

Abstract
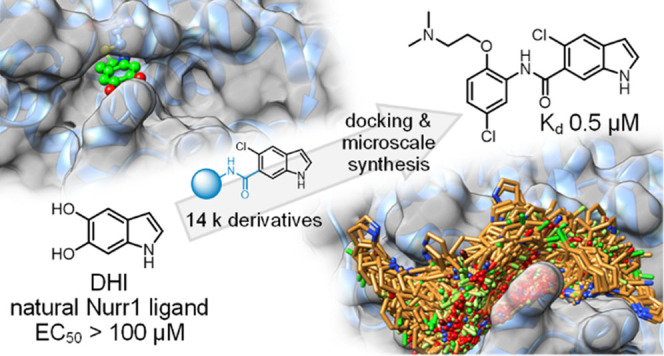
The neuroprotective transcription factor Nurr1 was recently found to bind the dopamine metabolite 5,6-dihydroxyindole (DHI) providing access to Nurr1 ligand design from a natural template. We screened a custom set of 14 k extended DHI analogues in silico for optimized descendants to select 24 candidates for microscale synthesis and in vitro testing. Three out of six primary hits were validated as novel Nurr1 agonists with up to sub-micromolar binding affinity, highlighting the druggability of the Nurr1 surface region lining helix 12. In vitro profiling confirmed cellular target engagement of DHI descendants and demonstrated remarkable additive effects of combined Nurr1 agonist treatment, indicating diverse binding sites mediating Nurr1 activation, which may open new avenues in Nurr1 modulation.
Introduction
Nuclear receptor-related 1 (Nurr1) is a neuronal ligand-activated transcription factor with neuroprotective and anti-neuroinflammatory roles.1,2 Animal models have characterized Nurr1 as a critical for (dopaminergic) neuron development and survival and link the transcription factor with neurodegenerative diseases.1−5 Diminished neuronal Nurr1 activity in mice caused a phenotype with features of Parkinson’s disease6 and exacerbated the pathology of Alzheimer’s disease models7 and experimental autoimmune encephalomyelitis.8 Moreover, Nurr1 levels were decreased in human patients of Parkinson’s and Alzheimer’s disease and in rodent models of these pathologies.9−13 The transcription factor may therefore provide new opportunities in the treatment of neurodegeneration.
Despite the remarkable therapeutic potential of Nurr1, modulators are still rare.14,15 The antimalarials amodiaquine (AQ) and chloroquine (CQ), identified as the first direct Nurr1 agonists,16 have limited potency but served for studies on Nurr1 biology7,16 and as lead structures.17,18 We have recently developed Nurr1 agonist 1 with significantly enhanced potency as a next-generation chemical tool to elucidate the therapeutic potential of Nurr1.19 Still, new and chemically diverse Nurr1 agonist scaffolds with improved potency are needed to enable target validation of this remarkable nuclear receptor.
Prostaglandins A1 and E120 and the oxidized dopamine metabolite 5,6-dihydroxyindole (DHI, 2a, Chart 1)21 have been discovered as potential endogenous Nurr1 ligands and were found to form covalent adducts with Cys566 of the Nurr1 ligand-binding domain (LBD). As a putative endogenous ligand, DHI is an attractive lead for Nurr1 agonist development. Structure–activity relationship (SAR) evaluation of DHI has yielded 5-chloro-1H-indole (2b) as a Nurr1 agonist with improved features and reduced toxicity,22 but an extension of the fragment-like 2a and 2b has not been studied.
Chart 1. Nurr1 Agonistsa.

a The blue arrow indicates the site for covalent interaction with Nurr1 (after oxidation of DHI).
Using DHI as lead and the Nurr1:DHI complex (PDB ID 6dda(21)) as a structural basis for docking-driven design and scoring, we have obtained derivatives of the natural Nurr1 ligand with enhanced affinity and potency. We employed a custom virtual library of 14,421 computationally generated DHI analogues, from which 24 top-scoring designs were prepared by microscale synthesis and tested for Nurr1 agonism. Six primary hits were prepared in batch and fully characterized to obtain three new validated and chemically diverse Nurr1 agonists. Additionally, structural fusion of the most active DHI analogue (5o, Kd 0.5 μM, EC50 3 μM) with the known ligand AQ produced another potent Nurr1 agonist (13, Kd 1.5 μM, EC50 3 μM). In vitro characterization confirmed cellular Nurr1 modulation by DHI descendants and interestingly revealed pronounced additive Nurr1 activation with the recently developed agonist 1.19 Our structure-driven design and microscale synthesis approach successfully generated innovative Nurr1 ligand scaffolds based on a natural ligand, demonstrating druggability of the Nurr1 surface region lining helix 12.
Results and Discussion
Structure-Guided Design
Despite recent progress in Nurr1 ligand discovery,1 only a few ligand-bound Nurr1 LBD cocrystal structures are available among which the Nurr1:DHI complex (PDB ID 6dda(21)) appeared best suitable for structure-based design due to the favorable features of the indole scaffold. Structure analysis suggested space for structural extension of DHI (2a) or the related 5-chloroindole (2b), especially in the 5- and 6-positions of the indole, while the five-membered ring was buried in a narrow pocket (Figure 1a). The indole 5- and 6-positions are solvent exposed with opportunities to explore adjacent grooves formed by helices H4/H12 and H10/H11 on the surface of the Nurr1 LBD. Previous structure–activity relationship analysis of the indole has demonstrated a preference for a chlorine substituent in the 5-position,22 prompting us to probe the extension of the indole in the 6-position to address the surface binding opportunities.
Figure 1.
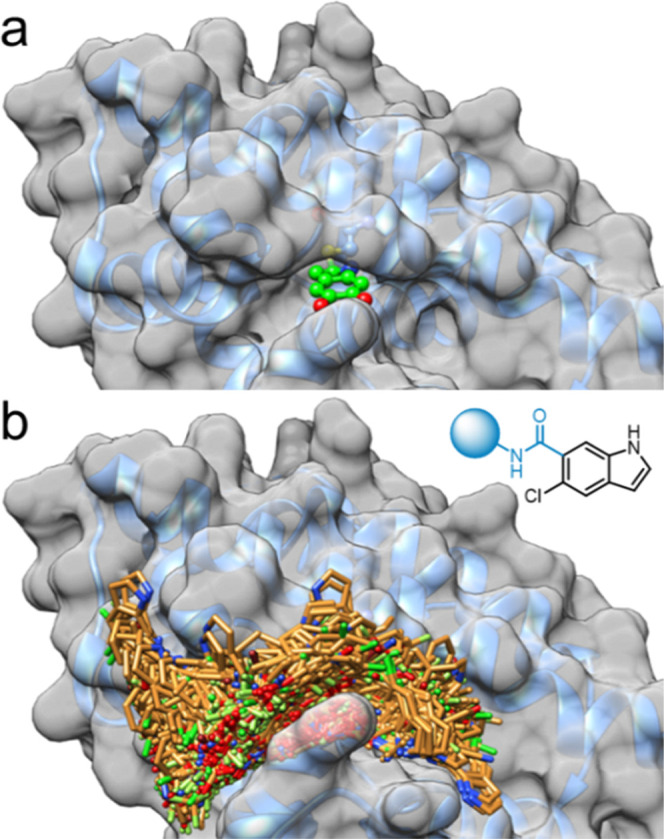
Structural basis for DHI-derived Nurr1 agonist design. (a) Cocrystal structure of the DHI-bound Nurr1 LBD (PDB ID 6dda,21 surface representation with bound DHI). DHI is bound in a narrow pocket with no space for extension around the five-membered ring, while the 5- and 6-positions are oriented toward the solvent. (b) General structure of the virtual amide library based on 5-chloro-1H-indole-6-carboxylic acid and 14,421 primary amines (MW ≤ 240) and docking of the virtual amide library to the Nurr1 LBD (PDB ID 6dda(21)). The virtual designs extended to the grooves on the Nurr1 LBD surface around the DHI-binding site.
For a broad virtual exploration of potential substituents, we generated a virtual library based on 5-chloroindole as a common motif, which was extended in the 6-position by an amide linker and fused with 14,421 commercially available primary amines (MW ≤ 240) to obtain a virtual library of amides (full list in the Supporting Information). Amide linkage was chosen since potential for H-bond formation between the carbonyl group and the His516 backbone amine and between the amide nitrogen and the backbone carbonyl of Pro597 was observed in a preliminary docking assessment (Figure S1). Additionally, this approach enabled rapid and economic preparation of computationally preferred designs in microscale format even from very expensive amine building blocks. The full virtual library was docked with Glide to the DHI-binding site of the Nurr1 LBD. In line with the design hypothesis, the molecules populated the two grooves on the protein surface (Figure 1b). Docking scores varied widely over the entire virtual library (−7.71 to +3.57 for the best pose of each molecule) suggesting that the model was suitable to distinguish promising designs. We inspected the binding modes of the 100 top-ranking designs and selected 24 structurally diverse molecules (Table 1) for synthesis and testing based on a binding mode addressing the DHI pocket and suitability for microscale amide synthesis.
Table 1. Gal4–Nurr1 Modulation by the Microscale DHI Analogue Librarya.
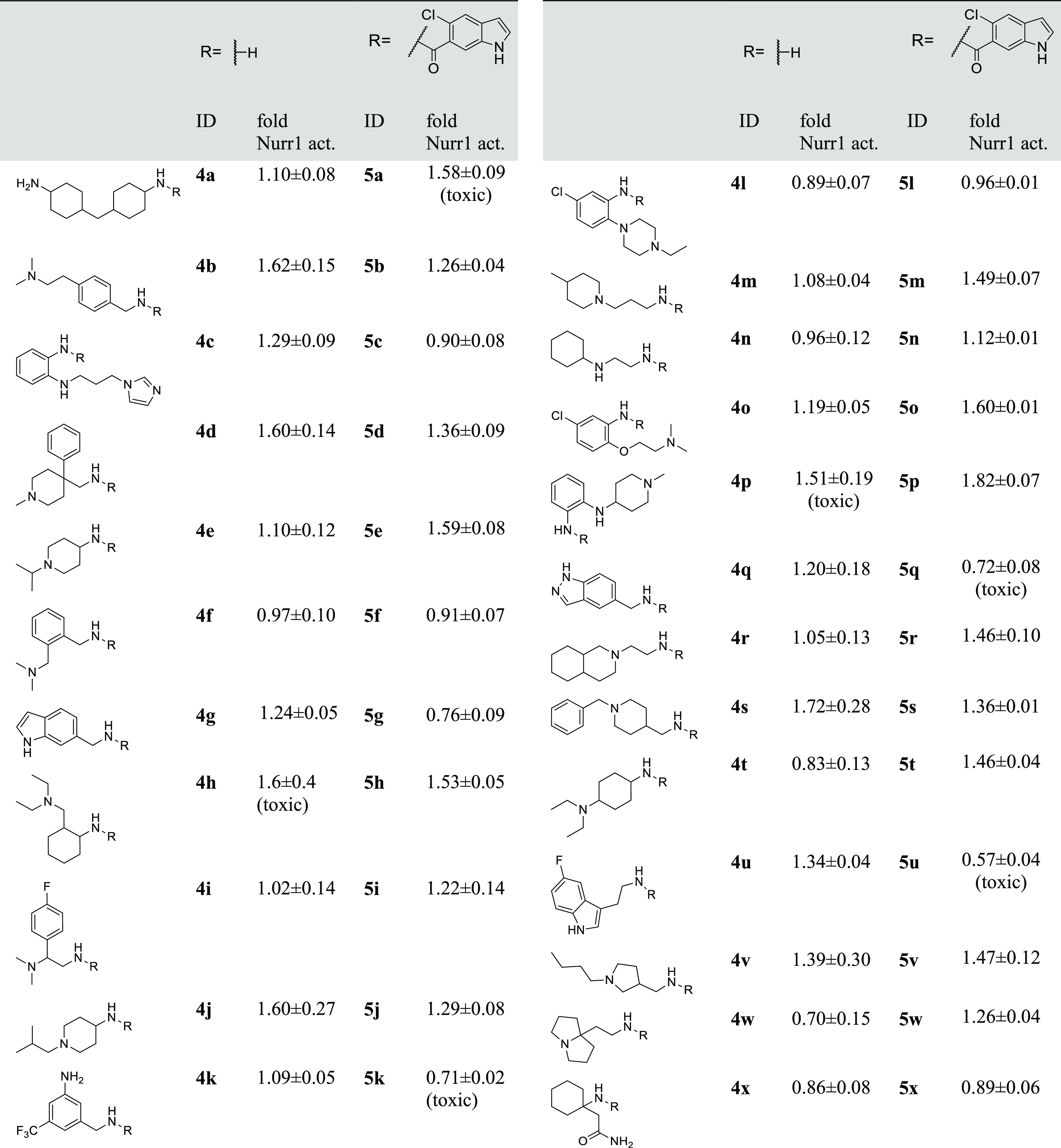
Compounds were screened at 100 μM assuming full conversion, actual concentrations were lower. Nurr1 activity data from the hybrid reporter gene assay are relative reporter activity vs DMSO-treated cells. Data are the mean ± SD, n = 3. Compounds causing a decrease of the Renilla luminescence to ≤50% of DMSO-treated cells were considered as potentially toxic (cf. Figure S2).
Microscale Library Preparation and Screening
To explore the Nurr1 agonist potential of the computationally preferred designs in a rapid and economical fashion, we initially prepared the compounds in a microscale format (100 μmol). Amide synthesis was performed in 1.5 mL reaction tubes from 5-chloro-1H-indole-6-carboxylic acid (3) and the respective amines (4a–x) with EDC·HCl in ethyl acetate to obtain 5a–x (Scheme 1a). Products from the microscale synthesis were roughly purified by a washing procedure with aqueous sodium bicarbonate, and amide formation was confirmed by mass spectrometry. The 5-chloro-1H-indole-6-carboxylic acid building block 3 was prepared from 2-chloro-4-methylbenzoic acid (6) by Batcho–Leimgruber indole synthesis (Scheme 1b) involving nitration to 7, esterification (8), cyclization with DMF dimethylacetal to indole 9, and ester hydrolysis to 3.
Scheme 1. Microscale Amide Synthesis (a) and Synthesis of Indole Building Block 3 (b).

Reagents and conditions: (i) EDC·HCl, EtOAc, rt, 36 h; (ii) HNO3/H2SO4, 5 °C, 0.5 h, 59%; (iii) acetyl chloride, MeOH, 50 °C, 4 h, 96%; (iv) DMF-DMA, DMF, 120 °C, 2 h; then Zn, AcOH/H2O, 80 °C, 2 h, 40%; (v) LiOH·H2O, EtOH/H2O, rt, 18 h, 94%.
A Gal4–Nurr123 hybrid reporter gene assay served for in vitro testing which is based on a chimeric receptor composed of the human Nurr1 LBD and the DNA-binding domain of Gal4 from yeast. A firefly luciferase construct with five tandem repeats of the Gal4 response element was used as the reporter gene and constitutively expressed (SV40 promoter) Renilla luciferase was employed for normalization and to capture potential test compound toxicity. The microscale synthesis products 5a–x were tested for Nurr1 modulation in the Gal4–Nurr1 hybrid reporter gene assay at 100 μM (assuming full conversion, actual concentrations were lower). To monitor the activity of the amine building blocks potentially remaining in the mixtures, the amines were also tested at 100 μM to reveal true positive Nurr1 ligand hits. This enabled the discovery of promising scaffolds for further evaluation from a very diverse set in a rapid and cost-efficient manner despite expensive building blocks. The possibility of false negatives resulting from the microscale synthesis format, purification procedure, and assay setting was accepted.
Of the 24 computationally favored designs 5a–x, six (5e, 5m, 5o, 5r, 5t, 5v; Table 1) were considered as primary hits as they activated Nurr1 by ≥140% compared to DMSO-treated cells, exhibited no pronounced effect on cell viability as observed by stable Renilla luciferase activity and in a WST-8 assay (Figure S2), and their corresponding amines were inactive/less active. These compounds were hence prepared in batch and isolated for full characterization.
Hit Validation
For orthogonal validation of the primary hits, we first employed isothermal titration calorimetry (ITC) to observe direct interaction and determine binding affinities (Table 2). Of the six hits in the primary screening, three (5e, 5m, 5t) exhibited no detectable interaction with the Nurr1 LBD and three revealed binding with Kd values of 0.5 μM (5o), 3.2 μM (5r), and 16 μM (5v), respectively. In accordance with these results, 5e, 5m, and 5t failed to activate Nurr1 up to 100 μM, while 5o, 5r, and 5v were confirmed as Nurr1 agonists (Table 2). 5o emerged as the most active DHI descendant with a Kd value of 0.5 μM and an EC50 value of 3 μM.
Table 2. Binding Affinity to the Nurr1 LBD and Nurr1 Modulation of the Primary Hits (2a and 2b for Comparison)a–c.

Binding affinity of 5e, 5m, 5o, 5r, 5t and 5v to the Nurr1 LBD was determined by ITC. Nurr1 modulation was determined in the Gal4–Nurr1 hybrid reporter gene assay; data are mean ± SD; n ≥ 3.
Covalent binder.
No interaction in ITC with 200 μM ligand and 30 μM protein.
Binding Site Evaluation
Inspection of the predicted binding modes of the three active hits to Nurr1 revealed a similar orientation of 5o, 5r, and 5v with binding to a hydrophobic groove on the Nurr1 LBD surface lining helix 12 (Figure 2a), suggesting that this surface region of Nurr1 is druggable. The common indole motif was bound facing Cys566 and sandwiched between Arg515 and Arg563. Binding of the amide substituents was mainly mediated by hydrophobic contacts. The most active compound 5o additionally formed a face-to-face interaction with His516 (Figure 2b), rationalizing its enhanced potency.
Figure 2.
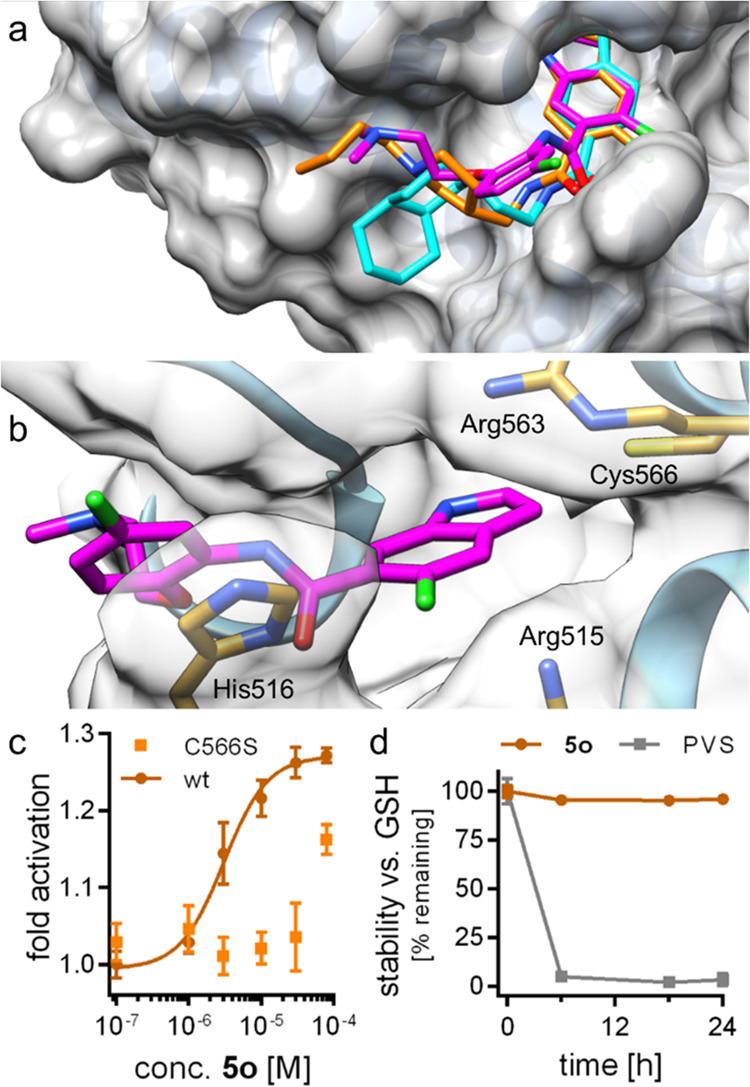
Predicted binding modes of 5o (magenta), 5r (cyan), and 5v (orange) to the Nurr1 LBD (PDB ID 6dda(21)). (a) The three active DHI descendants 5o, 5r, and 5v were predicted to bind to the DHI-binding site and extend toward a hydrophobic groove lining helix 12. (b) The most active Nurr1 agonist 5o formed a face-to-face contact with His516, which is not observed for 5r and 5v, supporting the higher affinity of 5o. (c) 5o was less active on the Nurr1-C566S mutant, supporting interaction with the proposed epitope. Data are the mean ± S.E.M.; n ≥ 3. (d) 5o was stable against reaction with glutathione (GSH). Phenyl vinyl sulfone (PVS) as positive control (125 μM 5o or PVS were incubated with 2.5 mM GSH in PBS at 37 °C). n = 3.
Mutagenesis of the binding site cysteine 566 to serine diminished the activity of 5o (Figure 2c), supporting interaction with this epitope and the importance of Cys566 in ligand–Nurr1 contacts. High stability of 5o against glutathione (Figure 2d) and no observable adduct formation between recombinant Nurr1 LBD and 5o in LCMS (Figures S3 and S4) indicated that the interaction was non-covalent despite the prominent role of Cys566.
Structure–Activity Relationship of DHI Descendant 5o
The DHI descendant 5o presented markedly enhanced activity and affinity (Figure 3a) compared to the natural template 2a and induced Nurr1-regulated expression of tyrosine hydroxylase (TH) and vesicular amino acid transporter 2 (VMAT2) in astrocytes (T98G), indicative of cellular target engagement (Figure 3b). Based on this promising profile, we explored the structure–activity relationship of this new Nurr1 agonist scaffold (Table 3). The inverted analogue 10 comprising the opposite indole regiochemistry exhibited similar activity as 5o, indicating, in line with the predicted binding mode (Figure 2), that the orientation of the indole scaffold in the binding site is not critical. In accordance with this, methylation of the indole nitrogen of 5o in 11 had also little effect on affinity, albeit a fivefold decrease in cellular potency was observed. Reduction of the amide in 5o to a secondary amine linker in 12 was also tolerated, but it led to no improvement in affinity or potency. These results thus suggest 6-chloro-1H-indole-5-carboxamide and 5-chloro-1H-indole-6-carboxamide as suitable and easily accessible scaffolds to develop Nurr1 ligands binding to the LBD surface.
Figure 3.

In vitro profiling of DHI-derived Nurr1 agonists. (a) Isothermal titration calorimetry (ITC) demonstrated binding of 5o and 13 to the recombinant Nurr1 LBD. The fitting of the heat of binding is shown, and the isotherms at 25 °C are shown as insets. (b) 5o (30 μM) induced Nurr1-regulated mRNA expression of tyrosine hydroxylase (TH) and vesicular amino acid transporter 2 (VMAT2) in T98G cells. Data are the mean ± S.E.M. rel. mRNA expression (2–ΔCt), n = 4. #p < 0.1, ** p < 0.01 (t-test vs DMSO ctrl). (c) 5o (EC50 = 2 ± 1 μM) and 13 (EC50 = 4 ± 1 μM) activated full-length human Nurr1 on the NBRE. Data are the mean ± S.E.M. fold act. vs DMSO ctrl, n ≥ 4. (d) 5o exhibited preference for Nurr1 over the related NR4A receptors (p < 0.001 vs NOR-1; p < 0.01 vs Nur77; two-way analysis of variance (ANOVA) with multiple comparisons test); 13 activated Nurr1 and Nur77 with equal potency but revealed preference over NOR-1 (p < 0.01). Data are the mean ± S.E.M. fold act. vs DMSO ctrl, n ≥ 4. (e) 5o and 13 did not modulate the activity of nuclear receptors outside the NR4A family. Data are the mean ± S.E.M. fold activation vs DMSO ctrl, n ≥ 3. (f) 5o and 13 enhanced max. Nurr1 activation by agonist 1 to 134% (5o) and 177% (13) of its max. effect alone. Data are the mean ± S.E.M. relative Nurr1 act. vs 1 μM 1, n ≥ 3. (g) DHI (2a), 5-chloroindole (5CI, 2b) and the descendants 5o and 13 modulated Nurr1-regulated gene expression in dopaminergic neural cells (N27). TH, tyrosine hydroxylase; VMAT2, vesicular amino acid transporter 2; SOD1/2, superoxide dismutase 1/2. Data are the mean ± S.E.M. relative mRNA expression compared to 0.1% DMSO; n = 7–8; * p < 0.05, ** p < 0.01, *** p < 0.001 (Wilcox test or t-test).
Table 3. In Vitro Characterization of 10–12 Derived from Screening Hit 5o and 5o/AQ-Hybrid 13a.

Binding affinity to the Nurr1 LBD was determined by ITC. Nurr1 modulation was determined in the Gal4–Nurr1 hybrid reporter gene assay; data are mean ± SD; n ≥ 3.
Several available Nurr1 ligands comprise a chloroquinoline motif derived from AQ,1,16,17 and we have observed that a scaffold hop from indole (fluvastatin, EC50 = 1.9 μM) to quinoline (pitavastatin, EC50 = 0.12 μM) produced a relevant improvement in potency.2 Thus, we evaluated the possibility of fusing the structural elements of 5o and AQ. Interestingly, the merged compound 13 composed of the 7-chloroquinolin-4-amine motif of AQ and the amide substituent of 5o acted as a Nurr1 agonist (Kd 1.5 μM, EC50 3 μM) with considerably higher potency than AQ (EC50 36 μM).
In Vitro Profiling of Optimized DHI Descendants
5o and its AQ-hybrid 13 emerged as the most potent DHI descendants from the structure-guided approach to Nurr1 modulator development. Their further in vitro profiling confirmed Nurr1 agonism also on the human full-length receptor on the response element NBRE with similar potency to that observed in the hybrid reporter gene assay (5o: EC50 = 2 ± 1 μM, 2.1 ± 0.2-fold activation; 13: EC50 = 4 ± 1 μM, 2.4 ± 0.2-fold activation; Figure 3c).
Selectivity profiling in Gal4 hybrid reporter gene assays (Figure 3d) revealed a preference of 5o for Nurr1 over Nur77 (NR4A1) and, interestingly, no NOR-1 (NR4A3) activation, suggesting that the binding site of the DHI descendants is not conserved in NOR-1. 13 activated Nurr1 and Nur77 with equal potency but was less active on NOR-1. No nuclear receptor modulation by 5o and 13 was observed outside the NR4A family (Figure 3e).
We also studied the response of Nurr1 to combined treatment with 5o/13 and the recently developed Nurr1 agonist 1 and, interestingly, detected additive Nurr1 activation (Figure 3f). The presence of 5o or 13 (20 μM each) did not alter the EC50 value of 1 but increased its max. effect by 34–77%. This non-competitive behavior with additive Nurr1 activation suggests different, non-overlapping binding sites of the ligands, opening new avenues to modulation of Nurr1.
To assess the effects of the DHI descendants in a native cellular setting, we evaluated their effects on Nurr1-regulated gene expression in the immortalized rat dopaminergic neural cell line N2724,25 (Figure 3g). 5-Chloroindole (2b), 5o, and 13 induced TH, VMAT, and superoxide dismutase 1 and 2 (SOD1/2) expression in a dose-dependent fashion. The efficacy of VMAT2 and SOD1 induction by 2b, 5o, and 13 was comparable, while TH and SOD2 upregulation by 5o and 13 was stronger compared to that by 2b. DHI (2a) had generally weaker effects and, interestingly, downregulated TH expression, suggesting that other regulatory mechanisms were involved.
Conclusions
Drug discovery based on natural ligands of nuclear receptors has been very fruitful in the past as exemplified by steroidal ligands for NR3 receptors or the FXR agonist obeticholic acid.15,26 Our results demonstrate that the binding site of the natural (covalent) Nurr1 ligand DHI (2a) is druggable and can be addressed by non-covalent binders with markedly enhanced potency compared to the natural template, opening a new avenue to Nurr1 agonist development.
Apart from the activity of 5-chloro-1H-indole (2b) and related fragment-like analogues,22 no SAR knowledge was available on DHI as Nurr1 agonist, prompting us to follow a structure-based virtual screening approach using a custom library of DHI derivatives. Modeling prioritized 24 molecules and experimental validation confirmed three DHI analogues as novel Nurr1 ligands. 5o emerged from these DHI analogues with sub-micromolar affinity to Nurr1, preference over Nur77, and selectivity over NOR-1. Additionally, 5o and its AQ-hybrid 13 caused additive Nurr1 activation with the recently discovered agonist 1,19 indicating that Nurr1 can be simultaneously modulated through diverse ligand-binding sites, which may have additional therapeutic potential.
Despite recent progress in ligand discovery, molecular understanding of ligand binding to Nurr1 is still limited, and binding sites are elusive.1 The binding site of DHI-derived Nurr1 ligands can be located with high confidence at the surface of the LBD close to helix 12 which opens new possibilities in Nurr1 ligand development. DHI descendants 5o and 13 valuably expand the scarce collection of validated Nurr1 modulators and can serve as tools for chemogenomics and functional studies on the role of DHI and its binding site in Nurr1 modulation.
Chemistry
The primary hits (5e, 5m, 5o, 5r, 5t, 5v) were prepared according to Scheme 2 by amide coupling of 3 and the corresponding amines 4e, 4m, 4o, 4r, 4t, and 4v, respectively. The synthesis of 10 comprising an inverted substitution pattern on the indole was performed by the same route using the corresponding 6-chloro-1H-indole-5-carboxylic acid (3a) for amide coupling with 4o. The N-methyl indole analogue 11 was obtained by N-methylation of the ester 9, followed by ester hydrolysis to 9a and amide coupling with 4o using TCFH-NMI as coupling agent.
Scheme 2. Batch Synthesis of 5e, 5m, 5o, 5r, 5t, 5v, and 10–13.

Reagents and conditions: (i) EDC·HCl, TEA, CHCl3, rt, 18 h, 8–32%; (ii) LiAlH4, THF, 0 °C, 1 h, 71%; (iii) Dess–Martin periodinane, DCM, DMF, 0 °C–rt, 1 h, 100%; (iv) NaBH(OAc)3, AcOH, DCM, DCE, rt, 2 h, 36%; (v) EDC·HCl, TEA, CHCl3, rt, 18 h, 5%; (vi) NaH, CH3I, DMF, 0 °C, 10 min, rt, 2 h, 33%; (vii) LiOH·H2O, EtOH, H2O, rt, 18 h, 99%; (viii) NMI, TCFH, DMF, 80 °C, 18 h, 26%; (ix) Pd(OAc)2, BINAP, K3PO4, dioxane, 90 °C, 24 h, 29%.
For the preparation of the secondary amine analogue 12, 9 was reduced to the corresponding alcohol 14 using LiAlH4 and reoxidized to its aldehyde 15 with Dess–Martin periodinane, which reacted with amine 4o in a reductive amination reaction to afford 12. The AQ/5o hybrid 13 was prepared from 4-bromo-7-chloroquinoline (16) by Buchwald–Hartwig amination with amine 4o.
Experimental Section
Chemistry
General
All chemicals were of reagent grade and used without further purification unless otherwise specified. All reactions were conducted in oven-dried Schlenk glassware under an argon atmosphere and in absolute solvents. Other solvents, especially for work-up procedures, were of reagent grade or purified by distillation (iso-hexane, ethyl acetate, ethanol). Reactions were monitored by thin-layer chromatography on TLC Silica gel 60 F254 aluminum sheets by Merck and visualized under ultraviolet light (254 nm). Purification by column chromatography (CC) was performed on a puriFlash XS520Plus system (Advion, Ithaca, NY) using high-performance spherical silica columns (SIHP, 50 μM) by Interchim and a gradient of iso-hexane to EtOAc; reversed-phase CC was performed on a puriFlash 5.250 system (Advion) using C18HP columns (SIHP, 15 μM) by Interchim and a gradient of 0.1% FA in H2O, 10–100% acetonitrile (HPLC gradient grade). Preparative HPLC was also performed on the puriFlash 5.250 system using a PREP-LC column (C18-HQ, 5 μM) by Interchim and a gradient of 0.1% FA in H2O, 10–100% methanol (HPLC gradient grade). Mass spectra were obtained on a puriFlash-CMS system (Advion) using atmospheric pressure chemical ionization (APCI). High-resolution mass spectrometry analysis (HRMS) was performed with a Thermo Finnigan LTQ FT instrument for electrospray ionization (ESI). NMR spectra were recorded on a Bruker Avance III HD 400 MHz or 500 MHz spectrometer equipped with a CryoProbe Prodigy broadband probe (Bruker). Chemical shifts are reported in δ values (ppm), and coupling constants (J) are reported in hertz (Hz). Purity was determined by quantitative 1H NMR (qH NMR) according to a method described by Pauli et al. with internal calibration.27 The qH NMR measurements were carried out under conditions allowing complete relaxation to assure the exact determination of peak area ratios. Used internal standards were ethyl 4-(dimethylamino)benzoate (LOT# BCCC6657, purity 99.63%) and maleic acid (LOT# BCBM8127V, purity 99.94%) in CDCl3, MeOD-d4, DMSO-d6 or acetone-d6. All compounds for biological testing had a purity ≥95% according to qH NMR.
2-Chloro-4-methyl-5-nitrobenzoic acid (7)28
2-Chloro-4-methylbenzoic acid (6, 10.0 g, 55.8 mmol, 1.00 equiv) was suspended in sulfuric acid (100 mL) and cooled to 5 °C. Over a period of 40 min nitric acid (3.39 mL, 52.7 mmol, 0.90 equiv) was added dropwise so that the reaction mixture did not exceed a temperature of 15 °C. After complete addition of nitric acid, the reaction mixture was stirred at rt for 30 min and then poured into ice water. The solid that subsequently precipitated was filtered off, washed with cold water, and dissolved in EtOH. Water was added dropwise to the solution, whereupon a colorless solid precipitated. The solid was filtered off, washed with cold water, and dried under reduced pressure, yielding compound 7 as a colorless solid (7.57 g, 59%). Rf (iso-hexane/EtOAc = 8:2 + 2% AcOH) = 0.33. MS (−APCI): m/z 214.5 ([M – H]−). 1H NMR (400 MHz, acetone-d6): δ = 8.54 (s, 1H), 7.72 (s, 1H), 2.65 (s, 3H) ppm. 13C NMR (101 MHz, acetone-d6): δ = 164.70, 148.50, 139.43, 138.57, 136.14, 129.76, 128.72, 20.03 ppm.
Methyl 2-Chloro-4-methyl-5-nitrobenzoate (8)28
Acetyl chloride (12.4 mL, 174 mmol, 5.00 equiv) was added dropwise to methanol (70 mL) at 5–10 °C. A solution of 2-chloro-4-methylnitrobenzoic acid (7, 7.50 g, 34.8 mmol, 1.00 equiv) in methanol (100 mL) was added in portions, and the reaction mixture was stirred at 50 °C for 4 h. After cooling to rt, the solvent was removed under reduced pressure. The yellow solid obtained was dissolved in DCM and washed with brine solution and water. The organic phase was dried over MgSO4, filtered, and evaporated to yield 8 as a colorless solid (7.66 g, 96%). Rf (iso-hexane/EtOAc = 8:2) = 0.41. MS (+APCI): m/z 229.5 ([M + H]+). 1H NMR (400 MHz, acetone-d6): δ = 8.48 (s, 1H), 7.73 (s, 1Hl), 3.95 (s, 3H), 2.65 (s, 3H) ppm. 13C NMR (101 MHz, acetone-d6): δ = 167.44, 148.34, 139.57, 138.30, 136.08, 129.51, 128.51, 53.18, 20.05 ppm.
Methyl 5-Chloro-1H-indole-6-carboxylate (9)28
Methyl 2-chloro-4-methyl-5-nitrobenzoate (8, 7.66 g, 33.4 mmol, 1.00 equiv) was dissolved in DMF (50 mL). N,N-dimethylformamide dimethylacetal (10.4 mL, 49.9 mmol, 1.50 equiv) was added to the solution, and the reaction mixture was stirred for 2 h at 130 °C. The reaction mixture was concentrated under vacuum, dissolved in EtOAc, and washed with brine and water. The aqueous phases were extracted with EtOAc, and the combined organic phases were dried over MgSO4. After removing the solvent, the obtained purple solid was dissolved in acetic acid (100 mL) and water (20 mL). Over a period of 40 min, zinc powder (13.06 g, 65.34 mmol, 6.0 equiv) was added in portions and the reaction mixture was heated to 80 °C for 2 h. After cooling to rt, the mixture was diluted with EtOAc and filtered. The organic phase was separated and washed with saturated NaHCO3 solution and brine. The organic phase was dried over MgSO4, filtered, and concentrated under reduced pressure. The brown oil was purified by CC and yielded compound 9 as a green solid (2.80 g, 40%). Rf (iso-hexane/EtOAc = 8:2) = 0.23. MS (+APCI): m/z 209.7 ([M + H]+). 1H NMR (400 MHz, acetone-d6): δ = 10.69 (s, 1H), 8.03 (s, 1H), 7.71 (s, 1H), 7.60 (t, J = 2.8 Hz, 1H), 6.54 (m, 1H), 3.87 (s, 3H) ppm. 13C NMR (126 MHz, acetone-d6): δ = 167.20, 134.86, 132.86, 130.68, 124.13, 123.34, 122.65, 116.02, 102.38, 52.26 ppm.
5-Chloro-1H-indole-6-carboxylic acid (3)
Methyl 5-chloro-1H-indole-6-carboxylate (9, 2.80 g, 13.4 mmol, 1.00 equiv) was dissolved in EtOH (25 mL) and water (25 mL). LiOH·H2O (1.69 g, 40.2 mmol, 3.00 equiv) was added, and the reaction mixture was stirred at rt for 18 h. After removal of the solvent, the resulting solid was dissolved in water. The alkaline solution (pH ≥ 11) was extracted with EtOAc. The aqueous layer was acidified with aqueous hydrochloric acid (10%) and extracted with EtOAc. The latter organic layers were combined, washed with brine, dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the residue was recrystallized from a mixture of DCM/diisopropylether (1:1), yielding compound 3 as a beige solid (2.47 g, 94%). Rf (iso-hexane/EtOAc = 8:2 + 2% AcOH) = 0.22. MS (+APCI): m/z 195.7 ([M + H]+). 1H NMR (400 MHz, acetone-d6): δ = 11.15 (s, 1H), 10.69 (s, 1H), 8.13 (s, 1H), 7.70 (s, 1H), 7.62–7.58 (m, 1H), 6.56–6.53 (m, 1H) ppm. 13C NMR (126 MHz, acetone-d6): δ = 168.58, 134.20, 131.45, 129.74, 123.62, 122.39, 121.82, 115.47, 101.46 ppm.
5-Chloro-N-{5-chloro-2-[2-(dimethylamino)ethoxy]phenyl}-1H-indole-6-carboxamide (5o)
5-Chloro-1H-indole-6-carboxylic acid (3, 150 mg, 767 μmol, 1.00 equiv) and oxalyl chloride (69.0 μL, 767 μmol, 1.00 equiv) were dissolved in 4 mL of a mixture of one drop of DMF in DCM (14 mL). After 2 h, the solvent was removed under reduced pressure. The remaining residue was dissolved in DCM (6 mL), and 5-chloro-2-[2-(dimethylamino)ethoxy]aniline (4o, 165 mg, 767 μmol, 1.00 equiv) was added to the solution. The reaction mixture was stirred for 18 h at rt. Then, DCM (10 mL) and saturated NaHCO3 solution (10 mL) were added. The separated organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by CC, and subsequent recrystallization from methanol yielded compound 5o as a colorless crystalline solid (23 mg, 8%). Rf (iso-hexane/EtOH = 1:1 + 2% TEA) = 0.24. MS (+APCI): m/z 391.3 ([M + H]+). HRMS (+EI): m/z calculated 391.08488 for [C19H19Cl2N3O2]+•, found: 391.08645 ([M]+•). 1H NMR (500 MHz, CDCl3) δ = 10.17 (s, 1H), 8.78 (s, 1H), 8.71–8.66 (m, 1H), 7.90–7.86 (m, 1H), 7.68 (s, 1H), 7.38–7.33 (m, 1H), 7.03 (dd, J = 8.6, 2.6 Hz, 1H), 6.92 (d, J = 8.7 Hz, 1H), 6.54–6.50 (m, 1H), 4.09 (t, J = 5.4 Hz, 2H), 2.55 (t, J = 5.0 Hz, 2H), 2.02 (s, 6H) ppm. 13C NMR (126 MHz, CDCl3) δ = 166.00, 146.62, 134.11, 131.66, 130.50, 129.03, 128.13, 128.09, 123.77, 122.13, 121.87, 120.81, 116.73, 113.63, 102.52, 69.45, 57.99, 45.18 ppm. qH NMR (400 MHz, MeOD-d4, maleic acid as reference): purity = 95.4%.
7-Chloro-N-{5-chloro-2-[2-(dimethylamino)ethoxy]phenyl}quinolin-4-amine (13)
4-Bromo-7-chloro-chloroquinoline (16, 170 mg, 699 μmol, 3.00 equiv), Pd(OAc)2 (5.3 mg, 23 μmol, 0.1 equiv), (+/−)-2,2′-Bis(diphenylphosphino)-1,1′-binaphthalene (29 mg, 47 μmol, 0.2 equiv), K3PO4 (82.6 mg, 389 μmol, 1.67 equiv) and 5-chloro-2-[2-(dimethylamino)ethoxy]aniline (5o, 50.0 mg, 233 μmol, 1.00 equiv) were dissolved in 1,4-dioxane (10 mL). The suspension was stirred at 90 °C for 24 h. After completion of the reaction, the mixture was filtered through Celite and purified by CC, yielding compound 13 as a yellow solid (25 mg, 29%). Rf (DCM/MeOH = 95:5) = 0.13. MS (+APCI): m/z 375.7 ([M + H]+). HRMS (+ESI): m/z calculated 376.09779 for [C19H20Cl2N3O]+, found 376.09885 ([M + H]+). 1H NMR (400 MHz, acetone-d6) δ = 8.57 (d, J = 5.2 Hz, 1H), 8.54 (s, 1H), 8.27 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 2.2 Hz, 1H), 7.55 (dd, J = 9.0, 2.2 Hz, 1H), 7.49 (d, J = 2.5 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 7.13 (dd, J = 8.7, 2.5 Hz, 1H), 7.01 (d, J = 5.2 Hz, 1H), 4.22 (t, J = 5.3 Hz, 2H), 2.57 (t, J = 5.4 Hz, 2H), 2.23 (s, 6H) ppm. 13C NMR (101 MHz, acetone-d6) δ = 152.13, 150.06, 149.79, 147.27, 134.39, 132.89, 128.44, 126.28, 125.43, 124.07, 123.28, 121.72, 119.01, 117.67, 102.99, 68.52, 57.84, 44.8 ppm. qH NMR (400 MHz, acetone-d6, Ethyl-4-(dimethylamino)benzoate as reference): purity = 95.2%.
Computational Methods
Preparation of the Virtual Screening Library
Commercially available primary amines were retrieved from Reaxys with a molecular weight cutoff of ≤240, yielding 14,421 molecules. The “RDKit Two Component Reaction” node in KNIME29 was used to generate a virtual library of amides by fusing compound 3 and the commercial amines. Three-dimensional (3D) structures of the resulting molecules were obtained using Schrödinger ligprep software with force field OPLS4. Protonation was calculated using Epik30 in a pH range of 6–8. Chiral information in the two-dimensional (2D) input structure was retained when present; otherwise, all possible stereoisomers were generated. The final amide library comprised 18,421 molecules for docking.
Docking
The 3D structure of Nurr1 LBD (PDB ID 6dda(21)) was prepared using the Schrödinger Protein Preparation Wizard in Maestro. The covalently bound ligand DHI was removed manually before docking. The center of the docking grid was centered at—40.62762552165796, 12.031786250745345, and 66.69510016401517. Docking was performed using Schrödinger GLIDE31 software in standard precision mode (SP). All parameters were left at the default values (SP; flexible ligands; sample ring conformations of ligand; add Epik state penalties to docking score; scaling of van der Waals radii = 0.8; postdocking minimization of five poses per ligand; write out of one pose per ligand). A total of 18,340 poses were generated with docking scores in the range between −7.71 and +5.73. A total of 18,313 poses had docking scores below zero. Structural analyses were visualized by UCSF Chimera.32
Biological Characterization
Hybrid Reporter Gene Assays
Nurr1 modulation was determined in HEK293T cells (German Collection of Microorganisms and Cell Culture GmbH, DSMZ) by Gal4 hybrid reporter gene assays using pFR-Luc (Stratagene, La Jolla, CA; reporter), pRL-SV40 (Promega, Madison, WI; internal control), and pFA-CMV-hNurr1-LBD, as described previously.23 Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose supplemented with 10% fetal calf serum (FCS), sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded in 96-well plates (3 × 104 cells/well). After 24 h, the medium was changed to Opti-MEM without supplements, and cells were transiently transfected using Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol. Five hours after transfection, cells were incubated with the test compounds in Opti-MEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.1% DMSO for 16 h before luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol on a Tecan Spark luminometer (Tecan Deutschland GmbH, Germany). Firefly luminescence was divided by Renilla luminescence and multiplied by 1000, resulting in relative light units (RLUs) to normalize for transfection efficiency and cell growth. Fold activation was obtained by dividing the mean RLU of the test compound by the mean RLU of the untreated control. All samples were tested in at least three biologically independent experiments in duplicate. For dose–response curve fitting and calculation of EC50 values, the equation “[Agonist] versus response (three parameters)” was used in GraphPad Prism (version 7.00, GraphPad Software, La Jolla, CA). Selectivity profiling was performed with identical procedures using pFA-CMV-Nur77-LBD,23 pFA-CMV-NOR-1-LBD,23 pFA-CMV-THRα-LBD,33 pFA-CMV-RARα-LBD,34 pFA-CMV-PPARγ-LBD,35 pFA-CMV-revERBα-LBD, pFA-CMV-RORγ-LBD,36 pFA-CMV-VDR-LBD,37 pFA-CMV-LXRα-LBD,37 pFA-CMV-FXR-LBD,38 and pFA-CMV-hRXRα-LBD.39 Activity of test compounds on the Nurr1-C566S mutant was determined as described for wild-type Gal4–Nurr1 using pFA-CMV-Nurr1-C566S-LBD, which was constructed by site-directed mutagenesis using the Phusion Site-Directed Mutagenesis Kit (Thermo Scientific) and the following mutagenesis primer sequences: Pho-5′-TCG TAC CCT TAG CAC ACA GGG-3′ and Pho-5′-AGT TCT GGG AGC TTC CCC AAC AGT TT-3′.
Reporter Gene Assay for Full-Length Human Nurr1
Activation of full-length human Nurr1 was studied in transiently transfected HEK293T cells as described previously23 using the reporter plasmid pFR-Luc-NBRE, the full-length human nuclear receptor Nurr1 encoded by pcDNA3.1-hNurr1-NE (Addgene plasmid #102363), and pRL-SV40 (Promega) for normalization of transfection efficacy and to observe test compound toxicity. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) high glucose supplemented with 10% fetal calf serum (FCS), sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded in 96-well plates (3 × 104 cells/well). After 24 h, The medium was changed to Opti-MEM without supplements, and cells were transiently transfected using Lipofectamine LTX reagent (Invitrogen) according to the manufacturer’s protocol. Five hours after transfection, cells were incubated with the test compounds in Opti-MEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.1% DMSO for 16 h before luciferase activity was measured using the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer’s protocol on a Tecan Spark luminometer (Tecan Deutschland GmbH, Germany). Firefly luminescence was divided by Renilla luminescence and multiplied by 1000, resulting in relative light units (RLUs) to normalize for transfection efficiency and cell growth. Fold activation was obtained by dividing the mean RLU of the test compound by the mean RLU of the untreated control. All samples were tested in at least three biologically independent experiments in duplicate. For dose–response curve fitting and calculation of EC50 values, the equation “[Agonist] versus response (three parameters)” was used in GraphPad Prism (version 7.00, GraphPad Software, La Jolla, CA).
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted as described previously19 on an Affinity ITC instrument (TA Instruments, New Castle, DE) at 25 °C and a stirring rate of 75 rpm. Nurr1 LBD protein (10 or 30 μM) in buffer (20 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol) containing 3–5% DMSO was titrated with the test compounds (100 or 200 μM in the same buffer containing 3–5% DMSO) in 21–26 injections (1 × 1 μL and 20–25 × 5 μL) with an injection interval of 150 s. As control experiments, the test compounds were titrated to the buffer, and the buffer was titrated to the Nurr1 LBD protein under otherwise identical conditions. The heat rates of the compound-Nurr1 LBD titrations were analyzed using NanoAnalyze software (TA Instruments, New Castle, DE) with an independent binding model.
Glutathione Reactivity Assay
A hundred microliters of test compound solution (5o or PVS, 250 μM each) in PBS buffer (pH 7.4 containing 2% DMSO) was mixed with 100 μL of 5 mM glutathione solution in PBS buffer (pH 7.4) to obtain a final test compound concentration of 125 μM and 2.5 mM glutathione in 200 μL of sample volume. The samples were shaken at 37 °C. At different time points (0, 6, 18, 24 h), a 20 μL aliquot of each reaction sample was diluted with 480 μL of mobile phase (0.1% formic acid/acetonitrile = 40/60, v/v) containing 10 μM 2-chloro-N-(5-chloro-2-(2-(dimethylamino)ethoxy)phenyl)benzamide as an internal standard. The resulting mixture was then analyzed by liquid chromatography–ultraviolet–electrospray ionization tandem mass spectrometry (LC–UV–ESI-MS) using an API 3200 QTrap triple quadrupole mass spectrometer (Sciex, Darmstadt, Germany) coupled to an Agilent 1100 HPLC system equipped with an Agilent 1100 diode array detector (G1315B, Agilent, Waldbronn, Germany) and a SIL-20A/HT autosampler (Shimadzu, Duisburg, Germany) controlled by Analyst software (v.1.6.3). A ZORBAX SB-Aq column (3.5 μm, 3.0 mm × 100 mm, Agilent, protected with a 0.5 μm and a 0.2 μm frit) was used as the stationary phase and 0.1% formic acid and acetonitrile (40:60, v/v) were used as the mobile phase at a flow rate of 400 μL/min. The injection volume was 15 μL per sample. MS detection was performed under positive ESI conditions in single-ion monitoring (SIM) mode recording m/z 392.1 (5o), m/z 353.0 (2-chloro-N-(5-chloro-2-(2-(dimethylamino)ethoxy)phenyl)benzamide), and m/z 169.1 (PVS). UV detection was performed at 254 nm. The AUC (area under the curve) values were determined by integration of LC–UV–ESI-MS chromatogram and then corrected using the internal standard for both MS and UV measurements. The experiment was repeated three times.
Evaluation of Covalent Nurr1 Ligand Adduct Formation
The Nurr1 LBD (10 μM) was incubated in buffer (20 mM Tris pH 7.5, 100 mM NaCl, 5% glycerol, 1% DMSO) with 5o (100 μM) or alone for 120 h at 4 °C and then analyzed by LC–UV/MS. LCMS spectra were recorded on a Bruker microTOF II in positive ionization mode. The instrument was calibrated in positive mode by direct infusion of a calibration solution (Agilent Technologies ESI-L Low Concentration Tuning Mix). The HPLC line was an Ultimate 3000 RP-HPLC System (Thermo Fisher Scientific) equipped with an Aeris C4 wide pore column (2.1 mm × 150 mm, 6 μm, Phenomenex) with 0.1% formic acid (A) and acetonitrile (B) as the mobile phase at a flow rate of 0.25 mL/min. The gradient was 0–3 min at 5% B and then 5–95% B in 7 min plus 2 min of washing at 95% B. The column eluent was monitored by UV detection at 214, 254, and 280 nm with a diode array detector.
Cytotoxicity Assay
HEK293T cells were cultured at 37 °C and 5% CO2 in DMEM high-glucose medium supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and 10% fetal calf serum (FCS) and seeded at a density of 10,000 cells in 96-well plates precoated with a 10 μg/mL collagen G solution (Merck KgaA, L7213) at 37 °C for 30 min. After 24 h, the cells were treated with the test compounds in Opti-MEM medium supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), and 0.1% DMSO or 0.1% DMSO alone as the untreated control. Each sample was prepared in four biologically independent repeats. After incubation for 24 h, the medium was removed and 10% water-soluble tetrazolinum salt (Cell Counting Kit-8, MedChemExpress) in Opti-MEM supplemented with penicillin (100 U/mL) and streptomycin (100 μg/mL) was added to assess metabolic activity. After 4 h, absorbance was measured at 450 nm using a Tecan Spark Cyto instrument (Tecan).
Evaluation of Nurr1-Regulated Gene Expression in T98G and N27 Cells
T98G cells (ATCC CRL-1690) were grown in DMEM high glucose supplemented with 10% FCS, sodium pyruvate (1 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded at a density of 250,000 cells per well in 12-well plates. After 24 h, the medium was changed to DMEM high glucose supplemented with 0.2% fetal calf serum (FCS), penicillin (100 U/mL), and streptomycin (100 μg/mL), and the cells were incubated for another 24 h before stimulation with test compound 5o (30 μM) solubilized with 0.1% DMSO or 0.1% DMSO as a negative control. After 16 h of incubation, the medium was removed and cells were washed with phosphate-buffered saline (PBS), and after full aspiration of residual liquids, cells were immediately frozen at −80 °C until further processing. N27 rat dopaminergic neural cells (SCC048, Sigma-Aldrich, Darmstadt, Germany) were grown in RPMI 1640 medium (Gibco, Thermo Fisher Scientific, Waltham) supplemented with 10% FCS, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5% CO2 and seeded at a density of 250,000 cells per well in 12-well plates. After 7 h, the medium was changed to RPMI 1640 medium supplemented with 0.2% FCS, penicillin (100 U/mL), and streptomycin (100 μg/mL), and the cells were incubated for another 24 h before stimulation with the test compound solubilized with 0.1% DMSO or with 0.1% DMSO in RPMI 1640 medium with 0.2% FCS as a negative control. After 16 h of incubation, the medium was removed and cells were washed with phosphate-buffered saline (PBS), and after full aspiration of the residual liquids, cells were immediately frozen at −80 °C until further processing. Total RNA was isolated from T98G or N27 cells using the E.Z.N.A. Total RNA Kit I (Omega Bio-tek, Norcross) following the manufacturer’s instructions. RNA concentration and purity were assessed using a NanoDrop One UV/VIS spectrophotometer (Thermo Fisher Scientific, Waltham) at 260/280 nm. Right before reverse transcription (RT), RNA was linearized at a concentration of 133 ng/μL (T98G) or 66.7 μg/μL (N27) at 65 °C for 10 min and then immediately incubated on ice for at least 1 min. Reverse transcription was performed using 2 μg (T98G) or 1 μg (N27) of total RNA, 20 U Recombinant RNasin Ribonuclease Inhibitor (Promega, Mannheim, Germany), 100 U SuperScript IV Reverse Transcriptase including 5× First Strand Buffer and 0.1 M dithiothreitol (Thermo Fisher Scientific, Waltham), 3.75 ng of linear acrylamide, 625 ng of random hexamer primers (#11277081001, Merck, Darmstadt, Germany), and 11.25 nmol deoxynucleoside triphosphate mix (2.8 nmol each ATP, TTP, CTP, GTP; #R0186, Thermo Fisher Scientific, Waltham) at a volume of 22.45 μL at 50 °C for 10 min and 80 °C for 10 min using a Thermal cycler XT96 (VWR International, Darmstadt, Germany). Quantitative polymerase chain reaction (qPCR) was conducted using an Applied Biosystems QuantStudio 1 (Waltham) and a SYBR green-based detection method. Appropriately diluted cDNA was added to 6 pmol forward and reverse primers, respectively, 0.8 U Taq DNA Polymerase (#M0267, New England Biolabs, Ipswich), 40 ppm of SYBR Green I (#S9430, Sigma-Aldrich, St. Louis), 15 nmol deoxynucleoside triphosphate mix (as indicated above), 60 nmol MgCl2, 4 μg of bovine serum albumin (#B14, Thermo Fisher Scientific, Waltham), 20% BioStab PCR Optimizer II (#53833, Merck, Darmstadt, Germany), and 10% Taq buffer without detergents (#B55, Thermo Fisher Scientific, Waltham) topped up to a final volume of 20 μL with ddH2O. Samples underwent 40 cycles of 15 s denaturation at 95 °C, 15 s of primer annealing at 59.4–62.4 °C (depending on the primer), and 20 s of elongation at 68 °C. PCR product specificity was evaluated using a melting curve analysis ranging from 65 to 95 °C. TH and VMAT2 mRNA expression was normalized to GAPDH mRNA expression per sample using the ΔCt method. The following primers for the human genes were used for T98G cell samples: hVMAT2 (SLC18A2), 5′-GCT ATG CCT TCC TGC TGA TTG C-3′ (fw) and 5′-CCA AGG CGA TTC CCA TGA CGT T-3′ (rev); hTH, 5′-GCT GGA CAA GTG TCA TCA CCT G-3′ (fw) and 5′-CCT GTA CTG GAA GGC GAT CTC A-3′ (rev); and hGAPDH, 5′-AGG TCG GAG TCA ACG GAT TT-3′ (fw) and 5′-TTC CCG TTC TCA GCC TTG AC-3′ (rev). The following primers for the rat genes were used for N27 cell samples: rVMAT2 (SLC18A2), 5′-CAG AGT GCA GCA GAG CCA T-3′ (fw) and 5′-CTG GGG ATG ATG GGA ACC AC-3′ (rev); rTH, 5′-TGG GGA GCT GAA GGC TTA TG-3′ (fw) and 5′-AGA GAA TGG GCG CTG GAT AC-3′ (rev); rSOD1, 5′-GAA GGC GAG CAT GGG TTC C-3′ (fw) and 5′-CAG GTC TCC AAC ATG CCT CTC T-3′ (rev); rSOD2, 5′-CGG GGG CCA TAT CAA TCA CA-3′ (fw) and 5′-TCC AGC AAC TCT CCT TTG GG-3′ (rev); and rGAPDH, 5′-CAG CCG CAT CTT CTT GTG C-3′ (fw) and 5′-AAC TTG CCG TGG GTA GAG TC-3′ (rev). For statistical analysis, data were evaluated for normal distribution (Shapiro–Wilk test) and outliers (Grubb′s test). Normally distributed data sets normalized to the respective vehicle control were tested for statistically significant differences to H0: μ = 1 with either a one-sample Wilcoxon rank-sum or a t-test depending on the respective Shapiro–Wilk test results. We considered a value of p < 0.05 to indicate statistical significance. P-values were indicated as * p <0.05, ** p <0.01, and *** p <0.001.
Acknowledgments
This research was co-funded by the European Union (ERC, NeuRoPROBE, 101040355). Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union or the European Research Council. Neither the European Union nor the granting authority can be held responsible for them. pcDNA3.1-hNurr1-NE (Addgene plasmid #102363) was a gift from Shu Leong Ho. Structural analyses were visualized with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco. The authors thank Vasily Morozov and Céline Douat for their support.
Glossary
Abbreviations Used
- AQ
amodiaquine
- CQ
chloroquine
- DHI
5,6-dihydroxyindole
- ITC
isothermal titration calorimetry
- LBD
ligand-binding domain
- Nurr1
nuclear receptor-related factor 1
- TH
tyrosine hydroxylase
- VMAT2
vesicular amino acid transporter 2
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.3c00852.
Initial docking experiments; toxicity evaluation of the screening compounds in a WST-8 assay; LC–UV chromatogram and MS pattern of the Nurr1 LBD with or without the ligand 5o; and synthesis procedures, analytical data, and purity analyses (PDF)
Molecular formula strings containing molecular structures of 5e, 5m, 5o, 5r, 5t, 5v, and 10–13 with associated activity data (CSV)
Virtual DHI analogue library containing the structures of virtually generated DHI analogues (CSV)
Docking containing the results of docking 5a–x to the Nurr1 LBD (ZIP)
Author Contributions
‡ M.S. and J.V. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Willems S.; Merk D. Medicinal Chemistry and Chemical Biology of Nurr1 Modulators: An Emerging Strategy in Neurodegeneration. J. Med. Chem. 2022, 65, 9548–9563. 10.1021/acs.jmedchem.2c00585. [DOI] [PubMed] [Google Scholar]
- Willems S.; Marschner J. A.; Kilu W.; Faudone G.; Busch R.; Duensing-Kropp S.; Heering J.; Merk D. Nurr1 Modulation Mediates Neuroprotective Effects of Statins. Adv. Sci. 2022, 9, 2104640 10.1002/advs.202104640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijo K.; Winner B.; Carson C. T.; Collier J. G.; Boyer L.; Rosenfeld M. G.; Gage F. H.; Glass C. K. A Nurr1/CoREST Pathway in Microglia and Astrocytes Protects Dopaminergic Neurons from Inflammation-Induced Death. Cell 2009, 137, 47–59. 10.1016/j.cell.2009.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterström R. H.; Solomin L.; Jansson L.; Hoffer B. J.; Olson L.; Perlmann T. Dopamine Neuron Agenesis in Nurr1-Deficient Mice. Science 1997, 276, 248–250. 10.1126/science.276.5310.248. [DOI] [PubMed] [Google Scholar]
- Willems S.; Zaienne D.; Merk D. Targeting Nuclear Receptors in Neurodegeneration and Neuroinflammation. J. Med. Chem. 2021, 64, 9592–9638. 10.1021/acs.jmedchem.1c00186. [DOI] [PubMed] [Google Scholar]
- Decressac M.; Volakakis N.; Björklund A.; Perlmann T. NURR1 in Parkinson Disease—from Pathogenesis to Therapeutic Potential. Nat. Rev. Neurol. 2013, 9, 629–636. 10.1038/nrneurol.2013.209. [DOI] [PubMed] [Google Scholar]
- Moon M.; Jung E. S.; Jeon S. G.; Cha M.-Y.; Jang Y.; Kim W.; Lopes C.; Mook-Jung I.; Kim K.-S. Nurr1 (NR4A2) Regulates Alzheimer’s Disease-Related Pathogenesis and Cognitive Function in the 5XFAD Mouse Model. Aging Cell 2019, 18, e12866 10.1111/acel.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montarolo F.; Perga S.; Martire S.; Bertolotto A. Nurr1 Reduction Influences the Onset of Chronic EAE in Mice. Inflammation Res. 2015, 64, 841–844. 10.1007/s00011-015-0871-4. [DOI] [PubMed] [Google Scholar]
- Decressac M.; Kadkhodaei B.; Mattsson B.; Laguna A.; Perlmann T.; Björklund A. α-Synuclein-Induced down-Regulation of Nurr1 Disrupts GDNF Signaling in Nigral Dopamine Neurons. Sci. Transl. Med. 2012, 4, 163ra156 10.1126/scitranslmed.3004676. [DOI] [PubMed] [Google Scholar]
- Liu W.; Gao Y.; Chang N. Nurr1 Overexpression Exerts Neuroprotective and Anti-Inflammatory Roles via down-Regulating CCL2 Expression in Both in Vivo and in Vitro Parkinson’s Disease Models. Biochem. Biophys. Res. Commun. 2017, 482, 1312–1319. 10.1016/j.bbrc.2016.12.034. [DOI] [PubMed] [Google Scholar]
- Parra-Damas A.; Valero J.; Chen M.; España J.; Martín E.; Ferrer I.; Rodríguez-Alvarez J.; Saura C. A. Crtc1 Activates a Transcriptional Program Deregulated at Early Alzheimer’s Disease-Related Stages. J. Neurosci. 2014, 34, 5776–5787. 10.1523/JNEUROSCI.5288-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y.; Le W.; Kompoliti K.; Jankovic J.; Mufson E. J.; Kordower J. H. Nurr1 in Parkinson’s Disease and Related Disorders. J. Comp. Neurol. 2006, 494, 495–514. 10.1002/cne.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J.-i.; Nakanishi M.; Koike F.; Miyake S.; Yamamoto T.; Kawai M.; Kikuchi S.; Nomura K.; Yokoyama K.; Ota K.; Kanda T.; Fukazawa T.; Yamamura T. Microarray Analysis Identifies an Aberrant Expression of Apoptosis and DNA Damage-Regulatory Genes in Multiple Sclerosis. Neurobiol. Dis. 2005, 18, 537–550. 10.1016/j.nbd.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Munoz-Tello P.; Lin H.; Khan P.; de Vera I. M. S.; Kamenecka T. M.; Kojetin D. J. Assessment of NR4A Ligands That Directly Bind and Modulate the Orphan Nuclear Receptor Nurr1. J. Med. Chem. 2020, 63, 15639–15654. 10.1021/acs.jmedchem.0c00894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isigkeit L.; Merk D. Opportunities and Challenges in Targeting Orphan Nuclear Receptors. Chem. Commun. 2023, 59, 4551–4561. 10.1039/D3CC00954H. [DOI] [PubMed] [Google Scholar]
- Kim C.-H.; Han B.-S.; Moon J.; Kim D.-J.; Shin J.; Rajan S.; Nguyen Q. T.; Sohn M.; Kim W.-G.; Han M.; Jeong I.; Kim K.-S.; Lee E.-H.; Tu Y.; Naffin-Olivos J. L.; Park C.-H.; Ringe D.; Yoon H. S.; Petsko G. A.; et al. Nuclear Receptor Nurr1 Agonists Enhance Its Dual Functions and Improve Behavioral Deficits in an Animal Model of Parkinson’s Disease. Proc. Natl. Acad. Sci. U.S.A. 2015, 112, 8756–8761. 10.1073/pnas.1509742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems S.; Ohrndorf J.; Kilu W.; Heering J.; Merk D. Fragment-like Chloroquinolineamines Activate the Orphan Nuclear Receptor Nurr1 and Elucidate Activation Mechanisms. J. Med. Chem. 2021, 64, 2659–2668. 10.1021/acs.jmedchem.0c01779. [DOI] [PubMed] [Google Scholar]
- Willems S.; Müller M.; Ohrndorf J.; Heering J.; Proschak E.; Merk D. Scaffold Hopping from Amodiaquine to Novel Nurr1 Agonist Chemotypes via Microscale Analogue Libraries. ChemMedChem 2022, 17, e2022000 10.1002/cmdc.202200026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vietor J.; Gege C.; Stiller T.; Busch R.; Schallmayer E.; Kohlhof H.; Höfner G.; Pabel J.; Marschner J. A.; Merk D. Development of a Potent Nurr1 Agonist Tool for in Vivo Applications. J. Med. Chem. 2023, 66, 6391–6402. 10.1021/acs.jmedchem.3c00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan S.; Jang Y.; Kim C. H.; Kim W.; Toh H. T.; Jeon J.; Song B.; Serra A.; Lescar J.; Yoo J. Y.; Beldar S.; Ye H.; Kang C.; Liu X. W.; Feitosa M.; Kim Y.; Hwang D.; Goh G.; Lim K. L.; et al. PGE1 and PGA1 Bind to Nurr1 and Activate Its Transcriptional Function. Nat. Chem. Biol. 2020, 16, 876–886. 10.1038/s41589-020-0553-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning J. M.; Wang Y.; Oltrabella F.; Tian B.; Kholodar S. A.; Liu H.; Bhattacharya P.; Guo S.; Holton J. M.; Fletterick R. J.; Jacobson M. P.; England P. M. Covalent Modification and Regulation of the Nuclear Receptor Nurr1 by a Dopamine Metabolite. Cell Chem. Biol. 2019, 26, 674–685.e6. 10.1016/j.chembiol.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kholodar S. A.; Lang G.; Cortopassi W. A.; Iizuka Y.; Brah H. S.; Jacobson M. P.; England P. M. Analogs of the Dopamine Metabolite 5,6-Dihydroxyindole Bind Directly to and Activate the Nuclear Receptor Nurr1. ACS Chem. Biol. 2021, 16, 1159–1163. 10.1021/acschembio.1c00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems S.; Kilu W.; Ni X.; Chaikuad A.; Knapp S.; Heering J.; Merk D. The Orphan Nuclear Receptor Nurr1 Is Responsive to Non-Steroidal Anti-Inflammatory Drugs. Commun. Chem. 2020, 3, 85 10.1038/s42004-020-0331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson E. D.; La Rosa F. G.; Edwards-Prasad J.; Weiland D. A.; Witta S. E.; Freed C. R.; Prasa K. N. Improvement of Neurological Deficits in 6-Hydroxydopamine-Lesioned Rats after Transplantation with Allogeneic Simian Virus 40 Large Tumor Antigen Gene-Induced Immortalized Dopamine Cells. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 1265–1270. 10.1073/pnas.95.3.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L.; Zhou W.; Symmes B.; Freed C. R. Re-Cloning the N27 Dopamine Cell Line to Improve a Cell Culture Model of Parkinson’s Disease. PLoS One 2016, 11, e0160847 10.1371/journal.pone.0160847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicciari R.; Fiorucci S.; Camaioni E.; Clerici C.; Costantino G.; Maloney P. R.; Morelli A.; Parks D. J.; Willson T. M. 6α-Ethyl-Chenodeoxycholic Acid (6-ECDCA), a Potent and Selective FXR Agonist Endowed with Anticholestatic Activity. J. Med. Chem. 2002, 45, 3569–3572. 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- Pauli G. F.; Chen S. N.; Simmler C.; Lankin D. C.; Gödecke T.; Jaki B. U.; Friesen J. B.; McAlpine J. B.; Napolitano J. G. Importance of Purity Evaluation and the Potential of Quantitative 1H NMR as a Purity Assay. J. Med. Chem. 2014, 57, 9220–9231. 10.1021/jm500734a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoull W.; Barton P.; Brown A. J. H.; Bowker S. S.; Cameron J.; Clarke D. S.; Davies R. D. M.; Dossetter A. G.; Ertan A.; Fenwick M.; Green C.; Holmes J. L.; Martin N.; Masters D.; Moore J. E.; Newcombe N. J.; Newton C.; Pointon H.; Robb G. R.; et al. Identification, Optimization, and Pharmacology of Acylurea GHS-R1a Inverse Agonists. J. Med. Chem. 2014, 57, 6128–6140. 10.1021/jm500610n. [DOI] [PubMed] [Google Scholar]
- Berthold M. R.; Cebron N.; Dill F.; Gabriel T. R.; Kötter T.; Meinl T.; Ohl P.; Thiel K.; Wiswedel B. KNIME - the Konstanz Information Miner: Version 2.0 and Beyond. SIGKDD Explor. Newsl. 2006, 11, 26–31. 10.1145/1656274.1656280. [DOI] [Google Scholar]
- Shelley J. C.; Cholleti A.; Frye L. L.; Greenwood J. R.; Timlin M. R.; Uchimaya M. Epik: A Software Program for PKa Prediction and Protonation State Generation for Drug-like Molecules. J. Comput. -Aided. Mol. Des. 2007, 21, 681–691. 10.1007/s10822-007-9133-z. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Gellrich L.; Heitel P.; Heering J.; Kilu W.; Pollinger J.; Goebel T.; Kahnt A.; Arifi S.; Pogoda W.; Paulke A.; Steinhilber D.; Proschak E.; Wurglics M.; Schubert-Zsilavecz M.; Chaikuad A.; Knapp S.; Bischoff I.; Fürst R.; Merk D. L-Thyroxin and the Nonclassical Thyroid Hormone TETRAC Are Potent Activators of PPARγ. J. Med. Chem. 2020, 63, 6727–6740. 10.1021/acs.jmedchem.9b02150. [DOI] [PubMed] [Google Scholar]
- Pollinger J.; Gellrich L.; Schierle S.; Kilu W.; Schmidt J.; Kalinowsky L.; Ohrndorf J.; Kaiser A.; Heering J.; Proschak E.; Merk D. Tuning Nuclear Receptor Selectivity of Wy14,643 towards Selective Retinoid X Receptor Modulation. J. Med. Chem. 2019, 62, 2112–2126. 10.1021/acs.jmedchem.8b01848. [DOI] [PubMed] [Google Scholar]
- Rau O.; Wurglics M.; Paulke A.; Zitzkowski J.; Meindl N.; Bock A.; Dingermann T.; Abdel-Tawab M.; Schubert-Zsilavecz M. Carnosic Acid and Carnosol, Phenolic Diterpene Compounds of the Labiate Herbs Rosemary and Sage, Are Activators of the Human Peroxisome Proliferator-Activated Receptor Gamma. Planta Med. 2006, 72, 881–887. 10.1055/s-2006-946680. [DOI] [PubMed] [Google Scholar]
- Moret M.; Helmstädter M.; Grisoni F.; Schneider G.; Merk D. Beam Search for Automated Design and Scoring of Novel ROR Ligands with Machine Intelligence**. Angew. Chem., Int. Ed. 2021, 60, 19477–19482. 10.1002/anie.202104405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flesch D.; Cheung S.-Y.; Schmidt J.; Gabler M.; Heitel P.; Kramer J. S.; Kaiser A.; Hartmann M.; Lindner M.; Lüddens-Dämgen K.; Heering J.; Lamers C.; Lüddens H.; Wurglics M.; Proschak E.; Schubert-Zsilavecz M.; Merk D.; Non-Acidic; Farnesoid X. Receptor Modulators. J. Med. Chem. 2017, 60, 7199–7205. 10.1021/acs.jmedchem.7b00903. [DOI] [PubMed] [Google Scholar]
- Schmidt J.; Klingler F.-M.; Proschak E.; Steinhilber D.; Schubert-Zsilavecz M.; Merk D. NSAIDs Ibuprofen, Indometacin, and Diclofenac Do Not Interact with Farnesoid X Receptor. Sci. Rep. 2015, 5, 14782 10.1038/srep14782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitel P.; Gellrich L.; Kalinowsky L.; Heering J.; Kaiser A.; Ohrndorf J.; Proschak E.; Merk D. Computer-Assisted Discovery and Structural Optimization of a Novel Retinoid X Receptor Agonist Chemotype. ACS Med. Chem. Lett. 2019, 10, 203–208. 10.1021/acsmedchemlett.8b00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.