

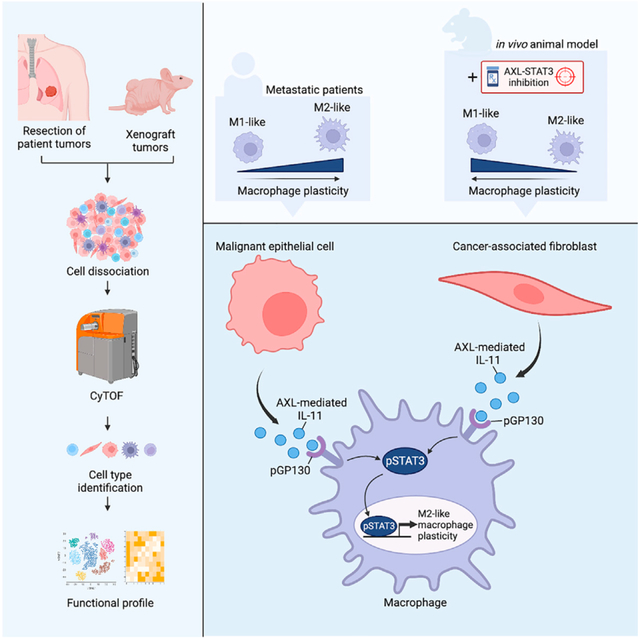
SUMMARY
Tumor-associated macrophages (TAMs) are integral to the development of complex tumor microenvironments (TMEs) and can execute disparate cellular programs in response to extracellular cues. However, upstream signaling processes underpinning this phenotypic plasticity remain to be elucidated. Here, we report that concordant AXL-STAT3 signaling in TAMs is triggered by lung cancer cells or cancer-associated fibroblasts in the cytokine milieu. This paracrine action drives TAM differentiation toward a tumor-promoting “M2-like” phenotype with upregulation of CD163 and putative mesenchymal markers, contributing to TAM heterogeneity and diverse cellular functions. One of the upregulated markers, CD44, mediated by AXL-IL-11-pSTAT3 signaling cascade, enhances macrophage ability to interact with endothelial cells and facilitate formation of primitive vascular networks. We also found that AXL-STAT3 inhibition can impede the recruitment of TAMs in a xenograft mouse model, thereby suppressing tumor growth. These findings suggest the potential application of AXL-STAT3-related markers to quantitatively assess metastatic potential and inform therapeutic strategies in lung cancer.
In brief
Hung et al. discuss that advanced lung adenocarcinoma (LAC) tumors harbor M2-polarized TAMs with concordant AXL-STAT3 signaling. LAC cells and fibroblasts secrete IL-11 in an AXL-dependent manner. Thus, IL-11 binds to GP130 complex and triggers pSTAT3 activation in M2-polarized macrophages. AXL-STAT3 targeting disrupts intercellular communication and suppresses tumor growth.
Graphical Abstract
INTRODUCTION
The tumor microenvironment (TME) is a highly diverse cellular network, embedding tumor cells, immune cells, cancer-associated fibroblasts, and newly developed blood vessels.1,2 Tumor-associated macrophages (TAMs), the major immune cell type infiltrating the TME, communicate with tumor cells, stromal fibroblasts, and other immune cells through the coordinated delivery of cytokines and other soluble factors.3,4 TAMs exhibit functional diversity and can participate in immune suppression, angiogenesis, tissue repair, and extracellular matrix (ECM) remodeling.5,6 In this way, TAMs maneuver an immunoevasive microenvironment that supports tumor cell proliferation and dissemination.7 Thus, targeting TAM heterogeneity and disrupting the intercellular communication between TAMs and supportive host cells in the TME represents a promising therapeutic strategy.
TAMs exhibit considerable transcriptomic diversity and phenotypic plasticity, contingent upon different microenvironmental stimuli.8,9 Macrophage polarization is a process whereby macrophages phenotypically mount a specific phenotype and functional response to surrounding lung TMEs.10,11 Macrophages undergo polarization toward an M1-like phenotype, systematizing Th1 responses and tumoricidal activities in nascent tumors.12 Conversely, tumor-promoting macrophages can become differentiated toward an M2-like phenotype that promotes tumorigenesis.13,14 TAM polarization is governed by a highly complex set of regulatory networks and coordinated signaling by T helper (Th) 1- or Th2-related cytokines, anti-inflammatory cytokine interleukin (IL)-10, transforming growth factor β (TGF-β), and toll-like receptor agonists.15,16 TAM reprogramming often involves signaling pathways frequently found in tumor cells, such as JNK-, PI3K/AKT-, Notch-, TGF-β-, nuclear factor κB (NF-κB)-, and hypoxia-dependent signaling pathways.7,17–21 This suggests that TAMs respond to microenvironmental cues by rewiring their signaling networks to mimic cancer cells in the vicinity. The intersection of these differential oncogenic pathways may also promote the diversification of TAMs and their corresponding roles in promoting tumor progression, angiogenesis, immune escape, and dissemination.
We previously reported a feedback loop between AXL and Janus kinase (JAK) 1/pSTAT3 (here referred to as STAT3) signaling in lung tumor cells.22 This coordinated signaling network reinforces the maintenance of epithelial-to-mesenchymal transition (EMT) and stemness states to promote lung cancer progression.22 Hybrid EMT and stemness traits may endow tumor cells with high cellular plasticity and motility for metastasis.23,24 While AXL-STAT3 in epithelial tumor cells is becoming delineated, the role of this signaling network in non-cancerous cells in the TME remains elusive. The present study explores whether AXL-STAT3 signaling reprograms TAMs to enhance their plasticity and tumor-promoting functions in lung TME. First, we demonstrate that concordant AXL-STAT3 signaling in TAMs is prevalent in pro-metastatic tumor niches. Second, we examine how this coordinated signaling in macrophages is initiated through paracrine actions exerted by stromal fibroblasts and tumor cells in co-culture systems. Third, we investigate how IL-11 cytokine activates the AXL-STAT3 signaling network in macrophages and enhances their ability to form primitive vasculogenic networks when co-cultured with endothelial cells. Last, we demonstrate the effect of AXL-STAT3 targeting on tumor growth and recruitment of TAMs and other host cells to the TME in a xenograft mouse model. The findings underscore the dependency of TAMs on AXL-STAT3 signaling and provide a therapeutic rationale for targeting this network in metastatic tumor niches.
RESULTS
Concordantly high expression of AXL and pSTAT3 in TAMs is commonly observed in advanced-stage lung tumors
To assess AXL-STAT3 signaling activation in TAMs and other cell types in lung TME, we conducted single-cell proteomic profiling of 15 lung tumors derived from 13 patients with lung adenocarcinoma, one patient with squamous cell lung cancer, and one patient with pleiomorphic carcinoma using cytometry by time of flight (CyTOF) (Figure 1A; Table S1). Principal-component analyses were first applied to confirm the consistency of sample acquisition and processing (Spearman’s coefficients >0.4; Figures S1A and S1B). The CyTOF panel included 20 protein markers related to AXL-STAT3 signaling, EMT, and stemness. Sixteen additional surface markers were used to differentiate 10 different cell types, including epithelial tumor cells, macrophages, stromal fibroblasts, endothelial cells, and other immune cells from among 619,200 cells (Figures 1B, 1C, and S1C–S1G; Table S2). Dominant cell types included T lymphocytes (30%), tumor cells (17%), B lymphocytes (14%), and macrophages (10%) (Figure 1C). Endothelial cells, tumor cells, fibroblasts, and macrophages expressed more AXL and pSTAT3 than the remaining six cell types (Figure 1D). Furthermore, the expression of AXL was significantly positively correlated with that of pSTAT3 in these four cell types, extending our previous observation that this concordant signaling is present not only in tumor cells but also in macrophages, fibroblasts, and endothelial cells (Figures 1E and S1H).22 Expression mean values of these markers were used as a cutoff to stratify individual TAMs into four categories: (I) AXLlo/pSTAT3lo, (II) AXLlo/pSTAT3hi, (III) AXLhi/pSTAT3lo, and (IV) AXLhi/pSTAT3hi (Figure 1E). Based on this categorization, lung cancer patients with lymph node and/or distant metastasis (group 2) frequently possessed category III/IV TAMs in their tumors as compared to lung cancer patients with no lymph node or distant metastasis (group 1) (Figures 1E and 1F). However, this trend was not perceptible in subpopulations of tumor cells, endothelial cells, or fibroblasts (Figures S1I and S1J). Although limited in the patient sample size, this initial observation suggests that concomitantly high AXL/pSTAT3 signaling in TAMs may contribute to the conditioning of pro-metastatic niches
Figure 1. Concordant activation of AXL-STAT3 signaling in macrophages in metastatic lung TMEs.
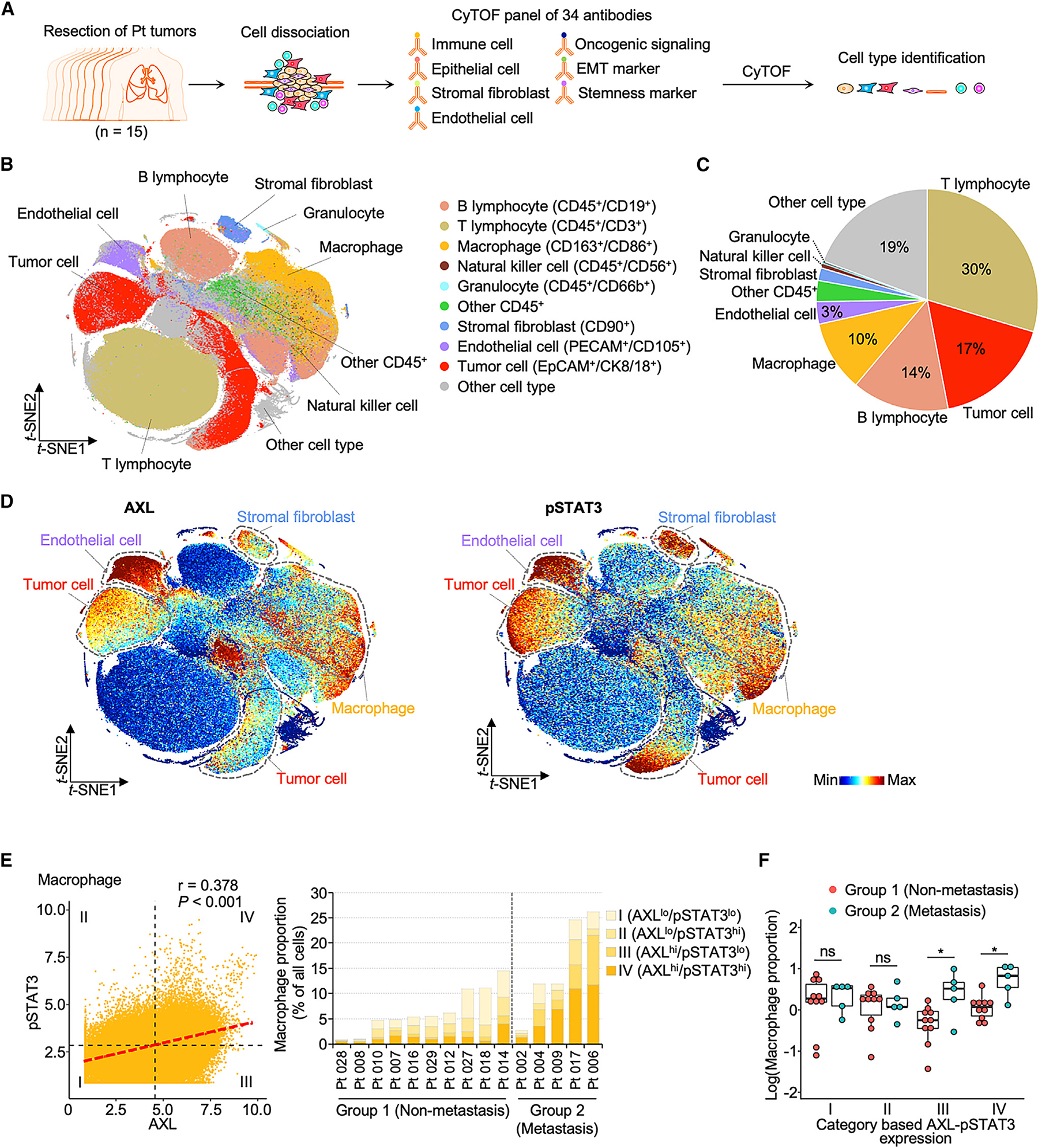
(A) Flow chart illustrates tissue processing and CyTOF analysis of 15 treatment-naive primary lung tumors.
(B) T-distributed stochastic neighbor embedding (t-SNE) scatterplot of immune, epithelial, and endothelial cells and stromal fibroblasts identified with corresponding markers in lung TMEs. See also Figure S1.
(C) Pie chart represents the proportion of cell subtypes.
(D) t-SNE scatterplots of expression levels of AXL and pSTAT3 in lung TMEs.
(E) AXL and pSTAT3 correlation scatterplot of macrophages and bar graph of four category proportions in individual patients. The four categories were stratified based on mean values of AXL and pSTAT3. Group 1 and 2 lung tumors were classified based on tumor-node-metastasis (TNM) staging of individual patients. Group 1 tumors were derived from patients with localized tumors. Group 2 tumors were derived from patients with lymph node and/or distant metastasis. See also Table S1
(F) Boxplot of category proportions between group 1 (n = 10) and group 2 (n = 5) tumors. Data are mean ± SD; *p < 0.05; Student’s t test for each category.
Concordant AXL-STAT3 enhances phenotypic diversity of TAMs
Concordantly high expression of AXL and pSTAT3 was associated with intermediate EMT and stemness features in lung tumor cells (Figures S2A–S2D). We then investigated whether TAMs similarly exploit AXL-STAT3 signaling to reproduce these oncogenic traits. Based on CyTOF data, TAMs abundantly expressed five of 12 EMT/stemness markers, namely vimentin, fibronectin, ZO-2, CD44, and CD133 (Figures S2A and S2C). These markers are mesenchymal, tight-junction, or cell-surface proteins that may contribute to intercellular communication and migration of TAMs.24–27 Thus, overexpression of these markers suggests macrophages share phenotypic characteristics with tumor cells, enhancing their pro-tumorigenic role in TMEs. In addition, category IV (AXLhi/pSTAT3hi) TAMs exhibited higher expression levels of the five signature markers than TAMs from remaining categories (Figure 2A). Approximately 54% of category IV TAMs highly expressed two polarization markers, CD86 and CD163 (Figures 2B and 2C). While CD163 is highly specific to M2 macrophages, CD86 is frequently associated with M1 macrophages and an M2b subtype.11,13,14 While the high expression of CD163 is most indicative of M2-like macrophage polarization, high expression of CD86 can be frequently associated with M1 macrophages and with M2-like TAMs.11,13,14,28,29 Concordantly high levels of CD86 and CD163 in TAMs suggests that these macrophages predominantly exhibit M2-like characteristics but still retain M1 traits for self-adaptation in dynamic TMEs.
Figure 2. Concordant AXL-STAT3 enhances pro-tumoral features of macrophages.
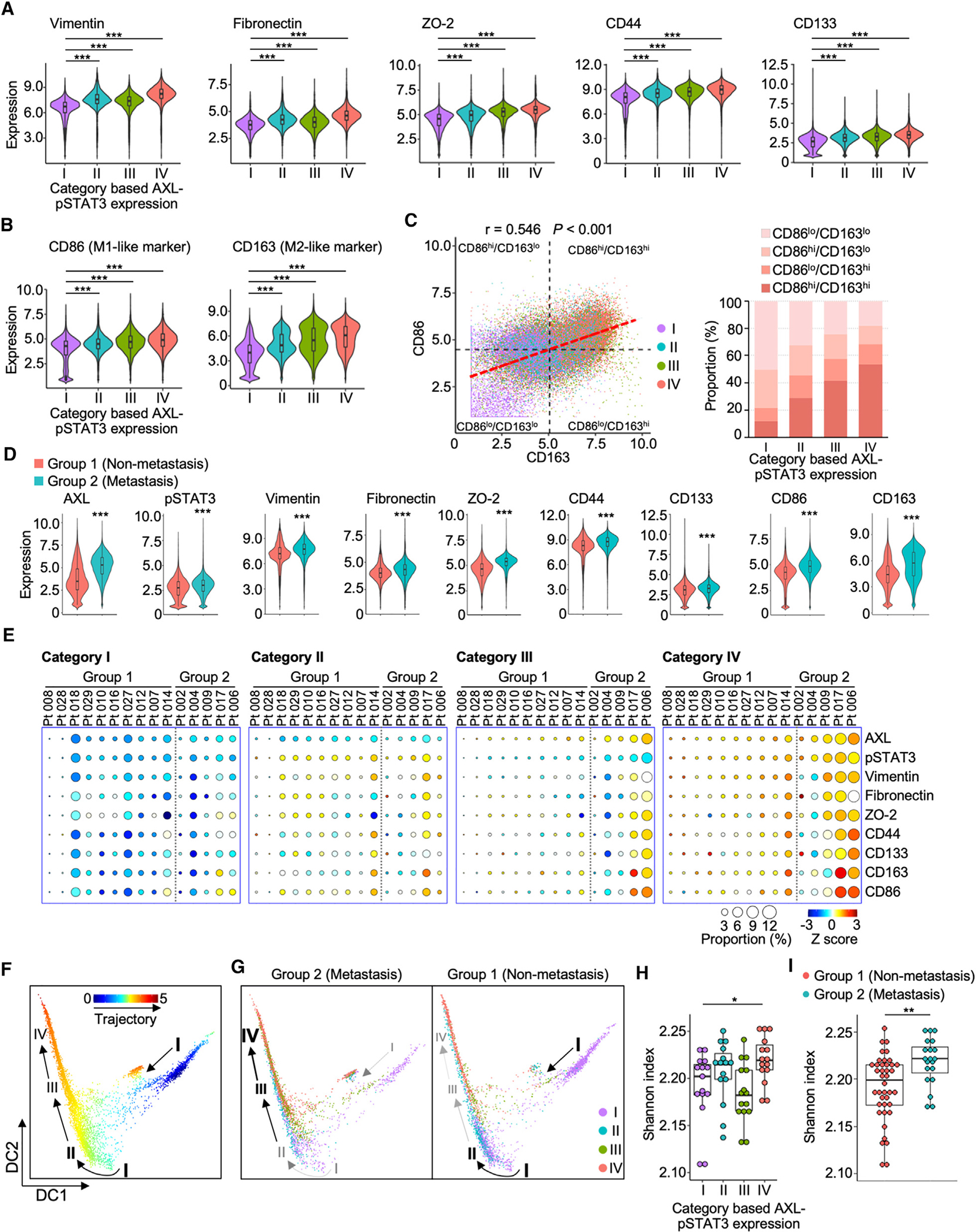
(A and B) Violin plots reveal expression levels of five EMT/stemness and two polarization markers of macrophages in the four AXL-pSTAT3 categories in lung TMEs. Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(C) CD86 and CD163 correlation scatterplot of macrophages demonstrates the four subtypes based on mean values of CD86 and CD163 and bar graph of the subtype proportions in the four AXL-pSTAT3 categories.
(D) Violin plots of expression levels of the seven markers in group 1 and 2 tumors. Data are mean ± SD; ***p < 0.001; Student’s t test.
(E) Circle plots showing the proportions and expression levels of AXL, pSTAT3, and the seven markers of the four AXL-pSTAT3 categories in individual tumors from group 1 and 2 patients.
(F and G) Diffusion maps of the pseudotime trajectories of the overall macrophages and macrophages from group 1 or 2 tumors of the four AXL-pSTAT3 categories. The arrows indicate the trajectory.
(H) Boxplot of Shannon indices in the four AXL-pSTAT3 categories (n = 15 for each category). Data are mean ± SD; *p < 0.05; one-way ANOVA followed by Duncan’s multiple range tests.
(I) Boxplot of Shannon indices in group 1 (n = 40) and 2 (n = 20) tumors. Data are mean ± SD; **p < 0.01; Student’s t test.
Next, we compared the expression levels of AXL-STAT3-associated markers (five EMT/stemness and two macrophage polarization markers) in TAM subpopulations derived from non-metastatic (group 1) and metastatic (group 2) lung tumors. It became evident that group 2 tumors expressed higher levels of these signature markers than in group 1 tumors (Figure 2D). Moreover, group 2 tumors had an abundance of category III/IV TAMs with highest expression of these signature markers. Conversely, group 1 tumors tended to display category I TAMs with lowest expression of these AXL-STAT3-related markers, which could account for their reduced pro-tumorigenic function (Figure 2E). Pseudotime analysis was used to infer the differentiation trajectory of TAMs and revealed two distinct patterns. The first was a linear trajectory where TAMs transitioned from category I/II (AXLlo/pSTAT3lo-hi) into III (AXLhi/pSTAT3lo) and IV (AXLhi/pSTAT3hi) (Figure 2F). The second trajectory was mainly confined within category I (AXLlo/pSTAT3lo) with a petering track. The first trajectory was commonly seen in group 2 TAMs, whereas the more confined trajectory was observed in group 1 TAMs (Figure 2G). Shannon diversity index was used to determine whether AXL-STAT3-related markers could be used as parameters to evaluate TAM diversity. Category IV TAMs were the most diverse and demonstrated the highest Shannon index as compared to other categories (Figure 2H). In addition, group 2 tumors exhibited more heterogeneous TAM populations than group 1 tumors (Figure 2I). The collective results imply that macrophages can be increasingly conditioned to co-express high levels of AXL-STAT3-related markers, transitioning to more plastic and heterogeneous states in the TME. Therefore, we proposed to assess the pro-tumoral features of macrophages based on expression of AXL, pSTAT3, and seven signature markers in subsequent in vitro and mouse studies.
Paracrine activation of AXL-STAT3 in macrophages exposed to lung cancer cells or cancer-associated fibroblasts
AXL and STAT3 activation in TAMs may be prompted by direct cellular communication with other cell types in the TME. Therefore, co-culture experiments were used to assess a causal relationship between macrophages and lung cancer cells or between macrophages and cancer-associated fibroblasts. First, U937-derived monocytes were differentiated into M0 macrophages with phorbol 12-myristate 13-acetate.30 Second, M0 macrophages were co-cultured with A549 lung cancer cells known to have metastatic potential in vivo (condition 1 vs. 3; Figure 3A).31,32 In a separate set of experiments, macrophages were co-cultured with MRC-5 lung fibroblasts known to overexpress CD90, a putative marker for cancer-associated fibroblasts (condition 1 vs. 5; Figure 3A).33 CyTOF profiling stratified 19 subpopulations of macrophages co-cultured with A549 or MRC-5 cells (Figure S3A), which revealed that macrophages co-cultured with A549 cells (condition 3) and MRC-5 cells (condition 5) were more M2-like and expressed higher levels of CD163, as compared to unexposed macrophages (condition 1) (Figure S3B). Enhanced expression of AXL and pSTAT3 was observed in macrophages exposed to A549 or MRC-5 cells (Figure 3B). Western blotting independently confirmed these findings in macrophages exposed to conditioned media of A549 or MRC-5 cells (Figure S3C). Consistent with those patient studies, AXL/pSTAT3 was significantly concordant in co-cultured macrophages (Figure 3C, left). Based on the four categories of macrophages described earlier, we observed an increase from 3% to 33% in the proportion of category IV (AXLhi/pSTAT3hi) macrophages following co-culture with A549 cells (Figure 3C, right). This increase appeared more dramatic (>70%) in macrophages exposed to MRC-5 fibroblasts. Macrophages co-cultured with A549 cells or MRC-5 fibroblasts displayed higher expression of four of the five EMT/stemness markers mentioned above (Figure 3D). The co-culture conditioning also enhanced the expression of both CD86 and CD163, facilitating polarization shift from M0-like (CD86lo/CD163lo) to more plastic states with hybrid M1/M2 phenotypes (Figures 3E and 3F). In addition, category IV macrophages exhibited higher expression levels of the seven signature markers (Figure 3G). Furthermore, higher Shannon diversity indices were observed in macrophages co-cultured with A549 cells or MRC-5 fibroblasts compared to those without the exposure (Figure 3H). Albeit less dramatic, M0 macrophages reciprocally influenced A549 cells and MRC-5 fibroblasts in co-culture systems and reduced the subpopulation of category IV A549 cells and MRC-5 fibroblasts (Figures S3D–S3I). These aggressive fibroblasts (category IV) could be subdued by M0 macrophages, resulting in a ~50% decrease in AXLhigh/pSTAT3high subpopulations (category IV in condition 5 vs. condition 4) (Figures S3G and S3H). The results demonstrate that lung cancer cells or cancer-associated fibroblasts induced the expression of AXL-STAT3 signaling components and signature markers in macrophages through paracrine actions, likely enhancing their plasticity and pro-tumoral phenotype.
Figure 3. Paracrine activation of AXL-STAT3 in macrophages exposed to lung cancer cells or cancer-associated fibroblasts.
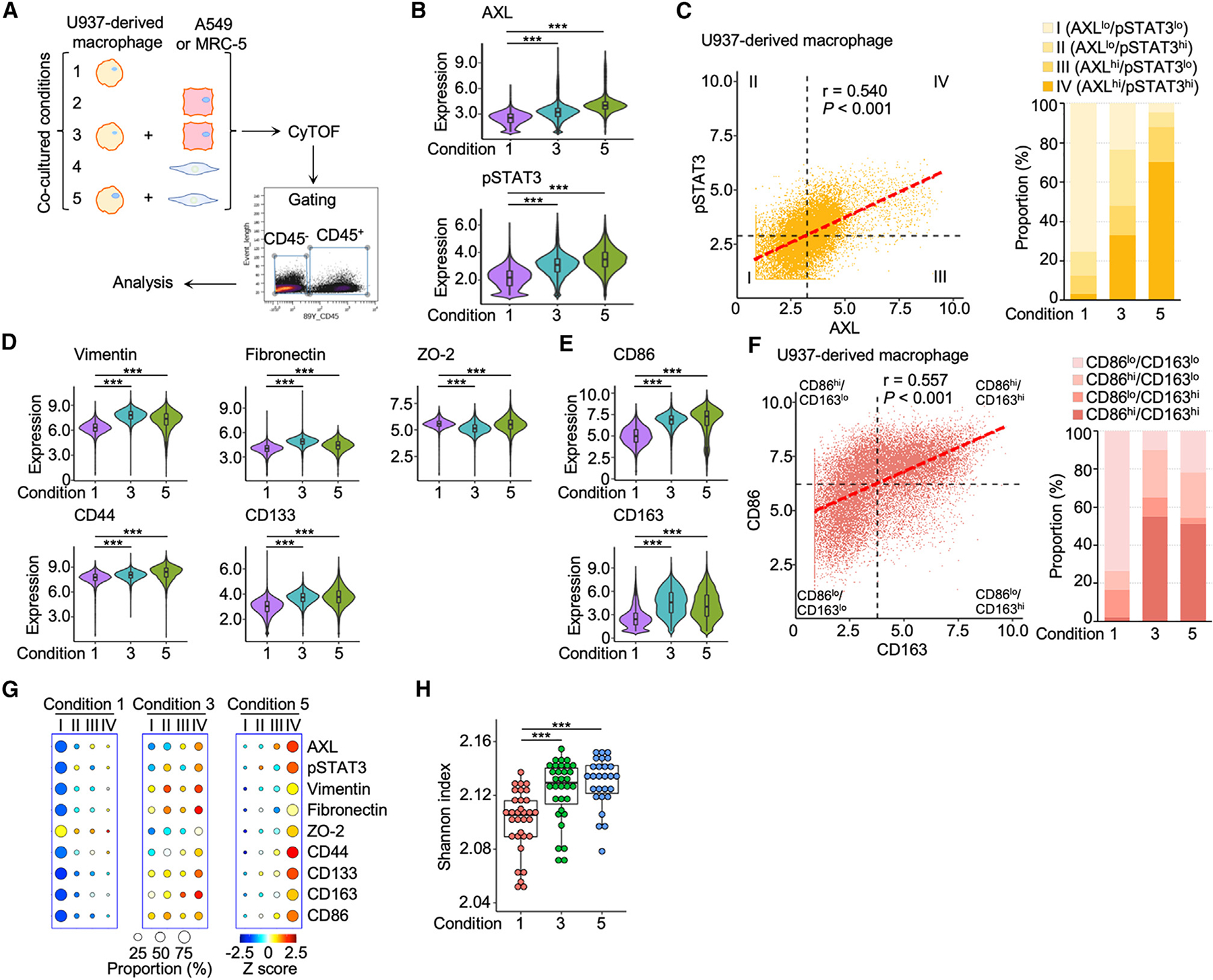
(A) Co-cultured conditions of U937-derived macrophages and A549 lung cancer cells or MRC-5 lung fibroblasts with cytometry by time-of-flight (CyTOF) workflow.
(B) Violin plots showing the expression levels of AXL and pSTAT3 of macrophages in condition 1, 3, and 5. Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(C) AXL and pSTAT3 correlation scatterplot of macrophages showing the four categories based on mean values of AXL and pSTAT3 and bar graph of category proportions in condition 1, 3, and 5.
(D and E) Violin plots of expression levels of the seven AXL-pSTAT3-related markers in macrophages of condition 1, 3, and 5. Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(F) CD86 and CD163 correlation scatterplot of macrophages showing the four subtypes based on mean values of CD86 and CD163 and bar graph of subtype proportions in condition 1, 3, and 5.
(G) Circle plots showing the proportions and expression levels of AXL, pSTAT3, and the seven markers of the four AXL-pSTAT3 categories in condition 1, 3, and 5.
(H) Boxplot of Shannon indices in condition 1, 3, and 5 of 32 PhenoGraph clusters (n = 32). Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
AXL-IL-11-STAT3-mediated CD44 enhances pro-angiogenic functions of macrophages
In search of paracrine factors that might activate AXL-STAT3 signaling in macrophages, we screened a panel of 39 cytokines and/or chemokines secreted in the media by A549 lung cancer cells untreated or treated with an AXL inhibitor (Figures S4A–S4C). Six pro-tumorigenic cytokines and soluble factors (vascular endothelial growth factor [VEGF], IL-8, IL-11, fibroblast growth factor [FGF]-2, granulocyte colony-stimulating factor [G-CSF], and IL-4) were determined to be regulated by AXL. Among these, IL-11 was an AXL-regulated cytokine that was abundantly secreted from lung cancer cells and MRC-5 fibroblasts (Figures 4A and 4B). IL11 was preferentially expressed in lung tumors of patients with poor survival outcomes in The Cancer Genome Atlas (TCGA) cohort (Figures 4C and 4D). Therefore, we investigated the effect of IL-11 on macrophage plasticity using CyTOF (Figure 4E). Concordant activation of AXL-STAT3 occurred in IL-11-treated U937-derived macrophages, although the effect appeared most pronounced with respect to pSTAT3 expression (Figures 4F and 4G). The CyTOF finding was independently validated by western blotting analysis, which revealed a more dramatic increase in phospho-STAT3 (pSTAT3) levels and a modest increase in the expression of a splice variant of phospho-AXL (pAXL) following IL-11 treatment in U937-derived macrophages (Figure 4H). Further elevation of AXL expression could be attributed to a downstream event of pSTAT3-mediated transcription (Figure S4D). Notably, IL-11 treatment upregulated three of the five signature markers (vimentin, fibronectin, and CD44) in U937-derived macrophages (Figure 4I). IL-11 also induced the expression of CD86 and CD163, producing an M2-like polarization in shift of these macrophages (Figures 4J and 4K). Because the U937-derived monocytic cell lines are derived from leukemia patients,30 a potential concern was that these cell lines may not adequately represent primary macrophages. Therefore, we repeated the IL-11 treatment experiments and CyTOF analysis using macrophage colony stimulating factor (M-CSF)-activated macrophages differentiated from primary monocytes of a healthy individual (Figures S4E–S4J).34 Flow cytometry results demonstrate increased expression levels of M2-like markers (CD163, CD206, and CD209) in primary macrophages treated with IL-11, which was dose dependent (Figures S4K and S4L).13,35,36 This result confirmed the original findings from U937-derived macrophages, suggesting IL-11 is an important paracrine factor promoting macrophage plasticity via pSTAT3 activation. Moreover, macrophage function, differentiation, and cytokine expression were attenuated following STAT3 knockdown in U937-derived macrophages based on transcriptomic analysis (Figures S5A–S5F).
Figure 4. AXL-mediated IL-11 secretion in lung cancer cells and lung stromal fibroblasts activates signaling cascades for pro-tumoral features of macrophages.
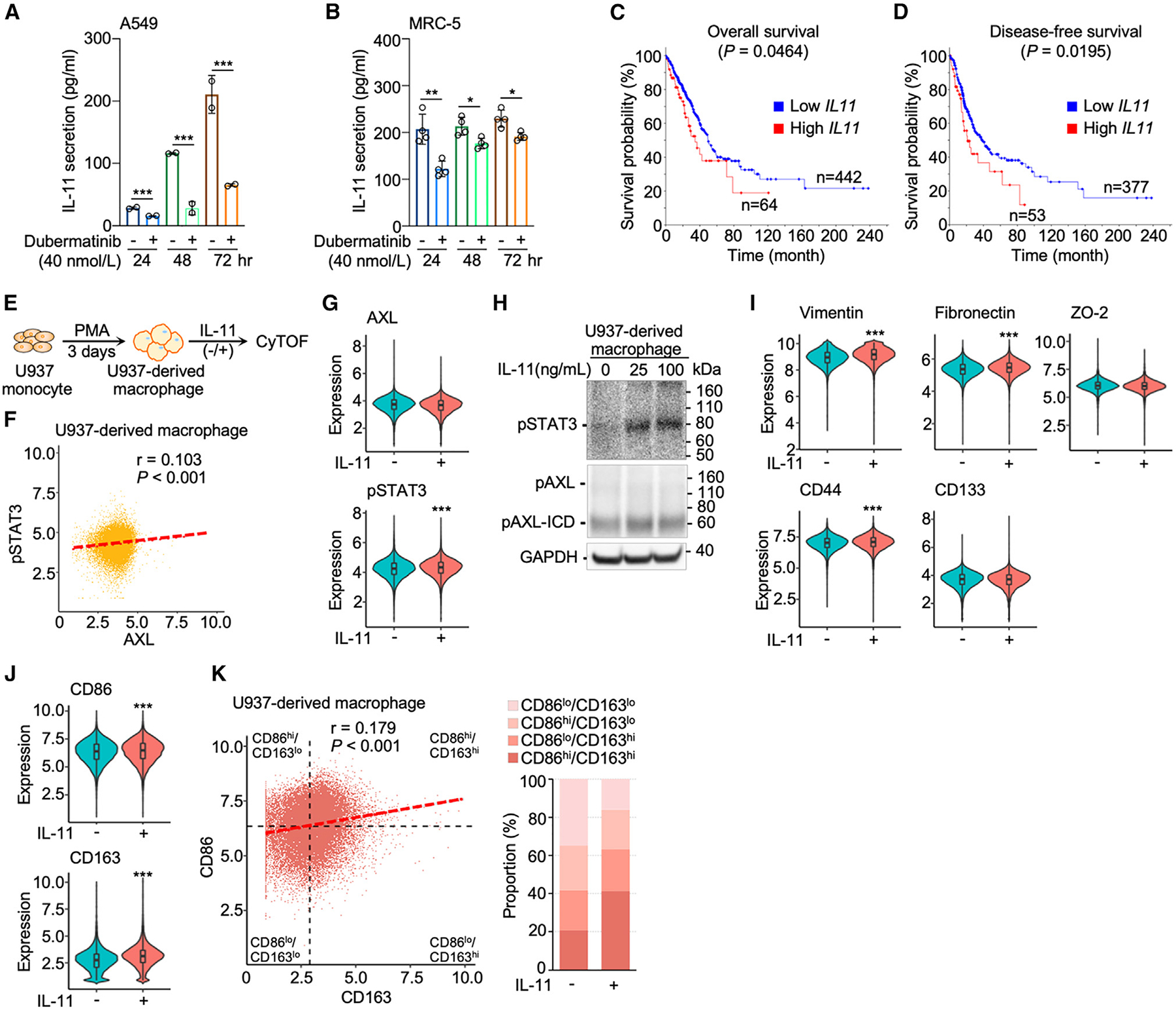
(A and B) IL-11 secretion was attenuated by dubermatinib (40 nmol/L) from A549 lung cancer cells (n = 2) and MRC-5 lung fibroblasts (n = 4). Data are mean ± SD; *p < 0.05, **p < 0.01, ***p < 0.001; Student’s t test for each time point.
(C and D) Kaplan-Meier curves depict overall and disease-free survival probability in TCGA lung adenocarcinoma cohort based on high (Z score > 1) and low (Z score < 1) IL11 expression of lung tumors.
(E) Flow chart of induction of U937-derived macrophages and IL-11 treatment for CyTOF analysis.
(F) AXL and STAT3 correlation scatterplot of U937-derived macrophages.
(G) Violin plots showing the expression levels of AXL and STAT3 without and with IL-11 treatment (25 ng/mL). Data are mean ± SD; ***p < 0.001; Student’s t test.
(H) Western blot analysis of IL-11-activated AXL-STAT3 signaling, i.e., phosphorylation of AXL and STAT3 (pSTAT3) in U937-derived macrophages. The cleavage product, phosphorylated AXL intracellular domain (pAXL-ICD), was observed.
(I and J) Violin plots of expression levels of the seven AXL-STAT3-related markers without and with IL-11 treatment (25 ng/mL). Data are mean ± SD; ***p < 0.001; Student’s t test.
(K) CD86 and CD163 correlation scatterplot of macrophages showing the four subtypes based on mean values of CD86 and CD163 and bar graph of subtype proportions of macrophages without and with IL-11 treatment (25 ng/mL).
To further elucidate the functional impact of these AXL-STAT3-related markers, we focused on CD44 that was prominently upregulated in U937-derived macrophages following IL-11 treatment (Figures 5A and S6A). We investigated the functional role of CD44 in macrophages, given previous studies that report macrophages engage in non-endothelial vascular formation in tumors and can express “cancer stem cell markers.”37–39 Proximity ligation assay (PLA) was used to determine how IL-11 contributes to extracellular and intracellular protein-protein interactions for the regulation of CD44 expression in macrophages (Figure 5B). A 2- to 4-fold increase in IL-11 and GP130 binding interactions occurred in U937-derived macrophages within 72 h following the addition of cytokine to the culture (Figure 5C).40 IL-11 engages with membrane-bound GP130 likely through binding of IL-11Rα, triggering intracellular activation of JAK kinases and STAT3 phosphorylation in macrophages (Figure 5D).41 Following this activation, pSTAT3 dimers can translocate to the nucleus to induce target gene transcription upon binding to specific motifs.41 We also demonstrated that increased binding of pSTAT3 to CD44 leads to an increase in its mRNA expression in macrophages (Figures 5E and 5F). To examine whether IL-11-mediated upregulation of CD44 enables macrophages to participate in the assembly of vascular networks with neighboring endothelial cells, human umbilical vein endothelial cells (HUVECs) were co-cultured with macrophages. Macrophages unexposed to IL-11 did not influence the vascular mesh networks of neighboring HUVECs (Figure 5G). However, the pretreatment with IL-11 enabled macrophages to engage with HUVECs and promote vascular mesh formation (Figures 5H and S6B). This macrophage-facilitated event was attenuated following treatment with anti-CD44 agent (Figures 5I and S6C). The results highlight the potential role of the AXL-IL-11-STAT3 axis to upregulate CD44 expression in macrophages and promote vasculogenesis in the TME.
Figure 5. AXL-IL-11-STAT3-mediated CD44 enhances macrophage ability to facilitate vasculogenesis.
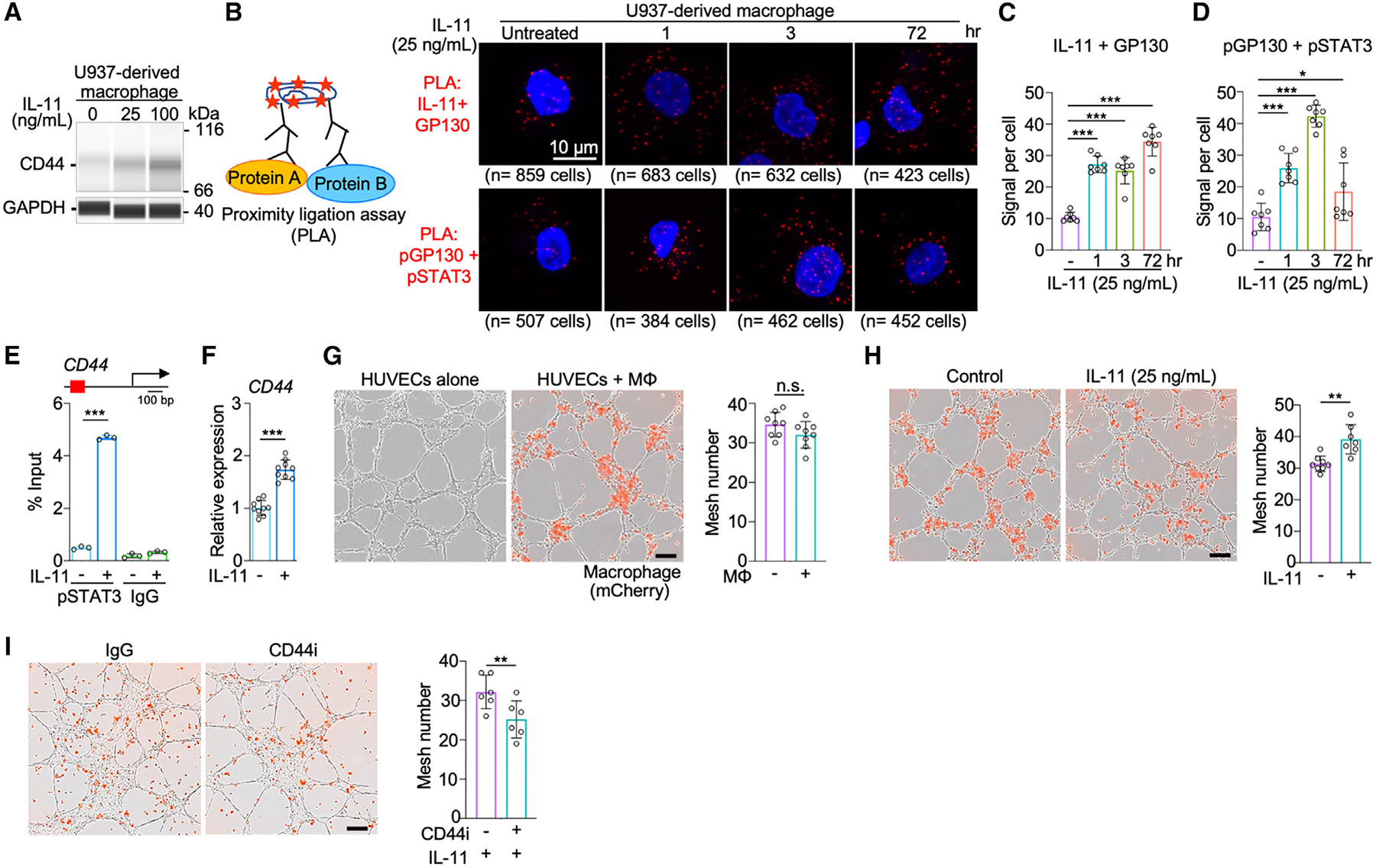
(A) Capillary western immunoassay (WES) of a stemness marker, CD44, in U937-derived macrophages untreated and treated with IL-11. Raw data of WES shown in Figure S6A.
(B) Diagram of proximity ligation assay (PLA) (left) and PLA images of protein-protein interactions between IL-11/GP130 and pGP130/pSTAT3 in macrophages untreated and treated with IL-11 at 1, 3, and 72 h (right). PLA utilizes a pair of oligonucleotide-conjugated secondary antibodies that correspond to antibodies targeting each interacting protein partner. The proximity of protein partners allows circular DNA amplification of oligonucleotide templates and detectable fluorescence signals upon in situ ligation.
(C and D) Quantitative analysis of PLA signals per cell indicating protein-protein interactions of IL-11 and the extracellular domain of GP130 (C) and pGP130 and pSTAT3 (D) in macrophages untreated and treated with IL-11 at 1, 3, and 72 h (n = 7). Data are mean ± SD; *p < 0.05, ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(E) Chromatin immunoprecipitation (ChIP)-qPCR analysis of pSTAT3 binding to promoter regions of CD44 in untreated and IL-11-treated U937-derived macrophages (n = 3). Data are mean ± SD; ***p < 0.001; Student’s t test for each treatment.
(F) qPCR analysis of CD44 expression in untreated and IL-11-treated U937-derived macrophages (n = 9). Data are mean ± SD; ***p < 0.001; Student’s t test.
(G) Images of vasculogenic mesh formation and bar graph of mesh number in human umbilical vein endothelial cells (HUVECs) co-cultured without or with U937-derived macrophages (n = 8). Scale bar, 200 μm. Data are mean ± SD; Student’s t test.
(H) Bar graph of mesh number in HUVECs co-cultured with U937-derived macrophages pre-treated without or with IL-11 (25 ng/mL) (n = 7). Data are mean ± SD; **p < 0.01; Student’s t test.
(I) Bar graph of mesh number in HUVECs co-cultured with U937-derived macrophages pre-treated with IL-11 (25 ng/mL) and without or with CD44 inhibitor (n = 6). Data are mean ± SD; **p < 0.01; Student’s t test.
AXL-STAT3 inhibition reduces TAM recruitment and tumor growth
To confirm activation of AXL-STAT3 signaling in macrophages within the host TME, immunofluorescence was conducted in EGFP-labeled A549 xenograft tumors subcutaneously transplanted in homozygous Nu/J nude mice that have a partial host immune system.42,43 CD90.2+ murine stromal fibroblasts and F4/80+ murine macrophages infiltrated the tumor and were in close proximity to EGFP+ A549 lung cancer cells (Figure 6A). Within the respective host TME, pAXL staining was most prominent in areas inhabited by lung cancer cells and occasionally present in regions occupied by murine fibroblasts and macrophages (Figures S7A and S7B). Interestingly, IL-11 staining was widespread in xenograft tumors, suggesting that this cytokine may serve as a conduit to maintain human AXL-STAT3 and murine Axl-Stat3 networks in the host microenvironment (Figure S7C). To disrupt the AXL-STAT3 signaling network, appropriate dosing schedules of dubermatinib (AXL inhibitor) and/or momelotinib (JAK1/2 inhibitor) were selected to minimize toxicity in mice (Figure S7D).22,44,45 Repression of pSTAT3 by momelotinib in cancer cells was previously determined in vitro and corroborated by western blotting (Figure S7E).44,46,47 Compared to the control group, the momelotinib group displayed slightly larger tumor volumes (Figures 6B and S7F). While tumor growth was initially suppressed by dubermatinib, it immediately rebounded after drug discontinuation on day 55 (Figure 6B). The findings suggest that single-agent treatments are ineffective against tumors that sustain an active AXL-STAT3/Axl-Stat3 network for proliferation and growth. Conversely, combination treatment substantially attenuated tumor growth, and this suppression persisted despite drug discontinuation (Figure 6B). A similar trend of tumor growth suppression by these treatments was observed in an H2009 mouse xenografts model (Figure S7G).
Figure 6. AXL-STAT3 inhibition attenuates macrophage plasticity and disrupts host cell conscription in xenograft TMEs.
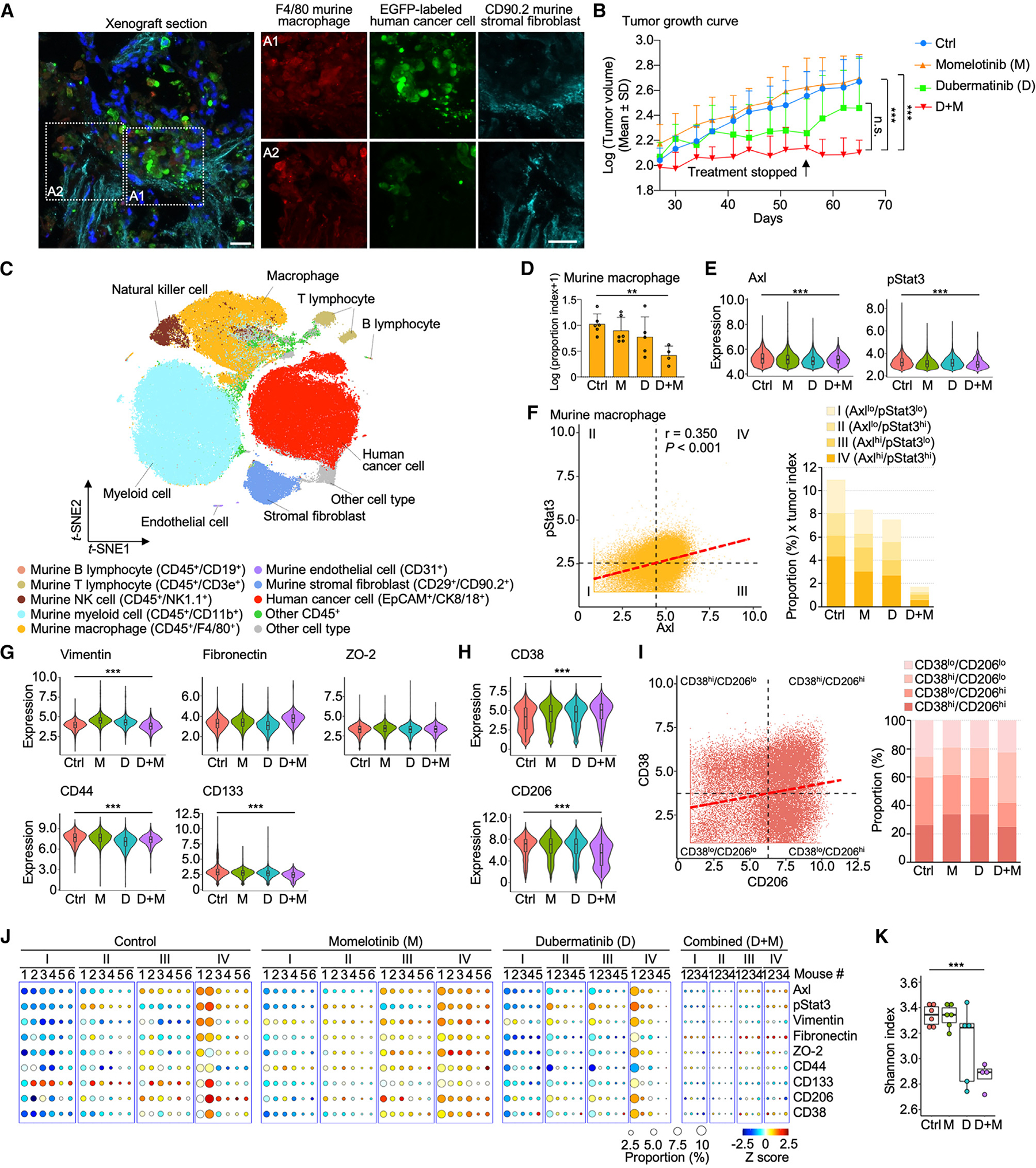
(A) Immunofluorescence images of a xenograft tumor section showing cell nuclei (DAPI, blue), human A549 lung cancer cells (EGFP labeled, green), murine stromal fibroblasts (CD90.2+, cyan), and murine macrophages (F4/80+, red). Scale bar = 20 μm.
(B) Growth curves of the average tumor size in log 10 base in the four treatment groups: vehicle control (n = 6); dubermatinib (AXL inhibitor), 120 mg/kg oral dose twice weekly for 28 days (n = 5); momelotinib (JAK/STAT3 inhibitor), 25 mg/kg orally once daily for 28 days (n = 6); combination treatment for 14 days (n = 4). Combination treatment resulted in 75% reduction in tumor volume compared with control. Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(C) t-SNE scatterplots of human cancer cells and murine fibroblasts, endothelial cells, macrophages, and other immune cells of xenograft tumors identified with corresponding markers. See also Figures S7H–S7K.
(D) Bar graph of murine macrophage proportion index in log 10 base of the four treatment groups (vehicle control, n = 6; dubermatinib, n = 5; momelotinib, n = 6; and combination treatment, n = 4). Data are mean ± SD; **p < 0.01; one-way ANOVA followed by Duncan’s multiple range tests.
(E) Violin plots showing the expression levels of AXL and STAT3 in the four treatment groups. Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(F) AXL and STAT3 correlation scatterplot of macrophages showing the four categories based on mean values of AXL and STAT3 and bar graph of category proportions in the four treatment groups.
(G and H) Violin plots of expression levels of the five AXL-STAT3-related EMT/stemness markers, M1-like marker CD38, and M2-like marker CD206 in the four treatment groups. Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
(I) CD38 and CD206 correlation scatterplot of macrophages showing the four subtypes based on mean values of CD38 and CD206 and bar graph of subtype proportions in the four treatment groups.
(J) Circle plots showing the proportions and expression levels of AXL, STAT3, and the seven related markers of the four AXL-STAT3 categories in individual xenograft tumors of the treatment groups.
(K) Boxplot of Shannon indices of the treatment groups (vehicle control, n = 6; dubermatinib, n = 5; momelotinib, n = 6; and combination treatment, n = 4). Data are mean ± SD; ***p < 0.001; one-way ANOVA followed by Duncan’s multiple range tests.
CyTOF was then used to determine treatment effects on the cellular composition of A549 xenograft tumors. A panel of 30 antibodies identified human and murine cells based on their respective cell-surface and oncogenic markers, totaling 129,926 cells from among 21 xenograft tumors (Table S3). Specifically, these cells comprised human-derived cancer cells, as well as murine-derived fibroblasts, endothelial cells, macrophages, and other immune cells (Figures 6C and S7H–S7K). Average proportions of cell subtypes were calculated for each treatment cohort (Figure S8A). In control mice, xenograft tumors comprised only 11.6% of human cancer cells, while the remaining cells (88.4%) were murine-derived host cells (Figure S8A). Notably, treatment groups displayed considerable variability in tumor size and cellular composition (Figure S8A). A proportion index was derived by factoring in the proportion of macrophages and tumor size across four treatment groups for direct comparisons (Figure 6D). There was a downward trend in the recruitment of host macrophages to a tumor site in single-agent groups relative to that of the control group. The combination treatment was most effective in hindering macrophage recruitment. We also observed that combination treatment resulted in the lowest proportion indices of human cancer cells, murine endothelial cells, T and B lymphocytes, myeloid cells, natural killer cells, stromal fibroblasts, and other CD45+ cells (Figure S8B). The finding suggests that targeting the AXL-STAT3/Axl-Stat3 network not only curbs macrophage recruitment but also prevents recruitment of other host cells to the TME.
Alterations in tumor size and cellular composition by these targeted drugs can be largely attributed to deregulated AXL-STAT3/Axl-Stat3 network in xenograft tumors. We found that concordance between Axl and pStat3 expression was reduced following drug treatments in mouse macrophages (Figures 6E and 6F). This deregulation appeared to reduce the expression of three of the five signature markers (vimentin, CD44, and CD133) in the combination treatment group (Figure 6G). Changes in the expression of murine M1-like marker CD38 and M2-like marker CD206 also occurred, shifting macrophage compositions from CD38lo/CD206hi predominant in the control group to CD38hi/CD206lo in the combination treatment group (Figures 6H and 6I). Overall, a reduction in macrophage subpopulation sizes was most pronounced in the combination treatment group relative to single-agent or control group (Figure 6J). Based on Shannon indices, alterations of cell complexity seen with single-agent treatments were more pronounced in the combination treatment group (Figure 6K). This reduced macrophage and TME heterogeneity suggests that the Axl-Stat3 network may be critical for the recruitment of plastic macrophages and other supportive host cells needed to form a symbiotic community for tumor growth and invasion (Figures 6K, S8A, and S8B).
DISCUSSION
The AXL-STAT3 network is known to promote hybrid EMT and stem-like phenotypes of lung tumor cells.48–50 These acquired phenotypic features enhance cellular plasticity in tumor ecosystems for self-adaptation, proliferation, and invasion.7,23,24 In this study, we report that this signaling network is concomitantly upregulated in macrophages and other non-cancerous cells in TME, likely orchestrating a symbiotic cellular community that promotes metastatic tumor niches. In lung tumors with high metastatic potential, concordant AXL-STAT3 signaling in TAMs leads to overexpression of five EMT/stemness markers based on CyTOF analysis. Category IV TAMs (AXLhi/STAT3hi) express high levels of mesenchymal proteins (vimentin and fibronectin) to facilitate their cellular movement in TME,51,52 tight-junction protein ZO-2 for intercellular communication,26 and stemness phenotypic markers (CD44 and CD133).53 We demonstrate that CD44 (adhesive protein) enables TAMs to engage with endothelial cells to form primitive vasculogenic networks, highlighting a potential role of TAMs to promote tumor vasculogenesis.37,54,55 In this manner, TAMs may engage with tumor cells and procure similar phenotypic traits (hybrid EMT and stemness traits) to enhance their pro-tumorigenic functions.
Additionally, the AXL-STAT3 network enhances polarization of these macrophages toward an M2-like phenotype while still retaining M1-like features. Unlike polarized macrophages, TAMs with M1/M2 hybrid phenotype demonstrate versatility and can sustain complex interactions with other pro- and anti-inflammatory immune cells. In this way, TAMs can maneuver an immune-evasive microenvironment by recruiting other anti-inflammatory immune cells such as regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs).3,56 One limitation of our study is that we did not investigate the role of AXL-STAT3 signaling in macrophage subsets (CD169+ macrophages and T cell receptor-positive macrophages).57 For future experiments, we plan to expand our CyTOF panel to include these cellular subtypes.
Directly targeting TAM plasticity in the TME using combined treatment with an AXL inhibitor and JAK1/2 inhibitor (STAT3 suppression) represents a novel therapeutic strategy for patients with metastatic lung adenocarcinoma. We found that AXL-STAT3 inhibition attenuated macrophage lineage transition states, reduced macrophage plasticity, and promoted M1-like polarization. Additionally, we found that combined targeting of the AXL-STAT3 network inhibited tumor growth and suppressed the recruitment of macrophages and other host cells to xenograft tumor site. Interestingly, the proportion of endothelial cells with high AXL expression was dramatically reduced and may impair the recruitment of host cells through the tumor vasculogenic network (Figures 1D and S8B). In this way, tumor cells are unable to conscript host cells, rendering the TME less heterogeneous.
In conclusion, our study provides a unique perspective of the AXL-STAT3 network in lung TME, revealing how lung tumor cells and stromal fibroblasts can reprogram TAMs via IL-11 paracrine effect. Moreover, combined targeting of AXL-STAT3 effectively attenuated tumor-promoting features of TAMs and restricted the recruitment of supportive host cells in xenograft nude mice, which harbor a partial host immune system. Future studies will involve CD34+ humanized hu-NSG-SGM3 mice model,58 which contains a functional repertoire of multi-lineage immune cell subpopulations, to study treatment effects using different combinations of therapeutics targeting AXL and/or STAT3. In addition, our current CyTOF panel will be further refined to incorporate protein markers for the identification of T cells, B cells, MDSCs, and macrophage subtypes to better assess the role of AXL-STAT3 in the tumor immune microenvironments. This comprehensive single-cell proteomic analysis of the TME could deepen our understanding of communication among multiple cell types, and treatment strategies that target co-dependent signaling networks of macrophages and tumor cells may improve survival outcomes for lung cancer patients.
Limitations of the study
This study has the following limitations. First, with the limited number of macrophage polarization markers used in CyTOF of patients’ tumor samples (Figure 2B), we cannot have better resolution of macrophage subtypes in the TME, even though we have performed flow cytometry in IL-11-treated primary macrophages with additional M2-like polarization markers (Figures S4K–S4J). Furthermore, the vascular network assay using HUVEC co-cultured with U937-derived macrophages only demonstrates the supportive role of IL-11-treated macrophages, but it cannot provide detailed machinery of how macrophages are involved in angiogenesis. In addition, while our study demonstrates that AXL-STAT3 targeting can reduce M2-like polarization and macrophage plasticity in the A549 xenograft mouse model, we are unable to fully understand cell-cell communication in the TME, as CyTOF does not present spatial information. Combining CyTOF with spatial transcriptomics and a spatial single-cell imaging platform could provide additional insights into cell-cell communication at different locations of the TME. Our future studies will integrate multiple platforms (e.g., CyTOF, spatial transcriptomics, spatial single-cell imaging, and single-cell RNA sequencing) to profile lung TMEs for better understanding of drug efficacy at the single-cell level in tumor cells and supportive host cells (stromal fibroblasts, endothelial cells, and immune cell subpopulations) and spatial interactions among these various cell types in the dynamic TME.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Josephine Taverna (TavernaJ@uthscsa.edu).
Materials availability
All unique/stable reagents can be requested from the lead contact.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
RNA-seq data for this study is available through the Gene Expression Omnibus (GEO) under accession number GSE231829.
EXPERIMENTAL MODEL AND SUBJECT PARTICIPANT DETAILS
Patients
The protocol was approved by the Institutional Review Board of the University of Texas Health Science Center at San Antonio (UTHSCSA). All patients were enrolled at UTHSCSA between October 2018 and January 2020 (Table S1). Lung tumor tissue samples were collected from 15 patients at the time of surgery (lobectomy or wedge resection). Clinicopathological characteristics of patients are provided in Table S1. Written informed consent was obtained from all patients in compliance with the Declaration of Helsinki, the Belmont Report, U.S. Common Rule following the U.S. Department of Health and Human Services and the FDA regulations and Good Clinical Practice guidelines. Patients in the study had not received any systemic treatment, and the site from which specimens were obtained had not been previously treated with radiotherapy.
Cell lines and macrophages
A549 lung cancer cells were obtained from the American Type Culture Collection (ATCC), and routinely cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS) and 100 units/mL penicillin-streptomycin (Gibco) aired with 5% CO2 at 37°C. For in vivo studies, A549 cells were labeled with luciferase. The pLenti-puro3/To/V5-GW/EGFP-Firefly luciferase plasmid59 was purified using QIAprep Spin Miniprep kit (Qiagen) and transfected 293T cells with the 3rd Generation Packaging Mix (Applied Biological Materials, Richmond, BC, Canada) for lentiviral production. The lentivirus-containing medium was harvested after 48 h of transfection, and A549 lung cancer cells were infected with lentivirus for 24 h. MRC-5 lung fibroblasts were obtained from the ATCC, and routinely cultured in RPMI 1640 supplemented with 10% FBS and 100 units/mL penicillin-streptomycin (Gibco) aired with 5% CO2 at 37°C.
U937 monocytic cell lines were obtained from the ATCC and routinely cultured with RPMI 1640 supplemented with 10% heat shocked FBS and 100 units/mL penicillin and 100 μg/mL streptomycin (Gibco) aired with 5% CO2 at 37°C. U937 monocytes were differentiated into macrophages by 100 ng/mL phorbol 12-myristate 13-acetate (PMA).30
Human peripheral blood CD14+ monocytes (Stemcell, Cat# 70035) were cultured in ImmunoCult-SF Macrophage Medium (Stemcell, Cat# 10961) containing 50 ng/mL of human recombinant M-CSF (Stemcell, Cat# 10961). On day 4, fresh media or fresh media containing 25 ng/mL of human IL-11 protein (R&D System, Cat# 218-IL) were replaced and cultured. On day six, the media was removed and the attached macrophages were harvested and incubated in 2.5 mM EDTA in PBS at 37°C for 15 min. Then 0.5 BSA in PBS medium was added and macrophages were collected through pipetting.
Animals
The protocol to obtain and process lung tumors were approved by the Institutional Animal Care and Use Committee (IACUC). Five week-old homozygous Nu/J mice were purchased from the Jackson Laboratory and quarantined for two weeks in the animal room. Luciferase-labeled A549 lung cancer cells (1 × 106) were mixed with 100 μL Corning Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix (Corning 354230) and injected subcutaneously in the 7 week-old Nu/J mice. After tumors were re-established (80–130 mm3), six mice per group were randomly assigned to four treatment groups: 1) vehicle control; 2) dubermatinib 120 mg/kg oral dose twice weekly; 3) momelotinib 25 mg/kg oral dose once daily46; and 4) dubermatinib 120 mg/kg oral dose twice weekly in combination with momelotinib 25 mg/kg oral dose once daily. Treatment duration was 28 days. Drugs were administered by plastic feeding tube (INSTECH) orally and given daily or twice weekly. To avoid spillage of the drug. All mice were treated and sacrificed according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the protocol IACUC approved on September 14th, 2020 (20190047AR).
METHOD DETAILS
Cytometry by time-of-flight (CyTOF) analysis
Single-cell suspensions derived from primary tumors, co-cultured cells, and xenograft tumors were incubated with Cell-ID Cisplatin (Fluidigm, Cat# 201064) to identify dead cells which were subsequently excluded from the analysis. Remaining cells were fixed using Maxpar Fix I Buffer (Fluidigm, Cat# 201065) following the manufacturer’s instructions. After washing, cells were incubated with antibodies for 1 h at room temperature for cell staining. These antibodies are listed in Table S2 (primary tumors and co-cultured cells) and Table S3 (xenograft tumors). Antibodies were purchased in conjugated form from Fluidigm or conjugated in-house according to the manufacturer’s instructions (Fluidigm). Phospho-AXL antibody was not available for use when we designed the CyTOF panel. Instead, total AXL antibody was used for single cell profiling. Cells were then incubated with Maxpar Fix and Perm Buffer (Fluidigm, Cat# 201067) containing Cell-ID Intercalator-Ir 191/193 (0.125 mM, Fluidigm, Cat# 201192A) at 4°C overnight to stain the nuclei. After signal beads were normalized by EQ Four Element Calibration, cell events were analyzed by Helios mass cytometer (Fluidigm). For CyTOF profiling of xenograft tumors, the staining processes were similar as primary tumors, except that xenograft tumors of each group were barcoded using Cell-ID 20-Plex Pd Barcoding Kit (Fluidigm, Cat# 201060) before incubation with the antibody cocktail.
Signals of samples were normalized using CyTOF software (Version 6.7.1014, Fluidigm). The generated files underwent signal cleanup and gating for live singlet cells in the following steps using Cytobank (https://www.cytobank.org/). 1) The calibration beads were gated out by selecting 140Ce− populations and singlet cells were gated based on 191Ir levels excluding higher 191Ir expressed doublets; 2) live singlet cells were then gated as 195Pt−/191Ir+/193Ir+ populations; 3) Live cell populations were validated using Gaussian parameters. The gated Flow Cytometry Standard (FCS) files were downloaded for further analysis using cytofkit2.68 Cell subpopulations were clustered using PhenoGraph algorithm embedded in cytofkit2 and visualized using t-distributed stochastic neighbor embedding (t-SNE) algorithm based on expression levels of selected markers for primary tumors, co-cultured cells, and xenograft tumors (Table S4).
Identification of cell types in primary tumors was based on the expression of lineage markers (Figure S2) as listed below:
T cell: CD45+/CD3+
B cell: CD45+/CD19+
NK cell: CD45+/CD56+
Macrophage: CD14+ and/or CD16+ and/or CD86+ and/or CD163+
Granulocyte: CD45+/CD66b+
CD45+ cell: remaining CD45+ cells negative of immune markers.
Endothelial cell: CD45−/PECAM+
Stromal cell: CD45−/EpCAM−/CK8/18−/CD90+
Tumor cell: CD45−/EpCAM+/CK8/18+
Other cells: negative of all lineage markers.
To evaluate potential batch effects of CyTOF datasets, we performed principal component analysis (PCA) on the protein expression data to calculate principal components (PC).60 We computed the Spearman’s correlation coefficients between the expression profiles of all proteins and the first two PCs which in total explained 51% of the variance. Other confounders, including tumor samples and processing dates, were similarly analyzed. Violin plots and scatterplots of protein expressions were generated by R package ggplot261 based on arcsinh-transformed CyTOF data output from cytofkit2. E-M indices were calculated based on the mean arcsinh-transformed expression values of epithelial (ZO-2, EpCAM, and CK8/18) and mesenchymal (SNAIL, VIMENTIN, TWIST, N-CADHERIN, FIBRONECTIN, and β-CATENIN) markers. Pseudotime analysis was performed with the destiny package69 in R using expression levels of AXL, STAT3 and seven plasticity markers of CyTOF data from individual patients to calculate dimensionality of data (DC1 and DC2) and diffusion pseudotime (DPT).69
Diversity values were calculated by Shannon index based on population proportion and expression level of AXL, STAT3, Vimentin, Fibronectin, ZO-2, CD44, CD133, CD86, and CD163.70
Cell type proportion index for xenograft tumors was calculated using the following equations:
Subpopulation index of xenograft tumors was computed using the following equation:
Co-culture assay
Five sets of the first co-culture study were: 1) U937-derived macrophages (2 × 106); 2) A549 lung cancer cells (2 × 106); 3) A549 lung cancer cells (2 × 106) + U937-derived macrophages (2 × 106); 4) MRC-5 lung fibroblasts (2 × 106); 5) MRC-5 lung fibroblasts (2 × 106)+ U937-derived macrophages (2 × 106). These cells were cultured in RPMI 1640 supplemented with 10% FBS and 100 units/mL penicillin-streptomycin (Gibco) for 72 h. After co-culture, cells were dissociated using TrypLE (Gibco) into single-cell suspensions for CyTOF analysis.
Western blotting
U937-derived macrophages were treated without or with IL-11 (25 and 100 ng/mL) for 72 h. Protein lysates were harvested in Pierce RIPA buffer (ThermoFisher Scientific, Cat# 89901) supplemented with Pierce proteinase and phosphatase inhibitor mini tablets (ThermoFisher Scientific, Cat# A32961). Protein concentrations were determined by Pierce BCA Protein Assay Kit (ThermoFisher Scientific). Fifty micrograms of protein lysates from each sample were loaded onto Bolt 4–12% Bis-Tris Mini Protein Gel and protein was transferred to nitrocellulose (NC) membrane. Phosphorylated STAT3 was detected with rabbit anti-pSTAT3 antibody (Cell Signaling Technology, 9131S) at 1:1000 dilution ratio.
U937-derived macrophages were treated with MRC-5-derived conditioned medium without or with IL-11 antibody (R&D Systems, Cat# MAB218) at 15 μg/mL. Protein lysates were harvested as described above. Thirty micrograms of protein lysates from each sample were loaded onto Bolt 4–12% Bis-Tris Mini Protein Gel and protein was transferred to NC membrane. CD163 was detected with rabbit anti-CD163 antibody (Abcam, Cat# AB87099) at 1:1000 dilution ratio.
A549 were cultured and treated with dubermatinib (20 nmol/L) and/or momelotinib (3 μmol/L) for 72 h. Protein lysates were harvested as described above. Fifty micrograms of protein lysates were separated by Bolt 4–12% Bis-Tris Mini Protein Gel (ThermoFisher Scientific) and transferred to polyvinylidene fluoride (PVDF) membrane. The membrane was then blocked with 5% of Blotting-Grade Blocker (BioRad) in TBST and probed using primary antibodies pSTAT3, (Cell Signaling Technology, Cat# 9131S) and loading control antibody GAPDH (Cell Signaling Technology, Cat# 2118S). Membrane was incubated in HRP-linked secondary antibodies following dilution with TBST (1:5000) at room temperature for 1 h. All the blots were developed using Western Lightning Plus-ECL Chemiluminescent Reagents (PerkinElmer, Waltham, MA) and Syngene G:BOX Imaging System.
Flow cytometry
Human primary macrophages treated with or without IL-11 were washed using PBS and then incubated in Zombie NIR solution (1:1000) for 20 min. After two washes, cells were incubated with Human TruStain FcX (BioLegend, Cat# 422301) for 10 min and stained with antibodies - Brilliant Violet 421 anti-human CD209 (BioLegend, Cat# 330117), Brilliant Violet 605 anti-human CD206 (BioLegend, Cat# 321140), Brilliant Violet 785 anti-human CD163 (BioLegend, Cat# 333632) and FITC anti-human CD86 (BioLegend, Cat# 374203) for 30 min. Cells were then analyzed by BD FACSCelesta Cell Analyzer (BD Biosciences), and data analyzed using FlowJo software v10.9.
In silico analyses
Clinical information and RNA-seq data of The Cancer Genome Atlas (TCGA) lung adenocarcinoma cohort were downloaded from the cBioPortal for Cancer Genomics (https://www.cbioportal.org/) hosted by the Center for Molecular Oncology at Memorial Sloan Kettering Cancer Center. High IL11 expression was defined as expression Z score > 1.62,71 Patients who were lack of disease-free data were removed in disease-free Kaplan-Meier curve. Kaplan-Meier curves were created in R with survival package63 for patients’ overall and disease-free survival outcomes. ChIP-seq raw data for STAT3 in H358 cells were from Gene Expression Omnibus (GEO) database (GSE79707) and mapped reads number of ChIP-seq dataset on the Galaxy (https://usegalaxy.org/) to generate bam files. All the bam files and reference genes were subsequently loaded into EaSeq software (http://easeq.net).64 ChIP-seq peaks plots were generated by EaSeq software.
Quantitative capillary Western immunoassay (WES)
Protein lysates of U937-derived macrophages treated without or with IL-11 (25 ng/mL and 100 ng/mL) for 72 h were prepared in radio-immunoprecipitation assay buffer (ThermoFisher Scientific) and then analyzed in 12–230 WES separation module (Protein Simple). CD44 (R&D Systems, Cat# BBA10) antibodies were used. Protein expression levels were normalized against GAPDH (Cell Signaling Technology, Cat# 2118S) as the loading control.
In situ proximity ligation assay (PLA) and analysis
Protein-protein interactions of IL-11-GP130 and pGP130-pSTAT3 were examined by PLA using the Duolink In Situ Red Starter kit following the manufacturer’s instructions (Sigma Aldrich, Cat# DUO92101 and DUO92105). For IL-11-GP130, U937-derived macrophages were permeabilized with 0.2% (v/v) Triton X-100 for 10 min after fixation and incubated with rabbit polyclonal anti-IL-11 antibody (1:200, ThermoFisher Scientific, Cat# PA595982) and mouse monoclonal anti-GP130 antibody (1:200, Abcam, Cat# ab27359), which were tagged with anti-rabbit PLUS and anti-mouse MINUS probes. For pGP130-pSTAT3, cells were permeabilized with 0.2% (v/v) Triton X-100 for 10 min after fixation and incubated with rabbit polyclonal anti-phospho GP130 antibody (1:200, ThermoFisher Scientific, Cat# BS-10122R) and mouse monoclonal anti-phospho STAT3 antibody (1:200, Cell Signaling Technology, Cat# 4113), which were tagged with anti-rabbit PLUS and anti-mouse MINUS probes. The cells were then mounted with DAPI after washing and signal amplification with Duolink reagents. Cell images were captured by Zeiss LSM 710 confocal microscope and exported by ZEN blue software. PLA analysis was conducted in two biological replicates and PLA signals from individual cells were counted using ImageJ.72
Chromatin immunoprecipitation (ChIP)-qPCR
U937-derived macrophages treated without or with IL-11 (25 ng/mL) were harvested for ChIP using Pierce Magnetic ChIP kit (ThermoFisher Scientific, Cat# 26157) following manufacturer’s protocol. Briefly, cells (2 × 106) were cross-linked using 1% formaldehyde for 10 min and treated with glycine for 5 min at room temperature. Cells were lysed and DNA was pre-digested by MNase at 37°C for 15 min. DNA were sheared to fragments of 300–500 bp in size by Q800R3 Sonicator at 20% amplitude for 4 min with 10 s on/20 s off cycles (Qsonica). DNA fragments were incubated with anti-pSTAT3 antibody (Cell Signaling Technology, Cat# 9131) or normal rabbit IgG (Pierce Magnetic ChIP Kit, ThermoFisher Scientific, Cat# 26157) at 4°C for 16 h. After proteinase K digestion, immunoprecipitated DNA fragments were eluted and amplified using quantitative real-time PCR (Roche). Antibody signals were normalized to the input signals and Student’s t test was performed to compare the results. Primer sequences were listed in Table S5.
Reverse transcription (RT)-qPCR
Total RNA was isolated using Direct-zol RNA Miniprep Kits (Zymo Research, Cat# R2053) in three biological replicates. Complementary DNA (cDNA) was synthesized from 1 μg of total RNA using High-Capacity cDNA Reverse Transcription kit (Applied Biosystems, Cat# 4368814). Real-time PCR quantification was performed by LightCycler 480 SYBR Green I Master (Roche, Cat# 04887352001), and each sample tested in triplicates. The relative expression levels were normalized by ACTB and calculated using the 2−ΔΔCt method. Primer sequences were listed in Table S6.
Vasculogenic assay and analysis
The tethering of U937-derived macrophages to human umbilical vein endothelial cells (HUVECs) (ATCC, Cat# CRL-1730, RRID:CVCL_2959) and formation of primitive vascular networks were examined using an angiogenesis assay kit (Abcam, Cat# ab204726). U937-derived macrophages were tagged with mCherry were utilized to distinguish them from HUVECs in the co-cultured system. Before cells were seeding, 50 μL of extracellular matrix solution was coated in a 96-well plate and incubated at 37°C for an hour to form a gel. HUVECs (2*104) were collected and mixed with pre-treated U937-derived macrophages (1*104) in 100 μL of RPMI-1640 supplemented with 10% heat-inactive FBS and seeded into the ECM coated wells. CD44 inhibitor (20 μg/mL; Novus Biologicals, Cat# NBP2–22530) was later applied to the wells. The vascular formation was observed for 16 h using IncuCyte ZOOM live cell analysis system (Sartorius). Images at 16-h were exported, and mesh numbers formed by HUVECs in the respective images were calculated with ImageJ.72
Bioluminescence imaging of tumor volumes in mice
Anesthetized mice received an intraperitoneal injection of 15 mg/mL D-luciferin (Goldbio, LUCK-1G) and imaged 10 min later with the IVIS Lumina system. Regions of interest (ROIs) were obtained to quantify bioluminescence utilizing an automated method by Living Image software (Caliper Life Science).
Immunofluorescence of xenograft tumor sections
Xenograft tumor tissues were dissected and embedded in optimal cutting temperature compound (OCT compound) at −20°C. Tissue section slides were obtained by sectioning frozen tissues with CryoStar NX50 (Epredia, MI). OCT compound was removed from section slides with Phosphate-Buffered Saline with 0.1% Tween 20 Detergent (PBST) for 10 min. Section slides were blocked with 1% BSA for 30 min at room temperature and then incubated at 4°C overnight with primary antibodies: rabbit monoclonal F4/80 antibody (1:100, ThermoFisher Scientific, Cat# MA5–16363, RRID:AB_2537882), rat monoclonal CD90.2 (Thy-1.2) (1:100, ThermoFisher Scientific, Cat# 14–0902-82, RRID:AB_467379), mouse monoclonal phospho-Axl (Tyr779) (1:100, ThermoFisher Scientific, Cat# MA5–24334, RRID:AB_2609001), and rabbit polyclonal IL-11 (1:200, ThermoFisher Scientific, Cat# PA595982, RRID:AB_2807784). Tissue section slides were then incubated with secondary antibodies for 1 h under room temperature and mounted by mounting medium with DAPI (Sigma Aldrich, DUO82040–5ML). Images were captured by Zeiss LSM 710 confocal microscope and exported by ZEN blue software (Zeiss).
Luminex multiplex assay
Fresh conditioned media were collected from A549 lung cancer cells culture dishes treated with or without dubermatinib over 24, 48 and 72 h. Debris and dead cells were removed by centrifugation and 0.45 μm filters. Thirty-nine cytokines were measured by MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel - Immunology Multiplex Assay (Millipore, Cat# HCYTOMAG-60K) following the manufacturer’s instructions. The protein analytes were detected suing the Luminex 200 multiplexing system. Thirty-nine cytokines were then categorized into anti-inflammatory, pro-inflammatory, and dual-function cytokines. Cytokine secretion level heatmaps of anti-inflammatory, pro-inflammatory, and dual-function were generated based on secretion level using the decadic logarithm (log base 10).
RNA sequencing
RNA was extracted from vehicle control and shSTAT3 knockdown U937-derived macrophages in three biological replicates by using the PureLink RNA Mini Kit (Thermo Fisher Scientific). shSTAT3 plasmid was purchased from Sigma (TRCN0000329887) and target sequence is GCACAATCTACGAAGAATCAA. Sequencing of cDNAs was performed with NovaSeq S4 100PE as per manufacturer’s instructions. Paired-end FASTQ files were generated and aligned with the human reference genome GRCh38 by using STAR alignment software.65 The RSEM software was applied to quantify expression levels, and fragments per kilo base of transcript per million (FPKM) mapped reads were calculated. Differential expressed genes were compared between control and STAT3 knockdown groups using DESeq2.66 Low expressed genes were filtered with low count values (<10) and candidate genes were divided into upregulated (≥2-fold) and downregulated (≤2-fold) groups with statistical significance (p < 0.001). Volcano plots were generated by enhancedvolcano R package (https://github.com/kevinblighe/EnhancedVolcano).67 Gene set enrichment analysis (GSEA) was performed based on the results from DESeq266 and GSEA plots were created using fgsea R package.73
QUANTIFICATION AND STATISTICAL ANALYSIS
Student’s t-tests or Duncan’s multiple range tests compared protein expressions between two or more groups. All statistical testing was 2-sided with a significance level of 5%. *p < 0.05, **p < 0.001, ***p < 0.0001.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-Human CD45 89Yb | Fluidigm | Cat#3089003B; RRID: AB_2661851 |
| Anti-Human CD3 141Pr | Fluidigm | Cat#3141019B; RRID: AB_2938864 |
| Anti-Human CD19 142ND | Fluidigm | Cat#3142001B; RRID: AB_2651155 |
| Anti-Human N-Cadherin 143ND | R&D Systems | Cat#AF6426; RRID: AB_10718850 |
| Anti-Human ALDH1A1 144ND | R&D Systems | Cat#MAB5869; RRID: AB_10973332 |
| Anti-Human CD163 145ND | Fluidigm | Cat#3145010B |
| Anti-Human ZO-2 146ND | Thermo Fisher Scientific | Cat#374700; RRID: AB_431446 |
| Anti-Human CD16 148ND | Fluidigm | Cat#3148004B; RRID: AB_2661791 |
| Anti-Human CD200 149Sm | Fluidigm | Cat#3149007B |
| Anti-Human CD86 150Ne | Fluidigm | Cat#3150020B; RRID: AB_2661798 |
| Anti-Human/Mouse CD133 151Eu | R&D Systems | Cat#MAB11331-100 |
| Anti-Human SMAD2 152Sm | Thermo Fisher Scientific | Cat#700048; RRID: AB_2532277 |
| Anti-Human JAK1 153Eu | R&D Systems | Cat#MAB4260; RRID: AB_2128403 |
| Anti-Human Fibronectin 155Gd | Thermo Fisher Scientific | Cat#MA517075; RRID: AB_2538546 |
| Anti-Human Vimentin 156Gd | R&D Systems | Cat#MAB2105; RRID: AB_2241653 |
| Anti-Human/Mouse pSTAT3 158Gd | Fluidigm | Cat#3158005A; RRID: AB_2811100 |
| Anti-Human CD90 159Tb | Fluidigm | Cat#3159007B; RRID: AB_2893063 |
| Anti-Human/Mouse OCT3/4 160Gd | R&D Systems | Cat#MAB1759; RRID: AB_2167713 |
| Anti-Human/Mouse AXL 161Dy | R&D Systems | Cat#AF154; RRID: AB_354852 |
| Anti-Human CD66b 162Dy | Fluidigm | Cat#3162023B |
| Anti-Human CD105 163Dy | Fluidigm | Cat#3163005B; RRID: AB_2893065 |
| Anti-Human SMAD4 164Dy | R&D Systems | Cat#MAB2097; RRID: AB_2286470 |
| Anti-Human TGFBR2 165Ho | R&D Systems | Cat#AF-241; RRID: AB_354416 |
| Anti-Human SNAIL 166Er | Sigma Aldrich | Cat#SAB 2108482; RRID: AB_2818978 |
| Anti-Human TWIST 167Er | R&D Systems | Cat#MAB6230; RRID: AB_2818958 |
| Anti-Human β-catenin 168Er | R&D Systems | Cat#MAB13292; RRID: AB_1207871 |
| Anti-Human STRO-1 170Er | R&D Systems | Cat#MAB1038; RRID: AB_357389 |
| Anti-Human/Mouse CD44 171Yb | Fluidigm | Cat#3171003B; RRID: AB_2895121 |
| Anti-Human PECAM 172Yb | Thermo Fisher Scientific | Cat#MA3100; RRID: AB_223516 |
| Anti-Human/Mouse EpCAM 173Yb | R&D Systems | Cat#AF960; RRID: AB_355745 |
| Anti-Human/Mouse CK8/18 174Yb | Fluidigm | Cat#3174014A |
| Anti-Human CD14 175Lu | Fluidigm | Cat#3175015B; RRID: AB_2811083 |
| Anti-Human CD56 176Yb | Fluidigm | Cat#3176003B; RRID: AB_2756430 |
| Anti-Mouse CD29 111Cd | Novus Biologicals | Cat#AF2405; RRID: AB_416591 |
| Anti-Human ZO2 112Cd | Thermo Fisher Scientific | Cat#374700; RRID: AB_431446 |
| Anti-Human VIMENTIN 141Pr | R&D Systems | Cat#MAB2105; RRID: AB_2241653 |
| Anti-Mouse CD11c 142ND | Fluidigm | Cat#3142003B; RRID: AB_2814737 |
| Anti-Mouse CD69 145ND | Fluidigm | Cat#3145005B; RRID: AB_2895115 |
| Anti-Mouse F4/80 146ND | Fluidigm | Cat#3146008B; RRID: AB_2895117 |
| Anti-Mouse CD45 147Sm | Fluidigm | Cat#3147003B; RRID: AB_2811243 |
| Anti-Mouse CD11b 148ND | Fluidigm | Cat#3148003B; RRID: AB_2814738 |
| Anti-Mouse CD19 149Sm | Fluidigm | Cat#3149002B; RRID: AB_2814679 |
| Anti-Mouse CD3e 152Sm | Fluidigm | Cat#3152004B; RRID: AB_2687836 |
| Anti-Mouse CD90.2 156Gd | Fluidigm | Cat#3156006B; RRID: AB_2801433 |
| Anti-Mouse CD163 162Dy | Abcam | Cat#ab182422; RRID: AB_2753196 |
| Anti-Mouse CD31 165Ho | Fluidigm | Cat#3165013B; RRID: AB_2801434 |
| Anti-Mouse CD206 169Tm | Fluidigm | Cat#3169021B; RRID: AB_2832249 |
| Anti-Mouse NK1.1 170Er | Fluidigm | Cat#3170002B; RRID: AB_2885023 |
| Anti-Mouse CD4 172Yb | Fluidigm | Cat#3172003B; RRID: AB_2811242 |
| Anti-Mouse CD38 175Lu | Fluidigm | Cat#3175014B RRID: AB_2895122 |
| Anti-phospho AXL | R&D Systems | Cat#AF2228; RRID: AB_2062560 |
| Anti-phospho STAT3 | Cell Signaling Technology | Cat#9131; RRID: AB_331586 |
| GAPDH | Cell Signaling Technology | Cat#2118S; RRID: AB_561053 |
| Anti-IL-11 | Thermo Fisher Scientific | Cat#PA595982; RRID: AB_2807784 |
| Anti-GP130 | Abcam | Cat#ab27359; RRID: AB_2125965 |
| Anti-phospho STAT3 antibody | Cell Signaling Technology | Cat#4113; RRID: AB_2198588 |
| Anti-phospho GP130 | Thermo Fisher Scientific | Cat#BS-10122R |
| Anti- F4/80 antibody | Thermo Fisher Scientific | Cat#MA5-16363; RRID:AB_2537882 |
| Anti- phospho-Axl (Tyr779) antibody | Thermo Fisher Scientific | Cat# MA5-24334; RRID:AB_2609001 |
| Anti- IL-11 antibody | Thermo Fisher Scientific | Cat#PA595982; RRID:AB_2807784 |
| Brilliant Violet 421™ anti-human CD209 | BioLegend | Cat#330117; RRID:AB_2734323 |
| FITC anti-human CD86 | BioLegend | Cat#374203; RRID:AB_2721573 |
| Brilliant Violet 605™ anti-human CD206 | BioLegend | Cat#321140; RRID:AB_2734300 |
| Brilliant Violet 785™ anti-human CD163 | BioLegend | Cat#333632; RRID:AB_2728288 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Cell-ID Cisplatin | Fluidigm | Cat#201064 |
| Cell-ID Intercalator-Ir | Fluidigm | Cat#201192A |
| Fix I Buffer | Fluidigm | Cat#201065 |
| RIPA buffer | Thermo Fisher Scientific | Cat#89901 |
| Dubermatinib | Selleckchem | Cat#S7846 |
| Momelotinib | Selleckchem | Cat#S2219 |
| D-luciferin | Goldbio | Cat#LUCK-1G |
| Zombie NIR™ Fixable Viability Kit | BioLegend | Cat#423106 |
| Human TruStain FcX™ | BioLegend | Cat#422301 |
|
| ||
| Critical commercial assays | ||
|
| ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| CellTiter-Glo® Luminescent Cell Viability Assay | Promega | Cat#G7571 |
| Cell-ID 20-Plex Pd Barcoding Kit | Fluidigm | Cat#201060 |
| Duolink™ In Situ Red Starter Kit Mouse/Rabbit | Sigma Aldrich | Cat#DUO92101 |
| Pierce Magnetic ChIP Kit | Thermo Fisher Scientific | Cat#26157 |
| LightCycler 480 SYBR Green I Master | Roche | Cat#04887352001 |
| 12-230 kDa Wes Separation Module, 8 × 25 capillary cartridges | ProteinSimple | Cat#SM-W004 |
| Anti-Rabbit Detection Module for Wes, Peggy Sue or Sally Sue | ProteinSimple | Cat#DM-001 |
| Anti-Mouse Detection Module for Wes, Peggy Sue or Sally Sue | ProteinSimple | Cat#DM-002 |
|
| ||
| Deposited data | ||
|
| ||
| RNA sequencing data | This paper | GEO: GSE231829 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| A549 | ATCC | ATCC CCL-185™ |
| U937 | ATCC | ATCC CRL-1593.2™ |
| Human: HUVEC cells | ATCC | Cat#PCS-100-013 |
| Human: peripheral blood CD14+ monocyte cells | Stemcell Technologies | Cat#70035 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: Nu/J | The Jackson Laboratory | JAX:002019, RRID: IMSR_JAX:002019 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for ChIP-qPCR | This paper | See Table S5 |
| Primers for quantitative RT-PCR | This paper | See Table S6 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLenti-puro3/To/V5-GW/EGFP-Firefly luciferase | Lorenzatti Hiles et al.59 | Addgene119816; RRID:Addgene_119816 |
| TRC-designed pLKO.1-puro | Sigma Aldrich | Cat#TRCN0000329887 |
|
| ||
| Software and algorithms | ||
|
| ||
| CyTOF (Version 7.0) | Fluidigm | RRID:SCR_021055 |
| R package: cytofkit2 | Chen et al.59 | https://github.com/JinmiaoChenLab/cytofkit2 |
| R package: ggplot2 | Wickham60 | http://ggplot2.tidyverse.org |
| R package: destiny | Angerer61 | https://github.com/theislab/destiny |
| R package: survival | Therneau et al.62 | https://github.com/therneau/survival |
| EaSeq software | Lerdrup et al.63 | http://easeq.net |
| ImageJ | Schneider et al.64 | https://imagej.nih.gov/ij/ |
| R package: DESeq2 | Love et al.65 | https://doi.org/10.1186/s13059-014-0550-8 |
| R package: EnhancedVolcano | Blighe et al.66 | https://github.com/kevinblighe/EnhancedVolcano |
| R package: fgsea | Korotkevich et al.67 | https://github.com/ctlab/fgsea |
| Compass for SW (Version: 3.1.7) | ProteinSimple | |
| ZEN blue software | Zeiss Company | N/A |
| MeV | N/A | http://mev.tm4.org/ |
| Living Image software | Caliper Life Science | |
| Prism 9 | GraphPad | https://www.graphpad.com/scientific-software/prism/ RRID:SCR_002798 |
| FlowJo™ v10.9 | BD Biosciences | RRID:SCR_008520 |
Highlights.
Concordant AXL-STAT3 enhances M2-like polarization and phenotypic diversity of TAMs
AXL-dependent IL-11 secretion by lung cancer cells and fibroblasts activates STAT3 in TAMs
TAMs promote vasculogenic network via IL-11/AXL-STAT3 manner to upregulate CD44 expression
AXL-STAT3 targeting attenuates M2 polarization and recruitment of TAMs in a mouse model
ACKNOWLEDGMENTS
We are grateful for the contribution made from patients to this study. We also thank the staff of the BioAnalytics and Single-Cell Core (BASiC) and the Next Generation Sequencing Shared Resource at the University of Texas Health Science Center at San Antonio for technical support in proteomic and transcriptomic analyses. This work was supported by the Max and Minnie Tomerlin Voelcker Fund Young Investigator Award (J.A.T.); NIH-NCATS grants KL2 TR001118 and KL2 TR002646 (J.A.T.); and NIH/NCI grants 1R01CA269766–01A1 (J.A.T.), U54CA217297 (T.H.M.H.), and P30CA54174 (NCI Cancer Center Support Grant). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
DECLARATION OF INTERESTS
S.W. is principal investigator at Sumitomo Dainippon Pharma Oncology.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113067.
REFERENCES
- 1.Altorki NK, Markowitz GJ, Gao D, Port JL, Saxena A, Stiles B, McGraw T, and Mittal V (2019). The lung microenvironment: an important regulator of tumour growth and metastasis. Nat. Rev. Cancer 19, 9–31. 10.1038/s41568-018-0081-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin MZ, and Jin WL (2020). The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Targeted Ther. 5, 166. 10.1038/s41392-020-00280-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Labani-Motlagh A, Ashja-Mahdavi M, and Loskog A (2020). The Tumor Microenvironment: A Milieu Hindering and Obstructing Antitumor Immune Responses. Front. Immunol. 11, 940. 10.3389/fimmu.2020.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quatromoni JG, and Eruslanov E (2012). Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am. J. Transl. Res. 4, 376–389. [PMC free article] [PubMed] [Google Scholar]
- 5.Corliss BA, Azimi MS, Munson JM, Peirce SM, and Murfee WL (2016). Macrophages: An Inflammatory Link Between Angiogenesis and Lymphangiogenesis. Microcirculation 23, 95–121. 10.1111/micc.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordon S, Plüddemann A, and Martinez Estrada F (2014). Macrophage heterogeneity in tissues: phenotypic diversity and functions. Immunol. Rev. 262, 36–55. 10.1111/imr.12223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bayik D, and Lathia JD (2021). Cancer stem cell-immune cell crosstalk in tumour progression. Nat. Rev. Cancer 21, 526–536. 10.1038/s41568-021-00366-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chambers M, Rees A, Cronin JG, Nair M, Jones N, and Thornton CA (2020). Macrophage Plasticity in Reproduction and Environmental Influences on Their Function. Front. Immunol. 11, 607328. 10.3389/fimmu.2020.607328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma RY, Black A, and Qian BZ (2022). Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 43, 546–563. 10.1016/j.it.2022.04.008. [DOI] [PubMed] [Google Scholar]
- 10.Macciò A, Gramignano G, Cherchi MC, Tanca L, Melis L, and Madeddu C (2020). Role of M1-polarized tumor-associated macrophages in the prognosis of advanced ovarian cancer patients. Sci. Rep. 10, 6096. 10.1038/s41598-020-63276-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Genin M, Clement F, Fattaccioli A, Raes M, and Michiels C (2015). M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 15, 577. 10.1186/s12885-015-1546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao X, Di Q, Liu H, Quan J, Ling J, Zhao Z, Xiao Y, Wu H, Wu Z, Song W, et al. (2022). MEF2C promotes M1 macrophage polarization and Th1 responses. Cell. Mol. Immunol. 19, 540–553. 10.1038/s41423-022-00841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jayasingam SD, Citartan M, Thang TH, Mat Zin AA, Ang KC, and Ch’ng ES (2019). Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 9, 1512. 10.3389/fonc.2019.01512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martinez FO, and Gordon S (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6, 13. 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orecchioni M, Ghosheh Y, Pramod AB, and Ley K (2019). Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front. Immunol. 10, 1084. 10.3389/fimmu.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das A, Yang CS, Arifuzzaman S, Kim S, Kim SY, Jung KH, Lee YS, and Chai YG (2018). High-Resolution Mapping and Dynamics of the Transcriptome, Transcription Factors, and Transcription Co-Factor Networks in Classically and Alternatively Activated Macrophages. Front. Immunol. 9, 22. 10.3389/fimmu.2018.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Himes SR, Sester DP, Ravasi T, Cronau SL, Sasmono T, and Hume DA (2006). The JNK are important for development and survival of macrophages. J. Immunol. 176, 2219–2228. 10.4049/jimmunol.176.4.2219. [DOI] [PubMed] [Google Scholar]
- 18.Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, and Tsatsanis C (2017). Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 198, 1006–1014. 10.4049/jimmunol.1601515. [DOI] [PubMed] [Google Scholar]
- 19.Wang YC, He F, Feng F, Liu XW, Dong GY, Qin HY, Hu XB, Zheng MH, Liang L, Feng L, et al. (2010). Notch signaling determines the M1 versus M2 polarization of macrophages in antitumor immune responses. Cancer Res. 70, 4840–4849. 10.1158/0008-5472.CAN-10-0269. [DOI] [PubMed] [Google Scholar]
- 20.Batlle E, and Massagué J (2019). Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 50, 924–940. 10.1016/j.immuni.2019.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malyshev I, and Malyshev Y (2015). Current Concept and Update of the Macrophage Plasticity Concept: Intracellular Mechanisms of Reprogramming and M3 Macrophage “Switch” Phenotype. BioMed Res. Int. 2015, 341308. 10.1155/2015/341308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taverna JA, Hung CN, DeArmond DT, Chen M, Lin CL, Osmulski PA, Gaczynska ME, Wang CM, Lucio ND, Chou CW, et al. (2020). Single-Cell Proteomic Profiling Identifies Combined AXL and JAK1 Inhibition as a Novel Therapeutic Strategy for Lung Cancer. Cancer Res. 80, 1551–1563. 10.1158/0008-5472.Can-19-3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiss F, Lauffenburger D, and Friedl P (2022). Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat. Rev. Cancer 22, 157–173. 10.1038/s41568-021-00427-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lambert AW, and Weinberg RA (2021). Linking EMT programmes to normal and neoplastic epithelial stem cells. Nat. Rev. Cancer 21, 325–338. 10.1038/s41568-021-00332-6. [DOI] [PubMed] [Google Scholar]
- 25.Chen L, Fu C, Zhang Q, He C, Zhang F, and Wei Q (2020). The role of CD44 in pathological angiogenesis. Faseb. J. 34, 13125–13139. 10.1096/fj.202000380RR. [DOI] [PubMed] [Google Scholar]
- 26.González-Mariscal L, Miranda J, Raya-Sandino A, Domínguez-Calderón A, and Cuellar-Perez F (2017). ZO-2, a tight junction protein involved in gene expression, proliferation, apoptosis, and cell size regulation. Ann. N. Y. Acad. Sci. 1397, 35–53. 10.1111/nyas.13334. [DOI] [PubMed] [Google Scholar]
- 27.Glumac PM, and LeBeau AM (2018). The role of CD133 in cancer: a concise review. Clin. Transl. Med. 7, 18. 10.1186/s40169-018-0198-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Duan Z, and Luo Y (2021). Targeting macrophages in cancer immunotherapy. Signal Transduct. Targeted Ther. 6, 127. 10.1038/s41392-021-00506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shrivastava R, and Shukla N (2019). Attributes of alternatively activated (M2) macrophages. Life Sci. 224, 222–231. 10.1016/j.lfs.2019.03.062. [DOI] [PubMed] [Google Scholar]
- 30.Baek YS, Haas S, Hackstein H, Bein G, Hernandez-Santana M, Lehrach H, Sauer S, and Seitz H (2009). Identification of novel transcriptional regulators involved in macrophage differentiation and activation in U937 cells. BMC Immunol. 10, 18. 10.1186/1471-2172-10-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu CS, Shiau AL, Su BH, Hsu TS, Wang CT, Su YC, Tsai MS, Feng YH, Tseng YL, Yen YT, et al. (2020). Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J. Hematol. Oncol. 13, 62. 10.1186/s13045-020-00887-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okazaki K, Anzawa H, Liu Z, Ota N, Kitamura H, Onodera Y, Alam MM, Matsumaru D, Suzuki T, Katsuoka F, et al. (2020). Enhancer remodeling promotes tumor-initiating activity in NRF2-activated non-small cell lung cancers. Nat. Commun. 11, 5911. 10.1038/s41467-020-19593-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huynh PT, Beswick EJ, Coronado YA, Johnson P, O’Connell MR, Watts T, Singh P, Qiu S, Morris K, Powell DW, and Pinchuk IV (2016). CD90(+) stromal cells are the major source of IL-6, which supports cancer stem-like cells and inflammation in colorectal cancer. Int. J. Cancer 138, 1971–1981. 10.1002/ijc.29939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin X, and Kruth HS (2016). Culture of Macrophage Colony-stimulating Factor Differentiated Human Monocyte-derived Macrophages. J. Vis. Exp. 10.3791/54244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li C, Xu X, Wei S, Jiang P, Xue L, and Wang J (2021). Tumor-associated macrophages: potential therapeutic strategies and future prospects in cancer. J. Immunother. Cancer 9, e001341. 10.1136/jitc-2020-001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yao Y, Xu XH, and Jin L (2019). Macrophage Polarization in Physiological and Pathological Pregnancy. Front. Immunol. 10, 792. 10.3389/fimmu.2019.00792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnett FH, Rosenfeld M, Wood M, Kiosses WB, Usui Y, Marchetti V, Aguilar E, and Friedlander M (2016). Macrophages form functional vascular mimicry channels in vivo. Sci. Rep. 6, 36659. 10.1038/srep36659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Graney PL, Ben-Shaul S, Landau S, Bajpai A, Singh B, Eager J, Cohen A, Levenberg S, and Spiller KL (2020). Macrophages of diverse phenotypes drive vascularization of engineered tissues. Sci. Adv. 6, eaay6391. 10.1126/sciadv.aay6391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paulis YWJ, Huijbers EJM, van der Schaft DWJ, Soetekouw PMMB, Pauwels P, Tjan-Heijnen VCG, and Griffioen AW (2015). CD44 enhances tumor aggressiveness by promoting tumor cell plasticity. Oncotarget 6, 19634–19646. 10.18632/oncotarget.3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grivennikov SI (2013). IL-11: a prominent pro-tumorigenic member of the IL-6 family. Cancer Cell 24, 145–147. 10.1016/j.ccr.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson DE, O’Keefe RA, and Grandis JR (2018). Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 15, 234–248. 10.1038/nrclinonc.2018.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.González M, Merino R, González AL, and Merino J (1995). The ability of B cells to participate in allogeneic cognate T-B cell interactions in vitro depends on the presence of CD4+ T cells during their development. J. Immunol. 155, 1091–1100. [PubMed] [Google Scholar]
- 43.Szadvari I, Krizanova O, and Babula P (2016). Athymic nude mice as an experimental model for cancer treatment. Physiol. Res. 65, S441–S453. 10.33549/physiolres.933526. [DOI] [PubMed] [Google Scholar]
- 44.Carniti C, Gimondi S, Vendramin A, Recordati C, Confalonieri D, Bermema A, Corradini P, and Mariotti J (2015). Pharmacologic Inhibition of JAK1/JAK2 Signaling Reduces Experimental Murine Acute GVHD While Preserving GVT Effects. Clin. Cancer Res. 21, 3740–3749. 10.1158/1078-0432.Ccr-14-2758. [DOI] [PubMed] [Google Scholar]
- 45.Asshoff M, Petzer V, Warr MR, Haschka D, Tymoszuk P, Demetz E, Seifert M, Posch W, Nairz M, Maciejewski P, et al. (2017). Momelotinib inhibits ACVR1/ALK2, decreases hepcidin production, and ameliorates anemia of chronic disease in rodents. Blood 129, 1823–1830. 10.1182/blood-2016-09-740092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chan E, Luwor R, Burns C, Kannourakis G, Findlay JK, and Ahmed N (2018). Momelotinib decreased cancer stem cell associated tumor burden and prolonged disease-free remission period in a mouse model of human ovarian cancer. Oncotarget 9, 16599–16618. 10.18632/oncotarget.24615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koblish H, Li YL, Shin N, Hall L, Wang Q, Wang K, Covington M, Marando C, Bowman K, Boer J, et al. (2018). Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies. PLoS One 13, e0199108. 10.1371/journal.pone.0199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Antony J, and Huang RYJ (2017). AXL-Driven EMT State as a Targetable Conduit in Cancer. Cancer Res. 77, 3725–3732. 10.1158/0008-5472.can-17-0392. [DOI] [PubMed] [Google Scholar]
- 49.Wu F, Li J, Jang C, Wang J, and Xiong J (2014). The role of Axl in drug resistance and epithelial-to-mesenchymal transition of non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 7, 6653–6661. [PMC free article] [PubMed] [Google Scholar]
- 50.Yu H, Lee H, Herrmann A, Buettner R, and Jove R (2014). Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat. Rev. Cancer 14, 736–746. 10.1038/nrc3818. [DOI] [PubMed] [Google Scholar]
- 51.Digiacomo G, Tusa I, Bacci M, Cipolleschi MG, Dello Sbarba P, and Rovida E (2017). Fibronectin induces macrophage migration through a SFK-FAK/CSF-1R pathway. Cell Adhes. Migrat. 11, 327–337. 10.1080/19336918.2016.1221566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Thalla DG, Rajwar AC, Laurent AM, Becher JE, Kainka L, and Lautenschläger F (2022). Extracellular vimentin is expressed at the rear of activated macrophage-like cells: Potential role in enhancement of migration and phagocytosis. Front. Cell Dev. Biol. 10, 891281. 10.3389/fcell.2022.891281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao W, Li Y, and Zhang X (2017). Stemness-Related Markers in Cancer. Cancer Transl. Med. 3, 87–95. 10.4103/ctm.ctm_69_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roh-Johnson M, Bravo-Cordero JJ, Patsialou A, Sharma VP, Guo P, Liu H, Hodgson L, and Condeelis J (2014). Macrophage contact induces RhoA GTPase signaling to trigger tumor cell intravasation. Oncogene 33, 4203–4212. 10.1038/onc.2013.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du Cheyne C, Tay H, and De Spiegelaere W (2020). The complex TIE between macrophages and angiogenesis. Anat. Histol. Embryol. 49, 585–596. 10.1111/ahe.12518. [DOI] [PubMed] [Google Scholar]
- 56.Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, Bercovici N, Guérin M, Biton J, Ouakrim H, et al. (2018). Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 115, E4041–E4050. 10.1073/pnas.1720948115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chávez-Galán L, Olleros ML, Vesin D, and Garcia I (2015). Much More than M1 and M2 Macrophages, There are also CD169(+) and TCR(+) Macrophages. Front. Immunol. 6, 263. 10.3389/fimmu.2015.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rios-Doria J, Stevens C, Maddage C, Lasky K, and Koblish HK (2020). Characterization of human cancer xenografts in humanized mice. J. Immunother. Cancer 8, e000416. 10.1136/jitc-2019-000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lorenzatti Hiles G, Cates AL, El-Sawy L, Day KC, Broses LJ, Han AL, Briggs HL, Emamdjomeh A, Chou A, Abel EV, et al. (2019). A surgical orthotopic approach for studying the invasive progression of human bladder cancer. Nat. Protoc. 14, 738–755. 10.1038/s41596-018-0112-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst S, Soysal SD, et al. (2019). A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 177, 1330–1345.e18. 10.1016/j.cell.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- 62.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. 10.1158/2159-8290.cd-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terry M Therneau PMG (2000). Modeling Survival Data: Extending the Cox Model (Springer; ). [Google Scholar]
- 64.Lerdrup M, Johansen JV, Agrawal-Singh S, and Hansen K (2016). An interactive environment for agile analysis and visualization of ChIP-sequencing data. Nat. Struct. Mol. Biol. 23, 349–357. 10.1038/nsmb.3180. [DOI] [PubMed] [Google Scholar]
- 65.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blighe K, Rana S, and Lewis M. (2018). EnhancedVolcano: Publication-ready volcano plots with enhanced colouring and labeling. https://github.com/kevinblighe/EnhancedVolcano. [Google Scholar]
- 68.Chen H, Lau MC, Wong MT, Newell EW, Poidinger M, and Chen J (2016). Cytofkit: A Bioconductor Package for an Integrated Mass Cytometry Data Analysis Pipeline. PLoS Comput. Biol. 12, e1005112. 10.1371/journal.pcbi.1005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Angerer P, Haghverdi L, Büttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- 70.Potts SJ, Krueger JS, Landis ND, Eberhard DA, Young GD, Schmechel SC, and Lange H (2012). Evaluating tumor heterogeneity in immunohistochemistry-stained breast cancer tissue. Lab. Invest. 92, 1342–1357. 10.1038/labinvest.2012.91. [DOI] [PubMed] [Google Scholar]
- 71.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1. 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2021). Fast gene set enrichment analysis. Preprint at bioRxiv, 060012. 10.1101/060012. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
RNA-seq data for this study is available through the Gene Expression Omnibus (GEO) under accession number GSE231829.