

SUMMARY
T-bet and FOXO1 are transcription factors canonically associated with effector and memory T cell fates, respectively. During an infectious response, these factors direct the development of CD8+ T cell fates, where T-bet deficiency leads to ablation of only short-lived effector cells, while FOXO1 deficiency results in selective loss of memory. In contrast, following adjuvanted subunit vaccination in mice, both effector- and memory-fated T cells are compromised in the absence of either T-bet or FOXO1. Thus, unlike responses to challenge with Listeria monocytogenes, productive CD8+ T cell responses to adjuvanted vaccination require coordinated regulation of FOXO1 and T-bet transcriptional programs. Single-cell RNA sequencing analysis confirms simultaneous T-bet, FOXO1, and TCF1 transcriptional activity in vaccine-elicited, but not infection-elicited, T cells undergoing clonal expansion. Collectively, our data show that subunit vaccine adjuvants elicit T cell responses dependent on transcription factors associated with effector and memory cell fates.
In brief
In T cells responding to infection, T-bet directs the formation of effector cells but not memory, while FOXO1 directs that of memory but not effectors. However, Ivanova et al. show that following adjuvanted vaccine administration, effector and memory T cell formation is compromised in the absence of T-bet or FOXO1.
Graphical abstract
INTRODUCTION
The primary goal of vaccination is the generation of protective long-term immune memory. This is optimally achieved by the use of live attenuated vaccines, which generate durable and potent cellular and humoral immunity.1–4 However, not all organisms can be properly and safely attenuated, and those that can are often plagued by production and storage issues, adverse reactions, and potential reversion to virulence. In contrast, subunit adjuvanted vaccines have relatively few manufacturing/virulence liabilities but often elicit an adaptive response with reduced breadth and durability. Thus, a better understanding of adjuvant-elicited responses is needed for developing vaccine strategies that generate more robust and durable adaptive responses.
A common justification for study of the adaptive immune response in animal models of infection, such as Listeria monocytogenes (LM), lymphocytic choriomeningitis virus (LCMV), or vaccinia virus (VV) is its application to subunit vaccine development and design. However, a growing body of evidence sup ports divergent mechanistic underpinnings for infection-elicited vs. adjuvant-elicited cellular immunity. This is exemplified in interleukin-27 (IL-27), a cytokine required for maximal adjuvant-elicited cellular responses5,6 but often restrictive of T cell responses to infectious challenge.7,8 Similar differential dependencies have been observed for IL-15, glycolysis vs. oxidative phosphorylation metabolism,5 and even B cells.9
In the present study, we reveal that one of the major reasons for this disconnect between adjuvant-elicited T cell (Tvac) immunity and infection-elicited T cell (Tinf) immunity is a fundamentally different utilization of T-bet, TCF1, and FOXO1, transcription factors canonically associated with either effector or memory cell fates. TCF1 has become the transcription factor most closely associated with formation of less differentiated stem cell-like memory T (Tscm) cells.10–13 Unlike terminally exhausted T cells, TCF1hi Tscm cells have maximal self-renewing capacity, longevity, and therapeutic potential because of their ability to replenish the pool of TCF1lo cells in response to checkpoint blockade therapies.10,14,15 Production of T cells expressing high TCF1 is therefore recognized as a major goal of immunotherapeutic regimens. Similarly, FOXO1 is broadly dedicated toward enforcement of an undifferentiated state and suppression of pathways associated with active proliferation.16 In multiple cell types, FOXO1 suppresses gene expression associated with activation, proliferation, and effector differentiation,17,18–22 competing directly against the activities of factors such as mTor and cMyc and their consequent augmentation of aerobic glycolysis.23 FOXO1 has numerous direct effects on mitochondria, primarily for maintaining mitochondrial homeostasis.24 Conversely, T-bet is directly wired into glycolytic metabolism and terminal differentiation, and its elevated/sustained expression is necessary for transcriptional and metabolic commitment to short-lived effector cells.25,26 The predominance of either FOXO1 or T-bet is thought to be largely controlled by the inflammatory cues surrounding the T cells during their initial phases of clonal expansion.27 Signaling through inflammatory cytokine receptors, as well as ongoing TCR engagement, increases the activity of phosphatidylinositol 3-kinase (PI3K)/Akt activity, which phosphorylates FOXO1, driving its export from the nucleus.28 These same signals augment mTor activity and consequent aerobic glycolysis, which contributes many metabolic intermediates necessary to fuel clonal expansion.16 Along with costimulatory signals (e.g., CD28 and CD27) and cytokine signaling (IL-2 and IL-15), mTor increases T-bet expression19 and prolonged cMyc stability,29,30 which supports the enormous biomass demands of the proliferating effector T cell. In contrast, reduced inflammatory cues allow persistent FOXO1 expression and loss of cMyc activity, mediating exit from clonal expansion and formation of long-term memory. The influence of these molecular signals, along with asymmetric allocation of metabolic and transcription factors during cell division,31,32 bifurcates the response into cells dedicated to either effector or memory, properly aligning cell fate and function.
In contrast to these published data, we show here that a productive CD8+ T cell response to adjuvant administration requires the transcriptional influence of T-bet, TCF1, and FOXO1, with TCF1/FOXO1 facilitating a durable commitment to proliferation and T-bet necessary for maximal T cell memory. Our data identify unprecedented roles of these factors in CD8+ T cell expansion and memory formation and continue to illustrate that Tvac immunity utilizes a host of pathways, factors, and cytokines distinct from their use in T cells responding to infection.
RESULTS
A memory phenotype predominates in vaccine adjuvant-elicited CD8+ T cells
In response to infection with LM expressing ovalbumin (OVA) protein, the majority of Tinfs differentiate into KLRG1+CD127− short-lived effector cells (SLECs) while giving rise to a smaller subset of CD127+KLRG1− memory precursor effector cells (MPECs) (Figures 1A and 1B).33 In contrast, subunit immunization (vaccine) against OVA with either single34 or combined adjuvant (poly(I:C)/αCD40),35 results in a Tvac response predominated by MPECs, not SLECs (Figures 1A and 1B).5,36 In conjunction with CD127, the majority of Tvacs also expressed high levels of TCF1 (Figures 1C and 1D).37,38 TCF1, FOXO1, and CD127, necessary for generation and maintenance of memory T cells,17,39,40 were expressed significantly higher on a percell basis in Tvacs compared with Tinfs (Figures 1E–1J). Although some variation in the frequency of TCF1hi was observed, depending on the adjuvant utilized, this phenotype applied to Tvacs elicited by a broad range of single adjuvants, including poly(I:C), monophosphoryl lipid A (MPLA), Pam3Cys, CpG, and even alum (Figure S1). Using adoptive transfer of a small number of congenically marked, CellTrace Violet (CTV)-labeled OT1 T cells (T cell receptor [TCR] transgenic T cells that are OVA specific) into vaccinated or LM-OVA-challenged mice, we observed that high TCF1 expression was apparent in Tvacs even in the earliest cell divisions (Figures 1K and 1L). Interestingly, Tvacs divided synchronously relative to Tinfs (Figure 1K), with the cells dividing 1–3 times by 36 h (Figure 1K, red), 3–6 times by 48 h (Figure 1K, green), and 5–8 times by 60 h (Figure 1K, orange). Infection-elicited cells actually decreased their TCF1 expression even before they divided for the first time, resulting in reduced TCF1 expression compared with naive T cells (Figure 1L, TCF1 0 divisions). In contrast, Tvacs rapidly upregulated TCF1, maintaining that expression over all cell divisions identified in the first 60 h (Figure 1L). Surprisingly, Tbet expression was similarly elevated in Tvacs over Tinfs for the majority of divisions analyzed (Figure 1L), providing our first glimpse of Tvacs coordinating their expression of transcription factors with canonically opposed fate designations. As predicted from their phenotype on day 7, the differences in memory/effector-associated protein expression were even more apparent in Tvacs at late time points after vaccination/challenge, with representation of CD127+ KLRG− T cells increasing further relative to Tinfs on day 36 (Figure 1M). In line with their elevated CD127 expression, almost all Tvacs co-expressed TCF1 and CD127 (Figure 1O), again with significantly more TCF1 (Figures 1O and 1P), FOXO1 (Figures 1Q and 1R), and CD127 (Figures 1S and 1T) on a per-cell basis even compared with TCF/FOXO1/CD127hi Tinfs. Elevated expression of these markers is strongly associated with long-lived memory T cell survival, suggesting that T cells derived from subunit vaccination might better survive the “crash” in T cell numbers typically observed following the peak of a primary response, ultimately forming a more stable memory pool than that observed after LM challenge. Because the actual numbers of T cells generated can vary significantly between the two challenges, we normalized each challenge to its own peak and then determined the fold contraction in memory T cells from the peak of the response. This revealed a significantly higher number of memory (CD127+TCF1+) cells retained after vaccination compared with infection (Figure 1U). This increased retention was observed even when evaluating memory T cells more than 100 days post vaccination/challenge (Figure 1V), when vaccine-elicited T cells demonstrated elevated cytokine production (Figure 1W) and target killing (Figure 1X) relative to their infection-elicited counterparts. Collectively, we conclude that Tvacs adopt a memory T cell phenotype and fate as early as a single division after vaccination; this phenotype predominates into extended memory time points and corresponds to an increased propensity for survival and host-protective effector functions.
Figure 1. A memory phenotype predominates in vaccine adjuvant-elicited CD8+ T cells.

C57BL/6 mice were harvested 7 days after adjuvanted (poly(I:C)+anti-CD40) vaccine administration or L. monocytogenes (LM)-OVA challenge (LM). Please note that (A–J) are from day 7, and (M–T) are from day 36.
(A) Representative CD127 versus KLRG1 staining.
(B) Percentage of CD127+ cells.
(C) Representative CD127 versus TCF1 staining.
(D) Percentage of TCF1+ cells from (C).
(E–J) Representative histograms and geometric mean fluorescent intensity (gMFI) quantification for TCF1 (E and F), FOXO1 (G and H), and CD127 (I and J) staining. Dashed lines represent staining of naive (CD44lo) T cells.
(K and L) CellTrace Violet (CTV)-labeled CD45.1+ OT1s were transferred into WT C57BL/6 recipients, followed by vaccination or LM-ova challenge. At the indicated times, splenic donor OT1 cells were evaluated for TCF1 or T-bet expression within each cell division regardless of harvest time point (K). TCF1 and Tbet gMFI are normalized to an undivided naive (transfer only) control (L). Error bars represent SEM. Data shown are representative of 2 experiments.
(M and N) The spleens from C57BL/6 mice were harvested 36 days post vaccination or LM-OVA challenge as in (A). Shown is representative CD127 versus KLRG1
(M) and TCF1 versus CD127 (N) staining.
(O–T) Representative histograms and gMFI quantification for TCF1 (O and P), FOXO1 (Q and R), and CD127 (S and T).
(U) Fold change in memory T cells from the peak determined by dividing the number of CD127+TCF1+ memory T cells more than 40 days post vaccination/challenge by the total number of antigen-specific T cells at the peak of the response on day 7. Data from 5 separate experiments are shown.
(V–X) OT1 T cell isolated 110 days post vaccination or LM-OVA challenge and evaluated for total cell numbers (U), frequency of IL-2/interferon γ (IFNγ) double-producing T cells (W), and antigen-specific target killing (X) as described in STAR Methods.
All data shown are mean ± SEM; n ≥ 4 mice per group, representative of 2–5 independent experiments. Significance was defined by unpaired t test with Welch’s correction and two-way ANOVA, where *p < 0.05, **p < 0.01, ***p < 0.001.
Optimal Tvac frequency and phenotype require the memory fate-associated transcription factors TCF1 and FOXO1
Because the peak of the T cell response to infection is dominated by an effector response, the number of T cells in the primary response is minimally affected by the absence of TCF1.41 Given their memory phenotype, we hypothesized that TCF1 might have more influence on the primary Tvac response compared with Tinfs. To evaluate this, we utilized OT1 T cells with a conditional deletion of TCF1 (tcf7fl/fl CD4-Cre, TCF1 knockout [KO])42 in co-transfer experiments with congenically distinct wild-type (WT) OT1 cells. Naive transfer recipients were either vaccinated or challenged with LM-OVA, and 7 days later, WT and TCF1 KO OT1 T cell responses were measured for their magnitude and evaluated for phenotype. A small difference between total numbers of WT and TCF1 KO OT1 was observed after LM-OVA infection (Figures 2A–2C),37 but this failed to meet statistical significance. In contrast, there was a significant impact on the total number of Tvacs in TCF1 KO vs. WT OT1s 7 days after vaccination (Figures 2A–2C). This loss in Tvac numbers was accompanied by a reduction in the per-cell expression of CD127 in Tvacs but not in Tinfs (Figure 2D). TCF1 deficiency results in a reduced memory T cell pool,37 which we also observed for Tvacs and Tinfs (Figures 2E–2G), although a more substantial impact was observed for Tvacs (Figure 2G). This was again accompanied by reductions in the expression of CD127 (Figure 2H) and Eomes (Figure 2I). We conclude from these data that expression of the memory fate-associated transcription factor TCF1 plays an unexpected role in conferring maximal CD8+ T cell responses, primary and memory, to subunit vaccination compared with infectious challenge.
Figure 2. Optimal Tvac frequency and phenotype require the memory fate-associated transcription factor TCF1.

WT and TCF1 KO OT1 cells were co-transferred into WT C57BL/6 recipients that were then vaccinated or challenged with LM-OVA.
The phenotype of WT and KO cells 7 days (A–D) or 77 days (E–J) post vaccination/challenge was characterized by flow cytometry.
(A) Representative contour plots for CD127 versus TCF1 staining of donor WT and KO cells.
(B) The number of total WT and KO OT1 cells split into CD127+ and CD127− subsets.
(C) The ratio of the number of WT cells divided by the number of KO cells in (B).
(D) The gMFI of CD127 in WT and KO OT1 cells.
(E) Representative contour plots for CD127 versus TCF1 staining.
(F) The number of total WT and KO OT1 cells split into CD127+ and CD127− subsets.
(G) The ratio of the number of WT cells divided by the number of KO cells in (F).
(H–J) Representative histograms and gMFIs for CD127 (H), EOMES (I), and FOXO1 (J) in WT and KO OT1 cells after vaccine or LM-OVA challenge.
Data shown are mean ± SEM; n ≥ 4 mice per group, representative of two experiments. Significance was defined by unpaired t test with Welch’s correction and two-way ANOVA, where *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
We noted that TCF1-deficient T cells expressed reduced levels of FOXO1 (Figure 2J), a transcription factor important for development and maintenance of memory T cells and known for sustaining TCF1+ memory-like cells during chronic infection.40 We therefore evaluated its role in Tvac responses using foxo1fl/fl mice43 crossed to OT1 mice expressing the Cre-recombinase gene driven by the distal promoter of Lck (dLck-Cre).44 As before, WT and FOXO1 KO OT1 cells were co-transferred into congenically distinct recipient mice that were either vaccinated or challenged with LM-OVA. Two features of the response of FOXO1 KO cells to either LM challenge or vaccine are noteworthy. First, although Tinf FOXO1 KO T cells were almost exclusively an effector (CD127−, TCF1−) phenotype, more than half of the responding FOXO1 KO Tvacs continued to express TCF1 (Figures 3A, 3B, and S2A). Thus, while CD127 expression appears to be a proxy for FOXO1 expression in activated T cells more generally, activated Tvacs maintain some FOXO1-independent regulation of TCF1. Second, unlike Tinfs, in which FOXO1 deficiency has little impact on the total number of responding T cells, FOXO1 is required for maximal Tvac expansion, having a far greater impact on the frequency of responding T cells than TCF1 deficiency (Figures 3C and 3D). Indeed, there was a more than 25-fold reduction in the total number of FOXO1 KO Tvacs (Figures 3C and 3D) that included loss of CD127+ (Figure 3C, closed bars) and CD127− (Figure 3C, open bars) T cells relative to the WT. This suggests that FOXO1 is unexpectedly critical for sustaining proliferation and/or survival of clonally expanding Tvacs. In spite of these differences in the primary response of FOXO1 KO Tvacs and Tinfs, both failed to form stable memory pools in the absence of FOXO1 (Figure S2B).40 Although TCF1 expression in memory Tvacs was affected surprisingly little by the absence of FOXO1 (Figures S2C and S2D), this sustained TCF1 expression was insufficient to maintain CD127 expression (Figure S2E). Collectively, we conclude that FOXO1 unexpectedly performs critical functions in Tvacs for controlling the magnitude of the primary T cell expansion and a sustained memory T cell pool, neither of which can be compensated for by expression of TCF1.
Figure 3. Tvacs require FOXO1 for peak and memory responses.

WT and FOXO1 KO OT1 cells were co-transferred into WT C57BL/6 recipients that were then vaccinated or challenged with LM-OVA. On day 7 post challenge, the splenic WT and KO OT1 phenotype was analyzed by flow cytometry.
(A) Representative contour plots for CD127 versus TCF1 staining.
(B) Representative contour plots for TCF1 versus FOXO1 staining.
(C) The number of total WT and KO OT1 cells split into CD127+ and CD127− subsets.
(D) The number of WT cells divided by the number of KO cells normalized to input.
(E) The percentage of TCF1+ in WT and KO OT1 cells.
Data shown are mean ± SEM; n ≥ 4 mice per group, representative of two experiments. Significance was defined by unpaired t test with Welch’s correction and two-way ANOVA, where *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Tvac peak and memory responses are compromised in the absence of T-bet
Having defined the importance of memory-associated transcription factors after adjuvant administration, we sought to better clarify the role of T-bet (tbx21), a transcription factor we previously associated with the Tvac response.5 We crossed tbx21 fl/fl to congenically marked OT1 × dLck-Cre mice (T-bet KO) and performed co-adoptive transfer experiments with WT OT1s into vaccinated or LM-OVA-challenged recipients. In accordance with the antagonistic relationship between T-bet and TCF1,45–47 the frequency of TCF1+ cells was increased in Tvacs and Tinfs in the absence of T-bet (Figure 4A). As expected, there was an ablation of effector-fated (TCF1−CD127−) cells 7 days after either vaccination or LM-OVA infection (Figures 4A and 4B). While the number of memory-fated cells (CD127+) was not significantly affected in the absence of T-bet after LM-OVA challenge, it was greatly reduced after vaccination (Figures 4B and 4C). In line with their elevated TCF1, FOXO1 expression was also elevated in T-bet KO Tinfs (Figures 4D and 4E). Again counter to Tinfs, FOXO1 expression was actually reduced in T-bet KO Tvacs (Figures 4D and 4E).
Figure 4. Tvac peak and memory responses are compromised in the absence of T-bet.

WT and T-bet KO OT1 cells were co-transferred into WT C57BL/6 recipients that were then vaccinated or challenged with LM-OVA. On day 7 (A–E) or 50 (F–J) post challenge, the splenic WT and KO OT1 phenotype was analyzed by flow cytometry.
(A) Representative contour plots for CD127 versus TCF1 expression in WT and KO OT1 cells.
(B) The number of WT and KO OT1 cells split into CD127+ and CD127− subsets.
(C) The ratio of WT:KO CD127+ cells.
(D) Representative histograms for FOXO1 staining of WT and KO OT1 cells.
(E) FOXO1 gMFI of WT and KO OT1 cells.
(F) Representative contour plots for CD127 versus TCF1 expression in WT and KO cells on day 50 post vaccination and infection.
(G) The number of WT and KO OT1 cells split into CD127+ and CD127− subsets.
(H) The ratio of WT:KO OT1 cells.
(I) Representative histograms for FOXO1 staining of WT and KO OT1 cells.
(J) The gMFI of FOXO1 in splenic WT and KO OT1 cells.
Data shown are mean ± SEM; n ≥ 4 mice per group, representative of two experiments. Significance was defined by unpaired t test with Welch’s correction and two-way ANOVA, where *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Evaluating WT and T-bet KO OT1s 50 days after initial vaccination/infection (Figure 4F) revealed no loss of T-bet KO Tinfs relative to the WT (Figures 4G and 4H) but an ~20-fold reduction in the number of T-bet KO OT1s relative to the WT after vaccination (Figure 4H). Memory T-bet KO Tinfs continued to express higher FOXO1 than their WT counterparts, while again FOXO1 expression was significantly reduced in T-bet KO Tvacs compared with WT (Figures 4I and 4J). Therefore, unique to an adjuvant-elicited response, T-bet displays an unusual positive regulation of FOXO1 expression and is required for optimal CD8+ T cell memory. Similar to FOXO1, T-bet performs critical functions in Tvacs for controlling the magnitude of the primary response as well as sustained memory T cell response to subunit vaccination.
Inflammation redirects Tvacs into a FOXO1-independent effector cell fate
All proposed models of T cell fate determination include a dominant role of inflammation in guiding cell fate decisions between effector and memory.25,26,48–51 The infrequency of effector-phenotype T cells derived from adjuvant administration suggested a reduced inflammatory environment relative to that found after infectious challenges. Gene set enrichment analysis (GSEA) from Tvac and Tinf RNA sequencing (RNA-seq) data was consistent with this hypothesis. Other than the Wnt/β-catenin signaling pathway (predicted by the high TCF1 expression in Tvacs), numerous inflammatory pathways were greatly underrepresented in Tvacs compared with Tinfs (Figure 5A).
Figure 5. Inflammation re-directs Tvacs into a FOXO1-independent effector cell fate.

(A) GSEA of bulk RNA-seq from day 3 post vaccination compared with day 4 post LM-OVA infection. These represent the peak proliferative time points for each immunological challenge.
(B–F) WT OT1 cells were transferred into congenically distinct C57BL/6 recipients and vaccinated with or without CpG (ODN1826) supplementation. On day 7 post vaccination, the spleens were harvested, and the phenotype of OT1 cells was analyzed by flow cytometry. (B) Representative contour plots for CD127 versus TCF1 staining. (C) The number of total WT OT1 cells split into CD127+ and CD127− subsets. (D) The number of total WT OT1 cells split into TCF1+ and TCF1− subsets. (E) The gMFI for TCF1 in TCF1+ OT1 cells. (F) The gMFI for FOXO1 in OT1 cells.
(G–K) WT and T-bet KO or WT and FOXO1 KO OT1 cells were co-transferred into WT C57BL/6 recipients, followed by vaccination with or without CpG (ODN1826). Splenic WT and KO OT1 T cells were evaluated by flow cytometry 7 days post vaccination. (G) Representative contour plots for CD127 versus TCF1 staining for control, FOXO1 KO, and T-bet KO responses with or without CpG. (H) The ratio of WT:FOXO1 KO OT1s. (I) The number of CD127− FOXO1 KO OT1 cells 7 days post vaccination with or without CpG. (J) The ratio of WT:T-bet KO OT1s. (K) The number of CD127+ T-bet KO OT1 cells 7 days post vaccination with or without CpG.
Data shown are mean ± SEM; n ≥ 4 mice per group, representative of two experiments. Significance was defined by unpaired t test with Welch’s correction and ordinary one-way ANOVA, where *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Based on these data, we hypothesized that the T-bet/FOXO1 transcriptional dependencies unique to Tvacs were the inadvertent result of the limited durability of inflammation following adjuvant administration compared with the prolonged inflammatory environment following infection. The TLR9 agonist CpG is a non-specific inducer of inflammation, and its use facilitates loss of TCF1 expression and reversal of memory T cell commitment.48,49,52,53 Adding CpG to our combined adjuvant, the TCF1 expression pattern reflected the more bimodal expression of TCF1 as seen in Tinfs (Figure 5B). Closer evaluation revealed that CpG addition had little effect on the total number of CD127+ memory phenotype cells, increasing only the number of effector CD127−/TCF1− cells relative to the combined adjuvant alone (Figures 5C and 5D). The similarity of the +CpG Tvacs to Tinfs was further noted in their reduced TCF1 and FOXO1 expression (Figures 5E and 5F). The influence of CpG on the Tvac phenotype occurred regardless of the base adjuvant utilized (Figure S3), confirming this as relevant to adjuvant-elicited responses more broadly. Importantly, addition of CpG substantially rescued the primary FOXO1 KO Tvac response (Figure 5G), significantly normalizing the ratio of WT:FOXO1 KO OT1s (Figure 5H) through expansion of CD127− effector-like T cells (Figure 5I). In contrast, CpG further exaggerated the ratio of WT:Tbet KO T cells at the peak of the response (Figure 5J), somewhat expected given the selective effect of CpG on increasing Tbet-dependent CD127−/TCF1− effector T cells (Figures 5C and 5D). Importantly, however, there was no corresponding rescue of the number of T-bet KO memory T cells (Figure 5K). Thus, addition of CpG only releases Tvacs from the constraints associated with FOXO1 deficiency, facilitating expansion of FOXO1-independent effector T cells.
Adjuvant-elicited T cells express dual T-bet and FOXO1/TCF1 transcriptional programs
With effector and memory Tvacs compromised in the absence of either FOXO1 or T-bet, this suggested cooperative functions for these factors, not their opposition, as described previously.17,45,47 To better understand the transcriptional heterogeneity within the responding CD8+ T cells, we performed single-cell RNA-seq (scRNA-seq) on WT Tvac and Tinf OT1s isolated from the spleens of mice 7 days after either vaccination or LM challenge, respectively. Using Seurat to identify cells with closely related transcriptional profiles, 4 major gene expression clusters were identified (Figures 6A–6C and S4). Cluster 1 was predominantly adjuvant elicited, with a small but significant population derived from LM challenge (Figure 6C). In contrast, cluster 2 was derived almost exclusively from LM challenge (Figure 6C). The majority of cells in clusters 3 and 4 were LM derived but still contained significant numbers of adjuvant-elicited cells (Figure 6C). Using the expression of cell-cycle-associated genes as an indicator of proliferation, only clusters 3 and 4 were actively engaged in clonal expansion (Figures 6B and 6D). Given the phenotype of the T cells generated by either LM or vaccine at this time point, we anticipated that the post-proliferative clusters 1 and 2 would show classical transcriptional bearings of memory (cluster 1, Tvacs) and effector cells (cluster 2, Tinfs), respectively. Indeed, assessment of each cluster for expression of markers of effector (KLRG1 and Zeb2) or memory (CD127 and TCF1) differentiation was consistent with this prediction (Figures 6E and S4A). By assessing the gene features defining each cluster more broadly (Figure S4B), we observed that Tvac cluster 1 and Tinf cluster 2 aligned well with gene sets identified previously as transcriptionally dedicated to either memory or effector (Figure 6F), respectively. Use of single-sell regulatory network inference and clustering (SCENIC) to infer transcription factor activity based on gene expression profile further confirmed these fate designations in clusters 1 and 2 specific to T-bet, FOXO1, and TCF1 transcriptional activity. Cluster 1 genes showed higher FOXO1, strong TCF1, but low T-bet transcriptional activity, whereas genes in cluster 2 showed the inverse transcriptional activity relationship (Figures 6G and S5A). Thus, for terminally differentiated (non-proliferative) cells, these results confirmed our phenotypic observations for Tvacs and Tinfs at the levels of gene transcription and transcription factor activity.
Figure 6. Adjuvant-elicited T cells express dual effector and memory transcriptional programs.

(A) Uniform Manifold Approximation and Projection (UMAP) Dimensionality reduction mapping analysis of single-cell gene expression data of OT1 cells isolated 7 days post infection with LM-OVA (LM, purple) or adjuvanted OVA vaccine (Adj, black).
(B) UMAP as in (A), with cells color coded by clusters derived from shared nearest neighbor analysis.
(C) Proportion of either Adj- or LM-elicited T cells within clusters depicted in (B).
(D) UMAP as in (A), with cells color coded by cell cycle status inferred from transcriptional profiles.
(E) Violin plots depicting expression of select genes associated with either the effector (left) or memory (right) phenotype within clusters 1 and 2.
(F) Violin plots depicting the module score of gene sets associated with effector (left) or memory (right) within clusters 1 and 2.
(G) Violin plots of SCENIC-inferred transcription factor activity within clusters 1 and 2.
(H) Violin plots depicting expression of select genes associated with either the effector (Klrg1 and Zeb2) or memory (Il7r and Tcf7) phenotype of Adj- or LM-elicited cells within clusters 3 and 4.
(I) Violin plots depicting the module score of gene sets associated with effector (left) or memory (right) of Adj or LM cells within clusters 3 and 4.
(J) Violin plots of SCENIC-inferred transcription factor activity of Adj- or LM-elicited cells within clusters 3 and 4.
The association between these gene expression profiles and transcriptional programs altered substantially when we focused our analysis only on cells actively undergoing clonal expansion. After filtering out S/G2/M-related genes to eliminate the influence of cell proliferation on fate designation, cells derived from LM challenge (in either cluster 3 or 4) expressed canonical effector-associated genes but not memory-associated genes (Figure 6H). As with cluster 2, the overall gene expression profile associated with Tinf cells in clusters 3 and 4 was that of effector and not memory (Figure 6I). Although reduced relative to cluster 2, the transcription factor activity scores for LM-derived cells were consistent with their “effector score,” with high activity for T-bet, surprisingly detectable but lower for FOXO1, and absent for TCF1 (Figures 6J and S5B). In sharp contrast, adjuvant-derived cells in clusters 3 and 4 expressed the same markers of memory as their non-proliferating counterparts in cluster 1 (Figure 6H). Counterintuitively, their overall pattern of gene expression aligned more robustly with an effector transcriptional program (Figure 6I), although the Tvac memory score was slightly elevated relative to Tinfs. In confirmation of their simultaneous expression of memory markers and high “effector” transcriptional score, Tvacs in clusters 3 and 4 showed clear activity for T-bet, FOXO1, and TCF1 (Figures 6J and S5B). The coexistence of these transcription factor activities would be unexpected based on classic definitions of clonally expanding effector T cells but is precisely the transcription factor activity profile predicted by our analysis of FOXO1 KO, TCF1 KO, and T-bet KO Tvacs. That numerous other memory/stem-associated factors (Lef1, Eomes, Pou2F1/Oct1, Foxp1, and FoxP4) show SCENIC activity scores differentially skewed toward Tvacs is additionally noteworthy (Figures 6J and S5B); their activity is generally unexpected in actively proliferating T cells.
mRNA-lipid nanoparticle (LNP)-induced cellular responses are Tbet/Foxo1 dependent
Mechanistic principles, such as IL-27/15 dependency and a predominating memory phenotype, are shared by all adjuvants examined so far.5,6 To verify the combined FOXO1/Tbet dependency in T cells elicited from a clinically relevant vaccine formulation/platform, we utilized an mRNA-LNP encoding whole OVA (OVA-mRNA-LNP). Mice were again co-transferred with either WT:FOXO1 KO or WT:Tbet KO OT1s, followed by immunization with OVA-mRNA-LNPs. T cell responses in the spleen were harvested 10 days post immunization because initial experiments determined this to represent the peak of the response to ovamRNA-LNP (Figure S6). WT OT1 T cells again dominated the response to ova-mRNA-LNPs (Figures 7A and 7B), with WT:KO ratios similar to those observed in response to the combined adjuvant (Figure 7C). The data affirm the combined FOXO1/Tbet-dependent mechanistic principles we identified as applicable to adjuvant-elicited responses more broadly, including that of mRNA/LNP vaccine formulations.
Figure 7. CD8 responses to mRNA-LNP vaccination are also T-bet and FOXO1 dependent.

WT (CD45.1/1) and KO (CD45.1/2) OT1 cells were co-transferred into CD45.2/2 C57BL/6 recipients. One day later, the mice were vaccinated with OVA-encoding mRNA-LNPs as described in the STAR Methods. On day 10 post vaccination, the spleens were harvested, and the WT:KO ratio and phenotype of the responding OT1s were was analyzed by flow cytometry.
(A) Representative dot plots of spleen cells, gated only on the transferred T cells, in mice co-transferred with WT:FOXO KO (left) or WT:T-bet KO (right) cells. Numbers represent percentages of WT or KO T cells of total transferred T cells in each panel.
(B) Quantification of WT and KO T cell numbers from (A).
(C) Ratio of WT:KO T cells in each recipient.
Data shown are mean ± SEM; representative of two experiments with n = 8–10 mice per group. Significance was defined by paired t test, where *p < 0.05, **p < 0.01.
DISCUSSION
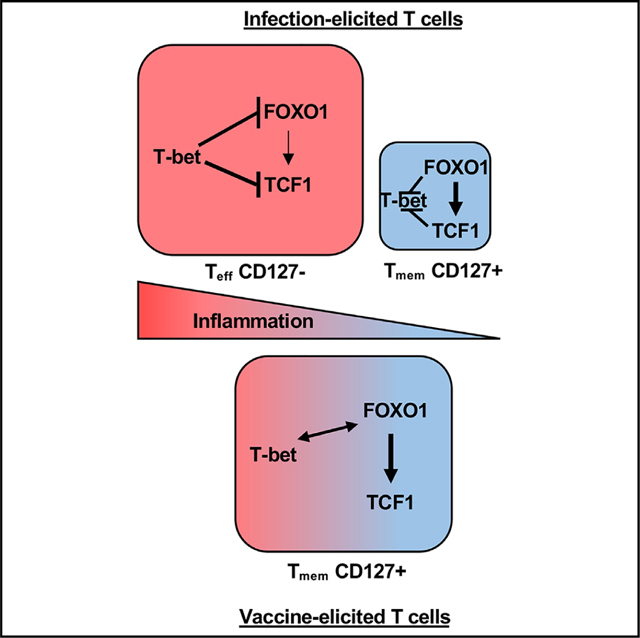
Increasing lines of evidence support the conclusion that immunological mechanisms relevant to adjuvant-elicited cellular immunity are distinct from those observed in response to infectious challenge. Our data here revealed an additional mechanistically unique feature of Tvacs: a cooperative interplay between T-bet and FOXO1/TCF1. In the current models of T cell fate determination, the conditions surrounding an infection ultimately contribute to formation of two distinct and non-overlapping cell types, effector- or memory-fated cells, dependent on either T-bet or FOXO1/TCF1, respectively. Our results with LM-OVA infection are consistent with this model. FOXO1 and TCF1 enact control over memory formation to LM challenge with little influence on effector generation (Figures 2 and 3), while T-bet deficiency yields the opposite phenotype, severely compromising the primary expansion of effector cells without long-term memory impact (Figure 4). Our scRNA transcriptional data also fully support this model, identifying a small number of LM-derived, non-proliferating memory T cells (cluster 1) and a larger number of proliferating (clusters 3 and 4) and non-proliferating (cluster 2) effector cells. Contrary to this paradigm, Tvacs require FOXO1/TCF1 and T-bet for optimal primary and memory T cell accumulation, indicating that Tvacs coordinate cooperative, not opposing, functions for T-bet and FOXO1/TCF1. Our scRNA-seq data again provided support for this model specific to adjuvant-elicited responses; non-proliferating Tvacs occupy essentially one population of canonical memory-fated T cells, while proliferating Tvacs display simultaneous transcriptional activity of T-bet, FOXO1, and TCF1, facilitating effector and memory transcriptional programs. This unusual integration of master regulators for effector and memory cell fates appears to be necessary to support clonal expansion in the absence of durable inflammation because, when added, it frees Tvac effector T cell generation from the regulatory control of FOXO1 (Figure 5).
The phenotype of CD8+ T cells after subunit vaccination is highly reminiscent of that observed from T cells responding to vaccination with peptide-pulsed dendritic cells (ppDCs).49,52 While these investigators evaluated the roles of different cytokines involved in the inflammatory response that reversed a memory fate, they did not investigate transcription factor dependencies of the T cells in the presence or absence of inflammation.52,54 Other studies have shown that ppDC-elicited T cells expressed high levels of TCF1 but again only mechanistically investigated the role of inflammatory cues in manipulating this TCF1hi phenotype.49 Our data extend these conclusions regarding the role of inflammation in effector cell generation in two important ways. First, previous studies made the assumption that effector and memory cell precursors generated under any condition (high or low inflammation) were transcriptionally identical to one another. Our data reveal that the response generated in the absence of inflammatory cues is unexpectedly co-dependent on FOXO1 and T-bet. Second, inflammatory cues free the expanding T cells from their FOXO1 dependency, allowing generation of the FOXO1-independent effectors traditionally observed in response to infectious challenge.
How Tvacs successfully integrate T-bet and FOXO1 transcriptional programs to support generation of primary and memory T cell pools has yet to be determined. In line with previous findings on the role of T-bet in suppressing CD127 and TCF1,25 Tvacs and Tinfs expressed higher levels of CD127 and TCF1 in the absence of T-bet. Interestingly, however, the absence of T-bet differentially affected FOXO1 expression in Tvacs and Tinfs. FOXO1 expression was increased in T-bet KO Tinfs, consistent with literature reports on their counter-regulation.17,18,55 In contrast, FOXO1 was actually reduced in T-bet KO Tvacs, suggesting that the link between T-bet expression and Tvac memory maintenance may be the result of a previously undetected positive regulatory loop, direct or indirect, between T-bet and FOXO1. More careful epigenetic analysis of these two factors are required, and specifically in the context of adjuvanted vaccination, to clarify the unexpected cooperative association between these two factors.
Similar to FOXO1, and perhaps for similar mechanistic reasons, TCF1 expression is also differentially regulated between Tinfs and Tvacs. Whereas naive T cells express TCF1 in a FOXO1-independent manner,18 expression of TCF1 in activated T cells generally relies on FOXO1 for its expression.18,39,56 In contrast, ~50% of FOXO1 KO Tvacs expressed TCF1 after subunit vaccination, indicating some means of FOXO1-independent regulation of TCF1 expression in activated T cells. That Wnt/b-catenin was the only “inflammatory” pathway over-represented in Tvacs vs. Tinfs in GSEA suggests that Tvacs, like naive T cells, somehow manage to utilize Wnt signaling mediators to regulate TCF1. However, this FOXO1-independent TCF1 expression was insufficient to compensate for the absence of FOXO1 in terms of memory formation, somewhat surprising given the similar transcriptional programs enacted by TCF1 and FOXO1.37,39,56 This again indicates the critical importance of FOXO1 as the primary memory-associ ated transcription factor in the Tvac response. This is supported by the minimal impact of TCF1-deficiency on the Tvac response (3- to 4-fold) compared with FOXO1 deficiency (20- to 30-fold). FOXO1 regulates genes like il7ra (encodes CD127) and sell (encodes CD62L), which promote Tinf memory formation and maintenance by increasing their survival and homing to lymphoid organs.57 While TCF1 KOs continued to express CD127 unabated, FOXO1 KO Tvacs did not and displayed early effector cell (CD127loKLRG1lo) and effector cell (CD127loKLRG1hi) phenotypes. Thus, despite the apparently central role of TCF1 in maintaining a more self-renewing potential in T cells responding to chronic infection or cancer, FOXO1 is clearly the more critical factor dictating long-lived, self-renewing memory in response to acute infection and adjuvanted subunit vaccines.
That said, TCF1 expression continues to be a reasonable marker for memory-fated T cells, and the fact that the TCF1hi/FOXO1hi phenotype for Tvacs is compromised through increasing degrees of inflammation likely reveals at least one reason why sustained CD8+ T cell immunity has been so difficult to achieve with existing adjuvants. Approved and experimental adjuvants were developed based on their capacity for generation of humoral immunity. Although formation of an “antigen depot” is not strictly required for adjuvant activity, a longer window of antigen availability does increase antibody maturation by prolonging the germinal center reaction.58,59 However, such depots dramatically compromise the cellular response,60,61 drawing in T cells after their expansion within secondary lymphoid tissues and driving their terminal differentiation and death within the smoldering, antigen-rich inflammatory environment of the injection site.62 The detrimental nature of precipitate/emulsion-based adjuvants on the cellular response has been known for nearly two decades, and yet the pursuit of adjuvants appropriate for immunotherapeutic application broadly fails to take these results into account and adapt adjuvant screens accordingly. The results we present here further emphasize the need to develop adjuvants with limited inflammatory durability because they run the risk of disrupting the unique capacity of Tvacs to cooperatively integrate memory (FOXO1) and effector (T-bet) transcriptional programs.
Limitations of the study
Our ability to fully extend these mechanistic conclusions to human immunology is obviously limited by our exclusive use of a mouse vaccine model system. Additionally, we intentionally chose to use the model antigen OVA because of its well-established ability to access antigen cross-presentation pathways. While we cannot extrapolate this degree of antigen presentation to virus/bacterium-specific antigens, our mechanistic conclusions are not unduly influenced by uncertainty in antigen presentation. There is no reason to suspect that T cells specific for other antigens, when engaged by said antigen, would not be subject to all of the mechanisms delineated in the present work. Indeed, previous assessments by us and others of the responses to such antigens are consistent with this prediction.62–64 Last, although we made every attempt to perform our mechanistic evaluation against a broad range of adjuvants, it is possible that ones we have not measured will not conform to the mechanisms we outlined here. Our use of the mRNA-LNP vaccine was performed in part to address this concern, and the fact that these mechanisms apply to a vaccine platform with such an obvious degree of clinical impact compels its continued investigation in ongoing adjuvant-related studies.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ross Kedl (Ross.Kedl@cuanschutz.edu).
Materials availability
This study did not generate unique reagents. All materials used are available upon request.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. RNAseq and single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Flow cytometeric data have been deposited at the Harvard Dataverse and are publicly available as of the date of publication. DOIs are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| αCD16/32 | hybridoma supernatent | 24G2 |
| anti-CD19 | Biolegend | clone 6D5 |
| anti-CD8α | Biolegend | clone 53-6.7 |
| anti-CD44 | Tonbo | clone IM-7 |
| anti-CD127 | Tonbo | clone A7R34 |
| anti-KLRG1 | Biolegend | clone 2F1/KLRG1 |
| anti-CD45.1 | Biolegend | Clone A20 |
| anti-CD45.2 | Biolegend | clone 104 |
| anti-CD122 | Biolegend | clone TM-b1 |
| anti-FOXO1 | Cell Signaling Technology | clone C29H4 |
| anti-Tbet | Biolegend | clone 4B10 |
| anti-TCF1 | Cell Signaling Technology | clone C63D9 |
| anti-Eomes | Biolegend | clone Dan11mag |
| anti-CD40 | BioXcell | clone FGK4.5 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| ova-expressing Listeria monocytogenes | Hao Shen | LM-OVA |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Kb-SIINFEKL tetramer | NIH Tetramer core facility | -PE or -APC conjugated |
| Fix/Perm solution | Tonbo | Cat# TNB1022L160 |
| Flow Cytometry Perm Buffer | Tonbo | Cat# TNB-1213-L150 |
| poly(I:C) | InvivoGen | cat# tlrl-pic |
| CpGODN 1826 | InvivoGen | Cat# tlrl-1826 |
| monophosphoryl lipid A (MPLA) | InvivoGen | cat# vac-mpla2 |
| Pam3Cys | InvivoGen | Cat# tlrl-pms |
| Alu-Gel-S | SERVA Electrophoresis GmbH | Cat# 12261.01 |
| Ovalbumin | Millipore-Sigma | Cat# A5503 |
| Neuro9 lipid mix | Precision Nanosystem | catalog#: NWS0001 |
|
| ||
| Deposited data | ||
|
| ||
| scRNA data | University of Colorado Cancer Center Genomics and Microarray Core Facility | GSE237416 |
| RNAseq data | University of Colorado Cancer Center Genomics and Microarray Core Facility | GSE237415 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson Laboratories | Strain #000664 |
| B6.SJL-Ptprca Pepcb/BoyJ mice | Jackson Laboratories | Strain #:002014 |
| OT1 (C57BL/6-Tg(TcraTcrb)1100Mjb/J) | Jackson Laboratories | Strain #:003831 |
| dLck-Cre (B6.Cg-Tg(Lck-icre)3779Nik/J) | Jackson Laboratories | Strain #012837 |
| Tcf7flox/flox × CD4-Cre | Tuoqi Wu | UT Southwestern |
| Foxo1flox/flox (Foxo1tmRdp) | Stephen Hedrick | UCSD |
| Tbx21flox/flox (B6.129-Tbx21tm2Srnr/J) | Christopher Hunter | U. Penn |
|
| ||
| Oligonucleotides | ||
|
| ||
| CleanCap AG capped mRNA, including N1-methylpseudouridine-5′-triphosphate substitutions, was produced by T7 RNA polymerase transcription from linearized plasmid templates and subsequently enzymatically polyadenylated with E. coli Poly(A) Polymerase | produced in-house, University of Colorado Anschutz Medical Campus | |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism | GraphPad | version 9.3.0 |
| FlowJo | BD Biosciences | V10.9 |
| Seurat package | Satija Lab, MIT | version 4.0.4 |
| Cell Ranger | 10x Genomics | version 2.1.0 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All experiments involving mice were conducted following protocols approved by the University of Colorado Institutional Animal Care and Use Committee (IACUC) according to guidelines provided by the Association for Assessment and Accreditation of Laboratory Animal Care. WT (C57BL/6J), congenic CD45.1 B6 (B6.SJL-Ptprca Pepcb/BoyJ), OVA-specific TCR-transgenic OT1 (C57BL/6-Tg(TcraTcrb) 1100Mjb/J) mice were originally obtained from the Jackson Laboratory and subsequently bred inhouse at the University of Colorado Anschutz Medical Campus. dLck-Cre (B6.Cg-Tg(Lck-icre)3779Nik/J) mice were provided by M. J. Bevan (originally from N. Killen).44 Tcf7flox/flox × CD4-Cre mice were kindly provided by Tuoqi Wu.65 Foxo1flox/flox (Foxo1tmRdp) and Tbx21flox/flox (B6.129-Tbx21tm2Srnr/J) mice were kindly provided by Stephen Hedrick (UCSD) and Christopher Hunter (U. Penn). Foxo1flox/flox and tbx21flox/flox were bred to dLck-Cre mice and OT1 mice. This study was carried out in accordance with protocols approved by The University of Colorado Anschutz Medical Campus Institutional Animal Care and Use Committee (IACUC), protocol #00172. Experiments were performed in 6 week-old or older male and female mice.
METHOD DETAILS
Immunization and infection
Mice were immunized via tail vein injection (i.v.) or intraperitoneal injection (i.p.) with the indicated innate receptor agonist(s), with or without αCD40 antibody (40 μg; clone FGK4.5, BioXcell), and detoxified whole chicken OVA protein (150 μg; Sigma). The following innate receptor agonist doses were used: poly(I:C) (40 μg; InvivoGen), CpG ODN 1826 (50–75 μg; InvivoGen), monophosphoryl lipid A (MPL; 40 μg; InvivoGen), Pam3Cys (40 μg; InvivoGen), or Alu-Gel-S (100 μL of 1.3% aluminum hydrochloride, SERVA Electrophoresis GmbH). Vaccines were made immediately prior to immunization with the exception of alum; OVA was emulsified in alum by gentle rocking at 4°C overnight. Unless otherwise specified, mice were immunized i.v. with combined-adjuvant subunit vaccine (αCD40, poly(I:C), and OVA). Mice were infected i.v. with 2 × 103 colony-forming units (CFU) of Listeria monocytogenes expressing whole OVA (LM-OVA).
mRNA-LNP formulation
The full-length DNA sequence of ovalbumin with optimized 5′ and 3′ UTRs and in-frame 3X FLAG tag was synthesized by Twist Bioscience in a pTwist Kan High Copy plasmid vector. CleanCap AG capped mRNA, including N1-methylpseudouridine-5′-triphosphate substitutions, was produced by T7 RNA polymerase transcription from linearized plasmid templates and subsequently enzymatically polyadenylated with E. coli Poly(A) Polymerase. Column-purified mRNA transcripts were incorporated into LNPs composed of Precision Nanosystem’s Neuro9 lipid mix (catalog#: NWS0001) on a microfluidic LNP mixer (e.g., Precision NanoSystems Spark) and dialyzed twice with PBS (pH 7.4 without Calcium and Magnesium, Corning #: 21–040-CV). Encapsulation efficiency and RNA content of LNPs was quantified using the ThermoFisher RiboGreen RNA Assay Kit (catalog# R32700) a dye-binding assay following detergent-based dissolution of the LNPs and LNPs at concentrations of 50–100 ng/μL were used for in vivo administration. Ova-LNPs containing 1–2μg of mRNA were injected IV into recipient mice.
Adoptive cell transfer
CD8+ T cells were magnetically purified from the spleens of indicated donor mice by negative selection using MojoSort Mouse CD8 T cell Isolation kit (Biolegend) to >95% purity. Where indicated, CD8+ T cells were CTV-labeled (Invitrogen) before transfer. OT1 cells were counted on a Vi-CELL automated cell counter (Beckman Counter) and indicated OT1 cell numbers (5 × 102 or 5 × 103) in 1 × PBS were transferred i.v. to recipient mice 1 day before or on the day of immunization or infection.
Cell isolation
Where indicated, donor OT1 cells that were present at low numbers post adoptive transfer were magnetically enriched with magnetic columns (LS Columns, Miltenyi Biotec). For early (≤72 h after immunization or infection) time points, spleens were removed and crushed between glass microscope slides into 6 well plate containing 2 mL Click’s Medium (FUJIFILM Irvine Scientific) and 30 μL of 5 mg/mL Collagenase D (Roche) and 100 μL of 2 mg/mL DNase I (Worthington Biochemical) per spleen. After a 15–20 min incubation at 37°C, 2 mL 0.1 M EDTA in 1 × PBS was added and cells were incubated at 37°C for additional 5 min. The cells were then washed with HBSS (Life Technologies) containing 5 mM EDTA and forced through 70 or 100 μm strainer. After wash, the cells were RBC lysed for 1 min in ACK buffer, then resusupended in complete RPMI and filtered through strainer. To enrich for CD45.1+ or CD45.2+ OT1, cells were resuspended in 500 μL RPMI and incubated with 10 μl of pulldown Ab (CD45.1 APC or CD45.2 APC) for 20 min at 4°C. After wash, cells were incubated in 500 μL RPMI with 5 μl anti-APC nanobeads (Biolegend) with rotation for 15 min at 4°C. After wash, cells were resuspended in 500 μl RPMI, filtered and purified through LS column (Miltenyi Biotec). For staining of spleenocytes at the later time points (>72 h after immunization or infection), whole organs were crashed through 70 or 100 μm strainers into complete RPMI to generate single cell suspensions. The cell suspensions were then RBC lysed in ACK buffer, washed in complete RPMI and counted on a Vi-CELL (Beckman Coulter) to determine total viable cell number. Blood was collected into tubes containing HBSS with 5 mM EDTA, RBC lysed in ACK buffer, washed with media and stained for flow cytometry.
Flow cytometry
Cells were incubated with αCD16/32 (clone 2.4G2; hybridoma supernatant) and plated on U-bottom 96-well plates at ≤ 3 × 106 cells/well in complete RPMI. Where indicated, cells were stained with Kb-SIINFEKL tetramer APC (NIH tetramer core) at 37°C for 30 min in the presence of αCD8α (53–6.7; Biolegend). For live-dead and surface staining, cells were washed with media, and stained in media for 20 min at RT with Fixable Viability Dye 780 (eBioscience) and surface antibodies for CD19 (6D5, Biolegend), CD8α (53–6.7, Biolegend), CD44 (IM-7; Tonbo), CD127 (A7R34, Tonbo), KLRG1 (2F1/KLRG1, Biolegend), CD45.1 (A20, Biolegend), CD45.2 (104, Biolegend), CD122 (TM-b1, Biolegend). After wash with RPMI, cells were fixed and permeabilized for 45 min at RT in Foxp3/Transcription Factor 1× Fix/Perm solution (Tonbo), followed by wash with 1× Flow Cytometry Perm Buffer (Tonbo) and intracellular staining for 45 min at RT with intracellular antibodies, including TCF1 (C63D9; Cell Signaling Technology), FOXO1 (C29H4, Cell Signaling Technology), T-bet (4B10, Biolegend), EOMES (Dan11mag; eBioscience). The cells were washed twice and resuspended in 1× Flow Cytometry Perm Buffer (Tonbo). Flow cytometry data was acquired on a four-laser (405, 488, 561, 638 nm) CytoFlex S (Beckman Coulter) and analyzed using FlowJo software (BD Biosciences).
In vivo target killing and cytokine production
To assess the target killing capacity of T cells from immunized or LM-ova challenged mice, an in vivo killing assay was performed as previously described.9 Briefly, splenocytes were peptide-pulsed with 1 μg/ml of OVA257–264 (target cells) or HSVgB498–505 (control cells), washed, then labeled with 0.05 mM or 0.5 mM CellTrace Violet (ThermoFisher) dye, respectively. Target and control splenocytes were mixed at a 1:1 ratio and transferred by tail vein injection into mice vaccinated or LM-ova challenged 110 days previously. Mice were euthanized after 2 h and the ratio of target:control was used to determine the percent killing compared to the same ratio observed for non-immunized control mice. Intracellular cytokine staining was performed after a 5-h stimulation with 1 mg/mL peptide of SIINFEKL for OVA immunizations and 3 mg/mL brefeldin A. After fixation and permeabilization, CD8 T cells were stained for intra-cellular IFNg (XMG1.2) and IL-2 (JES6–5H4) (BioLegend) and analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism (version 9.3.0, GraphPad) was used for all statistical analyses. Figure legends detail the number of experimental replicates and n-values. Unless noted, data presented are means ± SEM. Significance was defined using unpaired Student’s t-test with Welch’s correction or analysis of variance (ANOVA). Significance was denoted as follows: ns, not significant (p > 0.05) or significant with a p value less than 0.05.
Single-cell RNA sequencing analysis (scRNAseq)
scRNAseq sample preparation
A small number (500) of congenically marked (CD45.1) OT1 T cells were adoptively transferred into a naive B6 recipient (CD45.2). These recipients were then immunized intravenously (i.v.) against OVA using our combined adjuvant vaccine (poly I:C, anti-CD40) or infection with LM-OVA. T cells were isolated on day 7 post immunization and pooled from 5 mice each prior to cell sorting of CD45.1+ CD8+ cells. Cells from an individual condition (adjuvant or LM) were stained with an oligo-tagged anti-CD45 “hashtag” antibody (Biolegend). About 20,000 cells per were then loaded into Single Cell A chips (10x Genomics) and partitioned into Gel Bead In-Emulsions in a Chromium Controller (10x Genomics). Single-cell RNA libraries were prepared according to the 10x Genomics Chromium Single Cell 3′ Reagent Kits v2 User Guide and sequenced on a NovaSeq 6000 (Illumina).
scRNAseq mapping
Reads from scRNA-seq were aligned to mm10 or hashtag oligo sequences and collapsed into unique molecular identifier (UMI) counts using the 10x Genomics Cell Ranger software (version 2.1.0). The sample had appropriate numbers of genes detected (>1000), a high percentage of reads mapped to the genome (>70%), and a sufficient number of cells detected (>1000).
scRNA-seq dataset statistical analysis
The Seurat package (version 4.3.0) in R (version 4.0.4) was used for processing, analysis and visualization of the scRNAseq dataset. UMIs with gene counts less than 250, mitochondrial gene content of greater than 15% or expression of multiple hashtag sequences (doublets) were filtered out. Hashtag sequence counts were first normalized using the Seurat function NormalizeData and then demultiplexed using the Seurat function HTODemux which uses K-means clustering. Five mice per group were used for the single cell experiment. Cell read output from Cell Ranger was loaded into Seurat and then Adjuvant or LM-elicited cells were demultiplexed using the level of hashtag sequence. The scRNA-seq dataset was further filtered on the basis of gene numbers, mitochondria gene counts to total counts ratio, and expression of single hashtag sequences. Gene count data was then normalized using Seurat’s NormalizeData function. Top variable genes, principal components analysis (PCA), and uniform manifold approximation and projection for dimension reduction (UMAP) were calculated by the functions: FindVariableGenes, RunPCA, and RunUMAP. Only the top 2000 genes were considered in the PCA calculation and only the top 20 PCs were used in UMAP. Shared nearest-neighbor (SNN) identification and clustering were performed using the functions FindNeighbors and FindClusters using the top 20 PCs with resolution set to 0.3 and k set to 30. Memory and Effector scores were calculated using the AddModuleScore function with the gene sets GSE9650_EFFECTOR_VS._MEMORY_CD8_T cell_UP and GSE9650_EFFECTOR_VS._MEMORY_CD8_TCEL.
L_DN from the C7 immunologic signature collection of the molecular signature database
For transcription factor activity analysis the filtered count matrix from Seurat was exported for use with the SCENIC package (version 1.2.4) and pipeline in R. SCENIC was run with default parameters using the mm9–500bp-upstream-7species.mc9nr and mm9-tss-centered-10kb-7species.mc9nr databases for identification of transcription factor binding motifs. Using the Nextflow workflow from VSN-Pipelines (version 0.27.0) this process was iterated 10 times with with default threshold parameters for filtering genes and regulons. The resulting matrix of inferred transcription factor activity in each cell was then imported into Seurat for visualization and analysis within Seurat-calculated clusters.
Supplementary Material
Highlights.
A memory phenotype predominates in vaccine adjuvant-elicited CD8+ T cells
Adjuvant-elicited CD8+ T cells require both T-bet and FOXO1 transcription factors
Consequently, these T cells express dual effector and memory transcriptional programs
CD8 responses to mRNA-lipid nanoparticle vaccines also require T-bet and FOXO1
ACKNOWLEDGMENTS
We would like to specifically thank Stephen Hedrick for so freely sharing his mice (FOXO1fl/fl) and his advice, both of which were instrumental in the success of this project. We also thank Benjamin Willett, Cody Rester, Miguel Guerrero, John Manalastas, and Tonya Brunetti for technical assistance. This work was supported by NIH AI156456 and AI066121 (awarded to R.M.K.), AI148919 (awarded to R.M.K. and J.K.), and NIH U01 AI126899 (awarded to R.M.K. and C.A.H.). D.L.I. is a 2T32AI074491-11 postdoctoral fellowship recipient.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112911.
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Hansen SG, Piatak M Jr., Ventura AB, Hughes CM, Gilbride RM, Ford JC, Oswald K, Shoemaker R, Li Y, Lewis MS, et al. (2013). Immune clearance of highly pathogenic SIV infection. Nature 502, 100–104. 10.1038/nature12519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cox RJ, Brokstad KA, and Ogra P. (2004). Influenza virus: immunity and vaccination strategies. Comparison of the immune response to inactivated and live, attenuated influenza vaccines. Scand. J. Immunol. 59, 1–15. 10.1111/j.0300-9475.2004.01382.x. [DOI] [PubMed] [Google Scholar]
- 3.Hobson P, Barnfield C, Barnes A, and Klavinskis LS (2003). Mucosal immunization with DNA vaccines. Methods 31, 217–224. 10.1016/s1046-2023(03)00139-7. [DOI] [PubMed] [Google Scholar]
- 4.Polo JM, and Dubensky TW Jr. (2002). Virus-based vectors for human vaccine applications. Drug Discov. Today 7, 719–727. 10.1016/s1359-6446(02)02324-3. [DOI] [PubMed] [Google Scholar]
- 5.Klarquist J, Chitrakar A, Pennock ND, Kilgore AM, Blain T, Zheng C, Danhorn T, Walton K, Jiang L, Sun J, et al. (2018). Clonal expansion of vaccine-elicited T cells is independent of aerobic glycolysis. Sci. Immunol. 3, eaas9822. 10.1126/sciimmunol.aas9822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pennock ND, Gapin L, and Kedl RM (2014). IL-27 is required for shaping the magnitude, affinity distribution, and memory of T cells responding to subunit immunization. Proc. Natl. Acad. Sci. USA 111, 16472–16477. 10.1073/pnas.1407393111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hunter CA (2005). New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat. Rev. Immunol. 5, 521–531. 10.1038/nri1648. [DOI] [PubMed] [Google Scholar]
- 8.Kastelein RA, Hunter CA, and Cua DJ (2007). Discovery and biology of IL-23 and IL-27: related but functionally distinct regulators of inflammation. Annu. Rev. Immunol. 25, 221–242. [DOI] [PubMed] [Google Scholar]
- 9.Klarquist J, Cross EW, Thompson SB, Willett B, Aldridge DL, Caffrey-Carr AK, Xu Z, Hunter CA, Getahun A, and Kedl RM (2021). B cells promote CD8 T cell primary and memory responses to subunit vaccines. Cell Rep. 36, 109591. 10.1016/j.celrep.2021.109591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. 10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- 11.Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, Anderson SM, Handon R, Kenyon E, Elkahloun A, et al. (2016). The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 1, eaai8593. 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wieland D, Kemming J, Schuch A, Emmerich F, Knolle P, Neumann-Haefelin C, Held W, Zehn D, Hofmann M, and Thimme R. (2017). TCF1(+) hepatitis C virus-specific CD8(+) T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun. 8, 15050. 10.1038/ncomms15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kratchmarov R, Magun AM, and Reiner SL (2018). TCF1 expression marks self-renewing human CD8(+) T cells. Blood Adv. 2, 1685–1690. 10.1182/bloodadvances.2018016279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jadhav RR, Im SJ, Hu B, Hashimoto M, Li P, Lin JX, Leonard WJ, Greenleaf WJ, Ahmed R, and Goronzy JJ (2019). Epigenetic signature of PD-1+ TCF1+ CD8 T cells that act as resource cells during chronic viral infection and respond to PD-1 blockade. Proc. Natl. Acad. Sci. USA 116, 14113–14118. 10.1073/pnas.1903520116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siddiqui I, Schaeuble K, Chennupati V, Fuertes Marraco SA, Calderon-Copete S, Pais Ferreira D, Carmona SJ, Scarpellino L, Gfeller D, Pradervand S, et al. (2019). Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 50, 195–211.e10. 10.1016/j.immuni.2018.12.021. [DOI] [PubMed] [Google Scholar]
- 16.Zeng H, and Chi H. (2017). mTOR signaling in the differentiation and function of regulatory and effector T cells. Curr. Opin. Immunol. 46, 103–111. 10.1016/j.coi.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao RR, Li Q, Gubbels Bupp MR, and Shrikant PA (2012). Transcription factor Foxo1 represses T-bet-mediated effector functions and promotes memory CD8(+) T cell differentiation. Immunity 36, 374–387. 10.1016/j.immuni.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delpoux A, Lai CY, Hedrick SM, and Doedens AL (2017). FOXO1 opposition of CD8(+) T cell effector programming confers early memory properties and phenotypic diversity. Proc. Natl. Acad. Sci. USA 114, E8865-e8874. 10.1073/pnas.1618916114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao RR, Li Q, Odunsi K, and Shrikant PA (2010). The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32, 67–78. 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delpoux A, Michelini RH, Verma S, Lai CY, Omilusik KD, Utzschneider DT, Redwood AJ, Goldrath AW, Benedict CA, and Hedrick SM (2018). Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8(+) T cells. J. Exp. Med. 215, 575–594. 10.1084/jem.20170697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tejera MM, Kim EH, Sullivan JA, Plisch EH, and Suresh M. (2013). FoxO1 controls effector-to-memory transition and maintenance of functional CD8 T cell memory. J. Immunol. 191, 187–199. 10.4049/jimmunol.1300331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R, Zimmermann B, Aspalter IM, Franco CA, Boettger T, et al. (2016). FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature 529, 216–220. 10.1038/nature16498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saravia J, Raynor JL, Chapman NM, Lim SA, and Chi H. (2020). Signaling networks in immunometabolism. Cell Res. 30, 328–342. 10.1038/s41422-020-0301-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cheng Z. (2022). FoxO transcription factors in mitochondrial homeostasis. Biochem. J. 479, 525–536. 10.1042/BCJ20210777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, and Kaech SM (2007). Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295. 10.1016/j.immuni.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaech SM, and Cui W. (2012). Transcriptional control of effector and memory CD8+ T cell differentiation. Nat. Rev. Immunol. 12, 749–761. 10.1038/nri3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hedrick SM, Hess Michelini R, Doedens AL, Goldrath AW, and Stone EL (2012). FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol. 12, 649–661. 10.1038/nri3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benchoula K, Arya A, Parhar IS, and Hwa WE (2021). FoxO1 signaling as a therapeutic target for type 2 diabetes and obesity. Eur. J. Pharmacol. 891, 173758. 10.1016/j.ejphar.2020.173758. [DOI] [PubMed] [Google Scholar]
- 29.Marchingo JM, Sinclair LV, Howden AJ, and Cantrell DA (2020). Quantitative analysis of how Myc controls T cell proteomes and metabolic pathways during T cell activation. Elife 9, e53725. 10.7554/eLife.53725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, MacDonald HR, Cowling VH, and Cantrell DA (2015). Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 34, 2008–2024. 10.15252/embj.201490252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin WHW, Adams WC, Nish SA, Chen YH, Yen B, Rothman NJ, Kratchmarov R, Okada T, Klein U, and Reiner SL (2015). Asymmetric PI3K Signaling Driving Developmental and Regenerative Cell Fate Bifurcation. Cell Rep. 13, 2203–2218. 10.1016/j.celrep.2015.10.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang JT, Ciocca ML, Kinjyo I, Palanivel VR, McClurkin CE, Dejong CS, Mooney EC, Kim JS, Steinel NC, Oliaro J, et al. (2011). Asymmetric proteasome segregation as a mechanism for unequal partitioning of the transcription factor T-bet during T lymphocyte division. Immunity 34, 492–504. 10.1016/j.immuni.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kaech SM, and Wherry EJ (2007). Heterogeneity and cell-fate decisions in effector and memory CD8+ T cell differentiation during viral infection. Immunity 27, 393–405. 10.1016/j.immuni.2007.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kilgore AM, Pennock ND, and Kedl RM (2020). cDC1 IL-27p28 Production Predicts Vaccine-Elicited CD8(+) T Cell Memory and Protective Immunity. J. Immunol. 204, 510–517. 10.4049/jimmunol.1901357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, Vasilakos JP, Noelle RJ, and Kedl RM (2004). Combined TLR and CD40 triggering induces potent CD8+ T cell expansion with variable dependence on type I IFN. J. Exp. Med. 199, 775–784. 10.1084/jem.20031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edwards LE, Haluszczak C, and Kedl RM (2013). Phenotype and function of protective, CD4-independent CD8 T cell memory. Immunol. Res. 55, 135–145. 10.1007/s12026-012-8356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, and Xue HH (2010). Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–240. 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tiemessen MM, Baert MRM, Kok L, van Eggermond MCJA, van den Elsen PJ, Arens R, and Staal FJT (2014). T Cell factor 1 represses CD8+ effector T cell formation and function. J. Immunol. 193, 5480–5487. 10.4049/jimmunol.1303417. [DOI] [PubMed] [Google Scholar]
- 39.Hess Michelini R, Doedens AL, Goldrath AW, and Hedrick SM (2013). Differentiation of CD8 memory T cells depends on Foxo1. J. Exp. Med. 210, 1189–1200. 10.1084/jem.20130392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Utzschneider DT, Delpoux A, Wieland D, Huang X, Lai CY, Hofmann M, Thimme R, and Hedrick SM (2018). Active Maintenance of T Cell Memory in Acute and Chronic Viral Infection Depends on Continuous Expression of FOXO1. Cell Rep. 22, 3454–3467. 10.1016/j.celrep.2018.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jeannet G, Boudousquie C, Gardiol N, Kang J, Huelsken J, and Held W. (2010). Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA 107, 9777–9782. 10.1073/pnas.0914127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steinke FC, Yu S, Zhou X, He B, Yang W, Zhou B, Kawamoto H, Zhu J, Tan K, and Xue HH (2014). TCF-1 and LEF-1 act upstream of Th-POK to promote the CD4(+) T cell fate and interact with Runx3 to silence Cd4 in CD8(+) T cells. Nat. Immunol. 15, 646–656. 10.1038/ni.2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, et al. (2007). FoxOs are lineage-restricted redundant tumor suppressors and regulate endothelial cell homeostasis. Cell 128, 309–323. 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang DJ, Wang Q, Wei J, Baimukanova G, Buchholz F, Stewart AF, Mao X, and Killeen N. (2005). Selective expression of the Cre recombinase in late-stage thymocytes using the distal promoter of the Lck gene. J. Immunol. 174, 6725–6731. 10.4049/jimmunol.174.11.6725. [DOI] [PubMed] [Google Scholar]
- 45.Dominguez CX, Amezquita RA, Guan T, Marshall HD, Joshi NS, Kleinstein SH, and Kaech SM (2015). The transcription factors ZEB2 and T-bet cooperate to program cytotoxic T cell terminal differentiation in response to LCMV viral infection. J. Exp. Med. 212, 2041–2056. 10.1084/jem.20150186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, Casella V, Ngiow SF, Khan O, Huang YJ, et al. (2020). Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 52, 825–841.e8. 10.1016/j.immuni.2020.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oestreich KJ, Huang AC, and Weinmann AS (2011). The lineage-defining factors T-bet and Bcl-6 collaborate to regulate Th1 gene expression patterns. J. Exp. Med. 208, 1001–1013. 10.1084/jem.20102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, and Rao A. (2010). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90. 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Danilo M, Chennupati V, Silva JG, Siegert S, and Held W. (2018). Suppression of Tcf1 by Inflammatory Cytokines Facilitates Effector CD8 T Cell Differentiation. Cell Rep. 22, 2107–2117. 10.1016/j.celrep.2018.01.072. [DOI] [PubMed] [Google Scholar]
- 50.Wilson DC, Matthews S, and Yap GS (2008). IL-12 signaling drives CD8+ T cell IFN-gamma production and differentiation of KLRG1+ effector subpopulations during Toxoplasma gondii Infection. J. Immunol. 180, 5935–5945. 10.4049/jimmunol.180.9.5935. [DOI] [PubMed] [Google Scholar]
- 51.Agarwal P, Raghavan A, Nandiwada SL, Curtsinger JM, Bohjanen PR, Mueller DL, and Mescher MF (2009). Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 183, 1695–1704. 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Badovinac VP, Messingham KAN, Jabbari A, Haring JS, and Harty JT (2005). Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat. Med. 11, 748–756. 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- 53.Pham NLL, Badovinac VP, and Harty JT (2009). A default pathway of memory CD8 T cell differentiation after dendritic cell immunization is deflected by encounter with inflammatory cytokines during antigen-driven proliferation. J. Immunol. 183, 2337–2348. 10.4049/jimmunol.0901203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pham NLL, Badovinac VP, and Harty JT (2011). Differential role of “Signal 3” inflammatory cytokines in regulating CD8 T cell expansion and differentiation in vivo. Front. Immunol. 2, 4. 10.3389/fimmu.2011.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng Y, Kerdiles Y, Chu J, Yuan S, Wang Y, Chen X, Mao H, Zhang L, Zhang J, Hughes T, et al. (2015). Transcription factor Foxo1 is a negative regulator of natural killer cell maturation and function. Immunity 42, 457–470. 10.1016/j.immuni.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim MV, Ouyang W, Liao W, Zhang MQ, and Li MO (2013). The transcription factor Foxo1 controls central-memory CD8+ T cell responses to infection. Immunity 39, 286–297. 10.1016/j.immuni.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hills LB, Abdullah L, Lust HE, Degefu H, and Huang YH (2021). Foxo1 Serine 209 Is a Critical Regulatory Site of CD8 T Cell Differentiation and Survival. J. Immunol. 206, 89–100. 10.4049/jimmunol.2000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, et al. (2016). Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc. Natl. Acad. Sci. USA 113, E6639-e6648. 10.1073/pnas.1606050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee JH, Sutton HJ, Cottrell CA, Phung I, Ozorowski G, Sewall LM, Nedellec R, Nakao C, Silva M, Richey ST, et al. (2022). Long-primed germinal centres with enduring affinity maturation and clonal migration. Nature 609, 998–1004. 10.1038/s41586-022-05216-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burchill MA, Tamburini BA, Pennock ND, White JT, Kurche JS, and Kedl RM (2013). T cell vaccinology: exploring the known unknowns. Vaccine 31, 297–305. 10.1016/j.vaccine.2012.10.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Celis E. (2007). Toll-like receptor ligands energize peptide vaccines through multiple paths. Cancer Res. 67, 7945–7947. 10.1158/0008-5472.Can-07-1652. [DOI] [PubMed] [Google Scholar]
- 62.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang XF, Dorta-Estremera SM, Greeley NR, Nitti G, Peng W, et al. (2013). Persistent antigen at vaccination sites induces tumor-specific CD8⁺ T cell sequestration, dysfunction and deletion. Nat. Med. 19, 465–472. 10.1038/nm.3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Davenport BJ, Morrison TE, Kedl RM, and Klarquist J. (2021). Conserved and Novel Mouse CD8 T Cell Epitopes within SARS-CoV-2 Spike Receptor Binding Domain Protein Identified following Subunit Vaccination. J. Immunol. 206, 2503–2507. 10.4049/jimmunol.2100195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, Franci C, Cheung TK, Fritsche J, Weinschenk T, et al. (2014). Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576. 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 65.Wu T, Shin HM, Moseman EA, Ji Y, Huang B, Harly C, Sen JM, Berg LJ, Gattinoni L, McGavern DB, and Schwartzberg PL (2015). TCF1 Is Required for the T Follicular Helper Cell Response to Viral Infection. Cell Rep. 12, 2099–2110. 10.1016/j.celrep.2015.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. RNAseq and single-cell RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Flow cytometeric data have been deposited at the Harvard Dataverse and are publicly available as of the date of publication. DOIs are listed in the key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| αCD16/32 | hybridoma supernatent | 24G2 |
| anti-CD19 | Biolegend | clone 6D5 |
| anti-CD8α | Biolegend | clone 53-6.7 |
| anti-CD44 | Tonbo | clone IM-7 |
| anti-CD127 | Tonbo | clone A7R34 |
| anti-KLRG1 | Biolegend | clone 2F1/KLRG1 |
| anti-CD45.1 | Biolegend | Clone A20 |
| anti-CD45.2 | Biolegend | clone 104 |
| anti-CD122 | Biolegend | clone TM-b1 |
| anti-FOXO1 | Cell Signaling Technology | clone C29H4 |
| anti-Tbet | Biolegend | clone 4B10 |
| anti-TCF1 | Cell Signaling Technology | clone C63D9 |
| anti-Eomes | Biolegend | clone Dan11mag |
| anti-CD40 | BioXcell | clone FGK4.5 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| ova-expressing Listeria monocytogenes | Hao Shen | LM-OVA |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Kb-SIINFEKL tetramer | NIH Tetramer core facility | -PE or -APC conjugated |
| Fix/Perm solution | Tonbo | Cat# TNB1022L160 |
| Flow Cytometry Perm Buffer | Tonbo | Cat# TNB-1213-L150 |
| poly(I:C) | InvivoGen | cat# tlrl-pic |
| CpGODN 1826 | InvivoGen | Cat# tlrl-1826 |
| monophosphoryl lipid A (MPLA) | InvivoGen | cat# vac-mpla2 |
| Pam3Cys | InvivoGen | Cat# tlrl-pms |
| Alu-Gel-S | SERVA Electrophoresis GmbH | Cat# 12261.01 |
| Ovalbumin | Millipore-Sigma | Cat# A5503 |
| Neuro9 lipid mix | Precision Nanosystem | catalog#: NWS0001 |
|
| ||
| Deposited data | ||
|
| ||
| scRNA data | University of Colorado Cancer Center Genomics and Microarray Core Facility | GSE237416 |
| RNAseq data | University of Colorado Cancer Center Genomics and Microarray Core Facility | GSE237415 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J mice | Jackson Laboratories | Strain #000664 |
| B6.SJL-Ptprca Pepcb/BoyJ mice | Jackson Laboratories | Strain #:002014 |
| OT1 (C57BL/6-Tg(TcraTcrb)1100Mjb/J) | Jackson Laboratories | Strain #:003831 |
| dLck-Cre (B6.Cg-Tg(Lck-icre)3779Nik/J) | Jackson Laboratories | Strain #012837 |
| Tcf7flox/flox × CD4-Cre | Tuoqi Wu | UT Southwestern |
| Foxo1flox/flox (Foxo1tmRdp) | Stephen Hedrick | UCSD |
| Tbx21flox/flox (B6.129-Tbx21tm2Srnr/J) | Christopher Hunter | U. Penn |
|
| ||
| Oligonucleotides | ||
|
| ||
| CleanCap AG capped mRNA, including N1-methylpseudouridine-5′-triphosphate substitutions, was produced by T7 RNA polymerase transcription from linearized plasmid templates and subsequently enzymatically polyadenylated with E. coli Poly(A) Polymerase | produced in-house, University of Colorado Anschutz Medical Campus | |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism | GraphPad | version 9.3.0 |
| FlowJo | BD Biosciences | V10.9 |
| Seurat package | Satija Lab, MIT | version 4.0.4 |
| Cell Ranger | 10x Genomics | version 2.1.0 |