

SUMMARY
Recent Aβ-immunotherapy trials have yielded the first clear evidence that removing aggregated Aβ from the brains of symptomatic patients can slow the progression of Alzheimer’s disease. The clinical benefit achieved in these trials has been modest, however, highlighting the need for both a deeper understanding of disease mechanisms and the importance of intervening early in the pathogenic cascade. An immunoprevention strategy for Alzheimer’s disease is required that will integrate the findings from clinical trials with mechanistic insights from preclinical disease models to select promising antibodies, optimize the timing of intervention, identify early biomarkers, and mitigate potential side effects.
etoc
Recent Aβ-immunotherapy trials demonstrated that removing aggregated Aβ from the brains of symptomatic patients can slow progression of Alzheimer’s disease. This Perspective analyzes different immunoprevention strategies by integrating findings from clinical trials with mechanistic insights from preclinical disease models.
INTRODUCTION
The recent reports that monoclonal antibodies (lecanemab [Leqembi™] and donanemab) stimulate the removal of abnormal β-amyloid (Aβ) from the brain and slow the progression of early Alzheimer’s disease (AD)1,2 have given the research community the first clear clinicopathological indication that a disease-modifying treatment for AD is feasible. Together with evidence that another monoclonal antibody (aducanumab [Aduhelm™])3 also may be beneficial, the results provide clinical support for the importance of aberrant Aβ in the pathogenesis of AD. The findings also strengthen the ‘amyloid (Aβ) cascade’ hypothesis, which holds that the seminal event in the ontogeny of AD is the misfolding and aggregation of Aβ, followed by a host of sequelae that comprise the full clinical and pathological phenotype of the disease4,5
While there is now renewed hope for disease-modifying therapies, it is important to caution that the clinical benefit of the antibodies in the trials was limited and the disease still progressed in treated subjects, albeit at a slower pace. Removal of aberrant Aβ in symptomatic AD is unlikely to be a cure for the disease, which begins to germinate in the brain 20–30 years before the onset of obvious cognitive impairment6–8. By the time the signs and symptoms of AD first appear clinically, damage to the brain is considerable and at least partially beyond repair; hence a full return to baseline functionality is unlikely, in line with the limited efficacy of the antibody treatments. Hence, a prevention strategy is essential; chronic degenerative diseases such as AD are most effectively treated as early in their development as possible, preferably well before they become symptomatic9,10. Lessons learned from immunization treatment trials, combined with mechanistic insights from experimental and biomarker investigations, have now brought us a step closer to this goal.
THE PATHOBIOLOGY OF ALZHEIMER’S DISEASE
AD is defined histopathologically by the profusion of two proteinaceous lesions in the brain -Aβ plaques and neurofibrillary (tau) tangles. Burgeoning evidence indicates that the Aβ and tau proteins misfold, self-assemble, and propagate by an endogenous mechanism closely resembling the seeded aggregation and spread of the prion protein (PrP) in Creutzfeldt-Jakob disease and other prionopathies11–14. Aβ plaques disrupt circuits and neighboring brain cells15–17, but Aβ also forms small, soluble, oligomeric assemblies that impair the function of neurons and glia18,19. In addition, Aβ often accumulates in the walls of small to medium sized cerebral blood vessels manifesting as cerebral β-amyloid angiopathy (CAA)20 (Figure 1). Although the amount of CAA varies widely among AD patients, nearly half of end-stage AD patients exhibit moderate-to-severe CAA21,22.
Figure 1. The pathologic progression of AD, immunoprevention, and the impact of immunotherapy in symptomatic patients.
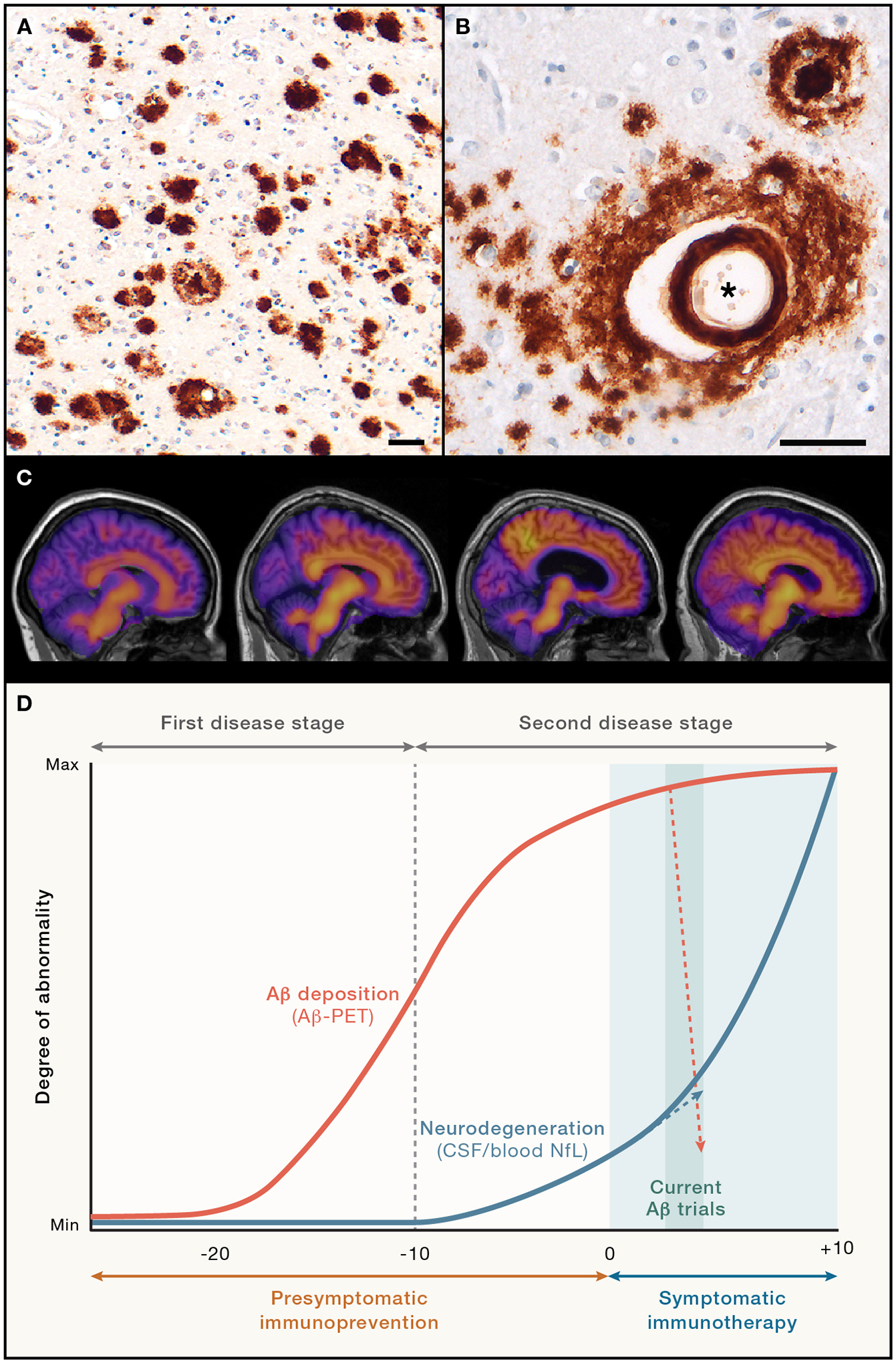
(A, B) Immunohistochemical detection of Aβ deposition in AD brain as plaques (A) and cerebral β-amyloid angiopathy (CAA; black asterisk in B); the affected vessel is surrounded by diffuse parenchymal Aβ deposits, and a dense-core plaque is in the upper right. CAA is moderate-to-severe in nearly half of all AD cases, and CAA has been linked to the side effects of Aβ-immunotherapy; antibody 4G8, Nissl counterstain; scale bars are 50 μm.
(C) Representative Aβ-PET images (left to right) from a control person (non-mutation carrier), a mutation carrier about 10 years before symptom onset, and two mutation carriers that are symptomatic (Pittsburgh compound B [PiB] tracer; shown are participants with familial AD23). The increase in PiB retention primarily occurs in the presymptomatic phase.
(D) In the two-stage model of AD,24–26 the first stage is dominated by Aβ deposition. The second stage commences approximately 10 years before symptom onset and becomes partly independent of Aβ deposition with the emergence of clear signs of neurodegeneration (as assessed, e.g., by NfL levels in CSF or blood) and, eventually, behavioral impairments.26 Targeting aberrant Aβ as immunoprevention (prevention of the disease) is likely to be most successful when initiated during or prior to the first stage. Given the growing pathologic complexity of the disease, it is not clear how much clinical benefit can be expected from Aβ-removing therapies alone beyond this time point. Indeed, Aβ-immunotherapy trials for 18 months with aducanumab, lecanemab, or donanemab removed >60% of the deposited Aβ, but NfL continued to rise (albeit at a reduced pace), paralleling the slowed - but not stopped - cognitive decline in treated subjects.
Aβ plaques and tau tangles both are abundant in advanced AD, but genetic, pathologic and biomarker findings show that Aβ-proteopathy is the crucial early impetus for the disease; widespread tauopathy and other sequelae are essential drivers of behavioral impairment that are downstream of Aβ7,27,28. AD thus is thought to progress in two stages; the first stage is characterized by the emergence and seeded propagation of aberrant Aβ and Aβ-associated pathologies, and the second stage includes a complex assortment of secondary changes that include tangles, inflammation, vascular abnormalities and neurodegeneration29. In the second stage, the disease appears to become at least partially independent of Aβ deposition24–26 (Figure 1). This bi-phasic trajectory of AD pathogenesis has important implications for both treatment and prevention strategies. As a defining pathologic feature of AD, tauopathy also has been the object of immunization strategies30,31, but the pathogenic primacy of Aβ makes it a particularly attractive target for early prevention.
Aβ-IMMUNOTHERAPY FOR AD
Immunization therapy for AD was launched in earnest in 1999, when active immunization of Aβ-precursor protein- (APP) transgenic mice with synthetic polymers of Aβ was shown to dramatically reduce cerebral plaque burden32. This report prompted a flurry of research on the potential of immune mechanisms to treat or prevent AD33. The initial clinical trial of active Aβ immunotherapy in humans (AN1792) was halted when a subset of the recipients developed aseptic meningoencephalitis34. A follow-up study of a small number of patients showed hints of slowed cognitive decline35 along with fairly compelling evidence in postmortem tissue for the clearance of Aβ plaques36,37. However, the meningoencephalitic side-effects, coupled with the inability to fully reverse the errant immune response, was a setback for active immunization. As a result, much research was steered toward passive immunotherapy with humanized monoclonal antibodies as a potentially safer alternative.
The antibodies that have advanced the farthest in clinical development include bapineuzumab, solanezumab, crenezumab, gantenerumab, aducanumab, lecanemab, and donanemab (Figure 2). These antibodies recognize partly different antigenic sites on Aβ, and they differ in their apparent clinical efficacy and in their ability to lower plaque load. All of them have been tested in phase 3 studies of patient cohorts with mild cognitive impairment or mild AD dementia2,38. Except for solanezumab and crenezumab, the antibodies reduced cerebral Aβ content as measured by positron-emission tomography (Aβ-PET)2,28. Although postmortem confirmation is largely lacking, based on previous comparisons of the Aβ-PET signal and postmortem Aβ load, a corresponding reduction of Aβ deposits is likely37,39. Thus far, the antibodies that yielded the greatest removal of Aβ deposition (>60% after 18 months of treatment) - lecanemab, donanemab and aducanumab - have shown evidence of slowed clinical decline1–3, though it is important to stress that direct comparison of clinical efficacy is hindered by differences in the trials such as dosage, treatment schedule, and the patient populations evaluated.
Figure 2. Aβ aggregation and the epitopes recognized by therapeutic antibodies.
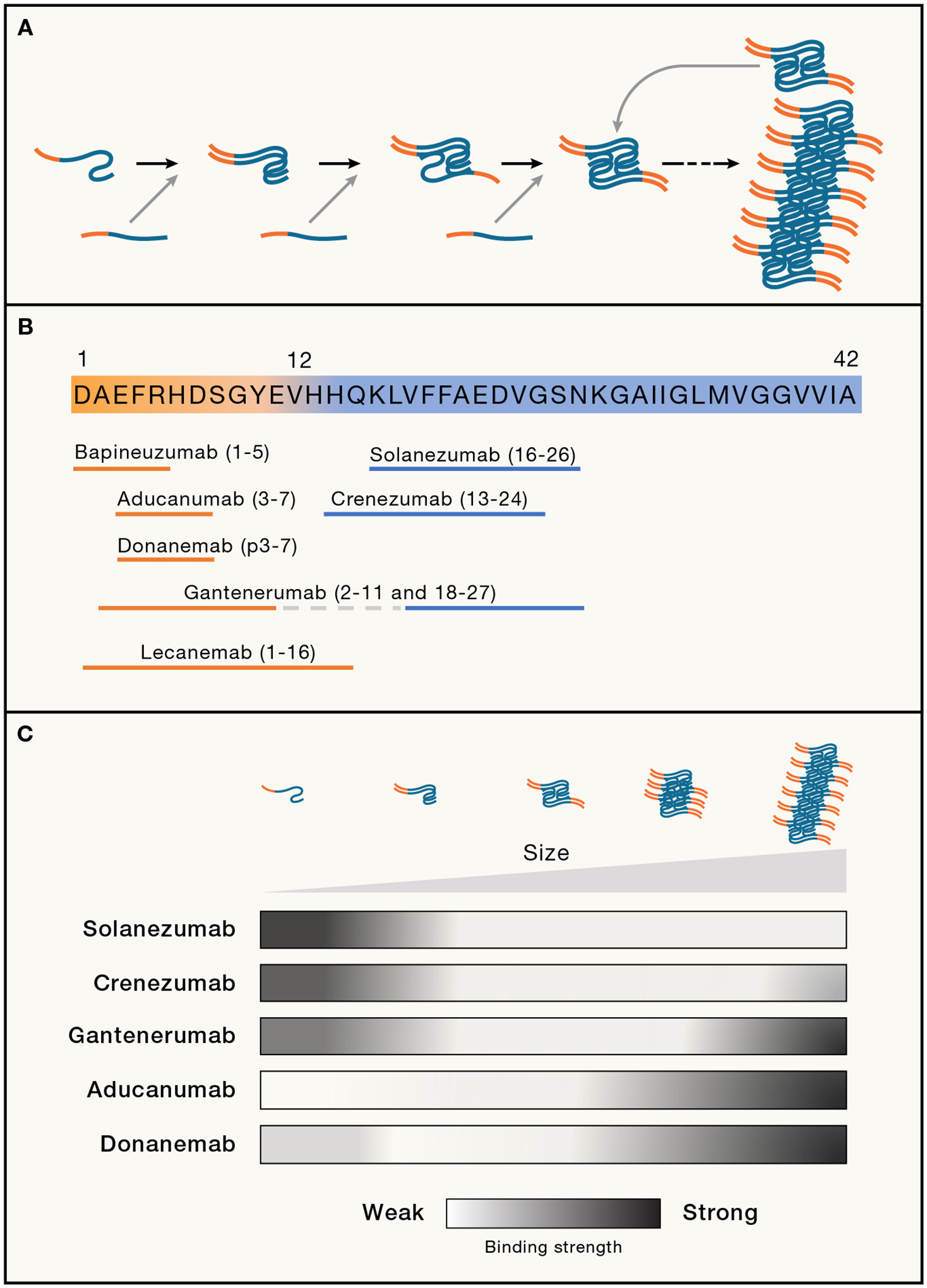
(A) Aβ aggregation starts with a slow nucleation phase during which Aβ assumes an alternative conformation that converts and binds to other Aβ molecules to form the initial segment of the amyloid fibril. With increasing length, the growing fibril eventually breaks and releases seeding-active Aβ multimers, at which stage the process becomes self-propagating (based on Jucker and Walker11). As deposition progresses, Aβ comprises a mixture of multimers, ranging from small soluble oligomers to long amyloid fibrils, which differ in their cytotoxicity and ability to seed further aggregation.18,19,40,41 The growing amyloid fibril schematically depicted here consists of two twisted protofilaments (based on Yang et al.42 for brain-derived Aβ42 fibrils). Note that the N-terminal amino acids (orange) are exposed from the hydrophobic amyloid core (blue).
(B) Diagram of Aβ42 showing the amino acid epitopes that therapeutic antibodies are thought to recognize (based on Plotkin and Cashman43). Common to the antibodies that cleared Aβ deposits in clinical trials (gantenerumab, aducanumab, donanemab, lecanemab) is that they recognize N-terminal amino acids (orange), i.e., epitopes that are exposed on mature amyloid fibrils. In contrast, solanezumab and crenezumab only recognize mid-sequence epitopes that are buried within the amyloid fibril; hence, these antibodies mainly recognize monomeric Aβ.
(C) Schematic illustration of the binding strength of five different antibodies to Aβ that was derived from native amyloid-laden brain samples (AD and mouse models) and fractionated according to size. The two antibodies that most effectively remove Aβ from the brain (donanemab, aducanumab) recognize predominantly large amyloid aggregates (based on Uhlmann et al.44).
Notably, reduction of Aβ burden was accompanied by decreased phosphorylated Tau (pTau) species and glial fibrillary acidic protein (GFAP) in the CSF or blood1,2,45–48. Thus, immunotherapy not only decreased β-amyloid load, but (based on fluid biomarkers) may also have decreased cerebral Aβ-associated tauopathy and astrocytic activation. In contrast, neurofilament light chain (NfL; a marker of neuronal abnormalities49–51 continued to rise in treated subjects, albeit somewhat more slowly than in controls, thereby mirroring the slowed (but not stopped) decline of cognitive changes1,2,45,46,48 (Figure 1).
Unfortunately, in some immunotherapy patients, the removal of aggregated Aβ has been associated with troublesome and sometimes serious side-effects known as amyloidrelated imaging abnormalities (ARIAs)1–3,45, which appear to be linked to the abundance of pre-existing Aβ deposition, especially as CAA. Aβ removal has been associated with an expansion of ventricular volume and an increased reduction of brain volume2,52,53, the functional significance of which remain uncertain. Overall, both the limited clinical benefit of antibody therapy and the risk of serious side-effects that are associated with the presence of a high amyloid burden underscore the importance of starting treatment much earlier in the pathogenic process.
FROM IMMUNOTHERAPY TO IMMUNOPREVENTION: WHAT DO WE NEED?
Below we consider four key research objectives that are needed to extend the results of past and ongoing clinical trials of anti-Aβ antibodies to the effective immunoprevention of AD: Define the best molecular target for Aβ-immunotherapy (epitopes), optimize the schedule of treatment (timing), establish early biological indicators of preventive efficacy (biomarkers), and identify and mitigate potential adverse reactions to antibody administration (side-effects).
Identify the optimal Aβ epitopes
Based on the results of the recent clinical trials, the most coherent (but still provisional) conclusion is that lowering cerebral Aβ load can slow the progression of AD. Solanezumab selectively binds Aβ monomers; although a meta-analysis suggests that it may have had some clinical efficacy54, solanezumab failed to reach primary endpoints in clinical trials55. Monomeric Aβ is abundant in brain, and its complete neutralization would require stoichiometric amounts of high-affinity antibodies able to compete with the binding of monomers to existing Aβ aggregates. Preclinical evidence indicates that the toxicity of Aβ is linked to its aggregated state26,27, and the antibodies that have shown the best evidence of clinical efficacy also achieved the largest reduction of aggregated Aβ1–3. For these reasons, an antibody that generally recognizes monomeric Aβ is unlikely to be the most favorable immunotherapeutic tool.
The multiple manifestations of aberrant Aβ could present challenges for immunoprevention. Aβ multimers range in size from small oligomers to protofibrils and long amyloid fibrils, and they differ in their cytotoxicity and ability to seed further aggregation (Figure 2). Moreover, the predominant species of Aβ can vary among patients, between the vasculature and parenchyma, and over the course of the disease12,20,42,56,57. Given the biochemical and structural complexity of Aβ aggregates, the best epitopic targets for the prevention or removal of Aβ multimers remain uncertain. As one example, the positive clinical outcome of the lecanemab trial might imply that Aβ-protofibrils are a particularly promising target58. Lecanemab was raised against recombinant ‘arctic’ mutant (E22G) Aβ, a form of the protein that is linked to a rare familial form of AD characterized by marked accumulation of protofibrils59. However, the protofibrillar nature of arctic Aβ in patients’ brains is incompletely understood60, and there is evidence that recombinant Aβ folds into a conformation that differs from that of Aβ that folds within the brain57,42,60. Hence, the clinical efficacy of lecanemab1 could result from the overall reduction of β-amyloid (at which the antibody is quite effective), and not from the neutralization of a specific type of multimer (i.e., protofibrils).
Similarly, in vitro studies suggest that aducanumab decreases Aβ oligomer generation from secondary nucleation61, but the general reduction of β-amyloid load in the aducanumab clinical trial3 precludes linking the clinical outcome to certain oligomeric species. To complicate things further, smaller, ‘soluble’ assemblies might be in a state of dynamic equilibrium with Aβ plaques18 such that eliminating plaques would indirectly reduce the population of oligomers and protofibrils, and vice versa (Figure 2). Imaging and fluid biomarkers for oligomeric Aβ are needed to meaningfully connect pathogenic molecular species to clinical efficacy (see ‘Optimize biomarker use’, below).
Pyroglutamate-modification of Aβ (e.g., AβN3pE) enhances the propensity of the protein to aggregate; AβN3pE emerges predominantly in later stages of cerebral β-amyloidosis62,63 and thus exemplifies how abnormal Aβ can change over the course of AD. Donanemab is directed at AβN3pE, and it has been shown to be highly effective at removing amyloid in symptomatic patients2. However, it is conceivable that donanemab’s specificity for a relatively late-arising epitope could diminish its ability to impede Aβ deposition at a much earlier stage of disease. Another molecular modification that occurs late in the maturation of Aβ plaques is phosphorylation at position 8 (AβpS8)64. Accordingly, Aβ-immunotherapy might need to be tailored to the characteristics of Aβ at different stages of AD in order to achieve optimal therapeutic and preventive efficacy (see ‘Establish the best timing for immunoprevention’, below).
To identify the most promising immunotherapeutic or immunopreventive Aβ epitope, clinical studies directly comparing several different antibodies would be informative. Moreover, postmortem biochemical analyses of the brain after treatment will then help to pinpoint changes induced by the antibodies that are most pertinent to effective prevention. In parallel, comparison of antibodies and the multimers they engage in preclinical models65, along with structural studies of the epitopes recognized by the various antibodies, are needed. Together with such properties of the antibodies as affinity, immunoglobulin subtype, posttranslational modifications, and half-life in blood (as well as the inherent immunogenicity of the antibodies themselves66), these data should help to facilitate the design of next-generation antibodies and define the most suitable molecular target for immunoprevention.
Establish the best timing for immunoprevention
Abnormal Aβ begins to accumulate in the brain two to three decades before the clinical signs and symptoms of AD become manifest6–8. True primary prevention - stopping Aβ aggregation before it begins - is a particularly attractive objective, but establishing an acceptable risk:benefit ratio for long-term administration of a preventive agent is a formidable task. From a practical standpoint, secondary prevention is a more likely scenario, i.e., initiating preventive measures in response to biomarker evidence that aberrant Aβ has begun to accumulate, but before the onset of the cognitive and behavioral changes of AD67 (Figure 1).
In the two-stage model of AD24–26, Aβ proteopathy initially drives the disease, but its relative influence diminishes concomitant with the emergence of myriad subsequent changes that include neurofibrillary tangle formation, inflammation, neurodegeneration and, eventually, behavioral impairments5. The transition from the first to the second stage is heralded by a steep rise in the CSF levels of NfL, and this has been estimated to occur around 10 years before the onset of symptoms26. At least in mouse models, the increase in NfL (and thus presumed neurodegeneration) coincides with saturated Aβ seeding activity of brain tissue26. Once the second stage is underway, it is not clear how much clinical benefit can be expected from Aβ-removing therapies alone (Figure 1); rather, treatments directed at both Aβ-proteopathy and its sequelae may be required. A clinical trial targeting Aβ in the first disease stage is being planned in carriers of dominant AD mutations 10 years or more prior to the estimated onset of symptoms (https://clinicaltrials.gov/ct2/show/NCT05552157). In addition, a clinical trial targeting both Aβ (lecanemab) and tau (antibody E2814) in the second disease stage (i.e., less than 10 years from the estimated disease onset) was recently launched (https://clinicaltrials.gov/ct2/show/NCT05269394).
For future investigations aiming to optimize the timing of immunoprevention, we need to determine whether the antibodies must be given continuously, or whether intermittent administration will suffice, and what the frequency and duration of treatment should be. When, and how often, treatment should be repeated for maximal efficacy in humans is not yet known, but insights into such mechanistic questions can be gleaned from studies in animal models. For example, experiments with mouse models suggest that acute removal of Aβ seeds at a very early stage of Aβ deposition delays both the accumulation of Aβ and the onset of downstream pathologies later in life68,44. Thus, it might not be necessary to continuously administer anti-Aβ antibodies to delay or prevent Aβ deposition (and, by extension, AD). Finally, we need to determine if methods to augment antibody entry into the brain, such as brain shuttle antibody constructs69 or ultrasound70, are necessary (or even desirable) to reduce the number of treatments required for immunoprevention.
Optimize biomarker use
Advances in Aβ-PET imaging and in the measurement of Aβ in biofluids have substantiated the decades-long presymptomatic development of AD6,71,72, and these technologies have been foundational for the implementation of recent clinical trials71 72. As sensitive and informative as these methods have become, they do not detect aberrant Aβ that might be revealed even earlier if the brains were to be analyzed with sensitive biochemical or immunohistological techniques44,73–75. Similarly, when Aβ-PET scans signal that immunotherapy has reduced the Aβ-load below detection levels, it is likely that a pathologically significant amount of aberrant Aβ remains. Moreover, current Aβ-PET imaging and biofluid Aβ measurements do not provide detailed information about the biochemical and structural characteristics of cerebral Aβ before and after immunotherapy, nor can they satisfactorily discriminate vascular from parenchymal amyloid to gauge the risk of side-effects (see ‘Mitigate the side-effects’, below). Thus, further improvements in assay sensitivity and specificity for early and different forms of aberrant brain Aβ are needed.
Multiple biomarkers in fluids now can track many of the sequelae of Aβ proteopathy. Major progress has been achieved in measuring Aβ-associated phosphorylated Tau (pTau 181, pTau217, pTau231) in CSF and blood76,77. More recently, tests have been developed to detect activated astrocytes and microglia (glial fibrillary acidic protein [GFAP] and soluble triggering receptor expressed on myeloid cells 2 [Trem2], respectively)72,78,79. In presymptomatic AD, the trajectories of these biomarkers closely parallel that of Aβ-deposition. In contrast, NfL in the CSF increases robustly only after half-maximal Aβ-deposition is reached, and this appears to mark the transition from the first to the second stage of AD progression26 (Figure 1).
Despite the considerable clinical utility of fluid biomarkers, the interpretation of clinical trial outcomes would be enriched by a better understanding of the molecular and cellular alterations that these biomarkers represent. For example, the paradoxical finding that pTau fluid biomarkers are strongly associated with the trajectories of Aβ-deposition but poorly with aberrant Tau as measured by PET currently lacks a persuasive explanation. In addition, the pathophysiologic basis of increased GFAP and Trem2 levels in biofluids is still uncertain, as is the means by which proteinaceous biomarkers, in particular intracellular structural proteins such as Tau, GFAP and NfL, make their way into the CSF and blood.
Many mechanistic questions about biomarkers now can be addressed in disease models (mainly genetically modified mice) that enable a direct and timely comparison of brain pathology to protein changes in biofluids. Animal models also can help to clarify issues that confound investigations of humans, including diagnostic uncertainty, comorbidities, perimortem irregularities (such as agonal state and postmortem interval), and lifestyle variability. For example, in APP-transgenic mouse models, Aβ deposition per se (i.e., in the absence of neurofibrillary tangles and neuron loss) is sufficient to induce increases in CSF pTau80. Moreover, transgenic mouse models manifesting distinct proteopathies enable the separation of Aβ-dependent secondary biomarker changes from those associated with possible co-morbid pathologies such as α-synucleinopathy81.
Finally, to optimize immunopreventive strategies, biomarkers of early pathogenesis with a robust effect size are needed to define both the inception and initial trajectory of the disease process. For instance, in both prion diseases14 and AD26, the respective seeding activities of PrP and Aβ in brain tissue rise steeply in the initial stage of protein aggregation before reaching a plateau around the time that neurodegeneration becomes apparent. Seeds of misfolded PrP have been amplified from the CSF of patients with Creutzfeldt-Jakob disease82, and α-synuclein seeds have been detected in CSF and blood from patients with α-synucleinopathies83,84). Similar novel biomarkers would help to characterize the earliest stages of AD, and could serve as informative readouts in immunoprevention trials.
Mitigate the side-effects
Amyloid-related imaging abnormalities (ARIAs) can be a serious side-effect of Aβ immunotherapy1,2,85. ARIAs include focal cerebral edema/effusion (ARIA-E) and hemosiderin accumulation (ARIA-H; a marker of previous microbleeds)20,86. Although ARIAs often are asymptomatic and can be managed in treated subjects by titration of the antibody dose1,85,87, in some instances severe reactions to treatment have occurred, raising concerns about the risk:benefit ratio of immunotherapy for AD38,88,89.
The mechanisms underlying ARIAs are incompletely understood, but the abnormalities are strongly associated with the presence of CAA20,90. CAA appears to be increased – at least temporarily – in response to Aβ immunotherapies that reduce parenchymal β-amyloid both in mouse models91 and in humans36,37,92,93. This increase may result from the translocation of Aβ from the parenchyma to the vascular wall94,95, although the precise mechanism is uncertain. As the immune system (e.g., perivascular macrophages) engages with Aβ in the vascular wall, the blood vessel is compromised and becomes prone to leakage and rupture20. In rare cases, Aβ-immunotherapy combined with blood thinners has caused fatal cerebral hemorrhage in subjects with CAA88, an adverse event that could have been predicted from preclinical studies in mouse models91,96.
ARIAs are most frequent in immunotherapy recipients bearing the ε4 allele of the gene for apolipoprotein E (APOEε4)20,38, consistent with APOEε4 being a prominent risk factor for CAA97. (CAA can be significant also in non-carriers of APOEε498, albeit less commonly than in carriers). Because CAA is moderate-to-severe in approximately 45% of AD cases21,22, Aβ-immunotherapy could put nearly half of symptomatic AD patients at greater risk of ARIAs. Experimental work with mouse models has found that Aβ-immunotherapy-related hemorrhages are evident only when substantial CAA is present91,99. It is therefore possible that the risk of ARIAs would be diminished or even eliminated if Aβ antibodies are administered before CAA becomes widespread in the brain, i.e., as a preventive measure.
The incidence of treatment-related ARIAs differed among past therapeutic trials of anti-Aβ-monoclonal antibodies38,86. It is uncertain whether this variation is related to characteristics of the antibodies (such as their antigen-recognition profiles), to differences in the trial participants (such as their disease stage or CAA load), or to methodological issues such as sensitivity in ARIA detection. CAA can be suspected based on certain clinical and biomarker signs20,100, but a definitive biomarker for CAA would enhance the prognostic precision for ARIAs in potential recipients of immunotherapeutics. CAA-specific PET ligands could emerge from recent findings that the 3-D architecture of β-amyloid fibrils in CAA differs from that in plaques57,42. The discovery that the protein medin co-deposits exclusively with Aβ in the vasculature101 also could enable the development of a biomarker specific for CAA.
Additional research is needed to delineate the fundamental mechanisms underlying the CAA-associated side effects of Aβ-immunotherapy. Recent insights into the clearance of Aβ by the vasculature-associated brain fluid drainage system102,103, the role of perivascular macrophages104, and neuro-immune interactions along CAA-prone meningeal and parenchymal blood vessels105 have set the stage for further research in preclinical models to understand and mitigate the side-effects of Aβ-immunotherapy. This work also is important for gauging any possible risks posed by immunopreventive measures initiated before CAA becomes widespread in the brain.
CONCLUDING REMARKS
Evidence of disease modification by monoclonal antibodies is a small but encouraging step forward in the campaign to subdue AD. We now need to extend the lessons learned from treatment trials to a new framework for the prevention of the disorder. A salient challenge for implementing early prevention is the need for robust prognostic indicators of incipient disease, whereas later preventive measures will be hindered by the relative complexity of advanced disease, which may necessitate combination therapy for multiple targets10,106.
To learn as much as possible from current and past treatment trials, an increasingly refined analysis is essential, not only of the behavioral and biomarker findings during the first 18 months of treatment, but more importantly the biomarker trajectories beyond this point. This applies to participants who continue antibody treatment past 18 months and those who discontinue treatment at any point in the trial. Ideally, such an analysis should be combined with a careful investigation of the biochemical and pathological status of the brain postmortem (similar to the long-term follow-up examination of actively immunized subjects37).
More research with experimental disease models is needed to address fundamental questions that have emerged from the immunotherapy trials65. For example, the interpretation of biomarker trajectories and treatment responses in humans is hampered by the incomplete mechanistic understanding of what these changes represent in the brain. Advances in analytic technologies now enable the measurement of low-abundance proteins in very small volumes of biofluids, making it feasible to measure the same analytes in the CSF and blood of mice and humans; such comparative analyses can inform the design and interpretation of therapeutic and prevention trials26,49,80,107. More broadly, the development of advanced disease models that more completely represent the complexity of later-stage AD would improve the translatability of basic research65.
Although passive immunization with anti-Aβ antibodies is especially promising as a preventive approach to AD, it is useful to consider alternative strategies. For instance, Aβ-proteopathy might be prevented indirectly by targeting proteins that co-deposit with Aβ (e.g., ApoE108 or β2-microglobulin109), or by delivering antibodies against Trem2, which stimulate microglial phagocytosis of Aβ assemblies110,111.
There are also good reasons to keep active immunization in play; current practical impediments to passive antibody treatment, such as cost and the mode and frequency of delivery, are likely to limit the widespread deployment of passive immunoprevention. The chief drawbacks of active immunization (vaccination) are the risks associated with an immune reaction to a ‘self’ antigen and the difficulty moderating an errant immune response112. Since the termination of the pioneering Aβ vaccine trial113, basic research has advanced the safety and efficacy of active immunization114,115. Although considerable challenges remain, if these can be overcome, active immunization could become a relatively simple, inexpensive, and accessible immunopreventive measure.
Finally, it should be emphasized that dementia can result from pathologies such as primary tauopathy, α-synucleinopathy, TDP-43 proteopathy, vascular disease106,116 and many others. In older people, several of these degenerative processes can be comorbid with AD29. Prevention of AD per se thus won’t completely eliminate the risk of dementia. Even so, since AD is the most common cause of dementia117, effective prevention would have considerable benefit for the world’s increasingly long-lived population.
ACKNOWLEDGMENTS
This work was supported by the Cure Alzheimer’s Foundation (M.J.); the German Research Foundation JU655/4-1 (M.J.); the Alexander von Humboldt Foundation (L.C.W.); and U.S. National Institutes of Health (NIH) grants P50 AG025688, ORIP/OD P51 OD011132 (L.C.W.). We thank Tammie Benzinger, Nicole McKay, and Flores Shaney (St. Louis) for the PET images. Harry LeVine (Lexington, Kentucky), Natalie de Souza (Zürich), Johannes Levin (Munich), and Nicolas Villain (Paris) for helpful comments, and Matthias Staufenbiel for many enlightening discussions and comments throughout the development of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests
REFERENCES
- 1.van Dyck CH, Swanson CJ, Aisen P, Bateman RJ, Chen C, Gee M, Kanekiyo M, Li D, Reyderman L, Cohen S, et al. (2023). Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med 388, 9–21. 10.1056/NEJMoa2212948. [DOI] [PubMed] [Google Scholar]
- 2.Sims JR, Zimmer JA, Evans CD, Lu M, Ardayfio P, Sparks J, Wessels AM, Shcherbinin S, Wang H, Monkul Nery ES, et al. (2023). Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA. 10.1001/jama.2023.13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Budd Haeberlein S, Aisen PS, Barkhof F, Chalkias S, Chen T, Cohen S, Dent G, Hansson O, Harrison K, von Hehn C, et al. (2022). Two Randomized Phase 3 Studies of Aducanumab in Early Alzheimer’s Disease. J Prev Alzheimers Dis 9, 197–210. 10.14283/jpad.2022.30. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J, and Selkoe DJ (2002). The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356. 10.1126/science.1072994 297/5580/353 [pii]. [DOI] [PubMed] [Google Scholar]
- 5.De Strooper B, and Karran E (2016). The Cellular Phase of Alzheimer’s Disease. Cell 164, 603–615. 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 6.Jansen WJ, Ossenkoppele R, Knol DL, Tijms BM, Scheltens P, Verhey FR, Visser PJ, Amyloid Biomarker Study G, Aalten P, Aarsland D, et al. (2015). Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 313, 1924–1938. 10.1001/jama.2015.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Dunn B, Haeberlein SB, Holtzman DM, Jagust W, Jessen F, Karlawish J, et al. (2018). NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 14, 535–562. 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDade E, Wang G, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, Fagan AM, Holtzman DM, Cairns NJ, Goate AM, et al. (2018). Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology 91, e1295–e1306. 10.1212/WNL.0000000000006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Selkoe DJ (2012). Preventing Alzheimer’s disease. Science 337, 1488–1492. 10.1126/science.1228541. [DOI] [PubMed] [Google Scholar]
- 10.McDade E, Llibre-Guerra JJ, Holtzman DM, Morris JC, and Bateman RJ (2021). The informed road map to prevention of Alzheimer Disease: A call to arms. Mol. Neurodegener 16, 49. 10.1186/s13024-021-00467-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jucker M, and Walker LC (2013). Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501, 45–51. 10.1038/nature12481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jucker M, and Walker LC (2018). Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci 21, 1341–1349. 10.1038/s41593-018-0238-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goedert M (2015). NEURODEGENERATION. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science 349, 1255555. 10.1126/science.1255555. [DOI] [PubMed] [Google Scholar]
- 14.Collinge J (2016). Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539, 217–226. 10.1038/nature20415. [DOI] [PubMed] [Google Scholar]
- 15.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, and Hyman BT (2008). Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 451, 720–724. 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, and Garaschuk O (2008). Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer’s disease. Science 321, 1686–1689. 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- 17.Yuan P, Zhang M, Tong L, Morse TM, McDougal RA, Ding H, Chan D, Cai Y, and Grutzendler J (2022). PLD3 affects axonal spheroids and network defects in Alzheimer’s disease. Nature 612, 328–337. 10.1038/s41586-022-05491-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Viola KL, and Klein WL (2015). Amyloid beta oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 129, 183–206. 10.1007/s00401-015-1386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tzioras M, McGeachan RI, Durrant CS, and Spires-Jones TL (2023). Synaptic degeneration in Alzheimer disease. Nat. Rev. Neurol 19, 19–38. 10.1038/s41582-022-00749-z. [DOI] [PubMed] [Google Scholar]
- 20.Greenberg SM, Bacskai BJ, Hernandez-Guillamon M, Pruzin J, Sperling R, and van Veluw SJ (2020). Cerebral amyloid angiopathy and Alzheimer disease - one peptide, two pathways. Nat. Rev. Neurol 16, 30–42. 10.1038/s41582-019-0281-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brenowitz WD, Nelson PT, Besser LM, Heller KB, and Kukull WA (2015). Cerebral amyloid angiopathy and its co-occurrence with Alzheimer’s disease and other cerebrovascular neuropathologic changes. Neurobiol. Aging 36, 2702–2708. 10.1016/j.neurobiolaging.2015.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jakel L, De Kort AM, Klijn CJM, Schreuder F, and Verbeek MM (2022). Prevalence of cerebral amyloid angiopathy: A systematic review and meta-analysis. Alzheimer’s & dementia : the journal of the Alzheimer’s Association 18, 10–28. 10.1002/alz.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McKay NS, Gordon BA, Hornbeck RC, Dincer A, Flores S, Keefe SJ, Joseph-Mathurin N, Jack CR, Koeppe R, Millar PR, et al. (2023). Positron emission tomography and magnetic resonance imaging methods and datasets within the Dominantly Inherited Alzheimer Network (DIAN). Nat. Neurosci 26, 1449–1460. 10.1038/s41593-023-01359-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zetterberg H, and Blennow K (2013). Biomarker evidence for uncoupling of amyloid build-up and toxicity in Alzheimer’s disease. Alzheimers. Dement 9, 459–462. 10.1016/j.jalz.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Karran E, Mercken M, and De Strooper B (2011). The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat. Rev. Drug Discov 10, 698–712. 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 26.Rother C, Uhlmann RE, Müller SA, Schelle J, Skodras A, Obermüller U, Häsler LM, Lambert M, Baumann F, Xu Y, et al. (2022). Experimental evidence for temporal uncoupling of brain Abeta deposition and neurodegenerative sequelae. Nat. Commun 13, 7333. 10.1038/s41467-022-34538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long JM, and Holtzman DM (2019). Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179, 312–339. 10.1016/j.cell.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karran E, and De Strooper B (2022). The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat. Rev. Drug Discov 21, 306–318. 10.1038/s41573-022-00391-w. [DOI] [PubMed] [Google Scholar]
- 29.DeTure MA, and Dickson DW (2019). The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 14, 32. 10.1186/s13024-019-0333-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji C, and Sigurdsson EM (2021). Current status of clinical trials on tau immunotherapies. Drugs 81, 1135–1152. 10.1007/s40265-021-01546-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imbimbo BP, Balducci C, Ippati S, and Watling M (2023). Initial failures of anti-tau antibodies in Alzheimer’s disease are reminiscent of the amyloid-beta story. Neural Regen. Res 18, 117–118. 10.4103/1673-5374.340409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173–177. 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 33.Brody DL, and Holtzman DM (2008). Active and passive immunotherapy for neurodegenerative disorders. Annu. Rev. Neurosci 31, 175–193. 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, et al. (2003). Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology 61, 46–54. 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 35.Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, et al. (2003). Antibodies against beta-amyloid slow cognitive decline in Alzheimer’s disease. Neuron 38, 547–554. 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 36.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, et al. (2006). Abeta species removal after abeta42 immunization. J. Neuropathol. Exp. Neurol 65, 1040–1048. 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 37.Nicoll JAR, Buckland GR, Harrison CH, Page A, Harris S, Love S, Neal JW, Holmes C, and Boche D (2019). Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain 142, 2113–2126. 10.1093/brain/awz142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villain N, Planche V, and Levy R (2022). High-clearance anti-amyloid immunotherapies in Alzheimer’s disease. Part 1: meta-analysis and review of efficacy and safety data, and medico-economical aspects. Rev. Neurol. (Paris) 178, 1011–1030. 10.1016/j.neurol.2022.06.012. [DOI] [PubMed] [Google Scholar]
- 39.La Joie R, Ayakta N, Seeley WW, Borys E, Boxer AL, DeCarli C, Dore V, Grinberg LT, Huang E, Hwang JH, et al. (2019). Multisite study of the relationships between antemortem [11C]PIB-PET Centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimers Dement. 15, 205–216. 10.1016/j.jalz.2018.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fritschi SK, Langer F, Kaeser SA, Maia LF, Portelius E, Pinotsi D, Kaminski CF, Winkler DT, Maetzler W, Keyvani K, et al. (2014). Highly potent soluble amyloid-beta seeds in human Alzheimer brain but not cerebrospinal fluid. Brain 137, 2909–2915. 10.1093/brain/awu255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ye L, Rasmussen J, Kaeser SA, Marzesco AM, Obermüller U, Mahler J, Schelle J, Odenthal J, Krüger C, Fritschi SK, et al. (2017). Abeta seeding potency peaks in the early stages of cerebral beta-amyloidosis. EMBO Rep 18, 1536–1544. 10.15252/embr.201744067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang Y, Arseni D, Zhang W, Huang M, Lövestam S, Schweighauser M, Kotecha A, Murzin AG, Peak-Chew SY, Macdonald J, et al. (2022). Cryo-EM structures of amyloid-beta 42 filaments from human brains. Science 375, 167–172. 10.1126/science.abm7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plotkin SS, and Cashman NR (2020). Passive immunotherapies targeting Abeta and tau in Alzheimer’s disease. Neurobiol. Dis 144, 105010. 10.1016/j.nbd.2020.105010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uhlmann RE, Rother C, Rasmussen J, Schelle J, Bergmann C, Ullrich Gavilanes EM, Fritschi SK, Buehler A, Baumann F, Skodras A, et al. (2020). Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat. Neurosci 23, 1580–1588. 10.1038/s41593-020-00737-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Salloway S, Farlow M, McDade E, Clifford DB, Wang G, Llibre-Guerra JJ, Hitchcock JM, Mills SL, Santacruz AM, Aschenbrenner AJ, et al. (2021). A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat. Med 27, 1187–1196. 10.1038/s41591-021-01369-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pontecorvo MJ, Lu M, Burnham SC, Schade AE, Dage JL, Shcherbinin S, Collins EC, Sims JR, and Mintun MA (2022). Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: A secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurol 79, 1250– 1259. 10.1001/jamaneurol.2022.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.U.S. Food & Drug Administration Peripheral and Central Nervous System Drugs Advisory Committee (2020). Aducanumab for the TREATMENT of Alzheimer’s disease. https://www.fda.gov/media/143577/download.
- 48.Mintun MA, Lo AC, Duggan Evans C, Wessels AM, Ardayfio PA, Andersen SW, Shcherbinin S, Sparks J, Sims JR, Brys M, et al. (2021). Donanemab in early Alzheimer’s disease. N. Engl. J. Med 384, 1691–1704. 10.1056/NEJMoa2100708. [DOI] [PubMed] [Google Scholar]
- 49.Bacioglu M, Maia LF, Preische O, Schelle J, Apel A, Kaeser SA, Schweighauser M, Eninger T, Lambert M, Pilotto A, et al. (2016). Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 91, 56–66. 10.1016/j.neuron.2016.05.018. [DOI] [PubMed] [Google Scholar]
- 50.Gafson AR, Barthélemy NR, Bomont P, Carare RO, Durham HD, Julien JP, Kuhle J, Leppert D, Nixon RA, Weller RO, et al. (2020). Neurofilaments: Neurobiological foundations for biomarker applications. Brain 143, 1975–1998. 10.1093/brain/awaa098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Preische O, Schultz SA, Apel A, Kuhle J, Kaeser SA, Barro C, Gräber S, Kuder-Buletta E, LaFougere C, Laske C, et al. (2019). Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer’s disease. Nat. Med 25, 277–283. 10.1038/s41591-018-0304-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alves F, Kalinowski P, and Ayton S (2023). Accelerated brain volume loss caused by anti-beta-amyloid drugs: A systematic review and meta-analysis. Neurology 100, e2114–e2124. 10.1212/WNL.0000000000207156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barkhof F, and Knopman DS (2023). Brain shrinkage in anti-beta-amyloid Alzheimer trials: neurodegeneration or pseudoatrophy? Neurology 100, 941–942. 10.1212/WNL.0000000000207268. [DOI] [PubMed] [Google Scholar]
- 54.Holdridge KC, Yaari R, Hoban DB, Andersen S, and Sims JR (2023). Targeting amyloid beta in Alzheimer’s disease: Meta-analysis of low-dose solanezumab in Alzheimer’s disease with mild dementia studies. Alzheimers. Dement 10.1002/alz.13031. [DOI] [PubMed] [Google Scholar]
- 55.AlzForum (2023). Legacy of A4 secondary prevention study goes beyond negative result. https://www.alzforum.org/news/research-news/legacy-a4-secondary-prevention-study-goes-beyond-negative-result.
- 56.Herzig MC, Van Nostrand WE, and Jucker M (2006). Mechanism of cerebral beta-amyloid angiopathy: Murine and cellular models. Brain Pathol 16, 40–54. 10.1111/j.1750-3639.2006.tb00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kollmer M, Close W, Funk L, Rasmussen J, Bsoul A, Schierhorn A, Schmidt M, Sigurdson CJ, Jucker M, and Fändrich M (2019). Cryo-EM structure and polymorphism of Abeta amyloid fibrils purified from Alzheimer’s brain tissue. Nat. Commun 10, 4760. https://doi.org/ 10.1038/s41467-019-12683-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lannfelt L, Moller C, Basun H, Osswald G, Sehlin D, Satlin A, Logovinsky V, and Gellerfors P (2014). Perspectives on future Alzheimer therapies: Amyloid-beta protofibrils – a new target for immunotherapy with BAN2401 in Alzheimer’s disease. Alzheimers Res. Ther 6, 16. 10.1186/alzrt246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, et al. (2001). The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Abeta protofibril formation. Nat. Neurosci 4, 887– 893. 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 60.Yang Y, Zhang W, Murzin AG, Schweighauser M, Huang M, Lovestam S, Peak-Chew SY, Saito T, Saido TC, Macdonald J, et al. (2023). Cryo-EM structures of amyloid-beta filaments with the Arctic mutation (E22G) from human and mouse brains. Acta Neuropathol 145, 325–333. 10.1007/s00401-022-02533-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Linse S, Scheidt T, Bernfur K, Vendruscolo M, Dobson CM, Cohen SIA, Sileikis E, Lundqvist M, Qian F, O’Malley T, et al. (2020). Kinetic fingerprints differentiate the mechanisms of action of anti-Abeta antibodies. Nat. Struct. Mol. Biol 27, 1125–1133. 10.1038/s41594-020-0505-6. [DOI] [PubMed] [Google Scholar]
- 62.Frost JL, Le KX, Cynis H, Ekpo E, Kleinschmidt M, Palmour RM, Ervin FR, Snigdha S, Cotman CW, Saido TC, et al. (2013). Pyroglutamate-3 amyloid-beta deposition in the brains of humans, non-human primates, canines, and Alzheimer disease-like transgenic mouse models. Am. J. Pathol 183, 369–381. 10.1016/j.ajpath.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rijal Upadhaya A, Kosterin I, Kumar S, von Arnim CA, Yamaguchi H, Fandrich M, Walter J, and Thal DR (2014). Biochemical stages of amyloid-beta peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 137, 887–903. 10.1093/brain/awt362. [DOI] [PubMed] [Google Scholar]
- 64.Li X, Ospitalieri S, Robberechts T, Hofmann L, Schmid C, Rijal Upadhaya A, Koper MJ, von Arnim CAF, Kumar S, Willem M, et al. (2022). Seeding, maturation and propagation of amyloid beta-peptide aggregates in Alzheimer’s disease. Brain 145, 3558–3570. 10.1093/brain/awac202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scearce-Levie K, Sanchez PE, and Lewcock JW (2020). Leveraging preclinical models for the development of Alzheimer disease therapeutics. Nat. Rev. Drug Discov 19, 447–462. 10.1038/s41573-020-0065-9. [DOI] [PubMed] [Google Scholar]
- 66.Lowe SL, Willis BA, Hawdon A, Natanegara F, Chua L, Foster J, Shcherbinin S, Ardayfio P, and Sims JR (2021). Donanemab (LY3002813) dose-escalation study in Alzheimer’s disease. Alzheimers. Dement. (NY) 7, e12112. 10.1002/trc2.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McDade E, and Bateman RJ (2017). Stop Alzheimer’s before it starts. Nature 547, 153–155. 10.1038/547153a. [DOI] [PubMed] [Google Scholar]
- 68.Karlnoski RA, Rosenthal A, Kobayashi D, Pons J, Alamed J, Mercer M, Li Q, Gordon MN, Gottschall PE, and Morgan D (2009). Suppression of amyloid deposition leads to long-term reductions in Alzheimer’s pathologies in Tg2576 mice. J. Neurosci 29, 4964–4971. 10.1523/JNEUROSCI.4560-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Niewoehner J, Bohrmann B, Collin L, Urich E, Sade H, Maier P, Rueger P, Stracke JO, Lau W, Tissot AC, et al. (2014). Increased brain penetration and potency of a therapeutic antibody using a monovalent molecular shuttle. Neuron 81, 49–60. 10.1016/j.neuron.2013.10.061. [DOI] [PubMed] [Google Scholar]
- 70.Bathini P, Sun T, Schenk M, Schilling S, McDannold NJ, and Lemere CA (2022). Acute effects of focused ultrasound-induced blood-brain barrier opening on anti-Pyroglu3 Abeta antibody delivery and immune responses. Biomolecules 12. 10.3390/biom12070951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansson O (2021). Biomarkers for neurodegenerative diseases. Nat. Med 27, 954–963. 10.1038/s41591-021-01382-x. [DOI] [PubMed] [Google Scholar]
- 72.Teunissen CE, Verberk IMW, Thijssen EH, Vermunt L, Hansson O, Zetterberg H, van der Flier WM, Mielke MM, and Del Campo M (2022). Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol 21, 66–77. 10.1016/S1474-4422(21)00361-6. [DOI] [PubMed] [Google Scholar]
- 73.Ikonomovic MD, Abrahamson EE, Price JC, Hamilton RL, Mathis CA, Paljug WR, Debnath ML, Cohen AD, Mizukami K, DeKosky ST, et al. (2012). Early AD pathology in a [C-11]PiB-negative case: A PiB-amyloid imaging, biochemical, and immunohistochemical study. Acta Neuropathol 123, 433–447. 10.1007/s00401-012-0943-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pletnikova O, Rudow GL, Hyde TM, Kleinman JE, Ali SZ, Bharadwaj R, Gangadeen S, Crain BJ, Fowler DR, Rubio AI, et al. (2015). Alzheimer lesions in the autopsied brains of people 30 to 50 years of age. Cogn. Behav. Neurol 28, 144–152. 10.1097/WNN.0000000000000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jagust WJ, and Landau SM; Alzheimer’s Disease Neuroimaging Initiative (2021). Temporal dynamics of beta-amyloid accumulation in aging and Alzheimer disease. Neurology 96, e1347–e1357. 10.1212/WNL.0000000000011524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barthelemy NR, Li Y, Joseph-Mathurin N, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, Fagan AM, Perrin RJ, Goate AM, et al. (2020). A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat. Med 26, 398–407. 10.1038/s41591-020-0781-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ashton NJ, Janelidze S, Mattsson-Carlgren N, Binette AP, Strandberg O, Brum WS, Karikari TK, Gonzalez-Ortiz F, Di Molfetta G, Meda FJ, et al. (2022). Differential roles of Ab42/40, p-tau231 and p-tau217 for Alzheimer’s trial selection and disease monitoring. Nat. Med 28, 2555–2562. 10.1038/s41591-022-02074-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE, Zetterberg H, Stomrud E, Ashton NJ, Blennow K, et al. (2021). Plasma GFAP is an early marker of amyloid-beta but not tau pathology in Alzheimer’s disease. Brain 144, 3505–3516. 10.1093/brain/awab223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morenas-Rodriguez E, Li Y, Nuscher B, Franzmeier N, Xiong C, Suarez-Calvet M, Fagan AM, Schultz S, Gordon BA, Benzinger TLS, et al. (2022). Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: A longitudinal observational study. Lancet Neurol 21, 329–341. 10.1016/S1474-4422(22)00027-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kaeser SA, Hasler LM, Lambert M, Bergmann C, Bottelbergs A, Theunis C, Mercken M, and Jucker M (2022). CSF p-tau increase in response to Abeta-type and Danish-type cerebral amyloidosis and in the absence of neurofibrillary tangles. Acta Neuropathol 143, 287–290. 10.1007/s00401-021-02400-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eninger T, Muller SA, Bacioglu M, Schweighauser M, Lambert M, Maia LF, Neher JJ, Hornfeck SM, Obermuller U, Kleinberger G, et al. (2022). Signatures of glial activity can be detected in the CSF proteome. Proc. Natl. Acad. Sci. USA 119. e2119804119. 10.1073/pnas.2119804119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mok TH, Nihat A, Majbour N, Sequeira D, Holm-Mercer L, Coysh T, Darwent L, Batchelor M, Groveman BR, Orr CD, et al. (2023). Seed amplification and neurodegeneration marker trajectories in individuals at risk of prion disease. Brain 146, 2570–2583. 10.1093/brain/awad101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Siderowf A, Concha-Marambio L, Lafontant DE, Farris CM, Ma Y, Urenia PA, Nguyen H, Alcalay RN, Chahine LM, Foroud T, et al. (2023). Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using alpha-synuclein seed amplification: A cross-sectional study. Lancet Neurol 22, 407–417. 10.1016/S1474-4422(23)00109-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Okuzumi A, Hatano T, Matsumoto G, Nojiri S, Ueno SI, Imamichi-Tatano Y, Kimura H, Kakuta S, Kondo A, Fukuhara T, et al. (2023). Propagative alpha-synuclein seeds as serum biomarkers for synucleinopathies. Nat. Med 29, 1448–1455. 10.1038/s41591-023-02358-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Salloway S, Chalkias S, Barkhof F, Burkett P, Barakos J, Purcell D, Suhy J, Forrestal F, Tian Y, Umans K, et al. (2022). Amyloid-related imaging abnormalities in 2 phase 3 studies evaluating aducanumab in patients with early Alzheimer disease. JAMA Neurol 79, 13–21. 10.1001/jamaneurol.2021.4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roytman M, Mashriqi F, Al-Tawil K, Schulz PE, Zaharchuk G, Benzinger TLS, and Franceschi AM (2023). Amyloid-related imaging abnormalities: An update. AJR Am. J. Roentgenol 220, 562–574. 10.2214/AJR.22.28461. [DOI] [PubMed] [Google Scholar]
- 87.Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, et al. (2012). Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: A retrospective analysis. Lancet Neurol 11, 241–249. 10.1016/S1474-4422(12)70015-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reish NJ, Jamshidi P, Stamm B, Flanagan ME, Sugg E, Tang M, Donohue KL, McCord M, Krumpelman C, Mesulam MM, et al. (2023). Multiple cerebral hemorrhages in a patient receiving lecanemab and treated with t-PA for stroke. N. Engl. J. Med 388, 478–479. 10.1056/NEJMc2215148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Solopova E, Romero-Fernandez W, Harmsen H, Ventura-Antunes L, Wang E, Shostak A, Maldonado J, Donahue M, Schultz D, Coyne TM, et al. (2023). Fatal iatrogenic cerebral amyloid-related encephalitis in a patient treated with lecanemab for Alzheimer’s disease: Neuroimaging and neuropathology. Preprint at medRixiv 10.1101/2023.04.26.23289061. [DOI] [PMC free article] [PubMed]
- 90.Sveikata L, Charidimou A, and Viswanathan A (2022). Vessels sing their ARIAs: The role of vascular amyloid in the age of aducanumab. Stroke 53, 298–302. 10.1161/STROKEAHA.121.036873. [DOI] [PubMed] [Google Scholar]
- 91.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, and Jucker M (2002). Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 298, 1379. 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 92.Plowey ED, Bussiere T, Rajagovindan R, Sebalusky J, Hamann S, von Hehn C, Castrillo-Viguera C, Sandrock A, Budd Haeberlein S, van Dyck CH, et al. (2022). Alzheimer disease neuropathology in a patient previously treated with aducanumab. Acta Neuropathol 144, 143– 153. 10.1007/s00401-022-02433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Scherlek AA, Kozberg MG, Nicoll JAR, Perosa V, Freeze WM, van der Weerd L, Bacskai BJ, Greenberg SM, Frosch MP, Boche D, et al. (2022). Histopathological correlates of haemorrhagic lesions on ex vivo magnetic resonance imaging in immunized Alzheimer’s disease cases. Brain Commun 4, fcac021. 10.1093/brain-comms/fcac021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Boche D, Zotova E, Weller RO, Love S, Neal JW, Pickering RM, Wilkinson D, Holmes C, and Nicoll JA (2008). Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 131, 3299–3310. 10.1093/brain/awn261. [DOI] [PubMed] [Google Scholar]
- 95.Sakai K, Boche D, Carare R, Johnston D, Holmes C, Love S, and Nicoll JA (2014). Abeta immunotherapy for Alzheimer’s disease: Effects on apoE and cerebral vasculopathy. Acta Neuropathol 128, 777–789. 10.1007/s00401-014-1340-9. [DOI] [PubMed] [Google Scholar]
- 96.Winkler DT, Bondolfi L, Herzig MC, Jann L, Calhoun ME, Wiederhold KH, Tolnay M, Staufenbiel M, and Jucker M (2001). Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci 21, 1619–1627. 10.1523/JNEUROSCI.21-05-01619.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heffernan AL, Chidgey C, Peng P, Masters CL, and Roberts BR (2016). The neurobiology and age-related prevalence of the epsilon4 allele of Apolipoprotein E in Alzheimer’s disease cohorts. J. Mol. Neurosci 60, 316–324. 10.1007/s12031-016-0804-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nelson PT, Pious NM, Jicha GA, Wilcock DM, Fardo DW, Estus S, and Rebeck GW (2013). APOE-epsilon2 and APOE-epsilon4 correlate with increased amyloid accumulation in cerebral vasculature. J. Neuropathol. Exp. Neurol. 72, 708–715. 10.1097/NEN.0b013e31829a25b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Foley KE, and Wilcock DM (2022). Vascular considerations for amyloid immunotherapy. Curr. Neurol. Neurosci. Rep 22, 709–719. 10.1007/s11910-022-01235-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Charidimou A, Boulouis G, Frosch MP, Baron JC, Pasi M, Albucher JF, Banerjee G, Barbato C, Bonneville F, Brandner S, et al. (2022). The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI-neuropathology diagnostic accuracy study. Lancet Neurol 21, 714–725. 10.1016/S1474-4422(22)00208-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wagner J, Degenhardt K, Veit M, Louros N, Konstantoulea K, Skodras A, Wild K, Liu P, Obermuller U, Bansal V, et al. (2022). Medin co-aggregates with vascular amyloid-beta in Alzheimer’s disease. Nature 612, 123–131. 10.1038/s41586-022-05440-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rasmussen MK, Mestre H, and Nedergaard M (2018). The glymphatic pathway in neurological disorders. Lancet Neurol 17, 1016– 1024. 10.1016/S1474-4422(18)30318-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Agarwal N, and Carare RO (2020). Cerebral vessels: An overview of anatomy, physiology, and role in the drainage of fluids and solutes. Front. Neurol 11, 611485. 10.3389/fneur.2020.611485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Drieu A, Du S, Storck SE, Rustenhoven J, Papadopoulos Z, Dykstra T, Zhong F, Kim K, Blackburn S, Mamuladze T, et al. (2022). Parenchymal border macrophages regulate the flow dynamics of the cerebrospinal fluid. Nature 611, 585–593. 10.1038/s41586-022-05397-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rustenhoven J, and Kipnis J (2022). Brain borders at the central stage of neuroimmunology. Nature 612, 417–429. 10.1038/s41586-022-05474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wilson DM 3rd, Cookson MR, Van Den Bosch L, Zetterberg H, Holtzman DM, and Dewachter I (2023). Hallmarks of neurodegenerative diseases. Cell 186, 693–714. 10.1016/j.cell.2022.12.032. [DOI] [PubMed] [Google Scholar]
- 107.Andersson E, Schultz N, Saito T, Saido TC, Blennow K, Gouras GK, Zetterberg H, and Hansson O (2023). Cerebral Ab deposition precedes reduced cerebrospinal fluid and serum Ab42/Ab40 ratios in the AppNL-F/NL-F knock-in mouse model of Alzheimer’s disease. Alzheimers Res. Ther 15, 64. 10.1186/s13195-023-01196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiong M, Jiang H, Serrano JR, Gonzales ER, Wang C, Gratuze M, Hoyle R, Bien-Ly N, Silverman AP, Sullivan PM, et al. (2021). APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function. Sci. Transl. Med 13. 10.1126/scitranslmed.abd7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhao Y, Zheng Q, Hong Y, Gao Y, Hu J, Lang M, Zhang H, Zhou Y, Luo H, Zhang X, et al. (2023). b2-Microglobulin coaggregates with Abeta and contributes to amyloid pathology and cognitive deficits in Alzheimer’s disease model mice. Nat. Neurosci 26, 1170–1184. 10.1038/s41593-023-01352-1. [DOI] [PubMed] [Google Scholar]
- 110.Schlepckow K, Monroe KM, Kleinberger G, Cantuti-Castelvetri L, Parhizkar S, Xia D, Willem M, Werner G, Pettkus N, Brunner B, et al. (2020). Enhancing protective microglial activities with a dual function TREM2 antibody to the stalk region. EMBO Mol. Med 12, e11227. 10.15252/emmm.201911227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhao P, Xu Y, Jiang L, Fan X, Li L, Li X, Arase H, Zhao Y, Cao W, Zheng H, et al. (2022). A tetravalent TREM2 agonistic antibody reduced amyloid pathology in a mouse model of Alzheimer’s disease. Sci. Transl. Med 14, eabq0095. 10.1126/scitranslmed.abq0095. [DOI] [PubMed] [Google Scholar]
- 112.Wisniewski T, and Goni F (2015). Immunotherapeutic approaches for Alzheimer’s disease. Neuron 85, 1162–1176. 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Schenk D (2002). Amyloid-beta immunotherapy for Alzheimer’s disease: The end of the beginning. Nat. Rev. Neurosci 3, 824–828. 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- 114.Winblad B, Graf A, Riviere ME, Andreasen N, and Ryan JM (2014). Active immunotherapy options for Alzheimer’s disease. Alzheimers Res. Ther 6, 7. 10.1186/alzrt237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vogt AS, Jennings GT, Mohsen MO, Vogel M, and Bachmann MF (2023). Alzheimer’s disease: A brief history of immunotherapies targeting amyloid b. Int. J. Mol. Sci 24. 10.3390/ijms24043895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boyle PA, Wang T, Yu L, Wilson RS, Dawe R, Arfanakis K, Schneider JA, and Bennett DA (2021). To what degree is late life cognitive decline driven by age-related neuropathologies? Brain 144, 2166–2175. 10.1093/brain/awab092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, Cummings J, and van der Flier WM (2021). Alzheimer’s disease. Lancet 397, 1577–1590. 10.1016/S0140-6736(20)32205-4. [DOI] [PMC free article] [PubMed] [Google Scholar]