

Abstract
Tumor cell heterogeneity drives disease progression and response to therapy, and therefore, there is a need for single-cell analysis methods. In this paper, we present an integrated, scalable method to analyze enzymatic activity in many individual cancer cells at once. The reported method uses dielectrophoresis (DEP) to selectively capture tumor cells at wireless electrodes aligned to an overlying array of cell-sized micropockets. Following hydrodynamic transfer of the captured cells into microfluidic chambers, the chambers are fluidically isolated and sealed with a hydrophobic ionic liquid, which possesses sufficient conductivity to allow for subsequent electrical lysis of the cells to access their contents for enzymatic assay. The wireless electrodes have an interlocking spiral design that ensures successful electrical lysis regardless of the location of the cell within the chamber. Here, breast cancer cells are assessed for β-galactosidase through its activation of a fluorogenic substrate. A key point is that the fluorogenic assay solution was optimized to allow for dielectrophoretic cell capture, thereby obviating the need for a solution exchange step. Our approach has several distinct advantages including a high rate of single-cell capture, a capture efficiency that is independent of the dimensions of the reaction chambers, no need for mechanical closure of reaction volumes, and no observed cross-talk. In this study, first, the steps of cell capture, transfer, and lysis are established on this platform in the presence of the optimized assay solution. We then quantify the increase in fluorescence intensity obtained over the duration of the enzymatic assay of individual cells. Finally, this method is applied to the analysis of β-galactosidase activity in 258 individual MDA-MB-231 breast cancer cells, revealing heterogeneity in expression of this enzyme in this cell line. We expect that the adaptability of this method will allow for expanded studies of single cell enzymatic expression and activity. This will in turn open avenues of research into cancer cell heterogeneity in metabolism, invasiveness, and drug response. The ability to study these features of cancer at the single-cell level raises the possibility for treatment plans tailored to target the specific combinations of cell subpopulations present in tumors. Furthermore, we expect that this method can be adapted to uses outside of cancer research, such as studies of neuron metabolism, pathogenesis in bacteria, and stem cell development.
Graphical Abstract
Introduction
Cancer cells are highly heterogeneous owing to the accumulation of genetic mutations and the exposure of individual cells to distinct microenvironments and selective pressures.1 A high degree of cellular heterogeneity among tumor cells is linked with poor prognosis.2 A trait such as chemotherapeutic resistance may be harbored by a small subpopulation that is not detected by techniques that analyze cells in ensemble. Following treatment, such a subclone can resurge leading to relapse of disease that is refractory to the primary therapy. Therefore, the characterization of cell-to-cell differences is critical for determination of prognosis and has the potential to inform therapeutic decisions. This requirement has motivated the development of a wide range of single-cell analysis techniques that quantify molecular features.3,4 Activity of the cytosolic enzyme, β-galactosidase, is one such feature that is widely used as a biomarker indicative of cellular senescence.5,6 Here, we report an integrated device and method for selection, individual sequestration, and electrical lysis of tumor cells followed by an assay of the activity of a cytosolic enzyme (Scheme 1). To prevent crosstalk, single-cell enzymatic analysis requires fluidic isolation of cells individually in discrete reaction volumes. Typically, cells are isolated in an assay solution within droplets,7,8 valved microchambers,9 or microwells.10,11 Of these three modalities, droplets allow for the most rapid generation of a large number of reaction volumes, but cell encapsulation is both non-selective, requiring a pre-sorting step, and passive, relying on Poisson statistics for single-cell isolation, which leads to a high proportion of empty droplets. Furthermore, altering the composition of the droplets for assay or lysis requires sophisticated microfluidics such as droplet splitter/merger systems8 or microdroplet printers.12 Wang et al. conducted a β-galactosidase assay in single K562 cells by depositing cells into microwells, displacing the bulk fluid with an immiscible oil, and adding a combined lysis/assay buffer to the resulting droplets using an inkjet-style printer.12 This innovative approach addressed the limitation of Poisson-distribution-based systems by forming the droplets in-situ around passively captured cells to ensure high occupancy rates; the subsequent modifications to the droplet required a complex microdroplet printer, however, and the microwell-based isolation system places constraints on the geometry available for assays.
Scheme 1.
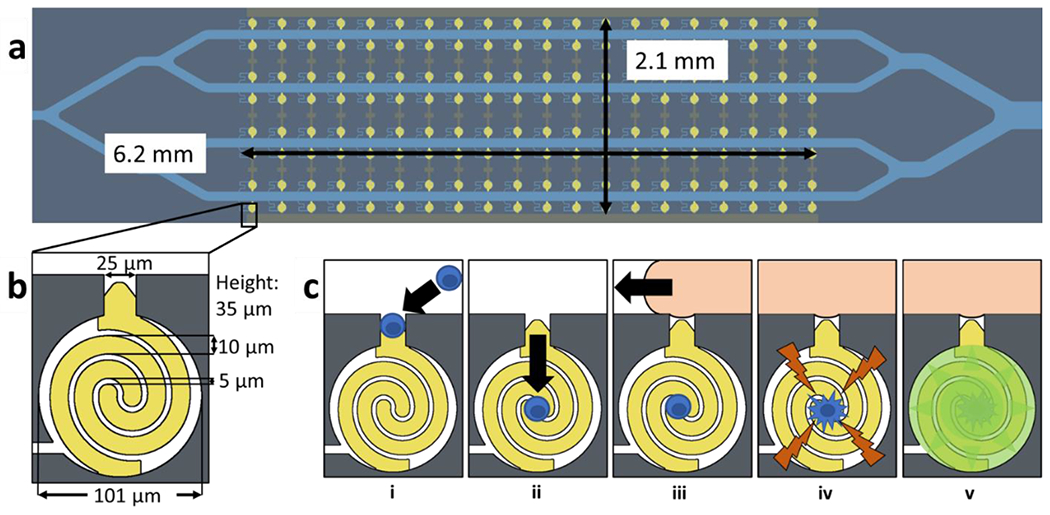
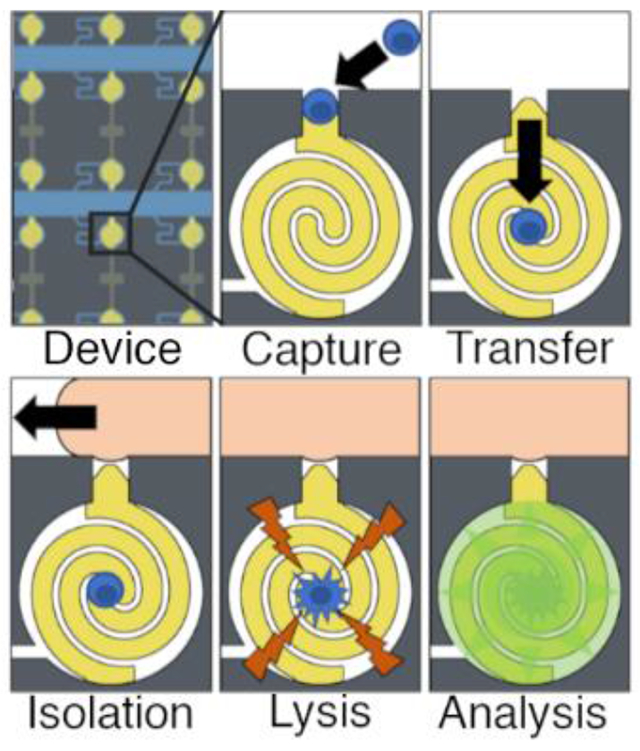
a) Illustration of the microfluidic device. The analysis area consists of a 6.2 × 2.1 mm array of 160 chambers b) scheme of electrode and chamber dimensions. Each chamber is a cylinder with a diameter of 101 μm and height of 35 μm. The chambers are connected to the main channels by a 25 × 25 μm capture pocket and a 7 μm wide leak channel. c) scheme of assay process i) DEP cell capture ii) fluidic cell transfer iii) ionic liquid isolation iv) electroporation v) analysis of fluorescence increase.
Single-cell enzymatic assays have also been carried out in chambers sealed by mechanical valves. Valve-based microfluidic systems utilize two layers – a microfluidic layer for cell analysis separated from an air pressure system by a thin membrane. When the air system is pressurized, the membrane distorts to selectively block portions of the microfluidic system, forming chambers. Solution exchange is more straightforward than in droplet-based systems. Eyer et al. designed a microvalve-based device to conduct ELISA on single HEK 293 cells. This device consisted of eight microfluidic channels overlaid with fifteen orthogonal air channels separated by a thin PDMS membrane. The air channels were designed to form microfluidic chambers around passively captured cells when pressurized. The orthogonal arrangement of the air pressure layer allowed for single chambers to be formed in each fluid channel, allowing for a high degree of control. Despite these advancements, the reliance of valved microchambers on multilayer device fabrication and air pressure systems is a disadvantage. Furthermore, the selection of tumor cells must be carried out in a previous step.
Microwell arrays facilitate single-cell analysis by physically restricting the number of cells that can be captured in each well. The microwells can be sealed to form separate reaction volumes by addition of a coverslip or by mechanical collapse of an overlying “ceiling”. Microwells are much simpler than microvalves and allow for facile fluid exchange, but lack the fine control afforded by valves. Critically, the single-cell capture fidelity in microwells is dependent on the volume of the microwell. The volume of the reaction chambers is thus dictated by the needs of the single-cell capture step rather than the needs of the assay. Liu et al. constructed an array of microwells for single-cell analysis by using a self-assembled monolayer of polystyrene microspheres as a mold for a PDMS device.13 The array consisted of densely packed hexagonal microwells with tunable size. Analysis of carboxylesterases in single HeLa cells was conducted by introducing a fluorogenic, membrane permeable substrate (calcein AM) to the array. The hexagonal microwell array achieved over 90% single-cell fidelity but lacked a selective mechanism. Kim et al. developed a selective microwell-based device for assaying β-galactosidase in single MDA-MB-231 cells using dielectrophoretic capture at interdigitated electrodes underlying an array of microwells.10,14 The captured cells were fluidically isolated by collapsing the PDMS microfluidic chamber over the wells to form a seal. The capture electrodes were employed for electrical lysis of the cells prior to the fluorometric assay. The addition of electrodes allowed for selective dielectrophoretic capture and lysis, but the need to collapse the micro chamber over the microwells adds to the complexity of operating the device. In addition, all microwell techniques suffer from restrictions on the microwell geometry due to the needs of single-cell capture. Single-cell fidelity depends on the presence of one cell excluding further cells from entering the well; this requirement places limitations on the dimensions of the microwell. Thus, the design of the microwell is dependent on the needs of the single-cell capture fidelity rather than the needs of the enzymatic assay.
Dielectrophoresis (DEP) is an electrokinetic phenomenon that is capable of selective manipulation of cells. DEP is a force exerted by an asymmetric electrical field on a dielectric particle, with the time-averaged force, , given by the following equation.15
| eq. 1 |
Here, is the radius of the particle, is the permittivity of the surrounding medium, is the magnitude of the applied electric field, and is the real part of the Clausius-Mossotti factor, given in eq. 2.
| eq. 2 |
The Clausius-Mossotti factor determines the direction and magnitude of DEP force as a function of applied field frequency.15 In this equation, and are the frequency-dependent complex permittivities of the particle and the medium, respectively. When the particle is more polarizable than the surrounding medium, is positive, which causes the DEP force to displace particles toward regions of high electric field strength (positive DEP or pDEP), whereas when the particle is less polarizable than the medium is negative, which causes the DEP force to displace particles toward regions of low electric field strength (negative DEP or nDEP). A frequency at which a particle transitions from nDEP to pDEP (or vice versa) is called a crossover frequency, cof.
In low conductivity media, cells exhibit a crossover from nDEP to pDEP that is dictated by membrane capacitance, which is governed by biologically relevant features such as membrane folding and glycosylation;16 thus, the frequency of the applied voltage can be used to cause some cell types to undergo pDEP while other cells types undergo nDEP. This mechanism makes DEP selective, and allows a given device to be adapted to new cell types without any alterations in the device geometry or fabrication. Scheme 2a illustrates pDEP and nDEP forces exerted on polarizable particles in a non-uniform electric field near the tip of an electrode (gold color). Scheme 2b depicts pDEP capture of cells at electrode tips aligned to cell-sized micropockets in a microfluidic device. DEP allows for single-cell isolation by combining the high spatial precision of dielectrophoretic capture with a microfluidic structure such as a microwell or a micropocket. Here, the presence of a cell in the micropocket discourages capture of further cells, and tangential fluid flow prevents “pearl chaining” of cells, which occurs via dipole-dipole interactions. Despite advantages in single-cell fidelity, selectivity, and facile imaging, integration of DEP isolation of single-cells with subsequent analysis remains an underdeveloped area of research.17
Scheme 2.
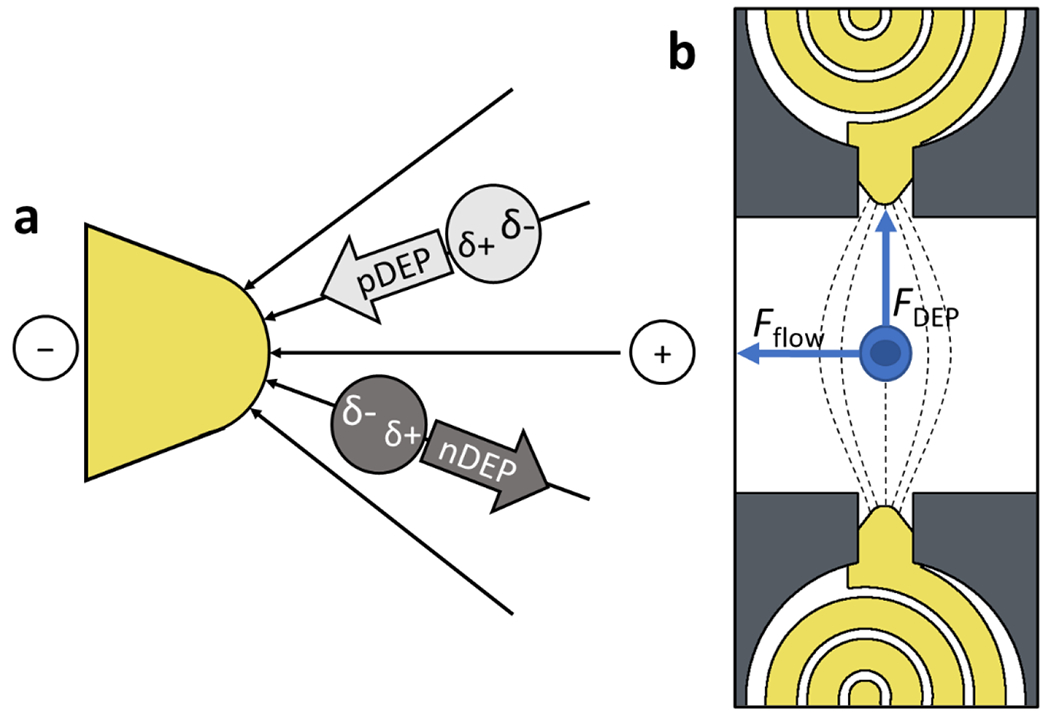
a) Principles of pDEP attraction and nDEP repulsion of dielectric particles b) Forces acting on a cell in a microfluidic channel. The force of the pDEP attraction toward the electrode tip is balanced by the force of the liquid flowing through the chamber. The dimensions of the capture pocket exclude additional cells to achieve single cell capture.
We previously reported a microfluidic platform that integrates dielectrophoretic capture, isolation, and electrical lysis of individual cells in parallel.18 In that design, an array of wireless bipolar electrodes (BPEs) was used to capture cells by DEP in cell-sized pockets lining several parallel channels. Each capture pocket in this DEP-BPE platform led to a nanoliter-scale chamber, into which a cell could be sequestered. Following cell capture and sequestration, the chambers were fluidically isolated by flowing a hydrophobic ionic liquid along the channels, thereby digitizing the aqueous chamber volumes. Finally, the cells were lysed by application of an AC voltage. This approach is similar to the self-digitization of nanoliter-scale chambers with oil reported by Chiu and coworkers19 and later exploited for cell analysis.20 However, the ionic liquid has the advantage of maintaining electrical communication for subsequent lysis.18 An advantage of this DEP-BPE platform over microwell arrays10 is that microfluidic capture pockets in plane with analysis chambers offer a simple method for isolating single cells. The dimensions of the capture pocket can be optimized to allow a single cell to enter the pocket, while the analysis chamber can be an arbitrary size to fit the needs of the chosen assay.
In this work, we integrate a quantitative fluorogenic β-galactosidase assay into the DEP-BPE platform. Several key changes to the device geometry include: the analysis chambers were made smaller to concentrate the cell contents in a smaller volume; second, the chambers were made circular to minimize surface area-to-volume ratio and, in turn, evaporation of chamber contents through the surrounding PDMS; third, the leak channel was made serpentine to make the device more compact; and finally, the electrodes were shaped into interlocking spirals occupying the full area of each chamber to prevent the cells from being displaced from the electrodes and becoming unavailable for electrical lysis. Scheme 1 shows the steps of operation of the device, in which cells suspended in a buffer containing a fluorogenic substrate are 1) captured by pDEP at BPE tips positioned in pockets, 2) transferred (with the voltage off) into the analysis chamber using hydrodynamic drag force created by a leak channel, 3) recaptured (with voltage on) on the interlocking spiral electrodes, 4) isolated using an ionic liquid, and 5) lysed (at higher voltage) to release the target enzyme. Finally, the fluorescent product of the enzymatic reaction is monitored using fluorescence microscopy. We developed a low conductivity assay solution to simultaneously address the requirement for a low-conductivity solution suitable for pDEP capture and to obviate the need for multiple fluid exchange steps. This optimized assay solution allowed for a 40% increase in enzymatic activity while decreasing conductivity below 180 μS/cm to allow for pDEP capture. 54% of chambers were occupied by a single cell, and <1.0% of chambers were occupied by two cells. The presence of empty chambers was leveraged as an internal control. Analysis of single MDA-MB-231 breast cancer cells had β-galactosidase activities up to 60 units/mL with a mean of 9.8 units/mL and a standard deviation of 10 units/mL, indicating cellular heterogeneity. A key point is that a control experiment performed with homogeneous solution of cell lysate (representative of the concentration achieved with one cell per chamber) yielded a signal corresponding to 6.0 units/mL and a standard deviation of 1.6 units/mL. This result demonstrates that the heterogeneity observed among cells is not an artifact of device operation. These advancements are significant because the platform will allow for new avenues of research into the heterogeneity of cancer cell metabolism, invasiveness, and drug response.
Materials and Methods
Chemicals.
The silicone elastomer and curing agent (Sylgard 184), bovine serum albumin (BSA) (biotech grade), 0.25% Trypsin EDTA (1X), sodium phosphate monobasic, sodium phosphate dibasic, fluorescein di-β-D-galactopyranoside (FDG), 2-mercaptoethanol, and 1-decyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ionic liquid were purchased from Fisher Scientific (Thermo Fisher Scientific, Inc., Waltham, MA). The DMEM/F12 cell culture medium, dextrose (D-glucose), sucrose, β-galactosidase, Pluronic F-108 were obtained from Sigma-Aldrich, Inc. (St. Louis, MO). PE/anti-EpCAM was obtained from BioLegend, San Diego, CA. All dilutions were conducted with Type 1 water (18.2 MΩ·cm). DEP buffer was comprised of 8.0% sucrose, 0.3% dextrose, and 0.1% BSA titrated to 60 μS/cm with 0.1 M phosphate buffer. The enzymatic assay buffer comprised 50 mM 2-mercaptoethanol and 1.0 mM fluorescein di-β-D-galactopyranoside (FDG) in 0.1 M phosphate buffer (pH 7.4). To optimize the cofactor concentration, magnesium chloride was added to distinct reaction volumes at 0.001 mM, 0.1 mM and 1.0 mM as described in the Results and Discussion section.
Cell Culture.
MDA-MB-231 cells (breast adenocarcinoma) were obtained from ATCC. They were cultured in DMEM/F12 with 10% fetal bovine serum supplementation at 37°C and 5% CO2. All cells were subcultured every 2-3 days to maintain the concentration of cells at less than 80% confluence. In preparation for DEP experiments, MDA-MB-231 cells were detached from culture flask using 0.25% Trypsin-EDTA (1X), followed by pelleting by centrifugation (1100 rpm, 5 min) and cells were resuspended in 2% PE/anti-EpCAM. The cells incubated on a cell rocker for 30 min before being centrifuged again and resuspended in 5.0 mL DEP buffer. Pelleting and resuspension was repeated to wash cells twice in DEP buffer before DEP capture experiments. The final cell concentration for each experiment was 1 × 106 cells/mL.
DEP Capture.
The device was designed to allow cells suspended in DEP buffer to flow through the channels and be attracted to the BPE tips in pDEP mode. AC voltage to drive the DEP response was applied to the driving electrodes (outer rows of electrodes) using a Tektronix AFG3011C waveform generator (Tektronix, Beaverton, OR) and Trek model 2205 amplifier (Trek, Lockport, NY). The AC frequency was maintained at 70 kHz to ensure the MDA-MB-231 cells experienced strong pDEP, based on crossover frequencies (25-50 kHz) reported by Shim et al.21 and corroborated by our previous work.22 The devices were imaged using a Nikon AZ100 microscope (Nikon, Tokyo, Japan). Flow was induced using a Pico Plus Elite syringe pump (Harvard Apparatus, Holliston, MA) paired with a 500 μL glass syringe (Hamilton Company, Reno, NV) in withdrawal mode. Cell counting was assisted through labeling of a cell surface antigen (EpCAM) with a phycoerythrin-linked antibody (PE/anti-EpCAM). The resolution of these fluorescence images, obtained over the entire array, is too low to determine cell volume. Higher resolution micrographs were obtained during DEP capture for limited sections of the array. Since cell volume influences the total quantity of enzyme contained in each cell, strategies for rapidly and accurately determining cell volume through imaging will be pursued in our future work.
Device Fabrication.
The microfluidic devices consisted of microchannels, formed in PDMS by a caste-mold process, bonded to a glass substrate with embedded thin-film gold electrodes. Briefly, glass slides coated with a 5-nm Cr adhesion layer followed by a 100-nm Au film were purchased commercially (Evaporated Metal Films, Ithaca, NY). Thin film electrodes were patterned by spin-coating each slide with a positive photoresist (AZP4620, Integrated Micro Materials (IMM), Argyle, TX) followed by UV exposure of the photoresist using a mask aligner (ABM-USA, San Jose CA). The unexposed photoresist was then removed with AZ 400K developer (IMM). The slides were then wet-etched with 4% KI/1% I2 followed by chrome etchant (Sigma Aldrich, St. Louis, MO). The remaining (patterned) photoresist was removed using N-methyl-2-pyrrolidone (VWR, Radnor, PA) and the slides were rinsed with 200 proof ethanol (Decon Labs, King of Prussia, PA) and dried using a N2 stream. The PDMS monolith was fabricated by pouring PDMS precursor (5:1 base:curing agent) over a photolithographically patterned 35 μm-thick SU-8 2050 film on a silicon wafer. The PDMS was cured at room temperature for 48 h before the monolith was cut from the mold and inlet and outlet reservoirs made using a biopsy punch. The patterned slide and PDMS monolith surfaces were activated using air plasma from a plasma cleaner (Harrick Plasma, Ithaca, NY) on medium power for 1 min. A drop of ethanol was placed on the glass slide to allow for alignment and the PDMS monolith place channel side down on the glass. After alignment, the device was dried in a 70 °C oven for 1 h to evaporate excess ethanol and promote bonding. Finally, the device was filled with 3.0 μM Pluronic and incubated overnight at 4 °C to prevent non-specific adhesion of the cells in subsequent experiments. Prior to an experiment, the solution in the inlet was replaced with DEP buffer, and the device was rinsed under a flow rate of 100 nL/min for 10 min.
Device Geometry.
The microfluidic device consisted of four parallel channels, which were each 100 μm wide and 7 mm long. Forty 25 × 25 μm capture pockets interconnected each channel to a 101-μm diameter circular analysis chamber. A serpentine leak channel (7 × 300 μm) interconnected each analysis chamber to the main channel to support slow fluid flow into the chamber, thereby facilitating hydrodynamic cell transfer.18 The height of the channels and chambers was 35 μm, and therefore, the chamber volume was 280 pL. The gold-on-glass wireless electrodes formed the base of the device. Each electrode consisted of a pair of interlocking spirals. One spiral terminated in a 25 μm-wide tip tapering to a point within the capture pocket, while the other spiral extended underneath the channel wall to form half of the interlocking spiral in the opposing chamber. Each spiral was 10.0 μm wide, with a spacing of 5.0 μm between the spirals.
Optimization of enzymatic assay conditions.
To determine the activity of the enzyme, a Biotek Synergy 2 microtiter plate reader (Winooski, Vermont) was used to measure the fluorescence of microwells containing a mixture of cell lysate, 1.0 mM FDG, 67 mM 2-mercaptoethanol, and varying concentrations of magnesium chloride in DEP buffer. The FDG was added to the solution immediately before the plate was read. The temperature of the plate reader was maintained at 37°C. Fluorescence measurements were obtained once per minute for 25 min.
Results and Discussion
Optimization of the enzymatic assay.
First, the solution employed for the β-galactosidase (β-gal) assay was optimized to minimize conductivity for pDEP cell capture while maximizing enzymatic activity. Initially, a typical composition for the assay solution was employed – 1.0 mM magnesium chloride, 50 mM 2-mercaptoethanol, and 0.88 mg/mL O-nitrophenyl-β-D-galactopyranoside (ONPG) in 0.1 M phosphate buffer (pH 7.4) with the exception that to make the assay more sensitive, the colorimetric ONPG substrate β-gal substrate was replaced with 1.0 mM FDG, a fluorogenic substrate. Further, the original assay was too conductive (22 mS/cm) to support pDEP capture of cells. Lowering the conductivity required replacing the 0.1 M phosphate buffer with the low conductivity DEP buffer described in the Materials and Methods section. Finally, the magnesium concentration was optimized. Mg2+ is reported22 as a cofactor for β-gal, but magnesium ion contributes to the high conductivity of the original assay solution. Therefore, we evaluated enzymatic activity as a function of Mg2+ concentration. Figure 1 is a plot of the fluorescence intensity measured by the microtiter plate reader as a function of time for each concentration of Mg2+. Contrary to expectations, the activity of the enzyme was inversely correlated with magnesium concentration. At 1.0 mM Mg2+, the slope of the fluorescence increase was 152 AU/min; at 0.1 mM the slope was 136 AU/min; and at 0.001 mM Mg2+, the slope was 218 AU/min. This increase is activity is 40% relative to that obtained at a typical assay solution composition (i.e., at 1.0 mM Mg2+), which makes the assay more rapid and sensitive. A key point is that this increased activity is accounted for by calibration as described in the subsection Assessing cell-to-cell variability in beta-galactosidase activity.
Figure 1.
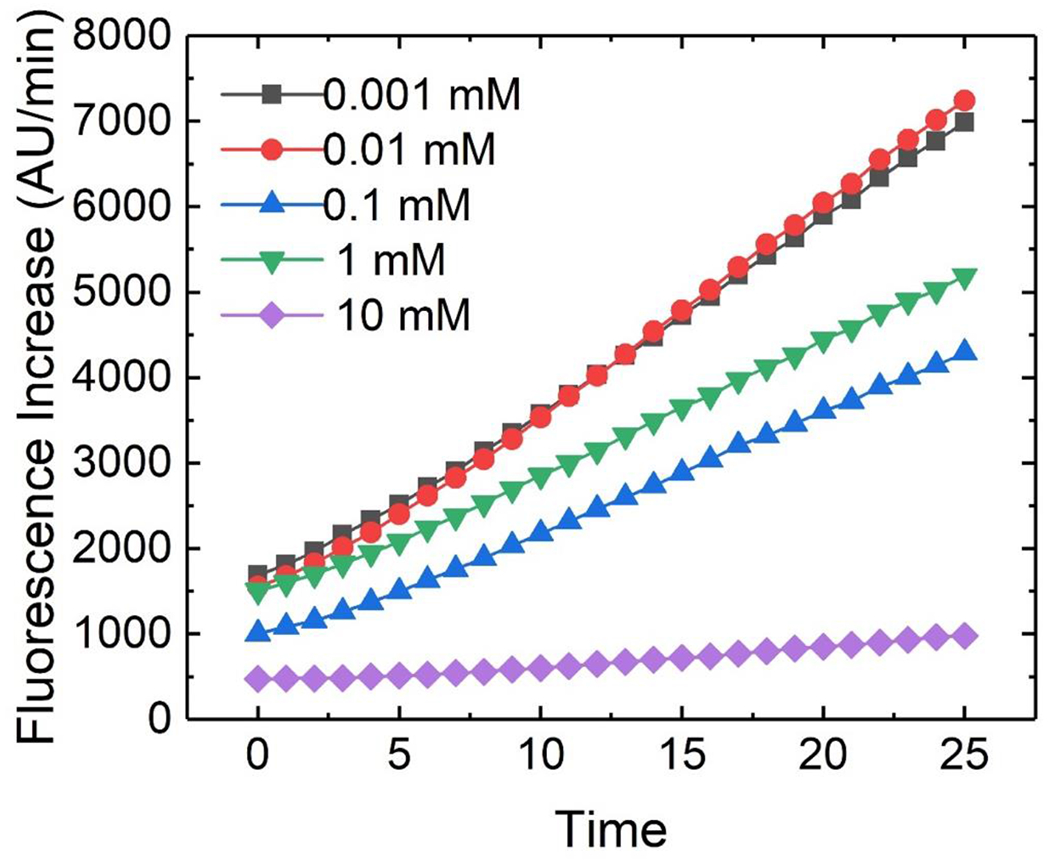
Microtitre plate reader data showing the fluorescence increase of the fluorogenic β-gal assay at several distinct magnesium concentrations. The slope of fluorescence increase was negatively correlated with magnesium concentration. This allowed for a low-conductivity assay mixture conducive to pDEP cell capture.
The reason for this observed negative correlation is unknown; the efficacy of Mg2+ as a cofactor for β-gal has been questioned24 but to our knowledge no study has demonstrated a negative correlation between β-gal activity and Mg2+ concentration. For all subsequent experiments, a Mg2+ concentration of 0.001 mM was used. The conductivity of the final solution was 180 μS/cm, which is higher than DEP buffer alone (60 μS/cm) but still sufficiently low to facilitate pDEP. As a result of the reduced conductivity, the cell capture step of the experiment could be conducted in a solution containing all the components of the assay, removing the need for a separate wash step. Furthermore, the possibility of nDEP repulsion of the cells (observed in the conventional high conductivity assay solution) during the application of lysis voltage was removed.
Cell capture, transfer, isolation and lysis.
We next identified conditions under which single-cell capture, transfer, isolation and lysis occurred in this DEP-BPE platform, which was modified significantly from the design employed in our previous work.18 We anticipated that the most impactful change was the electrode design (interlocking spiral) because electrode geometry and inter-electrode spacing impact the amplitude of the applied voltage required for cell capture and electrical lysis. Electrical lysis is valuable for two reasons: first, the cells’ metabolism is stopped and so variation in the cells over time does not affect the assay; second, the product of the enzymatic reaction is no longer confined to the cell, but instead diffuses over the entire chamber, which decouples the fluorometric analysis from cell morphology. Figure 2 shows micrographs of single-cell capture, transfer, and lysis steps. First, cells suspended in the optimized assay buffer were flowed into the device at a concentration of approximately 1.0×106 cells/mL at a linear velocity of 20 μm/s. An AC voltage of 14.8 Vpp at 70 kHz was applied to the driving electrodes for 5 min to facilitate pDEP capture of cells in the chamber openings (Figure 2a). Second, the cell solution in the inlet and channels was displaced with cell-free buffer, and then, the voltage was removed. The voltage remained off for 2 min to allow the cells to transfer into the analysis chambers and was then reapplied to recapture the cells (Figure 2b). Next, the ionic liquid was flowed into the inlet and channels to seal the chambers. Finally, an AC voltage of 148 Vpp at 70 kHz was applied in three 20-s pulses to lyse the cells, which is evidenced by fluorescence confined to each cell (Figure 2c) being dispersed to the full chamber volume (Figure 2d).
Figure 2.

Brightfield micrographs obtained using differential interference contrast (DIC) of a) two cells captured by DEP and b) the same cells transferred into the adjoining analysis chambers. Fluorescence micrographs of c) the captured cells stained with calcein AM dye and d) the same chambers following application of 148 Vpp lysis voltage. The calcein dye has diffused out of the electroporated cells to illuminate the chambers.
Beta-galactosidase fluorometric assay.
Following application of lysis voltage, we next measured the fluorescence increase over time to assess the activity of β-gal released from the lysed cells. Measuring the change in fluorescence over time offers two advantages: first, the total fluorescence due to enzymatic activity will increase over time, improving sensitivity; and second, measuring the increase over time eliminates error introduced by the initial background fluorescence in the chambers. Figures 3a–c show fluorescence micrographs of four adjacent chambers in a device following lysis, during the incubation step. Note that electrode dimensions appear diminished compared to previous schema due to variation in the extent of gold etching during device fabrication. This variation did not impact the voltage required for capture or lysis of MDA-MB-231 cells. The device was kept at 37°C using a transparent hotplate for the duration of the assay. Fluorescence micrographs of the array were collected once per minute for 25 min. The fluorescence intensity in each chamber was measured separately using ImageJ analysis software.25 Figure 3d shows the fluorescence traces obtained from the four chambers. Chamber 55 contains no cell and exhibits no fluorescence increase, while chambers 56, 75, and 76 each contain a single cell and exhibit three distinct rates of fluorescence increase. We attribute these differences in fluorescence increase to variability in β-gal activity among MDA-MB-231 cancer cells.
Figure 3.
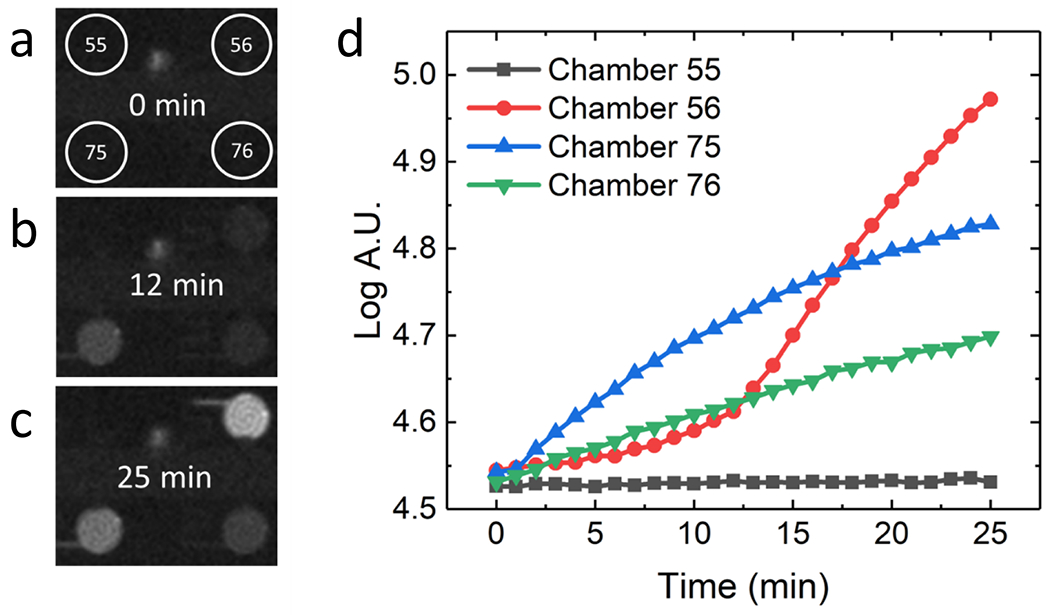
A series of fluorescence micrographs of four chambers at a) 0 min b) 12 min and c) 25 min. Chamber 55 did not contain a cell and exhibited no fluorescence. Chamber 56, 75, and 76 each contained a single cell and showed three different rates of fluorescence increase. d) Plot of the fluorescence increase in four selected chambers of the device over 25 min.
To verify that this variability in fluorescence increase is due to cell-to-cell differences in enzyme activity and not an artifact of the experimental design, we performed a control experiment in which a homogeneous solution of MDA-MB-231 cell lysate in assay solution was flowed into a DEP-BPE device, isolated in the chambers by flowing the ionic liquid through the channels, and then incubated for 25 min at 37°C. Over this time period, the rate of increase in fluorescence intensity was uniform across the array (Figure 4). This result indicates that variation in the reaction rate observed in single cells is representative of distinct enzyme concentrations and not an experimental artifact. Further, this experiment is useful because this homogenate contained the lysate from cells present at a concentration equivalent to one cell per chamber volume. Therefore, this rate can be compared to the average rate observed for a large population of cells as a test of internal consistency of the assay. This comparison is made in the following subsection for a population of 258 cells analyzed individually.
Figure 4.
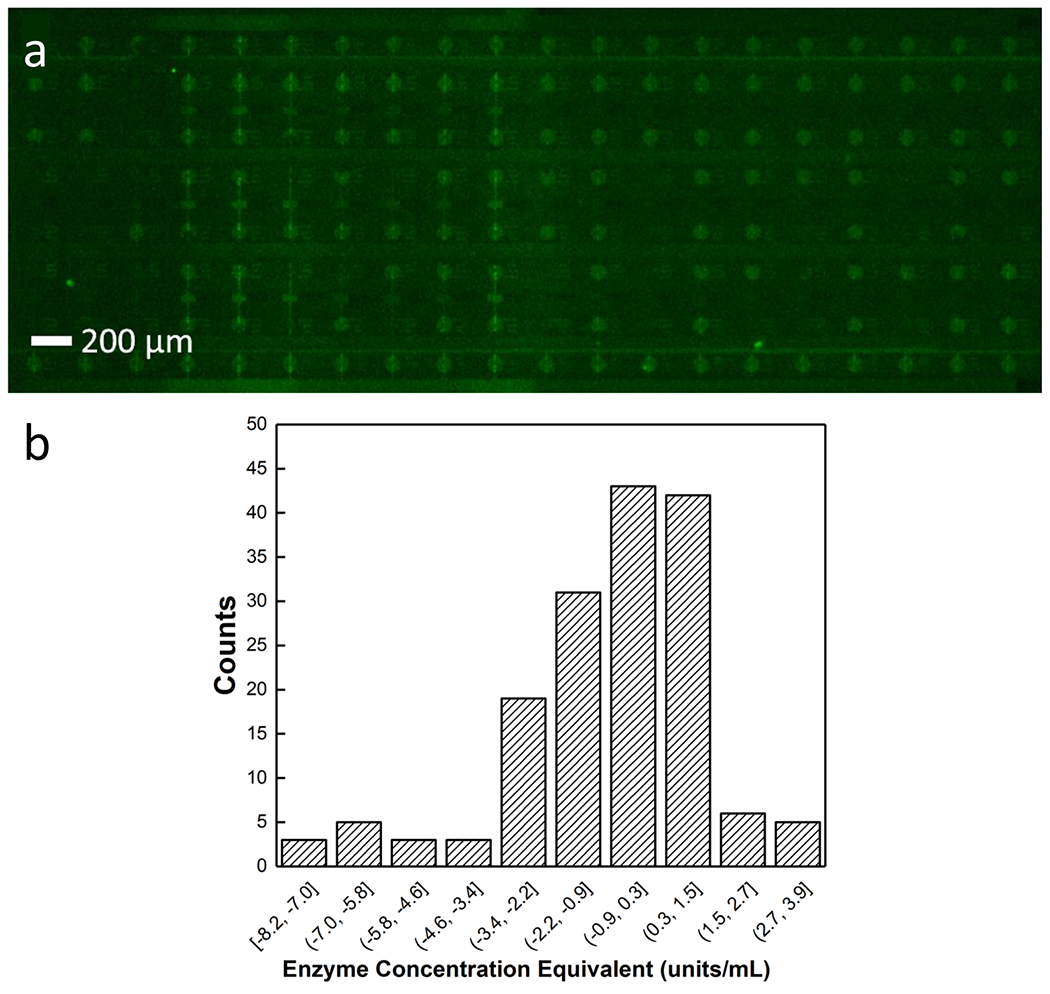
Distribution of fluorescence increases in a homogeneous solution of MDA-MB-231 cell lysate in assay solution in isolated chambers a) a fluorescence micrograph of the device 25 minutes after the chambers have been isolated with an ionic liquid b) histogram showing the distribution of fluorescence increase within the isolated bulk assay. The negative slopes are attributable to evaporation of the chamber contents over the course of the assay. The narrow distribution of fluorescence increase indicates that the heterogeneity is not an artifact of fluorescence measurements.
Assessing cell-to-cell variability in beta-galactosidase activity.
To assess the distribution of β-gal activities in MDA-MB-231 cells, we next assayed 258 isolated single cells. To directly correlate fluorescence increase to enzyme concentration, a standard curve was constructed by obtaining the slope in fluorescence during incubation of seven distinct concentrations of β-gal (provided at a known activity from the supplier) in assay solution. Figure 5a shows the standard curve. We utilized the curve to assess the variability in β-gal activity of MDA-MB-231 cells in terms of enzyme concentration, expressed as enzymatic activity units/mL. Figure 5b shows overlaid histograms of the distribution of activities observed in both singly occupied and empty chambers. The empty chambers served as a useful negative control to account for fluorescence due to potential autohydrolysis of the substrate or its hydrolysis due to application of the lysis voltage. The 258 chambers measured showed a wide distribution of enzymatic activities, from −4 to 60 units/mL with a mean of 9.8 units/mL and a standard deviation of 10. A homogeneous bulk assay, by comparison, had a range of 2.5-9.5 units/mL with a mean of 6.0 units/mL and a standard deviation of 1.6. The close agreement of means for the single-cell and homogeneous assays and the widely disparate ranges and standard deviations indicate a high degree of cellular heterogeneity.
Figure 5.
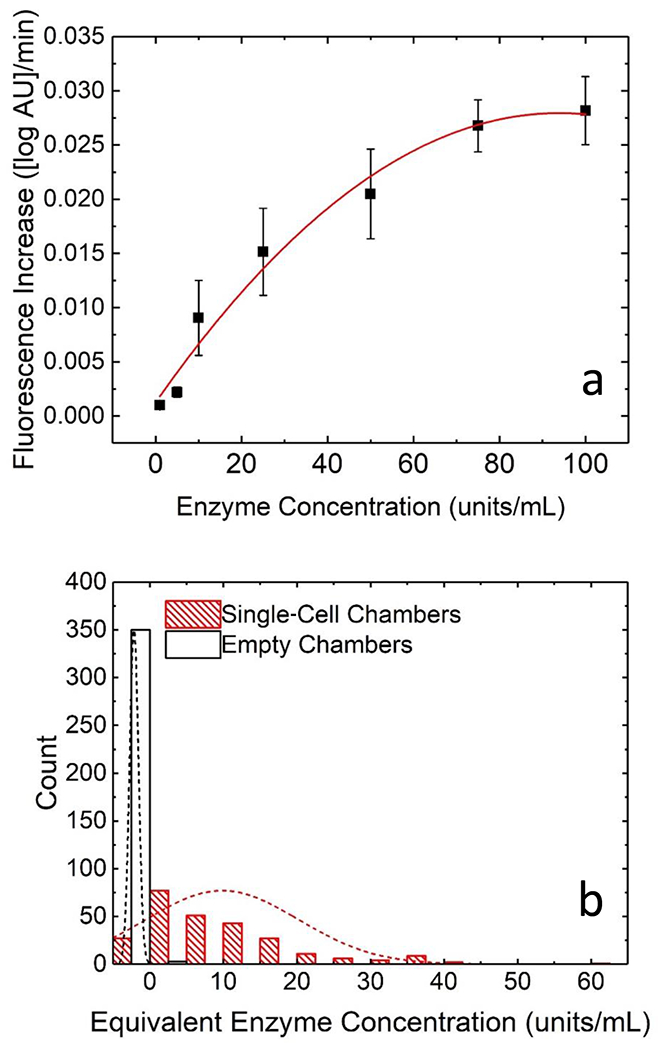
Plots demonstrating heterogeneity in β-gal activity in single MDA-MB-231 cells a) standard curve of fluorescence increase over time vs concentration of enzyme in units/mL b) histograms of the distribution of fluorescence increase in chambers containing single-MDA-MB-231 cells (red) and chambers containing no cell (black). The broad distribution of single-cell β-gal activities indicates a high degree of cellular heterogeneity.
If required by the application, the sensitivity of this assay can be improved still further by decreasing the chamber diameter or by increasing the assay duration (to accumulate more product). Note that decreasing the chamber height is detrimental to sensitivity due to a reduction in optical pathlength. There are many potential applications of this assay method for cytosolic enzymes (which require lysis) and for secreted enzymes. Further, β-gal is frequently utilized to tag antibodies to amplify signal in immunoassays, and therefore, this platform has the potential to enable quantification of low copy numbers of surface antigens on individual cells. If applied to the determination of cellular senescence, successful induction of senescence by a drug would lead to upregulation of β-gal and a concomitant increase in assay signal. Determining successful action of a drug would require comparison of the distribution of the activity of this enzyme in a population of cells relative to the population before drug challenge. Similarly, following drug challenge, an individual cell could be compared to the population of naïve cells to determine whether its activity in this enzyme is increased to an extent that is statistically significant. Ongoing studies in our laboratory are focused on developing these applications of the reported platform.
Conclusion
In conclusion, we have developed a microfluidic device that utilizes DEP capture with ionic liquid isolation and electrical lysis to analyze the β-gal activity in single MDA-MB-231 breast cancer cells. The device utilized an optimized assay solution to maximize enzymatic activity with minimal conductivity suited for pDEP capture. Analysis of 258 single MDA cells shows a wide range of fluorescence increases compared to the no-cell control, indicating a wide range of enzymatic activities in the cells. The device is user-friendly, allows for analysis of dozens of cells simultaneously, and is highly adaptable. The assay presented here can be used to analyze changes in senescence of cancer cells, for example following a drug challenge. The flexibility of the device design allows for a wide range of future capabilities. DEP capture allows for the device to be adapted to other cell types without modification. DEP capture allows for separation of desired cell types from a mixture by their electrophysiological properties, as shown in our previous work;22 we anticipate that this property could be used to analyze enzymatic activity in single circulating tumor cells. The device can also be readily adapted to analyze other enzymes for which there is a fluorogenic substrate, including matrix metalloproteases7 or telomerase.26,27 The enzymatic reaction can also be used as an amplification technique in single-cell immunoassays, for example in analysis of surface antigen markers expressed in low numbers (< 100 copies/cell).28 Extension of the reported methods and device to this wider range of applications is ongoing in our laboratory.
Acknowledgements
We would like to acknowledge the National Institute of Health for funding this project through the R21 Trailblazer award number EB 028583-01.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).Lu Y; Xue Q; Eisele MR; Sulistijo ES; Brower K; Han L; Amir ED; Pe D; Miller-jensen K; Fan R; Lu Y; Xue Q; Eisele MR; Sulistijo ES; Brower K; Han L; Amir ED; Pe D Highly Multiplexed Profiling of Single-Cell Effector Functions Reveals Deep Functional Heterogeneity in Response to Pathogenic Ligands. 2015. 10.1073/pnas.1416756112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bhatia S; Frangioni J. v.; Hoffman RM; Iafrate AJ; Polyak K The Challenges Posed by Cancer Heterogeneity. Nat Biotechnol 2012, 30 (7), 604–610. 10.1038/nbt.2294. [DOI] [PubMed] [Google Scholar]
- (3).Armbrecht L; Dittrich PS Recent Advances in the Analysis of Single Cells. Anal Chem 2017, 89 (1), 2–21. 10.1021/acs.analchem.6b04255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lin FR; Niparko JK; Ferrucci, and L. Advances in High-Throughput Single-Cell Microtechnologies. Bone 2014, 23 (1), 1–7. 10.1016/j.copbio.2013.09.005.Advances. [DOI] [Google Scholar]
- (5).Kurz DJ; Decary S; Hong Y; Erusalimsky JD Senescence-Associated (Beta)-Galactosidase Reflects an Increase in Lysosomal Mass during Replicative Ageing of Human Endothelial Cells. J Cell Sci 2000, 113 (20), 3613–3622. 10.1242/jcs.113.20.3613. [DOI] [PubMed] [Google Scholar]
- (6).Debacq-Chainiaux F; Erusalimsky JD; Campisi J; Toussaint O Protocols to Detect Senescence-Associated Beta-Galactosidase (SA-Bgal) Activity, a Biomarker of Senescent Cells in Culture and in Vivo. Nat Protoc 2009, 4 (12), 1798–1806. 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- (7).Yu Z; Zhou L; Zhang T; Shen R; Li C; Fang X; Liu J Sensitive Detection of MMP9 Enzymatic Activities in Single Cell- Encapsulated Microdroplets as an Assay of Cancer Cell Invasiveness. ACS Sens 2017, 2, 626–634. 10.1021/acssensors.6b00731. [DOI] [PubMed] [Google Scholar]
- (8).Ocvirk G; Salimi-Moosavi H; Szarka RJ; Arriaga EA; Andersson PE; Smith R; Dovichi NJ; Harrison DJ β-Galactosidase Assays of Single-Cell Lysates on a Microchip: A Complementary Method for Enzymatic Analysis of Single Cells. Proceedings of the IEEE 2004, 92 (1), 115–125. 10.1109/JPROC.2003.820551. [DOI] [Google Scholar]
- (9).Eyer K; Stratz S; Kuhn P; Küster SK; Dittrich PS Implementing Enzyme-Linked Immunosorbent Assays on a Microfluidic Chip to Quantify Intracellular Molecules in Single Cells. Anal Chem 2013, 85 (6), 3280–3287. 10.1021/ac303628j. [DOI] [PubMed] [Google Scholar]
- (10).Kim SH; Fujii T Efficient Analysis of a Small Number of Cancer Cells at the Single-Cell Level Using an Electroactive Double-Well Array. Lab Chip 2016, 16 (13), 2440–2449. 10.1039/c6lc00241b. [DOI] [PubMed] [Google Scholar]
- (11).Lu Y; Chen JJ; Mu L; Xue Q; Wu Y; Wu P; Li J; Vortmeyer AO; Miller-jensen K; Wirtz D; Fan R High-Throughput Secretomic Analysis of Single Cells to Assess Functional Cellular Heterogeneity. Anal Chem 2013, 85, 2548–2556. 10.1021/ac400082e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wang C; Liu W; Tan M; Sun H; Yu Y An Open-Pattern Droplet-in-Oil Planar Array for Single Cell Analysis Based on Sequential Inkjet Printing Technology. Biomicrofluidics 2017, 11 (4), 1–11. 10.1063/1.4995294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu C; Liu J; Gao D; Ding M; Lin JM Fabrication of Microwell Arrays Based on Two-Dimensional Ordered Polystyrene Microspheres for High-Throughput Single-Cell Analysis. Anal Chem 2010, 82 (22), 9418–9424. 10.1021/ac102094r. [DOI] [PubMed] [Google Scholar]
- (14).Kim SH; Fujii T Efficient Analysis of a Small Number of Cancer Cells at the Single-Cell Level Using an Electroactive Double-Well Array. Lab Chip 2016, 16 (13), 2440–2449. 10.1039/c6lc00241b. [DOI] [PubMed] [Google Scholar]
- (15).Gagnon ZR Cellular Dielectrophoresis: Applications to the Characterization, Manipulation, Separation and Patterning of Cells. Electrophoresis 2011, 32 (18), 2466–2487. 10.1002/elps.201100060. [DOI] [PubMed] [Google Scholar]
- (16).Yale AR; Nourse JL; Lee KR; Ahmed SN; Arulmoli J; Jiang AYL; McDonnell LP; Botten GA; Lee AP; Monuki ES; Demetriou M; Flanagan LA Cell Surface N-Glycans Influence Electrophysiological Properties and Fate Potential of Neural Stem Cells. Stem Cell Reports 2018, 11 (4), 869–882. 10.1016/j.stemcr.2018.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li M; Anand RK Cellular Dielectrophoresis Coupled with Single-Cell Analysis. Anal Bioanal Chem 2018, 410 (10), 2499–2515. 10.1007/s00216-018-0896-y. [DOI] [PubMed] [Google Scholar]
- (18).Li M; Anand RK Integration of Marker-Free Selection of Single Cells at a Wireless Electrode Array with Parallel Fluidic Isolation and Electrical Lysis. Chem Sci 2019, 10 (5), 1506–1513. 10.1039/c8sc04804e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Cohen DE; Schneider T; Wang M; Chiu DT Self-Digitization of Sample Volumes. Anal Chem 2010, 82 (13), 5707–5717. 10.1021/ac100713u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Qin Y; Wu L; Schneider T; Yen GS; Wang J; Xu S; Li M; Paguirigan AL; Smith JL; Radich JP; Anand RK; Chiu DT A Self-Digitization Dielectrophoretic (SD-DEP) Chip for High-Efficiency Single-Cell Capture, On-Demand Compartmentalization, and Downstream Nucleic Acid Analysis. Angewandte Chemie - International Edition 2018, 57 (35), 11378–11383. 10.1002/anie.201807314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Shim S; Stemke-Hale K; Noshari J; Becker FF; Gascoyne PRC Dielectrophoresis has Broad Applicability to Marker-Free Isolation of Tumor Cells from Blood by Microfluidic Systems. Biomicrofluidics 2013, 7 (1), 0011808. 10.1063/1.4774307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li M; Anand RK High-Throughput Selective Capture of Single Circulating Tumor Cells by Dielectrophoresis at a Wireless Electrode Array. J Am Chem Soc 2017, 139 (26), 8950–8959. 10.1021/jacs.7b03288. [DOI] [PubMed] [Google Scholar]
- (23).Smale ST β-Galactosidase Assay. Cold Spring Harb Protoc 2010, 2010 (5), 1–3. 10.1101/pdb.prot5423. [DOI] [PubMed] [Google Scholar]
- (24).Banerjee G; Ray A; Hasan KN Is Divalent Magnesium Cation the Best Cofactor for Bacterial β-Galactosidase? J Biosci 2018, 43 (5), 941–945. 10.1007/s12038-018-9814-x. [DOI] [PubMed] [Google Scholar]
- (25).Schneider CA; Rasband WS; Eliceiri KW NIH Image to ImageJ: 25 Years of Image Analysis. Nat Methods 2012, 9 (7), 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).He C; Liu Z; Wu Q; Zhao J; Liu R; Liu B; Zhao T Ratiometric Fluorescent Biosensor for Visual Discrimination of Cancer Cells with Di Ff Erent Telomerase Expression Levels. ACS Sens 2018, 3, 757–762. 10.1021/acssensors.8b00059. [DOI] [PubMed] [Google Scholar]
- (27).Li X; Cui Y; Du Y; Tang A; Kong D Label-Free Telomerase Detection in Single Cell Using a Five-Base Telomerase Product-Triggered Exponential Rolling Circle Amplification Strategy. ACS Sens 2019, 4, 1090–1096. 10.1021/acssensors.9b00334. [DOI] [PubMed] [Google Scholar]
- (28).Johnson ES; Anand RK; Chiu DT Improved Detection by Ensemble-Decision Aliquot Ranking of Circulating Tumor Cells with Low Numbers of a Targeted Surface Antigen. Anal Chem 2015, 87 (18), 9389–9395. 10.1021/acs.analchem.5b02241. [DOI] [PubMed] [Google Scholar]