAuxin‐inducible degradation of Rim4 causes Ama1‐dependent proteolysis in P HSL1 ‐CDC20 cells. At t = 7.5 h (arrows), CuSO4 and the auxin 5‐Ph‐IAA were added to induce expression and activation of OsTir1, resulting in degradation of AID‐tagged Rim4‐mNG. Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
-
B, CImaging of Rim4‐mNG and SPBs (Cnm67‐Tomato). Top, time‐lapse series. Bottom, the presence of Rim4‐mNG and SPB reduplication quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0). (B) While control cells degrade Rim4 at meiosis II (four SPBs), spo13Δ mutants do so at meiosis I (two SPBs). Spo13‐mD delays Rim4 degradation by 64 min (95% CI, 58–69 min; P < 0.0001, Welch's t‐test). (C) Deletion of SPO13 causes P HSL1 ‐CDC20 cells to degrade Rim4.
Data information: Data are representative of two (B) or three (C) independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Spo13 prevents degradation of the translational repressor Rim4 at metaphase I
The consequences of removing Rim4 from Cdc20‐depleted cells resemble those of deleting SPO13. Thus, Spo13 might restrain the accumulation of Ama1 by preventing the degradation of Rim4. To test this, we imaged Rim4‐mNG together with SPBs carrying Cnm67‐tdTomato. In control cells, Rim4 disappears shortly after SPBs reduplicate at entry into metaphase II (Fig 3B). While spo13Δ cells prolong metaphase I and do not reduplicate SPBs, they nevertheless degrade Rim4. Thus, spo13Δ cells degrade Rim4 at meiosis I. In spo13‐mD cells, on the other hand, Rim4 degradation is delayed and therefore occurs well after entry into metaphase II (Fig 3B). These data suggest that Rim4 degradation is inhibited until Spo13 is subjected to APC/C‐dependent proteolysis. Accordingly, Rim4 persists in P HSL1 ‐CDC20 cells in which Spo13 is stable, but is rapidly degraded in P HSL1 ‐CDC20 spo13Δ cells (Fig 3C). In fact, Rim4 is degraded with similar kinetics in spo13Δ cells containing or lacking Cdc20 (Fig 3B and C). While P HSL1 ‐CDC20 spo13Δ double mutants undergo nuclear division in an Ama1‐dependent manner, nuclear division is not required for Rim4 degradation. The SPO13 deletion also elicits Rim4 degradation in P HSL1 ‐CDC20 ama1Δ cells, which stably arrest at metaphase I (Fig EV1C). We conclude that APC/CCdc20 induces Rim4 degradation by mediating the proteolysis of its canonical substrate Spo13, which in turn unleashes the mechanism directly responsible for Rim4 degradation. Does Spo13 restrain Ama1‐dependent proteolysis solely by repressing AMA1 translation via Rim4? If so, expression of Ama1 from a foreign promoter should eliminate the effect of the SPO13 deletion. While the SPO13 deletion has no effect on the accumulation of Ama1 expressed from an estradiol‐inducible GAL promoter, it nevertheless results in degradation of APC/C substrates and nuclear division (Fig EV1D). The implication is that Spo13 not only represses Ama1 synthesis but additionally employs a post‐translational mechanism to inhibit Ama1 activity. We shall return to this issue further below.
Spo13 prevents Ime2 from promoting the degradation of Rim4 at metaphase I
Degradation of Rim4 at entry into metaphase II depends on the activity of the meiosis‐specific kinase Ime2 (Berchowitz et al, 2013; Carpenter et al, 2018). Ime2 activity is also required for premature Rim4 degradation in spo13Δ cells, as revealed by inhibition of analogue‐sensitive Ime2‐as (Benjamin et al, 2003) in the P HSL1 ‐CDC20 ama1Δ spo13Δ triple mutant (Fig 4A). Next, we asked whether Ime2 activity is required for Ama1 translation in metaphase I‐arrested spo13Δ cells. However, Ime2 is required for the accumulation of Ndt80 and thereby for entry into metaphase I (Sopko et al, 2002). On the other hand, cdc20 spo13Δ cells start to produce Ama1 soon after entry into metaphase I. Thus, we used estradiol‐dependent expression of NDT80 (P EST ‐NDT80; Benjamin et al, 2003) to arrest cells at prophase and subsequently release them into a metaphase I‐arrest imposed by depletion of Cdc20. 1Na‐PP1 was added shortly after cells had accumulated Ndt80 and entered metaphase I. While P EST ‐NDT80 P HSL1 ‐CDC20 control cells produce little Ama1, deletion of SPO13 causes robust accumulation of Ama1, which results in degradation of APC/C substrates and nuclear division (Fig 4B). Importantly, inhibition of Ime2 prevents accumulation of Ama1, degradation of APC/C substrates, and nuclear division. Ime2 activity is also required for the transient accumulation of Clb3 elicited by the SPO13 deletion. We conclude that Spo13 prevents Ime2 from promoting the degradation of Rim4 and thereby translation of AMA1 as well as CLB3 at metaphase I. Our data predict that hyperactivation of Ime2 promotes Rim4 degradation even in the presence of Spo13. To test this, we used Ime2‐ΔC, a kinase that is more stable and active due to the removal of the C‐terminal, regulatory domain (Sari et al, 2008). Indeed, Ime2‐ΔC induces Rim4 degradation in P HSL1 ‐CDC20 ama1Δ cells, albeit with slower kinetics than the SPO13 deletion (Fig 4C, middle). Furthermore, Ime2‐ΔC causes P HSL1 ‐CDC20 cells to translate ama1 1‐30 ‐mNG mRNA into mNG protein (Appendix Fig S3A). Thus, Ime2‐ΔC uncouples Rim4 degradation from the inactivation of Spo13, suggesting that wild‐type Ime2 functions downstream of and is inhibited by Spo13.
Figure 4. Ime2 activity is required for Rim4 degradation upon removal of Spo13.
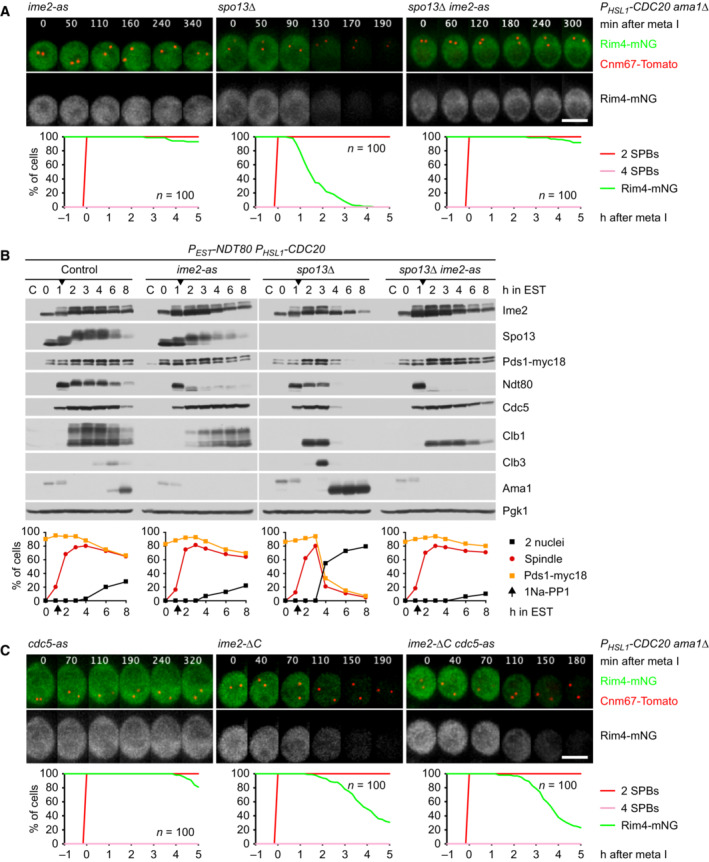
- Inhibition of Ime2 activity prevents Rim4 degradation elicited by the SPO13 deletion in metaphase I‐arrested P HSL1 ‐CDC20 ama1Δ cells. Ime2‐as was inhibited with 1Na‐PP1 at 7 h in SPM. Top, time‐lapse series from the imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato). Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
- Inhibition of Ime2 activity blocks degradation of APC/C substrates and nuclear division in Cdc20‐depleted spo13Δ cells. P EST ‐NDT80 P HSL1 ‐CDC20 strains were released from prophase with estradiol (EST, t = 0), and Ime2‐as was inhibited at metaphase I (1Na‐PP1 at t = 75 min, arrows). Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
- Ime2‐ΔC induces Rim4 degradation in metaphase I‐arrested P HSL1 ‐CDC20 ama1Δ cells even when Cdc5 is inhibited. Cdc5‐as was inhibited with CMK at 8 h in SPM. Top, time‐lapse series from the imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato). Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0). Rim4 is degraded with similar timing in ime2‐ΔC cells with active or inactive Cdc5 (P = 0.88, Welch's t‐test).
Data information: Data are representative of three (A) or two (C) independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Ime2 is inhibited by Cdc5‐Spo13 and activated by free Cdc5
How does Spo13 inhibit Ime2's function in Rim4 degradation? Spo13 binds to the polo‐box domain (PBD) of Cdc5. A mutation in Spo13's PBD‐binding motif, called spo13‐m2, weakens this interaction and shows similar but less severe defects than the SPO13 deletion (Matos et al, 2008; Oz et al, 2022). While the spo13‐m2 mutation prolongs metaphase I to 60 min, it results in a lower incidence (~38%) of cells undergoing a single round of Cdc14 release than the SPO13 deletion (> 90%; Fig EV2A). Nevertheless, the spo13‐m2 mutation causes P HSL1 ‐CDC20 cells to advance the accumulation of Ama1 by 2 h (Fig EV4B). As a result, these cells degrade APC/C substrates and undergo nuclear division in an Ama1‐dependent manner. Furthermore, P HSL1 ‐CDC20 spo13‐m2 cells degrade Rim4, albeit with slower kinetics than the spo13Δ mutant (Fig EV2C). We conclude that binding to Cdc5 is required for Spo13's ability to prevent Ime2 from promoting the degradation of Rim4 at metaphase I.
Figure EV2. Analysis of the spo13‐m2 mutation.
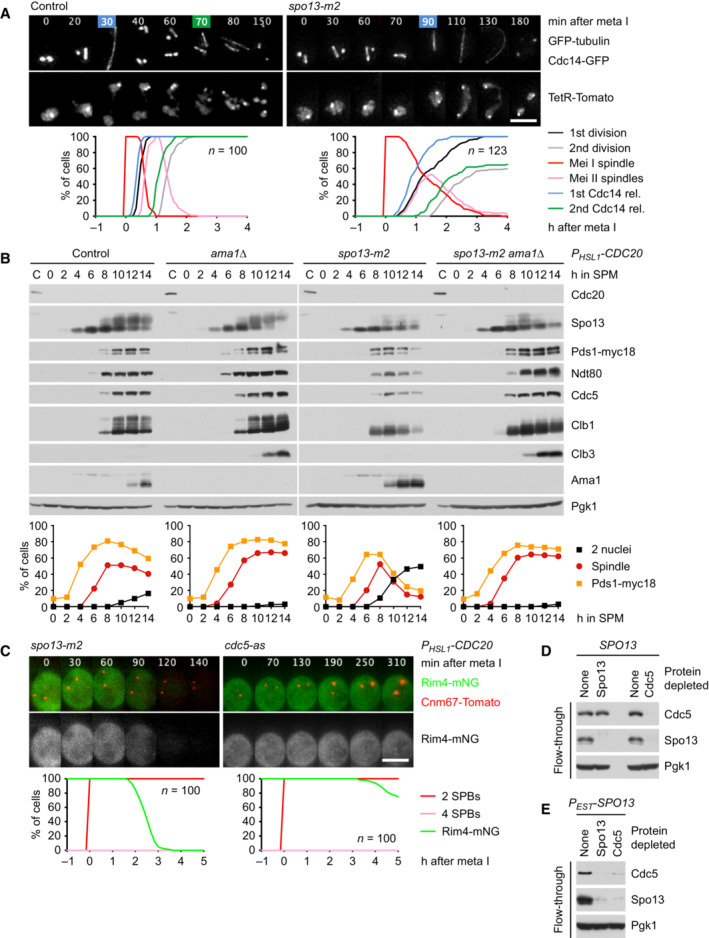
-
AMeiosis in control and spo13‐m2 cells. Top, imaging of spindles (GFP‐tubulin), nucleolar release of Cdc14‐GFP, and TetR‐Tomato, which labels the nucleoplasm (diffuse signal) and the URA3 locus of one copy of chromosome V (dots). First (blue) and second (green) Cdc14 release are marked. Bottom, meiotic events quantified in cells synchronized in silico to spindle formation at metaphase I (t = 0). spo13‐m2 prolongs metaphase I to 60 ± 26 min (control, 23 ± 6 min; P < 0.0001, Welch's t‐test) and causes ~38% of cells to undergo a single round of spindle formation and Cdc14 release.
-
Bspo13‐m2 causes Ama1‐dependent proteolysis and nuclear division in P HSL1 ‐CDC20 cells. Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
-
CImaging of Rim4‐mNG and SPBs (Cnm67‐Tomato) in P HSL1 ‐CDC20 cells carrying spo13‐m2 or cdc5‐as. Cdc5‐as was inhibited with CMK at 7.5 h in SPM. Top, time‐lapse series. Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
-
D, EExtracts from metaphase I‐arrested P HSL1 ‐CDC20 cells (8 h in SPM) were applied to columns carrying no antibody or antibodies to either Spo13 or Cdc5. Flow‐throughs were analyzed by immunoblotting. (D) Depletion of Spo13 from extracts of control cells (SPO13) removes little Cdc5 (10%). (E) Depletion of Spo13 from extracts of cells overexpressing Spo13 (P EST ‐SPO13, estradiol for 45 min) removes most of Cdc5 (97%).
Data information: (B–D) are representative of two independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Figure EV4. Identification of proteins copurifying with Ama1.
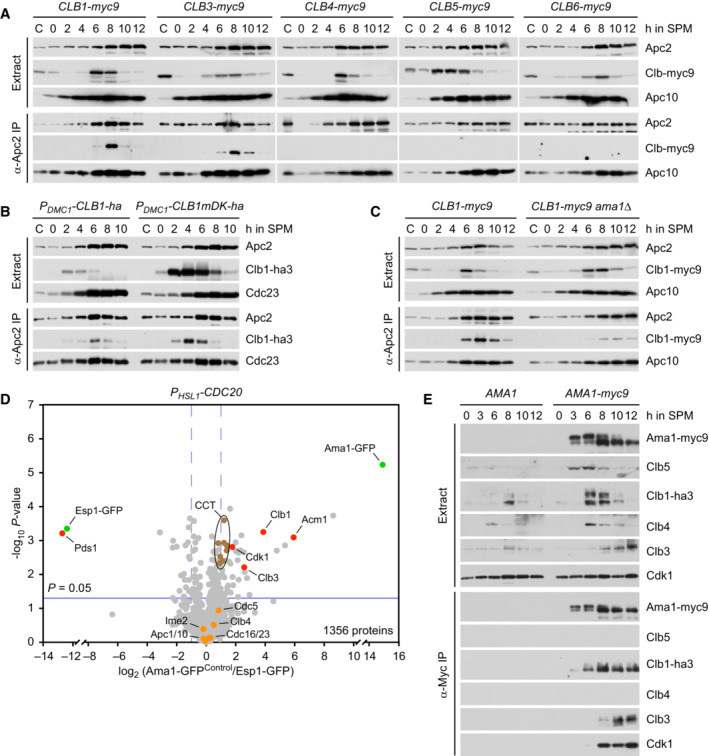
-
A–CBinding of cyclins to the APC/C immunoprecipitated with α‐Apc2 antibodies. (A) Clb1 and Clb3 but not the other Clbs copurify with the APC/C. (B) The degrons of Clb1 are not required for binding to the APC/C. Ha3‐tagged Clb1 and Clb1‐mDK were expressed from the DMC1 promoter. (C) Deletion of AMA1 reduces Clb1's interaction with the APC/C.
-
DProteins interacting with Ama1 at metaphase I. Ama1‐GFP and Esp1‐GFP (negative control) were purified from P HSL1 ‐CDC20 strains at 8 h in SPM, digested with trypsin and Lys‐C, and subjected to LC–MS/MS. ‐log10‐transformed P‐values and mean log2‐transformed label‐free quantifications of copurifying proteins were obtained from MaxQuant and displayed as a volcano plot. Baits (green), selected proteins with P < 0.05 (red) or P > 0.05 (orange), and the eight subunits of the CCT chaperonin (brown) are labeled. CCT is known to encapsulate and fold APC/C activators (Camasses et al, 2003). Only a subset of APC/C subunits is marked. Note that C‐terminally tagged Ama1 cannot bind the APC/C.
-
EBinding of Clb1, Clb3, and Cdk1 to Ama1‐myc9 immunoprecipitated with α‐Myc antibodies.
Data information: (D) is based on α‐GFP purifications from three independent cultures per strain.
Source data are available online for this figure.
By contrast, inhibition of analogue‐sensitive Cdc5‐as (Snead et al, 2007) in P HSL1 ‐CDC20 cells does not cause Rim4 degradation (Fig EV2C), Ama1 accumulation, or nuclear division (Fig 5A). Importantly, inhibition of Cdc5 blocks Ama1 accumulation in P HSL1 ‐CDC20 spo13Δ cells (Fig 5A). From these data, one might presume that Spo13 functions as an inhibitor of Cdc5. However, we have recently shown that at metaphase I, the levels of Cdc5 greatly exceed those of Spo13 (Oz et al, 2022). Accordingly, depleting metaphase I‐extracts of Cdc5 co‐depletes most of Spo13, whereas Spo13 depletion removes only a small fraction of Cdc5 (Fig EV2D). However, Cdc5 is co‐depleted with Spo13 when Spo13 is overexpressed (Fig EV2E). These data argue against Spo13 being a stoichiometric inhibitor of Cdc5. Indeed, we show further below that Cdc5‐Spo13 is an active kinase, which coexists with free Cdc5 at metaphase I. What is the function of free Cdc5? Ime2‐ΔC restores Rim4 degradation in P HSL1 ‐CDC20 ama1Δ cells even when Cdc5 is inhibited (Fig 4C), implying that wild‐type Ime2 promotes Rim4 degradation in a manner dependent on Cdc5 activity. To test this, we used ndt80Δ spo13Δ cells. ndt80Δ mutants arrest at prophase and express Ime2 but not Cdc5 (Benjamin et al, 2003; Clyne et al, 2003). SPO13 was deleted to exclude any inhibitory effect on Ime2. Nevertheless, Rim4 accumulates in these cells, demonstrating that Ime2 is inactive towards Rim4 (Fig 5B). However, expression of Cdc5 causes degradation of Rim4, which in turn requires Ime2 activity. Our data suggest that while Cdc5‐Spo13 and free Cdc5 coexist at metaphase I, the former prevents Ime2 from promoting the degradation of Rim4. This enables Rim4 to repress the translation of AMA1. Proteolysis of Spo13 at anaphase I then allows Cdc5 to activate Ime2's function in Rim4 degradation (Fig 5C).
Figure 5. The role of Cdc5 in the control of Ama1 accumulation.
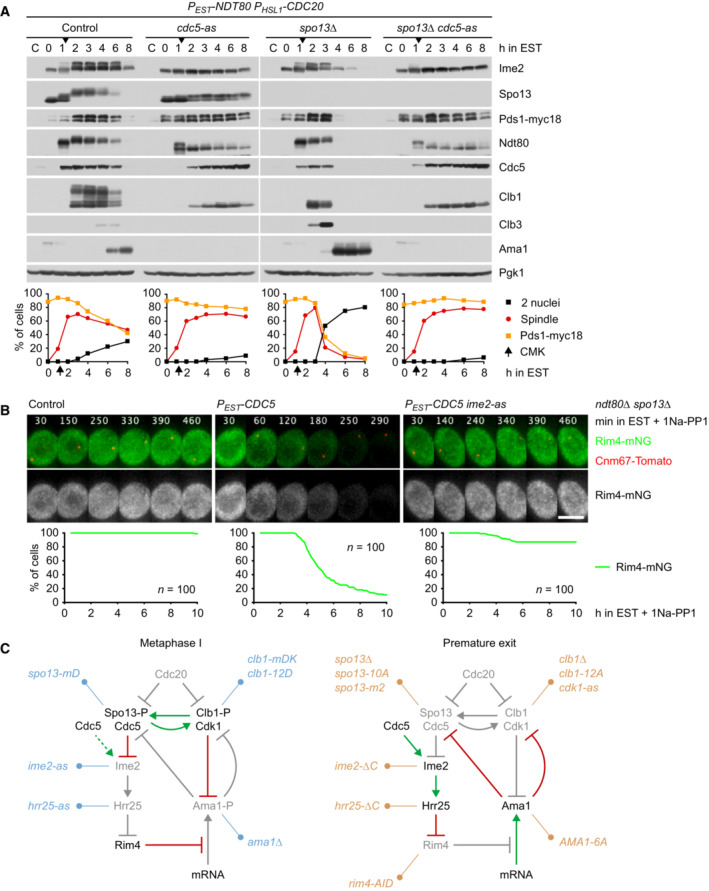
- Inhibition of Cdc5 activity blocks degradation of APC/C substrates and nuclear division in Cdc20‐depleted spo13Δ cells. P EST ‐NDT80 P HSL1 ‐CDC20 strains were released from prophase with estradiol (EST, t = 0), and Cdc5‐as was inhibited at metaphase I (CMK at t = 75 min, arrows). Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
- Cdc5 activity is required for Ime2‐dependent degradation of Rim4. ndt80Δ spo13Δ strains were allowed to arrest at prophase. At 4 h in SPM (t = 0), P EST ‐CDC5 was induced with EST, and Ime2‐as was inhibited with 1Na‐PP1. Top, time‐lapse series from the imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato). Bottom, quantification of the presence of Rim4‐mNG.
- The regulatory network controlling Ama1 synthesis and activity. Left, the low‐Ama1/high‐Cdk1 state of metaphase I. Right, the high‐Ama1/low‐Cdk1 state that induces premature exit from metaphase I. Green arrows: activation. Dashed, green arrow: latent activation overwhelmed by inhibition. Red, bar‐headed lines: inhibition. Gray items are inactive or absent. P denotes phosphorylation. Blue, mutations that stabilize metaphase I. Ochre, mutations causing exit from metaphase I. See text for details.
Data information: (B) is representative of two independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Rim4 degradation depends on the activity of Hrr25/CK1δ
Our data raise a conundrum: In spo13Δ mutants, Cdc5 is required for the ability of Ime2 to promote Rim4 degradation and Ama1 accumulation (Figs 4A and 5A). However, as Ama1 accumulates, it mediates the degradation of Cdc5 (Fig 2A). This raises the question of how AMA1 translation is sustained when Cdc5 levels decline. A related question arises at meiosis II when Ama1 accumulates despite mediating the degradation of Cdc5 (Arguello‐Miranda et al, 2017). To investigate this, we used CDC20‐meiotic‐arrest/release (CDC20‐mAR) cells, which are first arrested at metaphase I through the depletion of Cdc20 and then released into anaphase I by a copper‐inducible CDC20 gene (Arguello‐Miranda et al, 2017). Inhibition of Cdc5 at anaphase I, shortly after the degradation of Spo13, reduces subsequent nuclear division due to the collapse of anaphase II spindles (Arguello‐Miranda et al, 2017) but has no effect on the accumulation of Ama1 (Appendix Fig S4). Similarly, inhibition of Ime2 at anaphase I reduces the stability of Ndt80 and hinders the meiosis II‐division but has little effect on the accumulation of Ama1 (Appendix Fig S4). Thus, while Rim4 degradation and Ama1 accumulation initially depend on Cdc5 and Ime2, they subsequently become independent of these two kinases. One possibility is that Ime2 induces an activity that promotes Rim4 degradation in a self‐sustaining manner.
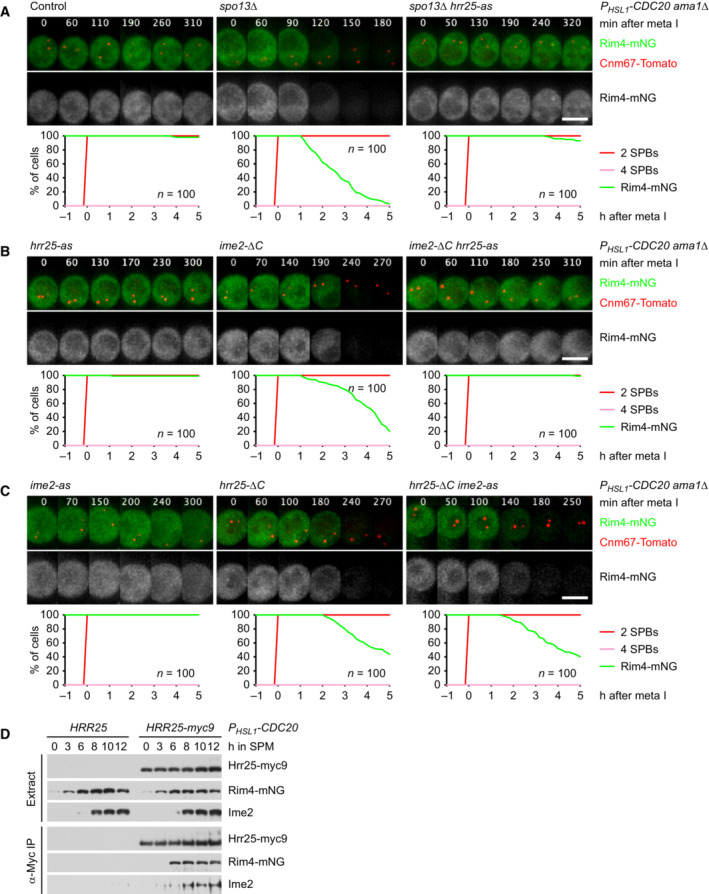
Consistent with previous work (Arguello‐Miranda et al, 2017), we find that accumulation of Ama1 and Clb3 at meiosis II depends on the activity of the casein kinase 1δ (CK1δ) Hrr25 (Appendix Fig S4). Accordingly, inhibition of Hrr25 blocks Rim4 degradation in metaphase I‐arrested spo13Δ mutants (Fig EV3A). This raises the question of where Hrr25 resides in the pathway controlling Rim4 degradation. While Ime2‐ΔC induces Rim4 degradation even when Cdc5 is inhibited (Fig 4C), it fails to do so when Hrr25 is inhibited (Fig EV3B). Thus, Hrr25 might function together with or downstream of Ime2. To test this, we generated a hyperactive Hrr25 kinase by removing the C‐terminal, autoinhibitory region common to CK1δ‐orthologues (Graves & Roach, 1995; Cegielska et al, 1998; Elmore et al, 2018). Hrr25‐ΔC has little effect on proliferation or meiotic nuclear division in otherwise normal cells (Appendix Fig S3B). On the other hand, it induces AMA1 translation in metaphase I‐arrested cells (Appendix Fig S3C). Importantly, Hrr25‐ΔC induces Rim4 degradation even when Ime2 is inhibited (Fig EV3C), placing Hrr25 downstream of Ime2. Consistent with this, Hrr25 binds to Ime2 and Rim4 (Fig EV3D). Taken together, our data suggest that Cdc5‐Spo13 inhibits AMA1 translation by preventing the Cdc5‐Ime2‐Hrr25 kinase cascade from inducing the degradation of Rim4 (Fig 5C). Recent work suggests that Rim4 is removed by autophagy (Wang et al, 2020; Herod et al, 2022), and that Hrr25 is required for different pathways of selective autophagy (Farre & Subramani, 2016). Interestingly, Hrr25 binds the Rab‐family GTPase Ypt1/Rab1, which activates Hrr25 and recruits it to the phagophore assembly site (PAS; Wang et al, 2015). Once activated, Hrr25 might sustain autophagic degradation of Rim4 independently of Ime2 and Cdc5.
Figure EV3. Regulation of Rim4 degradation by Hrr25.
-
A–CImaging of Rim4‐mNG and SPBs (Cnm67‐Tomato) in metaphase I‐arrested P HSL1 ‐CDC20 ama1Δ strains. Top, time‐lapse series. Bottom, the presence of mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0). (A) Inhibition of Hrr25 activity prevents Rim4 degradation elicited by the SPO13 deletion. Hrr25‐as was inhibited with 1NM‐PP1 at 7 h in SPM. (B) Inhibition of Hrr25 prevents Rim4 degradation induced by hyperactive Ime2‐ΔC. Hrr25‐as was inhibited with 1NM‐PP1 at 7 h in SPM. (C) Rim4 is degraded with similar timing in hrr25‐ΔC cells with active or inactive Ime2 (P = 0.91, Welch's t‐test). Ime2‐as was inhibited with 1Na‐PP1 at 7 h in SPM.
-
DBinding of Rim4‐mNG and Ime2 to Hrr25‐myc9 immunoprecipitated with α‐Myc antibodies from extracts of P HSL1 ‐CDC20 cells.
Data information: Data are representative of two (A and B) or three (C) independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Cdk1‐Clb1 is required for the persistence of Rim4 at metaphase I
Cells lacking the M‐phase cyclin Clb1 resemble spo13Δ mutants in several aspects: First, the CLB1 deletion prolongs metaphase I, which lasts for up to 200 min (mean, 81 min; control, 25 min), probably due to inefficient activation of APC/CCdc20 (Rudner & Murray, 2000; Appendix Fig S5A). clb1Δ cells undergo either one (47%) or two (53%) rounds of Cdc14 release and nuclear division, whereby cells with a long metaphase I tend to undergo only one round (Appendix Fig S5B). Second, deletion of CLB1, but not of CLB3 or CLB4, results in nuclear division in Cdc20‐depleted cells (Oz et al, 2022). Indeed, the CLB1 deletion causes P HSL1 ‐CDC20 cells to advance the accumulation of Ama1 by 2 h, and the degradation of APC/C substrates as well as nuclear division depends on Ama1 (Fig 6A). The CLB1 deletion also advances the appearance of Clb3, which is more obvious in P HSL1 ‐CDC20 ama1Δ strains. Third, consistent with Ama1‐dependent proteolysis of Spo13, P HSL1 ‐CDC20 clb1Δ cells degrade Rim4 (Fig 6B, left). We conclude that Clb1 as well as Spo13 is required for suppressing the accumulation of Ama1 and Clb3 at metaphase I.
Figure 6. Cdk1‐Clb1 is required for preventing Ama1 accumulation at metaphase I.
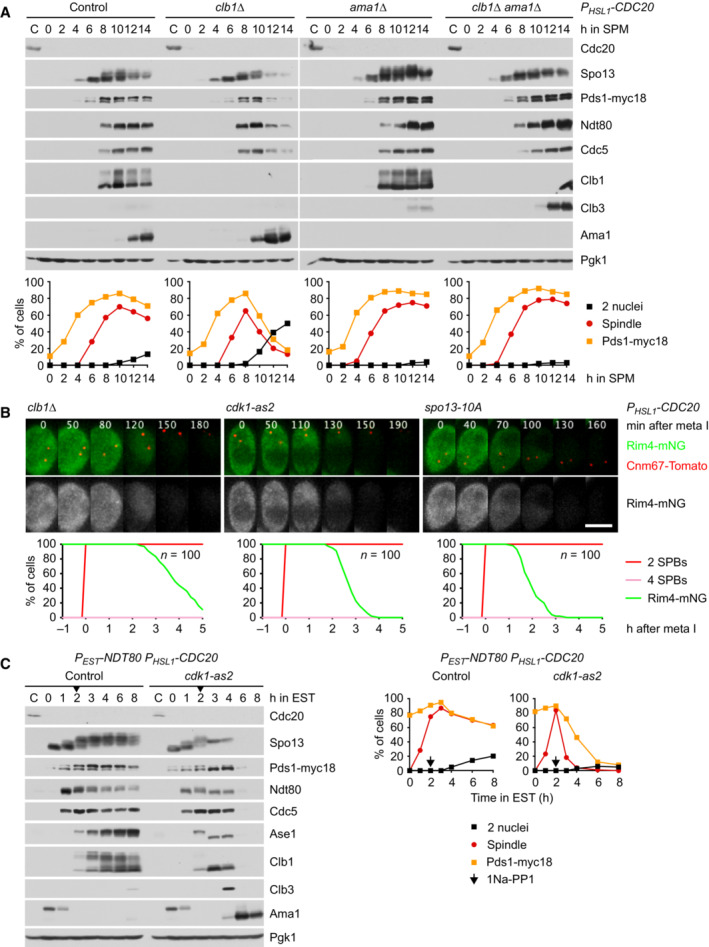
- Deletion of CLB1 causes Ama1‐dependent proteolysis and nuclear division in P HSL1 ‐CDC20 cells. Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
- Imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato) in P HSL1 ‐CDC20 cells carrying clb1Δ, cdk1‐as2, or spo13‐10A. Cdk1‐as2 was inhibited with 1Na‐PP1 at 7.5 h in SPM. Top, time‐lapse series. Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
- Inhibition of Cdk1 at metaphase I causes accumulation of Ama1 and degradation of APC/C substrates. P EST ‐NDT80 P HSL1 ‐CDC20 strains were released from prophase with estradiol (EST, t = 0), and Cdk1‐as2 was inhibited at metaphase I (1Na‐PP1 at t = 120 min, arrows). Left, immunoblot detection of proteins. Right, progression of meiosis quantified in fixed cells.
Data information: (B) is representative of two independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
To test whether the function of Clb1 depends on Cdk1 activity, we inhibited analogue‐sensitive Cdk1‐as2 (Bishop et al, 2000) shortly after NDT80 P HSL1 ‐CDC20 cells had entered metaphase I. This causes accumulation of Ama1 and Clb3, which is followed by the degradation of APC/C substrates, while spindle disassembly prevents nuclear division (Fig 6C). Live‐imaging revealed that inhibition of Cdk1 causes P HSL1 ‐CDC20 cells to degrade Rim4 (Fig 6B, middle). Thus, the consequences of inactivating Cdk1‐Clb1 resemble those of deleting SPO13. One possibility is that Cdk1 activity is required for an aspect of Spo13 function. We have recently shown that Spo13 requires phosphorylation by Cdk1 in order to suppress spore formation in Cdc20‐depleted cells. A non‐phosphorylatable mutant, called spo13‐10A, results in premature degradation of Spc72, the repressor of spore formation (Oz et al, 2022). Indeed, P HSL1 ‐CDC20 spo13‐10A cells also degrade Rim4 (Fig 6B, right). While Rim4 degradation leads to Ama1 accumulation and Spo13 degradation, deletion of CLB1, inhibition of Cdk1, and the spo13‐10A mutation also cause Rim4 degradation in P HSL1 ‐CDC20 ama1Δ cells (Appendix Fig S5C and D). This confirms that Cdk1‐Clb1 promotes not only the stability but also the activity of Spo13. We conclude that phosphorylation by Cdk1‐Clb1 is required for Spo13's ability to prevent Rim4 degradation and thereby AMA1 translation at metaphase I (Fig 5C). Cdk1‐Clb1 might be assisted in this task by Cdk1‐Clb3 since Clb3 is translated together with Ama1. However, we found no evidence for such a mechanism: Deletion of CLB3 does not affect Rim4 degradation, even in clb1Δ cells (Appendix Fig S5C).
Cdk1‐Clb1 inhibits the function of Ama1 as an APC/C activator
In addition to repressing the translation of AMA1, Spo13 also inhibits the activity of the Ama1 protein (Fig EV1D). Similarly, Cdk1 can inhibit Ama1‐dependent proteolysis independently of its effect on Ama1 synthesis. For instance, expression of Clb1 in prophase‐arrested cells represses Ama1‐dependent proteolysis without affecting Ama1 levels (Okaz et al, 2012). This raises the question of how inhibition of Ama1 by Cdk1 is integrated with that by Spo13. Below, we first investigate the mechanism of Ama1's inhibition by Cdk1 and then turn to the role of Spo13.
APC/C activators carry two motifs essential for binding to the APC/C: the C‐box in the N‐terminal region and the Ile‐Arg tail at the C‐terminus. Cdc20 and Cdh1 dissociate from the APC/C upon Cdk1‐dependent phosphorylation of multiple sites around the C‐box (Zachariae et al, 1998; Schwab et al, 2001; Labit et al, 2012; Chang et al, 2015). Thus, we mutated the six Ser/Thr‐Pro motifs in Ama1's N‐terminal region to non‐phosphorylatable Ala‐Pro. AMA1‐6A single mutants produce four spores (viability, 96%), implying that the mutation has little effect in otherwise normal cells. In P HSL1 ‐CDC20 cells, however, Ama1‐6A elicits degradation of APC/C substrates and nuclear division, while similar levels of Ama1 have little effect (Fig 7A). Thus, Ama1‐6A is a more potent activator than the wild‐type protein. Ama1‐6A also induces Rim4 degradation and therefore accumulates 2 h earlier than Ama1 (Fig 7A and B). Ama1‐6A mediates Rim4 degradation by targeting for proteolysis not only Spo13 but also Clb1; Rim4 is only stable in the presence of non‐degradable versions of both Spo13 and Clb1 (Fig 7C), which is consistent with the notion that Spo13 function requires Cdk1‐Clb1 activity. Rim4 degradation induced by Ama1‐6A depends on the activities of Cdc5, Ime2, and Hrr25 (Fig 7D), indicating that it requires signalling through the Cdc5‐Ime2‐Hrr25 kinase cascade. These data suggest that phosphorylation restrains the synthesis as well as the activity of Ama1 (Fig 5C).
Figure 7. Analysis of AMA1‐6A mutants.
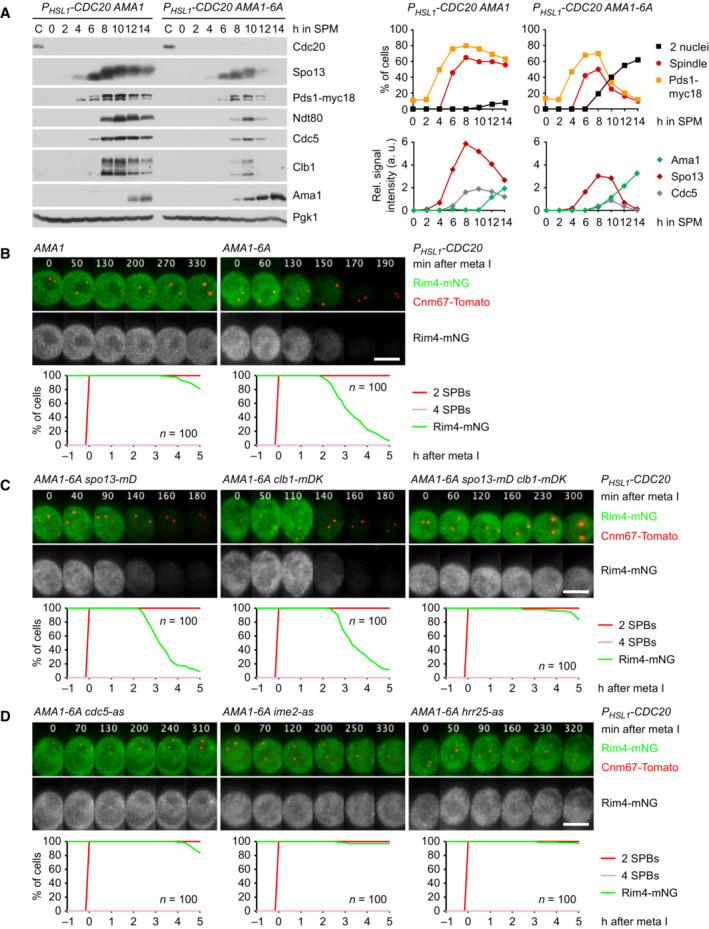
-
AAma1‐6A causes degradation of APC/C substrates and nuclear division in P HSL1 ‐CDC20 cells. Left, immunoblot detection of proteins. Top right, progression of meiosis quantified in fixed cells. Bottom right, relative signal intensities of proteins.
-
B–DAnalysis of Rim4 degradation in P HSL1 ‐CDC20 cells carrying AMA1‐6A. Top, time‐lapse series from the filming of Rim4‐mNG and SPBs (Cnm67‐Tomato). Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0). (B) Ama1‐6A causes Rim4 degradation. (C) Rim4 degradation is not affected by either spo13‐mD (P = 0.86) or P DMC1 ‐clb1‐mDK (P = 0.20) alone, but is blocked in the presence of both alleles (P < 0.0001, Welch's t‐test). Cells in (B) and (C) were filmed together. (D) Rim4 degradation depends on the activities of Cdc5, Ime2, and Hrr25. Analogue‐sensitive kinases were inhibited at 8 h in SPM.
Data information: Data are representative of four (B), two (C), or three (D) independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
To investigate whether inhibition of APC/CAma1 involves an interaction with CDKs, we immunoprecipitated the APC/C with antibodies to Apc2. Of the five B‐type cyclins expressed at meiosis (Clb1, 3, 4, 5, 6; Chu & Herskowitz, 1998), only Clb1 and Clb3 copurify with the APC/C (Fig EV4A). These interactions are unlikely to reflect substrate recognition by the APC/C because the degrons of Clb1 are dispensable for binding to the APC/C (Fig EV4B). On the other hand, deletion of AMA1 reduces Clb1's association with the APC/C (Fig EV4C), suggesting that most of Clb1/3 copurifies with the APC/C via binding to Ama1. To investigate this, we purified Ama1‐GFP from extracts of Cdc20‐depleted cells. Ama1‐GFP does not bind the APC/C because C‐terminal tags prevent the Ile‐Arg tail from accessing its receptor (Vodermaier et al, 2003; Matyskiela & Morgan, 2009; Chang et al, 2015). Mass spectrometry identified Clb1, Clb3, and Cdk1 as high‐scoring interactors of Ama1‐GFP, whereas Clb4 and APC/C subunits show little or no binding (Fig EV4D). Specific binding of Cdk1‐Clb1/3 was also observed in immunoprecipitations of Ama1‐myc9 (Fig EV4E). These interactions do not depend on Spo13 (Appendix Fig S6A and B), raising the question of whether Spo13 is required for the ability of Cdk1‐Clb1 to inhibit Ama1. When expressed in prophase‐arrested ndt80Δ cells, Clb1 inhibits Ama1 in a Cdk1‐dependent manner, and cells produce M phase regulators under the control of the mitosis‐specific transcription factor Ndd1 (Okaz et al, 2012). Clb1 also inhibits Ama1 in ndt80Δ spo13Δ cells (Appendix Fig S6C), suggesting that Cdk1‐Clb1 can inhibit Ama1 independently of Spo13, provided that AMA1 transcription is low (i.e., not induced by Ndt80). Despite binding to Ama1, Clb3 does not inhibit Ama1‐dependent proteolysis when expressed in ndt80Δ cells (Appendix Fig S6D). Accordingly, deletion of CLB3 does not elicit nuclear division in Cdc20‐depleted cells (Oz et al, 2022). Ama1‐GFP also binds Acm1 (Fig EV4D), which has been shown to inhibit Cdh1 at mitosis (Dial et al, 2007; Enquist‐Newman et al, 2008). However, Acm1 does not play a role in suppressing Ama1's activity and synthesis at metaphase I since deletion of ACM1 does not affect Rim4 stability in P HSL1 ‐CDC20 or P HSL1 ‐CDC20 AMA1‐6A cells (Appendix Fig S6E). Taken together, our data suggest that Cdk1‐Clb1 employs two mechanisms to restrain Ama1‐dependent proteolysis at metaphase I (Fig 5C): First, by activating Spo13, it prevents the degradation of Rim4 and thereby the translation of AMA1. Second, by phosphorylating Ama1, it inhibits Ama1's activity. Consistent with this idea, APC/C only binds the unmodified form of Ama1 (Appendix Fig S2B). This brings back the question of how Spo13 inhibits the activity of Ama1.
Inhibition of APC/CAma1 requires phosphorylation of Clb1 by Cdc5‐Spo13
While Cdk1‐Clb1 inhibits the synthesis and activity of Ama1, Clb1 is also a substrate of APC/CAma1 (Okaz et al, 2012; Arguello‐Miranda et al, 2017). This constitutes a double‐negative feedback loop, which generates two, mutually exclusive states: a state of low APC/CAma1 and high Cdk1 activity that is characteristic of metaphase I and a state of high APC/CAma1 and low Cdk1 activity, which resembles anaphase II. The low‐Ama1/high‐Cdk1 state could be stabilized by rendering Clb1 less susceptible to Ama1‐dependent proteolysis (Fig 5C). It is noteworthy, therefore, that Clb1 shows multiple bands of reduced electrophoretic mobility at metaphase I but not at metaphase II or mitosis (Attner et al, 2013; Tibbles et al, 2013). This modification correlates with the levels of Spo13 since it is enhanced and persists longer in spo13‐mD cells (Fig 2B). Indeed, Clb1's modification depends on Spo13 (Fig 2A and C), the binding of Spo13 to Cdc5 (Fig EV2B), and the catalytic activity of Cdc5 (Fig 5A). It also requires the activity of Cdk1, the kinase that activates Spo13 (Fig 6C). Remarkably, Cdc5 still binds but cannot modify Clb1 in the absence of Spo13 (Fig EV5A). We conclude that the Cdc5‐Spo13 complex is an active kinase that phosphorylates Clb1.
Figure EV5. Analysis of Cdc5/Spo13‐dependent phosphorylation of Clb1.
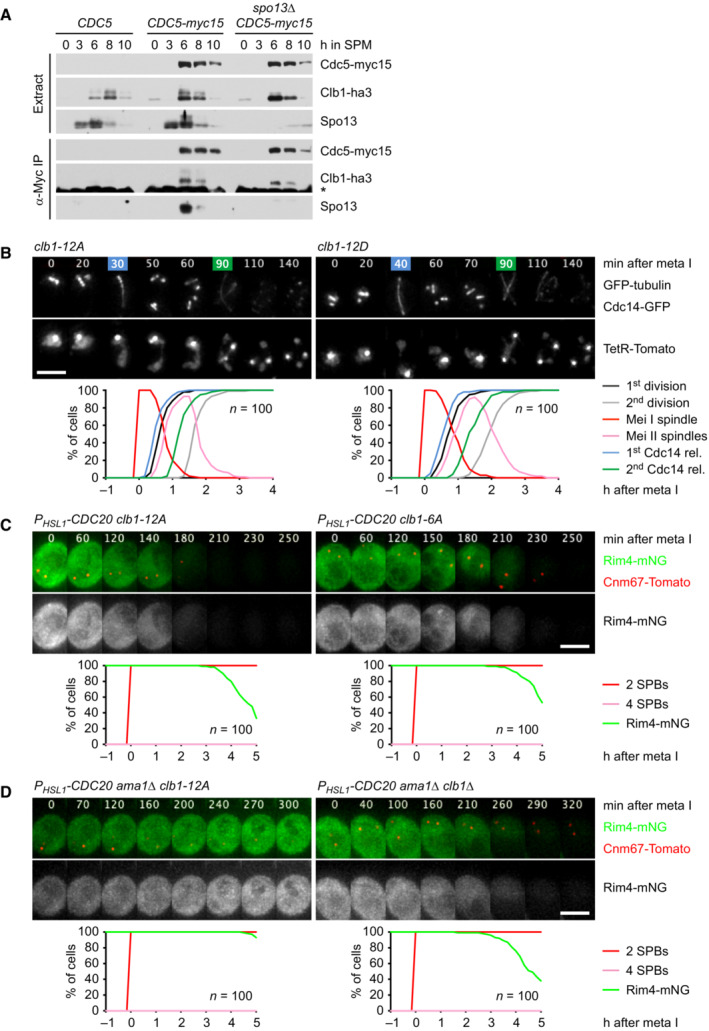
- Clb1‐ha3 binds to Cdc15‐myc15 in α‐Myc immunoprecipitations from extracts of control and spo13Δ cells.
- Meiosis in clb1‐12A and clb1‐12D cells. Top, time lapse series from the imaging of spindles (GFP‐tubulin), nucleolar release of Cdc14‐GFP, and TetR‐Tomato, which labels the nucleoplasm (diffuse signal) and the centromeres of one copy of chromosome V (dots). First (blue) and second (green) Cdc14 release are marked. Bottom, meiotic events quantified in cells synchronized in silico to spindle formation at metaphase I (t = 0). The duration of metaphase I is similar in clb1‐12A and clb1‐12D cells (P = 0.33).
- P HSL1 ‐CDC20 clb1‐12A cells degrade Rim4 earlier than P HSL1 ‐CDC20 clb1‐6A cells (P < 0.0001). Top, time‐lapse series from the imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato). Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
- Clb1 phosphorylation is not required for the activation of Spo13. Top, time‐lapse series from the imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato) in P HSL1 ‐CDC20 ama1Δ cells carrying clb1‐12A or clb1Δ. Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
Data information: Data are representative of two (B and C) or three (D) independent experiments. Means were compared using Welch's t‐test. Scale bar, 4 μm.
Source data are available online for this figure.
To identify phosphorylation sites, we purified GFP‐tagged Clb1 from P SCC1 ‐CDC20 ama1Δ strains arrested at metaphase I (Appendix Fig S7A). Mass spectrometry revealed phosphorylation of eight serines in Clb1‐GFP from control cells. These phosphorylation sites were not detected or only at low intensity in Clb1‐GFP from cells lacking Spo13 or Cdc5, although the amounts of Clb1‐GFP were similar to those obtained from control cells (Appendix Fig S7B–D). Seven of these serines and four additional serines were also found to be phosphorylated in Clb1‐GFP purified from P EST ‐NDT80 cells progressing into metaphase I (Appendix Fig S8A). Phosphorylation site intensities are strongly reduced in Clb1‐GFP from spo13Δ cells at metaphase I and control cells at metaphase II (Appendix Fig S8B–D). The 12 phospho‐serines are located in Clb1's N‐terminal, intrinsically disordered region, but not the conserved cyclin‐box domain (Fig 8A; Varadi et al, 2022). The N‐terminal region contains 22 serines and 10 threonines (Appendix Fig S8E), indicating that at least in Clb1, Cdc5‐Spo13 prefers serine to threonine. We mutated the phosphorylated serines to either alanine or aspartate to generate non‐phosphorylatable Clb1‐12A and phospho‐mimicking Clb1‐12D, respectively. clb1‐12A and clb1‐12D mutants markedly differ from clb1Δ cells in that they undergo two divisions and produce four spores with high viability (99%; Fig EV5B). This suggests that Clb1‐12A and Clb1‐12D are capable of activating Cdk1.
Figure 8. Analysis of CLB1 phosphorylation site mutants.
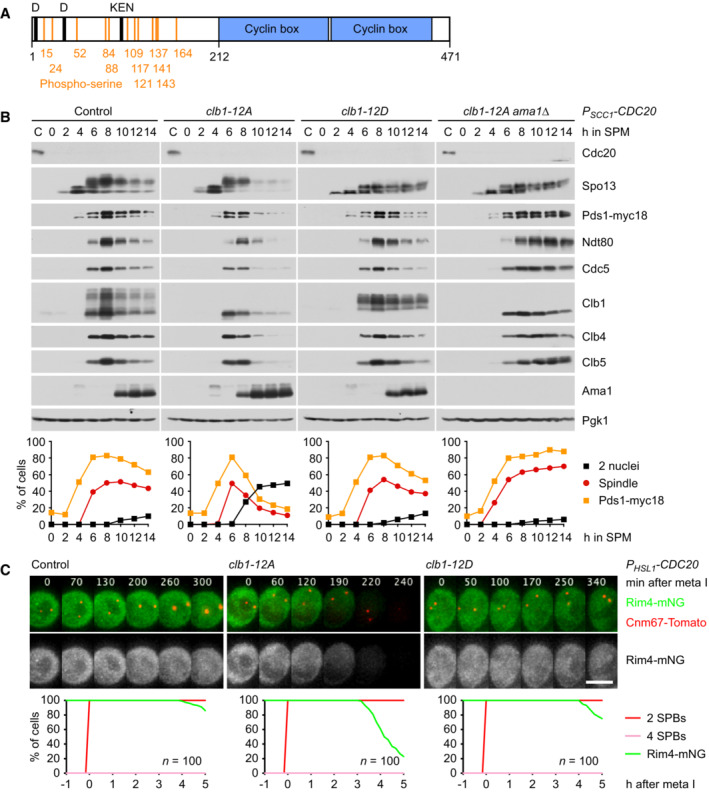
- Domain structure of Clb1 showing D‐ and KEN‐boxes (black), phospho‐serines (orange), and cyclin boxes (blue).
- clb1‐12A but not clb1‐12D causes Ama1‐dependent proteolysis and nuclear division in Cdc20‐depleted cells. Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
- Imaging of Rim4‐mNG and SPBs (Cnm67‐Tomato) in P HSL1 ‐CDC20 control cells and cells carrying clb1‐12A or clb1‐12D. Top, time‐lapse series. Bottom, the presence of Rim4‐mNG quantified in cells synchronized in silico to SPB separation at metaphase I (t = 0).
Data information: (C) is representative of three independent experiments. Scale bar, 4 μm.
Source data are available online for this figure.
Next, we introduced clb1‐12A and clb1‐12D into P HSL1 ‐CDC20 cells. Immunoblotting revealed that mutating these serines either eliminates (Clb1‐12A) or strongly reduces (Clb1‐12D) the modification of Clb1, confirming that phosphorylation is confined to Clb1's N‐terminal region (Fig 8B). However, Clb1‐12A and Clb1‐12D show strikingly different properties: Clb1‐12A is more sensitive to APC/CAma1 activity than wild‐type Clb1, being degraded as soon as Ama1 appears. As a result, P HSL1 ‐CDC20 clb1‐12A cells degrade APC/C substrates and undergo nuclear division in an Ama1‐dependent manner (Fig 8B). Furthermore, these cells degrade Rim4, which in turn advances the accumulation of Ama1 (Fig 8C). Thus, Ama1's ability to target Clb1‐12A for proteolysis unleashes feedback loops, which cause Ama1 to increase its own activity and synthesis through the degradation of Clb1 and Spo13 (Fig 5C). We also analyzed clb1‐6A in which only six high‐confidence phosphorylation sites are replaced by alanine (the first six sites in Appendix Fig S7C). P HSL1 ‐CDC20 clb1‐6A cells degrade Rim4 more slowly than cells carrying clb1‐12A (Fig EV5C), indicating that most, if not all, of the 12 phospho‐serines contribute to suppressing Ama1's activity and synthesis. Indeed, phospho‐mimetic Clb1‐12D persists in the presence of Ama1, similar to wild‐type Clb1 (Fig 8B). Clb1‐12D is therefore able to inhibit Ama1‐dependent proteolysis, so that APC/C substrates persist and nuclear division is blocked. P HSL1 ‐CDC20 clb1‐12D cells also maintain high levels of Rim4, which prevents premature synthesis of Ama1 (Fig 8C). These data suggest that phosphorylation of Clb1 by Cdc5‐Spo13 restrains Ama1's ability to increase its own activity and synthesis at metaphase I (Fig 5C). Phosphorylation of Clb1 might enable Cdk1‐Clb1 to inhibit Ama1's function as an APC/C activator or it might allow Cdk1‐Clb1 to activate Spo13. To test the latter possibility, we imaged Rim4‐mNG in P HSL1 ‐CDC20 ama1Δ strains (Fig EV5D). Rim4 persists in cells carrying clb1‐12A but is degraded in the clb1Δ mutant, suggesting that activation of Spo13 requires Cdk1‐Clb1 activity but not the phosphorylation of Clb1. This finding indicates that phosphorylation enables Clb1 to inhibit Ama1's activity, which in turn stabilizes not only Clb1 but also Spo13.
Phosphorylation of Clb1 reduces its Ama1‐dependent degradation
Our data imply that phosphorylation, or the phospho‐mimicking mutations, protects Clb1 from Ama1‐dependent proteolysis. To investigate the extent of this protection, we arrested ndt80Δ cdk1‐as1 strains at prophase and added 1NM‐PP1 to inhibit Cdk1. Under these conditions, APC/CAma1 is active towards Clb1 and other M phase regulators (Okaz et al, 2012). We expressed Clb1 from the early meiosis‐specific DMC1 promoter and followed its degradation upon inhibition of translation by cycloheximide (Fig 9A). Wild‐type Clb1 and Clb1‐12A are degraded with a half‐life of 5–6 min (P = 0.14). Thus, at prophase, when Cdc5‐Spo13 is absent, wild‐type Clb1 behaves similarly to Clb1‐12A. However, Clb1‐12D is degraded more slowly, showing a half‐life of 12 min (P < 0.001). Upon deletion of AMA1, all Clb1 variants are stabilized, displaying half‐lives of ~57 min. These data suggest that phosphorylation of Clb1 by Cdc5‐Spo13 results in a 2.5‐fold reduction in Clb1's degradation rate. While modest, the increase in Clb1's stability supports feedback loops that reinforce the stability and phosphorylation of Spo13 and thereby inhibit the synthesis as well as the activity of Ama1 (Fig 5C). Accordingly, deletion of SPO13 advances the accumulation of Ama1 in P HSL1 ‐CDC20 clb1‐12D cells, resulting in the degradation of APC/C substrates and nuclear division (Fig 9B). In fact, spo13Δ cells degrade Clb1‐12D as fast as Clb1‐12A, suggesting that the phospho‐mimicking mutations provide little protection from Ama1‐dependent degradation once Ama1 synthesis has been induced (Fig 9B). Taken together, our data show that mutual phosphorylation of Cdc5‐Spo13 and Cdk1‐Clb1 integrates and amplifies the pathways that repress Ama1 synthesis and activity at metaphase I (Fig 5C).
Figure 9. Protection of Clb1 and Spo13 from Ama1‐dependent proteolysis.
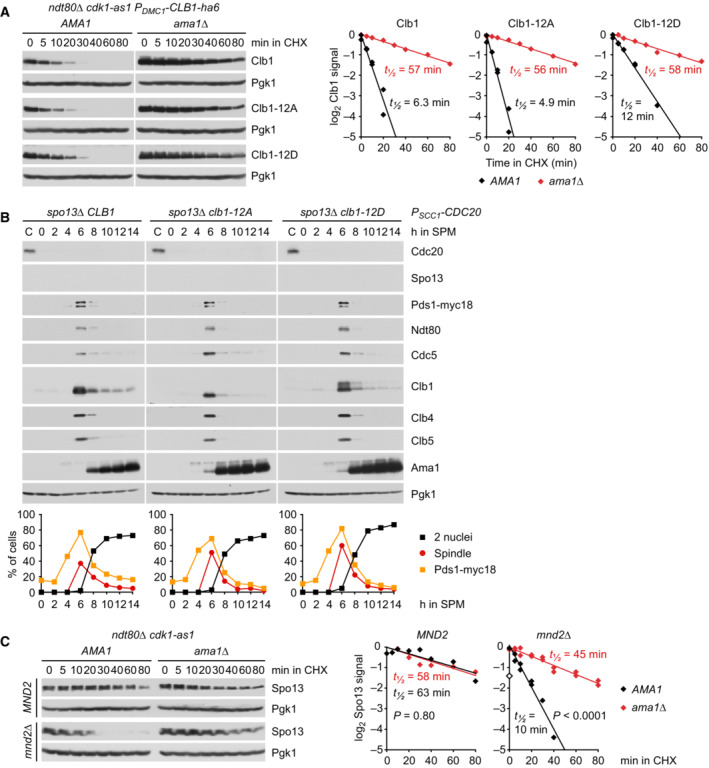
- Stability of Clb1, Clb1‐12A, and Clb1‐12D. CLB1 alleles were expressed from the DMC1 promoter in ndt80Δ cdk1‐as1 strains carrying AMA1 or ama1Δ. Strains were arrested at prophase in the presence of 1NM‐PP1 to inhibit Cdk1‐as1. At 6 h in SPM, translation was inhibited with cycloheximide (CHX, t = 0). Left, immunoblot detection of Clb1. Right, log2‐transformed Clb1 signal intensities (normalized to t = 0) plotted over time in CHX. Half‐lives were calculated by linear regression.
- Analysis of APC/C substrates in P SCC1 ‐CDC20 cells carrying spo13Δ and CLB1, clb1‐12A, or clb1‐12D. Top, immunoblot detection of proteins. Bottom, progression of meiosis quantified in fixed cells.
- Spo13 stability at prophase. ndt80Δ cdk1‐as1 strains carrying AMA1 or ama1Δ were arrested at prophase in the presence of 1NM‐PP1 to inhibit Cdk1‐as1. At 6 h in SPM, translation was inhibited with CHX (t = 0). mnd2Δ cells expressing Spo13 from the DMC1 promoter were processed in parallel. Left, immunoblot detection of Spo13. Right, log2‐transformed Spo13 signal intensities (normalized to t = 0) plotted over time in CHX. Half‐lives were calculated by linear regression. Open diamond, data point omitted from regression.
Data information: (A) AMA1 data are from two independent experiments. R 2 values of AMA1 and ama1Δ regression lines: Clb1, 0.92 and 0.99; Clb1‐12A, 0.95 and 0.99; Clb1‐12D, 0.99 and 0.97. (C) mnd2Δ data are from two independent experiments. R 2 values of AMA1 and ama1Δ regression lines: MND2, 0.75 and 0.84; mnd2Δ, 0.98 and 0.95. Slopes were compared using ANCOVA.
Source data are available online for this figure.
Restoring a two‐division meiosis in spo13Δ mutants
Deletion of AMA1 might be expected to restore a second division in spo13Δ cells, which implies, however, that Ama1 differs from Cdc20 by its regulation but not its range of substrates. This is not the case: spo13Δ ama1Δ cells undergo a single division (Appendix Fig S9A). Indeed, deletion of AMA1 causes accumulation of Ndt80, which results in higher levels of M phase regulators, including Cdc5 and Cdc20 (Okaz et al, 2012; Appendix Fig S9C). This might increase the activity of APC/CCdc20, in particular after a prolonged metaphase I. While APC/CCdc20 mediates a division in spo13Δ ama1Δ cells, it might subsequently prevent a second one. To test this idea, we tagged Ndt80 with AID but found that the tag reduces Ndt80 levels about tenfold, even in the absence of OsTir1 (Appendix Fig S9D). While Ndt80‐AID has little effect in otherwise normal cells, it causes 70% of spo13Δ ama1Δ cells to undergo two divisions (Appendix Fig S9B). Ndt80‐AID also restores two divisions in 50% of spo13Δ cells, probably because it reduces the activity of APC/CCdc20 as well as APC/CAma1. Thus, a two‐division meiosis depends on the mutual regulation between APC/CCdc20 and APC/CAma1 via Spo13 and Ndt80.
Mnd2 protects Spo13 from Ama1‐dependent degradation at prophase
At entry into metaphase I, Ndt80 induces the transcription of AMA1 and CDC5 (Fig 2C). To suppress the translation of AMA1 mRNA, Cdc5‐Spo13 must prevent free Cdc5 from inducing the degradation of Rim4. Indeed, Spo13 accumulates at prophase, that is, before Cdc5 (Fig 2A; Matos et al, 2008). However, APC/CAma1 is active, albeit at low levels, at this stage of meiosis (Okaz et al, 2012). Similar to Pds1 and Clb5, Spo13 might be protected from Ama1‐dependent proteolysis by the inhibitory APC/C subunit Mnd2 (Oelschlaegel et al, 2005; Penkner et al, 2005). To test this idea, we measured Spo13's stability in prophase‐arrested cells. In cells containing Mnd2, Spo13 shows a half‐life of ~60 min in the presence and absence of Ama1 (Fig 9C). In mnd2Δ cells, however, Spo13 is targeted for degradation by Ama1 (half‐life, 10 min). Accordingly, Mnd2 is required for normal levels of Spo13 at prophase (Appendix Fig S10). By protecting Spo13, but not Cdc5, from Ama1‐dependent proteolysis at prophase, Mnd2 ensures that Cdc5‐Spo13 is formed as soon as Cdc5 appears at entry into metaphase I.
Discussion
Meiosis I and ‐II must have different outcomes in order to achieve genome haploidization and gamete differentiation. For instance, exit from meiosis must be initiated at meiosis II to generate a two‐division meiosis. However, mRNAs for cell cycle regulators and differentiation factors are produced by a transcriptional program shortly before the first division. This raises the question of how proteins involved in meiosis II‐specific events are translated or activated not at meiosis I but at meiosis II. We argue that the spo13 mutant is key to answering this question because it fails to repress meiosis II‐specific events at meiosis I. Indeed, spo13 cells perform only one division because they prematurely activate the meiotic exit machinery.
The spo13 mutant fails to repress meiosis II‐specific processes at meiosis I
The spo13 mutant was among the first single‐division mutants to be discovered more than 40 years ago (Klapholz & Esposito, 1980a). It has attracted attention mainly because defects in sister kinetochore mono‐orientation and centromeric cohesin protection suggest that Spo13 coordinates centromere‐related processes unique to meiosis I (Katis et al, 2004; Lee et al, 2004; Matos et al, 2008). However, disabling these processes does not result in a single‐division meiosis (Toth et al, 2000; Rabitsch et al, 2003), suggesting that Spo13 has additional functions. A clue to these functions is the observation that despite of undergoing a single wave of Cdk1 and APC/C activity, spo13Δ cells show aspects of meiosis I and ‐II. For instance, the spo13Δ‐division is preceded by recombination, a process associated with meiosis I, but concluded by exit from meiosis and spore formation, which are meiosis II‐specific processes (Klapholz & Esposito, 1980b). We show that spo13Δ cells prematurely degrade the translational repressor Rim4 (Fig 3) and therefore synthesize meiosis II‐specific proteins, including Ama1 and sporulation factors (Figs 2 and EV1). spo13Δ cells also fail to produce phosphorylated Clb1, an inhibitor of Ama1 activity (Figs 2 and 8). Thus, activation of APC/CAma1 at metaphase I causes spo13Δ cells to exit from meiosis after a single division. Furthermore, spo13Δ cells prematurely degrade Spc72, the repressor of meiotic plaque (MP) assembly, which, together with the translation of sporulation factors, leads to spore formation at meiosis I (Oz et al, 2022). A major defect of spo13Δ cells is therefore the inability to repress meiosis II‐specific events at meiosis I.
This hypothesis can also be applied to chromosome segregation. Sister kinetochore orientation is governed by two processes: Monopolin's loading onto kinetochores at prophase is required for mono‐orientation at meiosis I, while its removal at the onset of metaphase II allows bi‐orientation at meiosis II (Toth et al, 2000). spo13Δ cells fail to retain monopolin at kinetochores at metaphase I (Katis et al, 2004; Lee et al, 2004), which can be interpreted as a failure to repress the removal of monopolin. Likewise, the stepwise cleavage of cohesin depends on two processes: the protection of centromeric cohesin by Sgo1 at anaphase I and its de‐protection through APC/C‐dependent removal of Sgo1 from centromeres at anaphase II (Riedel et al, 2006; Mengoli et al, 2021). spo13Δ cells loose Sgo1 from centromeres at metaphase I (Katis et al, 2004; Lee et al, 2004), which can be viewed as a defect in repressing the removal of Sgo1. Therefore, most if not all phenotypes of the spo13Δ mutant can be explained by a single, testable hypothesis, namely premature inactivation of mechanisms that repress meiosis II‐specific events at meiosis I.
A striking feature of the spo13Δ phenotype is its dependence on the duration of metaphase I. spo13Δ cells undergo two divisions when metaphase I is short but a single division when metaphase I is prolonged due to inhibition of Cdc20. In spo13Δ cells, Cdc20 is inhibited by the SAC. The SAC might respond to bivalent chromosomes that cannot balance spindle forces because they carry a bioriented and a mono‐oriented pair of sister kinetochores (Nerusheva et al, 2014). In clb1Δ mutants, Spo13 is largely inactive, while activation of APC/CCdc20 is compromised. Thus, the single‐division meiosis of spo13 mutants results from two conditions: reduced Spo13 function and delayed APC/CCdc20 activation. The prolonged metaphase I of spo13Δ cells (66 min) allows meiosis II‐specific events to unfold. By contrast, metaphase I is very short in spo13Δ mad2Δ cells (10 min). While ‘quick’ meiosis II‐events might still occur at meiosis I, more elaborate processes, such as Ama1 accumulation or spore formation, are only completed by the time these cells have progressed into meiosis II. Therefore, spo13Δ mad2Δ cells exit from meiosis and sporulate after the second division (Shonn et al, 2002). The time‐dependence of the spo13 phenotype highlights a problem unique to meiotic cells: To ensure a two‐division meiosis, SAC activity at metaphase I has to halt not only entry into anaphase I but also meiosis II‐specific events. This is achieved by Spo13, a meiosis I‐specific substrate of APC/CCdc20. Upon activation of the SAC at meiosis I, Spo13 is stabilized, which in turn prevents the inactivation of repressors of meiosis II‐specific events.
Ama1: a Cdh1‐related APC/C activator tailored to the regulation of meiosis
In yeast, exit from meiosis involves the APC/C activator Ama1, which is related to the G1‐specific activator Cdh1 but shows features tailored to meiotic cell cycle control. At prophase, low levels of Ama1 suppress the expression of M phase regulators by the mitosis‐specific transcription factor Ndd1, which renders entry into metaphase I dependent on Ndt80 and the completion of recombination (Okaz et al, 2012). At meiosis II, Ama1 accumulates to high levels and promotes exit from meiosis through the degradation of M phase regulators, such as Cdc5, B‐type cyclins, and Ndt80. Similar to Cdh1, Ama1 is phosphorylated at Cdk1 sites around the C‐box, which prevents binding of the activator to the APC/C. While Cdh1 is inhibited by different Cdk1‐Clb kinases and Ime2 (Zachariae et al, 1998; Jaspersen et al, 1999; Bolte et al, 2002; Holt et al, 2007), Ama1 is specifically inhibited by Cdk1‐Clb1, which allows Ama1 to be active at prophase. The mutual inhibition between Cdk1‐Clb1 and Ama1, known as a double‐negative feedback loop, results in two distinct states: a state of high Cdk1‐Clb1 and low Ama1 activity and a state where the reverse is true (Okaz et al, 2012). Entry into metaphase I requires conditions that enable Cdk1‐Clb1 to inhibit APC/CAma1, which entails induction of CLB1 transcription by Ndt80 when Ama1 levels are low. Conversely, the transition from the high‐ to the low‐Cdk1 state is initiated by Cdc20‐dependent proteolysis of Clb1 and an increase in Ama1 synthesis. A two‐division meiosis depends on this transition occurring at meiosis II but not at meiosis I. While AMA1 transcription is induced by Ndt80 (Chu et al, 1998; Primig et al, 2000), its translation is repressed at metaphase I by Rim4 (Fig 3A). However, translational repression becomes increasingly leaky because AMA1 mRNA accumulates throughout metaphase I (Fig 2C). The appearance of Ama1 poses a threat to the high‐Cdk1 state of metaphase I since APC/CAma1 eventually gains the ability to overwhelm its inhibitor Cdk1‐Clb1, resulting in premature exit from meiosis I. To stabilize metaphase I and allow its prolongation by the SAC, meiotic cells rely on the coordinated repression of Ama1 synthesis and activity by Spo13 (Fig 5C).
Spo13: activator or inhibitor of Cdc5/PLK1?
We show that Spo13 prevents the Ime2 kinase from promoting Rim4 degradation and thereby AMA1 translation at metaphase I. While Spo13's function depends on its ability to bind Cdc5, inhibition of Cdc5 activity blocks rather than elicits Rim4 degradation (Fig EV2C). At first glance, this result seems to imply that Spo13 inhibits Cdc5. We find, however, that Cdc5 exists in two forms at metaphase I, free Cdc5 and Cdc5 bound to Spo13, whereby the level of free Cdc5 greatly exceeds that of Cdc5‐Spo13 (Fig EV2D; Oz et al, 2022). Analysis of spo13Δ mutants shows that free Cdc5 is required for the ability of Ime2 to promote Rim4 degradation and Ama1 accumulation. Accordingly, Cdc5 binds and phosphorylates Ime2 independently of Spo13 (Oz et al, 2022; Fig 5A). The function of Cdc5‐Spo13 as an active kinase was revealed through the identification of a substrate, namely Clb1 (Appendix Figs S7 and S8). Phosphorylation of Clb1 depends on Spo13, Spo13's binding to Cdc5, and the kinase activity of Cdc5. Remarkably, Cdc5 binds, but cannot phosphorylate, Clb1 in the absence of Spo13. Thus, Clb1 is a substrate of the Cdc5‐Spo13 kinase but not of free Cdc5. Ime2 promotes another meiosis II‐specific process, namely the degradation of Spc72, which is a prerequisite for MP assembly and spore formation (Knop & Strasser, 2000; Oz et al, 2022). Ime2 activity requires Cdc5 and is inhibited by Spo13 in a manner dependent on its binding to Cdc5. Were it not for sub‐stoichiometric amounts of Spo13, it could be argued, again, that Spo13 inhibits Cdc5. However, MP assembly is also inhibited by a second, Spc72‐independent pathway, which requires Cdc5 activity as well as Spo13 and therefore depends on an active Cdc5‐Spo13 kinase (Oz et al, 2022). Thus, a simple model posits that Ime2 is inhibited by Cdc5‐Spo13 and activated by free Cdc5 (Fig 5C). At metaphase I, inhibition by Cdc5‐Spo13 dominates over activation by free Cdc5. At anaphase I, the activating function of free Cdc5 is unleashed by the degradation of Spo13. This model for the regulation of Ime2 is reminiscent of the phospho‐regulation of Cdk1‐cyclin B. Cdk1 is subject to inhibitory phosphorylation by the Wee1 kinase and activating phosphorylation by Cdk1‐activating kinase (CAK; Perry & Kornbluth, 2007). Cdk1 is inhibited until the inhibitory phosphorylation is removed upon activation of the Cdc25 phosphatase and the inactivation of Wee1.
How does Cdc5‐Spo13 recognizes its substrates? Cdc5/PLK1 binds to a phosphopeptide motif within the substrate via its C‐terminal PBD, which releases an autoinhibitory interaction between kinase domain and PBD (Elia et al, 2003a, 2003b; Xu et al, 2013). Interestingly, Spo13 itself binds to the PBD (Matos et al, 2008), which blocks the recognition of conventional substrates. However, recent work has identified a surface on the back of the PBD that binds substrates even when the phosphopeptide‐binding groove is occupied (Chen & Weinreich, 2010; Almawi et al, 2020). This interaction is inhibited rather than promoted by phosphorylation. Thus, Spo13 might both activate Cdc5 and enforce the unconventional mode of substrate recognition. Spo13 might also alter the specificity of Cdc5‐dependent phosphorylation. Cdc5‐Spo13 phosphorylates at least 12 out of 22 serines but none of the 10 threonines in Clb1's N‐terminal region (Appendix Figs S7 and S8). This suggests that at least on Clb1, Cdc5‐Spo13 prefers serine to threonine, whereas Cdc5/PLK1 phosphorylates both residues (Mok et al, 2010; Santamaria et al, 2011). However, more substrates need to be identified to establish the consensus motif for phosphorylation by the Cdc5‐Spo13 kinase. Another question concerns the phosphatases that remove Cdc5/Spo13‐dependent phosphorylation when Spo13 is degraded at anaphase I. It is conceivable that Cdc5‐Spo13 confines the activity of such phosphatases to meiosis II. We note that Cdc5‐Spo13 represses the translation of GIP1 (Fig EV1B), which encodes a meiosis‐specific targeting subunit of the Glc7/PP1 phosphatase involved in spore formation (Tachikawa et al, 2001).
Regulation of Rim4 degradation by Cdc5, Ime2, and Hrr25
Our data suggest that free Cdc5 promotes whereas Cdc5‐Spo13 inhibits Ime2's function in Rim4 degradation. Accordingly, hyperactive Ime2‐ΔC induces Rim4 degradation even when Cdc5 is inhibited. However, Ime2‐ΔC also induces Rim4 degradation in cdc20 ama1Δ cells, that is, in cells containing stable Spo13. We propose that both free Cdc5 and Cdc5‐Spo13 control Ime2 activity by targeting its C‐terminal regulatory domain. How does Ime2 promote Rim4 degradation? Interestingly, Rim4 inhibits the translation of mRNAs while adopting an amyloid‐like structure (Berchowitz et al, 2015). Furthermore, Rim4 is phosphorylated on more than 30 sites in an Ime2‐dependent manner. It has been proposed that this phosphorylation dissolves the amyloid‐like structure, thereby releasing mRNAs for translation and subjecting soluble Rim4 to ubiquitinylation and proteasomal degradation (Carpenter et al, 2018). However, we also show that Hrr25 functions downstream of Ime2 in promoting the degradation of Rim4. The ability of Ime2‐ΔC to induce Rim4 degradation is blocked upon inhibition of Hrr25, whereas hyperactive Hrr25‐ΔC induces Rim4 degradation even when Ime2 is inhibited (Fig EV3). Formally, our data are consistent with a linear Cdc5‐Ime2‐Hrr25 kinase cascade, whereby Hrr25, rather than Ime2, elicits the degradation of Rim4 (Fig 5C). In this scenario, Ime2 might activate Hrr25 by targeting the C‐terminal, autoinhibitory domain of Hrr25. Indeed, Ime2 and Rim4 co‐immunoprecipitate with Hrr25 (Fig EV3D). The idea that Cdc5‐Spo13 inhibits signalling through the Cdc5‐Ime2‐Hrr25 cascade at metaphase I is consistent with the observation that Ime2 activity increases as cells enter meiosis II (Berchowitz et al, 2013). It might also explain why Hrr25 promotes events, such as MP assembly or Sgo1's removal from centromeres, at meiosis II but not at meiosis I (Arguello‐Miranda et al, 2017).
Recent work suggests another, mutually not exclusive scenario whereby Rim4 degradation and exit from meiosis depend on autophagy, a proteolytic system capable of removing large aggregates and organelles from the cytosol (Farre & Subramani, 2016; Wang et al, 2020; Herod et al, 2022). Indeed, Hrr25 is required for different pathways of selective autophagy (Mochida et al, 2014; Pfaffenwimmer et al, 2014; Tanaka et al, 2014; Meguro et al, 2020). Hrr25 phosphorylates autophagy receptors to promote their binding to Atg11, the organizer of the PAS. A simple hypothesis is that upon activation by Ime2, Hrr25 induces several pathways of selective autophagy, one of which mediates the degradation of Rim4. In this scenario, mRNAs to be translated must be released from Rim4 before its enclosure by autophagosomes. This release might be elicited by Hrr25 since Hrr25‐ΔC is capable of inducing AMA1 translation at metaphase I (Appendix Fig S3C).
Hrr25's prominent role in autophagy could explain a perplexing change in the regulation of Rim4 degradation and Ama1 accumulation: On the one hand, Rim4 degradation elicited by the SPO13 deletion or the AMA1‐6A mutation depends on Cdc5 and Ime2 activity. On the other hand, Ama1 accumulation results in degradation of Cdc5 and inactivation of Ime2 (Figs 2A and 7A). While initially dependent on Cdc5 and Ime2, Rim4 degradation and Ama1 accumulation become independent of these kinases at some time after the inactivation of Spo13. Accordingly, inhibition of Cdc5 or Ime2 at late anaphase I has no effect on subsequent Ama1 accumulation at meiosis II. By contrast, Hrr25 remains essential for Rim4 degradation and Ama1 accumulation (Appendix Fig S4; Arguello‐Miranda et al, 2017). This is indicative of an irreversible transition whereby Hrr25‐dependent removal of Rim4 becomes self‐sustaining. An important step in the induction of selective autophagy is binding of Hrr25 to the Rab GTPase Ypt1/Rab1, which activates Hrr25 and recruits it to Atg11 (Wang et al, 2015). Once activated at the PAS, Hrr25 might sustain autophagic degradation of Rim4 independently of Cdc5 and Ime2.
Regulation of Ama1 activity by Cdc5‐Spo13
Spo13 also employs a post‐translational mechanism to inhibit Ama1‐dependent proteolysis (Fig EV1D). We show that in essence, Cdc5‐Spo13 converts Cdk1‐Clb1 from a target into an inhibitor of Ama1. Cdk1‐Clb1 binds to free as well as APC/C‐bound Ama1 (Fig EV4). This interaction does not require the degrons of Clb1 but might be mediated by its hydrophobic patch, a surface involved in targeting Cdk‐cyclin complexes to their substrates (Ord et al, 2019). In addition, phosphorylation of Ama1's N‐terminal Thr‐Pro motif might create a docking site for Cks1, the phospho‐adaptor subunit of CDKs, thereby facilitating the phosphorylation of downstream Ser‐Pro motifs (Koivomagi et al, 2013; McGrath et al, 2013). We propose that Cdk1‐Clb1 bound to Ama1 has two alternative fates: Clb1 can be ubiquitinylated, which requires binding of Ama1 to the APC/C. Alternatively, Cdk1‐Clb1 phosphorylates Ama1, which prevents Ama1's binding to the APC/C and thereby ubiquitinylation of Clb1. By phosphorylating Clb1, Cdc5‐Spo13 shifts the balance from Clb1 ubiquitinylation towards Ama1 phosphorylation. Multisite phosphorylation of Clb1's N‐terminal region might hinder ubiquitinylation by obstructing ubiquitin‐accepting lysine residues, impeding the binding of D‐ and KEN‐boxes to their receptors, or reducing Ama1's affinity for the APC/C. Cellular polyanions, such as polyphosphate, weaken the activator‐APC/C interaction (Mizrak & Morgan, 2019). While conventional substrates can revert this effect, phosphorylated Clb1 is covalently linked to a polyanion. Phosphorylation causes a modest reduction in Clb1's degradation rate (Fig 9A), which is, however, amplified by its effects on Ama1 activity and synthesis (Fig 5C). In this way, phosphorylated Clb1 can inhibit Ama1 but still be targeted by Cdc20. Phosphorylation also inhibits degradation of the APC/C substrates Pds1, Cdc6, and KIF1C (Wang et al, 2001; Mailand & Diffley, 2005; Singh et al, 2014).
Integration of APC/CAma1 regulation with the Cdk1‐APC/CCdc20 oscillator
Progression through the two M phases of meiosis might be divided into three phases: entry into and stabilization of metaphase I, transition from meiosis I to ‐II, and exit from meiosis. At entry into metaphase I, Cdc5‐Spo13 and Cdk1‐Clb1 repress the activity of APC/CAma1 despite being susceptible to Ama1‐dependent proteolysis. While the synthesis of Clb1 and Cdc5 depends on Ndt80, Spo13 accumulates already at prophase. This is possible because Spo13, like Pds1, is protected from Ama1‐dependent proteolysis at prophase by the APC/C subunit Mnd2 (Fig 9C; Oelschlaegel et al, 2005; Penkner et al, 2005). The Cdc5‐Spo13 kinase therefore forms as soon as Cdc5 appears and prevents free Cdc5 from inducing Rim4 degradation and subsequent AMA1 translation. Furthermore, by phosphorylating Clb1, Cdc5‐Spo13 generates an inhibitor of Ama1. As Cdc5‐Spo13 and Cdk1‐Clb1 accumulate, they phosphorylate and activate each other. Phosphorylation of Clb1 enables Cdk1‐Clb1 to inhibit Ama1, which in turn stabilizes Clb1 and Spo13 (Fig 8B). Phosphorylation of Spo13 activates the Cdc5‐Spo13 kinase (Fig 6B and Appendix Fig S5D; Oz et al, 2022), which promotes the phosphorylation of Clb1 and the repression of AMA1 translation. Thus, Cdc5‐Spo13 and Cdk1‐Clb1 are connected by direct and indirect positive feedback loops, which stabilize the low‐Ama1/high‐Cdk1 state of metaphase I (Fig 5C). Since Clb3 is translated together with and binds to Ama1, it seems well suited to assist Clb1 in repressing Ama1‐dependent proteolysis. However, we found no evidence for Cdk1‐Clb3 inhibiting the degradation of Rim4 or the activity of Ama1 (Appendix Figs S5C and S6D).
At anaphase I, Spo13 and Clb1 are substrates of APC/CCdc20. Ama1 synthesis and activity are therefore unleashed in a coordinated manner when the SAC is silenced upon biorientation of bivalent chromosomes on the meiosis I‐spindle. APC/CCdc20 employs a robust mechanism, known as a coherent feedforward loop, to inactivate Spo13 and the phosphorylated form of Clb1: APC/CCdc20 not only targets Spo13 for proteolysis but also destroys the Spo13‐activating kinase Cdk1‐Clb1. Similarly, APC/CCdc20 targets Clb1 for degradation and additionally inactivates Cdc5‐Spo13, the kinase that phosphorylates Clb1. While Spo13 is degraded at anaphase I, APC/CAma1 must only be activated at anaphase II to allow the re‐accumulation of M phase regulators required for entry into metaphase II. Indeed, Rim4 degradation is not complete until cells reach metaphase II. Furthermore, degradation of Ama1‐specific substrates at anaphase II depends on high levels of Ama1 (Arguello‐Miranda et al, 2017; Fig 2B). This requires a high rate of translation since Ama1 is an unstable protein (Jonak et al, 2017). Meanwhile, Clb1 is not completely degraded at anaphase I (Buonomo et al, 2003), giving Cdk1‐Clb1 a head‐start on Ama1 as both accumulate at meiosis II.
Upon silencing of the SAC at meiosis II, APC/CCdc20 induces the degradation of Clb1 and the concomitant activation of high levels of Ama1. While the activity of APC/CCdc20 declines as cyclins are degraded, that of APC/CAma1 increases, resulting in irreversible exit from meiosis. Ama1 accumulates at meiosis II despite terminating Ndt80‐dependent transcription through the degradation of Ndt80 (Okaz et al, 2012). Similarly, cdc20 spo13Δ and cdc20 AMA1‐6A cells accumulate Ama1 and sporulation factors while degrading Ndt80 (Figs 2A and 7A). These data imply that mRNAs encoding meiosis II‐specific proteins are protected from degradation until translation is unleashed. How mRNA protection is related to translational repression remains to be investigated.
A conserved role for Spo13/MEIKIN in preventing premature exit from meiosis
While budding and fission yeast express meiosis‐specific APC/C activators dedicated to exit from meiosis, mammalian genomes encode only two APC/C activators, namely Cdh1 (also known as Fzr1) and Cdc20. Nevertheless, spermatocytes of Meikin knock‐out mice resemble the spo13Δ mutant in that they undergo a single division, which corresponds to meiosis I (Kim et al, 2015). Remarkably, the only other mutation known to date showing a similar phenotype is a non‐phosphorylatable version of Cdh1. Whereas Cdh1‐9A knock‐in mice develop normally, males are infertile because spermatocytes exit from meiosis after a single division, similar to the yeast AMA1‐6A mutant (Tanno et al, 2020). Thus, phosphorylation at CDK motifs is dispensable for the inhibition of Cdh1 in somatic cells. In spermatocytes, however, it is essential for entry into meiosis II, even though Cdh1‐9A is compatible with entry into meiosis I. The implication is that Cdh1‐9A amplifies its activity during meiosis I by, for instance, increased translation or degradation of an inhibitor. We therefore speculate that PLK1‐MEIKIN counteracts the Cdh1‐activating mechanisms that cause Cdh1‐9A spermatocytes to prematurely exit from meiosis I. Thus, Spo13 and MEIKIN might ensure a two‐division meiosis through mechanisms conserved from yeast to male mammals.
Materials and Methods
Construction of Saccharomyces cerevisiae strains
This study was performed with diploid SK1 strains (Kane & Roth, 1974) generated by mating of the appropriate haploids. Genotypes are listed in Appendix Table S1. We used a Gal4‐estrogen receptor fusion for estradiol‐inducible expression from the GAL promoter (called P EST herein; Benjamin et al, 2003), the CUP1 promoter (450 bp) for expression induced by CuSO4, and the DMC1 promoter (340 bp) for expression at early prophase. Meiotic depletion was achieved with the mitosis‐specific promoters of HSL1 (Okaz et al, 2012) or SCC1 (Clyne et al, 2003). P EST ‐NDT80 P HSL1 ‐CDC20 strains were constructed to release cells from prophase into an arrest at metaphase I. Arrest/release at metaphase I was performed with CDC20‐mAR cells containing endogenous CDC20 controlled by a mitosis‐specific promoter and an additional P CUP1 ‐CDC20 gene (Arguello‐Miranda et al, 2017). SK1 strains carrying ime2‐as (M146G; Benjamin et al, 2003), cdc28/cdk1‐as1 (F88G; Oelschlaegel et al, 2005), cdc28/cdk1‐as2 (F88A; Oz et al, 2022), cdc5‐as (L158G; Okaz et al, 2012), or hrr25‐as (I82G; Petronczki et al, 2006) have been described. Hyperactive kinases were generated by replacing the C‐terminal 241 (ime2‐ΔC) or 189 residues (hrr25‐ΔC) with an Ha3 tag. The alleles spo13‐mD (L26A; Sullivan & Morgan, 2007), spo13‐m2 (S132T, S134T; Matos et al, 2008), and spo13‐10A (all S/T–P to A‐P; Oz et al, 2022) have been described. For auxin‐inducible degradation, Rim4‐mNG was tagged with AID* (residues 71–114 of IAA17; Morawska & Ulrich, 2013) and crossed into a strain expressing OsTir1‐F74G (Yesbolatova et al, 2020) from the CUP1 promoter (a gift from Joao Matos). To construct CLB1 mutants, Ser 15, 24, 52, 84, 88, 109, 117, 121, 137, 141, 143, and 164 were mutated to Ala (clb1‐12A) or Asp (clb1‐12D). In clb1‐6A, Ser 15, 84, 109, 137, 141, and 143 are mutated to Ala. CLB1 alleles (−462 to +1811, ATG = +1) were cloned into YIplac128 (Gietz & Sugino, 1988) and integrated at the clb1Δ locus. To measure degradation rates, CLB1‐ha6 and SPO13 were cloned behind the DMC1 promoter into YIplac vectors and integrated at leu2 or ura3 of ndt80Δ cdk1‐as1 cells. In AMA1‐6A, Ser/Thr 3, 61, 63, 69, 182, and 199 are mutated to Ala. Wild‐type AMA1 and AMA1‐6A (−497 to +2004, ATG = +1) were cloned into YIplac128 and integrated at the ama1Δ locus. For live‐imaging, we used Cnm67‐tdTomato, Htb1‐mCherry (Matos et al, 2008), Cdc5‐eGFP (Okaz et al, 2012), and Rim4 tagged with mNeonGreen (mNG; Shaner et al, 2013). Strains expressing Cdc14‐eGFP, eGFP‐tubulin, and TetR‐tdTomato labelling CENV‐tetO and the nucleoplasm have been described (Arguello‐Miranda et al, 2017). To monitor translation, coding sequences downstream of codon 30 (AMA1) or codon 15 (GAT4, GIP1, SPS4, SSP2) were replaced with mNG. The AurCMX4 cassette conferring resistance to aureobasidin A carries S. cerevisiae AUR1‐C (F158Y, A240C; Hashida‐Okado et al, 1998) synthesized with alternative codons and flanked by A. gossypii TEF1 promoter/terminator. To assess spore viability, 36 tetrads per strain were dissected on YPD plates.
Meiotic cultures
Meiosis was induced at 30°C (Arguello‐Miranda et al, 2017). Briefly, respiration‐competent cells grown on YPD medium for 24 h were evenly spread on YPD plates and allowed to form a lawn (24 h). Cells were inoculated (OD600 = 0.3; 5 × 106 cells/ml) into liquid YPA medium (YP plus 2% K‐acetate) and grown for 12 h until entry into a transient G1 arrest. Cells were washed with sporulation medium (SPM, 2% K‐acetate), inoculated (OD600 = 2.4) into 100 ml of SPM in a 2.8 l‐Fernbach flask, and rotated on an orbital shaker (200 rpm). P EST ‐NDT80 P HSL1 ‐CDC20 cells were released into metaphase I with estradiol (5 μM) at 7 h in SPM, and CDC20‐mAR cells were released into anaphase I with CuSO4 (10 μM) at 8 h in SPM. Kinases were inhibited with 1NM‐PP1 (Cayman Chemical; Cdk1‐as1 and Hrr25‐as, 5 μM), 1Na‐PP1 (Cayman Chemical; Cdk1‐as2, 10 μM; Ime2‐as, 20 μM), or CMK (MedChemExpress; Cdc5‐as, 20 μM). The auxin analogue 5‐Ph‐IAA (BioAcademia, Osaka, Japan) was used at 0.1 mM and cycloheximide at 0.5 mg/ml.
Live‐cell imaging
Cells diluted to OD600 = 0.4 were applied to an 8‐well glass bottom μ‐slide (ibidi, Gräfelfing, Germany) coated with Concanavalin A (Sigma‐Aldrich; 0.5 mg/ml) in 250 μl of SPM per well to give a density of 20–30 cells per field of view. Drugs and CuSO4 were added in 125 μl of SPM. Cells were imaged on a DeltaVision Elite system (Cytiva) consisting of an Olympus IX71 microscope with autofocus (Ultimate Focus) and solid‐state illumination (InsightISS) attenuated by an ND filter (25% T), a PlanApo 100×/1.4 NA oil objective, DeltaVision filter sets, a CoolSnap HQ2 camera (Photometrics), and an environmental chamber set to 30°C. We acquired Z‐stacks (8 × 1 μm) from 8 to 10 fields of view per strain in the GFP and the RFP channel every 10 min for 14 h. Exposure times were 100–200 ms with the adjustable ND filter set to 10% (GFP) or 32% (RFP). Z‐stacks were deconvolved and projected to a single 2D‐image with softWoRx 6.1 (standard projection). Time‐lapse series (frame width, 5 μm) were produced in Fiji (http://fiji.sc/). For quantification, time series of individual cells (n = 100) were aligned to a reference event (e.g., SPB separation at metaphase I) set to t = 0 in each cell. Percentages of other features (e.g., presence of Rim4) were calculated at 10 min‐intervals relative to the reference event using Microsoft Excel.
Immunofluorescence microscopy
Cells (1 ml) were fixed overnight at 4°C in formaldehyde (3.7%), converted to spheroplasts with Zymolyase 100T (amsbio; 10 μg/ml), and placed on 15‐well multi‐test slides (MP Biomedicals; Salah & Nasmyth, 2000). We used monoclonal antibodies to Myc (mouse 9E10, Invitrogen; 1:100) and α‐tubulin (rat YOL1/34, Serotec; 1:300) as primary antibodies, which were detected with Alexa Fluor‐conjugated secondary antibodies from donkey (Invitrogen). DNA was stained with DAPI. Cells were observed on a Zeiss Axio Imager M2 with Colibri 7 LED light source and a Plan‐Apochromat 100×/1.4 NA oil objective. Cellular features were scored in ≥ 100 cells per timepoint.
Analysis of mRNA
RNA was extracted with the RNeasy Mini Kit (Qiagen) from cells (2 ml) converted to spheroplasts with Zymolyase 100T (amsbio; 1.4 mg/ml). Residual DNA was removed by on‐column digestion with RNase‐free DNase I. We used a Qubit 4 fluorometer (Invitrogen) to measure RNA concentration and an Agilent 2100 Bioanalyzer for quality control. cDNA was synthesized from 1 μg of total RNA using SuperScript III First‐Strand Synthesis SuperMix (Invitrogen) and random hexamers. qPCR was performed in triplicate on a StepOnePlus real‐time PCR system (Applied Biosystems) using Takyon Rox SYBR MasterMix blue dTTP (Eurogentec) and primers at 100 nM. mRNA levels were normalized to the ACT1 transcript by calculating the 2−ΔCt value for each time point (Livak & Schmittgen, 2001). We used the following primers:
ACT1‐fw, 5′‐gaaatgcaaaccgctctca‐3′; ACT1‐rv, 5′‐taccggcagattccaaaccc‐3′;
mNG‐fw, 5′‐ctacgagggaagccacatca‐3′; mNG‐rv, 5′‐agtcttcttcgacctgcacc‐3′;
CDC5‐fw, 5′‐acaccatatgcggaacacct‐3′; CDC5‐rv, 5′‐tctcttgcttggaagggtgg‐3′.
Analysis of proteins
For immunoprecipitations, cells (40 ml) were treated with 2 mM PMSF, washed with 2 mM PMSF in cold water, and frozen in liquid nitrogen. Cells were broken with 0.5 mm‐glass beads, and lysates were cleared by centrifugation and incubation with protein A‐agarose beads (Roche; 10% v/v). Proteins were captured from extracts (0.5 ml, 12 mg/ml) with antibodies to Apc2 (Camasses et al, 2003; 1:200) or Myc (mouse 9E11, Invitrogen; 1:200) and protein A‐agarose beads. For immunodepletions, cleared extracts (0.5 ml; 7 mg/ml) were passed (3×) through 0.1 ml‐protein A‐agarose columns loaded with antibodies. To analyze protein levels, cells (9 ml) were collected by centrifugation, frozen in liquid nitrogen, and broken with glass beads in 10% trichloroacetic acid (Okaz et al, 2012). Proteins (60 μg) were separated in SDS‐8% PAA gels and transferred to Immobilon P membranes (Millipore) by semi‐dry blotting. Membranes were horizontally cut into 2–3 slices and incubated with primary antibodies for 2 h. Antibodies had been raised in rabbits to Apc10/Doc1 (1:2,000), Cdc23 (1:2,000), Cdh1 (1:5,000), and Clb3 (1:3,000) (Schwickart et al, 2004), Ama1 (1:2,000) and Mnd2 (1:2,000) (Oelschlaegel et al, 2005), Cdc5 (1:5,000) and Spo13 (1:5,000) (Matos et al, 2008), Apc2 (1:2,000) and Cdc20 (1:2,000) (Camasses et al, 2003), Ase1 (a gift from David Pellman; Juang et al, 1997; 1:1,000), Cdk1/Cdc28 (Petronczki et al, 2006); 1:5,000), Ndt80 (a gift from Kirsten Benjamin; Benjamin et al, 2003; 1:5,000), and Tub2/β‐tubulin (a gift from Wolfgang Seufert; Schwab et al, 2001; 1:20,000). We purchased rabbit antibodies to OsTir1 (Medical & Biological Laboratories PD048; 1:1,000), Myc (Sigma c3956; 1:1,000), mNG (Cell Signaling Technology 53061; 1:1,000), and Zip1 (Santa Cruz sc‐33733; 1:3,000) and goat antibodies (Santa Cruz) to Clb1 (sc‐7647; 1:300), Clb4 (sc‐6702; 1:400), Clb5 (sc‐6704; 1:200), and Ime2 (sc‐26444; 1:300). We used monoclonal antibodies to Ha (rat 3F10, Roche; 1:2,000), Act1/actin (mouse AC‐40, Sigma‐Aldrich; 1:500), GFP (mouse 7.1 and 13.1, Roche; 1:500), Myc (mouse 9E10, Invitrogen; 1:1,000), and Pgk1 (mouse 22C5D8, Invitrogen; 1:20,000). HRP‐conjugated secondary antibodies were detected on X‐ray films using ECL start reagents (Cytiva). Signals were digitized on an Epson Perfection V750 Pro scanner and quantified in Fiji (http://fiji.sc/).
Affinity‐enrichment of GFP‐tagged proteins
Strains were cultured in 15 l‐glass beakers placed in a 30°C‐water bath and aerated through 120 mm‐air stone discs with pressurized air, which was prewarmed and humidified with a gas‐washing bottle. Cells from 12 YPD plates were inoculated (OD600 = 0.3) into 8 l of YPA plus antifoam SE‐15 (Sigma‐Aldrich; 1:1,000) and grown for 12 h. Cells were washed with SPM and inoculated (OD600 = 3) into 4 l of SPM. P HSL1 ‐CDC20 and P SCC1 ‐CDC20 strains arresting at metaphase I were cultured for 8 h. P EST ‐NDT80 strains were released from prophase at 7 h in SPM and harvested 80 min (metaphase I) or 120 min (metaphase II) thereafter. Cultures were poured through crushed ice, washed with 2 mM PMSF in cold water, and resuspended in 15 ml of buffer B70 containing protease and phosphatase inhibitors (2 mM PMSF, EDTA‐free Complete, PhosSTOP; Roche). B70 is buffer B (50 mM Hepes/KOH pH 7.4, 40 mM β‐glycerophosphate, 2.5 mM Mg‐acetate, 1 mM DTT, 0.1% Triton X100, 10% glycerol) plus 70 mM K‐acetate. Subsequent steps were performed at 4°C. Cells were broken with 0.5 mm‐glass beads in a mixer mill (Retsch MM 400, 4 × 4 min, 30 Hz), and lysates were cleared by centrifugation (27,000 g; 45 min). Extracts (23 ml, 23 mg/ml) were incubated with Sepharose 4B (Cytiva, 13 ml of 75% slurry) for 30 min and then with agarose beads carrying anti‐GFP nanobodies (GFP‐Trap, Chromotek; 0.5 ml of 50% slurry) for 60 min. Beads were washed with 45 ml‐aliquots of buffer B70 (2 washes), B150, B200, and W (50 mM Tris/HCl pH 7.5, 70 mM NaCl, 5% glycerol; 2 washes). Beads were resuspended in 0.5 ml of buffer E (50 mM Tris/HCl pH 7.5, 2 M urea, 1 mM DTT), divided into two samples, and subjected to on‐bead digestion with modified trypsin (Promega, sequencing grade; 5 ng/μl) or Lys‐C (Promega, mass spec grade; 5 ng/μl) for 40 min at 22°C. Digests were collected by centrifugation and the beads washed with 2 × 125 μl of buffer E plus iodoacetamide (5.5 mM). Trypsin and Lys‐C digests were combined with the corresponding washes, incubated overnight at 22°C, and quenched with trifluoroacetic acid (1% final). Peptide mixtures were desalted on C18‐StageTips (Rappsilber et al, 2003), vacuum‐dried, and stored at −20°C.
LC–MS/MS data acquisition
Trypsin and Lys‐C digests were analyzed in separate LC–MS/MS runs. A 30 cm × 75 μm‐column packed in‐house with 1.9 μm‐silica beads (ReproSil‐Pur C18‐AQ; Dr Maisch GmbH) was operated at 60°C by an Easy‐nLC 1200 system (Thermo Fisher Scientific). Peptides were loaded at 250 nl/min in buffer A (0.1% formic acid) plus 2% buffer B (80% acetonitrile, 0.1% formic acid) and eluted by a gradient to 30% B (120 min), 60% B (10 min), 95% B (5 min), and 95% B (5 min). Eluting peptides were transferred via the nano‐electrospray interface into a Q Exactive HF Orbitrap instrument (Thermo Fisher Scientific). Precursor ions were selected by a data‐dependent top‐12 method for high‐energy collisional dissociation with a normalized collision energy of 28 (Scheltema et al, 2014). MS/MS parameters and instrument settings are given in the source data.
MS/MS data analysis
Raw MS files of corresponding trypsin and Lys‐C digests were merged and searched in MaxQuant (version 2.0.1.0) against the S. cerevisiae UniProt database (UP000002311) combined with the relevant GFP‐tagged bait protein and 247 common contaminants (Tyanova et al, 2016a). Minimum peptide length was set to seven amino acids and maximum mass to 4,600 Da. Strict trypsin or Lys‐C specificity was required, allowing up to two missed cleavages. Carbamidomethylation of Cys was set as fixed modification and N‐terminal protein acetylation, oxidation of Met, deamination of Asn or Gln, and phosphorylation of Ser, Thr, or Tyr as variable modifications. The maximum number of modifications per peptide was set to five. Relative, label‐free quantifications (LFQ) were obtained from the MaxLFQ algorithm using default parameters and further analyzed in Perseus (version 1.6.15.0; Tyanova et al, 2016b). Contaminants and matches to the reversed‐sequence decoy database were removed, and LFQ intensities were log2‐transformed. For interaction proteomics, proteins showing at least two valid values in at least one group were retained. Missing values were imputed with values from a normal distribution at the detection limit of the mass spectrometer (width, 0.3 SD; down‐shift, 1.8 SD). For Clb1 phosphorylation analysis, phospho‐sites with a localization probability of less than 0.75 were removed. Phospho‐sites with at least two valid values in the control were retained, and phosphorylation site intensities were log2‐transformed. Missing values were imputed as above.
Statistical analysis
Mean values are given ± 1 SD. Distributions of the durations of metaphase I and the times from SPB separation to Rim4 degradation tend to be symmetrical or slightly skewed. Thus, we used Welch's t‐test (unpaired, two‐tailed) to compare means and provide 95% confidence intervals for the difference between means. With a balanced design and a sample size of n = 100, Welch's t‐test is robust against deviations from normality. Volcano plots were generated in GraphPad Prism 9 using log2‐transformed LFQs and Benjamini‐Hochberg‐corrected P‐values calculated by Perseus (Tyanova et al, 2016b). We used Perseus to generate heatmaps and to perform hierarchical clustering (distance metric, Euclidian; linkage criterion, Average). To compare degradation rates, linear regression was applied to log2‐transformed signal intensities over time, and slopes were compared with ANCOVA in GraphPad Prism 9.
Author contributions
Wolfgang Zachariae: Conceptualization; formal analysis; supervision; funding acquisition; validation; investigation; visualization; writing – original draft; project administration; writing – review and editing. Julie Rojas: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Tugce Oz: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Katarzyna Jonak: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Oleksii Lyzak: Data curation; formal analysis; validation; investigation; visualization; methodology; writing – review and editing. Vinal Massaad: Data curation; formal analysis; investigation; visualization; writing – review and editing. Olha Biriuk: Data curation; formal analysis; investigation; visualization; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Acknowledgements
We thank former group members Martin Schwickart, Tobias Oelschlaegel, Jesse Lipp, and Joao Matos for initial experiments on Ama1 and Spo13, Saskia Moser for contributions to Fig EV1B, Valentina Mengoli for the hrr25‐ΔC strain, and Orlando Argüello‐Miranda for data in Appendix Figs S4, and S6C and D. We are grateful to Barbara Steigenberger and the MPIB mass spectrometry facility for LC–MS/MS. We thank Kirsten Benjamin, David Pellman, Joao Matos, and Wolfgang Seufert for antibodies and yeast strains. We are grateful to Isabella Mathes and Albena Bergsoy for tetrad dissection. This work was supported by the Max Planck Society. Open Access funding enabled and organized by Projekt DEAL.
The EMBO Journal (2023) 42: e114288
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD044597 (http://www.ebi.ac.uk/pride/archive/projects/PXD044597).
References
- Almawi AW, Langlois‐Lemay L, Boulton S, Rodriguez Gonzalez J, Melacini G, D'Amours D, Guarne A (2020) Distinct surfaces on Cdc5/PLK Polo‐box domain orchestrate combinatorial substrate recognition during cell division. Sci Rep 10: 3379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arguello‐Miranda O, Zagoriy I, Mengoli V, Rojas J, Jonak K, Oz T, Graf P, Zachariae W (2017) Casein kinase 1 coordinates cohesin cleavage, gametogenesis, and exit from M phase in meiosis II. Dev Cell 40: 37–52 [DOI] [PubMed] [Google Scholar]
- Asakawa H, Kitamura K, Shimoda C (2001) A novel Cdc20‐related WD‐repeat protein, Fzr1, is required for spore formation in Schizosaccharomyces pombe . Mol Genet Genomics 265: 424–435 [DOI] [PubMed] [Google Scholar]
- Attner MA, Miller MP, Ee LS, Elkin SK, Amon A (2013) Polo kinase Cdc5 is a central regulator of meiosis I. Proc Natl Acad Sci USA 110: 14278–14283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin KR, Zhang C, Shokat KM, Herskowitz I (2003) Control of landmark events in meiosis by the CDK Cdc28 and the meiosis‐specific kinase Ime2. Genes Dev 17: 1524–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchowitz LE, Gajadhar AS, van Werven FJ, De Rosa AA, Samoylova ML, Brar GA, Xu Y, Xiao C, Futcher B, Weissman JS et al (2013) A developmentally regulated translational control pathway establishes the meiotic chromosome segregation pattern. Genes Dev 27: 2147–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berchowitz LE, Kabachinski G, Walker MR, Carlile TM, Gilbert WV, Schwartz TU, Amon A (2015) Regulated formation of an amyloid‐like translational repressor governs gametogenesis. Cell 163: 406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD et al (2000) A chemical switch for inhibitor‐sensitive alleles of any protein kinase. Nature 407: 395–401 [DOI] [PubMed] [Google Scholar]
- Blanco MA, Pelloquin L, Moreno S (2001) Fission yeast mfr1 activates APC and coordinates meiotic nuclear division with sporulation. J Cell Sci 114: 2135–2143 [DOI] [PubMed] [Google Scholar]
- Bolte M, Steigemann P, Braus GH, Irniger S (2002) Inhibition of APC‐mediated proteolysis by the meiosis‐specific protein kinase Ime2. Proc Natl Acad Sci USA 99: 4385–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS (2012) High‐resolution view of the yeast meiotic program revealed by ribosome profiling. Science 335: 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buonomo SB, Clyne RK, Fuchs J, Loidl J, Uhlmann F, Nasmyth K (2000) Disjunction of homologous chromosomes in meiosis I depends on proteolytic cleavage of the meiotic cohesin Rec8 by separin. Cell 103: 387–398 [DOI] [PubMed] [Google Scholar]
- Buonomo SB, Rabitsch KP, Fuchs J, Gruber S, Sullivan M, Uhlmann F, Petronczki M, Toth A, Nasmyth K (2003) Division of the nucleolus and its release of CDC14 during anaphase of meiosis I depends on separase, SPO12, and SLK19. Dev Cell 4: 727–739 [DOI] [PubMed] [Google Scholar]
- Camasses A, Bogdanova A, Shevchenko A, Zachariae W (2003) The CCT chaperonin promotes activation of the anaphase‐promoting complex through the generation of functional Cdc20. Mol Cell 12: 87–100 [DOI] [PubMed] [Google Scholar]
- Carlile TM, Amon A (2008) Meiosis I is established through division‐specific translational control of a cyclin. Cell 133: 280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter K, Bell RB, Yunus J, Amon A, Berchowitz LE (2018) Phosphorylation‐mediated clearance of amyloid‐like assemblies in meiosis. Dev Cell 45: 392–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cegielska A, Gietzen KF, Rivers A, Virshup DM (1998) Autoinhibition of casein kinase I epsilon (CKI epsilon) is relieved by protein phosphatases and limited proteolysis. J Biol Chem 273: 1357–1364 [DOI] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Yang J, McLaughlin SH, Barford D (2015) Atomic structure of the APC/C and its mechanism of protein ubiquitination. Nature 522: 450–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Weinreich M (2010) Dbf4 regulates the Cdc5 Polo‐like kinase through a distinct non‐canonical binding interaction. J Biol Chem 285: 41244–41254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu S, Herskowitz I (1998) Gametogenesis in yeast is regulated by a transcriptional cascade dependent on Ndt80. Mol Cell 1: 685–696 [DOI] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, Herskowitz I (1998) The transcriptional program of sporulation in budding yeast. Science 282: 699–705 [DOI] [PubMed] [Google Scholar]
- Clyne RK, Katis VL, Jessop L, Benjamin KR, Herskowitz I, Lichten M, Nasmyth K (2003) Polo‐like kinase Cdc5 promotes chiasmata formation and cosegregation of sister centromeres at meiosis I. Nat Cell Biol 5: 480–485 [DOI] [PubMed] [Google Scholar]
- Cooper KF, Mallory MJ, Egeland DB, Jarnik M, Strich R (2000) Ama1p is a meiosis‐specific regulator of the anaphase promoting complex/cyclosome in yeast. Proc Natl Acad Sci USA 97: 14548–14553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dial JM, Petrotchenko EV, Borchers CH (2007) Inhibition of APCCdh1 activity by Cdh1/Acm1/Bmh1 ternary complex formation. J Biol Chem 282: 5237–5248 [DOI] [PubMed] [Google Scholar]
- Elia AE, Cantley LC, Yaffe MB (2003a) Proteomic screen finds pSer/pThr‐binding domain localizing Plk1 to mitotic substrates. Science 299: 1228–1231 [DOI] [PubMed] [Google Scholar]
- Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB (2003b) The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo‐box domain. Cell 115: 83–95 [DOI] [PubMed] [Google Scholar]
- Elmore ZC, Guillen RX, Gould KL (2018) The kinase domain of CK1 enzymes contains the localization cue essential for compartmentalized signaling at the spindle pole. Mol Biol Cell 29: 1664–1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enquist‐Newman M, Sullivan M, Morgan DO (2008) Modulation of the mitotic regulatory network by APC‐dependent destruction of the Cdh1 inhibitor Acm1. Mol Cell 30: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farre JC, Subramani S (2016) Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol 17: 537–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galander S, Barton RE, Borek WE, Spanos C, Kelly DA, Robertson D, Rappsilber J, Marston AL (2019) Reductional meiosis I chromosome segregation is established by coordination of key meiotic kinases. Dev Cell 49: 526–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Sugino A (1988) New yeast‐Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six‐base pair restriction sites. Gene 74: 527–534 [DOI] [PubMed] [Google Scholar]
- Graves PR, Roach PJ (1995) Role of COOH‐terminal phosphorylation in the regulation of casein kinase I delta. J Biol Chem 270: 21689–21694 [DOI] [PubMed] [Google Scholar]
- Hashida‐Okado T, Ogawa A, Kato I, Takesako K (1998) Transformation system for prototrophic industrial yeasts using the AUR1 gene as a dominant selection marker. FEBS Lett 425: 117–122 [DOI] [PubMed] [Google Scholar]
- Hepworth SR, Friesen H, Segall J (1998) NDT80 and the meiotic recombination checkpoint regulate expression of middle sporulation‐specific genes in Saccharomyces cerevisiae . Mol Cell Biol 18: 5750–5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herod SG, Dyatel A, Hodapp S, Jovanovic M, Berchowitz LE (2022) Clearance of an amyloid‐like translational repressor is governed by 14‐3‐3 proteins. Cell Rep 39: 110753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt LJ, Hutti JE, Cantley LC, Morgan DO (2007) Evolution of Ime2 phosphorylation sites on Cdk1 substrates provides a mechanism to limit the effects of the phosphatase Cdc14 in meiosis. Mol Cell 25: 689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt JE, Tran SM, Stewart JL, Minahan K, Garcia‐Higuera I, Moreno S, Jones KT (2011) The APC/C activator FZR1 coordinates the timing of meiotic resumption during prophase I arrest in mammalian oocytes. Development 138: 905–913 [DOI] [PubMed] [Google Scholar]
- Holt JE, Pye V, Boon E, Stewart JL, Garcia‐Higuera I, Moreno S, Rodriguez R, Jones KT, McLaughlin EA (2014) The APC/C activator FZR1 is essential for meiotic prophase I in mice. Development 141: 1354–1365 [DOI] [PubMed] [Google Scholar]
- Horie S, Watanabe Y, Tanaka K, Nishiwaki S, Fujioka H, Abe H, Yamamoto M, Shimoda C (1998) The Schizosaccharomyces pombe mei4+ gene encodes a meiosis‐specific transcription factor containing a forkhead DNA‐binding domain. Mol Cell Biol 18: 2118–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaspersen SL, Charles JF, Morgan DO (1999) Inhibitory phosphorylation of the APC regulator Hct1 is controlled by the kinase Cdc28 and the phosphatase Cdc14. Curr Biol 9: 227–236 [DOI] [PubMed] [Google Scholar]
- Jin L, Zhang K, Xu Y, Sternglanz R, Neiman AM (2015) Sequestration of mRNAs modulates the timing of translation during meiosis in budding yeast. Mol Cell Biol 35: 3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonak K, Zagoriy I, Oz T, Graf P, Rojas J, Mengoli V, Zachariae W (2017) APC/C‐Cdc20 mediates deprotection of centromeric cohesin at meiosis II in yeast. Cell Cycle 16: 1145–1152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juang YL, Huang J, Peters JM, McLaughlin ME, Tai CY, Pellman D (1997) APC‐mediated proteolysis of Ase1 and the morphogenesis of the mitotic spindle. Science 275: 1311–1314 [DOI] [PubMed] [Google Scholar]
- Kane SM, Roth R (1974) Carbohydrate metabolism during ascospore development in yeast. J Bacteriol 118: 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katis VL, Matos J, Mori S, Shirahige K, Zachariae W, Nasmyth K (2004) Spo13 facilitates monopolin recruitment to kinetochores and regulates maintenance of centromeric cohesion during yeast meiosis. Curr Biol 14: 2183–2196 [DOI] [PubMed] [Google Scholar]
- Katis VL, Lipp JJ, Imre R, Bogdanova A, Okaz E, Habermann B, Mechtler K, Nasmyth K, Zachariae W (2010) Rec8 phosphorylation by casein kinase 1 and Cdc7‐Dbf4 kinase regulates cohesin cleavage by separase during meiosis. Dev Cell 18: 397–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Ishiguro K, Nambu A, Akiyoshi B, Yokobayashi S, Kagami A, Ishiguro T, Pendas AM, Takeda N, Sakakibara Y et al (2015) Meikin is a conserved regulator of meiosis‐I‐specific kinetochore function. Nature 517: 466–471 [DOI] [PubMed] [Google Scholar]
- Klapholz S, Esposito RE (1980a) Isolation of Spo12‐1 and Spo13‐1 from a natural variant of yeast that undergoes a single meiotic division. Genetics 96: 567–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klapholz S, Esposito RE (1980b) Recombination and chromosome segregation during the single division meiosis in Spo12‐1 and Spo13‐1 diploids. Genetics 96: 589–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Mahr P, Galova M, Buonomo SB, Michaelis C, Nairz K, Nasmyth K (1999) A central role for cohesins in sister chromatid cohesion, formation of axial elements, and recombination during yeast meiosis. Cell 98: 91–103 [DOI] [PubMed] [Google Scholar]
- Knop M, Strasser K (2000) Role of the spindle pole body of yeast in mediating assembly of the prospore membrane during meiosis. EMBO J 19: 3657–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivomagi M, Ord M, Iofik A, Valk E, Venta R, Faustova I, Kivi R, Balog ER, Rubin SM, Loog M (2013) Multisite phosphorylation networks as signal processors for Cdk1. Nat Struct Mol Biol 20: 1415–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labit H, Fujimitsu K, Bayin NS, Takaki T, Gannon J, Yamano H (2012) Dephosphorylation of Cdc20 is required for its C‐box‐dependent activation of the APC/C. EMBO J 31: 3351–3362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Kiburz BM, Amon A (2004) Spo13 maintains centromeric cohesion and kinetochore coorientation during meiosis I. Curr Biol 14: 2168–2182 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- Ma W, Zhou J, Chen J, Carr AM, Watanabe Y (2021) Meikin synergizes with shugoshin to protect cohesin Rec8 during meiosis I. Genes Dev 35: 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailand N, Diffley JF (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C‐dependent proteolysis. Cell 122: 915–926 [DOI] [PubMed] [Google Scholar]
- Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W (2008) Dbf4‐dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135: 662–678 [DOI] [PubMed] [Google Scholar]
- Matyskiela ME, Morgan DO (2009) Analysis of activator‐binding sites on the APC/C supports a cooperative substrate‐binding mechanism. Mol Cell 34: 68–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath DA, Balog ER, Koivomagi M, Lucena R, Mai MV, Hirschi A, Kellogg DR, Loog M, Rubin SM (2013) Cks confers specificity to phosphorylation‐dependent CDK signaling pathways. Nat Struct Mol Biol 20: 1407–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meguro S, Zhuang X, Kirisako H, Nakatogawa H (2020) Pex3 confines pexophagy receptor activity of Atg36 to peroxisomes by regulating Hrr25‐mediated phosphorylation and proteasomal degradation. J Biol Chem 295: 16292–16298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengoli V, Jonak K, Lyzak O, Lamb M, Lister LM, Lodge C, Rojas J, Zagoriy I, Herbert M, Zachariae W (2021) Deprotection of centromeric cohesin at meiosis II requires APC/C activity but not kinetochore tension. EMBO J 40: e106812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizrak A, Morgan DO (2019) Polyanions provide selective control of APC/C interactions with the activator subunit. Nat Commun 10: 5807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochida K, Ohsumi Y, Nakatogawa H (2014) Hrr25 phosphorylates the autophagic receptor Atg34 to promote vacuolar transport of alpha‐mannosidase under nitrogen starvation conditions. FEBS Lett 588: 3862–3869 [DOI] [PubMed] [Google Scholar]
- Moens PB, Rapport E (1971) Spindles, spindle plaques, and meiosis in yeast Saccharomyces cerevisiae (Hansen). J Cell Biol 50: 344–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok J, Kim PM, Lam HY, Piccirillo S, Zhou X, Jeschke GR, Sheridan DL, Parker SA, Desai V, Jwa M et al (2010) Deciphering protein kinase specificity through large‐scale analysis of yeast phosphorylation site motifs. Sci Signal 3: ra12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morawska M, Ulrich HD (2013) An expanded tool kit for the auxin‐inducible degron system in budding yeast. Yeast 30: 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami‐Tonami Y, Yamada‐Namikawa C, Tochigi A, Hasegawa N, Kojima H, Kunimatsu M, Nakanishi M, Murakami H (2007) Mei4p coordinates the onset of meiosis I by regulating cdc25+ in fission yeast. Proc Natl Acad Sci USA 104: 14688–14693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A (2015) The molecular biology of spindle assembly checkpoint signaling dynamics. Curr Biol 25: R1002–R1018 [DOI] [PubMed] [Google Scholar]
- Nasmyth K (2015) A meiotic mystery: how sister kinetochores avoid being pulled in opposite directions during the first division. Bioessays 37: 657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nerusheva OO, Galander S, Fernius J, Kelly D, Marston AL (2014) Tension‐dependent removal of pericentromeric shugoshin is an indicator of sister chromosome biorientation. Genes Dev 28: 1291–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oelschlaegel T, Schwickart M, Matos J, Bogdanova A, Camasses A, Havlis J, Shevchenko A, Zachariae W (2005) The yeast APC/C subunit Mnd2 prevents premature sister chromatid separation triggered by the meiosis‐specific APC/C‐Ama1. Cell 120: 773–788 [DOI] [PubMed] [Google Scholar]
- Okaz E, Arguello‐Miranda O, Bogdanova A, Vinod PK, Lipp JJ, Markova Z, Zagoriy I, Novak B, Zachariae W (2012) Meiotic prophase requires proteolysis of M phase regulators mediated by the meiosis‐specific APC/CAma1. Cell 151: 603–618 [DOI] [PubMed] [Google Scholar]
- Ord M, Venta R, Moll K, Valk E, Loog M (2019) Cyclin‐specific docking mechanisms reveal the complexity of M‐CDK function in the cell cycle. Mol Cell 75: 76–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz T, Mengoli V, Rojas J, Jonak K, Braun M, Zagoriy I, Zachariae W (2022) The Spo13/Meikin pathway confines the onset of gamete differentiation to meiosis II in yeast. EMBO J 41: e109446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkner AM, Prinz S, Ferscha S, Klein F (2005) Mnd2, an essential antagonist of the anaphase‐promoting complex during meiotic prophase. Cell 120: 789–801 [DOI] [PubMed] [Google Scholar]
- Perry JA, Kornbluth S (2007) Cdc25 and Wee1: analogous opposites? Cell Div 2: 12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronczki M, Siomos MF, Nasmyth K (2003) Un ménage à quatre: the molecular biology of chromosome segregation in meiosis. Cell 112: 423–440 [DOI] [PubMed] [Google Scholar]
- Petronczki M, Matos J, Mori S, Gregan J, Bogdanova A, Schwickart M, Mechtler K, Shirahige K, Zachariae W, Nasmyth K (2006) Monopolar attachment of sister kinetochores at meiosis I requires casein kinase 1. Cell 126: 1049–1064 [DOI] [PubMed] [Google Scholar]
- Pfaffenwimmer T, Reiter W, Brach T, Nogellova V, Papinski D, Schuschnig M, Abert C, Ammerer G, Martens S, Kraft C (2014) Hrr25 kinase promotes selective autophagy by phosphorylating the cargo receptor Atg19. EMBO Rep 15: 862–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primig M, Williams RM, Winzeler EA, Tevzadze GG, Conway AR, Hwang SY, Davis RW, Esposito RE (2000) The core meiotic transcriptome in budding yeasts. Nat Genet 26: 415–423 [DOI] [PubMed] [Google Scholar]
- Rabitsch KP, Petronczki M, Javerzat JP, Genier S, Chwalla B, Schleiffer A, Tanaka TU, Nasmyth K (2003) Kinetochore recruitment of two nucleolar proteins is required for homolog segregation in meiosis I. Dev Cell 4: 535–548 [DOI] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix‐assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Riedel CG, Katis VL, Katou Y, Mori S, Itoh T, Helmhart W, Galova M, Petronczki M, Gregan J, Cetin B et al (2006) Protein phosphatase 2A protects centromeric sister chromatid cohesion during meiosis I. Nature 441: 53–61 [DOI] [PubMed] [Google Scholar]
- Rudner AD, Murray AW (2000) Phosphorylation by Cdc28 activates the Cdc20‐dependent activity of the anaphase‐promoting complex. J Cell Biol 149: 1377–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah SM, Nasmyth K (2000) Destruction of the securin Pds1p occurs at the onset of anaphase during both meiotic divisions in yeast. Chromosoma 109: 27–34 [DOI] [PubMed] [Google Scholar]
- Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, Schmidt A, Sillje HH, Korner R, Nigg EA (2011) The Plk1‐dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics 10: M110.004457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sari F, Heinrich M, Meyer W, Braus GH, Irniger S (2008) The C‐terminal region of the meiosis‐specific protein kinase Ime2 mediates protein instability and is required for normal spore formation in budding yeast. J Mol Biol 378: 31–43 [DOI] [PubMed] [Google Scholar]
- Scheltema RA, Hauschild JP, Lange O, Hornburg D, Denisov E, Damoc E, Kuehn A, Makarov A, Mann M (2014) The Q Exactive HF, a Benchtop mass spectrometer with a pre‐filter, high‐performance quadrupole and an ultra‐high‐field Orbitrap analyzer. Mol Cell Proteomics 13: 3698–3708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab M, Neutzner M, Möcker D, Seufert W (2001) Yeast Hct1 recognizes the mitotic cyclin Clb2 and other substrates of the ubiquitin ligase APC. EMBO J 20: 5165–5175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwickart M, Havlis J, Habermann B, Bogdanova A, Camasses A, Oelschlaegel T, Shevchenko A, Zachariae W (2004) Swm1/Apc13 is an evolutionarily conserved subunit of the anaphase‐promoting complex stabilizing the association of Cdc16 and Cdc27. Mol Cell Biol 24: 3562–3576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ, Sible JC (2003) Hysteresis drives cell‐cycle transitions in Xenopus laevis egg extracts. Proc Natl Acad Sci USA 100: 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M et al (2013) A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat Methods 10: 407–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shonn MA, McCarroll R, Murray AW (2002) Spo13 protects meiotic cohesin at centromeres in meiosis I. Genes Dev 16: 1659–1671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SA, Winter D, Kirchner M, Chauhan R, Ahmed S, Ozlu N, Tzur A, Steen JA, Steen H (2014) Co‐regulation proteomics reveals substrates and mechanisms of APC/C‐dependent degradation. EMBO J 33: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead JL, Sullivan M, Lowery DM, Cohen MS, Zhang C, Randle DH, Taunton J, Yaffe MB, Morgan DO, Shokat KM (2007) A coupled chemical‐genetic and bioinformatic approach to Polo‐like kinase pathway exploration. Chem Biol 14: 1261–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sopko R, Raithatha S, Stuart D (2002) Phosphorylation and maximal activity of Saccharomyces cerevisiae meiosis‐specific transcription factor Ndt80 is dependent on Ime2. Mol Cell Biol 22: 7024–7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M, Morgan DO (2007) A novel destruction sequence targets the meiotic regulator Spo13 for anaphase‐promoting complex‐dependent degradation in anaphase I. J Biol Chem 282: 19710–19715 [DOI] [PubMed] [Google Scholar]
- Tachikawa H, Bloecher A, Tatchell K, Neiman AM (2001) A Gip1p‐Glc7p phosphatase complex regulates septin organization and spore wall formation. J Cell Biol 155: 797–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka C, Tan LJ, Mochida K, Kirisako H, Koizumi M, Asai E, Sakoh‐Nakatogawa M, Ohsumi Y, Nakatogawa H (2014) Hrr25 triggers selective autophagy‐related pathways by phosphorylating receptor proteins. J Cell Biol 207: 91–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno N, Kuninaka S, Fujimura S, Takemoto K, Okamura K, Takeda N, Araki K, Araki M, Saya H, Ishiguro KI (2020) Phosphorylation of the anaphase promoting complex activator FZR1/CDH1 is required for meiosis II entry in mouse male germ cell. Sci Rep 10: 10094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbles KL, Sarkar S, Novak B, Arumugam P (2013) CDK‐dependent nuclear localization of B‐cyclin Clb1 promotes FEAR activation during meiosis I in budding yeast. PLoS ONE 8: e79001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A, Rabitsch KP, Galova M, Schleiffer A, Buonomo SB, Nasmyth K (2000) Functional genomics identifies monopolin: a kinetochore protein required for segregation of homologs during meiosis I. Cell 103: 1155–1168 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Cox J (2016a) The MaxQuant computational platform for mass spectrometry‐based shotgun proteomics. Nat Protoc 11: 2301–2319 [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J (2016b) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740 [DOI] [PubMed] [Google Scholar]
- Tyson JJ, Novak B (2008) Temporal organization of the cell cycle. Curr Biol 18: R759–R768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, Yuan D, Stroe O, Wood G, Laydon A et al (2022) AlphaFold Protein Structure Database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res 50: D439–D444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodermaier HC, Gieffers C, Maurer‐Stroh S, Eisenhaber F, Peters JM (2003) TPR subunits of the anaphase‐promoting complex mediate binding to the activator protein CDH1. Curr Biol 13: 1459–1468 [DOI] [PubMed] [Google Scholar]
- Wang H, Liu D, Wang Y, Qin J, Elledge SJ (2001) Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev 15: 1361–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Davis S, Menon S, Zhang J, Ding J, Cervantes S, Miller E, Jiang Y, Ferro‐Novick S (2015) Ypt1/Rab1 regulates Hrr25/CK1delta kinase activity in ER‐Golgi traffic and macroautophagy. J Cell Biol 210: 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Zhang R, Feng W, Tsuchiya D, Ballew O, Li J, Denic V, Lacefield S (2020) Autophagy of an amyloid‐like translational repressor regulates meiotic exit. Dev Cell 52: 141–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whinston E, Omerza G, Singh A, Tio CW, Winter E (2013) Activation of the Smk1 mitogen‐activated protein kinase by developmentally regulated autophosphorylation. Mol Cell Biol 33: 688–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Shen C, Wang T, Quan J (2013) Structural basis for the inhibition of Polo‐like kinase 1. Nat Struct Mol Biol 20: 1047–1053 [DOI] [PubMed] [Google Scholar]
- Yesbolatova A, Saito Y, Kitamoto N, Makino‐Itou H, Ajima R, Nakano R, Nakaoka H, Fukui K, Gamo K, Tominari Y et al (2020) The auxin‐inducible degron 2 technology provides sharp degradation control in yeast, mammalian cells, and mice. Nat Commun 11: 5701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachariae W, Schwab M, Nasmyth K, Seufert W (1998) Control of cyclin ubiquitination by CDK‐regulated binding of Hct1 to the anaphase promoting complex. Science 282: 1721–1724 [DOI] [PubMed] [Google Scholar]