

Abstract
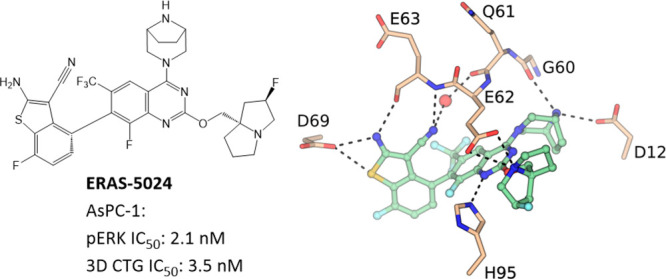
KRAS G12D mutation has been found in approximately 45% of pancreatic ductal adenocarcinoma (PDAC) cases, making it an attractive therapeutic target. Through structure-based drug design, a series of potent and selective KRAS G12D inhibitors were designed. The lead compound, ERAS-5024, inhibited ERK1/2 phosphorylation and cell proliferation in three-dimensional Cell-Titer Glo assays in AsPC-1 PDAC cells with single-digit nanomolar potency and caused tumor regression in the in vivo efficacy studies. We describe here the details of the design and synthesis program that led to the discovery of ERAS-5024.
Keywords: KRAS, G12D selective inhibitor, ERAS-5024, SBDD, PDAC, anti-tumor efficacy
KRAS cycles between the inactive (GDP) and active (GTP) states to regulate the RAS/MAPK pathway. Oncogenic mutations in KRAS lead to activation of the RAS/MAPK signaling pathways, which promotes cell proliferation, survival, and migration. G12D mutation is one of the most common KRAS mutations, accounting for approximately 45% of KRAS mutations in pancreatic cancer.1,2
Directly targeting the GTP binding pocket of KRAS has proven challenging due to the strong binding affinity of GTP to KRAS and the high concentration of GTP present in the cell.3 KRAS was considered undruggable until the recent FDA approval of two KRAS G12C inhibitors, sotarasib4 and adagrasib,5 for the treatment of G12C-positive non-small-cell lung cancer. These inhibitors were developed by utilizing a switch II pocket discovered in the pioneering work from Shokat et al.6 Following the success of drugging G12C, efforts from both academia and the pharmaceutical industry have been directed to drug other KRAS mutants, including G12D. Recent work by Wang et al.7 and Kemp et al.8 described the successful targeting of the KRAS G12D GDP state and the identification of the small-molecule inhibitor, MRTX1133 (Figure 1), which recently entered testing in a Phase I clinical trial. In this paper, structure-based drug design (SBDD) and characterization of a series of potent and selective G12D inhibitors exemplified by ERAS-5024 are described.
Figure 1.

Structures of MRTX1133 and ERAS-5024.
When our KRAS G12D small-molecule inhibitor project was initiated, there was no published literature on G12D-selective inhibitors. KRAS G12C inhibitors utilize an acrylamide warhead to react with the Cys residue at codon 12 to achieve the desired potency. To identify chemical matter for our medicinal chemistry effort, we screened the penultimate intermediates from both in-house and literature G12C programs, and compound 2,9 with a quinazoline scaffold, was identified as a bona fide hit. In RAS-RAF binding (RRB) assays, measuring the inhibition of formation of KRAS G12D or KRAS wild-type (WT) complex with RAF-RBD (RAS binding domain of Raf1 kinase), compound 2 demonstrated a G12D IC50 of 76.9 nM and a WT IC50 of >1000 nM (Table 1), suggesting it is selective against WT KRAS. AsPC-1, a pancreatic ductal adenocarcinoma cell line with a homozygous G12D mutation, was selected for our primary cellular assay. In the AsPC-1 cell-based assay, measuring the inhibition of ERK1/2 phosphorylation (pERK) formation, compound 2 was not active when tested at the highest concentration of 10 μM.
Table 1. SAR Summary for Lead Identificationa.

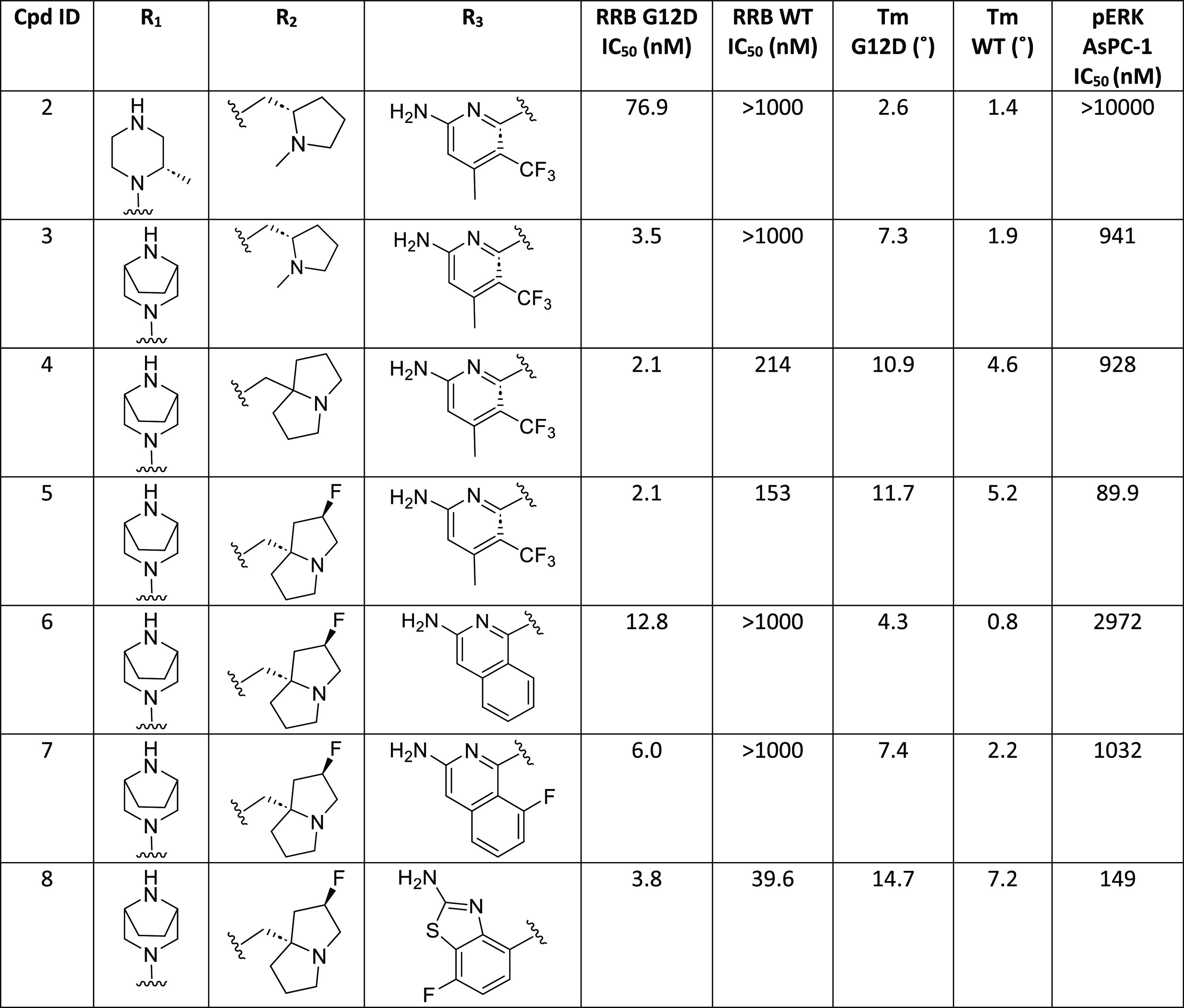
At that time in our research, a patent that described G12D-selective inhibitors was published.10 The structures of this series of compounds included a bridged amine. Modeling studies suggested that the bridged amine provides a stronger interaction with Asp12 compared to the piperazine moiety in compound 2. Indeed, when the methyl piperazine in compound 2 was replaced with the bridged amine, compound 3 exhibited an RRB G12D IC50 of 3.5 nM, a 22-fold increase in potency. Compound 3 demonstrated an RRB WT IC50 of >1000 nM, yielding a WT selectivity of >286-fold. Compound 3 exhibited a pERK IC50 of 941 nM in the AsPC-1 cell line. The in vitro WT selectivity and cell-based activity from compound 3 validated utilizing a quinazoline core to design G12D-selective inhibitors. For the subsequent structure–activity relationship (SAR) development, we decided to maintain the bridged amine at the C4 position of the quinazoline scaffold.
Next, the monocyclic amine tail in compound 3 was replaced with a fused bicyclic amine (butterfly amine tail). Compound 4 exhibited a significantly higher thermal shift (Tm) than compound 3, but it had a similar cellular potency. Tm is determined in a differential scanning fluorimetry assay measuring the stabilization of KRAS-G12D GDP or KRAS-WT GDP upon binding to the inhibitors and used as a surrogate for direct binding affinity. The higher Tm value for compound 4 suggested an increased binding affinity. When the butterfly amine tail was substituted with the same chiral fluorine atom as used in MRTX1133, the resultant compound 5 showed a cellular IC50 of 89.9 nM, a 10-fold increase over that of compound 4. The significant boost in cellular potency for compound 5 is likely due to increased permeability when the basicity of the butterfly amine is reduced by F substitution.
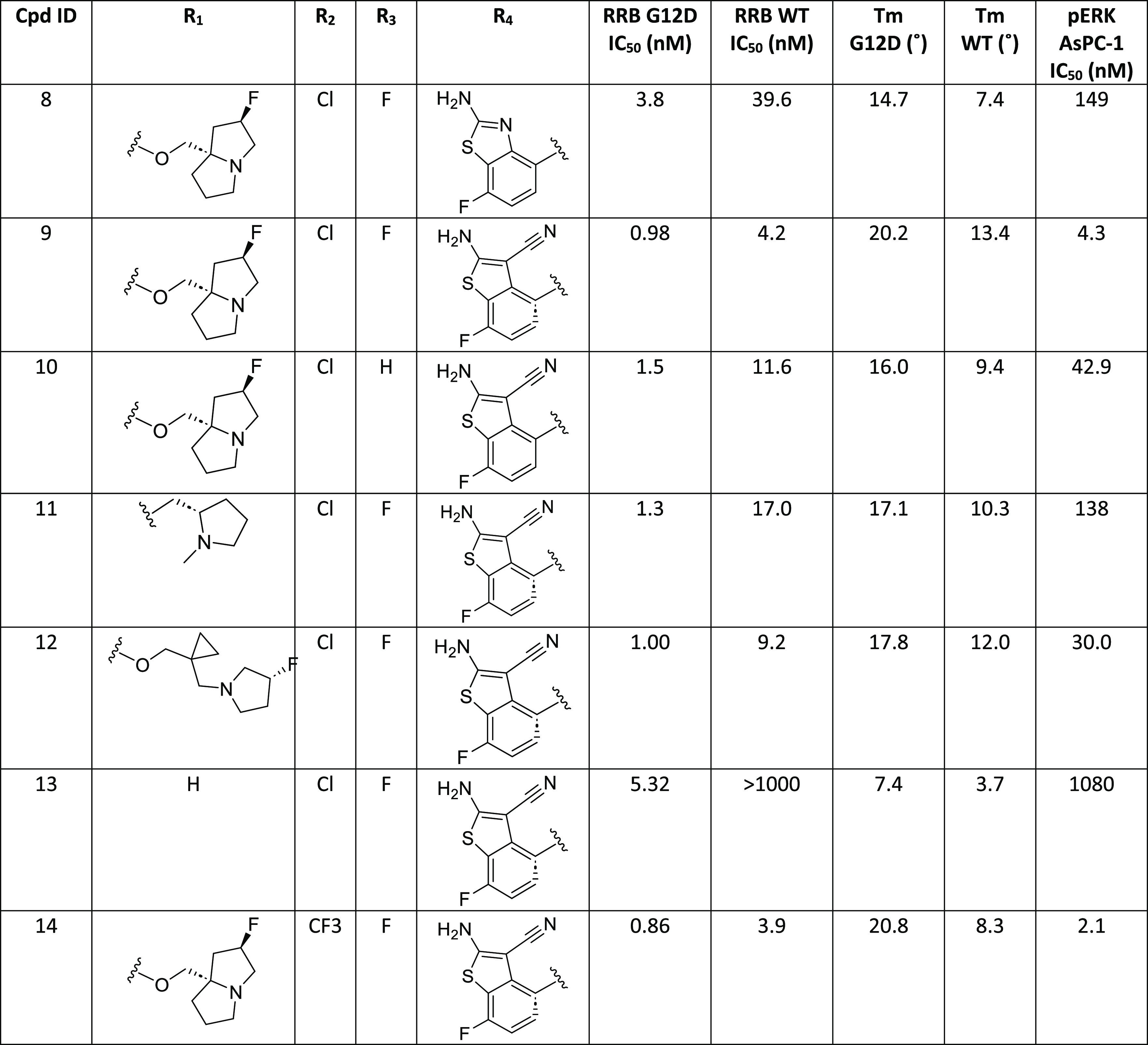
A crystal structure of compound 5 bound in the G12D protein was determined (Figure 2). As suggested by our modeling studies, the bridged amine takes a conformation with low strain energy and forms H bond interactions with both the Asp12 side-chain carboxylate and the Gly60 backbone carbonyl in region 1. The substituted aminopyridine binds in the SWII pocket, which is referred to as region 2. CF3 and Me on the pyridine ring bind in the bottom of the SWII pocket; this part of the SWII pocket is hydrophobic and formed by the side chains of Val9, Met72, Tyr96, and Ile100. The amino group on the pyridine forms H bond interactions with the side chain of Asp69 and the backbone carbonyl of Glu63 from the SWII loop. The pyridine ring N forms a H bond interaction with a conserved water molecule, which is within the H bond interaction range from both the Arg68 side chain and the backbone NH of Glu63. We named the substituent binding in region 2 as the “A ring”. In region 3, the F butterfly tail takes a conformation with low strain energy and interacts with the Glu62 side-chain carboxylate, and the core N at position 1 forms a H bond interaction with the His95 side chain. The crystal structure of 5 suggests that both the bridged amine and the F-substituted butterfly amine tail bind optimally in region 1 and region 3 of G12D, respectively.
Figure 2.
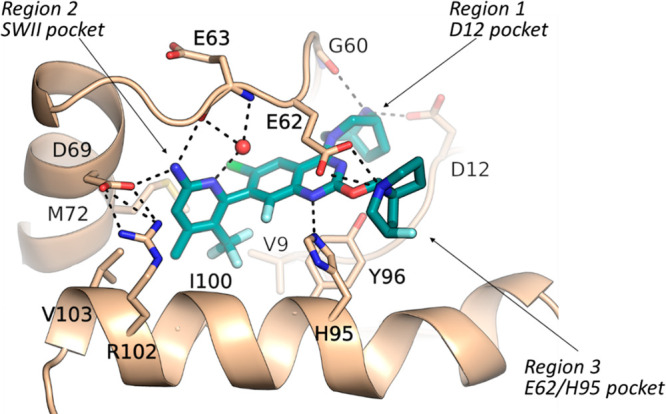
Crystal structure for compound 5 bound in KRAS G12D. Resolution is 1.35 Å. PDB code: 8TXE. Dashed lines represent H bond interactions.
In order to further increase the potency, we turned our attention to finding optimal A rings. First, we evaluated the isosteres of the A ring in compound 5. Compounds 6 and 7, with aminoquinoline and F-substituted aminoquinoline A rings, took a significant hit in both the binding affinity, measured by Tm, and the cellular potency. Compound 7 is 11-fold less active than compound 5 in the AcPC-1 cellular assay. When F-substituted aminobenzothiazole11 was placed at the 7 position, compound 8 exhibited comparable cell potency but significantly higher Tm in comparison with compound 5.
The high Tm value for compound 8 prompted us to evaluate its binding mode by crystallography; subsequently, a crystal structure of compound 8 bound in G12D protein was determined (Figure 3). The interactions of compound 8 with G12D in region 1 and region 3 are the same as those of compound 5. For the A ring interaction with the SWII pocket, the amino group forms H bond interactions with both the Asp69 side-chain carboxylate and the Glu63 backbone carbonyl, and the sulfur atom of the benzothiazole also forms a direct interaction with the Asp69 side-chain carboxylate. The nitrogen of the benzothiazole forms a H bond with a water molecule, which is within the H bond interaction range with both the Arg68 side chain and the backbone NH of the Glu63. We reasoned that a direct interaction between the A ring and the Glu63 backbone NH would further increase potency. Modeling studies suggested that a nitrile group substituted at the 3 position of the aminobenzothiazole could replace the water molecule, enabling a direct H bond interaction with the Glu63 backbone NH.
Figure 3.
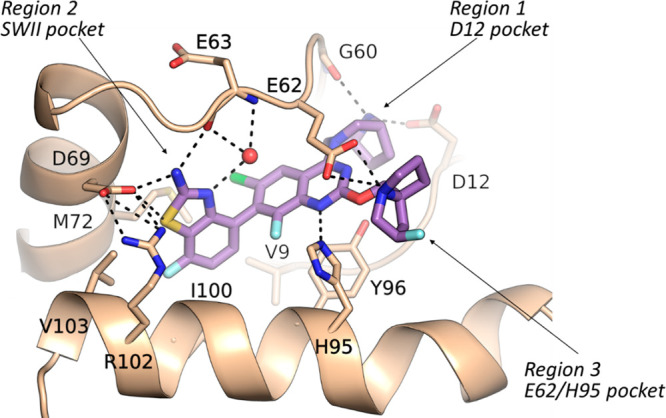
Crystal structure for compound 8 bound in KRAS G12D. Resolution is 1.5 Å. PDB code: 8TXG.
Compound 9 (ERAS-4693) was synthesized and tested.12 In comparison with compound 8, compound 9 demonstrated a G12D Tm of 20.2 °C, significantly higher than the 14.7 °C observed for compound 8. In the G12D RRB assay, compound 9 exhibited an IC50 of 0.98 nM, reaching the lower detection limit of the assay (Table 2). In the pERK assay in the AsPC-1 cell line, compound 9 demonstrated an IC50 of 4.3 nM, which is a 35-fold increase over the pERK IC50 for compound 8.
Table 2. SAR Summary for Lead Optimizationa.
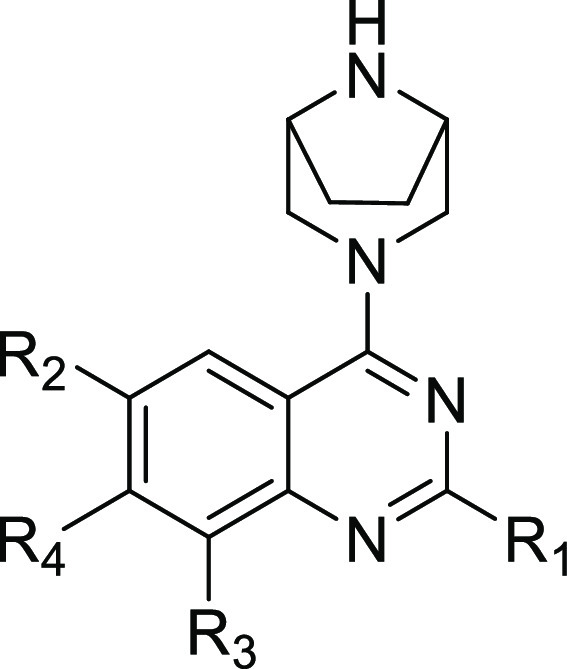
To further understand the SAR, compounds 10 through 13 were designed and synthesized. Removal of the F atom at the C8 position of the core in compound 9 yielded compound 10, which showed a significant decrease in G12D Tm by 4.1 °C and a 10-fold loss in cellular potency. The removal of F at C8 position makes the A ring conformationally more flexible, which is likely the reason for the significant decrease in G12D binding affinity, which translated to reduced cellular potency. Replacement of the F butterfly amine tail with a pyrrolidine tail afforded compound 11. This structural change also caused a significant decrease in G12D Tm by 3.1 °C and a 32-fold loss of cellular potency. Similarly, when the F butterfly amine tail was replaced by a cyclopropyl-derived tail, compound 12 exhibited decreases in both G12D Tm and pERK IC50. When the F butterfly amine tail is absent, compound 13 demonstrated a decrease in G12D Tm by 12.9 °C and a 251-fold loss of pERK IC50. In the RRB assays, compound 13 exhibited 188-fold selectivity over WT KRAS. Since the structural difference between WT and G12D is the residue at codon 12, it is reasonable to assume that the strong interaction between the Asp12 side-chain carboxylate and the bridged amine is the main reason for the observed 188-fold selectivity.
Finally, to optimize the interactions between the substituent at the C6 position and G12D, the Cl atom in compound 9 was replaced by a CF3 group. The resultant compound 14 (ERAS-5024)12 demonstrated the highest G12D Tm (20.8 °C) and the most potent pERK IC50 (2.1 nM) out of this quinazoline series.
A crystal structure of compound 14 bound in KRAS G12D was determined (Figure 4). The interactions of compound 14 with G12D in region 1 and region 3 are the same as those for compound 8. In region 2, compound 14 also maintained the interactions between the A ring in compound 8 and G12D. In addition, the cyano substituent on the A ring forms a direct H bond interaction with the backbone NH of the Glu63, and CF3 at the C6 position optimally binds in the pocket formed by the side chains of Thr58, Arg68, and Met72.
Figure 4.
Structure for compound 14 bound in KRAS G12D. Resolution is 1.2 Å. PDB code: 8TXH.
Overall, for the quinazoline series, the bridged amine interacts with both the Gly60 backbone carbonyl and the Asp12 side-chain carboxylate, conferring high G12D selectivity for the series. The cyano-substituted aminobenzothiophene A ring forms extensive H bond interactions in the SWII pocket and is the most potent A ring investigated in our G12D program. In addition, substitutions at both the C6 and the C8 positions of the quinazoline core restrict the conformation of the A ring, contributing to the observed single-digit nanomolar pERK IC50s for the series.
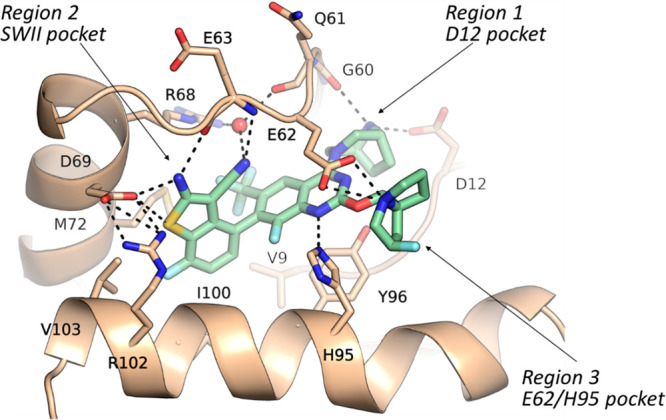
Illustrated in Scheme 1 is a general synthetic scheme for the compounds discussed in this paper. The experimental procedures for preparing compound 14 are included in the Supporting Information (SI), and detailed syntheses for the key compounds discussed herein are included in patent WO/2023018699.12
Scheme 1. General Synthetic Route.
In step A, compound 14 undergoes an SNAr reaction with mono-Boc-protected diazabicyclo[3.3.1]octane in a solvent such as dichloromethane and in the presence of a base such as triethylamine to afford compound 15. In step B, compound 15 is treated with KF in a solvent such as N,N-dimethylacetamide to generate compound 16. In step C, compound 16 is treated with a boronic acid under Suzuki reaction conditions to afford compound 17. In step D, the substituent -R1 is introduced by substitution of the fluoride with a nucleophile with the formula H-R1 in a solvent such as acetonitrile in the presence of a base such as diisopropylethylamine to provide compound 18. In step E, the Boc group of compound 18 is removed using conditions known in the art, for example, with trifluoroacetic acid, to provide compound of formula 19. Atropisomers, when present, may be separated by conventional methods such as chiral HPLC.
Anti-proliferative activities of 9, 14, and MRTX1133 were evaluated in a panel of cell lines ranging from the sensitive cell line GP2d to the insensitive cell line SNU-1033, and the results are summarized in Table 3. Selectivity against WT KRAS was assessed in RAS initiative (RI) G12D and RI WT cell lines, and all three compounds exhibited >100-fold cellular selectivity.
Table 3. Summary of Anti-proliferative Activities for ERAS-4693, ERAS-5024, and MRTX1133.
| 3D
CTG IC50 (nM) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cpd ID | AsPC-1 | GP2d | AGS | SK-LU-1 | HPAC | LS513 | HPAF-II | Panc04.03 | SNU1033 | RI G12D | RI WT |
| ERAS-4693 (9) | 15.1 | 0.2a | 1.2 | 1.4 | 3.4 | 7.5a | 14.2a | 58.6 | 1892a | 33.4 | 3435 |
| ERAS-5024 (14) | 3.5 | 0.2 | 0.7 | 0.5 | 1.8 | 2.0 | 7.2 | 10.6 | 623 | 5.8 | 978 |
| MRTX1133 | 11.0 | 0.4 | 3.2 | 2.7 | 4.2 | 8.1 | 14.9 | 27.6 | 4981 | 16.4 | 6468 |
From a single experiment. All others are average values with n ≥ 2.
Compound 14 demonstrated a robust pharmacokinetics (PK) profile in mouse in vivo PK studies when dosed by intraperitoneal (IP) administration (data discussed in the SI). Subsequently, compound 14 was progressed to an in vivo efficacy study, along with MRTX1133 as a reference compound. In the KRAS G12D mutant pancreatic ductal adenocarcinoma (PDAC) xenograft model AsPC-1, both ERAS-5024 and MRTX1133 were dosed BID at 3, 10, and 20 mg/kg. Compared with vehicle control, repeated dosing of ERAS-5024 at 3 and 10 mg/kg inhibited tumor growth by 51% and 77%, respectively, and induced tumor regression by 54% at 20 mg/kg (Table 4 and Figure 5). In comparison, repeated administration of MRTX1133 at 3, 10, and 20 mg/kg inhibited tumor growth by 61%, 83%, and 94%, respectively. ERAS-5024 is 3-fold more potent than MRTX1133 in the in vitro three-dimensional Cell-Titer Glo (3D CTG) assay with the AsPC-1 cell line, and ERAS-5024 also demonstrates more potent in vivo activity than MRTX1133. Both ERAS-5024 and MRTX1133 were tolerated in mice during the treatment period in this study.
Table 4. Anti-tumor Efficacy in the KRAS G12D Mutant PDAC Xenograft Model AsPC-1.
| Treatment group | Dose level (mg/kg/dose BID) | Anti-tumor efficacy | P values vs vehicle control |
|---|---|---|---|
| Vehicle | 0 | – | – |
| ERAS-5024 (14) | 3 | 51% TGI | 2.8 × 10–4 |
| 10 | 77% TGI | 1.1 × 10–5 | |
| 20 | 54% tumor regression | 2.3 × 10–6 | |
| MRTX1133 | 3 | 61% TGI | 4.6 × 10–5 |
| 10 | 83% TGI | 2.0 × 10–6 | |
| 20 | 94% TGI | 1.2 × 10–6 | |
Figure 5.

Anti-tumor efficacy and body weight change in the KRAS G12D mutant PDAC xenograft model AsPC-1. Mice were treated with indicated KRAS G12D inhibitors and dose levels. A: antitumor efficacy; B: body weight change. Graphed data points represent group means (n = 8), and error bars represent SEM.
In summary, through SBDD, a series of potent and selective G12D inhibitors were designed. The cyano substituent on the aminobenzothiophene A ring formed a novel H bond interaction with SWII loop residue Glu63, driving the pERK cellular potency to the single-digit nM range for the series. The lead compound, ERAS-5024, demonstrated potent in vitro anti-proliferative activities in a panel of cell lines harboring G12D mutation and exhibited robust in vivo efficacy with regression activity in a PDAC CDX model.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00245.
Synthesis of ERAS-5024; PK profile of ERAS-5024 and MRTX1133; RAS-RAF binding assay; differential scanning fluorimetry (DSC); inhibition of KRAS G12D-mediated ERK1/2 phosphorylation; inhibition of KRAS G12D-mediated cell viability by KRAS G12D inhibitors; protein expression and purification; crystallization, data collection, structure determination, and refinement (PDF)
Author Contributions
The manuscript was prepared by H.C. with contributions from all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Punekar S. R.; Velcheti V.; Neel B. G.; Wong K. The Current State of the Art and Future Trends in RAS-Targeted Cancer Therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. 10.1038/s41571-022-00671-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J. KRAS Mutation in Pancreatic Cancer. Seminars Oncol. 2021, 48, 10–18. 10.1053/j.seminoncol.2021.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannoura S. F.; Khan H. Y.; Azmi A. S. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered?. Front. Oncol. 2022, 12, 1013902. 10.3389/fonc.2022.1013902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanman B. A.; Allen J. R.; Allen J. G.; Amegadzie A. K.; Ashton K. S.; Booker S. K.; Chen J. J.; Chen N.; Frohn M. J.; Goodman G.; Kopecky D. J.; Liu L.; Lopez P.; Low J. D.; Ma V.; Minatti A. E.; Nguyen T. T.; Nishimura N.; Pickrell A. J.; Reed A. B.; Shin Y.; Siegmund A. C.; Tamayo N. A.; Tegley C. M.; Walton M. C.; Wang H.-L.; Wurz R. P.; Xue M.; Yang K. C.; Achanta P.; Bartberger M. D.; Canon J.; Hollis L. S.; McCarter J. D.; Mohr C.; Rex K.; Saiki A. Y.; San Miguel T.; Volak L. P.; Wang K. H.; Whittington D. A.; Zech S. G.; Lipford J. R.; Cee V. J. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J. Med. Chem. 2020, 63, 52–65. 10.1021/acs.jmedchem.9b01180. [DOI] [PubMed] [Google Scholar]
- Fell J. B.; Fischer J. P.; Baer B. R.; Blake J. F.; Bouhana K.; Briere D. M.; Brown K. D.; Burgess L. E.; Burns A. C.; Burkard M. R.; Chiang H.; Chicarelli M. J.; Cook A. W.; Gaudino J. J.; Hallin J.; Hanson L.; Hartley D. P.; Hicken E. J.; Hingorani G. P.; Hinklin R. J.; Mejia M. J.; Olson P.; Otten J. N.; Rhodes S. P.; Rodriguez M. E.; Savechenkov P.; Smith D. J.; Sudhakar N.; Sullivan F. X.; Tang T. P.; Vigers G. P.; Wollenberg L.; Christensen J. G.; Marx M. A. Identification of the clinical development candidate MRTX849, a covalent KRASG12C inhibitor for the treatment of cancer. J. Med. Chem. 2020, 63, 6679–6693. 10.1021/acs.jmedchem.9b02052. [DOI] [PubMed] [Google Scholar]
- Ostrem J. M.; Peters U.; Sos M. L.; Wells J. A.; Shokat K. M. K-Ras (G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503, 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Allen S.; Blake J.; Bowcut V.; Briere D.; Calinisan A.; Dahlke J. R.; Fell J. B.; Fischer J. P.; Gunn R. J.; Hallin J.; Laguer J.; Lawson J. D.; Medwid J.; Newhouse B.; Nguyen P.; O’Leary J. M.; Olson P.; Pajk S.; Rahbaek L.; Rodriguez M.; Smith C. R.; Tang T. P.; Thomas N. C.; Vanderpool D.; Vigers G. P.; Christensen J. G.; Marx A. M. Identification of MRTX1133, a Noncovalent, Potent and Selective KRASG12D Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. 10.1021/acs.jmedchem.1c01688. [DOI] [PubMed] [Google Scholar]
- Kemp S. B.; Cheng N.; Markosyan N.; Sor R.; Kim I.; Hallin J.; Shoush J.; Quinones L.; Brown N. V.; Bassett J. B.; Joshi N.; Yuan S.; Smith M.; Vostrejs W. P.; Perez-Vale K. Z.; Kahn B.; Mo F.; Donahue T. R.; Radu C. G.; Clendenin C.; Christensen J. G.; Vonderheide R. H.; Stanger B. Z. Efficacy of a Small-Molecule Inhibitor of KrasG12D in Immunocompetent Models of Pancreatic Cancer. Cancer Discov. 2023, 13, 298–311. 10.1158/2159-8290.CD-22-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra S.; Xin J.. Fused ring compounds. WO/2030097537, May 14, 2020.
- Wang X.; Burns A. C.; Christensen J. G.; Ketcham J. M.; Lawson J. D.; Marx M. A.; Smith C. R.; Allen S.; Blake J. F.; Chicarelli M. J.; Dahlke J. R.; Dai D.; Fell J. B.; Fischer J. P.; Mejia M. J.; Newhouse B.; Nguyen P.; O’leary J. M.; Pajk S.; Rodriguez M. E.; Savechenkov P.; Tang T. P.; Vigers G. P. A.; Zhao Q.. Kras G12D Inhibitors. WO/2021041571, March 4, 2021.
- Barda D. A.; Coates D. A.; Linder R. J.; Peng S.; Zia-Ebrahimi M. S.. KRAS G12C inhibitors. US/20200115375, April 16, 2020.
- Cheng H.; Vernier J.; Li P.. Preparation of quinazoline-based selective KRAS inhibitors. WO/2023018699, Feb 16, 2023.; Provisional patent that contains synthetic procedure for ERAS-4693 was filed on Aug 10, 2021. Provisional patent that contains synthetic procedure for ERAS-5024 was filed on Dec 20, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.