

Abstract

Diacylglycerol O-acyltransferase 2 (DGAT2) inhibitors have been shown to lower liver triglyceride content and are being explored clinically as a treatment for non-alcoholic steatohepatitis (NASH). This work details efforts to find an extended-half-life DGAT2 inhibitor. A basic moiety was added to a known inhibitor template, and the basicity and lipophilicity were fine-tuned by the addition of electrophilic fluorines. A weakly basic profile was required to find an appropriate balance of potency, clearance, and permeability. This work culminated in the discovery of PF-07202954 (12), a weakly basic DGAT2 inhibitor that has advanced to clinical studies. This molecule displays a higher volume of distribution and longer half-life in preclinical species, in keeping with its physicochemical profile, and lowers liver triglyceride content in a Western-diet-fed rat model.
Keywords: DGAT2, Diacylglycerol O-acyltransferase 2, NAFLD, NASH, Half-life, pKa, Fluorination
Non-alcoholic fatty liver disease (NAFLD) is considered to be the most common liver disease in Western countries and is characterized by an excess of triglycerides in the liver.1,2 Non-alcoholic steatohepatitis (NASH) is a more severe form of NAFLD, which also includes inflammatory-driven liver injury and often fibrosis. In the liver, two enzymes catalyze the majority of triglyceride synthesis from fatty acyl-CoA and diacylglycerol, termed diacylglycerol O-acyltransferase 1 and 2 (DGAT1 and DGAT2).3 While these two enzymes share identical catalytic functions, they differ in tissue distribution, with DGAT1 being highly expressed in the small and large intestines. In clinical studies, DGAT1 inhibition impaired lipid absorption, leading to diarrhea and gastrointestinal discomfort. DGAT2, which is enriched in hepatocytes, has minimal intestinal expression, and thus, clinical inhibition of DGAT2 has not caused the gastrointestinal side effects observed with DGAT1 inhibition.4 Furthermore, these two acyl transferases utilize different lipid sources: DGAT2 prefers de novo-synthesized fatty acids, while DGAT1 prefers fatty acids derived from diet and lipolysis.3 It is hypothesized that by inhibiting DGAT2, liver fat content can be reduced and the progression of NASH can be slowed.5−7 There are currently no approved therapies for NASH, although several mechanisms are in advanced clinical development.8 DGAT2 inhibitors have been previously described (Figure 1)9−15 and shown to reduce hepatic steatosis in human subjects.16 At least one small-molecule DGAT2 inhibitor, ervogastat (PF-06865571),17 is currently in clinical studies, as well as LG203003.18 In addition, an antisense therapy, ION224, designed to decrease levels of DGAT2 protein, is under clinical investigation for NASH.19,20
Figure 1.

Structures of clinical candidate DGAT2 inhibitors.
The goal of this discovery effort was to find a DGAT2 inhibitor with a longer predicted human half-life (t1/2) as a backup to the clinical candidate ervogastat, which showed a human half-life of approximately 1.5–5 h in the single ascending dose phase 1 study.4 Extending half-life can have certain advantages in some cases, such as reducing the daily tablet burden, improving patient compliance, and reducing the peak-to-trough ratio.21,22 This last attribute may have advantages when considering selectivity over off-targets, safety margins, and the drug–drug interaction potential.
Our work to extend the predicted half-life began with the clinical DGAT2 inhibitor ervogastat. This is a neutral molecule, which accordingly has a moderate volume of distribution in preclinical species (Vss = 0.71 and 0.91 L/kg in rat and monkey, respectively). Our approach to extending the half-life within this chemotype was to increase the volume of distribution by adding a basic center.23 Basic compounds generally have higher volumes of distribution than acidic or neutral species, and this is hypothesized to occur due to their low affinity for plasma albumin and their electrostatic attraction to negatively charged phospholipid membranes.21 Since the half-life is directly proportional to the steady-state volume of distribution (Vss) according to eq 1,
| 1 |
this can be a successful strategy if a basic center is tolerated in the target binding site and clearance (CL) is controlled. Bases can also show greater solubility compared to neutrals. Other strategies to increase half-life by focusing on reducing clearance (eliminating soft spots, deuteration, and increasing LipMetE) were pursued but did not result in success.
Based on the previous structure–activity relationship (SAR) explorations during the discovery of ervogastat, the amide vector offered the most productive optimization options. Hence, the initial effort to install a basic center on this scaffold was focused on the amide vector (Table 1). All analogues were synthesized according to the methods shown in Scheme 1 and the Supporting Information.24 As in previous publications, a biochemical assay using human DGAT2 was used as the primary driver to explore potency. Compounds 1–5 include several unsubstituted pyrrolidine and piperidine amides. While these analogues were less potent, they did confirm that a basic nitrogen was somewhat tolerated, and the large decrease in logD compared to that of ervogastat may be partly responsible for the loss of potency. Each of these pyrrolidines and piperidines have pKa > 9 and, as expected, displayed poor passive permeability. Consistent with its basicity and as shown in Table 3, 2 did have a higher volume of distribution and longer half-life in rat compared to ervogastat (2, Vss = 4.1 L/kg and t1/2 = 1.9 h; ervogastat, Vss = 0.91 L/kg and t1/2 = 0.3 h) when dosed intravenously.
Table 1. SAR of Strongly Basic Amines.

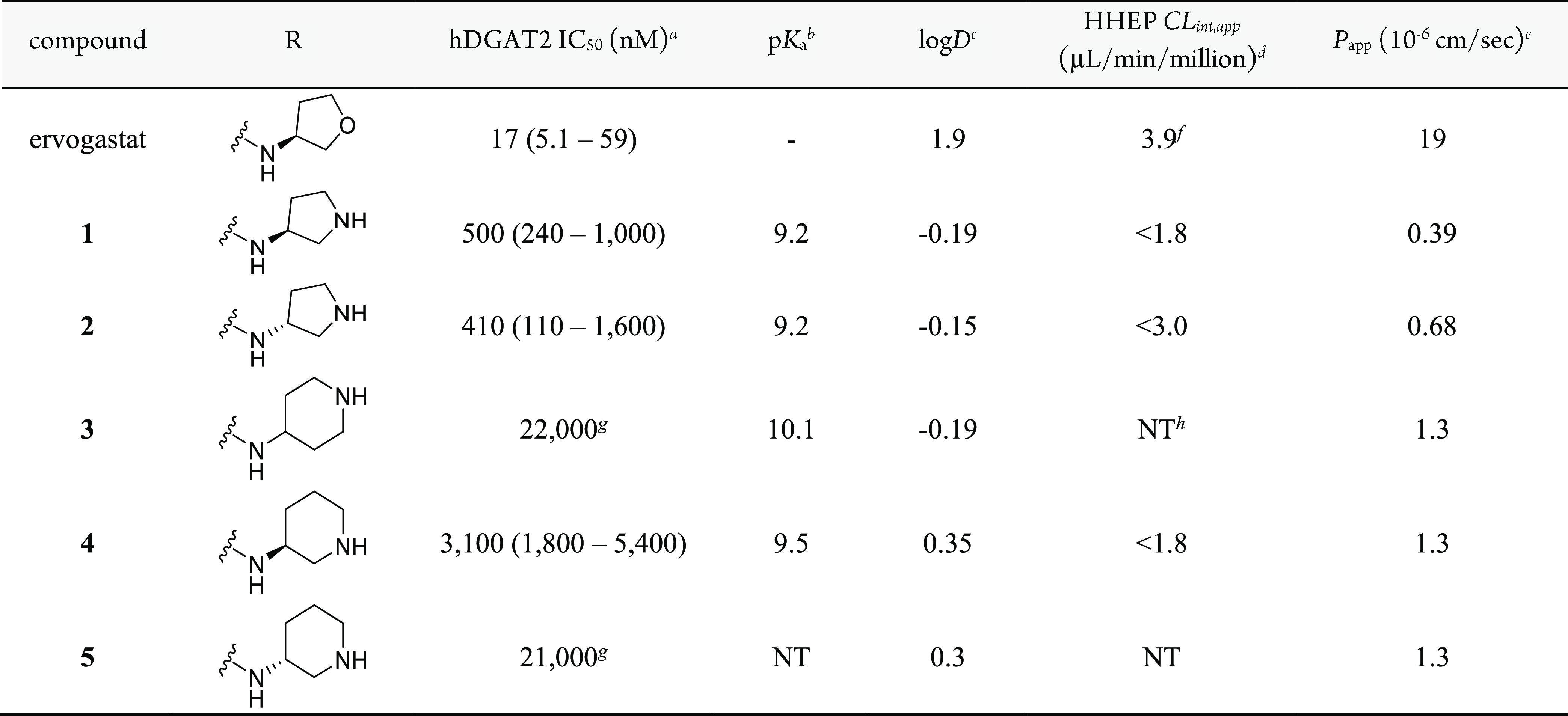
IC50 values are reported as the geometric mean of n ≥ 3 with the 95% confidence interval (CI) in parentheses, except where otherwise noted.
Experimental pKa.
logD measured with the shake-flask method.34
Apparent intrinsic clearance (CLint,app) obtained from human hepatocyte cells.35
Permeability determined from an assay using low-efflux MDCKII cells.36
CLint,app obtained from human hepatocyte cells using the relay method for low-clearance compounds.37
n = 2.
NT = not tested.
Scheme 1. Synthesis of Analogues 1–13.

(a) Amidation conditions include HATU, DMC, or T3P; (b) where needed, Cbz deprotection using HCl or Boc deprotection using TFA.
Table 3. In Vivo Pharmacokinetic Profile in Preclinical Speciesa.
| compound | species | CL (mL min–1 kg–1) | CLr (mL min–1 kg–1) | Vss (L/kg) | t1/2, i.v./p.o. (h) | F (%) |
|---|---|---|---|---|---|---|
| ervogastat | rat | 61 | <0.1 | 0.91 | 0.3/1.4e | 31 |
| ervogastat | monkeyb | 10 | <0.1 | 0.71 | 1.0/3.9 | 48 |
| 2 | rat | 51 | 5.2 | 4.1 | 1.9/NR | <1 |
| 12 | ratc | 64 | 7.5 | 2.7 | 0.9/NR | 12f |
| 12 | monkeyd | 17 | 0.29 | 1.6 | 1.4/6.5 | 21 |
| 12 | dog | 22 | 5.5 | 2.1 | 1.5/ND | ND |
Pharmacokinetic parameters were calculated from plasma concentration–time data and are reported as mean values. Intravenous (iv) PK (1 mg/kg) was conducted in male sex of each species (Wistar Han rats, beagle dogs and cynomolgus monkey, n = 2, except where otherwise noted). Solution formulation for the iv dose included 10% dimethyl sulfoxide (DMSO)/30% poly(ethylene glycol) (PEG) 400/60% water for rat and 23% hydroxypropyl-β-cyclodextrin (HPBCD) in water in dog and monkey except where noted. Oral (po) studies were administered as suspensions in 0.5% methylcellulose, except where noted. F was calculated using AUC0–∞, except where noted. NR = not reported due to insufficient regression points. ND = not determined.
Solution formulation for iv monkey study included 2% dimethylacetamide/98% (20% sulfobutylether-β-cyclodextrin (SBECD) in water).
Solution formulation for po rat study included 0.5% methylcellulose.
Oral (po) study administered as a solution in 23% w/v HPBCD in water.
n = 5.
AUC0–tlast was used since regression points were not selected.
Measurable potency and the proof of concept that increased volume of distribution leads to increased half-life were encouraging first steps. Further efforts to alter pKa were undertaken to determine whether optimal properties could be found within this basic series. Modification of the pyrrolidine and piperidine motifs was straightforward using readily available substituted building blocks. While the unsubstituted pyrrolidines were more potent than the corresponding piperidines, the greatest optimization potential was found with 3-aminopiperidines. Optimization of the piperidine motif with fluorine substitution is shown in Table 2. Fluorine substitution is known to alter the pKa of a nearby nitrogen atom and can offer the advantage of improving permeability and metabolic stability in certain cases.25
Table 2. SAR of Weakly Basic Amines.

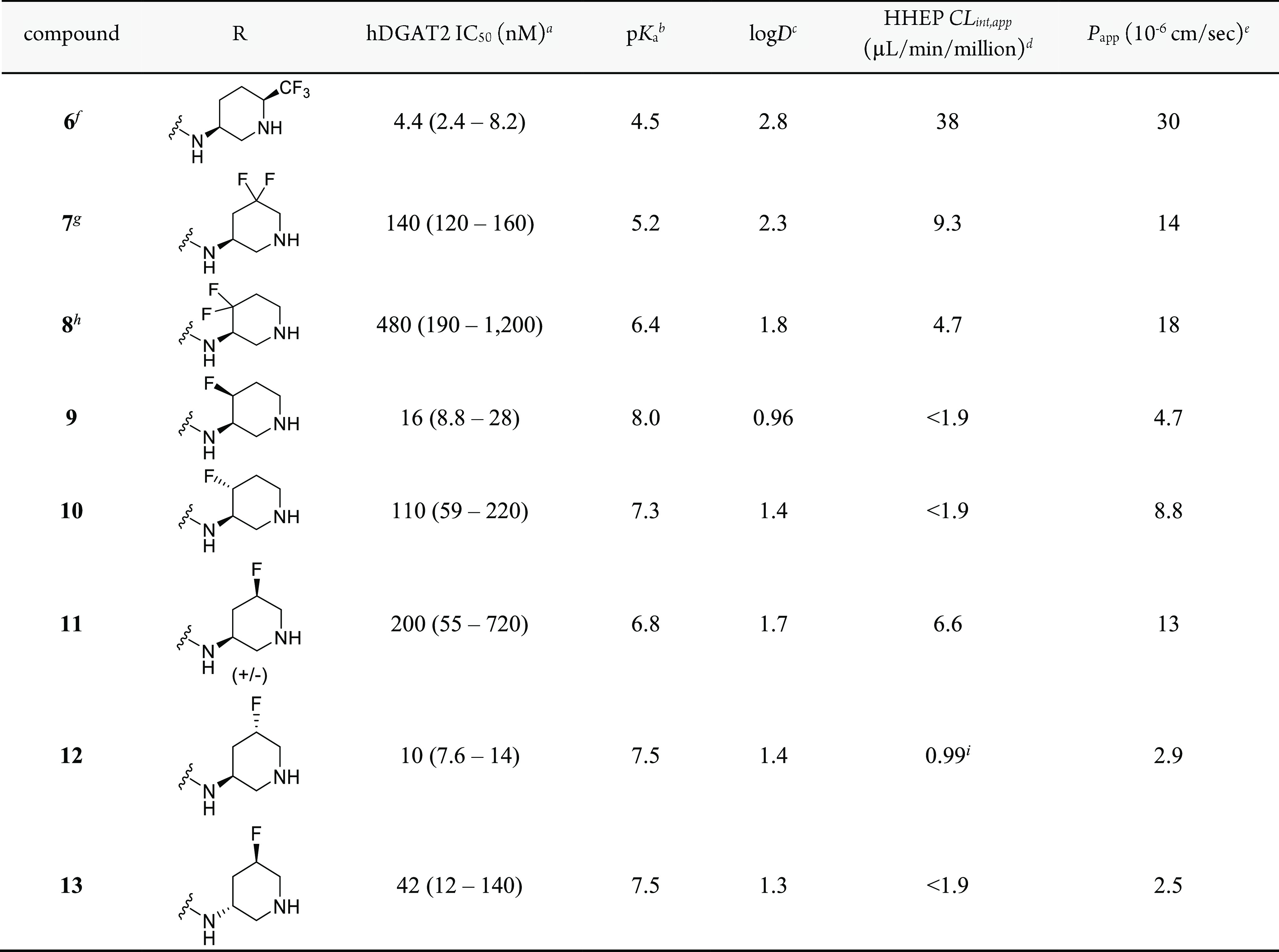
IC50 values are reported as the geometric mean of n ≥ 3 with the 95% CI in parentheses, except where otherwise noted.
Experimental pKa.
Experimental logD using the shake-flask method at pH 7.4.34
Apparent intrinsic clearance (CLint,app) obtained from human hepatocyte cells.35
Permeability determined from an assay using low-efflux MDCKII cells.36
6 is the more potent cis enantiomer; stereochemistry arbitrarily assigned.
7 is the more potent enantiomer; stereochemistry arbitrarily assigned.
8 is the more potent enantiomer; stereochemistry arbitrarily assigned.
CLint,app obtained from human hepatocyte cells using the relay method for low-clearance compounds37
The addition of an α-trifluoromethyl group (6) lowered the pKa by ∼5 units and raised the logD by ∼2.5 units compared to unsubstituted comparators 4 and 5. A significant improvement in DGAT2 potency was observed, along with increased permeability. Unfortunately, human hepatocyte clearance was markedly increased. Difluorination of the β-position (7) relative to the basic center increased the pKa, and γ-difluorination (8) increased the pKa further, relative to 6. These analogues showed relatively lower lipophilicity, permeability, and clearance, with each of these properties trending with basicity.
To further improve potency and reduce clearance, monofluoro substitution was also examined. Compounds 9 and 10 include isomers with γ-substitution. These inhibitors showed the expected higher pKa and lower lipophilicity relative to the difluoro version. Biochemical potency was improved, especially for 9, and both isomers showed lower clearance and good permeability. Examining the β-substituted monofluoro isomers (11–13), the basicity was moderately reduced and the lipophilicity was moderately increased, on average, as expected compared to the more distal γ-fluorinated analogues 9 and 10. There are also notable differences in basicity between β-substituted monofluoro isomers. Presumed axially substituted fluorine enantiomers 12 and 13 are more basic than the presumed equatorial racemic mixture of 11. This has also been observed previously25−30 and is likely due to the antiparallel dipole interaction that stabilizes the protonated form in only the axial case. It is noticeable that metabolic clearance and permeability showed some correlation with the observed pKa of the piperidine, with markedly higher values for both parameters as pKa moved below physiological values. However, the correlation of potency with pKa was much less clear, suggesting more subtle influences of specific fluorine placement. The S,S isomer, 12, also showed excellent potency (>300-fold improvement relative to 2), displayed very low hepatocyte clearance, and had acceptable permeability. This isomer was selected for further characterization.
Potency was also assessed in cryopreserved human hepatocytes by measuring [14C]glycerol incorporation into triglycerides using thin-layer chromatography. A selective DGAT1 inhibitor was added to this cellular assay to enhance the window to observe DGAT2-mediated triglyceride synthesis. In this assay, 12 showed an IC50 (95% CI) of 11 nM (8.6–15 nM). Compound 9 was somewhat less potent in the cryopreserved human hepatocyte assay with an IC50 (95% CI) of 23 nM (19–28 nM), strengthening the decision to pursue 12. Selectivity was demonstrated by testing 12 against related acyltransferases (DGAT1, MGAT1, MGAT2, and MGAT3: all IC50 > 50,000 nM) and a broad off-target panel (Table S1).
Table 3 shows the pharmacokinetic parameters observed in both rat and monkey after iv administration of both 12 and ervogastat as a comparison. The observed volume of distribution of weakly basic 12 is 2- to 3-fold higher than that observed for neutral ervogastat in both rat and monkey. In addition, since the clearance appears comparable across compounds, this leads to a longer half-life for 12 in the species examined: 0.9 h in rat and 1.4 h in monkey. The bioavailability of 12 is 2- to 3-fold lower in rat and monkey compared to ervogastat, likely due to the lower in vitro passive permeability of 12.
In keeping with the Extended Clearance Classification System (ECCS)31 class 4 designation of 12, hepatic metabolism and renal clearance are predicted to be major clearance mechanisms. Preliminary human clearance for 12 was estimated by scaling the in vitro CLint,app in human hepatocytes (Table 2) using the well-stirred model. Renal clearance in humans was estimated using simple allometry from rat and dog.32 The human volume of distribution of 12 was estimated to be moderate (2.3 L/kg) utilizing the Rogers and Rowland33 tissue Kp calculation with predicted preclinical Vss being within 2-fold of the observed value using the same method. Single-species scaling across preclinical species predicted a moderate Vss (1.5–2.4 L/kg). In comparison, for ervogastat, the Rogers and Rowland tissue Kp calculation predicted a Vss of 1.2 L/kg, and single-species scaling predicted a low to moderate Vss (0.7–1.5 L/kg). For 12, along with adequate oral absorption, a projected human half-life of approximately 8 h was considered acceptably longer than ervogastat to progress to further preclinical characterization.
Compound 12 was next examined for metabolic effects in vivo. The rat DGAT2 in vitro potency was similar to that of human (rat DGAT2 IC50 (95% CI) = 17 nM (9.1–33 nM)). Rats placed on a high-fat, high-sucrose, high cholesterol (Western) diet were treated orally twice daily for 8 days with 12. An observed dose-dependent reduction in plasma and liver triglycerides resulted, as shown in Figure 2, with the high dose achieving a 71% reduction in plasma triglycerides and 48% reduction in liver triglycerides. While plasma triglycerides returned to chow-vehicle control levels at the top doses, a partial reduction was observed for hepatic triglycerides, similar to results observed with ervogastat in this same assay.12 The partial reduction of hepatic triglycerides could be due to the inhibition of lipoprotein secretion from blunted hepatic SREBP signaling, a known regulator of lipoprotein secretion that we have demonstrated to be suppressed with DGAT2 inhibition.16 Compound 12 also reduced multiple SREBP target genes (Table S2), which also contributes to reduced de novo lipogenesis. Dose-dependent reductions in hepatic and circulating triglycerides, along with SREBP target gene expression reduction, were also noted after single-dose administration to high-sucrose-diet-fed rats (Figures S2 and S1 and Tables S4 and S3).
Figure 2.

Plasma and hepatic triglycerides in Western-diet-fed rats after an 8-day treatment with 12. Data are mean ± standard deviation from eight animals per group. The difference between group means relative to the Western diet vehicle was performed by one-way ANOVA taking account for unequal variance followed by a Dunnett’s multiple comparisons test. ****, p < 0.0001 relative to Western diet vehicle-treated animals; ***, p < 0.001 relative to Western diet vehicle-treated animals; *, p < 0.05 relative to Western diet vehicle-treated animals. BID dose administration was separated by 8 h. #On day 8 a single dose was administered in the morning, and animals were sacrificed 2 h after treatment.
Adding a basic center in this case was a successful strategy to identify a DGAT2 inhibitor with an extended preclinical and predicted human half-life. A balanced basicity and lipophilicity profile was needed to achieve an acceptable suite of attributes, including potency, permeability, and clearance. Ultimately an ideal balance of basicity and lipophilicity was achieved through the strategic introduction of fluorine to the piperidine ring, to arrive at monofluorinated derivative 12. This molecule, PF-07202954, was nominated as a development candidate and advanced to phase 1 clinical trials in order to determine its human pharmacokinetic profile.
Acknowledgments
We thank Junshan Luo, Juan Shi, and Wenjuan Wong at Wuxi AppTec as well as Kim Huard and Esther Lee for their contributions.
Glossary
Abbreviations
- NAFLD
non-alcoholic fatty liver disease
- NASH
non-alcoholic steatohepatitis
- DGAT2
diacylglycerol O-acyltransferase 2
- DGAT1
diacylglycerol O-acyltransferase 1
- t1/2
half-life
- Vss
steady-state volume of distribution
- ECCS
Extended Clearance Classification System
- D
distribution
- HHEP
human hepatocyte
- Papp
apparent permeability
- CL
clearance
- SREBP
sterol regulatory element-binding protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00330.
Synthetic procedures and characterization data for analogues 1–13, activity in a broad off-target panel, in vitro and hepatocyte assay procedures, in vivo protocols and results for 8-day treatment of Western-diet-fed rats and single-dose treatment of sucrose-diet-fed rats, both with PF-07202954 (12) (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
All experiments involving animals were conducted in our AAALAC-accredited facilities and were reviewed and approved by Pfizer Institutional Animal Care and Use Committee (IACUC).
The authors declare the following competing financial interest(s): All authors were employees of Pfizer, Inc. at the time that work was done and may own Pfizer stock. This work was financially supported by Pfizer, Inc.
Supplementary Material
References
- Fon Tacer K.; Rozman D. Nonalcoholic Fatty liver disease: focus on lipoprotein and lipid deregulation. J. Lipids 2011, 2011, 783976. 10.1155/2011/783976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas J. T.; Francque S.; Staels B. Pathophysiology and Mechanisms of Nonalcoholic Fatty Liver Disease. Annu. Rev. Physiol. 2016, 78, 181–205. 10.1146/annurev-physiol-021115-105331. [DOI] [PubMed] [Google Scholar]
- Yen C. L.; Stone S. J.; Koliwad S.; Harris C.; Farese R. V. Jr. Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J. Lipid Res. 2008, 49 (11), 2283–2301. 10.1194/jlr.R800018-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin N. B.; Saxena A. R.; Somayaji V.; Dullea R. Inhibition of Diacylglycerol Acyltransferase 2 Versus Diacylglycerol Acyltransferase 1: Potential Therapeutic Implications of Pharmacology. Clin. Ther. 2023, 45 (1), 55–70. 10.1016/j.clinthera.2022.12.008. [DOI] [PubMed] [Google Scholar]
- Choi S. H.; Ginsberg H. N. Increased very low density lipoprotein (VLDL) secretion, hepatic steatosis, and insulin resistance. Trends Endocrinol. Metab. 2011, 22 (9), 353–363. 10.1016/j.tem.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y.; Millar J. S.; Cromley D. A.; Graham M.; Crooke R.; Billheimer J. T.; Rader D. J. Knockdown of acyl-CoA:diacylglycerol acyltransferase 2 with antisense oligonucleotide reduces VLDL TG and ApoB secretion in mice. Biochim. Biophys. Acta 2008, 1781 (3), 97–104. 10.1016/j.bbalip.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Yu X. X.; Murray S. F.; Pandey S. K.; Booten S. L.; Bao D.; Song X. Z.; Kelly S.; Chen S.; McKay R.; Monia B. P.; Bhanot S. Antisense oligonucleotide reduction of DGAT2 expression improves hepatic steatosis and hyperlipidemia in obese mice. Hepatology 2005, 42 (2), 362–371. 10.1002/hep.20783. [DOI] [PubMed] [Google Scholar]
- Vuppalanchi R.; Noureddin M.; Alkhouri N.; Sanyal A. J. Therapeutic pipeline in nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18 (6), 373–392. 10.1038/s41575-020-00408-y. [DOI] [PubMed] [Google Scholar]
- Futatsugi K.; Kung D. W.; Orr S. T.; Cabral S.; Hepworth D.; Aspnes G.; Bader S.; Bian J.; Boehm M.; Carpino P. A.; Coffey S. B.; Dowling M. S.; Herr M.; Jiao W.; Lavergne S. Y.; Li Q.; Clark R. W.; Erion D. M.; Kou K.; Lee K.; Pabst B. A.; Perez S. M.; Purkal J.; Jorgensen C. C.; Goosen T. C.; Gosset J. R.; Niosi M.; Pettersen J. C.; Pfefferkorn J. A.; Ahn K.; Goodwin B. Discovery and optimization of imidazopyridine-based inhibitors of diacylglycerol acyltransferase 2 (DGAT2). J. Med. Chem. 2015, 58 (18), 7173–7185. 10.1021/acs.jmedchem.5b01006. [DOI] [PubMed] [Google Scholar]
- Futatsugi K.; Huard K.; Kung D. W.; Pettersen J. C.; Flynn D. A.; Gosset J. R.; Aspnes G. E.; Barnes R. J.; Cabral S.; Dowling M. S.; Fernando D. P.; Goosen T. C.; Gorczyca W. P.; Hepworth D.; Herr M.; Lavergne S.; Li Q.; Niosi M.; Orr S. T. M.; Pardo I. D.; Perez S. M.; Purkal J.; Schmahai T. J.; Shirai N.; Shoieb A. M.; Zhou J.; Goodwin B. Small structural changes of the imidazopyridine diacylglycerol acyltransferase 2 (DGAT2) inhibitors produce an improved safety profile. MedChemComm 2017, 8 (4), 771–779. 10.1039/C6MD00564K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren D. G.; Han S.; Murphy B. A.; Wilsie L.; Stout S. J.; Zhou H.; Roddy T. P.; Gorski J. N.; Metzger D. E.; Shin M. K.; Reilly D. F.; Zhou H. H.; Tadin-Strapps M.; Bartz S. R.; Cumiskey A. M.; Graham T. H.; Shen D. M.; Akinsanya K. O.; Previs S. F.; Imbriglio J. E.; Pinto S. DGAT2 inhibition alters aspects of triglyceride metabolism in rodents but not in non-human primates. Cell Metab. 2018, 27 (6), 1236–1248. 10.1016/j.cmet.2018.04.004. [DOI] [PubMed] [Google Scholar]
- Futatsugi K.; Cabral S.; Kung D. W.; Huard K.; Lee E.; Boehm M.; Bauman J.; Clark R. W.; Coffey S. B.; Crowley C.; Dechert-Schmitt A. M.; Dowling M. S.; Dullea R.; Gosset J. R.; Kalgutkar A. S.; Kou K.; Li Q.; Lian Y.; Loria P. M.; Londregan A. T.; Niosi M.; Orozco C.; Pettersen J. C.; Pfefferkorn J. A.; Polivkova J.; Ross T. T.; Sharma R.; Stock I. A.; Tesz G.; Wisniewska H.; Goodwin B.; Price D. A. Discovery of Ervogastat (PF-06865571): A Potent and Selective Inhibitor of Diacylglycerol Acyltransferase 2 for the Treatment of Non-alcoholic Steatohepatitis. J. Med. Chem. 2022, 65 (22), 15000–15013. 10.1021/acs.jmedchem.2c01200. [DOI] [PubMed] [Google Scholar]
- Imbriglio J. E.; Shen D. M.; Liang R.; Marby K.; You M.; Youm H. W.; Feng Z.; London C.; Xiong Y.; Tata J.; Verras A.; Garcia-Calvo M.; Song X.; Addona G. H.; McLaren D. G.; He T.; Murphy B.; Metzger D. E.; Salituro G.; Deckman D.; Chen Q.; Jin X.; Stout S. J.; Wang S. P.; Wilsie L.; Palyha O.; Han S.; Hubbard B. K.; Previs S. F.; Pinto S.; Taggart A. Discovery and pharmacology of a novel class of diacylglycerol acyltransferase 2 inhibitors. J. Med. Chem. 2015, 58 (23), 9345–9353. 10.1021/acs.jmedchem.5b01345. [DOI] [PubMed] [Google Scholar]
- Camp N. P.; Naik M.. Novel DGAT2 inhibitors. WO 2015077299, 2015.
- Escribano A. M.; Gonzalez M. R.; Lafuente Blanco C.; Martin-Ortega Finger M. D.; Wiley M. R.. Novel DGAT2 inhibitors. WO 2016187384, 2016.
- Amin N. B.; Carvajal-Gonzalez S.; Purkal J.; Zhu T.; Crowley C.; Perez S.; Chidsey K.; Kim A. M.; Goodwin B. Targeting diacylglycerol acyltransferase 2 for the treatment of nonalcoholic steatohepatitis. Sci. Transl. Med. 2019, 11 (520), eaav9701 10.1126/scitranslmed.aav9701. [DOI] [PubMed] [Google Scholar]
- Pfizer Product Pipeline. https://www.pfizer.com/science/drug-product-pipeline (accessed 2022-10-24).
- LG Chem Business Areas: Life Science. https://www.lgchem.com/company/company-information/business-domain/biology (accessed 2022-11-21).
- Loomba R.; Morgan E.; Watts L.; Xia S.; Hannan L. A.; Geary R. S.; Baker B. F.; Bhanot S. Novel antisense inhibition of diacylglycerol O-acyltransferase 2 for treatment of non-alcoholic fatty liver disease: a multicentre, double-blind, randomised, placebo-controlled phase 2 trial. Lancet Gastroenterol. Hepatol. 2020, 5 (9), 829–838. 10.1016/S2468-1253(20)30186-2. [DOI] [PubMed] [Google Scholar]
- Ionis Pipeline. https://www.ionispharma.com/ionis-innovation/pipeline/ (accessed 2022-10-31).
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Relevance of half-life in drug design. J. Med. Chem. 2018, 61 (10), 4273–4282. 10.1021/acs.jmedchem.7b00969. [DOI] [PubMed] [Google Scholar]
- Gunaydin H.; Altman M. D.; Ellis J. M.; Fuller P.; Johnson S. A.; Lahue B.; Lapointe B. Strategy for extending half-life in drug design and its significance. ACS Med. Chem. Lett. 2018, 9 (6), 528–533. 10.1021/acsmedchemlett.8b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. A.; Beaumont K.; Maurer T. S.; Di L. Volume of distribution in drug design. J. Med. Chem. 2015, 58 (15), 5691–5698. 10.1021/acs.jmedchem.5b00201. [DOI] [PubMed] [Google Scholar]
- Edmonds D. J.; Filipski K. J.; Futatsugi K.; Garnsey M. R.; Lee J. C. H.; Smaltz D. J.. Preparation of piperidinyl pyrimidines as diacylglycerol acyltransferase 2 inhibitors. WO 2021064590, 2021.
- Gillis E. P.; Eastman K. J.; Hill M. D.; Donnelly D. J.; Meanwell N. A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58 (21), 8315–8359. 10.1021/acs.jmedchem.5b00258. [DOI] [PubMed] [Google Scholar]
- van Niel M. B.; Collins I.; Beer M. S.; Broughton H. B.; Cheng S. K.; Goodacre S. C.; Heald A.; Locker K. L.; MacLeod A. M.; Morrison D.; Moyes C. R.; O’Connor D.; Pike A.; Rowley M.; Russell M. G.; Sohal B.; Stanton J. A.; Thomas S.; Verrier H.; Watt A. P.; Castro J. L. Fluorination of 3-(3-(piperidin-1-yl)propyl)indoles and 3-(3-(piperazin-1-yl)propyl)indoles gives selective human 5-HT1D receptor ligands with improved pharmacokinetic profiles. J. Med. Chem. 1999, 42 (12), 2087–2104. 10.1021/jm981133m. [DOI] [PubMed] [Google Scholar]
- Cox C. D.; Coleman P. J.; Breslin M. J.; Whitman D. B.; Garbaccio R. M.; Fraley M. E.; Buser C. A.; Walsh E. S.; Hamilton K.; Schaber M. D.; Lobell R. B.; Tao W.; Davide J. P.; Diehl R. E.; Abrams M. T.; South V. J.; Huber H. E.; Torrent M.; Prueksaritanont T.; Li C.; Slaughter D. E.; Mahan E.; Fernandez-Metzler C.; Yan Y.; Kuo L. C.; Kohl N. E.; Hartman G. D. Kinesin spindle protein (KSP) inhibitors. 9. Discovery of (2S)-4-(2,5-difluorophenyl)-N-[(3R,4S)-3-fluoro-1-methylpiperidin-4-yl]-2-(hydroxymethyl)-N-methyl-2-phenyl-2,5-dihydro-1H-pyrrole-1-carboxamide (MK-0731) for the treatment of taxane-refractory cancer. J. Med. Chem. 2008, 51 (14), 4239–4252. 10.1021/jm800386y. [DOI] [PubMed] [Google Scholar]
- Nairoukh Z.; Strieth-Kalthoff F.; Bergander K.; Glorius F. Understanding the conformational behavior of fluorinated piperidines: the origin of the axial-F preference. Chem. - Eur. J. 2020, 26 (28), 6141–6146. 10.1002/chem.202001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairoukh Z.; Strieth-Kalthoff F.; Bergander K.; Glorius F. Corrigendum: Understanding the conformational behavior of fluorinated piperidines: the origin of the axial-F preference. Chem. - Eur. J. 2020, 26 (61), 14018–14019. 10.1002/chem.202004224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenthaler M.; Schweizer E.; Hoffmann-Roder A.; Benini F.; Martin R. E.; Jaeschke G.; Wagner B.; Fischer H.; Bendels S.; Zimmerli D.; Schneider J.; Diederich F.; Kansy M.; Muller K. Predicting and tuning physicochemical properties in lead optimization: amine basicities. ChemMedChem 2007, 2 (8), 1100–1115. 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- Varma M. V.; Steyn S. J.; Allerton C.; El-Kattan A. F. Predicting clearance mechanism in drug discovery: Extended Clearance Classification System (ECCS). Pharm. Res. 2015, 32 (12), 3785–3802. 10.1007/s11095-015-1749-4. [DOI] [PubMed] [Google Scholar]
- Paine S. W.; Menochet K.; Denton R.; McGinnity D. F.; Riley R. J. Prediction of human renal clearance from preclinical species for a diverse set of drugs that exhibit both active secretion and net reabsorption. Drug Metab. Dispos. 2011, 39 (6), 1008–1013. 10.1124/dmd.110.037267. [DOI] [PubMed] [Google Scholar]
- Rodgers T.; Leahy D.; Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate-to-strong bases. J. Pharm. Sci. 2005, 94 (6), 1259–1276. 10.1002/jps.20322. [DOI] [PubMed] [Google Scholar]
- Stopher D.; McClean S. An improved method for the determination of distribution coefficients. J. Pharm. Pharmacol. 1990, 42 (2), 144. 10.1111/j.2042-7158.1990.tb05373.x. [DOI] [PubMed] [Google Scholar]
- Keefer C.; Chang G.; Carlo A.; Novak J. J.; Banker M.; Carey J.; Cianfrogna J.; Eng H.; Jagla C.; Johnson N.; Jones R.; Jordan S.; Lazzaro S.; Liu J.; Scott Obach R.; Riccardi K.; Tess D.; Umland J.; Racich J.; Varma M.; Visswanathan R.; Di L. Mechanistic insights on clearance and inhibition discordance between liver microsomes and hepatocytes when clearance in liver microsomes is higher than in hepatocytes. Eur. J. Pharm. Sci. 2020, 155, 105541. 10.1016/j.ejps.2020.105541. [DOI] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J. 3rd; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100 (11), 4974–4985. 10.1002/jps.22674. [DOI] [PubMed] [Google Scholar]
- Di L.; Trapa P.; Obach R. S.; Atkinson K.; Bi Y. A.; Wolford A. C.; Tan B.; McDonald T. S.; Lai Y.; Tremaine L. M. A novel relay method for determining low-clearance values. Drug Metab. Dispos. 2012, 40 (9), 1860–1865. 10.1124/dmd.112.046425. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.