

Abstract
Mitochondrial DNA (mtDNA) leakage into the cytoplasm can occur when cells are exposed to noxious stimuli. Specific sensors recognize cytoplasmic mtDNA to promote cytokine production. Cytoplasmic mtDNA can also be secreted extracellularly, leading to sterile inflammation. However, the mode of secretion of mtDNA out of cells upon noxious stimuli and its relevance to human disease remain unclear. Here, we show that pyroptotic cells secrete mtDNA encapsulated within exosomes. Activation of caspase‐1 leads to mtDNA leakage from the mitochondria into the cytoplasm via gasdermin‐D. Caspase‐1 also induces intraluminal membrane vesicle formation, allowing for cellular mtDNA to be taken up and secreted as exosomes. Encapsulation of mtDNA within exosomes promotes a strong inflammatory response that is ameliorated upon exosome biosynthesis inhibition in vivo. We further show that monocytes derived from patients with Behçet's syndrome (BS), a chronic systemic inflammatory disorder, show enhanced caspase‐1 activation, leading to exosome‐mediated mtDNA secretion and similar inflammation pathology as seen in BS patients. Collectively, our findings support that mtDNA‐containing exosomes promote inflammation, providing new insights into the propagation and exacerbation of inflammation in human inflammatory diseases.
Keywords: Behçet's syndrome, caspase‐1, exosome, mitochondrial DNA, pyroptosis
Subject Categories: Autophagy & Cell Death, Immunology, Organelles
The secretion of extracellular mtDNA in response to caspase‐1 activation is mediated via exosomes and leads to excessive inflammation.
Introduction
When cells receive noxious stimuli, inflammasome complex formation is induced to activate the inflammatory caspase, caspase‐1. Subsequently, active caspase‐1 cleaves pro‐interleukin (IL)‐1β and IL‐18 and gasdermin‐D to form a plasma membrane pore, resulting in the secretion of mature IL‐1β and IL‐18, and pyroptosis, a form of cytolytic inflammatogenic cell death (Broz & Dixit, 2016). Pyroptosis is a fundamental defense mechanism that deprives invading pathogens of their essential survival niche and induces a robust inflammatory response (Miao et al, 2010; Jorgensen et al, 2017). However, even if the inflammasome is activated, cells do not necessarily undergo pyroptosis; there is an activated state in which cells secrete IL‐1β without cell death and a homeostatic state in which cells adapt to survival by inducing autophagy (Evavold & Kagan, 2019). Thus, there may be other modes of secretion of pathogen‐ and damage‐associated molecular patterns (PAMPs/DAMPs) between the activated state of secreting IL‐1β and the fate of cells that end up in pyroptosis.
Mitochondria serve as a platform that connects cell death and inflammation (Bock & Tait, 2020). PAMPs/DAMPs stimuli induce mitochondrial outer membrane permeabilization (MOMP) and cytochrome c secretion from the mitochondria, leading to the activation of apoptotic caspases. PAMPs/DAMPs also produce mitochondrial reactive oxygen species (mROS) and activate caspase‐1 by inducing inflammasome formation, leading to the induction of pyroptosis by cleaving gasdermin‐D, whose amino‐terminal fragment (Gsdmd‐NT) forms pores in the cell membrane (Kayagaki et al, 2015; Shi et al, 2015; Ding et al, 2016; Liu et al, 2016). Additionally, increased MOMP triggered by PAMP/DAMP stimulation allows mitochondrial DNA (mtDNA) leakage into the cytoplasm, causing a strong inflammation by inducing the production of various cytokines, including type I interferon recognized via cyclic GMP‐AMP synthase (Rongvaux et al, 2014; White et al, 2014), proinflammatory cytokines via toll‐like receptor (TLR) 9 (Zhang et al, 2010; Oka et al, 2012), and IL‐1β and IL‐18 via NLR family pyrin domain containing 3 (NLRP3) inflammasome activation (Broz & Dixit, 2016). Moreover, extracellular mitochondria‐derived DAMPs, including mtDNA, can drive severe inflammation when leaked during large‐scale cell destruction (Zhang et al, 2010; Galluzzi et al, 2012). Additionally, leaked mtDNA has been reported to be involved in several human diseases, including trauma‐related acute respiratory disease syndrome, heart failure, and systemic lupus erythematosus (SLE) (Zhang et al, 2010; Oka et al, 2012; Lood et al, 2016; Melki et al, 2021). However, the mode of secretion of mtDNA outside the cells during the inflammasome activation through the cell death process remains to be elucidated.
Extracellular membrane vesicles (EVs) of different sizes and origins are released from a variety of cells, including cancer, inflammatory, and dying cells (van Niel et al, 2018). Apoptotic cells release EVs (ApoEVs) with a diameter of 0.4–5 μm by shedding the plasma membrane. ApoEVs are usually endocytosed by phagocytic cells and degraded to maintain immunological silence (Doran et al, 2020). Exosomes are 40–150 nm EVs derived from intraluminal vesicles (ILVs) produced by the inward budding of the endosome membrane to form multivesicular bodies (MVBs) (Colombo et al, 2014). In the steady‐state, MVBs fuse with lysosomes, and their content is thereby degraded, whereas, under cellular stress conditions, MVBs fuse with the plasma membrane, resulting in the extracellular release of ILVs as exosomes. EVs contain different types of proteins, mRNAs, miRNAs, DNAs, and lipids (Valadi et al, 2007; Okoye et al, 2014; Jeppesen et al, 2019) and transfer their content to the acceptor cells through endocytosis or plasma membrane fusion, thus affecting the immune response (Buzas et al, 2014; Okoye et al, 2014; Kalluri & LeBleu, 2020). However, whether EVs contribute to the release of mtDNA and the relevance of mtDNA‐containing EVs to human autoimmune diseases remain to be fully elucidated.
Behçet's syndrome (BS) is a chronic systemic inflammatory disorder with transient, periodic, and recurrent symptoms, including ocular signs, oral and genital ulcers, and skin lesions (Sakane et al, 1999; Suzuki Kurokawa & Suzuki, 2004), and has been recognized as a disease at the crossroads between autoimmune and autoinflammatory syndromes (Masters et al, 2009). Cumulative evidence has associated BS pathogenesis with aberrant adaptive immunity, including the involvement of a certain class I human leukocyte antigens (Ohno et al, 1982), autoreactive lymphocytes such as type 1 T helper (Th1) and Th17 cells (Frassanito et al, 1999; Chi et al, 2008), cross‐reactivity between human and bacterial heat shock proteins 60/65 (Pervin et al, 1993; Hu et al, 1998), and the IL12/IL23RB gene polymorphism observed in a genome‐wide association study (Mizuki et al, 2010). BS is also genetically associated with polymorphisms in TLRs and inflammasome‐related genes, such as IL‐1 and the familial Mediterranean fever gene (MEFV) (Kirino et al, 2013; Takeuchi et al, 2017). Indeed, BS has been suggested to have clinical similarities to familial Mediterranean fever, the most prevalent monogenic autoinflammatory disease, in which missense mutations in the MEFV gene result in the constant activation of the pyrin inflammasome and the increased secretion of IL‐1β. Additionally, an aberrant response to PAMP/DAMP stimuli in the innate immune system has been reported to trigger excessive inflammation in BS. Some microorganisms have also been associated with the development of BS (Mumcu et al, 2009; Mumcu & Direskeneli, 2019; van der Houwen et al, 2020), as well as the infiltration of activated neutrophils and macrophages, and the overactivation of TLRs and inflammasome signaling (Liang et al, 2013; Nakano et al, 2018; Le Joncour et al, 2019). However, the pathogenesis of BS caused by innate immune disorders has not been determined.
Here, we showed that the activation of caspase‐1 induced mtDNA leakage from the mitochondria into the cytoplasm via gasdermin‐D and that active caspase‐1 also induces the gasdermin‐D‐independent formation of ILVs, allowing cytoplasmic mtDNA to be taken up and secreted into the extracellular space as exosomes. Additionally, mtDNA promoted a robust sterile inflammation by being encapsulated in exosomes, and the inhibition of the formation of mtDNA‐containing exosomes ameliorated inflammasome‐associated inflammation. Furthermore, caspase‐1 was activated in BS monocytes, resulting in increased secretion of mtDNA‐containing exosomes, and these mtDNA‐rich BS exosomes promoted inflammation characteristics in BS manifestations.
Results
Mitochondrial DNA is released by pyroptotic cells via exosomes
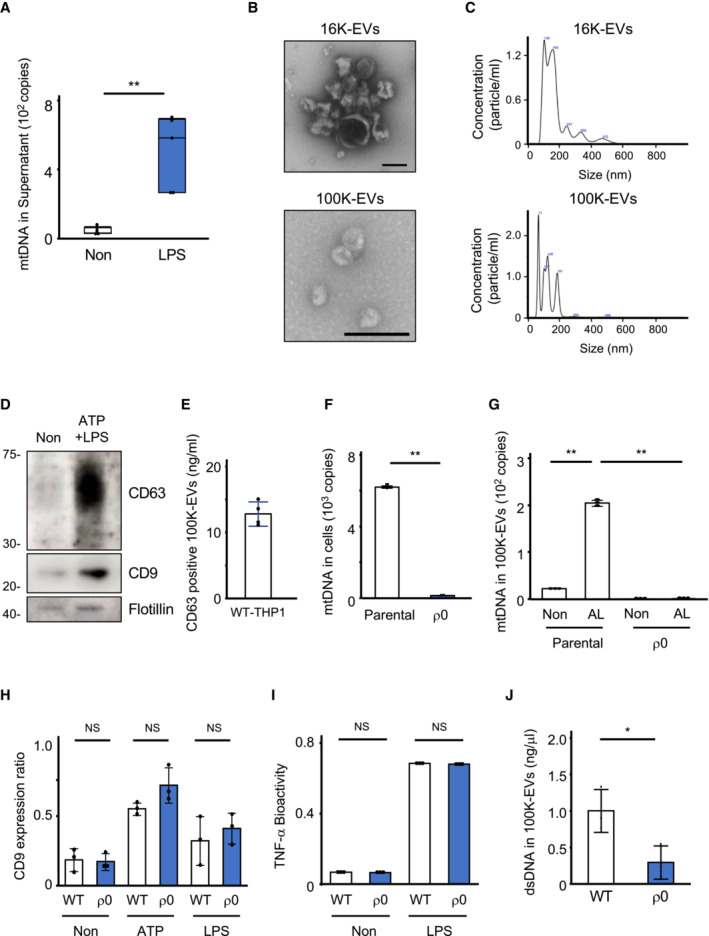
We first determined the increased mtDNA levels in the culture supernatant (CS) of lipopolysaccharide (LPS)‐stimulated human peripheral blood mononuclear cells (PBMCs) via quantitative PCR (Fig EV1A). We subsequently investigated which leukocytes released mtDNA by activating healthy donor neutrophils, CD14+ monocytes, and CD14− PBMCs (lymphocyte‐rich population) with various stimulators. mtDNA was extracellularly released upon cytolytic stimulation, and mtDNA released from neutrophils or lymphocytes was digested with DNase I, whereas mtDNA released from CD14+ monocytes was protected from DNase I digestion unless the membranes were permeabilized with Triton‐X100 (Fig 1A–C). We then induced human monocytic THP‐1 cells to undergo different types of cell deaths, including necrosis, apoptosis, and pyroptosis, to determine whether the mode of mtDNA release varied between the death processes by characterizing the sensitivity of mtDNA to DNase I and Triton‐X100 digestion. Extracellular mtDNA released from THP‐1 cells via necrosis or secondary necrosis was digested with DNase I; conversely, mtDNA released via apoptosis or pyroptosis could not be digested with DNase I unless the samples were treated with Triton‐X100 (Fig 1D). We previously showed that dsDNA contained in ApoEVs is not digested by DNase I unless treated with a detergent (Kato et al, 2018). Based on these, we hypothesized that when monocytes undergo apoptosis or pyroptosis, mtDNA is sequestered in EVs and released to the extracellular space. To investigate whether mtDNA sorting into EVs differed between apoptosis and pyroptosis, we collected EVs via centrifugation at 16,000 g to isolate ApoEVs (16K‐EVs) and ultracentrifugation at 100,000 g to isolate exosomes (100K‐EVs) (Fig EV1B–E). 16K‐EVs contained higher mtDNA levels upon apoptosis induction, whereas mtDNA was only detected in 100K‐EVs from both THP‐1 cells and human CD14+ monocytes upon pyroptosis induction (Fig 1E and F). Higher mtDNA levels were detected in 100K‐EVs when pyroptosis was induced by LPS plus ATP or intracellular LPS stimulation (Fig 1G and H). In addition, dsDNA deposition was detected in the 100K‐EVs from pyroptotic THP‐1 cells using immunoelectron microscopy (Fig 1I). Since the 100K‐EVs may contain both mtDNA and nuclear DNA (nDNA), we generated mtDNA‐defective ρ0 THP‐1 cells (Fig EV1F and G) (Hashiguchi & Zhang‐Akiyama, 2009) and compared the amount of dsDNA in 100K‐EVs between parental and ρ0 THP1 cells. Although ρ0 THP1 cells secreted as much CD9‐positive 100K‐EVs and produced TNF‐α as the parental THP1 cells (Fig EV1H and I), the 100K‐EVs of ρ0 THP1 cells carried lower amounts of dsDNA (Fig EV1J), suggesting that mtDNA is preferentially sorted into the 100K‐EVs. Taken together, these observations suggest that mtDNA is released via 100K‐EVs from monocytic cells when pyroptosis is induced.
Figure EV1. Characteristics of 100K‐EVs isolated from THP‐1 cells.
-
AExtracellular mtDNA leakage from human peripheral blood leukocytes following LPS stimulation. White blood cells isolated from peripheral blood by removing red blood cells were stimulated with LPS (500 ng/ml) for 4 h. DNA was purified from the CS, and the levels of mtDNA were measured using qPCR.
-
BTransmission electron microscopic image of 100K‐EVs. 100K‐EVs isolated from the CS of pyroptotic THP‐1 cells were attached to the coverslip and visualized using transmission electron microscopy. Scale bar, 200 nm.
-
CParticle sizes of 16K‐EVs (upper) and 100K‐EVs (lower) isolated from CS of WT THP1 cells.
-
DExpression of representative exosome markers in 100K‐EVs. 100K‐EVs isolated from the CS of pyroptotic THP‐1 cells were lysed, and the levels of CD9, CD63, and Flotillin were evaluated using western blotting.
-
EAmount of 100K‐EVs used in each experiment. 100K‐EVs isolated from CS of confluent WT THP1 cells in 10 cm culture dishes were quantified by CD63‐specific ELISA.
-
F, GGeneration of mtDNA‐defective THP‐1 (ρ0 THP‐1) cells. ρ0 THP‐1 cells were established by culturing WT THP‐1 cells with medium containing a low concentration of Ethidium Bromide. The absence of mtDNA in ρ0 THP‐1 cells (F) and 100K‐EVs released upon LPS plus ATP stimulation (G) was confirmed using qPCR.
-
H, IEquivalent secretion of 100K‐EVs and TNF‐α in ρ0 THP1 cells. The amount of 100K‐EVs and TNF‐α in CS of WT and ρ0 THP1 cells stimulated with ATP or LPS were measured by western blotting (H) and the reporter cells for TNF‐α (I).
-
JThe level of dsDNA in 100K‐EVs. dsDNA was purified from 100K‐EVs of WT and ρ0 THP1 cells, and the amount of dsDNA was quantified using the Quant‐iT dsDNA Assay Kit.
Data information: Statistical analyses were performed using a Mann–Whitney U test (A: median; 25th and 75th percentile; minimum and maximum value excluding population outliers; **P < 0.01), a Student's t‐test (F: mean ± SD; **P < 0.01), a Mann–Whitney U test (J: mean ± SD; *P < 0.05), or an ANOVA with Tukey's post‐hoc test (G–I) (mean ± SD; *P < 0.05, **P < 0.01; NS, not statistically significant). For panel E, (mean ± SD). The data are representative of two (B, C) three (A, D–J) independent experiments.
Source data are available online for this figure.
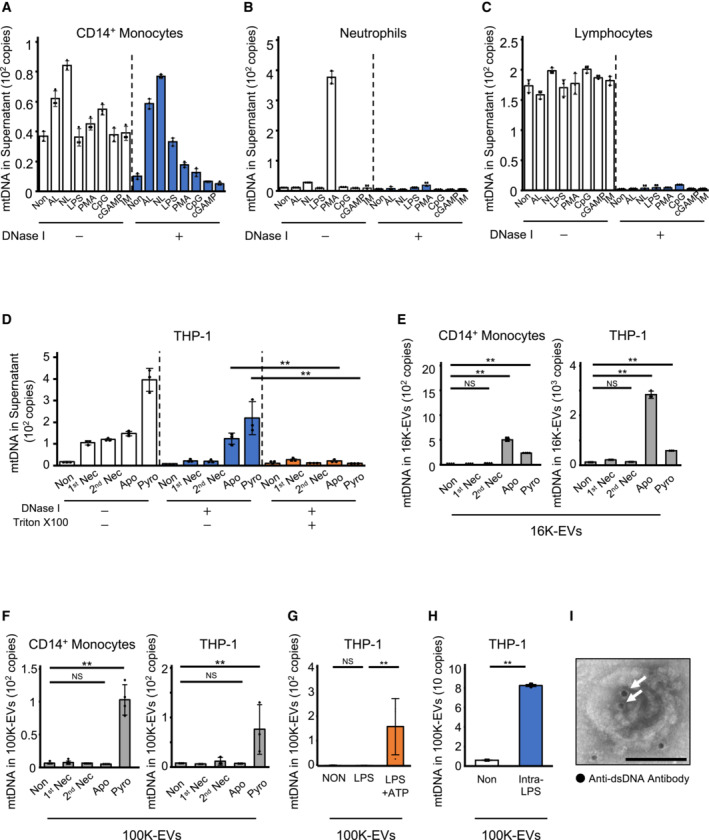
Figure 1. Extracellular secretion of mtDNA encapsulated within EVs by pyroptotic cells.
-
A–CmtDNA levels in the CS of HC‐derived leukocytes following PAMP/DAMP stimulation, with or without DNase I treatment. CD14+ monocytes (A), neutrophils (B), and lymphocytes (C) isolated from the peripheral blood of HCs were PAMP/DAMP stimulated utilizing: AL (ATP and LPS), NIG (nigericin and LPS), LPS, PMA, fMLP, CpG (ODN2006), cGAMP, or IM (ionomycin). The COXIII levels in purified DNA in CS were measured using qPCR after removing cell debris and DNase I treatment or mock digestion.
-
DmtDNA levels in the CS of THP‐1 cells induced to undergo primary necrosis (1st Nec), secondary necrosis (2nd Nec), apoptosis (Apo), or pyroptosis (Pyro). After removal of cell debris, the supernatant was untreated (left, middle) or treated with Triton‐X100 (right), then undigested (left) or digested (middle, right) with DNase I. mtDNA levels were measured in purified DNA.
-
E, FmtDNA levels in 16K‐ (E) and 100K‐EV (F) fractions in the CS of THP‐1 cells and CD14+ monocytes induced to undergo 1st Nec, 2nd Nec, Apo, or Pyro. 16K‐EVs and 100K‐EVs were separated using centrifugation at 16,000 g and 100,000 g, respectively. mtDNA levels in these EVs were measured by qPCR.
-
G, HSecretion of mtDNA in 100K‐EVs by pyroptotic stimulation. THP1 cells were stimulated with LPS alone or LPS + ATP (G). HC–derived CD14+ monocytes were intracellularly stimulated with LPS enclosed in liposomes utilizing Avalanche‐Omni (H). mtDNA levels in 100K‐EVs were measured after 100K‐EVs were isolated from CS.
-
IDeposition of dsDNA in 100K‐EVs. 100K‐EVs isolated from the CS of THP‐1 cells after LPS/ATP stimulation were fixed and stained with an anti‐dsDNA antibody, and then visualized with a secondary antibody conjugated to 10‐nm gold particles. Images were obtained using transmission immunoelectron microscopy. Black dots (white arrows) indicate the dsDNA deposition. Scale bar, 100 nm.
Data information: Statistical analyses were performed using an ANOVA with Tukey's post‐hoc test (D–G) (mean ± SD; **P < 0.01; NS, not statistically significant) and a Student's t‐test (H) (mean ± SD; **P < 0.01). For panel A‐C (mean ± SD). The data are representative of three independent experiments.
Source data are available online for this figure.
Caspase‐1 and gasdermin‐D are required for mtDNA secretion via exosomes
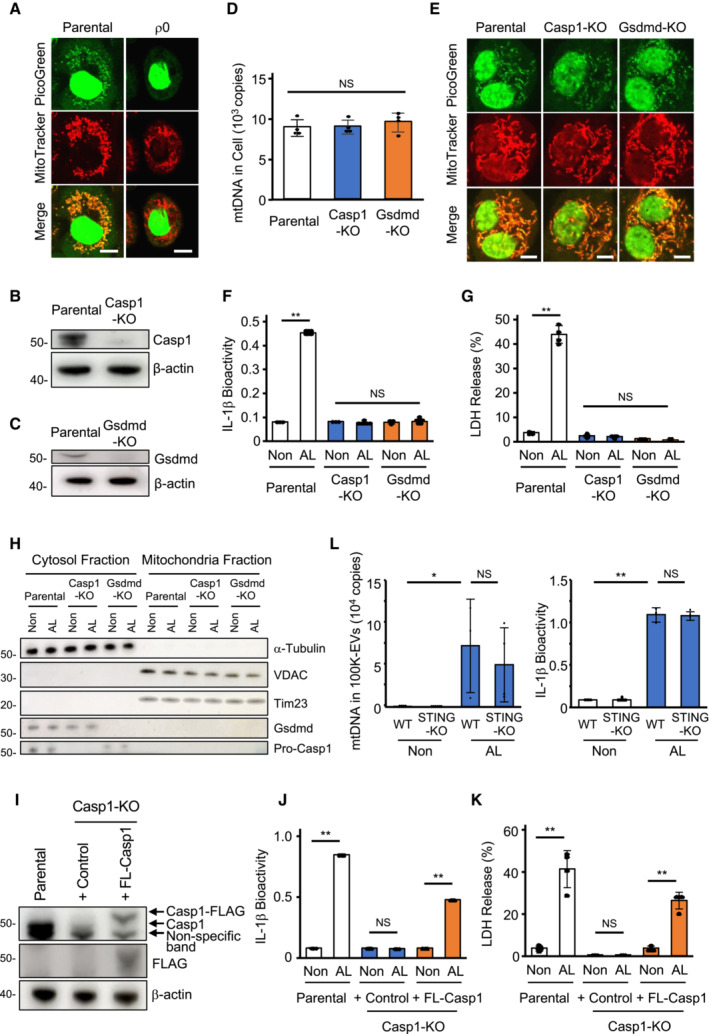
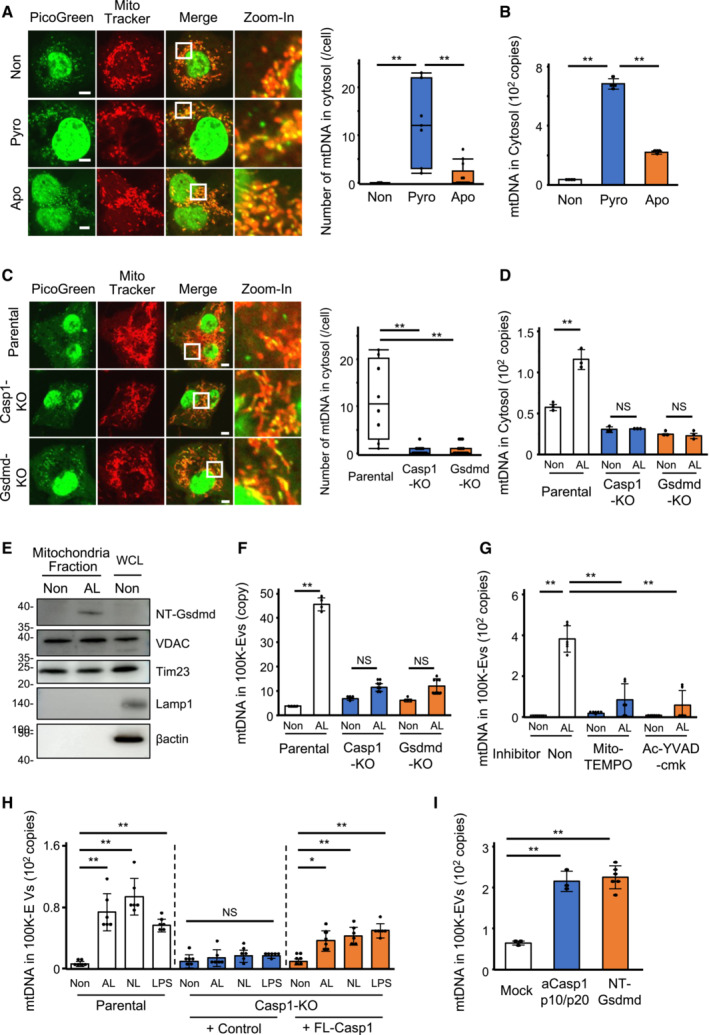
Subsequently, we investigated mtDNA leakage during cell death and confirmed that mtDNA could be visualized as PicoGreen‐positive dots in the cytoplasm (White et al, 2014) (Fig EV2A). PicoGreen‐positive dots were observed in the non‐mitochondrial region of the cytoplasm of pyroptotic cells, whereas very few PicoGreen‐positive dots were observed in the cytoplasm of apoptotic cells (Fig 2A). Quantitatively, the level of mtDNA in the mitochondria‐removed cytoplasmic fraction was higher in pyroptotic cells than in apoptotic cells, as evaluated by qPCR (Fig 2B), suggesting that mtDNA preferentially leaked from the mitochondria into the cytosol during pyroptosis. Since pyroptosis is mediated by mROS and the activation of caspase‐1 and subsequent gasdermin‐D activation (Kayagaki et al, 2015; Shi et al, 2015; Liu et al, 2016), we examined the involvement of these molecules in the secretion of mtDNA‐containing exosomes by generating caspase‐1‐knockout (Casp1‐KO) and gasdermin‐D‐knockout (Gsdmd‐KO) THP‐1 cells (Fig EV2B and C). These cells showed comparable levels and distribution patterns of mtDNA at the steady‐state and decreased IL‐1β and lactate dehydrogenase (LDH) release compared to parental wild‐type (WT) THP‐1 cells (Fig EV2D–G). We then stimulated WT, Casp1‐KO, or Gsdmd‐KO‐THP‐1 cells with LPS and ATP and evaluated mtDNA leakage by confocal microscopy and qPCR after the removal of the mitochondria fraction (Fig EV2H). WT THP‐1 cells, but not Casp1‐KO or Gsdmd‐KO‐THP‐1 cells, exhibited mtDNA leakage from the mitochondria into the cytoplasm (Fig 2C and D). Notably, the N‐terminal region of gasdermin‐D (NT‐Gsdmd) was detected in the mitochondria fraction upon LPS + ATP stimulation (Fig 2E). Additionally, mtDNA extracellular secretion mediated by 100K‐EVs was reduced in Casp1‐KO and Gsdmd‐KO THP1 cells (Fig 2F). Consistently, mtDNA leakage into the extracellular space via 100K‐EVs was reduced when the cells were treated with the Mito‐TEMPO, an mROS chelator, or Ac‐YVAD‐cmk, a caspase‐1 inhibitor, upon pyroptosis induction (Fig 2G). We subsequently conducted gain‐of‐function experiments by restoring full‐length caspase‐1 to Casp1‐KO (FL‐Casp1‐Casp1‐KO) THP‐1 cells and transducing an active form of caspase‐1 (aCasp1) (p10/p20) or NT‐Gsdmd into WT THP‐1 cells (Fig EV2I–K). Release of extracellular mtDNA via 100K‐EVs was rescued in FL‐Casp1‐Casp1‐KO‐THP‐1 cells (Fig 2H) and was increased in WT cells transfected with aCasp1 or NT‐Gsdmd (Fig 2I). On the other hand, mtDNA is recognized by cGAS, but cGAS–STING is not involved in 100K‐EV‐mediated mtDNA secretion or IL‐1β production during pyroptosis induction (Fig EV2L). These results suggested that mtDNA leakage into the cytoplasm and its subsequent extracellular secretion require the activation of the mROS–caspase‐1–gasdermin‐D pathway.
Figure EV2. Establishment of mtDNA‐defective, caspase‐1‐deficient, or gasdermin‐D‐deficient THP‐1 cells.
-
ALocalization of mtDNA and mitochondria. Parental and ρ0 THP‐1 cells were stained with the PicoGreen (green) and MitoTracker‐Deep Red (red) dyes and visualized using confocal microscopy. Scale bar, 10 μm.
-
B, CEstablishment of Casp1‐KO and Gsdmd‐KO‐THP‐1 cells. Caspase‐1 levels in parental WT and Casp1‐KO‐THP‐1 cells (B) and gasdermin‐D in parental WT and Gsdmd‐KO‐THP‐1 cells (C) were evaluated using western blotting using anti‐caspase‐1 (B) and anti‐gasdermin‐D (C) antibodies.
-
D, EEquivalent mitochondrial homeostasis in steady‐state Casp1‐KO and Gsdmd‐KO‐THP‐1 cells. Cellular mtDNA levels (D) and distribution of mtDNA and mitochondria (E) in parental WT, Casp1‐KO, and Gsdmd‐KO‐THP‐1 cells at the steady‐state. Scale bar, 5 μm.
-
F, GIL‐1β and LDH release by Casp1‐KO and Gsdmd‐KO‐THP‐1 cells. Release of IL‐1β (F) and LDH (G) from parental WT, Casp1‐KO, and Gsdmd‐KO‐THP‐1 cells upon pyroptosis‐inducing stimulation.
-
HSeparation of cytoplasmic and mitochondrial fractions. After the removal of nuclei from WT, Caps1‐KO, and Gsdmd‐KO THP1 cells stimulated with LPS and ATP, the mitochondrial fraction was separated. The remaining fraction was designated the cytosol fraction. The expression of the proteins shown in the figure in each fraction was evaluated by western blotting.
-
I–KRestoration of full‐length caspase‐1 in Casp1‐KO‐THP‐1 cells. FLAG‐tagged full‐length caspase‐1 was transduced via lentivirus into Casp1‐KO (FL‐Casp1‐Casp1‐KO) THP‐1 cells. Pro‐caspase‐1 and FLAG expression levels in parental WT, Casp1‐KO, and FL‐Casp1‐Casp1‐KO‐THP‐1 cells were evaluated using western blotting (I). The levels of IL‐1β (J) and LDH (K) in the supernatant were also evaluated.
-
LEvaluation of STING's involvement. The level of mtDNA in 100K‐EVs (left) and IL‐1β (right) in CS of WT and STING‐KO THP1 upon LPS plus ATP simulation was determined.
Data information: Statistical analyses were performed using an ANOVA with Tukey's post‐hoc test (D, F, G, J–L) (mean ± SD; *P < 0.05, **P < 0.01; NS, not statistically significant). The data are representative of two (A–H, I–K) and three (L) independent experiments.
Source data are available online for this figure.
Figure 2. Leaked cytosolic mtDNA is taken up by ILVs.
-
ALeakage of mtDNA from the mitochondria into the cytosol. THP‐1 cells under normal, apoptotic, or pyroptotic conditions were stained with the PicoGreen (green) and MitoTracker‐Deep Red (red) dyes and visualized using confocal microscopy. Scale bar, 5 μm (left). Quantification of the number of PicoGreen‐positive dots in the cytosol (right).
-
BLeaked cytosolic mtDNA levels following cell death induction. After removing the nuclei, the mitochondrial and cytosolic fractions from THP‐1 cells were isolated under normal, apoptotic, or pyroptotic conditions. DNA was purified from the cytosolic fraction, and the mtDNA levels were measured.
-
C, DCytosolic mtDNA levels in Casp1‐KO or Gsdmd‐KO cells undergoing pyroptosis. mtDNA and mitochondria in WT, Casp1‐KO, and Gsdmd‐KO‐THP‐1 cells induced to undergo pyroptosis were stained with the PicoGreen (green) and MitoTracker‐Deep Red (red) dyes and visualized using confocal microscopy. Scale bar, 5 μm (left). Quantification of the number of PicoGreen‐positive dots in the cytosol (right) (C). mtDNA level in the cytosolic fraction was measured by qPCR (D).
-
EMitochondria localization of NT‐gasdermin‐D. Ten minutes after THP1 cells were induced pyroptosis, mitochondria fraction was isolated and Western blotting was performed using anti‐gasdermin‐D antibody.
-
FmtDNA level in 100K‐EVs. 100K‐EVs were isolated from CS of WT, Casp1‐KO, Gsdmd‐KO THP1 cells induced to undergo pyroptosis, and mtDNA level in 100K‐EVs was measured.
-
GmtDNA levels in 100K‐EVs from pyroptotic cells with inhibited caspase‐1 or mROS. CD14+ monocytes were pre‐treated with Mito‐TEMPO or Ac‐YVAD‐cmk and then stimulated with LPS and ATP. The levels of mtDNA in 100K‐EVs in CS were measured.
-
HmtDNA levels in 100K‐EVs from caspase‐1‐restored Casp1‐KO‐THP‐1 cells. The levels of mtDNA were measured in 100K‐EVs secreted by pyroptotic WT, Casp1‐KO, and FL‐Casp1‐Casp1‐KO‐THP‐1 cells.
-
ImtDNA levels in 100K‐EVs from cells overexpressing aCasp1 or NT‐Gsdmd. aCasp1‐p10/p20 or NT‐Gsdmd were introduced into WT THP‐1 cells, and the extracellular mtDNA levels within 100K‐Evs were measured.
Data information: Statistical analyses were performed using a Steel–Dwass test (A, C) (median; 25th and 75th percentiles; minimum and maximum value of a population; **P < 0.01), and an ANOVA with Tukey's post‐hoc test (B, D, F–I) (mean ± SD; *P < 0.05, **P < 0.01; NS, not statistically significant). The data are representative of two (E) and three (A–D, F–I) independent experiments.
Source data are available online for this figure.
Involvement of caspase‐1 in ILV formation
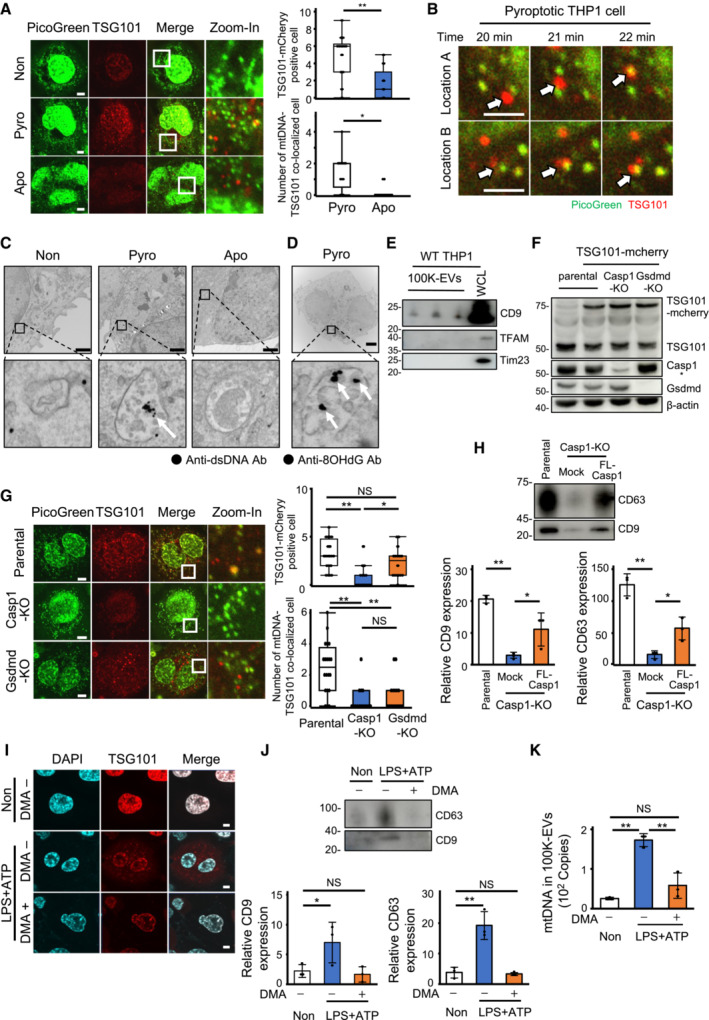
We next observed the formation of ILVs by transducing mCherry‐tagged Tsg101, a component of the endosomal sorting complex required for transport (ESCRT)‐I and a representative ILV marker (Banfer et al, 2018), into THP‐1 cells. The formation of Tsg101 puncta was observed in pyroptotic THP‐1 cells but less in apoptotic ones (Fig 3A). In addition, co‐localization of Tsg101 puncta and mtDNA was observed approximately 20 min after LPS and nigericin stimulation (Fig 3B). Consistently, transmission immunoelectron microscopy revealed dsDNA deposition in the MVBs of pyroptotic THP‐1 cells but not in those of apoptotic THP‐1 cells (Fig 3C). In particular, deposition of mROS‐induced 8‐OHdG‐modified DNA was observed in the MVBs of pyroptotic THP‐1 cells (Fig 3D), It has been reported that mtDNA is packed with TFAM (Alam et al, 2003) and released out of mitochondria by herniation of the mitochondrial membrane via BAK/BIX‐macropore (McArthur et al, 2018). It has also been shown that mitochondrial proteins including TFAM are detected in 100K‐EVs isolated from T cell culture supernatants (Torralba et al, 2018). Therefore, we examined whether mitochondrial proteins such as Tim23 and TFAM could be detected in purified 100K‐EVs. However, these proteins were not detected (Fig 3E). However, it could not be completely determined whether mitochondrial proteins are present in 100K‐EVs from pyroptotic cells; since there is a possibility that mtDNA is trapped in 100K‐EVs that do not contain mitochondrial proteins, or that mtDNA and mitochondrial proteins are trapped simultaneously in 100K‐EVs but mitochondrial proteins are not detected, perhaps due to insufficient antibody affinity. However, the present study indicate that mtDNA leaking into the cytoplasm was taken up by ILVs and subsequently released into the extracellular space within exosomes. We further investigated the involvement of caspase‐1 and gasdermin‐D in ILV formation by transducing Tsg101‐mCherry into Casp1‐KO and Gsdmd‐KO‐THP‐1 cells (Fig 3F). Formation of Tsg101 puncta upon LPS and ATP stimulation was not observed in Casp1‐KO‐THP‐1 cells, but it was detected in Gsdmd‐KO‐THP‐1 cells (Fig 3G). Additionally, the secretion of 100K‐EVs expressing CD9/CD63 upon LPS and ATP stimulation was consistently diminished in Casp1‐KO‐THP‐1 cells, and it was ameliorated by the restoration of full‐length caspase‐1 (Fig 3H). Moreover, we investigated whether the inhibition of exosome biosynthesis suppressed mtDNA secretion. To this end, THP‐1 cells undergoing pyroptosis were treated with dimethyl amiloride (DMA), a sodium/potassium exchanger inhibitor (Savina et al, 2003). DMA inhibited the induction of the formation of Tsg101 puncta and the production of CD9/CD63‐expressing 100K‐EVs (Fig 3I and J) and suppressed the secretion of 100K‐EVs containing mtDNA as with Ac‐YVAD‐cmk (Fig 3K). These results indicated that caspase‐1, but not gasdermin‐D, played an essential role in ILV formation and that exosome biosynthesis was important for extracellular mtDNA secretion during pyroptosis.
Figure 3. Involvement of caspase‐1 in ILV formation.
-
A, BFormation of Tsg101 puncta and co‐localization with mtDNA. THP‐1 cells transduced with Tsg101‐mCherry (red) were stained with PicoGreen (green) and induced to undergo cell death (no death, pyroptosis, or apoptosis). Co‐localization of mtDNA with Tsg101 was visualized using confocal microscopy (left). The number of cells expressing Tsg101 puncta (upper) and the number of cells mtDNA and Tsg101 co‐localized cells (lower) in around 20 randomly observed fields are shown on the right. Scale bar, 5 μm (A). The cells were imaged over a time course of pyroptosis induction. White arrows indicate the Tsg101‐mcherry positive ILVs. Scale bar, 2.5 μm (B).
-
CDeposition of dsDNA in multivesicular endosomes following cell death induction. After induction of pyroptosis and apoptosis, transmission immunoelectron microscopy was performed using an anti‐dsDNA antibody and a secondary antibody conjugated to 10‐nm gold particles. Black dots (white arrow) indicate dsDNA deposition; scale bar, 1 μm.
-
D8‐OHdG‐positive deposition in MVBs of pyroptotic cells. After induction of pyroptosis using LPS and ATP, transmission immunoelectron microscopy was performed using an anti‐8‐OHdG antibody and a secondary antibody conjugated to 10‐nm gold particles. Black dots (white arrows) indicate 8‐OHdG‐positive depositions. Scale bar, 2 μm.
-
ETFAM and Tim23 in 100K‐EVs. 100K‐EVs were isolated from CS of WT THP1 cells induced to undergo pyroptosis; western blotting was performed using the indicating antibodies.
-
F, GInduction of ILV formation in pyroptotic cells. Tsg101‐mCherry‐transduced WT, Casp1‐KO, and Gsdmd‐KO‐THP‐1 cells (F) were stimulated with LPS and ATP. The co‐localization of mtDNA (green) and Tsg101 (red) was visualized using confocal microscopy. Scale bar, 5 μm (left). The number of cells expressing Tsg101 puncta (upper) and the number of cells mtDNA and Tsg101 co‐localized cells (lower) in around 20 randomly observed fields are shown on the right (G).
-
HLevels of 100K‐EVs secreted from caspase‐1‐restored Casp1‐KO‐THP‐1 cells. The levels of CD63‐ and CD9‐expressing 100K‐EVs from pyroptosis‐induced FL‐Casp1‐Casp1‐KO‐THP‐1 cells were assessed using western blotting (upper). Quantitative data of CD9 (left) and CD63 (right) are shown at the lower.
-
I, JInhibition of ILV formation by DMA. THP‐1 cells were treated with DMA (1.5 μM) for 12 h and subsequently stimulated with LPS and ATP. The formation of Tsg101 puncta (red) was visualized using confocal microscopy. Scale bar, 5 μm (I). 100K‐EVs were isolated from the CS, and their CD9 and CD63 expression levels were determined using western blotting (upper). Quantitative data are shown on the lower (J).
-
KReduced secretion of mtDNA in 100K‐EVs by DMA. THP‐1 cells were treated with DMA (1.5 μM) for 12 h and subsequently stimulated with LPS and ATP. After the isolation of 100K‐EVs from the CS, the mtDNA levels were measured.
Data information: Statistical analyses were performed using a Mann–Whitney U test (A) (median; 25th and 75th percentiles; minimum and maximum value of a population; *P < 0.05, **P < 0.01; NS, not statistically significant), a Steel–Dwass test (G) (median; 25th and 75th percentiles; minimum and maximum value of a population; *P < 0.05, **P < 0.01), and an ANOVA with Tukey's post‐hoc test (H, J, K) (mean ± SD; *P < 0.05, **P < 0.01). The data are representative of two (C–F) and three (A, B, G–K) independent experiments.
Source data are available online for this figure.
Encapsulation of mtDNA within exosomes promoted strong inflammation
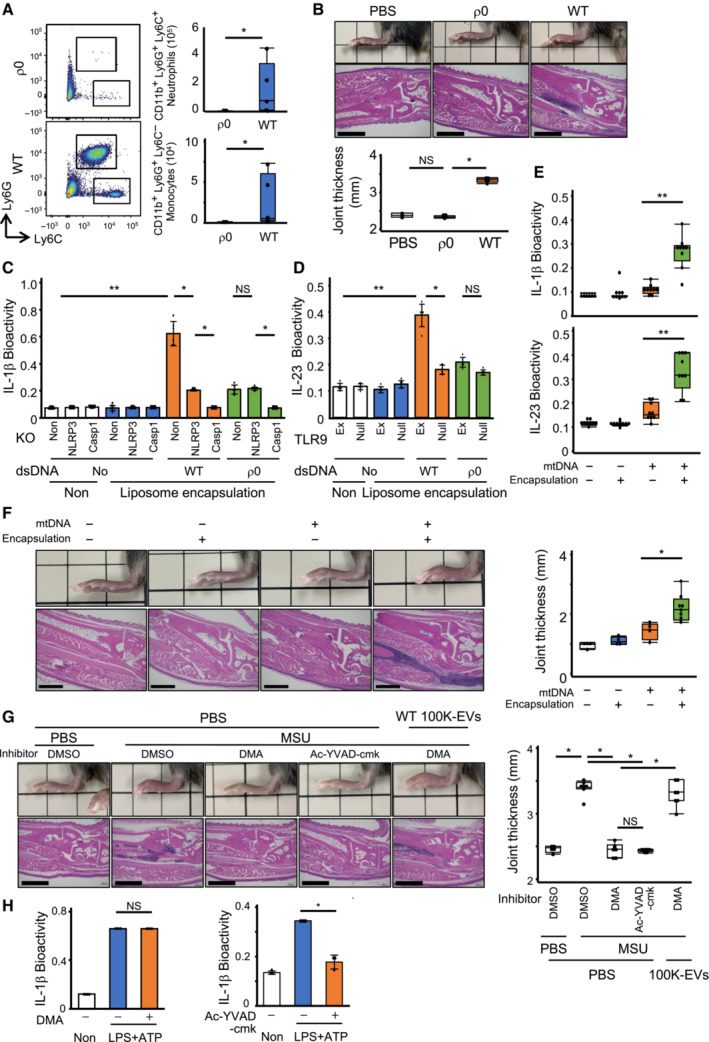
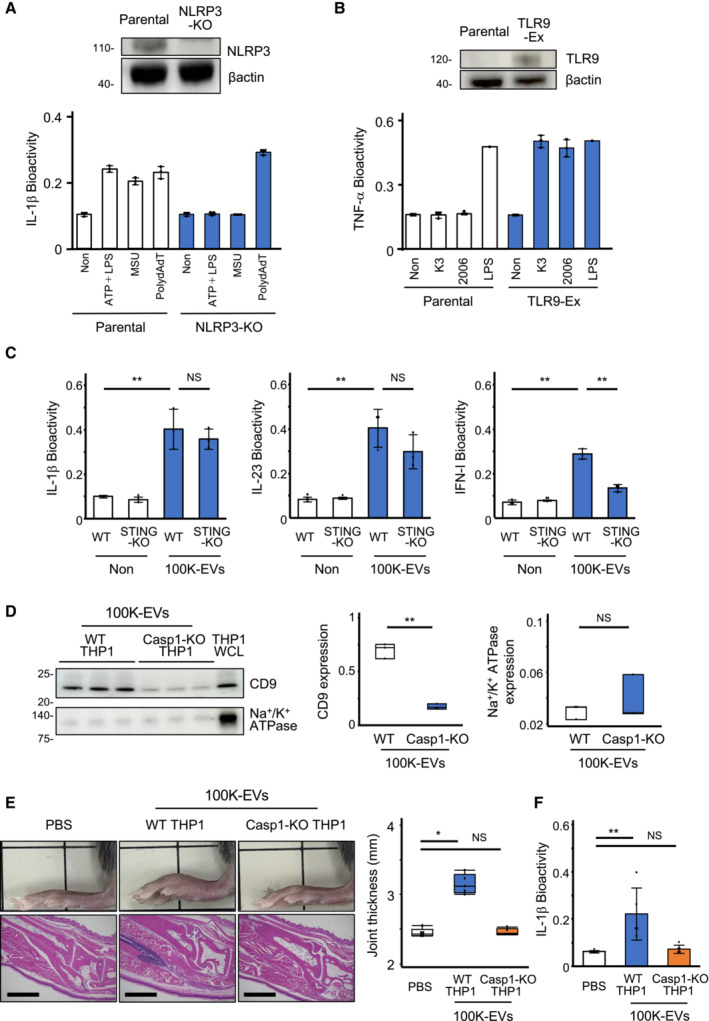
Since mtDNA is known to be a strong proinflammatory factor, we further investigated the effect of mtDNA‐encapsulated exosomes on the promotion of inflammation. First, 100K‐EVs isolated from WT and mtDNA‐defective ρ0 THP‐1 cells were injected into the peritoneum or ankle of C57BL6/J mice. 100K‐EVs derived from WT THP‐1 cells, but not those from ρ0 THP‐1 cells, promoted the infiltration of neutrophils (CD11b+Ly6C+Ly6G+) and monocytes (CD11b+Ly6C+Ly6G−) in the peritoneal cavity and induced ankle swelling (Fig 4A and B). Next, to confirm the contribution of mtDNA to 100K‐EVs‐induced inflammation, NLRP3‐deficient THP1 cells (NLRP3‐KO) and TLR9‐expressing THP1 cells (TLR9‐Ex) were generated (Fig EV3A and B), as the parental THP1 cells are not responsive to TLR ligands (Kato et al, 2018). The liposome‐encapsulated dsDNA purified from 100K‐EVs of WT THP1 cells, but not from ρ0 THP1 cells, induced the secretion of large amounts of IL‐1β and IL‐23 via NLRP3 and TLR9, respectively. On the other hand, dsDNA from ρ0 THP1 cells induced the secretion of small amounts of IL‐1β in an NLRP3‐independent manner, suggesting that nDNA may induce IL‐1β via NLRP3‐independent mechanisms such as that through AIM2 and IFI16 (Fig 4C and D). Regarding the involvement of the cGAS–STING pathway in 100K‐EVs‐containing dsDNA recognition, STING‐KO THP1 cells showed reduced type I IFN (IFN‐I) production upon WT 100K‐EVs stimulation, but comparable IL‐1β and IL‐23 production (Fig EV3C). These results indicated that exosome‐encapsulated dsDNA promoted sterile inflammation and that mtDNA is a major factor for 100K‐EV‐induced inflammation. Subsequently, we purified liposome‐encapsulated mtDNA from the mitochondria to compare the cytokine production levels and degree of ankle inflammation between liposome‐encapsulated and non‐encapsulated mtDNA, and we observed that liposome‐encapsulated mtDNA resulted in greater IL‐1β and IL‐23 production by PBMCs and ankle swelling than non‐encapsulated mtDNA (Fig 4E and F). Furthermore, to investigate the significance of mtDNA encapsulation in exosomes with regard to the induction of in vivo inflammation, we inhibited the in vivo production of exosomes via DMA. DMA was administered to the ankle of the mice, where it was injected with sodium urate (MSU) crystals, an inflammasome activator. Treatment with DMA reduced MSU‐induced ankle swelling by suppressing leukocyte infiltration to similar levels to those following treatment with Ac‐YVAD‐cmk, and administration of WT 100K‐EVs restored the ankle inflammation in DMA‐treated mice (Fig 4G). Of note, in contrast to Ac‐YVAD‐cmk, DMA did not suppress IL‐1β production following LPS and ATP stimulation (Fig 4H), suggesting that DMA inhibited the production of exosomes but not the activation of the inflammasome. Furthermore, 100K‐EVs isolated from Casp1‐KO THP1 cells, which contained fewer mtDNA and CD9‐positive exosomes (Fig EV3D), failed to induce ankle arthritis and IL‐1β production (Fig EV3E and F). These results indicated that mtDNA promoted a stronger sterile inflammation in vivo when encapsulated within exosomes.
Figure 4. Encapsulation of mtDNA in membrane vesicles induces strong inflammation.
-
ALeukocyte mobilization by 100K‐EVs. 100K‐EVs isolated from the CS of pyroptotic WT or ρ0 THP‐1 cells were adoptively injected into the peritoneal cavity of WT mice. Four hours later, cells in the peritoneal cavity were isolated and stained for CD11b, Ly6C, and Ly6G. CD11b+Ly6G+ neutrophils and CD11b+Ly6C+ monocytes were counted using FACS. Population (left) and absolute number (right) of cells are shown.
-
B100K‐EV‐induced ankle swelling. 100K‐EVs isolated from the CS of pyroptotic WT or ρ0 THP‐1 cells were adoptively injected into the ankle of WT mice. The ankle thickness was measured 24 h later. Histological imaging, scale bar, 1 mm (upper), swollen ankle thickness (lower).
-
C, DEncapsulation of dsDNA by liposomes induces cytokine production via NLRP3 and TLR9. dsDNA isolated from CS of WT or ρ0 THP1 cells were encapsulated into liposomes using Avalanche‐Omni and added to WT, NLRP3‐KO, Casp‐1‐KO, parental TLR9‐null, or TLR9‐Ex THP‐1 cells. The levels of IL‐1β (C) and IL‐23 (D) in the CS were measured.
-
E, FEncapsulation of mtDNA by liposomes induces strong inflammation. mtDNA was encapsulated or non‐encapsulated into liposomes using Avalanche‐Omni and added to WT THP1 cells (E), and injected into the ankle of mice (F). The levels of IL‐1β (upper) and IL‐23 (lower) in the CS were measured. The thickness of the swollen ankles was evaluated after 24 h. Histological imaging, scale bar, 1 mm (left) and ankle thickness (right) (F).
-
GMSU‐induced arthritis under DMA treatment. The ankles of mice were pre‐treated with DMA or Ac‐YVAD‐cmk for 2 h, and then MSU was injected. After 24 h, ankle thickness was measured. Swollen ankle thickness, scale bar, 1 mm (left) and histological imaging (right).
-
HEffect of DMA on IL‐1β production. THP‐1 cells were treated with DMA (top) for 12 h or Ac‐YVAD‐cmk (bottom) for 2 h and subsequently stimulated with LPS and ATP. The levels of IL‐1β were measured.
Data information: Statistical analyses were performed using a Mann–Whitney U test (A) (mean ± SD; *P < 0.05), a Steel–Dwass test (B, E–G) (median; 25th and 75th percentiles; minimum and maximum value of a population; *P < 0.05), and an ANOVA with Tukey's post‐hoc test (C, D, H) (mean ± SD; *P < 0.05, **P < 0.01; NS, not statistically significant). The data are representative of two (B, F, G) and three (A, C–E, H) independent experiments.
Source data are available online for this figure.
Figure EV3. Assessment of nuclear DNA and microvesicles' involvement in 100K‐EV‐induced inflammation.
-
AEstablishment of NLRP3‐KO‐THP‐1 cells. NLRP3‐KO‐THP‐1 cells were generated using the CRISPR/Cas9 system. Expression of NLRP3 was assessed using western blotting with an anti‐NLRP3 antibody (upper). IL‐1β production by parental WT‐ and NLRP3‐KO‐THP‐1 cells in response to LPS, ATP, MSU, and poly:dAdT (lower).
-
BEstablishment of TLR9‐expressing THP‐1 cells. Human TLR9 was introduced into THP‐1 cells by lentiviral transduction (TLR9‐Ex). Expression of TLR9 was assessed using western blotting with an anti‐TLR9 antibody (upper). TNF‐α production induced by TLR9 ligands, such as K3 and ODN2006, was elevated in TLR9‐Ex THP‐1 cells (lower).
-
CAssessment of cGAS–STING involvement in 100K‐EV‐induced cytokine production. 100K‐EVs isolated from CS of WT THP1 cells undergoing pyroptosis were added to WT or STING‐KO THP1 cells. The level of IL‐1β (left), IL‐23 (middle), and IFN‐I (right) was quantified using the specific reporter cells for these cytokines.
-
DExosomes and microvesicles in 100K‐EVs. 100K‐EVs were isolated from CS of WT and Casp1‐KO THP1 cells undergoing pyroptosis, the level of exosomes and microvesicles were evaluated by Western blotting (left) using anti‐CD9 (middle) and anti‐Na+/K+ ATPase (right) antibodies.
-
E, FInflammation caused by 100K‐EVs of Casp1‐KO THP1 cells. 100K‐EVs isolated from CS of WT and Casp1‐KO THP1 cells undergoing pyroptosis were injected into the ankle of WT mice (E) and added to WT THP1 cells (F). The evaluation of ankle swelling (upper) and histological analysis (lower) was performed after 18 h. Scale bar, 1 mm. Quantitative data from the swollen ankle are shown on the right (E). IL‐1β bioactivity in CS was measured using reporter cells for IL‐1β (F).
Data information: Statistical analyses were performed using a Mann–Whitney U test (D), (median; minimum and maximum value excluding population outliers; **P < 0.01; NS, not statistically significant), an ANOVA with Tukey's post‐hoc test (C, F) (mean ± SD; *P < 0.05, **P < 0.01; NS, not statistically significant), or a Steel–Dwass test (E) (median; 25th and 75th percentile; minimum and maximum value excluding population outliers; *P < 0.05; NS, not statistically significant). For panel A and B, mean ± SD. The data are representative of two (A, B, D–F) and three (C) independent experiments.
Source data are available online for this figure.
Patients with BS presented increased mtDNA in blood circulation
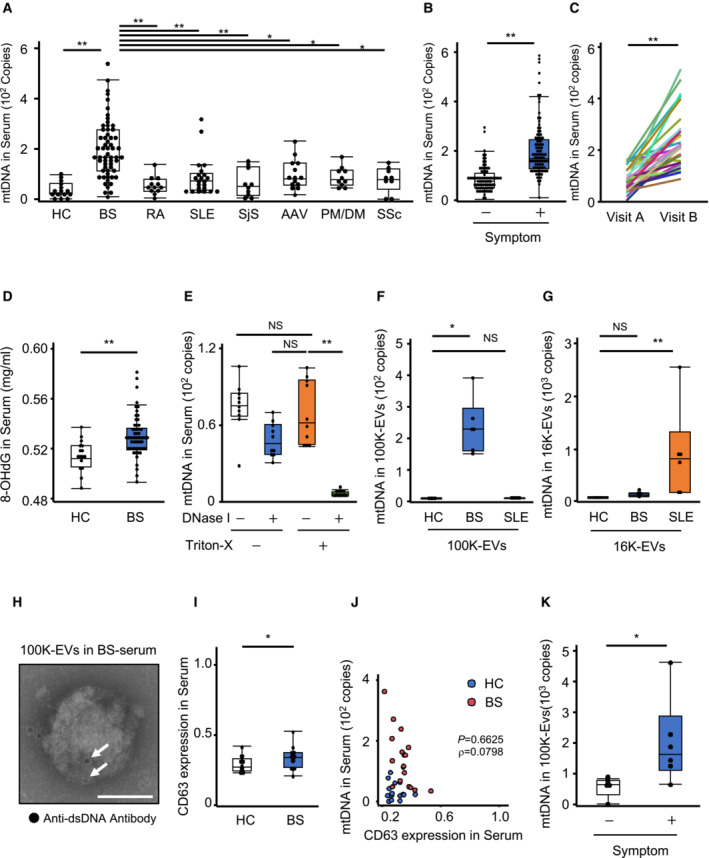
We further investigated the clinical significance of exosome‐encapsulated mtDNA using human blood samples. Since inflammasomes and pyroptosis have been implicated in the etiology of autoinflammatory diseases, special attention was paid to BS, which exhibits autoinflammatory disease‐like clinical symptoms. To this end, we conducted a retrospective study that included various collagen diseases and measured the mtDNA levels after purification of DNA from the serum of patients with BS, n = 59; rheumatoid arthritis (RA), n = 12; SLE, n = 22; Sjögren syndrome (SjS), n = 10; ANCA‐associated vasculitis (AAV), n = 14; polymyositis/dermatomyositis (PM/DM), n = 10; systemic sclerosis (SSc), n = 9; and healthy volunteer donors (HCs), n = 15; the characteristics of the patients and HCs are provided in Appendix Table S1. The levels of mtDNA were elevated in BS compared to the other collagen diseases (Fig 5A), although the serum IL‐1β levels were comparable between BS, RA, and SjS (Fig EV4A). Additionally, serum mtDNA levels were higher in patients showing BS symptoms, including cutaneous signs, uveitis, genital ulcers, arthralgias, and intestinal lesions, than in asymptomatic patients (Figs 5B and EV4B–G). Notably, longitudinal evaluation of these patients showed that serum mtDNA levels were higher during symptomatic than during asymptomatic periods (Fig 5C). Additionally, the levels of 8‐OHdG‐modified DNA were elevated in BS sera compared to HC sera (Fig 5D). Since the mtDNA spiked into the human serum was completely digested by DNase I treatment (Fig EV4H), the serum was subsequently treated with DNase I and Triton‐X100 to confirm the encapsulation of mtDNA into EVs. We observed that the mtDNA was not digested by DNase I if the serum was not pre‐treated with Triton‐X100 (Fig 5E). We further isolated 16K‐EVs and 100K‐EVs (Fig EV4I–L) from the DNase I‐treated serum from BS, SLE, and HC individuals and measured the mtDNA levels in each EV type. The mtDNA levels in 16K‐EVs were high in SLE patients, while the BS and HC individuals had comparable mtDNA levels in 16K‐EVs. However, the mtDNA levels in 100K‐EVs were significantly higher in BS than in HC and SLE patients (Fig 5F and G). Additionally, transmission immunoelectron microscopy revealed dsDNA deposition in the 100K‐EVs purified from the serum of BS patients (Fig 5H), even when they were isolated from plasma; and 100K‐EVs from BS patients contained more mtDNA than those from HCs (Fig EV4M). CD63‐positive 100K‐EVs were slightly increased in BS serum, but the amount of mtDNA showed a more significant increase compared to the increase in the 100K‐EVs themselves (Fig 5I and J). Consistently, the mtDNA levels in 100K‐EVs were higher in patients with BS symptoms (Fig 5K). These results indicated that BS exhibited increased extracellular exosome‐encapsulated mtDNA.
Figure 5. Increased mtDNA in exosomes in BS blood circulation.
-
ASerum mtDNA levels in patients with various autoimmune diseases. After the purification of DNA from the serum, the mtDNA levels were determined using qPCR targeting COXIII, which is encoded by mtDNA. Subjects: HCs (n = 15), and patients with BS (n = 59), RA (n = 12), SLE (n = 22), SjS (n = 10), AAV (n = 14), PM/DM (n = 10), or SSc (n = 9).
-
BRelationship between the mtDNA levels and the presence of BS symptoms. Serum mtDNA levels were compared between BS patients with any symptoms (oral ulcer, uveitis, skin lesion, genital ulcer, arthralgia, or intestinal lesion) (n = 99) and asymptomatic (n = 83) patients.
-
CTime‐series study of the serum mtDNA levels in a BS patient. Serum mtDNA levels were compared between two visits (n = 32): Just before the onset of symptoms (Visit A) and when the patient had already presented overt symptoms (Visit B).
-
DLevels of 8‐OHdG‐modified DNA in serum. After the purification of DNA from HC (n = 16) or BS (n = 57) serum, the levels of 8‐OHdG‐modified DNA were evaluated using ELISA.
-
EmtDNA in BS serum could only be digested by DNase I when the sample had been treated with Triton X‐100. After the purification of DNA from the serum, the mtDNA levels were measured (n = 10).
-
F, GmtDNA levels in 100K‐Evs and 16K‐Evs. 100K‐Evs (F) and 16K‐Evs (G) were isolated from the serum of the same HC (n = 4), BS (n = 5), or SLE (n = 5) patients, then DNA was subsequently purified from each of them, and the mtDNA levels were measured.
-
HDeposition of dsDNA in 100K‐Evs from BS serum. 100K‐Evs isolated from BS serum after DNase I treatment were fixed and stained with an anti‐dsDNA antibody, then visualized with a secondary antibody conjugated to 10‐nm gold particles. Images were obtained using transmission immunoelectron microscopy. Black dots (white arrows) indicate dsDNA deposition. Scale bar, 100 nm.
-
ICD63 level in HC and BS serum. The levels of CD63 in HC (n = 15) and BS (n = 18) serum were evaluated by CD63‐specific ELISA.
-
JSerum mtDNA level vs. serum CD63 level. Scatter plots of serum mtDNA level measured by qPCR (Y‐axis) and serum CD63 level determined by ELISA (X‐axis) in HC (blue circle) (n = 15) and BS (red circle) (n = 18).
-
KmtDNA levels in 100K‐EVs depending on the presence or absence of BS symptoms. mtDNA levels in 100K‐EVs from symptomatic (n = 6) and asymptomatic (n = 5) BS patients were compared.
Data information: Statistical analyses were performed using a Steel–Dwass test (A, E–G) or a Mann–Whitney U test (B, D, I, K) (median; 25th and 75th percentiles; minimum and maximum value excluding outliers; *P < 0.05, **P < 0.01; NS: not statistically significant), a Wilcoxon signed‐rank test (C) (**P < 0.01), or a non‐parametric Spearman's rank test (J).
Source data are available online for this figure.
Figure EV4. Relationship between serum mtDNA levels and BS symptoms and characteristics of 100K‐EVs purified from human sera.
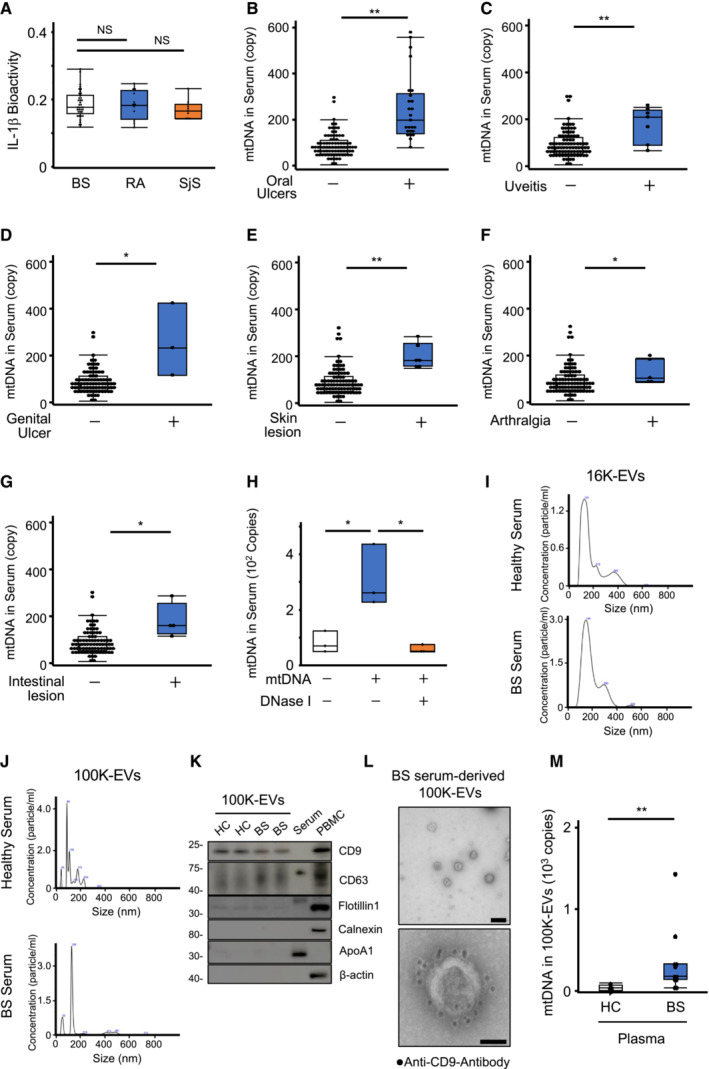
-
ASerum IL‐1β bioactivity. Serum IL‐1β levels in BS, RA, and SjS were evaluated using the reporter cells for IL‐1β.
-
B–GRelationship between serum mtDNA levels and BS symptoms. Serum mtDNA levels in BS patients were compared between asymptomatic patients and patients who exhibited any of the following symptoms on the sampling date: oral ulcers (B), uveitis (C), genital ulcers (D), skin lesions (E), arthralgia (F), and intestinal lesions (G).
-
HDegradation of mtDNA added to the serum by DNase I. mtDNA was spiked into the BS serum and digested by DNase I.
-
I–LCharacterization of EVs isolated from serum. Particle sizes of 16K‐EVs (I) and 100K‐EVs (J) isolated from HC (upper) or BS serum (lower). Representative exosome markers, such as CD9, CD63, and Flotillin 1, but not calnexin, apolipoprotein‐A1, or β‐actin, were present in 100K‐EVs isolated from serum (K). 100K‐EVs isolated from BS serum were attached to a coverslip and visualized through transmission immunoelectron microscopy using an anti‐CD9 primary antibody and a secondary antibody conjugated to 10‐nm gold particles. Scale bar, 200 nm (upper) and 50 nm (lower) (L).
-
MPlasma mtDNA levels in 100K‐EVs. 100K‐EVs were isolated from HC and BS plasma, and DNA was purified. The mtDNA levels in 100K‐EVs were measured using qPCR.
Data information: Statistical analyses were performed using a Mann–Whitney U test (B–G, M) (median; 25th and 75th percentiles; minimum and maximum value excluding outliers; *P < 0.05, **P < 0.01) or a Steel–Dwass test (A, H) (median; 25th and 75th percentile; minimum and maximum value excluding population outliers; *P < 0.05; NS, not statistically significant). The data are representative of two (M) and three (H) independent experiments.
Source data are available online for this figure.
Mitochondrial damage due to caspase‐1 overactivation in BS monocytes
We then investigated the activation state of the inflammasome in BS‐derived CD14+ monocytes. BS monocytes secreted more IL‐1β and LDH and exhibited higher levels of active caspase‐1 (p10) and IL‐1β (p17) (Fig 6A–C), as well as increased mROS levels and a greater reduction in MMP upon LPS and ATP stimulation compared to HC monocytes (Fig EV5A and B). BS monocytes, but not neutrophils, secreted more mtDNA‐containing exosomes (Fig 6D), and the increased secretion of mtDNA in 100K‐EVs and reduced MMP induced by LPS and ATP stimulation was abrogated by the Ac‐YVAD‐cmk (Figs 6E and EV5C). Furthermore, transmission immunoelectron microscopic imaging showed deposition of dsDNA and 8‐OHdG‐positive DNA in the ILVs of MVBs of LPS‐ and ATP‐stimulated cells (Fig 6F). Furthermore, we investigated whether an NLRP3 mutant (V200M‐NLRP3) observed in familial BS caused increased exosome‐mediated mtDNA secretion via V200M‐NLRP3 and WT‐NLRP3 reconstitution into NLRP3‐KO‐THP‐1 cells (Yuksel et al, 2014) (Fig 6G). Following LPS and ATP stimulation, V200M‐NLRP3 reconstituted NLRP3‐KO‐THP‐1 cells secreted more mtDNA in 100K‐EVs and IL‐1β than WT‐NLRP3 reconstituted NLRP3‐KO‐THP‐1 cells (Fig 6H and I). These results indicated that BS monocytes exhibited inflammasome overactivation and mitochondrial damage, leading to increased exosome‐mediated mtDNA secretion.
Figure 6. Increased secretion of mtDNA via exosomes due to overactivation of caspase‐1 in BS monocytes.
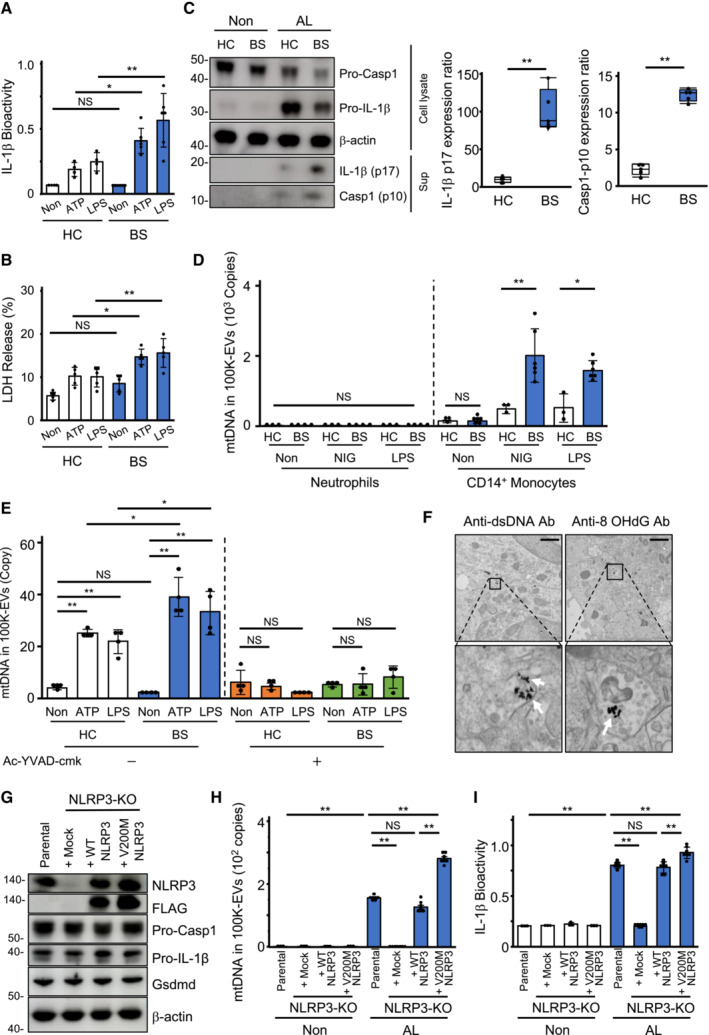
-
A–CIncreased inflammasome activation and pyroptosis in BS monocytes. HC (n = 4)‐ and BS (n = 6) ‐derived CD14+ monocytes were stimulated with LPS or ATP. The bioactivity of IL‐1β (Α) and the levels of LDH (B) in CS were measured. The levels of active caspase‐1 (p10) and mature IL‐1β (p17) in CS of HC (n = 6)‐ and BS (n = 6)‐CD14+ monocytes were evaluated through western blotting using anti‐Casp1 (p10) and anti–IL‐1β (p17) antibodies (left), respectively. The concentrations of caspase‐1 (p10) and IL‐1β (p17) in gel bands were determined using the ImageJ software, and statistical analyses were performed (right) (C).
-
DSecretion levels of mtDNA via 100K‐EVs in BS monocytes. Neutrophils and CD14+ monocytes isolated from the peripheral blood of HC or BS patients were stimulated with nigericin or LPS. After DNA was purified from 100K‐EVs in the CS, the mtDNA levels were measured using qPCR.
-
ECaspase‐1‐dependent over‐secretion of mtDNA in BS monocytes. HC and BS monocytes were pre‐treated with or without Ac‐YVAD‐cmk, and the cells were stimulated with LPS or ATP. The levels of mtDNA in 100K‐EVs in the CS were measured using qPCR.
-
FDeposition of dsDNA and 8‐OHdG in MVBs of BS monocytes. After inducing pyroptosis via LPS and ATP, transmission immunoelectron microscopy was performed using an anti‐dsDNA (upper) antibody and an anti‐8‐OHdG (lower) antibody and a secondary antibody conjugated to 10‐nm gold particles. Black dots (white arrows) indicate dsDNA‐ (left) or 8‐OHdG‐ (right) positive depositions. Scale bar, 1 μm.
-
G–IIncreased secretion of mtDNA in 100K‐EVs by an NLRP3‐mutant derivative. Full‐length NLRP3 (WT‐NLRP3) and an NLRP3 mutant (V200M‐NLRP3) were restored into NLRP3‐KO (FL‐NLRP3‐NLRP3‐KO and V200M‐NLRP3‐NLRP3‐KO) THP‐1 cells (G). The cells were subsequently stimulated with LPS and ATP. The levels of mtDNA in 100K‐EVs (H) and IL‐1β in the CS (I) were measured.
Data information: Statistical analyses were performed using an ANOVA with Tukey's post‐hoc test (A, C–E, H, I) (mean ± SD; *P < 0.05, **P < 0.01; NS, not statistically significant) and a Mann–Whitney U test (B) (median; 25th and 75th percentile; minimum and maximum value of a population; **P < 0.01). The data are representative of two (A–F) and three (G–I) independent experiments.
Source data are available online for this figure.
Figure EV5. Increased mitochondrial damage in BS monocytes.
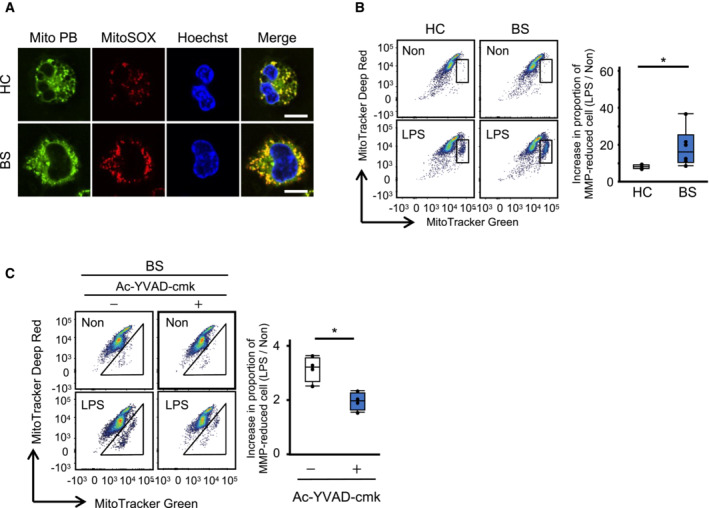
- mROS production in CD14+ monocytes. HC and BS monocytes were stained with MitoPB (green) and MitoSox (red), and mROS production was visualized using confocal microscopy. Scale bar, 5 μm.
- Mitochondrial membrane potential in CD14+ monocytes. HC‐ and BS‐derived CD14+ monocytes were stimulated with LPS, and the reduction in the MMP was monitored by FACS using MitoTracker Green (X‐axis) and MitoTracker‐Deep Red (Y‐axis) (left). The increase in the proportion of MMP‐reduced cells (gated population) following LPS stimulation was quantified (right).
- Effect of caspase‐1 on the increasing MMP of BS monocytes. BS‐derived CD14+ monocytes were stimulated with LPS in the presence or absence of Ac‐YVAD‐cmk, and the reduction in the MMP was monitored using FACS.
Data information: Statistical analyses were performed using a Mann–Whitney U test (B, C) (median; 25th and 75th percentiles; minimum and maximum value excluding outliers; *P < 0.05). The data are representative of two (A) and three (B, C) independent experiments.
Source data are available online for this figure.
mtDNA in BS exosomes induced the production of proinflammatory cytokines via NLRP3 and TLR9
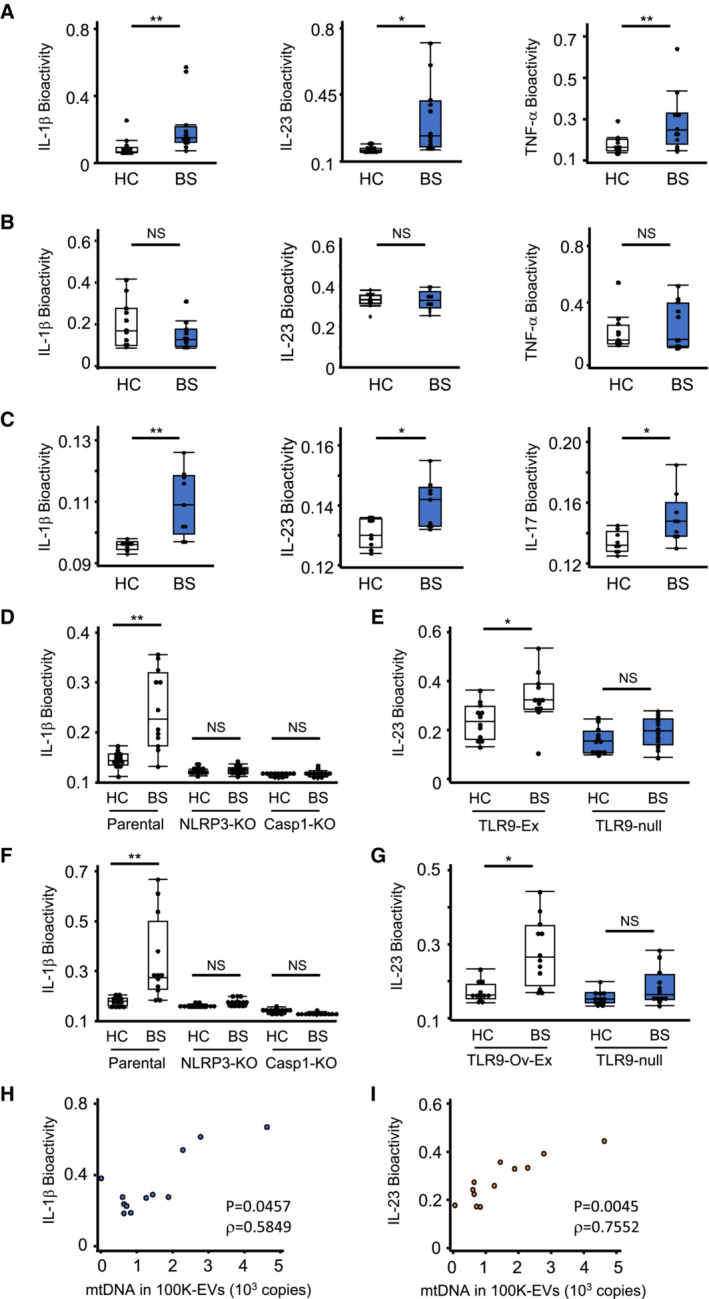
We next investigated whether exosomes isolated from BS serum induced inflammation after confirming the absence of the TLR2, 3, 4, 7, and 8 ligands using TLR ligand‐reporter cells (Appendix Fig S1A–F). BS 100K‐EVs induced the production of cytokines, including IL‐1β, IL‐23, and TNF‐α in healthy PBMCs, while BS 16K‐EVs induced cytokine production similar to that of HC 16K‐EVs (Fig 7A and B). Additionally, when HC and BS 100K‐EVs were injected into the peritoneum of C57BL6/J mice, BS 100K‐EVs induced significantly greater production of proinflammatory cytokines, such as IL‐1β, IL‐23, and IL‐17, in ascites compared to that induced by HC 100K‐EVs (Fig 7C). These results suggested that BS exosomes had the potential to induce proinflammatory cytokine production. We subsequently investigated whether mtDNA could be the leading cause of BS exosome‐induced inflammation by adding HC or BS 100K‐EVs to NLRP3‐KO, Casp1‐KO, and TLR9‐Ex THP‐1 cells. BS 100K‐EVs induced IL‐1β and IL‐23 production in WT THP‐1 cells, but IL‐1β production was completely abolished in NLRP3‐KO, almost to the same extent as that in Casp1‐KO THP1 cells, and IL‐23 production was abolished in TLR9‐null THP‐1 cells (Fig 7D and E). Furthermore, we purified dsDNA from HC and BS 100K‐EVs, encapsulated them within liposomes, and added them to WT, NLRP3‐KO, Casp1‐KO, and TLR9‐Ex THP‐1 cells. Liposomes containing BS 100K‐EV‐derived dsDNA induced IL‐1β and IL‐23 production via NLRP3 and TLR9, respectively (Fig 7F and G). Consistently, higher levels of liposomal mtDNA purified from BS 100K‐EVs resulted in a greater production of IL‐1β and IL‐23 in THP‐1 cells (Fig 7H and I). These results indicated that mtDNA was the primary proinflammatory component of BS exosomes.
Figure 7. BS exosomes promote cytokine production via NLRP3 and TLR9.
-
A, BCytokine production by EVs. 100K‐EVs (A) or 16K‐EVs (B) isolated from HC or BS serum were added to human PBMCs. Cytokines from these PBMCs were evaluated via reporter cells for IL‐1β, IL‐23, and TNF‐α.
-
CCytokine levels in ascites via intraperitoneal injection of 100K‐EVs. After injection of 100K‐EVs isolated from HC and BS serum, the ascites levels of IL‐1β and IL‐23 at 24 h, and of IL‐17 at 72 h, were determined using reporter cells.
-
D–GmtDNA in BS 100K‐EVs induce cytokine production through NLRP3 and TLR9. 100K‐EVs isolated from HC or BS serum were added to WT, NLRP3‐KO, and Casp1‐KO‐THP‐1 cells (D) or WT (TLR9‐null) and TLR9‐Ex THP‐1 cells (E). DNA purified from HC or BS 100K‐EVs was re‐enclosed in liposomes using Avalanche‐Omni and added to WT, NLRP3‐KO, and Casp1‐KO‐THP‐1 cells (F) or WT (TLR9‐null) and TLR9‐Ex THP‐1 cells (G). After 24 h, the levels of IL‐1β (D, F) and IL‐23 (E, G) were measured using reporter cells.
-
H, ICorrelation between cytokine production and mtDNA levels in 100K‐EVs. Levels of mtDNA in 100K‐EVs and IL‐1β (H) and IL‐23 (I) production in THP‐1 cells upon liposome‐enclosed mtDNA stimulation.
Data information: Statistical analyses were performed using a Mann–Whitney U test (A–C) or a Steel–Dwass test (D–G) (median; 25th and 75th percentile; minimum and maximum value excluding population outliers; *P < 0.05, **P < 0.01; NS, not statistically significant) or a non‐parametric Spearman's rank test (H, I). Correlation coefficient (R), *P < 0.05. The data are representative of two (A–C, H, I) and three (D–G) independent experiments.
Source data are available online for this figure.
BS exosomes induced pathological inflammation resembling that of BS
We finally investigated whether BS‐derived exosomes could induce the inflammation characteristics of BS symptoms by injecting 100K‐EVs into the peritoneum or ankle of C57BL6/J mice. 100K‐EVs isolated from BS serum, but not those isolated from HC serum, promoted the infiltration of neutrophils (CD11b+Ly6C+Ly6G+) and monocytes (CD11b+Ly6C+Ly6G−) and ankle swelling with increased infiltration of immune cells (Fig 8A and B). Additionally, since uveitis is an important complication of BS in which IL‐17‐producing cells are reportedly involved (Chi et al, 2008; Letko et al, 2015), we developed an experimental autoimmune uveitis (EAU) model by immunizing C57BL/6J mice with an IRBP1‐20 peptide and complete Freund adjuvant (Agarwal et al, 2012), followed by intravenous administration of 100K‐EVs to the immunized mice. We performed a histological analysis of the retina on day 18, and observed that the disease score of mice treated with BS 100K‐EVs was worse than that of mice treated with HC 100K‐EVs (Fig 8C). BS 100K‐EVs increased IL‐17 production in PBMCs, which was reduced with a combination treatment including an IL‐1β antagonist and an IL‐23 neutralizing antibody (Fig 8D) (Sutton et al, 2009; El‐Behi et al, 2011), suggesting that BS exosomes promoted IL‐1β and IL‐23 production, thus facilitating the production of IL‐17, which exacerbated uveitis. Taken together, BS exosomes exacerbated and propagated the pathological inflammation characteristics of BS by promoting cytokine production and leukocyte mobilization.
Figure 8. BS exosomes drive pathogenic inflammation.
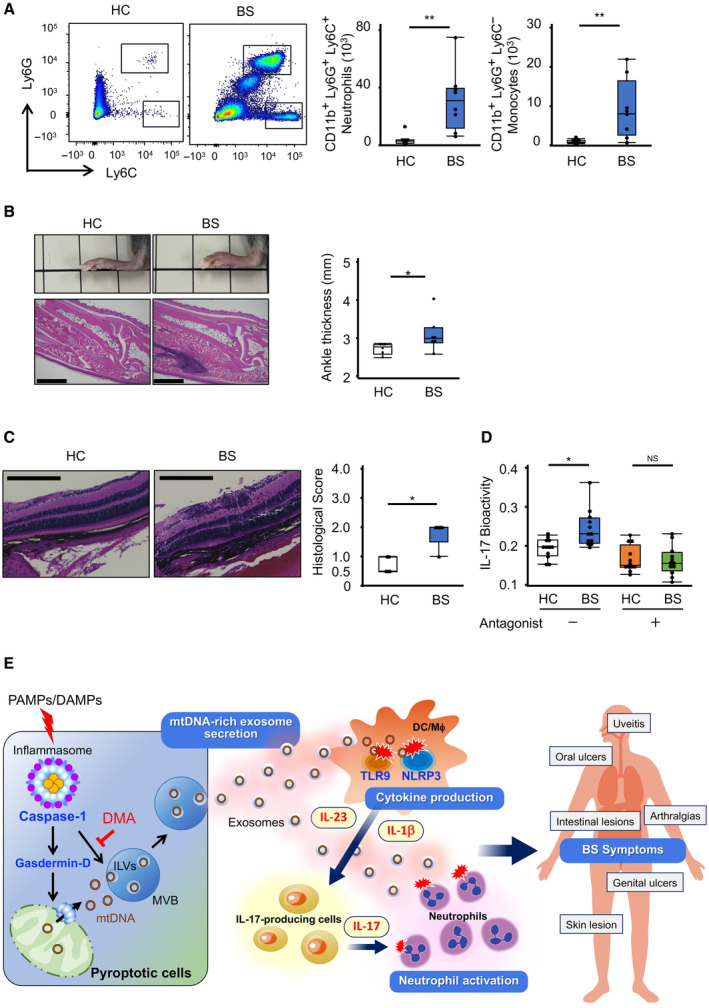
- In vivo leukocyte mobilization by 100K‐EVs. 100K‐EVs isolated from HC (n = 7) or BS (n = 8) serum were adoptively injected into the peritoneal cavity of WT mice. Cells in the peritoneal cavity at 4 h were stained for CD11b, Ly6C, and Ly6G, and CD11b+Ly6G+ neutrophils and CD11b+Ly6C+ monocytes were counted using FACS. Population (left) and absolute number of cells (right) are shown.
- BS‐100K‐induced ankle swelling. 100K‐EVs isolated from HC (n = 6) or BS (n = 7) serum were adoptively injected into the ankle of WT mice. After 24 h, ankle thickness was measured. Swollen ankle and histological imaging (left) and ankle thickness (right) Scale bar, 1 mm.
- Effect of 100K‐EVs on experimental EAU. WT mice were immunized with IRBP1‐20 peptide and complete Freund adjuvant at day 0, and then injected intravenously with 100K‐EVs isolated from HC (n = 5) or BS (n = 5) serum (days 0, 1, 2, and 7). On day 18, a histological analysis of the eyes was performed (left), and the EAU disease score was evaluated (right) Scale bar, 200 μm.
- IL‐17 production by 100K‐EVs via IL‐1β and IL‐23. 100K‐EVs isolated from HC (n = 12) and BS (n = 12) serum were added to human PBMCs in the presence or absence of an IL‐1β antagonist and an IL‐23 antibody. IL‐17 production was evaluated using IL‐17 reporter cells.
- Schematic summary of this study. Inflammasome activation and subsequent induction of pyroptosis increase the release of mtDNA‐containing exosomes to facilitate inflammation (left). When caspase‐1 is activated by noxious stimuli, gasdermin‐D is cleaved, and the localization of NT‐gasdermin‐D to mitochondria results in mtDNA leakage from the mitochondria. Then, the cytosolic mtDNA is taken up by ILVs and released into the extracellular space as exosomes; Caspase‐1 is also involved in this process. Exosomes encapsulating mtDNA promote excessive inflammation via NLRP3 and TLR9. BS monocytes exhibit hyperactivation of Caspase‐1 for an undetermined reason, increasing extracellular mtDNA release via exosomes (right). The mtDNA‐rich exosomes facilitate, propagate, and exacerbate inflammation, increasing leukocyte mobilization and promoting the production of cytokines such as IL‐1β and IL‐23, which leads to IL‐17 production. As a result, neutrophils are activated, causing the characteristic symptoms of BS.
Data information: Statistical analyses were performed using a Mann–Whitney U test (A–C) or a Steel–Dwass test (D) (median; 25th and 75th percentile; minimum and maximum value excluding population outliers; *P < 0.05, **P < 0.01; NS, not statistically significant). The data are representative of two (B, C) and three (A) independent experiments.
Source data are available online for this figure.
Discussion
In this study, we revealed that pyroptotic cells secrete mtDNA encapsulated within exosomes, which promotes a strong inflammation in vivo. Mechanistically, the activation of caspase‐1 and gasdermin‐D promotes mtDNA leakage from the mitochondria into the cytoplasm; furthermore, caspase‐1 induces ILV formation, capturing cytoplasmic mtDNA and releasing it out of the cell as exosomes. Additionally, we found that the overactivation of caspase‐1 in monocytes of BS leads to an increase in mtDNA‐containing exosomes in serum and causes pathological inflammation in BS (Fig 8E).
Cell death is a defense mechanism that deprives invading pathogens of their survival niche, but severe cell death causes serious damage to both neighboring cells and the host. In addition, the effects of cytokines are limited, as they only act on the cells expressing their receptors; however, exosomes can communicate between cells by transmitting their content without specific receptors (van Niel et al, 2018), and thus releasing exosomes containing mtDNA would have the advantage of quickly transmitting information to the neighboring cells and throughout the body. We showed that the specific inhibition of exosome biosynthesis via DMA ameliorated inflammation by suppressing the release of mtDNA‐containing exosomes without affecting the production of IL‐1β, indicating that mtDNA‐rich exosomes can promote inflammation on their own in vivo. Additionally, encapsulation of mtDNA in exosomes caused a stronger inflammatory response, probably due to mtDNA potentially gaining access to cytoplasmic mtDNA sensors. Monocytes and their derivatives are specific pathogen detectors that express pattern recognition receptors (Kawai & Akira, 2011; Okude et al, 2021). Thus, it seems reasonable that this exosome‐mediated mechanism of mtDNA secretion is built into these cells to “raise the alarm” and promptly transmit the danger to the adjacent cells, promoting an inflammatory response.
We extracted 100K‐EVs via a standard ultracentrifugation method, as described in the Materials and Methods section. According to the MISEV 2018 international guidelines for EV extraction and evaluation (Théry et al, 2018), our extracted 100K‐EVs can be considered functional exosomes based on the following observations: (i) the 100K‐EVs were 40–150 nm in diameter and expressed markers in the 1a (CD63), 1b (CD9), and 2a (Flotillin) categories; (ii) dsDNA deposition was observed in ILVs in MVBs using transmission electron microscopy; (iii) Tsg101‐positive ILVs took up cytosolic mtDNA; and (iv) mtDNA secretion decreased upon DMA inhibition of exosome biosynthesis. It is possible that nDNA and mtDNA are present in the 100K‐EVs, and that microvesicles and exosomes might also be present in 100K‐EVs. To evaluate this possibility, cytokine productions by WT 100K‐EVs (with both mtDNA and nDNA present) and ρ0 100K‐EVs (with only nDNA present) were compared using WT, NLRP3‐KO, Casp1‐KO, TLR9‐expressing, and STING‐KO THP1 cells since mtDNA is known to be recognized by NLRP3, AIM2, TLR9, and cGAS. WT 100K‐EVs, but not ρ0 100K‐EVs, induced IL‐1β and IL‐23 via NLRP3 and TLR9, respectively, and IFN‐I via cGAS–STING dependent pathway and both WT and ρ0 100K‐EVs induced IL‐1β production in an NLRP3 independent mechanism probably via AIM2. In addition, nDNA isolated from ρ0 THP1 cells and 100K‐EV purified from Casp1‐KO THP1 cells, which contain few exosomes, induced lower cytokine secretion. These results suggest that the inflammation caused by nDNA and microvesicles is not so significant, even if the 100K‐EVs are somewhat contaminated with nDNA and microvesicles. Taken together, we concluded that the 100K‐EVs we isolated were exosomes with the potential to facilitate, propagate, and exacerbate inflammation by encapsulating mtDNA.
Cytoplasmic mtDNA leakage occurs in apoptotic and pyroptotic cells through BAX/BAK‐dependent MOMP and the voltage‐dependent anion channel protein, which forms a permeability transition pore in the mitochondrial outer membrane (Zhou et al, 2011; Bock & Tait, 2020). We showed that the activation of caspase‐1 and gasdermin‐D plays a role in the extracellular release of mtDNA via exosomes. Gsdmd‐NT forms pores in the plasma membrane by binding to the membrane‐forming lipids phosphatidylserine and cardiolipin (Shi et al, 2015; Liu et al, 2016). Stress‐exposed mitochondria present flipped‐out phosphatidylserines, co‐localization of cardiolipin with permeability transition pores in the mitochondrial outer membrane (Dudek, 2017), and mtDNA leakage into the cytoplasm via Gsdmd‐NT‐forming pores in the mitochondrial membrane (Rogers et al, 2019; Huang et al, 2020). We observed reduced cytoplasmic mtDNA leakage in Gsdmd‐KO‐THP‐1 cells and the localization of Gsdmd‐NT at mitochondria upon inflammasome activation, suggesting that gasdermin‐D possibly forms pores in the mitochondrial membrane and promotes mtDNA leakage. Since pyroptosis is also induced by intracellular LPS through caspases 4, 5, and 11 (Kayagaki et al, 2015; Shi et al, 2015; Liu et al, 2016), we confirmed that intracellular LPS could promote the secretion of mtDNA‐containing exosomes, but we could not examine the involvement of non‐canonical pyroptosis in depth as the cells were too fragile to be evaluated.
ILVs are thought to be produced by the clustering of lipids and membrane‐related cargo proteins in the microdomains of the MVBs, where adapter molecules such as ESCRT are sequentially mobilized to bud into the lumen, thus resulting in the uptake of cargo proteins and cytoplasmic molecules (van Niel et al, 2018). Additionally, MVBs are decomposed by lysosomes at the steady state and secreted by fusion with the plasma membrane under certain conditions to eliminate waste products (Hessvik & Llorente, 2018). We demonstrated that caspase‐1 plays an essential role in the ILV‐mediated uptake of leaked cytoplasmic mtDNA by showing impaired Tsg101 puncta formation and CD9‐positive exosome secretion in Casp1‐KO cells. Therefore, caspase‐1 may be involved in the initiation of ILV formation; however, we have not been able to determine its substrates so far. Therefore, future studies are expected to elucidate the mechanisms of exosome biosynthesis and spatiotemporal regulation, especially those mediated by caspase‐1. It would also be required to elucidate the qualitative differences in mtDNA since nucleic acid sequences and modifications can reportedly affect preferential sorting into ILVs (Villarroya‐Beltri et al, 2013).
mtDNA, inflammasomes, and pyroptosis are reportedly involved in various human diseases, including infection (Kuriakose & Kanneganti, 2019), cancer (Xia et al, 2019), vascular diseases (Zhaolin et al, 2019), and autoinflammatory diseases (Xue et al, 2019), in which DAMPs and cytokines such as IL‐1β are thought to play crucial roles. However, the clinical significance of inflammation mediated by mtDNA‐containing exosomes in human diseases has not been determined. Here, we showed that BS monocytes present caspase‐1 overactivation, leading to increased secretion of exosome‐encapsulated mtDNA and that mtDNA‐rich exosomes promote the sterile inflammation characteristics of BS manifestations. The involvement of extracellular mtDNA in human autoimmune diseases has been reported (West & Shadel, 2017): extracellular mtDNA is released via neutrophil extracellular traps during cell death (NETosis), and mtDNA from platelets is also released via FcγRIIA in SLE pathophysiology (Lood et al, 2016; Melki et al, 2021). We observed that mtDNA was preferentially included in 100K‐EVs in BS sera, whereas it was present in 16K‐EVs in SLE sera. Therefore, the mtDNA release mode seems to differ between these diseases. We also observed that mtDNA‐containing 100K‐EVs induced IFN‐I production by in vitro experiments. IFN‐I is thought to be involved in SLE pathogenesis (Kato et al, 2018; Morand et al, 2020), while the inflammasome is thought to be involved in BS pathogenesis (Emmi et al, 2016; Yazici et al, 2021). Therefore, the cGAS–STING pathway is more likely to be activated in SLE because IFN‐I induces cGAS and STING, whereas NLRP3 may be easily activated by inflammasome activation in BS. Furthermore, activation of caspase‐1 has been reported to suppress cGAS–STING signaling by cleaving cGAS (Wang et al, 2017; Evavold et al, 2018). Hence, the role of mtDNA in producing cytokines may vary from disease to disease.
More than 90% of the patients with BS were outpatients, while almost all collagen disease patients, including patients with RA, SLE, and SjS, were active patients who required hospitalization and had serum samples taken prior to treatment, in which serum IL‐1β levels were similar. Nevertheless, serum mtDNA levels in BS were significantly higher than in RA, SLE, and SjS, although inflammasome activation is reportedly involved in the pathogenesis of RA and SjS. Hence, increased mtDNA in exosomes could be a pathological feature of BS.
Typical BS symptoms appear at the host–microorganism interfaces, such as the oral cavity, skin, and intestines, suggesting the involvement of aberrant inflammasome responses against the invading microorganisms. Indeed, it has been suggested that the microbial colonization in the skin, oral cavity, and intestines of BS patients is different from that of healthy subjects (The Behçet's Disease Research Committee of Japan, 1989; Nian et al, 2012; Yuksel et al, 2014; Nakano et al, 2018). Recent genome‐wide association studies on BS have shown polymorphisms in TLRs and inflammasome‐related genes (Kirino et al, 2013; Takeuchi et al, 2017). Additionally, we showed that THP‐1 cells reconstituted with a disease‐related NLRP3 mutant (V200M), which has been reported in the BS family (Yuksel et al, 2014), secreted more mtDNA‐containing exosomes than WT‐NLRP3‐reconstituted cells. Thus, the genetic and environmental features of BS seem responsible for the characteristic phenotype of the disease. However, pyroptosis has been suggested in relation to monogenic autoinflammatory diseases, including familial Mediterranean fever and cryopyrin‐associated periodic syndrome, caused by a missense mutation in the MEFV gene and a gain‐of‐function mutation in the NLRP3 gene, respectively (Xue et al, 2019). BS clinical manifestations are similar to those of autoinflammatory diseases, suggesting that the etiology caused by mtDNA‐rich exosome‐mediated inflammation may be shared with these diseases. Future studies are needed to clarify the involvement of mtDNA‐rich exosomes in these autoinflammatory diseases.
Collectively, we revealed that exosome‐mediated mtDNA secretion is another alarm transduction pathway that can promote a strong inflammatory response in adjacent cells and that the excessive secretion of mtDNA‐rich exosomes causes the pathogenesis of BS. Therefore, the secretion of mtDNA via exosomes may be a viable therapeutic target for inflammasome‐related inflammation.
Materials and Methods
Reagents and Tools table
| Name | Vender | Catalog number | Clone |
|---|---|---|---|
| Antibodies for western blotting | |||
| ASC (B‐3) | Santa Cruz | sc‐514414 | G0716 |
| Anti‐TLR9 (26C593) | Santa Cruz | sc‐52966 | E0818 |
| Rb mAb to ProCaspase1 + p10 + p12 | Abcam | ab179515 | GR3232708‐2 |
| Rb mAb to Gasdermin D | Abcam | ab210070 | |
| Anti‐cleaved N‐terminal GSDMD Antibody | Abcam | ab215203 | |
| Anti‐TSG101 Ms mAb | Abcam | ab83 | 4A10 |
| Anti‐Calnexin (C5C9) Rabbit mAb | Cell Signaling | 2679S | |
| Anti‐Lamp1 Rabbit mAb | Abcam | ab24170 | |
| Anti‐βactin Rabbit mAb (HRP Conjugate) | Cell Signaling | #5125S | |
| Anti‐p38 MAPK Rabbit Ab | Cell Signaling | #9212S | |
| Anti‐pp38 MAPK Rabbit Ab | Cell Signaling | #9211S | |
| Anti‐p42/44 MAPK (Erk1/2) Rabbit Ab | Cell Signaling | 137F5 | |
| Anti‐pp42/44 MAPK (Erk1/2) Rabbit Ab | Cell Signaling | T202/Y204 | |
| VADC Rabbit Ab | Cell Signaling | #4866S | |
| Purified Mouse Anti‐Tom20 | BD BioScience | 612278 | |
| Purified Mouse Anti‐Tim23 | BD BioScience | 611222 | |
| Purified Mouse Anti‐Flotillin‐1 | BD BioScience | 610821 | |
| mAb anti‐FLAG M2‐Peroxidase (HRP) antibody | Sigma | A8592‐1MG | 046K6075 |
| Monoclonal Anti‐acetylated Tubllin antibody produced in mouse | Sigma | 109M4831V | |
| pAb anti‐NLRP3/NALP3 | NOVUS | NBP2‐12446 | B‐3 |
| Anti‐hIL‐1b Affinity Purified Goat IgG | RSD | AF‐201‐NA | QM1018101 |
| Ms mAb Anti‐Hu CD9 | Invivogen | AHS0902 | |
| Anti‐CD63 (LAMP3) mAb | MBL | MEX002‐3 | |
| Cleaved Caspase‐8 Rabbit mAb | Cell Signaling Technology | #9496 | |
| TFAM antibody | Cell Signaling Technology | #7495 | |
| Anti‐Sodium Potassium ATPase antibody | Abcam | ab76020 | |
| Apolipoprotein A1 antibody | GeneTex | GTX112692 | |
| Antibodies for flow cytometry and confocal microscopy | |||
| Mito Tracker Green | InvitroGen | M7514 | |
| Mito Tracker DeepRed | InvitroGen | M22426 | |
| MitoSox Red Mitochondrial Superoxide Indicator | InvitroGen | M36008 | 2127454 |
| Quanti‐iT PicoGreen dsDNA regent | InvitroGen | 1094619 | |
| Anti‐CD11b PE (clone M1/70) | BD BioScience | 553311 | 64266 |
| Anti‐Ly6G APC‐Cy7 (clone 1A8) | Bio Legend | 128031 | B250488 |
| Anti‐Ly6C Briliant Violet (clone HK1.4) | Sirigen | 127623 | B238261 |
| Antibodies for Immune electron microscopy | |||
| Anti‐8‐hydroxy‐deoxyguanosine monoclonal antibody | JaLCA | MOG‐020P | |
| Anti‐dsDNA antibody | Abcam | ab27156 | |
| Anti‐mouse IgG (H + L) 10 nm Gold | BBI Solutions | EM.GMHL10 | |
| Nanogold®‐Fab' Goat anti‐Mouse IgG (H + L) | Nanoprobes | #2002 | |
| Beads for AutoMACS | |||
| Human CD14 microbeads | Miltenyi Biotec | 130‐050‐201 | |
| Reagents | |||
| Adenosine5‐triphosphate (ATP) disodium salt hydrate | Sigma | A1852 | |
| Nigericin sodium salt Ready Made Solution (NIG) | Sigma | SML1779 | |
| Lipopolysaccharides from Escherichia coli O111:B4 (LPS) | Sigma | L3024 | |
| Phorbol 12‐myristate 13‐acetate (PMA) | Sigma | P1585‐1MG | |
| N‐Formyl‐Met‐Leu‐Phe | Sigma | F3506‐5MG | |
| DNase1 Amplification Grade | Sigma | AMPD1‐1KT | |
| IL‐1R antagonist | Sigma | 407616‐25MGCN | |
| ODN2006 | InvivoGen | tlr‐2006 | |
| 2,3‐cGAMP | InvivoGen | tlrl‐cga23 | |
| Ac‐YVAD‐cmk | InvivoGen | 178603‐78‐6 | |
| Monosodium urate crystals | InvivoGen | tlrl‐msu‐25 | |
| Recombinant human IL‐12/p70 | PeproTech | 200‐12 | |
| Recombinant human IL‐17 | PeproTech | 200‐17 | |
| Recombinant human TNFα | PeproTech | 300‐01A | |
| Recombinant human IL1beta/IL1F2 | R&D | 201‐LB‐025 | |
| Recombinant human IL‐23 | R&D | 1290‐IL‐010 | |
| Recombinant human IL‐18/IL‐1F4 | R&D | 9124‐IL‐010 | |
| IL‐23R blocking peptide | BD BioScience | MBS9222188 | |
| Purified Anti‐human CD28 (clone CD28.2) | BD BioScience | 14‐0289‐82 | E025695 |
| Purified Anti‐human CD3 (clone OKT3) | BD BioScience | 16‐0037‐85 | E019164 |
| Staurosporine | KOM | AG‐CN‐0022‐C100 | |
| 5‐(N.N‐dimethyl)‐Amirolide (DMA) | CAY | 19100 | |
| Mito‐TEMPO | ENZO | ALX‐430‐150‐M005 | |
| Pertussis toxin (PTx) | List Biological Laboratories | LBL‐0901‐80 | |
| Ionomycin Ca2+ salt | Abcam | ab120116 | |
| Complete Freunds Adjuvant (CFA) | Chondrex | #7023 | |
| IRBP peptide (1‐20) human | ANASPEC | AS‐62297 | |
| Triton (R)X‐100 | Nacalai | 35501‐02 | |
| Q‐Vai‐Asp‐Oph | R&D | OPH001‐01M | |
| Birinapant | CAY | 19699 | |
| Amicon Ultra‐15 | Millipore | UFC901024 | R9CA90257 |
| ELISA | |||
| Human exosome CD63 ELISA kit | CosmoBio | HAK‐HEL6363‐1 | |
| Highly Sensitive ELISA kit for 8‐OHdG | JaICA | KOG‐HS10/E | |
| LDH Cytotoxicity assay | |||
| CytoSelect LDH Cytotoxicity Assay kit | CosmoBio | CBA‐241 | |
| immune cells isolation | |||
| Ficoll‐Paque plus | Cytiva (GE) | 17144003 | |
| Polymorphprep | CosmoBio | 1114683 | |
| DNA isolation | |||
| MagNA Pure Compact | Roche Life Science | ||
| DNA Extractor SP Kit | WAKO | 296‐60501 | |
| DNA Extractor WB kit | WAKO | 291‐50502 | |
| Quant‐iT™ dsDNA Assay Kits, high sensitivity (HS) and broad range (BR) | InvitroGen | Q33120 | |
| RNA isolation | |||
| Rneasy Mini kit (250) | QIAGEN | 74106 | |
| QIA shredder (250) | QIAGEN | 79656 | |
| Real‐Time PCR | |||
| TB Green Fast qPCR Mix | TaKaRa | RR430A | |
| mtDNA primer for Real‐Time PCR | |||
| Human cytochrome C oxidase subunit III (CoxIII) | Fw | 5′‐ATGACCCACCAATCACATGC‐3′ | |
| Rv | 5′‐ATCACATGGCTAGGCCGGAG‐3′ | ||
| Human NADH dehydrogenase (NADH) | Fw | 5′‐ATACCCATGGCCAACCTCCT‐3′ | |
| Rv | 5′‐GGGCCTTTGCGTAGTTGTAT‐3′ | ||
| Primers and vector for gene cloning | |||
| Human TLR9 cloning primer (infusion) | Fw | 5′‐GTGACAGATCCAAGGTGAAGT‐3′ | |
| Rv | 5′‐CTTCCTCTACAAATGCATCACT‐3′ | ||
| 3*FLAG Oligo promer | Fw | 5′‐GACTACAAAGACCATGACGGTGATTATAAAGATCATGACATCGACTACAAGGATGACGATGACAAGTAGTGA‐3′ | |
| Rv | 5′‐TCACTACTTGTCATCGTCATCCTTGTAGTCGATGTCATGATCTTTATAATCACCGTCATGGTCTTTGTAGTC‐3′ | ||
| Human caspase1 + 3*FLAG cloning primer (infusion) | Fw (lenti‐Casp) | 5′‐AATTCTGCAGCGGCCGCGCCACCATGGCCGACAAG‐3′ | |
| Rv (Casp‐FLAG) | 5′‐CCGTCATGGTCTTTGTAGTCATGTCCTGGGAAGAGGTAGA‐3′ | ||
| Rv (FLAG‐Lenti) | 5′‐GGAGAGGGGCGGATCCCTACCAAGAAGGCTCAAAG‐3′ | ||
| Human NLRP3 + 3*FLAG cloning primer (infusion) | Fw (Lenti‐NLRP3) | 5′‐AATTCTGCAGCGGCCGCGCCACCATGAAGATGGCAAGCACCCG‐3′ | |
| Rv (NLRP3‐FLAG) | 5′‐CCGTCATGGTCTTTGTAGTCCCAAGAAGGCTCAAAGACGA‐3′ | ||
| Rv (FLAG‐Lenti) | 5′‐GGAGAGGGGCGGATCCCTACCAAGAAGGCTCAAAG‐3′ | ||
| Human mutant NLRP3 (Val200Met) cloning primer | Fw (Lenti‐NLRP3) | 5′‐AATTCTGCAGCGGCCGCGCCACCATGAAGATGGCAAGCACCCG‐3′ | |
| Rv1 (mutation sequence) | 5′‐AATGGGACTCATGGGGCTCT‐3′ | ||
| Fw2 (mutation sequence) | 5′‐AGAGCCCCATGAGTCCCATT‐3′ | ||
| Rv (FLAG‐Lenti) | 5′‐GGAGAGGGGCGGATCCCTACCAAGAAGGCTCAAAG‐3′ | ||
| Caspase1 p10 + 3*FLAG cloning primer (infusion) | Fw | 5′‐AATTCTGCAGCGGCCGCGCCACCATGGCTATTAAGA‐3′ | |
| Rv | 5′‐CCGTCATGGTCTTTGTAGTCATGTCCTGGGAAGAGGTAGA‐3′ | ||
| Caspase1 p20 + 3*FLAG cloning primer (infusion) | Fw | 5′‐AATTCTGCAGCGGCCGCGCCACCATGAACCCAGCT‐3′ | |
| Rv | 5′‐CCGTCATGGTCTTTGTAGTCATCTTTAAACCACACCACAC‐3′ | ||
| Gasmd‐NT‐3*FLAG | Eurofins Genomics | ||
| TSG101‐mCherry | VectorBuilder | pLV[Exp]‐Puro‐EF1A>mCherry/hTSG101[NM_006292.4] | |
| Target seqence for Generation of ko‐cell‐lines | |||
| Generation of KO‐NLRP3 THP1 cell | sgRNA sequence | 5′‐CGAAGCAGCACTCATGCGAG‐3′ | |
| 5′‐GTCTGATTCCGAAGTCACCG‐3′ | |||
| 5′‐ATTCTCCCCCACATAGAGTC‐3′ | |||
| Generation of KO‐Caspase1 THP1 cell | sgRNA sequence | 5′‐GACAGTATTCCTAGAAGAAC‐3′ | |
| 5′‐TAATGAGAGCAAGACGTGTG‐3′ | |||
| 5′‐GCTCCCTAGAAGAAGCTCAA‐3′ | |||
| Generation of KO‐GasderminD THP1 cell | sgRNA sequence | 5′‐CGGCTCTCACCTGTCGCGGG‐3′ | |
| 5′‐CTTGCTTTAGACGTGCAGCG‐3′ | |||
| 5′‐ACCTTCCAGCCACCCGCGAC‐3′ | |||
| Reporter cells for measurement of cytokine bioactivity and TLR signaling | |||
| HEK‐Blue IL‐1β | InvivoGen | hkb‐il1b | |
| HEK‐Blue IL‐12 | InvivoGen | hkb‐il12 | |
| HEK‐Blue IL‐17 | InvivoGen | hkb‐il17 | |
| HEK‐Blue IL‐18 | InvivoGen | hkb‐il18 | |
| HEK‐Blue IL‐23 | InvivoGen | hkb‐il23 | |
| HEK‐Blue TNFα | InvivoGen | hkb‐tnfadmyd | |
| HEK‐Blue TLR2 | InvivoGen | hkb‐htlr2 | |
| HEK‐Blue TLR3 | InvivoGen | hkb‐htlr3 | |
| HEK‐Blue TLR4 | InvivoGen | hkb‐htlr4 | |
| HEK‐Blue TLR7 | InvivoGen | hkb‐htlr7 | |
| HEK‐Blue TLR8 | InvivoGen | hkb‐htlr8 | |
| HEK‐Blue TLR9 | InvivoGen | hkb‐htlr9 | |
| Transfection Reagent | |||
| LentiGuide‐Puro | Addgene | ||
| Avalanche‐Omni Transfection Reagent | EZ biosystems | EZT‐OMNI‐1‐1.5 | |
| Gene Editing Reagent | |||
| In‐Fusion HD cloning kit | TaKaRa | Z9649N | |
| DNA Ligation Kit (Mighty Mix) | TaKaRa | 6023 | |
| PrimeSTAR GXL DNA polymerase | TaKaRa | R050A | |
| CUGA7 gRNA Synthesis kit | Nippon Gene | 314‐08691 | |
| ISOSPIN PCR Product | Nippon Gene | 315‐08001 | |
| Establishment of p0 cell Reagent | |||
| L‐Glutamine (200 mM) | Thermo Fisher | A2916801 | |
| Sodium Pyruvate (100 mM) | Thermo Fisher | 11360070 | |
| Uridine > 99% | Sigma | U3750‐1G | |
| Ethidium bromide solition (EtBr) | Sigma | E1385‐5ML | |
| Isolation of Mitochondria | |||
| Mitochondria/Cytosol Fractionation Kit | Bio Vision | #K256‐25 | |
Methods and Protocols
Mice
C57BL6/J mice were obtained from SLC. Mice were maintained in a specific pathogen‐free environment and used at 6–8 weeks of age. All animal experimental procedures were approved by Osaka University (28‐008‐034) and are consistent with the university's institutional guidelines.
Clinical samples
Subjects were Japanese patients who were admitted to the Department of Clinical Immunology, Osaka University, from 2015 to 2020. Serum samples were collected from patients. BD was diagnosed by the criteria established by the BD Research Committee in Japan (Suzuki Kurokawa & Suzuki, 2004); RA by the Updated American College of Rheumatology revised criteria (1987) (Arnett et al, 1988) and ACR/EULAR rheumatoid arthritis classification criteria (2010) (Aletaha et al, 2010); SLE by the New American College of Rheumatology criteria for SLE (1997) (Hochberg, 1997); SjS by the Classification criteria for Sjögren's syndrome: a revised version of the European criteria proposed by the American‐European Consensus Group (2002) (Vitali et al, 2002); PM/DM by the criteria of the Research Committees of Japanese Ministry of Health; and SSc by the Classification criteria for systemic sclerosis: an ACR/EULAR collaborative initiative (2013) (van den Hoogen et al, 2013). Patients with AAV were diagnosed according to the criteria of the Research Committees of the Japanese Ministry of Health. Microscopic polyangiitis (MPA) and granulomatosis with polyangiitis (GPA) were defined according to the 2012 Chapel Hill Consensus Conference nomenclature and definitions (Jennette et al, 2013). Normal control samples were obtained from healthy volunteer donors. A total of 59 BD patients, 12 RA patients, 22 SLE patients, 10 SjS patients, 10 PM/DM patients, 9 SSc patients, 14 AAV patients, and 15 healthy control volunteer donors were enrolled in this study, which was approved by the institutional review boards of Osaka University (11366, 11122, and 20019). We obtained informed consent from all study subjects. Samples were collected by blood sampling from the outpatients, regardless of treatment procedure. To avoid the possibility of contamination from bacteria‐derived substances, we excluded patients who had massive infections. Medical records of patients were reviewed and analyzed retrospectively.
Regents, antibodies, fluorescent dyes, and reporter cells
Reagents were obtained from the indicated suppliers: ultrapure LPS, phorbol‐12‐myristate‐13‐acetate (PMA), ionomycin, f‐Met‐Leu‐Phe (fMLP), nigericin, ATP, and DNase I (Sigma‐Aldrich); Triton X‐100 (Nacalai Tesque); ODN2006, 2.3‐cGAMP, MSU, and Ac‐YVAD‐cmk (Invivogen); staurosporine (KOM); DMA (CAY); anti‐CD11b (clone M1/70) antibody (BD BioScience); anti‐Ly6G (clone 1A8) antibody (BioLegend); anti‐Ly6C (clone HK1.4) antibody (Sirigen); Mitotracker Green and Mitotracker‐Deep Red (ThermoFisher); MitoSox red mitochondrial superoxide indicator and Quanti‐iT PicoGreen dsDNA regent (Invitrogen); and Mito‐TEMPO (ENZO). Reporter cell lines HEK‐Blue‐IL‐1β, ‐IL‐23 were purchased from InvivoGen.
Isolation of human leukocytes
Neutrophils and peripheral blood mononuclear cells (PBMCs) were isolated using Polymorphprep (Axis‐Shield). CD14‐positive monocytes were separated from PBMCs using human CD14 microbeads and AutoMACS (Miltenyi Biotec), and the remaining CD14‐negative fraction was collected as the lymphocyte fraction.
DNA purification
DNA was purified from 200 μl serum using the MagNA Pure Compact (Roche Life Science). For the purification of DNA from 16K‐ and 100K‐EVs, the DNA Extractor SP Kit (WAKO) was used. In brief, EVs isolated from 100 μl serum or culture supernatants were enzymatically digested at 56°C for 10 min and then treated with sodium iodide to remove proteins. After purification and alcohol precipitation, the DNA was resuspended in DNase‐free water. If necessary, the serum and culture supernatant were digested with DNase I at 37°C for 30 min before DNA purification.
Measurement of mtDNA
We validated the measurement values of mtDNA by qPCR by measuring two different mtDNA‐encoded genes, human cytochrome C oxidase subunit III (COXIII) and NADH dehydrogenase (NADH). Primers were as follows: COXIII, forward 5′‐ATGACCCACCAATCACATGC‐3′ and reverse 5 ′‐ATCACATGGCTAGGCCGGAG‐3′; NADH, forward 5′‐ATACCCATGGCCAACCTCCT‐3′ and reverse 5′‐GGGCCTTTGCGTAGTTGTAT‐3′. Quantitative real‐time PCR was performed using TB Green Fast qPCR Mix (Takara Bio). Real‐time standard curves were generated using 5‐fold serial dilutions of purified DNA from human PBMC whole‐cell lysate. Samples that produced no PCR products after 40 cycles were considered ‘undetectable.’
Establishment of mtDNA‐defective ρ0 THP1
Clonal mtDNA‐defective ρ0‐THP1 cells were generated as described elsewhere (Hashiguchi & Zhang‐Akiyama, 2009). In brief, THP1 cells were cultured with RPMI medium containing uridine, pyruvate, and 50 ng ml−1 EtBr for 1 month after single cells were isolated under a microscope. The absence of mtDNA was confirmed by qPCR.
Separation of the cytosol and mitochondria fractions
The cytosol and mitochondria fractions were separated using the Mitochondria/Cytosol fraction Kit (BioVision). Briefly, cells were thoroughly washed by centrifugation at 600 g for 5 min at 4°C, resuspended in cytosol extraction buffer mix containing DTT and protease inhibitors, incubated on ice for 10 min, and homogenized. After the homogenate was centrifuged at 700 g for 10 min at 4°C to remove debris, the supernatant was centrifuged at 10,000 g for 30 min at 4°C to collect the supernatant as the cytosol fraction. After washing with PBS, the pellets were collected as the mitochondria fraction.
Purification of 16K‐ and 100K‐EVs
For the purification of 16K‐ and 100K‐EVs, EVs were separated by centrifugation with a gradual increase in centrifugal force following a modified version of a protocol described elsewhere (Théry et al, 2018). In brief, cells were centrifuged twice at 600 g for 30 min each to remove whole cells and debris. After the supernatant (4K supernatant) was treated with DNase I at 37°C for 30 min, it was centrifuged at 16,000 g for 40 min at 4°C to obtain the 16K‐EV fraction. The 16K supernatant was filtered through a 0.22‐μm filter, and the resultant filtrate was ultra‐centrifuged at 100,000 g for 70 min at 4°C. To eliminate contaminating proteins, the pellet was resuspended in 2.2 ml PBS and centrifuged at 100,000 g for 70 min at 4°C to obtain 100K‐EVs; the 100K‐EVs were suspended in PBS.
Western blotting
For evaluation of cell lysates, cells were harvested, lysed in lysis buffer (50 mM Tris–HCl [pH 8.0], 250 mM NaCl, 5 mM EDTA, 1% Nonidet P‐40, 0.25% Na‐deoxycholate, and 1 mM NaF), and centrifuged at 16,000 g for 20 min at 4°C. The supernatant was mixed with 5× Laemmli sample buffer containing 2‐mercaptoethanol and denatured at 95°C for 5 min. The reduced samples were electrophoresed on 4–12% Bis‐Tris gels (Life Technologies), transferred to nitrocellulose membranes, and blotted with antibodies listed in Appendix Table S2. To evaluate EV lysates (CD9 and CD63), EVs isolated from serum, plasma, and culture supernatant were mixed with 5× Laemmli sample buffer without 2‐mercaptoethanol and denatured at 70°C for 5 min. The reduced samples were electrophoresed on 12% Bis‐Tris gels (Life Technologies), transferred to nitrocellulose membranes, and blotted with the antibodies listed in Appendix Table S2. Protein bands in SDS‐PAGE gels were quantified using the ImageJ software and statistically analyzed using Student's t‐test.
Measuring the amount of 100K‐EVs
For the evaluation of 100K‐EVs in CS, after purification of 100K‐EVs by ultracentrifugation, the level of 100K‐EVs was evaluated by western blotting using anti‐CD9 or ‐CD63 antibodies. Protein bands on SDS‐PAGE gels were quantified using ImageJ.
Nanoparticle tracking analysis (NTA)
Analyses of the size distribution and the number of EVs were carried out on a NanoSight LM10HS with a blue laser system (NanoSight, Amesbury, UK), as previously described. Briefly, nanoparticle tracking analysis (NTA) was performed on isolated EVs diluted 20‐fold with PBS. All events were recorded in a 60‐s video for further analysis using the NTA software. The Brownian motion of each particle was tracked between frames to calculate its size using the Stokes–Einstein equation.
Induction of cell death
To induce cell death, THP‐1 cells were seeded at a concentration of 1 × 106 ml−1 in RPMI medium supplemented with exosome‐free FCS. To induce primary necrosis, cells were incubated at 56°C for 1 h after incubation at 37°C for 3 h. For apoptosis, cells were stimulated with 1 μM staurosporine at 37°C for 4 h. To induce pyroptosis, cells were stimulated with 10 mM ATP for 1 h after priming with 0.5 μg ml−1 LPS at 37°C for 3 h. To induce secondary necrosis, apoptotic cells were incubated with 1 μM staurosporine at 37°C for 24 h after thorough washing. To induce necroptosis, cells were stimulated with a combination of TNF‐α, Smac, and Qvad for 24 h. Unstimulated control samples were incubated at 37°C for 4 h. To evaluate extracellular mtDNA release by pyroptosis, human primary leukocytes were stimulated with ATP (5 mM), LPS (500 ng ml−1), nigericin (10 ng ml−1), ODN2006 (250 nM), cGAMP (5 μg ml−1), ionomycin (250 ng ml−1), and fMLP (1 nM) for 5 h. THP‐1 was cultured with PMA (50 nM) at 37°C for 3 days and stimulated with ATP (5 mM), nigericin (10 ng ml −1), and LPS (500 ng ml−1) for 5 h.
Measurement of the released LDH
Lactate dehydrogenase (LDH) released from permeabilized cells was measured using the CytoSelect LDH Cytotoxicity Assay Kit (CELL BIOLABS). Briefly, the LDH assay reagent was mixed with culture supernatant of cells induced to undergo cell death. Absorbance was measured at 450 nm using a spectrophotometer. Culture supernatant of cells lysed with Triton X‐100 was used as a positive control, and the culture supernatant of unstimulated cells was used as a negative control.
Cell‐based reporter assay
We used HEK‐Blue IL‐1β, IL‐23 (reporter cells) to measure cytokines. To determine the cytokine activities of serum or culture supernatant, reporter cells were incubated for 24 h at 37°C with medium containing 20% serum or 20% culture supernatant. To determine the cytokine activity of mouse lavage fluid, peritoneal fluid was concentrated using an Amicon Ultra‐15 (Millipore). Embryonic alkaline phosphatase (SEAP) secreted from the reporter cells was detected with QUANTI‐Blue (InvivoGen) and quantified by measuring optical density at 620 nm.
Generation of KO‐THP‐1 using the CRISPR‐Cas9 system
NLRP3‐, Caspase‐1–, and gasdermin‐D–deficient THP‐1 were generated by transducing Cas9‐expressing THP‐1 cells (THP‐1‐Cas9) with cgRNA, as described elsewhere. Briefly, cgRNA was synthesized using the CUGA7 cgRNA synthesis kit (Nippon Gene). THP‐1‐Cas9 cells (5 × 104 cells/well) were transduced with 20 ng synthetic cgRNA using 0.14 μl Avalanche‐Omni and cultured in 96‐well plates for about 5 days. After single clones were picked under the microscope, defects in target genes were confirmed by western blotting. Target sequences were as follows: Caspase‐1 knockout, 5′‐GACAGTATTCCTAGAAGAAC‐3′, 5′‐TAATGAGAGCAAGACGTGTG‐3′, and 5′‐GCTCCCTAGAAGAAGCTCAA‐3′; and Gasdermin‐D knockout, 5′‐CGGCTCTCACCTGTCGCGGG‐3′, 5′‐CTTGCTTTAGACGTGCAGCG‐3′, and 5′‐ACCTTCCAGCCACCCGCGAC‐3′.
Expression vector cloning
cDNA encoding full‐length Caspase‐1, p10‐Caspase‐1, and p20‐Caspase‐1 were fused at the C‐terminus to a 3× FLAG tag and cloned into the lentiviral vector CSII‐EF‐MCS‐IRES2‐Venus provided by Dr. Hiroyuki Miyoshi, Keio University. The N‐terminal sequence of Gasdermin‐D was synthesized by Eurofins genomics and cloned into the NotI–BamHI site of vector CSII‐EF‐IRES2‐Venus using the In‐Fusion HD Cloning kit. mCherry‐tagged TSG‐101 in a lentivirus backbone vector was produced using Vector Builder.
Imaging by microscopy
For imaging mtDNA, PicoGreen, which intercalates dsDNA, was used as described elsewhere (White et al, 2014). Briefly, PMA‐treated THP‐1 was placed on 4‐well chamber glass‐bottom dishes and incubated for 1 h at 37°C with 3 μl/ml PicoGreen dye (InvivoGen) and 250 nM Mitotracker‐Deep Red dye (InvivoGen). For imaging of ILVs, WT‐, Casp1‐KO‐, and GsdmD‐KO‐THP‐1 cells were lentivirally transduced with TSG101‐mCherry. To analyze the co‐localization of cytosolic leakage of mtDNA and TSG101, cells were stimulated with 500 ng/ml LPS plus 10 mM ATP to induce pyroptosis. After tight‐washing by PBS, the distribution of PicoGreen was visualized by confocal microscopy (Olympus, FV3000).
Imaging by transmission immunoelectron microscopy
THP1 cells were cultured overnight with PMA (100 nM) on Cell Desk polystyrene coverslips (Sumitomo Bakelite). After fixation with 4% PFA for 1 h, cells were permeabilized with 0.2% saponin for 30 min and blocked with 10% BSA, 10% normal goat serum, and 0.1% cold‐water fish gelatin in sodium‐phosphate buffer. Cells were stained with anti‐DNA antibody (Abcam) overnight at 4°C plus 1.4‐nm gold‐particle–conjugated anti‐mouse IgG (Nanogold Fab' fragment of goat anti‐mouse IgG, Nanoprobes) for 2 h at room temperature. Then, the cells were fixed with 1% glutaraldehyde for 10 min and treated with a gold‐enhancement mixture, GoldEnhance‐EM (Nanoprobes). For post‐fixation, cells were treated with 1% osmium tetroxide and 1.5% potassium ferrocyanide in 0.1 M sodium phosphate buffer (pH 7.4) for 1 h and dehydrated in a graded series of ethanol. After being embedded in Epon812 (TAAB), ultra‐thin (80‐nm) sections of cells were stained with saturated uranyl acetate and Reynolds lead citrate solution. Electron micrographs were obtained with a JEM‐1011 transmission electron microscope (JEOL).
In vivo peritonitis model
Five hours after the administration of 100K‐EVs into the peritoneal cavity of mice, cells collected from lavage fluid in the peritoneal cavity were stained with anti‐CD11b‐PE (BD Bioscience), anti‐Ly6G‐APC‐Cy7 (BioLegend), and anti‐Ly6C‐Brilliant Violet (Sirigen), and then analyzed by flow cytometry (FACS Canto). Neutrophils were defined as the CD11b+Ly6C+Ly6G+ population and monocytes as the CD11b+Ly6C+Ly6G− population. The absolute number of cells in the whole lavage was calculated as (total infiltrating cell number) × (% of each population).
In vivo arthritis model
For the exosome‐induced arthritis model, 100K‐EVs or DNA purified from 100K‐EVs were administered to the ankle of C57BL6/J mice. 100K‐EVs were collected from THP1 cells by ultracentrifugation. DNA purified from 100K‐EVs was enclosed in liposomes using Avalanche‐Omni. For the MSU‐induced arthritis model, C57BL6/J mice were pre‐treated with 10 μl of DMSO or 0.25 mg of c‐YVAD‐cmk‐ or DMA‐dissolved DMSO via the ankle joint injection. Two hours later, 10 μl of PBS or 0.25 mg of MSU crystal–dissolved PBS was injected into each ankle using a 29‐gauge needle. Twenty‐four hours later, the ankle thickness was measured with precision calipers (Fisherbrand Traceable Digital Calipers; Fisher Scientific), and the tissue of the ankle joint was analyzed histologically by hematoxylin–eosin staining.
In vivo experimental autoimmune uveitis (EAU) model
C57BL6/J mice were immunized via s.c. injection with 200 μl of emulsion containing 200 μg of IRBP1–20 in Complete Freund's Adjuvant (CFA), and 0.2 μg of pertussis toxin was added i.p. on the same day. 100K‐EVs were administrated via i.v. injection on days 0, 1, 2, and 7 and mice were sacrificed at day 18. After whole eyes were removed and fixed in 4% paraformaldehyde (PFA), sections were cut through the pupillary‐optic nerve plane, and H&E staining was performed. The severity of retinal inflammation was assessed by experimenters who were not informed of the experimental conditions and graded as follows: 0: no change; 0.5: mild inflammatory cell infiltration and no tissue damage; 1: infiltration, retinal fold and focal retinal detachment, few and small granulomas, and peri‐vasculitis; 2: moderate infiltration, retinal fold and detachment, focal photoreceptor cell damage, small‐ to medium‐size granulomas, and vasculitis; 3: medium to heavy infiltration, severe retinal fold with detachment, moderate photoreceptor cell damage, large granulomas, and neovascularization; 4: heavy infiltration, diffuse retinal detachment with bleeding, severe photoreceptor cell damage, large granuloma lesions, and neovascularization.
Statistical analysis
Logarithmic transformation was performed to achieve a normal distribution. Normally distributed data are presented as means ± SD and were compared using two‐way Student's t‐test or two‐way ANOVA followed by post‐hoc multiple comparisons using the Tukey–Kramer test. Non‐normally distributed data are expressed as medians, 25th and 75th percentiles, and minimum and maximum values excluding outliers, and were compared using the two‐way Mann–Whitney U test. The two paired groups were analyzed by Wilcoxon signed‐rank test. Scatter plot data are expressed as Spearman's correlation (r) and analyzed by Spearman's correlation test. A value of P < 0.05 was considered statistically significant.
Author contributions
Hachiro Konaka: Conceptualization; formal analysis; investigation; methodology; writing – original draft; project administration; writing – review and editing. Yasuhiro Kato: Methodology; project administration. Toru Hirano: Methodology; project administration. Kohei Tsujimoto: Investigation; methodology. JeongHoon Park: Investigation; methodology. Taro Koba: Investigation; methodology. Wataru Aoki: Investigation; methodology. Yusei Matsuzaki: Investigation; methodology. Masayasu Taki: Methodology. Shohei Koyama: Methodology. Eri Itotagawa: Investigation. Tatsunori Jo: Investigation. Takehiro Hirayama: Investigation. Taro Kawai: Methodology. Ken J Ishii: Methodology. Mitsuyoshi Ueda: Methodology. Shigehiro Yamaguchi: Methodology. Shizuo Akira: Methodology. Takayoshi Morita: Methodology. Yuichi Maeda: Methodology. Masayuki Nishide: Methodology. Sumiyuki Nishida: Methodology. Yoshihito Shima: Methodology. Masashi Narazaki: Methodology. Hyota Takamatsu: Conceptualization; data curation; funding acquisition; investigation; methodology; writing – original draft; writing – review and editing. Atsushi Kumanogoh: Supervision; funding acquisition; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We are grateful to Dr. Hiroyuki Miyoshi (Keio University, Japan) and RIKEN BRC for providing materials. This study was supported in part by research grants from JSPS KAKENHI (15K09550 to HT; 18K08386 to HT; 22H03111 to HT); a Core Research for Evolutionary Science and Technology (CREST) grant from the Japan Science and Technology Agency (JST) (JPMJCR16G2 to HT); The Naito Foundation (to HT); the Center of Innovation program (COI‐STREAM) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (to AK); the Japan Agency for Medical Research and Development (AMED)‐CREST (15652237 to AK); the Japan Society for the Promotion of Science (JSPS) KAKENHI (JP18H05282 to AK); the Japan Agency for Medical Research and Development (AMED) (J200705023, J200705710, J200705049, JP18cm016335, and JP18cm059042 to AK); and Grants from Mitsubishi Zaidan (to AK). The authors have no conflicting financial interests.
The EMBO Journal (2023) 42: e112573
Data availability
Source Image Data in this paper was deposited in the BioImage Archive (Accession number: S‐BIAD808; S‐BIAD807; S‐BIAD777) (https://www.ebi.ac.uk/biostudies/bioimages/studies/S‐BIAD808?key=5f96944a‐1272‐4094‐8e44‐0111da741fa1; https://www.ebi.ac.uk/biostudies/bioimages/studies/S‐BIAD807?key=c6869915‐6a88‐49cf‐b59b‐497cf3004dd8; https://www.ebi.ac.uk/biostudies/bioimages/studies/S‐BIAD777?key=41c8e9a5‐c75f‐4413‐95ce‐d6145290cf6d). Cells and other reagents that were used in this research will be available upon reasonable request.
References
- Agarwal RK, Silver PB, Caspi RR (2012) Rodent models of experimental autoimmune uveitis. Methods Mol Biol 900: 443–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam TI, Kanki T, Muta T, Ukaji K, Abe Y, Nakayama H, Takio K, Hamasaki N, Kang D (2003) Human mitochondrial DNA is packaged with TFAM. Nucleic Acids Res 31: 1640–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aletaha D, Neogi T, Silman AJ, Funovits J, Felson DT, Bingham CO 3rd, Birnbaum NS, Burmester GR, Bykerk VP, Cohen MD et al (2010) 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 69: 1580–1588 [DOI] [PubMed] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS et al (1988) The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 31: 315–324 [DOI] [PubMed] [Google Scholar]
- Banfer S, Schneider D, Dewes J, Strauss MT, Freibert SA, Heimerl T, Maier UG, Elsasser HP, Jungmann R, Jacob R (2018) Molecular mechanism to recruit galectin‐3 into multivesicular bodies for polarized exosomal secretion. Proc Natl Acad Sci U S A 115: E4396–E4405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock FJ, Tait SWG (2020) Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21: 85–100 [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420 [DOI] [PubMed] [Google Scholar]
- Buzas EI, Gyorgy B, Nagy G, Falus A, Gay S (2014) Emerging role of extracellular vesicles in inflammatory diseases. Nat Rev Rheumatol 10: 356–364 [DOI] [PubMed] [Google Scholar]
- Chi W, Zhu X, Yang P, Liu X, Lin X, Zhou H, Huang X, Kijlstra A (2008) Upregulated IL‐23 and IL‐17 in Behcet patients with active uveitis. Invest Ophthalmol Vis Sci 49: 3058–3064 [DOI] [PubMed] [Google Scholar]
- Colombo M, Raposo G, Thery C (2014) Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol 30: 255–289 [DOI] [PubMed] [Google Scholar]
- Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F (2016) Pore‐forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116 [DOI] [PubMed] [Google Scholar]
- Doran AC, Yurdagul A Jr, Tabas I (2020) Efferocytosis in health and disease. Nat Rev Immunol 20: 254–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek J (2017) Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol 5: 90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El‐Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, Rostami A (2011) The encephalitogenicity of T(H)17 cells is dependent on IL‐1‐ and IL‐23‐induced production of the cytokine GM‐CSF. Nat Immunol 12: 568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmi G, Talarico R, Lopalco G, Cimaz R, Cantini F, Viapiana O, Olivieri I, Goldoni M, Vitale A, Silvestri E et al (2016) Efficacy and safety profile of anti‐interleukin‐1 treatment in Behcet's disease: a multicenter retrospective study. Clin Rheumatol 35: 1281–1286 [DOI] [PubMed] [Google Scholar]
- Evavold CL, Kagan JC (2019) Inflammasomes: threat‐assessment organelles of the innate immune system. Immunity 51: 609–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC (2018) The pore‐forming protein gasdermin D regulates interleukin‐1 secretion from living macrophages. Immunity 48: 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassanito MA, Dammacco R, Cafforio P, Dammacco F (1999) Th1 polarization of the immune response in Behçet's disease: A putative pathogenetic role of interleukin‐12. Arthritis Rheum 42: 1967–1974 [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G (2012) Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol 13: 780–788 [DOI] [PubMed] [Google Scholar]
- Hashiguchi K, Zhang‐Akiyama QM (2009) Establishment of human cell lines lacking mitochondrial DNA. Methods Mol Biol 554: 383–391 [DOI] [PubMed] [Google Scholar]
- Hessvik NP, Llorente A (2018) Current knowledge on exosome biogenesis and release. Cell Mol Life Sci 75: 193–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg MC (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 40: 1725 [DOI] [PubMed] [Google Scholar]
- van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, Matucci‐Cerinic M, Naden RP, Medsger TA Jr, Carreira PE et al (2013) 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum 65: 2737–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Houwen TB, van Laar JAM, Kappen JH, van Hagen PM, de Zoete MR, van Muijlwijk GH, Berbers RM, Fluit AC, Rogers M, Groot J et al (2020) Behcet's disease under microbiotic surveillance? A combined analysis of two cohorts of Behcet's disease patients. Front Immunol 11: 1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Hasan A, Wilson A, Stanford MR, Li‐Yang Y, Todryk S, Whiston R, Shinnick T, Mizushima Y, van der Zee R et al (1998) Experimental mucosal induction of uveitis with the 60‐kDa heat shock protein‐derived peptide 336‐351. Eur J Immunol 28: 2444–2455 [DOI] [PubMed] [Google Scholar]
- Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, Malik AB (2020) mtDNA activates cGAS signaling and suppresses the YAP‐mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52: 475–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores‐Suarez LF, Gross WL, Guillevin L, Hagen EC et al (2013) 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65: 1–11 [DOI] [PubMed] [Google Scholar]
- Jeppesen DK, Fenix AM, Franklin JL, Higginbotham JN, Zhang Q, Zimmerman LJ, Liebler DC, Ping J, Liu Q, Evans R et al (2019) Reassessment of exosome composition. Cell 177: 428–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, Miao EA (2017) Programmed cell death as a defence against infection. Nat Rev Immunol 17: 151–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R, LeBleu VS (2020) The biology, function, and biomedical applications of exosomes. Science 367: eaau6977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, Ueda M, Nishide M, Koyama S, Hayama Y et al (2018) Apoptosis‐derived membrane vesicles drive the cGAS‐STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis 77: 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S (2011) Toll‐like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34: 637–650 [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- Kirino Y, Zhou Q, Ishigatsubo Y, Mizuki N, Tugal‐Tutkun I, Seyahi E, Ozyazgan Y, Ugurlu S, Erer B, Abaci N et al (2013) Targeted resequencing implicates the familial Mediterranean fever gene MEFV and the toll‐like receptor 4 gene TLR4 in Behcet disease. Proc Natl Acad Sci U S A 110: 8134–8139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriakose T, Kanneganti TD (2019) Pyroptosis in antiviral immunity. Curr Top Microbiol Immunol 10.1007/82_2019_189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Joncour A, Martos R, Loyau S, Lelay N, Dossier A, Cazes A, Fouret P, Domont F, Papo T, Jandrot‐Perrus M et al (2019) Critical role of neutrophil extracellular traps (NETs) in patients with Behcet's disease. Ann Rheum Dis 78: 1274–1282 [DOI] [PubMed] [Google Scholar]
- Letko E, Yeh S, Foster CS, Pleyer U, Brigell M, Grosskreutz CL, AIN457A2208 Study Group (2015) Efficacy and safety of intravenous secukinumab in noninfectious uveitis requiring steroid‐sparing immunosuppressive therapy. Ophthalmology 122: 939–948 [DOI] [PubMed] [Google Scholar]
- Liang L, Tan X, Zhou Q, Zhu Y, Tian Y, Yu H, Kijlstra A, Yang P (2013) IL‐1beta triggered by peptidoglycan and lipopolysaccharide through TLR2/4 and ROS‐NLRP3 inflammasome‐dependent pathways is involved in ocular Behcet's disease. Invest Ophthalmol Vis Sci 54: 402–414 [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lood C, Blanco LP, Purmalek MM, Carmona‐Rivera C, De Ravin SS, Smith CK, Malech HL, Ledbetter JA, Elkon KB, Kaplan MJ (2016) Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus‐like disease. Nat Med 22: 146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters SL, Simon A, Aksentijevich I, Kastner DL (2009) Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease (*). Annu Rev Immunol 27: 621–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur K, Whitehead LW, Heddleston JM, Li L, Padman BS, Oorschot V, Geoghegan ND, Chappaz S, Davidson S, San Chin H et al (2018) BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 359: eaao6047 [DOI] [PubMed] [Google Scholar]
- Melki I, Allaeys I, Tessandier N, Levesque T, Cloutier N, Laroche A, Vernoux N, Becker Y, Benk‐Fortin H, Zufferey A et al (2021) Platelets release mitochondrial antigens in systemic lupus erythematosus. Sci Transl Med 13: eaav5928 [DOI] [PubMed] [Google Scholar]
- Miao EA, Leaf IA, Treuting PM, Mao DP, Dors M, Sarkar A, Warren SE, Wewers MD, Aderem A (2010) Caspase‐1‐induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat Immunol 11: 1136–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuki N, Meguro A, Ota M, Ohno S, Shiota T, Kawagoe T, Ito N, Kera J, Okada E, Yatsu K et al (2010) Genome‐wide association studies identify IL23R‐IL12RB2 and IL10 as Behcet's disease susceptibility loci. Nat Genet 42: 703–706 [DOI] [PubMed] [Google Scholar]
- Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A et al (2020) Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med 382: 211–221 [DOI] [PubMed] [Google Scholar]
- Mumcu G, Direskeneli H (2019) Triggering agents and microbiome as environmental factors on Behcet's syndrome. Intern Emerg Med 14: 653–660 [DOI] [PubMed] [Google Scholar]
- Mumcu G, Inanc N, Aydin SZ, Ergun T, Direskeneli H (2009) Association of salivary S. mutans colonisation and mannose‐binding lectin deficiency with gender in Behcet's disease. Clin Exp Rheumatol 27: S32–S36 [PubMed] [Google Scholar]
- Nakano H, Kirino Y, Takeno M, Higashitani K, Nagai H, Yoshimi R, Yamaguchi Y, Kato I, Aoki I, Nakajima H (2018) GWAS‐identified CCR1 and IL10 loci contribute to M1 macrophage‐predominant inflammation in Behcet's disease. Arthritis Res Ther 20: 124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nian H, Liang D, Zuo A, Wei R, Shao H, Born WK, Kaplan HJ, Sun D (2012) Characterization of autoreactive and bystander IL‐17+ T cells induced in immunized C57BL/6 mice. Invest Ophthalmol Vis Sci 53: 897–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Niel G, D'Angelo G, Raposo G (2018) Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol 19: 213–228 [DOI] [PubMed] [Google Scholar]
- Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M (1982) Close association of HLA‐Bw51 with Behcet's disease. Arch Ophthalmol 100: 1455–1458 [DOI] [PubMed] [Google Scholar]
- Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K et al (2012) Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 485: 251–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okoye IS, Coomes SM, Pelly VS, Czieso S, Papayannopoulos V, Tolmachova T, Seabra MC, Wilson MS (2014) MicroRNA‐containing T‐regulatory‐cell‐derived exosomes suppress pathogenic T helper 1 cells. Immunity 41: 89–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okude H, Ori D, Kawai T (2021) Signaling through nucleic acid sensors and their roles in inflammatory diseases. Front Immunol 11: 625833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervin K, Childerstone A, Shinnick T, Mizushima Y, van der Zee R, Hasan A, Vaughan R, Lehner T (1993) T cell epitope expression of mycobacterial and homologous human 65‐kilodalton heat shock protein peptides in short term cell lines from patients with Behcet's disease. J Immunol 151: 2273–2282 [PubMed] [Google Scholar]
- Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes‐Alnemri T, Alnemri ES (2019) Gasdermin pores permeabilize mitochondria to augment caspase‐3 activation during apoptosis and inflammasome activation. Nat Commun 10: 1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY et al (2014) Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell 159: 1563–1577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakane T, Takeno M, Suzuki N, Inaba G (1999) Behcet's disease. N Engl J Med 341: 1284–1291 [DOI] [PubMed] [Google Scholar]
- Savina A, Furlan M, Vidal M, Colombo MI (2003) Exosome release is regulated by a calcium‐dependent mechanism in K562 cells. J Biol Chem 278: 20083–20090 [DOI] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH (2009) Interleukin‐1 and IL‐23 induce innate IL‐17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 31: 331–341 [DOI] [PubMed] [Google Scholar]
- Suzuki Kurokawa M, Suzuki N (2004) Behcet's disease. Clin Exp Med 4: 10–20 [DOI] [PubMed] [Google Scholar]
- Takeuchi M, Mizuki N, Meguro A, Ombrello MJ, Kirino Y, Satorius C, Le J, Blake M, Erer B, Kawagoe T et al (2017) Dense genotyping of immune‐related loci implicates host responses to microbial exposure in Behcet's disease susceptibility. Nat Genet 49: 438–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Behcet's Disease Research Committee of Japan (1989) Skin hypersensitivity to streptococcal antigens and the induction of systemic symptoms by the antigens in Behcet's disease–a multicenter study. J Rheumatol 16: 506–511 [PubMed] [Google Scholar]
- Théry C, Witwer KW, Aikawa E, Alcaraz MJ, Anderson JD, Andriantsitohaina R, Antoniou A, Arab T, Archer F, Atkin‐Smith GK et al (2018) Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles 7: 1535750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torralba D, Baixauli F, Villarroya-Beltri C, Fernandez-Delgado I, Latorre-Pellicer A, Acin-Perez R, Martin-Cofreces NB, Jaso-Tamame AL, Iborra S, Jorge I, et al (2018) Priming of dendritic cells by DNA-containing extracellular vesicles from activated T cells through antigen-driven contacts. Nat Commun 9: 2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO (2007) Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9: 654–659 [DOI] [PubMed] [Google Scholar]
- Villarroya‐Beltri C, Gutierrez‐Vazquez C, Sanchez‐Cabo F, Perez‐Hernandez D, Vazquez J, Martin‐Cofreces N, Martinez‐Herrera DJ, Pascual‐Montano A, Mittelbrunn M, Sanchez‐Madrid F (2013) Sumoylated hnRNPA2B1 controls the sorting of miRNAs into exosomes through binding to specific motifs. Nat Commun 4: 2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitali C, Bombardieri S, Jonsson R, Moutsopoulos HM, Alexander EL, Carsons SE, Daniels TE, Fox PC, Fox RI, Kassan SS et al (2002) Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American‐European Consensus Group. Ann Rheum Dis 61: 554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ning X, Gao P, Wu S, Sha M, Lv M, Zhou X, Gao J, Fang R, Meng G et al (2017) Inflammasome activation triggers caspase‐1‐mediated cleavage of cGAS to regulate responses to DNA virus infection. Immunity 46: 393–404 [DOI] [PubMed] [Google Scholar]
- West AP, Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17: 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MJ, McArthur K, Metcalf D, Lane RM, Cambier JC, Herold MJ, van Delft MF, Bedoui S, Lessene G, Ritchie ME et al (2014) Apoptotic caspases suppress mtDNA‐induced STING‐mediated type I IFN production. Cell 159: 1549–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X, Wang X, Cheng Z, Qin W, Lei L, Jiang J, Hu J (2019) The role of pyroptosis in cancer: pro‐cancer or pro‐“host”? Cell Death Dis 10: 650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM (2019) Emerging activators and regulators of inflammasomes and pyroptosis. Trends Immunol 40: 1035–1052 [DOI] [PubMed] [Google Scholar]
- Yazici Y, Hatemi G, Bodaghi B, Cheon JH, Suzuki N, Ambrose N, Yazici H (2021) Behçet syndrome. Nat Rev Dis Primers 7: 67 [DOI] [PubMed] [Google Scholar]
- Yuksel S, Eren E, Hatemi G, Sahillioglu AC, Gultekin Y, Demiroz D, Akdis C, Fresko I, Ozoren N (2014) Novel NLRP3/cryopyrin mutations and pro‐inflammatory cytokine profiles in Behcet's syndrome patients. Int Immunol 26: 71–81 [DOI] [PubMed] [Google Scholar]
- Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K, Hauser CJ (2010) Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464: 104–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhaolin Z, Guohua L, Shiyuan W, Zuo W (2019) Role of pyroptosis in cardiovascular disease. Cell Prolif 52: e12563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]