

Abstract

TLR7 agonists have significant therapeutic potential in a variety of oncology and autoimmune applications. We recently reported a potent TLR7 selective agonist 1 that could be delivered by antibody–drug conjugate (ADC) technology to elicit potent anticancer activity. Herein we report synthetic chemistry and structure–activity relationship studies to develop TLR7 agonists with improved potency for next-generation ADC efforts. We found that the addition of hydrophobic acyl tails to parent compound 1 generally resulted in retained or improved TLR7 agonist activity without sacrificing the permeability or the selectivity over TLR8. In contrast, the addition of a simple alkyl tail at the same position resulted in a dramatic loss in potency. Molecular modeling was performed to provide a rationale for this dramatic loss in potency. We ultimately identified compounds 17b, 16b, and 16d as highly potent TLR7 agonists that potently induced the activation of mouse macrophages and hPBMCs at low-nanomolar concentrations.
Keywords: Toll-like receptor, TLR7, TLR8, Cytokine, TNFα, IFNα
Toll-like receptors (TLRs) play a critical role in the innate immune system by virtue of their ability to recognize danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs). TLRs are widely found on the surface of various leukocytes but are particularly prevalent on myeloid cells such as monocytes, dendritic cells, and macrophages. When a TLR engages a DAMP or PAMP, it activates various downstream pathways (particularly the NFκB and IRF pathways) that result in cytokine release and increased proliferation. There are 10 human TLRs that have been identified to date, and each one recognizes a specific type of PAMP or DAMP. For example, TLR4 recognizes lipopolysaccharide (LPS) found on the outer surface of Gram-negative bacteria, TLR9 recognizes unmethylated CpG DNA found in various bacteria and viruses, and TLR7 and TLR8 recognize single-stranded viral RNA.1−3
Given the crucial role that TLRs play in both innate and adaptive immunity, it is not surprising that they are also important in the pathophysiology of various disease states and have also served as important targets for drug development. Dysregulation of TLR signaling has been linked to a number of autoimmune and inflammatory diseases. For example, a classical hallmark of lupus, rheumatoid arthritis, and multiple sclerosis is the overactivation of TLR signaling pathways.1,4,5 On the other hand, activation of TLR pathways has also been receiving increased attention in recent years due to therapeutic potential in the generation of vaccine adjuvants, antiviral agents, and cancer immunotherapies.6−9
Among the TLRs, TLR7 and TLR8 agonists have been a particular focus of various drug discovery efforts in recent years. Both of these TLRs are involved in the recognition of single-stranded RNA and play an important role in antiviral immune responses. Unlike most TLRs, which generally respond only to large peptides and lipids, TLR7 and TLR8 are known to be modulated by a variety of small “drug-like” agonists that are conceptually derived from purines and pyrimidines. Numerous small-molecule TLR7/8 agonists have been evaluated in clinical and preclinical studies (Figure 1). Indeed, imiquimod (a modestly potent dual TLR7/8 agonist) is FDA-approved as a topical antiviral agent and is also used for the treatment of basal cell carcinoma.10,11 Other more potent TLR7/8 agonists have also been evaluated in the clinic, including resiquimod (R848) and motolimod (VTX-2337). TLR7/8 agonists are known to activate both the innate and adaptive immune systems through their induction of TH1-polarizing cytokines such as interferon-α (IFNα) and tumor necrosis factor α (TNFα).
Figure 1.

Structures of known small-molecule TLR7/8 agonists.
Unfortunately, the therapeutic utility of TLR7/8 agonists has been greatly impeded by the risk associated with cytokine release syndrome (CRS). Several strategies have been employed to overcome this limitation, including topical delivery, antedrug design, and, most recently, antibody-mediated delivery.12−16 Our team recently reported an antibody–drug conjugate (ADC)-mediated approach to deliver TLR7 agonists selectively to cancer tissue.17 This approach relies on a tumor-targeting antibody that is covalently bound to a highly permeable TLR7 agonist via a lysosomally cleavable linker. Upon internalization into the tumor cells, the ADC linker is cleaved, thereby releasing the TLR7 agonist to diffuse into nearby tumor-associated immune cells. (Figure 2) The focus of these initial ADC design efforts was on a particular imidazo[4,5-c]quinoline known as E104 (1). A focus of the work presented herein is to increase the potency and permeability of compound 1 in order to facilitate more robust antitumor activity in future ADC designs.
Figure 2.
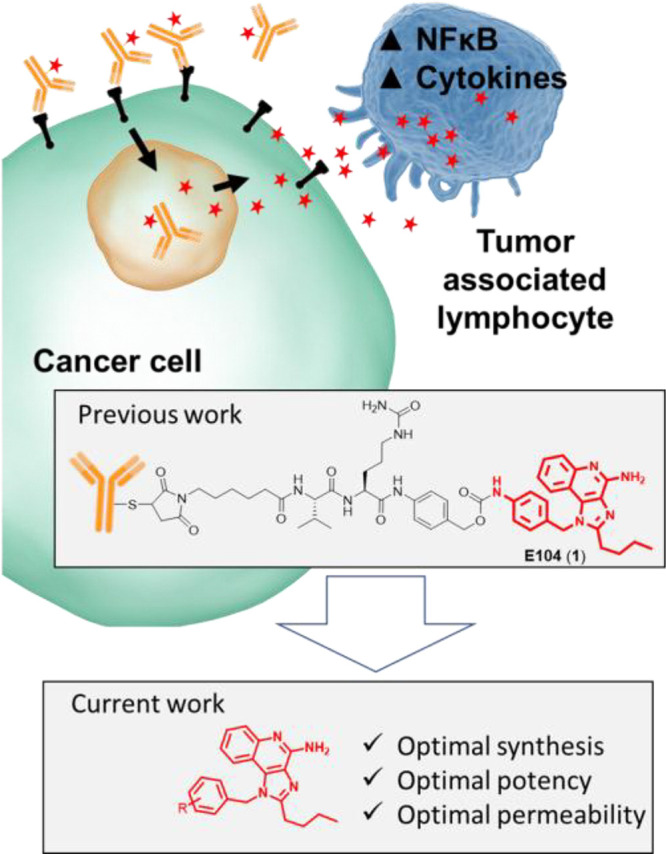
Overview of previous and current work.
One of the limitations of ADC therapeutics is their reliance on cell-surface antigens for uptake. As the antigen–ADC complex is internalized, the antigen density on the cell surface is reduced, thereby limiting the number of ADC payloads that can be processed. For this reason, ADC design typically requires exceptionally high potency payloads, often in the single-digit-nanomolar to picomolar range. As we previously reported, the design of 1 was based on the premise that our ADC technology required a selective TLR7 agonist with low-nanomolar potency, high permeability, and an amine handle that could be used for linker attachment.17 With this in mind, we wish to report our efforts to understand the structure–activity relationship (SAR) of the anilino portion of 1 with the goal of identifying second-generation TLR7 agonists that may be useful for new ADC designs.
Previously published synthetic routes for indolo[4,5-c]quinolines typically relied on the introduction of the benzylic group in the first step of the sequence.18 Inspired by a 2014 patent by Vernejoul,19 we designed a synthetic route (Scheme 1) that introduced the diversity element of interest (the benzyl group) late in the synthetic pathway, thus facilitating SAR studies. This route relied on the preparation of an imidazolo[4,5-c]quinoline intermediate 5 that could (reportedly) be alkylated with 4-nitrobenzyl bromide at the 1-position of the imidazolo ring, resulting in compound 7. While a previously published alkylation with 4-methoxybenzyl chloride favored alkylation at the 3-position,20 we were optimistic that a change in the electronics of the alkylating agent would favor our desired regioisomer, as reported by Vernejoul. The first three steps of the sequence proceeded without issue. Compound 2 underwent an SNAr reaction with ammonia resulting in nitroquinoline 3 in high yield, which in turn was reduced with iron to give diamine 4. Thermal-promoted dehydration with valeric acid resulted in the formation of the core imidazolo[4,5-c]quinoline intermediate 5. Unfortunately, we found that the alkylation of 5 proceeded with undesired regiochemistry, providing 3-alkylated product 6 as the major isomer. Only trace amounts of the desired regioisomer 7 were formed.
Scheme 1. Synthetic Route to 1 via Intermediate 7.
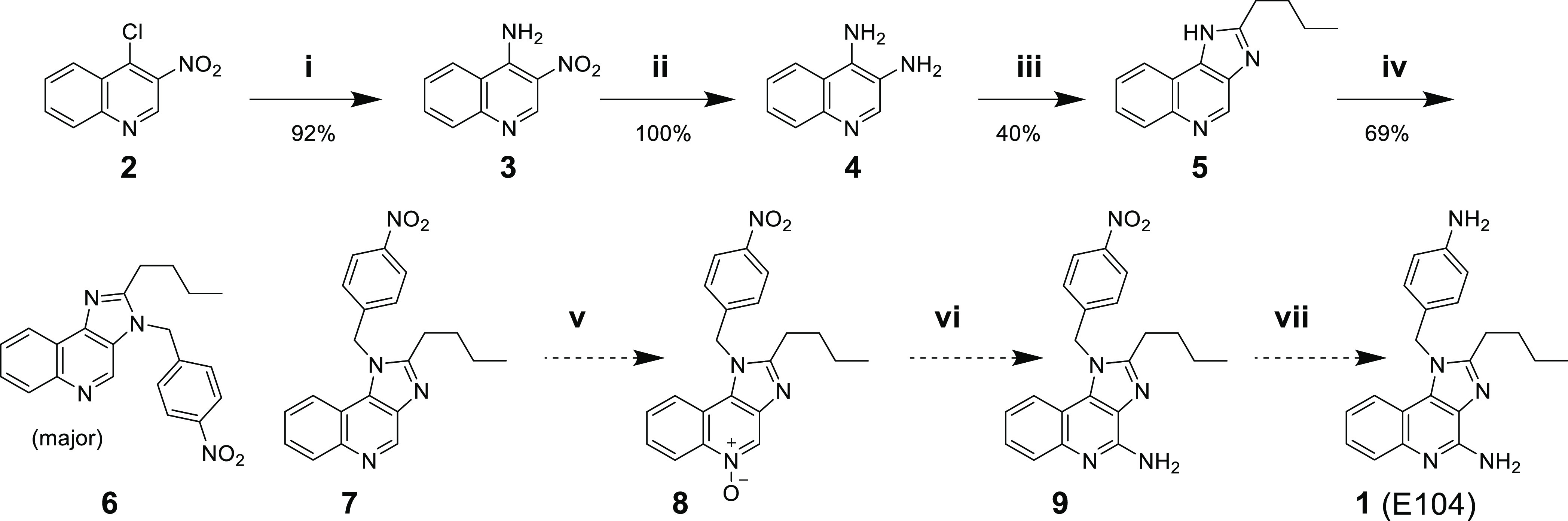
Reagents and conditions: (i) NH4OH, dioxane, 120 °C; (ii) NH4Cl, Fe, EtOH, H2O; (iii) valeric acid, 130 °C; (iv) p-nitrobenzyl bromide, DMF; (v) mCPBA, CHCl3; (vi) NH4OH, CHCl3; (vii) NH4COOH, Zn, MeOH.
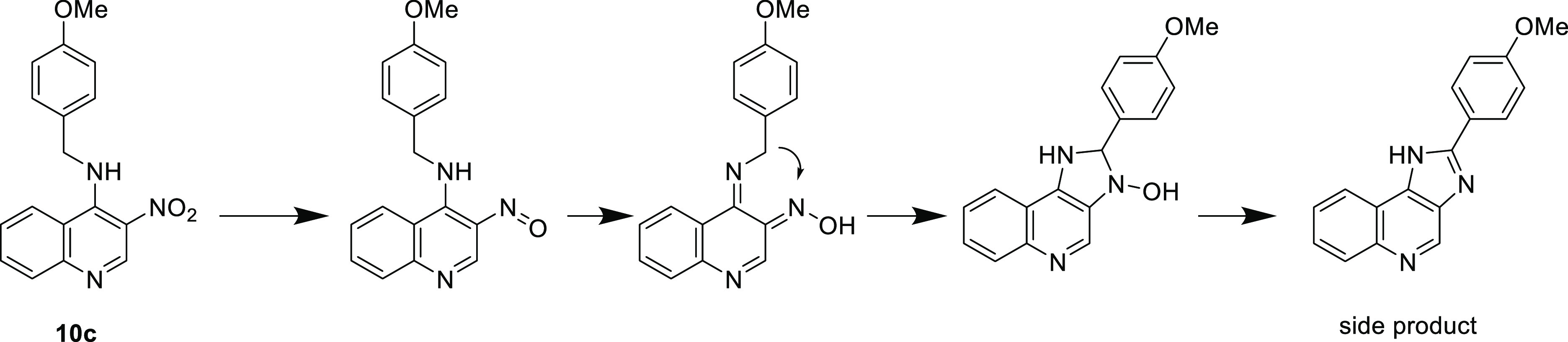
Based on the unsuccessful attempts to introduce the benzylic element late in the sequence, we reverted to a slight modification of the published synthetic scheme wherein the benzylic amine is added as the first step in the sequence. (Scheme 2) SNAr displacement of the 4-chloro moiety of 2 proceeded in high yield with little difficulty, giving 10a–d. The zinc-mediated reduction of the 3-nitro group resulted in the production of a side product that severely impacted the yield. The molecular weight and polarity of this byproduct were consistent with 2-(4-methoxyphenyl)-1H-imidazo[4,5-c]quinoline resulting from a rearrangement through a possible mechanism shown in Scheme 3. Performing the reaction at increased temperature exacerbated this problem. However, we found that using an excess of reagents (10 equiv each of Zn and ammonium chloride), keeping the reaction at 0 °C, and strictly controlling the reaction time (∼25 min) resulted in minimal formation of the byproduct while maintaining reproducible and high yields of compounds 11a–d.
Scheme 2. Synthetic Route to 1 and Derivatives via Intermediates 10.
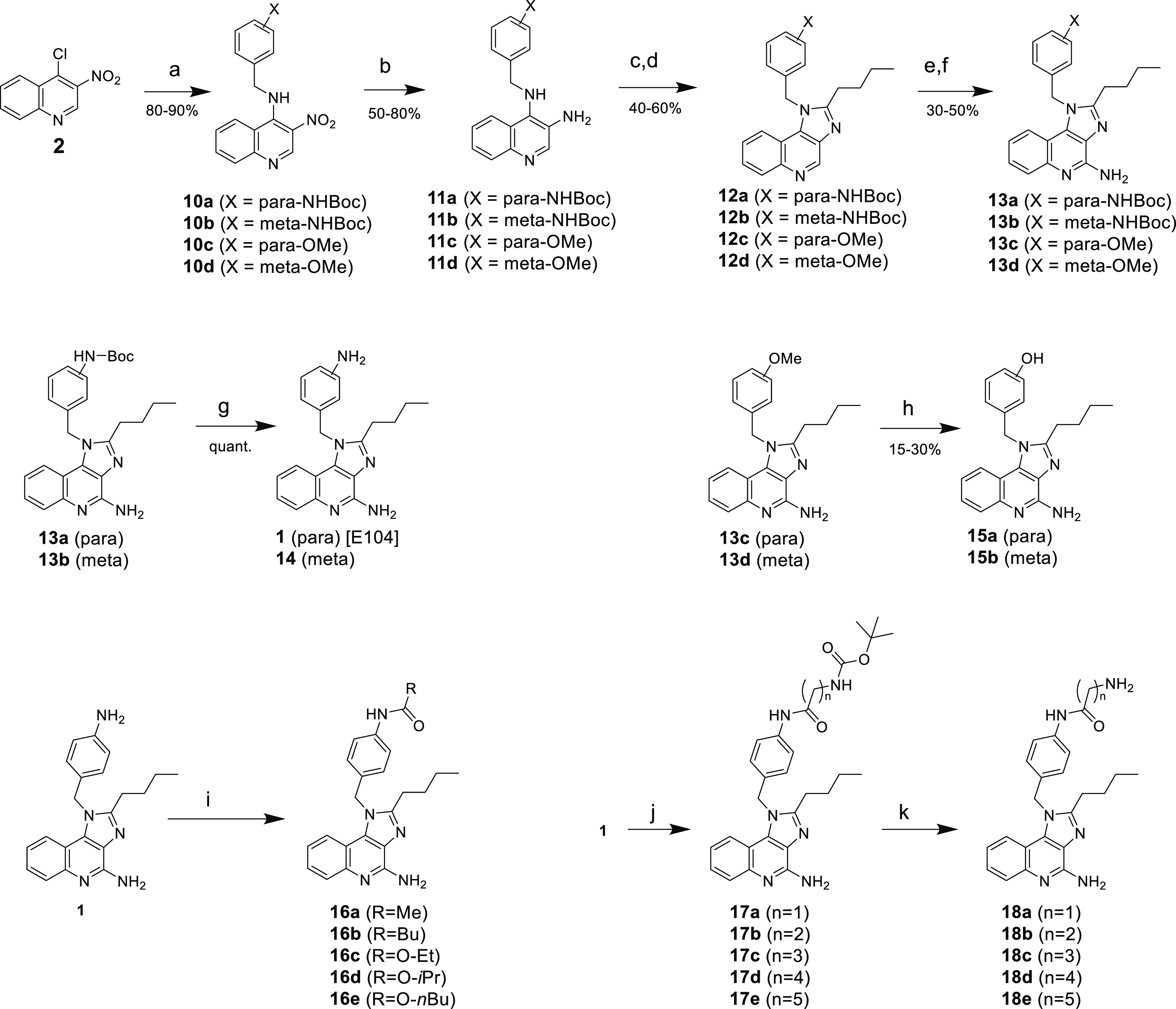
Reagents and conditions: (a) Et3N, substituted benzyl amine, DCM, 30–40 °C; (b) Zn, NH4Cl, MeOH, 0 °C; (c) C5H9ClO, Et3N, EtOAc, 0 °C; (d) NaOH, EtOH, H2O, 80 °C; (e) mCPBA, CHCl3, 40 °C; (f) NH4OH, TsCl, CHCl3; (g) TFA, DCM; (h) BBr3, DCM, 0 °C → RT; (i) RCOCl, Et3N, DCM; (j) RCO2H, HOBt, DIEA, EDC; (k) TFA, DCM.
Scheme 3. Proposed Structure of a Byproduct That Complicated the Zinc Reduction of 10c and 10d.
The sequence continued with acylation of the 3-amino group with valeryl chloride followed by base-promoted cyclization, giving imidazo[4,5-c]quinolines 12a–d. Amination of the core quinoline took place in a two-step process involving N-oxidation with mCPBA followed by treatment with TsCl and ammonia, giving 13a–d. We found that it was essential that this reaction be performed in a “two-phase” system wherein the N-oxide reactant was rapidly stirred in CHCl3 and treated with excess aqueous ammonium hydroxide prior to the addition of the TsCl. The best yields were obtained when the reaction mixture was gently heated and the reaction time was kept under 2 h. Finally, the Boc protecting group was removed from 13a and 13b with TFA, providing compounds 1 and 14, respectively. Likewise, the demethylation of 13c and 13d with BBr3 resulted in the clean formation of 15a and 15b, respectively, albeit in low yield.
In comparing the functional activity of these derivatives (13c, 13d, 1, 14, 15a, and 15b), it became apparent that the most potent derivative was originally identified as compound 1 (see below). Therefore, compound 1 was used as the starting material to explore various analogs that incorporated hydrophobic tails attached to the anilino moiety. We initially presumed that acylation would selectively occur on the anilino nitrogen rather the amidine of the imidazolo[4,5-c]quinoline due to the electron-withdrawing effect of the quinoline nitrogen. However, during EDC couplings to prepare 17a–e, we frequently observed trace to (sometimes) substantial amounts of the alternative acylation product in which the amidine of the core was also acylated (Figure 3, isomer B) While the byproduct was readily removable by preparative HPLC, the adverse impact on product yield (particularly at larger scale) prompted us to investigate whether the selection of the coupling agent may minimize the formation of this byproduct. Interestingly, we found that the reactions promoted by T3P and EEDQ were extremely slow, with minimal product formation observed after 24 h. On the other hand, we found that HATU rapidly promoted the desired acylation while resulting in considerably lower levels of the alternative acylation product.
Figure 3.

LCMS/MS fragmentation was used to confirm the site of acylation.
The presence of the two acylated isomers in some of the reactions prompted us to develop an MS/MS method to conclusively verify which amino group of 1 underwent acylation. Specifically, we relied on the fragmentation of the weak benzylic bond, as shown in Figure 3. Fragmentation of isomer A (the anticipated major product) should result in fragments A and A′, both of which were observed in the major product. On the other hand, fragmentation of isomer B should result in fragments B and B′, both of which were observed in the minor product. An example of one such structural determination using this LC–MS/MS fragmentation method (for compound 17b) is shown in Figure 3. Additional examples of these structural determinations are shown in Figure S1.
The various analogs described in Scheme 2 were evaluated in a dose–response assay using a Ramos Blue reporter assay that has been widely used for assessment of TLR7 agonists. Ramos cells are a human B-cell lymphoma line that endogenously expresses TLR1, -2, -4, -7, and -9. The Ramos Blue cell line (InvivoGen) has been engineered to produce secreted embryonic alkaline phosphatase (SEAP) in response to NFκB activation, thereby allowing rapid colorimetric assessment of TLR7 activation. We previously reported using this assay system to demonstrate that compound 1 potently activated the NFκB pathway in response to TLR7 stimulation.17 Accordingly, we performed a dose titration of Ramos Blue cells in order to evaluate the potency and efficacy of the compounds prepared above (Figure 4). Most compounds elicited a 5–15-fold activation of SEAP compared to baseline levels. The efficacies of the compounds were compared using the EC50, as determined by a four-parameter nonlinear regression model, and the Cmax, defined to be the lowest concentration of agonist that resulted in a maximal NFκB response (Table 1).
Figure 4.

Ramos Blue cells were treated with the shown compounds for 72 h. The dotted blue line represents results for our previously published parent compound E104 (1). For the sake of clarity, some of the high-concentration data points that elicited negative feedback (or cell toxicity) were eliminated in order to allow for improved curve fitting. The full curves without any deleted points can be found in the Supporting Information.
Table 1. Results of the Ramos Blue NFκB Reporter Assay and PAMPA Permeability Screeninga.
| compd | R1 | R2 | EC50 (nM) | Cmax (nM) | Papp (10–6 cm/s) | log Papp |
|---|---|---|---|---|---|---|
| R848 | (see Figure 1) | 616 ± 24 | 1870 | 5.30 | –5.29 | |
| 1 | –NH2 | –H | 69 ± 10 | 620 | 8.51 | –5.08 |
| 14 | –H | –NH2 | 199 ± 26 | 620 | 5.26 | –5.28 |
| 13b | –H | –NHBoc | 1310 ± 230 | 5600 | 1.24 | –5.91 |
| 13c | –OMe | –H | 232 ± 0.9 | 620 | 4.01 | –5.58 |
| 13d | –H | –OMe | 540 ± 14 | 1870 | 2.50 | –5.62 |
| 15a | –OH | –H | 275 ± 29 | 1870 | 3.06 | –5.52 |
| 15b | –H | –OH | 231 ± 7 | 620 | 0.98 | –6.02 |
| 16a | –NHAc | –H | 107 ± 10 | 620 | 3.45 | –5.46 |
| 16b | –NHC(O)nBu | –H | 59 ± 4 | 210 | 1.59 | –5.80 |
| 16c | –NHC(O)OEt | –H | 84 ± 4 | 210 | 8.78 | –5.07 |
| 16d | –NHC(O)OiPr | –H | 168 ± 7 | 620 | 5.12 | –5.29 |
| 16e | –NHC(O)OnBu | –H | 344 ± 9 | 1870 | 14.2 | –4.85 |
| 17a | –NHC(O)CH2NHBoc | –H | 44 ± 2 | 210 | 4.48 | –5.35 |
| 17b | –NHC(O)(CH2)2NHBoc | –H | 18 ± 1 | 69 | 0.434 | –6.36 |
| 17c | –NHC(O)(CH2)3NHBoc | –H | 39 ± 3 | 210 | 1.33 | –5.88 |
| 17d | –NHC(O)(CH2)4NHBoc | –H | 62 ± 5 | 210 | 1.95 | –5.73 |
| 17e | –NHC(O)(CH2)5NHBoc | –H | 176 ± 9 | 620 | 1.91 | –5.73 |
| 18a | –NHC(O)CH2NH2 | –H | 209 ± 12 | 620 | 0.834 | –6.08 |
| 18b | –NHC(O)(CH2)2NH2 | –H | 2060 ± 92 | 5600 | 0.131b | –6.89b |
| 18c | –NHC(O)(CH2)3NH2 | –H | 840 ± 60 | 1870 | 0.159 | –6.80 |
| 18d | –NHC(O)(CH2)4NH2 | –H | 1340 ± 51 | 5600 | 0.838 | –6.11 |
| 18e | –NHC(O)(CH2)5NH2 | –H | 485 ± 14 | 5600 | 0.436 | –6.36 |
All experiments were performed in triplicate, and the numbers shown represent averages of three experiments.
Permeability was assessed using the absorbance at 320 nm instead of 280 nm.
One of the particular points of interest based on our previous publication was the role of the anilino nitrogen in driving the potency of compound 1. We found that replacement of the anilino moiety with a hydroxyl (13c) or methoxy (15a) resulted in an ∼3-fold increase in EC50, from 69 to ∼250 nM. Likewise, moving the amine to the meta position (14) or incorporation of a hydroxyl (15b) or methoxy (13d) group at the meta position resulted in a modest drop in potency. Interestingly, acetylation of aniline (16a) had a minimal impact on the EC50. Lengthening the amide chain (16b) or replacement with a small carbamate (16c) was well-tolerated, giving activity in the 50–80 nM range. However, more bulky carbamates resulted in a loss of potency (16d, 16e). Given our ultimate interest in incorporating these agonists into antibody conjugates, we specifically sought to introduce an amine “handle” onto the side chain (18a–e). Unfortunately, we found that the primary amine dramatically reduced the potency of the agonists. Interestingly, however, the Boc-protected intermediates (17a–e) retained very high potency—in most cases even exceeding that of the parent molecule 1. Compound 17b proved to be the most potent compound of the set, eliciting an EC50 of 18 nM and a Cmax of 69 nM.
In parallel with the Ramos Blue functional assay, we also assessed the apparent permeability (Papp) of each compound using a parallel artificial membrane permeability assay (PAMPA).21 We employed a commercial PAMPA kit (Corning 353015) that uses an artificial cell membrane composed of a lipid/oil/lipid trilayer arranged in a 96-well plate format with donor and acceptor wells. As can be seen in Figure 5, compound 1 exhibited permeability comparable to that of R848 in spite of its 10-fold improved potency. As anticipated, incorporation of primary alkyl amines into the TLR7 agonists (18a–e) tended to result in reduced permeability, while compounds which incorporated a hydrophobic side chain (16a–17e) tended to exhibit comparable or improved permeability. There was a slight apparent correlation between permeability and potency (Figure 5), which is perhaps not surprising given that TLRs are endosomal proteins.
Figure 5.
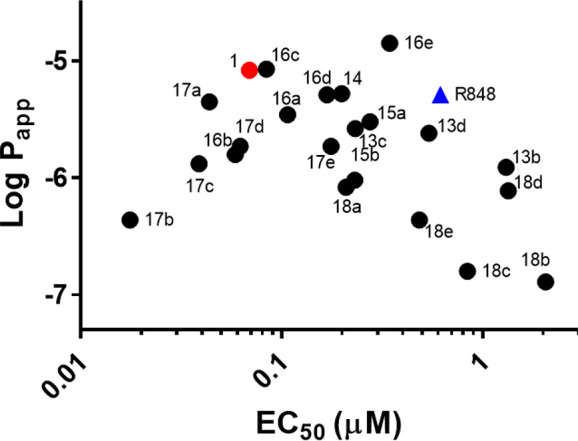
Plot of potency vs permeability. For comparison, compound 1 (E104) is shown with a red star, and R848 is shown with a blue triangle.
While the identification of derivatives with retained/increased potency (i.e., 16a–c and 17a–c) was encouraging, these analogs did not have a readily available site for attachment of an ADC linker. Acylation of the aniline nitrogen made it nonreactive for the addition of a linker, while initial attempts to add a linker to the amidine nitrogen of the imidazole[4,5-c]quinoline core proved to be challenging due to its poor nucleophilicity (Figure 6). Therefore, we attempted to retain the aniline nitrogen by introducing a hydrophobic tail to compound 1 using reductive amination, hoping to see a similar gain in potency as was observed for 16a–c/17a–c. Unexpectedly, the resulting compounds (19a–d) exhibited dramatically reduced potency against TLR7. This prompted us to perform ligand docking studies using the published crystal structure of R848 bound to TLR7 (PDB entry 5GMH) in order to understand the reduced activity.22
Figure 6.

Reductive amination of 1 provided analogs 19a–d that retained a nucleophilic aniline group for linker attachment.
TLR7 is an m-shaped dimeric dual receptor that has two distinct ligand-binding sites, one of which binds to small ligands such as guanosine and imidazoquinolines. This conserved binding site (found in both TLR7 and TLR8) is embedded at the dimerization interface of the two TLR7 monomers (Figure 7a, shown in green and cyan). Four representative ligands were selected for the comparative ligand-docking study (R848, 16b, 17b, and 19b). R848 is a well-known TLR7/8 ligand and was selected as a control for our comparative docking analysis. It is reported that R848 interacts with residues F349, F351, Y356, F408, V381, D555*, L557*, G584*, and T586* (note that the asterisk differentiates between the two TLR7 monomers). We constructed the grid for targeted docking based on these residues. Using this constrained model, we validated our approach by docking R848 to the guanosine binding site. Encouragingly, our model predicted a nearly identical binding mode as found in the reported crystal structure (Figure S6).
Figure 7.

Molecular docking was performed to better understand the potency. (A) Crystal structure of monkey TLR7 bound to R848 (PDB entry 5GMH). (B) Docking results for R848, 16b, 17b, and 19b compared to the experimental binding affinity as shown in Table 1. (C) Predicted interactions of compound 17b with the TLR7 guanosine binding site. (D) Illustration of the key interactions observed between compound 17b and the binding site.
Having successfully recapitulated the published binding mode of R848, we next performed the same docking exercise using 16b, 17b, and 19b to understand the results obtained in the in vitro assays. As shown in Figure 7b, the docking scores for 16b and 17b were significantly lower than the docking score for R848 (−9.3 and −9.7 kcal/mol vs −8.9 kcal/mol). This is consistent with the significantly improved EC50 for compounds 16b and 17b compared to R848 (Figure 7b and Table 1). In contrast, compound 19b was found to have a far higher docking score (−6.2 kcal/mol), consistent with the very weak potency observed for this compound (>30 μM).
To better understand the results, we looked at the binding site interactions of these molecules compared to R848. Similar to R848, the cores of 16b and 17b form π–π interactions with F408 and Y356. Likewise, the amidines of both molecules form hydrogen-bonding interactions with D555* and T586*. The acyl side chain of 17b reaches into a part of the pocket unoccupied by R848, forming interactions with N534, H558, and F507, perhaps explaining the increased potency for this molecule (Figure 7C,D). Likewise, the acyl side chain from compound 16b reaches into the same region of the binding pocket but is unable to make the additional hydrogen-bonding interactions observed for 17b. In contrast, compound 19b, which replaces the more rigid acyl group with a fully rotatable single bond, exhibited a far lower docking score, perhaps by virtue of the increased entropic penalty associated with the binding of the flexible hydrophobic tail. Consistent with this hypothesis, the only alkyl analogue that had measurable activation of the TLR7 receptor was compound 19a, which had the shortest alkyl tail, thus paying the lowest entropic penalty. Interestingly, the predicted lowest-energy docking structure of 19b exhibited a conformation completely different from that of the other three inhibitors and made completely different interactions with the TLR7 binding site (see Figure S6).
A subset of three highly permeable and highly potent derivatives of 1 were selected for more detailed functional studies (16b, 16d, and 17b). We were particularly interested in compound 17b given that it was ∼35-fold more potent than R848 and ∼4–10 times more potent than compound 1 without exhibiting a loss of permeability. Compounds 16b, 16d, and 17b were found to potently induce the release of IFNα upon incubation with human PBMCs, likely due to stimulation of plasmacytoid dendritic cells (PDCs)23 (Figure 8A, left). The compounds also induced the release of TNFα from PBMCs (Figure 8A, right). Consistent with the SAR described above, compounds 17b and 16b were both 5–15-fold more potent than compound 1 in this PBMC assay, while compound 16d had equivalent or marginally lower activity than 1.
Figure 8.

Lead agonists were evaluated in a variety of functional assays. (A) Induction of IFNα and TNFα in human PBMCs. (B) Activation of hTLR7 and hTLR8 reporter cells. (C) Activation of hMDMs at 80, 400, and 2000 nM. (D) Activation of a mouse macrophage cell line (RAW dual).
The induction of TNFα from the PBMCs was unanticipated. While the absolute amount of TNFα release was relatively small (LPS often elicits the release of ng/mL concentrations of TNFα in PBMCs), the dose response was quite clear. Monocytes, the main circulating cell type involved in TNFα release, are well-known to release TNFα in response to TLR8 agonists. Therefore, we speculated that the addition of the hydrophobic tailpiece onto the amine handle had inadvertently resulted in a gain of TLR8 activity. However, this proved not to be the case. As shown in Figure 8B, we directly compared compound 17b in TLR7 and TLR8 reporter cell lines and found that the TLR7-overexpressing reporter line was ∼500–1000 times more responsive than the TLR8-overexpressing cell line. Indeed, no activation of hTLR8 was observed below concentrations of ∼10 μM, while concentrations as low as 16 nM elicited full activation of hTLR7. Consistent with the Ramos Blue data (Table 1), compound 17b was >5-fold more active against TLR7 than compound 1 (Figure 8B, left), In order to further confirm the lack of TLR8 involvement, we looked for cytokine release from monocyte-derived macrophages (hMDMs). PBMC-derived monocytes were cultured with 50 ng/mL recombinant human granulocyte-macrophage colony-stimulating factor (hGMCSF) for 6 days in order to induce differentiation into a “neutral macrophage” cell type. Macrophages were treated with various TLR agonists at 80, 400, or 2000 nM, as shown in Figure 8C. Interestingly, only the selective TLR8 agonist (motolimod) and the dual TLR7/8 agonist (R848) induced significant release of TNFα and IL6. Compound 17b induced a slight release of TNFα, but only at 2000 nM—a dose which, according to Figure 8B, may result in slight TLR8 activation. No release of IL-1b, IL-12 p70, IL-10, IL-2, or IFNγ was observed for any of the treatment groups (data not shown). These results suggest that the TNFα release in PBMCs (Figure 8B) is not derived from monocytes or macrophages.
Importantly, all the studies shown in Figures 4 and 8A–C involve human cells. Our aforementioned ADC study showed that ADCs releasing compound 1 induced tumor regression in a mouse xenograft model. Thus, in order to pave the way for future in vivo studies, we wanted to show that these compounds can also activate mouse lymphocyte cell lines. RAW 264.7 cells are a widely used mouse macrophage line that responds to various TLR agonists.24 Unlike human macrophages, mouse macrophages are known to express high levels of TLR7. Using a commercial NFκB reporter version of this cell line (Invivogen), we demonstrated that compounds 16b and 16d exhibit equivalent or slightly improved potency compared to compound 1. Unexpectedly, compound 17b was slightly less potent than compound 1 (Figure 8D). The reason for this discrepancy is not clear at this time but may simply be due to slight sequence differences between the mouse and human receptors.
TLR7 agonists have significant therapeutic potential in a variety of oncology, infectious disease, and autoimmune disease states. However, to date, the immunological risks associated with TLR7 agonism have prevented therapeutic programs from progressing, thereby illustrating the importance of targeted delivery approaches. We recently reported a potent TLR7-selective agonist 1 that could be delivered by ADC technology to elicit potent anticancer activity. Herein we have described synthetic chemistry and SAR studies to understand and improve the potency and permeability of 1 for incorporation into next-generation immune-stimulating ADCs. We designed a small set of 25 analogs of compound 1 with a particular focus on understanding the SAR around the benzylic “headpiece” of the molecule. Each analogue was initially evaluated in a TLR7-expressing B-cell reporter assay and a PAMPA permeability assay. We found that addition of hydrophobic acyl tails onto the aniline moiety of parent compound 1 generally resulted in retained or improved TLR7 agonist activity without sacrificing the permeability or the selectivity over TLR8. The addition of hydrophobic alkyl groups to the same site unexpectedly resulted in a dramatic loss in potency. We ultimately identified compounds 17b, 16b, and 16d as highly potent TLR7 agonists, which were further profiled in a variety of functional assays. Specifically, we showed that the three lead compounds potently induce the release of IFNα and TNFα from hPBMCs at low-nanomolar concentrations. One of these compounds (17b) was shown to be >500-fold selective for TLR7 over TLR8. As anticipated, these compounds were shown to result in activation of the NFκB pathway in a mouse macrophage cell line, thus supporting their further exploration in immuno-oncology applications. We believe that these leads will serve as useful starting points for next-generation immune-stimulating ADC technology.
Acknowledgments
This work was financially supported by the NIH (1R15AI149755-01 and R01GM140026) and also by Heidelberg Pharma AG. Additionally, we also acknowledge the Dr. G. Clifford and Florence B. Decker Foundation for the purchase of instrumentation used in this research.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.3c00260.
Supplemental figures, synthetic procedures, compound characterization, and assay conditions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Elshabrawy H. A.; Essani A. E.; Szekanecz Z.; Fox D. A.; Shahrara S. TLRs, Future Potential Therapeutic Targets for RA. Autoimmun. Rev. 2017, 16, 103–113. 10.1016/j.autrev.2016.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D. Toll-like Receptors: Activation, Signalling and Transcriptional Modulation. Cytokine 2015, 74 (2), 181–189. 10.1016/j.cyto.2015.02.025. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhang S.; Li H.; Wang H.; Zhang T.; Hutchinson M. R.; Yin H.; Wang X. Small-Molecule Modulators of Toll-like Receptors. Acc. Chem. Res. 2020, 53 (5), 1046–1055. 10.1021/acs.accounts.9b00631. [DOI] [PubMed] [Google Scholar]
- Wu Y.-w.; Tang W.; Zuo J.-p. Toll-like Receptors: Potential Targets for Lupus Treatment. Acta Pharm. Sin. 2015, 36 (12), 1395–1407. 10.1038/aps.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racke M. K.; Drew P. D. Toll-like Receptors in Multiple Sclerosis. Curr. Top. Microbiol. Immunol. 2009, 336, 155–168. 10.1007/978-3-642-00549-7_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M.; García-Martínez E.; Pitter M. R.; Fucikova J.; Spisek R.; Zitvogel L.; Kroemer G.; Galluzzi L. Trial Watch: Toll-like Receptor Agonists in Cancer Immunotherapy. OncoImmunology 2018, 7 (12), e1526250. 10.1080/2162402X.2018.1526250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horscroft N. J.; Pryde D. C.; Bright H. Antiviral Applications of Toll-like Receptor Agonists. J. Antimicrob. Chemother. 2012, 67 (4), 789–801. 10.1093/jac/dkr588. [DOI] [PubMed] [Google Scholar]
- Bam R. A.; Hansen D.; Irrinki A.; Mulato A.; Jones G. S.; Hesselgesser J.; Frey C. R.; Cihlar T.; Yant S. R. TLR7 Agonist GS-9620 Is a Potent Inhibitor of Acute HIV-1 Infection in Human Peripheral Blood Mononuclear Cells. Antimicrob. Agents Chemother. 2017, 61 (1), e01369-16. 10.1128/AAC.01369-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan D. C.; Herschke F.; Pauwels F.; Stoops B.; Smyej I.; Last S.; Pieters S.; Embrechts W.; Khamlichi M. D.; Thoné T.; Van Schoubroeck B.; Mostmans W.; Wuyts D.; Verstappen D.; Scholliers A.; De Pooter D.; Dhuyvetter D.; Borghys H.; Tuefferd M.; Arnoult E.; Hong J.; Fanning G.; Bollekens J.; Urmaliya V.; Teisman A.; Horton H.; Jonckers T. H. M.; Raboisson P. Identification and Optimization of Pyrrolo[3,2-d]Pyrimidine Toll-like Receptor 7 (TLR7) Selective Agonists for the Treatment of Hepatitis B. J. Med. Chem. 2017, 60 (14), 6137–6151. 10.1021/acs.jmedchem.7b00365. [DOI] [PubMed] [Google Scholar]
- Yoo E.; Crall B. M.; Balakrishna R.; Malladi S. S.; Fox L. M.; Hermanson A. R.; David S. A. Structure–Activity Relationships in Toll-like Receptor 7 Agonistic 1H-Imidazo[4,5-c]Pyridines. Org. Biomol. Chem. 2013, 11 (38), 6526–6545. 10.1039/c3ob40816g. [DOI] [PubMed] [Google Scholar]
- Fan Q.; Cohen S.; John B.; Riker A. I. Melanoma in Situ Treated with Topical Imiquimod for Management of Persistently Positive Margins: A Review of Treatment Methods. Ochsner J. 2015, 15 (4), 443–447. [PMC free article] [PubMed] [Google Scholar]
- Ackerman S. E.; Pearson C. I.; Gregorio J. D.; Gonzalez J. C.; Kenkel J. A.; Hartmann F. J.; Luo A.; Ho P. Y.; LeBlanc H.; Blum L. K.; Kimmey S. C.; Luo A.; Nguyen M. L.; Paik J. C.; Sheu L. Y.; Ackerman B.; Lee A.; Li H.; Melrose J.; Laura R. P.; Ramani V. C.; Henning K. A.; Jackson D. Y.; Safina B. S.; Yonehiro G.; Devens B. H.; Carmi Y.; Chapin S. J.; Bendall S. C.; Kowanetz M.; Dornan D.; Engleman E. G.; Alonso M. N. Immune-Stimulating Antibody Conjugates Elicit Robust Myeloid Activation and Durable Antitumor Immunity. Nat. Cancer 2021, 2 (1), 18–33. 10.1038/s43018-020-00136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brant M. G.; Garnett G. A. E.; Guedia J.; Lasalle M.; Lawn S.; Petersen M. E.; Duan R.; Mendez-Campos J.; Hirkala-Schaefer T.; Winters G. C.; Barnscher S. D. Generation and Structure-Activity Relationships of Novel Imidazo-Thienopyridine Based TLR7 Agonists: Application as Payloads for Immunostimulatory Antibody Drug-Conjugates. Bioorg. Med. Chem. Lett. 2023, 91, 129348 10.1016/j.bmcl.2023.129348. [DOI] [PubMed] [Google Scholar]
- Biffen M.; Matsui H.; Edwards S.; Leishman A. J.; Eiho K.; Holness E.; Satterthwaite G.; Doyle I.; Wada H.; Fraser N. J.; Hawkins S. L.; Aoki M.; Tomizawa H.; Benjamin A. D.; Takaku H.; McInally T.; Murray C. M. Biological Characterization of a Novel Class of Toll-like Receptor 7 Agonists Designed to Have Reduced Systemic Activity. Br. J. Pharmacol. 2012, 166 (2), 573–586. 10.1111/j.1476-5381.2011.01790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurimoto A.; Hashimoto K.; Nakamura T.; Norimura K.; Ogita H.; Takaku H.; Bonnert R.; McInally T.; Wada H.; Isobe Y. Synthesis and Biological Evaluation of 8-Oxoadenine Derivatives as Toll-like Receptor 7 Agonists Introducing the Antedrug Concept. J. Med. Chem. 2010, 53 (7), 2964–2972. 10.1021/jm100070n. [DOI] [PubMed] [Google Scholar]
- Tom J. K.; Dotsey E. Y.; Wong H. Y.; Stutts L.; Moore T.; Davies D. H.; Felgner P. L.; Esser-Kahn A. P. Modulation of Innate Immune Responses via Covalently Linked TLR Agonists. ACS Cent. Sci. 2015, 1 (8), 439–448. 10.1021/acscentsci.5b00274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S.; Brems B. M.; Olawode E. O.; Miller J. T.; Brooks T. A.; Tumey L. N. Design and Characterization of Immune-Stimulating Imidazo[4,5-c]Quinoline Antibody–Drug Conjugates. Mol. Pharmaceutics 2022, 19 (9), 3228–3241. 10.1021/acs.molpharmaceut.2c00392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla N. M.; Mutz C. A.; Ukani R.; Warshakoon H. J.; Moore D. S.; David S. A. Syntheses of Fluorescent Imidazoquinoline Conjugates as Probes of Toll-like Receptor 7. Bioorg. Med. Chem. Lett. 2010, 20 (22), 6384–6386. 10.1016/j.bmcl.2010.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernejoul F.; Debin A.; Drocourt D.; Perouzel E.; Tiraby G.; Liouz T.. Conjugated tlr7 and/or tlr8 and tlr2 agonists. US 20140141033 A1, 2014. https://patents.google.com/patent/US20140141033A1/en (accessed 2022-12-26).
- Shukla N. M.; Malladi S. S.; Day V.; David S. A. Preliminary Evaluation of a 3H Imidazoquinoline Library as Dual TLR7/TLR8 Antagonists. Bioorg. Med. Chem. 2011, 19 (12), 3801–3811. 10.1016/j.bmc.2011.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller B. Artificial Membrane Assays to Assess Permeability. Curr. Drug Metab. 2008, 9 (9), 886–892. 10.2174/138920008786485227. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Ohto U.; Shibata T.; Krayukhina E.; Taoka M.; Yamauchi Y.; Tanji H.; Isobe T.; Uchiyama S.; Miyake K.; Shimizu T. Structural Analysis Reveals That Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45 (4), 737–748. 10.1016/j.immuni.2016.09.011. [DOI] [PubMed] [Google Scholar]
- Li S.; Wu J.; Zhu S.; Liu Y.-J.; Chen J. Disease-Associated Plasmacytoid Dendritic Cells. Front. Immunol 2017, 8, 1268. 10.3389/fimmu.2017.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F.; Ahmad-Nejad P.; Hemmi H.; Hochrein H.; Ampenberger F.; Gellert T.; Dietrich H.; Lipford G.; Takeda K.; Akira S.; Wagner H.; Bauer S. The Toll-like Receptor 7 (TLR7)-Specific Stimulus Loxoribine Uncovers a Strong Relationship within the TLR7, 8 and 9 Subfamily. Eur. J. Immunol. 2003, 33 (11), 2987. 10.1002/eji.200324238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.