

Abstract
Recent studies suggest that dopamine D3 receptors (D3R) may be a therapeutic target for opioid use disorders (OUD). This study examined the effects of the D3R partial agonist (±)VK4-40 and the D3R-selective antagonist (±)VK4-116, compared to the mu-opioid receptor antagonist naltrexone (NTX), in nonhuman primate models of OUD and antinociception. Adult male and female (N = 4/sex) cynomolgus monkeys were trained to self-administer oxycodone (0.003–0.1 mg/kg/injection) first under a fixed-ratio (FR) and then a progressive-ratio (PR) schedule of reinforcement during daily 1- and 4-hr sessions, respectively. Under the FR schedule, intravenous NTX (0.01–0.1 mg/kg), (±)VK4-116 (1.0–10 mg/kg), and (±)VK4-40 (1.0–10 mg/kg) were studied in combination with the peak oxycodone dose and a dose on the descending limb of the dose-effect curve; NTX and (±)VK4-40 were also studied at the peak of the PR dose-response curve (N = 4). Following saline extinction, each compound was examined on oxycodone-induced reinstatement. Finally, these compounds were assessed in adult male rhesus monkeys (N = 3) in a warm-water (38 °C, 50 °C, 54 °C) tail withdrawal assay. NTX decreased responding on the peak of the FR oxycodone dose-response curve, but increased responding on the descending limb. (±)VK4-40, but not (±)VK4-116, significantly decreased peak oxycodone self-administration; (±)VK4-40 did not increase responding on the descending limb. NTX and (±)VK4-40, but not (±)VK4-116, attenuated oxycodone-induced reinstatement. Under PR responding, NTX and (±)VK4-40 decreased breakpoints. Oxycodone-induced antinociception was attenuated by NTX, but not by (±)VK4-40 or (±)VK4-116. Together, these results suggest that further research evaluating the effects of (±)VK4-40 as a novel pharmacotherapy for OUD is warranted.
Subject terms: Pharmacology, Reward
Introduction
The prevalence of substance use disorders (SUDs) in the United States has become a national problem; the estimated cost of SUDs in the United States is more than $800 billion a year due to loss of workplace productivity, health care costs, and criminal justice expenses [1, 2]. In 2020, nearly 92,000 people in the U.S. died from drug-involved overdoses and 75% of these overdose deaths (~69,000) involved opioids [3, 4]. Moreover, this figure continues to increase and in 2021 75,000 people died from opioid overdoses [5]. The initial increase in opioid misuse and overdose-related deaths was primarily caused by a high rate of opioid prescriptions for the treatment of pain by medical practitioners [6]. Despite the prevalence of opioid misuse, there are only three FDA-approved medications for OUD, and these include naltrexone, methadone, and buprenorphine [7]. One goal identified by the National Institute on Drug Abuse is to identify abuse deterrents – drugs that can decrease the addictive liability of opioids, without affecting opioid-induced analgesia [7]. Most drugs of abuse, including opioids, alter dopamine (DA) concentrations and recent studies have identified the DA D3 receptor (D3R) as a potential therapeutic target for substance use disorders, including OUD [8]; for reviews, see [9] and [10].
While drugs that bind to D3R have shown some promise in animal studies, translating these effects to humans has proven difficult due to either the lack of appropriate ADME (absorption, distribution, metabolism, and excretion) or predicted cardiotoxicity, notably when administered in combination with cocaine [11, 12]. Efforts to develop a new generation of D3R compounds that are devoid of these undesirable attributes have led to the discovery of the highly selective D3R antagonist (±)VK4-116 (D3R Ki = 6.8 nM; D2R Ki = 11,400 nM) and the D3R partial agonist (±)VK4-40 (D3R Ki = 0.36 nM; D2R Ki = 151 nM) ([13]). (±)VK4-116 has high metabolic stability in mouse microsomes and has been shown to block the acquisition of oxycodone-induced locomotor sensitization in mice and dose-dependently attenuate oxycodone-induced conditioned-place preference (CPP) in rats [13]. In another study, pretreatment with (±)VK4-116 dose-dependently decreased oxycodone self-administration and reduced withdrawal-induced hyperalgesia in opioid-dependent rats [14]. Recent studies with the D3R antagonist R-VK4-40 [15] reported dose-dependent decreases in oxycodone breakpoints in rats responding under a progressive-ratio (PR) schedule of reinforcement, without affecting opioid-induced antinociceptive or locomotor effects in rats [15]. Nevertheless, behavioral investigation conducted on the D3R partial agonist (±)VK4-40 has been limited to two studies in rodent models of cocaine use disorder [16, 17]. These promising results in rodent studies (see also [18]) warrant further research on the effect of these D3R-selective compounds with varying selectivity and efficacy at D3R/D2R, in nonhuman primate models of oxycodone self-administration.
The primary goal of the present study was to compare (±)VK4-116 and (±)VK4-40 to the opioid receptor antagonist naltrexone, in non-physically dependent monkeys self-administering the opioid agonist oxycodone. In one experiment, oxycodone was available under a fixed-ratio (FR) schedule of reinforcement and both D3R compounds were tested at two parts of the oxycodone dose-response curve. The effects of (±)VK4-40 were also examined in the same monkeys responding under a PR schedule of reinforcement. The use of both schedules of reinforcement allowed for the comparison of D3R compounds on the reinforcing effects of oxycodone (under the FR schedule) and the reinforcing strength of oxycodone (under the PR schedule). In addition to studying oxycodone self-administration, relapse to drug use during abstinence is especially high in patients with OUD [19]. However, little research has specifically characterized oxycodone relapse [20]. Thus, a second goal of this study was to examine both (±)VK4-116 and (±)VK4-40 on oxycodone-induced reinstatement. Finally, as stated above [7], the efficacy of a treatment for OUD would ideally not affect the antinociceptive effects of the opioid. Thus, a final study was conducted in monkeys to examine the effects of both D3R compounds on oxycodone-induced antinociception using a tail-withdrawal assay.
The present study was also designed to detect sex differences in the behavioral effects of these D3R compounds in an animal model of OUD. While there are significant differences between men and women in every stage of the addiction cycle [21–23], it is also clear that the experimental conditions can yield potentially different outcomes and interpretations. In preclinical studies, it has been found that female rats self-administered opioids at higher response rates and greater intakes compared with male rats when responding under an FR schedule of reinforcement [24, 25]. Similarly, when the schedule of reinforcement was a PR schedule, these same investigators reported that female rats had higher breakpoints compared with male rats. However, Townsend et al. (2019) extended the study of sex differences in opioid self-administration to a concurrent choice schedule of fentanyl vs. diluted Ensure and found that male rats chose 3.2 µg/kg/injection fentanyl over 18% Ensure at a higher percentage than female rats [25]. Consistent with this finding, Comer et al. (2010) reported in human subjects that the analgesic effects of morphine were not different in women and men, but men showed greater positive subjective effects and women reported greater negative effects after morphine administration [26]. Very few preclinical studies have addressed sex differences in treatment interventions. For the present self-administration and reinstatement studies, the effects of both the D3R-selective antagonist ((±)VK4-116) and the D3R partial agonist ((±)VK4-40), as well as the mu opioid receptor antagonist naltrexone, were studied in male and female cynomolgus monkeys.
Materials and methods
Subjects
For the FR self-administration study (Experiment 1), eight adult cynomolgus macaques (Macaca fascicularis) (n = 4/sex) served as subjects. One male monkey was experimentally naïve at the start of this study (M-8261), three males (M-7903, M-7962, M-8103) had a history of cocaine self-administration and pretreatments with cannabinoids (unpublished studies); two females (F-7438, F-7579) had a brief history of acute drug pretreatments on food-maintained responding (unpublished) and two females (F-7453, F-7440) had a history of cocaine self-administration. All females were drug-free for 2.5 years before the start of this study. For the reinstatement study (Experiment 2), six macaques from Experiment 1 served as subjects (n = 3/sex) and for the PR study (Experiment 3), four cynomolgus macaques from Experiments 1 and 2 served as subjects. This included one female (F-7440) and three males (M-7962, M-7903, M-8103). In all experiments, the number of monkeys used was based on availability, with a minimum of four animals.
Monkeys in all three self-administration experiments were pair-housed in same-sex groups, in a temperature- and humidity-controlled room maintained on a 14-h light/10-hour dark cycle (lights on between 6:00 AM and 8:00 PM) in stainless-steel cages with ad libitum access to water. Monkeys were weighed weekly and fed enough food daily (Purina LabDiet 5045, St Louis, MO, USA and fresh fruit and vegetables) to maintain a healthy body weight and appearance as determined by daily inspection and periodic veterinary examinations. Environmental enrichment was provided as outlined in the Animal Care and Use Committee of Wake Forest University Non-Human Primate Environmental Enrichment Plan, including chew toys, mirrors, music, and foraging feeders. The menstrual cycle in all females was monitored daily, except for one monkey (F-7579) because she had prior bilateral ovariectomy and hysterectomy procedures. Animal housing, handling, and experimental protocols were performed per the 2011 National Research Council Guidelines for the Care and Use of Mammals in Neuroscience and Behavioral Research and were approved by the Animal Care and Use Committee of Wake Forest University.
For the tail withdrawal antinociception study (Experiment 4), three individually housed adult male rhesus macaques (Macaca mulatta) of either Indian or Chinese origin and weighing between 10–14 kg served as subjects. All animals had experimental histories involving opioid ligands, monoamine transporter ligands, and NMDA antagonists ([27]). Diet consisted of laboratory monkey chow (#5049, Purina, Framingham, MA, USA) supplemented with fresh fruits, vegetables, and fish oil. Monkeys had ad libitum water access in the housing chamber. Housing facilities were on a 12 h light/dark cycle (lights on from 6:00 AM to 6:00 PM) and were licensed by the United States Department of Agriculture and accredited by AAALAC International. Research and husbandry were conducted in accordance with the 2011 Guide for the Care and Use of Laboratory Animals. The Virginia Commonwealth University Institutional Animal Care and Use Committee approved both research and enrichment protocols.
Catheter implantation
For the oxycodone self-administration studies, monkeys were surgically implanted with both a chronic indwelling intravenous catheter and a subcutaneous vascular access port (VAP) under sterile conditions. Before surgery, each animal received an antibiotic (30 mg/kg Kefzol, i.m.; cefazolin sodium, Marsam Pharmaceuticals Inc., Cherry Hill, NJ, USA). Anesthesia was induced using ketamine (10–15 mg/kg, i.m.) and maintained during the procedure with 1.5% isoflurane gas. The proximal end of the catheter was passed through a major vein (femoral, internal, or external jugular) to the level of the posterior vena cava. An incision was then made slightly off the midline of the monkey’s back, and through blunt dissection, a subcutaneous pocket was created for the VAP. The distal end of the catheter was subcutaneously threaded to the incision site and connected to the VAP. During post-operative recovery, monkeys received either ketoprofen (5 mg/kg, i.m.) or Metacam (meloxicam; 1.5 mg.kg, p.o.), and starting the following day, Naxcel (ceftiofur sodium; 2.2 mg/kg, i.m., SID) for 7–14 days. Additionally, each port and catheter were filled with a solution of heparinized saline (100 U/mL) between experimental sessions to prolong patency and prevent clotting.
Apparatus
All monkeys, fitted with aluminum collars, were trained to sit in a primate restraint chair (Primate Products, Redwood City, CA). Self-administration and reinstatement experiments (Experiments 1–3) were conducted in ventilated, sound-attenuating operant chambers (1.5 × 0.76 × 0.76 m; Med Associates, St. Albans, VT, USA). During each session, white noise was also played to mask any potentially obstructing sounds from outside the experimental room. Inside each chamber was an operant panel with three horizontally aligned photo-optic switches (Model 117-1007; Stewart Ergonomics, Inc., Furlong, PA, USA) that were positioned within reach when monkeys were seated in a primate chair, and above each switch were two stimulus lights. Only one switch was active, signaled by the illumination of a white light above the switch; for five monkeys, it was the right switch and for the other three monkeys it was the left switch. A food receptacle, located in the middle of the operant panel below the middle switch, was connected to a pellet dispenser (Med Associates) on the top of the chamber with a Tygon tube for delivery of 1.0-g banana-flavored food pellets (Bio-Serv, Frenchtown, NJ, USA). A peristaltic infusion pump (Cole-Parmer Instrument Co., Niles, IL, USA) was also located on top of the chamber for intravenous drug delivery at a rate of approximately 1.5 mL per 10 sec. Illumination of a red light inside the chamber accompanied food presentations and drug injections.
Before every session, the area on the monkey’s back containing the VAP was prepared with an antiseptic povidone-iodine (PVP) scrub (Medline Industries, Mundelein, IL, USA) followed by isopropyl alcohol (Fisher Scientific, Fair Lawn, NJ, USA). The area was given a final preparation with PVP scrub before a 22-gauge needle (Access Technologies) was inserted into the monkey’s port, connecting the catheter to the infusion pump on the chamber. Before the start of each session, the pump was run for approximately 3 sec to fill each monkey’s port with saline or concentration of drug available. All pretreatment drug administration was performed outside the chamber before placing the monkey into the operant chamber.
Experiment 1
Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone self-administration under a fixed-ratio schedule of reinforcement. All monkeys were trained to respond under an FR schedule of food presentation followed by a timeout (TO) period. Females responded under an FR 30 with a 10-s TO, and males responded under an FR 50 with a 60-s TO. Sessions ended after 30 reinforcers or 60 min, whichever occurred first. When responding was stable, defined as three consecutive sessions in which response rates did not vary by more than 20% of the 3-day mean, saline was substituted for food until responding declined to 20% of baseline (~5 sessions). After a return to food-reinforced responding, a dose of oxycodone was substituted for food for at least 5 consecutive sessions and until responding was stable; a complete oxycodone dose-response curve (0.003-0.1 mg/kg/injection) was determined in each monkey, with doses available for at least 5 consecutive sessions. Doses were tested in random order and saline was occasionally retested throughout the experiment.
After determining the oxycodone dose-response curve, which was characterized as an inverted U-shaped function of dose in each monkey, drug pretreatment studies began. For these studies, two oxycodone doses were examined – the dose that was associated with peak response rates and a dose on the descending limb of the oxycodone baseline dose-effect curve; these doses were individually determined in each monkey. If there were large differences between rates at the peak of the curve and the descending limb, we tested pretreatment drugs on the lowest oxycodone dose on the descending limb; if two doses on the descending limb were relatively flat, we tested the higher oxycodone dose. The effects of acute pretreatments with naltrexone (0.001–0.1 mg/kg, i.v.), administered 5 min before sessions, were studied first in all monkeys; it varied in a non-systematic fashion as to whether this occurred first with the dose of oxycodone on the peak or descending limb. Next, (±)VK4-116 (1.0–10 mg/kg, i.v.) and (±)VK4-40 (1.0–10 mg/kg, i.v.) were administered 5 min before the start of a session. Pretreatment times were based, in part, on pharmacokinetic studies in rodents that showed the R-enantiomer of VK4-40 was stable in liver microsomes at 60 min [14]. Pretreatment drugs were typically tested twice weekly, ensuring at least one day between test sessions. Each pretreatment dose was determined at least twice unless an adverse event (e.g., vomiting or body shaking) was observed following administration of a given dose.
Experiment 2
Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone-induced reinstatement. After Experiment 1, saline was substituted for oxycodone, until responding declined by at least 80%. Next, an acute dose of oxycodone (0.03-0.17 mg/kg, i.v.) was administered 1 min before a saline self-administration session. The oxycodone dose that resulted in the most saline injections (0.1 mg/kg in five monkeys and 0.17 mg/kg in one monkey) was chosen to evaluate the effects of naltrexone (0.01–0.1 mg/kg; 5 min pretreatment), (±)VK4-116 (3–10 mg/kg; 5 min pretreatment), and (±)VK4-40 (3–10 mg/kg; 5 min pretreatment) on oxycodone-induced reinstatement. Oxycodone alone was tested frequently to assure reinstatement; if the effect of oxycodone alone was lower than previous determinations, the prior pretreatment data were excluded from analyses. After several saline sessions, the self-administered dose of oxycodone that resulted in peak response rates or a dose on the descending limb was used to re-establish self-administration before continuing saline extinction and reinstatement. As in Experiment 1, test sessions were generally twice per week and each dose was typically tested twice. M-8261 was found deceased in his cage due to gas distention in the abdomen (i.e., bloat) and did not complete the study.
Experiment 3
Effects of (±)VK4-40 on oxycodone self-administration under a progressive-ratio schedule of reinforcement. Following Experiment 2, a subset of monkeys (n = 4) were trained to self-administer oxycodone under a PR schedule of reinforcement, in which the response requirement increased after every drug infusion (see Supplemental Table S1). Daily behavioral sessions were comprised of a multiple schedule, in which the first component was an FR 20 schedule of food availability (available on the right switch). Each food reinforcer was followed by 5-sec time-out and the component ended after delivery of 10 banana-flavored pellets or 5 min, whichever occurred first. Following a 5-min TO, the second component of the multiple schedule was a PR schedule of oxycodone self-administration (available on the left switch). The first response requirement under the PR schedule was 20, and each available oxycodone injection was determined by the formula of Richardson and Roberts [28]; there was a 10-sec TO after each injection. Sessions ended after a maximum 30 injections, the 30-min limited hold (LH) expired, or 4 h elapsed. The LH corresponds to the maximum time allotted for monkeys to receive an oxycodone injection. The number of injections earned in the session was the breakpoint (BP). After determining an oxycodone dose-response curve (0.001-0.17 mg/kg/injection) in each monkey, with doses tested in random order and for at least 5 consecutive sessions, acute drug pretreatment studies were conducted at the peak of the oxycodone dose-response curve (i.e., highest BP). During this, (±)VK4-40 (0.3–5.6 mg/kg, i.v.) was administered 5-min prior to the start of the session, typically on Tuesdays and Fridays, if responding was stable. Stability was defined as oxycodone injections not varying by more than 20% of the 3-day mean at each dose. For comparison, single determinations with naltrexone (0.003–0.03 mg/kg) were also evaluated.
Experiment 4
Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone-induced antinociception. Drug interactions were examined in a warm-water tail withdrawal procedure as previously described [27]. Rhesus monkeys were trained to comfortably sit in an acrylic chair using the pole-and-collar technique. The subject’s tail was shaved 10–12 cm from the distal end weekly and immersed in a thermal container containing warm water. If the subject did not remove its tail by 20 s, the experimenter removed the animal’s tail and a latency of 20 sec was assigned. All latencies were recorded using a stopwatch. During each 15-min cycle, tail-withdrawal latencies were recorded from water warmed to 38 °C, 50 °C, and 54 °C. In each successive cycle, the presentation of the warm-water stimulus was randomized. Baseline latencies at each temperature were determined before each test session and test sessions only continued if latencies at 38 °C did not occur before the 20-s cutoff. This criterion was met in all monkeys during every test session. (±)VK4-116 (10 mg/kg) and vehicle (250 mg/mL beta-cyclodextrin) were administered intravenously as a 60-min pretreatment through a temporary catheter in a saphenous vein. In addition, 1.0 and 3.2 mg/kg (±)VK4-40 and vehicle were also administered intravenously as a 5-min pretreatment through a temporary catheter. Pilot experiments demonstrated these were the maximal tolerated (±)VK4-116 and (±)VK4-40 doses in these monkeys. Larger doses were evaluated in a single monkey and resulted in overt sedation and ataxia to a sufficient degree that precluded further testing in all monkeys. Naltrexone (0.032 mg/kg) was administered intramuscularly (i.m.) as a 30-min pretreatment. Oxycodone (0.01–3.2 mg/kg, IM) was administered using a cumulative dosing procedure consisting of five to six 15-min cycles composed of a 10-min drug pretreatment phase and a 5-min testing phase. Each drug dose increased the total cumulative oxycodone dose by one-half log units. Tail-withdrawal latencies were re-determined during the 5-min testing phase as described above. (±)VK4-116 and vehicle pretreatments were counter-balanced across monkeys, such that two monkeys received (±)VK4-116 first and one monkey received the vehicle first. (±)VK4-40 and vehicle pretreatment effects were determined next and also counterbalanced. Naltrexone effects were determined last in all monkeys. Doses were only tested once in each monkey due to limited drug supply and test sessions were conducted once per week.
Data analysis
Experiments 1 and 2: The primary dependent variable was response rate. Using a 3-day mean, initial dose-effect functions were determined for each monkey. Following this, dose-effect functions for naltrexone, (±)VK4-40, and (±)VK4-116 were analyzed when tested at the oxycodone dose that resulted in peak response rates and at an oxycodone dose on the descending limb of the baseline oxycodone dose-response curve. To be included in the dose-effect function and in statistical analyses, a test must have been double determined. Dose-effect curves were analyzed using a mixed-effect analysis of variance (ANOVA) with dose and sex as the main factors. For each animal, response rates at the dose of drug (naltrexone, (±)VK4-40, and (±)VK4-116) that produced the greatest effect, either an increase or decrease from baseline, were used in the analysis. A significant ANOVA was followed by paired t-tests to compare response rates on the peak at baseline and with the test drug, as well as to compare response rates on the descending limb at baseline and with the test drug. Mauchly’s Test of Sphericity was used to indicate if the assumption of sphericity was violated and if it was, the Greenhouse-Geisser Correction was applied. If there was no statistically significant result (i.e., dose, sex, and interaction were non-significant), a one-way repeated measures ANOVA was performed using combined male and female data (n = 8 in Experiment 1 and n = 6 in Experiment 2). All statistical tests were analyzed with SPSS. Any dose of drug that resulted in an adverse event (AE) was excluded from data analysis.
Experiment 3
The primary dependent variable was the number of drug injections as a function of dose for both males and females. The effects of (±)VK4-40 (0.3-5.6 mg/kg) on peak oxycodone BP were analyzed using a one-way repeated measures ANOVA using combined male and female data. A significant ANOVA was followed by pairwise multiple comparisons (Holm-Sidak) post-hoc tests to compare baseline oxycodone injections and pretreatment conditions. Furthermore, the effect of each pretreatment on food-maintained responding was analyzed using a one-way repeated measures ANOVA. Because only single determinations of a limited dose range of naltrexone (0.003-0.03 mg/kg) were evaluated, statistical analyses were not conducted. Any dose of drug that resulted in an adverse event (AE) was excluded from data analysis.
Experiment 4
Raw tail withdrawal latencies were converted to percent maximal possible effect (% MPE) for all analyses. % MPE was defined as [(Test latency – Control latency) divided by (20 – Control latency) * 100] where “test latency” was the latency from 50 °C or 54 °C water at each dose during the cumulative dosing procedure, and “control latency” was the latency from 50 °C or 54 °C water taken during the baseline period prior to oxycodone administration. Statistical analyses of tail withdrawal data were conducted using a repeated-measures two-way ANOVA with pretreatment and oxycodone dose as the main effects.
Additionally, the effective dose (ED50) that produced 50% MPE under each pretreatment condition at 50 °C water was determined by interpolation when only 2 data points were available (one below and one >50% effect) or by linear regression when at least 3 data points on the linear portion of the dose-effect function were available. Individual ED50 values were subsequently averaged to yield group mean ED50 values. ED50 values were considered to be significantly different if the 95% confidence intervals did not overlap.
For all statistical analyses, significance was set at an alpha of 0.05.
Drugs
(-)-Oxycodone HCl and (-)-naltrexone were supplied by the National Institute on Drug Abuse (Bethesda, MD) and dissolved in 0.9% saline. (±)VK4-116 and (±)VK4-40 were synthesized by Ms. Jianjing Cao in the Medicinal Chemistry Section, NIDA-Intramural Research Program, as described in Kumar et al. [13], and dissolved in 25% beta-cyclodextrin (CAS: 128446-35-5; Acros Organics, New Jersey) and sterile water. Heat and sonication were used for solubility purposes and all drug solutions were passed through a sterile 0.22 μm filter (Millipore Sigma, Burlington, MA) for intravenous administration. This route was chosen to assure accurate dosage and shorter pretreatment times. All drug doses are expressed as the salt form. For self-administration studies in Experiment 1, oxycodone doses were based on 3-sec pump duration for females and 10-sec for males. In Experiment 3, the pump duration was 10-sec for all four monkeys.
Results
Experiment 1
Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone self-administration under a fixed-ratio schedule of reinforcement. Oxycodone self-administration varied as a function of dose in female and male monkeys [F(1.09,6.58) = 7.32, p = 0.031], with no sex differences noted [F(1.09,6.58) = 0.948, p > 0.05]. Although there was evidence of individual-subject variability in response rates in both females and males, all monkeys’ oxycodone self-administration dose-response curves represented an inverted-U shaped function of dose; there were no sex differences noted [all p > 0.05] in the effects of any of these pretreatments on oxycodone self-administration (Supplemental Fig. S1-S3).
When tested at the dose of oxycodone that maintained peak rates of responding, naltrexone (Fig. 1A) dose-dependently decreased responding in all monkeys [t(7) = 2.67, p = 0.031]. In the females, the naltrexone doses used in Fig. 1A were 0.01-0.1 mg/kg; for the males, the doses were 0.003-0.056 mg/kg (Supplemental Fig. S1). When tested with a dose of oxycodone on the descending limb (Fig. 1A), naltrexone increased response rates [t(7) = −3.01, p = 0.020], shifting the oxycodone dose-response curve to the right. The D3R antagonist ±VK4-116 (1.0–10 mg/kg) did not affect oxycodone self-administration [F(3,4) = 1.50, p > 0.05] at either the peak oxycodone dose or in combination with a dose on the descending limb of the dose-response curve (Fig. 1B and Supplemental Fig. S2). In contrast, the D3R partial agonist (±)VK4-40 (Fig. 1C and Supplemental Fig. S3) significantly reduced oxycodone response rates when tested at the dose of oxycodone that maintained peak rates of responding [t(7) = 2.63, p = 0.034]. In the females, the (±)VK4-40 doses used in Fig. 1C were 1.0–5.6 mg/kg; for the males, the doses were 3.0–10 mg/kg. However, (±)VK4-40 had no effect on response rates when tested with a dose on the descending limb of the oxycodone dose-response curve [t(7) = 0.196, p > 0.05].
Fig. 1. Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone self-administration under a fixed-ratio schedule of reinforcement.

This figure shows the effects of (A) naltrexone (NLX, striped bars), (B) ±VK4-116 (gray bars), and (C) (±)VK4-40 (white bars) on oxycodone self-administration (black bars) under an FR schedule of reinforcement. Each bar represents mean (N = 8) ± the standard error of the mean (SEM). *p < 0.05; **p < 0.01.
Experiment 2
Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone-induced reinstatement. When saline was substituted for oxycodone, responding extinguished (i.e., decreased by at least 80% of baseline for 3 consecutive sessions) typically within 5–7 sessions. Oxycodone (0.1 or 0.17 mg/kg, i.v.) reinstated oxycodone seeking in all monkeys; there was no evidence of sex differences, so data for females and males were averaged (Fig. 2, filled bar; F(3,3) = 56.83, p = 0.004). Similarly, for the pretreatment studies, there were no significant effects of sex on response rates (p > 0.05), so data for the females and males (N = 3/sex) were combined (Fig. 2). Doses that produced the largest effect were 0.01-0.1 mg/kg naltrexone, 3.0–10 mg/kg (±)VK4-116 and 5.6 mg/kg (±)VK4-40, for females and males. Both naltrexone (p < 0.001) and (±)VK4-40 (p < 0.001), but not (±)VK4-116, significantly attenuated the effects of 0.1 or 0.17 mg/kg oxycodone-induced reinstatement (Fig. 2, striped and open bars, respectively).
Fig. 2. Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone reinstatement.
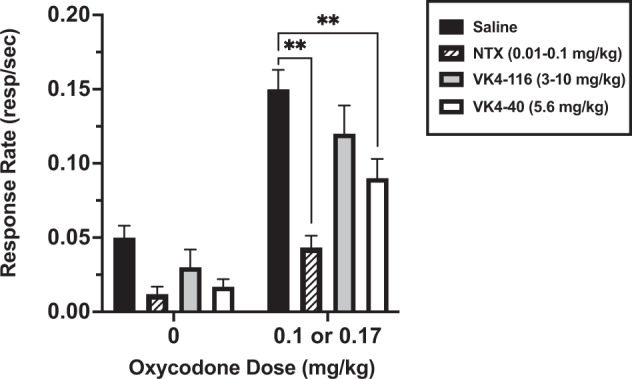
This figure shows the effects of naltrexone (NTX; striped bars), (±)VK4-116 (gray bars), and (±)VK4-40 (white bars) on oxycodone reinstatement (black bars). The oxycodone dose was 0.1 mg/kg in all monkeys, except F-7453, in which 0.17 mg/kg oxycodone was used. Each bar represents the mean (N = 6) ± standard error of the mean (SEM). *p < 0.05; **p < 0.01.
Experiment 3
Effects of (±)VK4-40 on oxycodone self-administration under a progressive-ratio schedule of reinforcement. Oxycodone self-administration under a PR schedule of reinforcement varied with dose and all monkeys’ oxycodone dose-response curves (n = 4) represented an inverted-U shaped function (Supplemental Fig. S4). Peak BPs were at 0.01 and 0.1 mg/kg/injection oxycodone. The doses of (±)VK4-40 used for statistical analyses ranged from 3.0–5.6 mg/kg. Statistical analyses revealed a significant main effect of treatment [F(2,2) = 69.45, p = 0.014] and post-hoc analyses showed that (±)VK4-40 (Fig. 3) significantly reduced the number of oxycodone injections received when compared to baseline levels (p = 0.003). In addition, at these doses, (±)VK4-40 had no significant effect on food-maintained responding (data not shown) [F(2,2) = 1.00, p = 0.5]. As a comparison, naltrexone was also tested and decreased peak PR responding (Fig. 3 and Supplemental Fig. S4). Neither naltrexone nor VK4-40 had any effect on food-maintained responding (data not shown) [F(2,6) = 0.429, p = 0.670].
Fig. 3. Effects of (±)VK4-40 on oxycodone self-administration under a PR schedule of reinforcement.
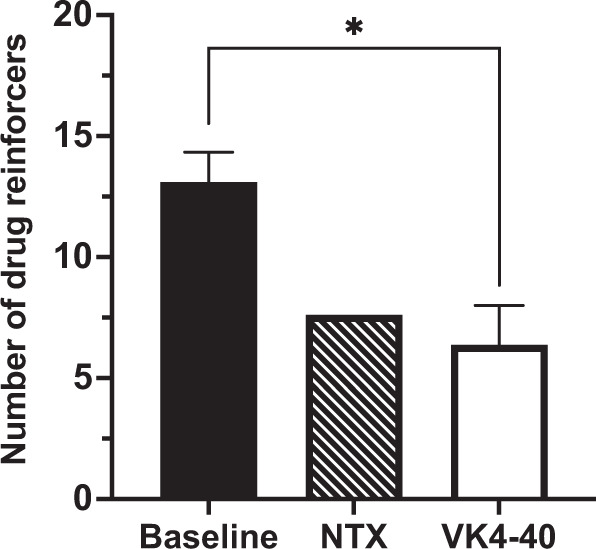
This figure depicts the effects of naltrexone (0.01–0.03 mg/kg; striped bar) and (±)VK4-40 (3.0–5.6 mg/kg; white bar) on the peak number of oxycodone injections (0.01–0.1 mg/kg; black bar) in monkeys responding under a PR schedule of reinforcement Each bar represents the mean (N = 4) ± standard error of the mean (SEM). *p < 0.05; **p < 0.01.
Experiment 4
Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone-induced antinociception. Baseline mean ± SEM tail withdrawal latencies were 0.70 ± 0.1 and 0.60 ± 0.1 s for 50 °C and 54 °C, respectively. Oxycodone produced dose-dependent antinociception at both 50 °C Fig. 4A [F(1, 2.1) = 123.3, p = 0.0007] and Fig. 4B [F(1.1, 2.3) = 32.2, p = 0.023] and 54 °C Fig. 4C [F(1, 2) = 62.0, p = 0.015] warmed water. Following either 60-min or 5-min vehicle pretreatment, the group mean oxycodone ED50 values at 50 °C were 0.17 mg/kg and 0.14 mg/kg, respectively (Table 1). Pretreatment with (±)VK4-116 (60-min) or (±)VK4-40 (5-min or 60-min) did not significantly alter oxycodone antinociception following ANOVAs (Fig. 4A, B); however, (±)VK4-116 pretreatment did enhance oxycodone antinociceptive potency based on non-overlapping confidence limits (Table 1). In contrast, naltrexone produced an approximate 10-fold potency shift in the oxycodone ED50 value at 50 °C (Table 1).
Fig. 4. Effects of naltrexone, (±)VK4-116, and (±)VK4-40 on oxycodone-induced antinociception.

This figure demonstrates the antinociceptive effects of oxycodone in an assay of thermal nociception at 50 °C and 54 °C in male rhesus monkeys following either vehicle, (±)VK4-116, or (±)VK4-40. Top panels show effects of vehicle, 10 mg/kg (±)VK4-116 (60-min) pretreatment; (A) or 1–3.2 mg/kg (±)VK4-40 (5-min) pretreatment; (B) administered intravenously (IV) to oxycodone cumulative dosing at 50 °C. Bottom panels show vehicle, (±)VK4-116 (C), or (±)VK4-40 (D) pretreatment effects to oxycodone cumulative dosing at 54 °C thermal intensity. Abscissae: cumulative intramuscular oxycodone dose (milligrams per kilogram). Ordinates: % maximum possible effect (%MPE). Each point represents the mean (N = 3) ± standard error of the mean (SEM).
Table 1.
Individual and group mean ED50 values (95% confidence limits) for oxycodone following either a 60-min or 5-min pretreatment of vehicle, (±)VK4-116, or (±)VK4-40 in an assay of thermal nociception at 50 °C.
| 50 °C (60-min for vehicle, (±)VK4-116 and (±)VK4-40) | M1473 ED50 | M1503 ED50 | M1414 ED50 | Group ED50 values (95% CL) |
|---|---|---|---|---|
| Vehicle | 0.17 | 0.17 | 0.16 | 0.17 (0.16, 0.18) |
| (±)VK4-116 (10 mg/kg, IV) | 0.11 | 0.06 | 0.11 | 0.09 (0.06, 0.14) |
| (±)VK4-40 (3.2 mg/kg, IV) | 0.06 | 0.06 | > | >0.06 (0.06, 0.06) |
| Naltrexone (0.032 mg/kg, IM) | 2.26 | 1.34 | 2.26 | 1.9 (1.35, 2.67) |
| 50 °C (5-min for vehicle and (±)VK4-40) | ||||
| Vehicle | 0.06 | 0.17 | 0.3 | 0.14 (0.06, 0.38) |
| (±)VK4-40 (1 mg/kg, IV) | 0.17 | 0.18 | 0.26 | 0.2 (0.15, 0.26) |
| (±)VK4-40 (3.2 mg/kg, IV) | 0.1 | 0.06 | 0.56 | 0.15 (0.04, 0.57) |
> denotes an ED50 value could not be calculated because no oxycodone dose produced >50%MPE.
Naltrexone was administered as a 30-min pretreatment.
Discussion
The goal of the present study was to evaluate the effect of two dopamine D3 receptor-selective compounds on several behaviors related to oxycodone misuse in translational nonhuman primate models of OUD. Importantly, binding affinities, selectivity and functional efficacies of (±)VK4-116 and (±)VK4-40 were previously measured in vitro in human D3R-transfected cell based assays [13]. Thus, identifying a pharmacological profile that best translated behaviors from rodent to nonhuman primate models of OUD was desirable. The efficacy of the opioid receptor antagonist naltrexone, an FDA-approved treatment for opioid use disorder, was also assessed for comparison. The present findings demonstrate that under an FR schedule of reinforcement, naltrexone decreased the reinforcing potency of oxycodone as demonstrated by rightward shifts in the oxycodone dose-response curves. Although the D3R antagonist (±)VK4-116 did not affect oxycodone self-administration under an FR schedule of reinforcement, the D3R partial agonist (±)VK4-40 significantly reduced oxycodone response rates when tested at the dose of oxycodone that maintained peak rates of responding. Moreover, both naltrexone and (±)VK4-40, but not (±)VK4-116, attenuated the effects of oxycodone-induced reinstatement. Given these findings, the effects of (±)VK4-40 on oxycodone self-administration were also tested in monkeys responding under a PR schedule of reinforcement and significantly reduced the reinforcing strength of oxycodone. Importantly, (±)VK4-40 did not attenuate food-maintained responding or oxycodone-induced antinociception, suggesting that the acute effects of this D3R partial agonist were selective for oxycodone reinforcement. Together, these studies suggest that D3R-selective partial agonists, specifically (±)VK4-40, may attenuate the abuse liability of opioids without affecting their clinical utility in pain management and warrant further study, including chronic dosing conditions.
Previous studies in rodents responding under an FR schedule of reinforcement found that (±)VK4-116 dose-dependently reduced oxycodone self-administration and oxycodone-induced reinstatement [8, 14]. Moreover, (±)VK4-116 was shown to enhance the antinociceptive effects of oxycodone, as well as reduce withdrawal-induced hyperalgesia and irritability in rats [8, 14]. In the present nonhuman primate study, (±)VK4-116 did not attenuate oxycodone self-administration under an FR schedule of reinforcement, nor did it block oxycodone-induced reinstatement. (±)VK4-116 pretreatment slightly enhanced the antinociceptive potency of oxycodone in monkeys in the present study, consistent with and extending these previous rodent studies. Potential explanations for the differential (±)VK4-116 results in rodents vs. monkeys is discussed below.
One study in rodents examining R-VK4-40 did correspond to the findings of the present study where systemic administration of R-VK4-40 dose-dependently decreased FR oxycodone self-administration and shifted the oxycodone dose-effect curve downward [15]. It must be noted that the R-enantiomer of VK4-40 is a selective D3R antagonist, like (±)VK4-116, used in the present study. Nevertheless, (±)VK4-40 was shown to dose-dependently block cocaine self-administration and reinstatement in rats [16], further supporting the effectiveness of D3R-selective partial agonists in models of SUD but also highlighting potential species differences.
Recently, Galaj et al. (2022) examined the effects of (S)- and (R)-enantiomers of (±)-ABS01-113, two structural analogs of (±)-VK4-40, in several preclinical models of OUD, including in heroin self-administration and reinstatement [18]. In in vitro functional assays, it was reported that (R)-ABS01-113 was a D3R antagonist, while (S)-ABS01-113 was a D3R partial agonist [18, 29]. Importantly, both the (S)- and (R)-ABS01-113 decreased heroin self-administration, and heroin + cue-induced reinstatement, suggesting that the D3R partial agonists are potentially viable as therapeutics for SUD but also may be useful in the treatment of comorbid affective disorders.
The present study extended the previous rodent findings to nonhuman primates and the reinforcing strength of oxycodone under a PR schedule of reinforcement. Although FR schedules of reinforcement are primarily used for abuse liability assessment, PR schedules provide crucial information regarding reinforcer strength [30, 31]. Earlier rodent studies examined the effectiveness of the D3R antagonists (±)VK4-116 and R-VK4-40 on oxycodone self-administration using a PR schedule of reinforcement and reported that both compounds significantly decreased oxycodone breakpoints [8, 15]. Because (±)VK4-116 did not significantly affect FR oxycodone self-administration in the nonhuman primates in this study, we only examined the effect of (±)VK4-40 on oxycodone self-administration under a PR schedule of reinforcement. Using a within-subjects design, we showed that (±)VK4-40 decreased oxycodone self-administration under both the FR vs. PR schedules of reinforcement (see Supplemental Figs. S3 and S4). Furthermore, acute (±)VK4-40 did not significantly alter food-maintained responding or oxycodone thermal antinociception, demonstrating a degree of behavioral selectivity.
There are several possible explanations for the discrepancy between the efficacy of (±)VK4-116 in rodent models compared to the present study in nonhuman primates. There are noted species differences in D3R distribution [32]. In fact, studies have shown that while humans and nonhuman primates have low D3R levels in the dorsal striatum, a brain region implicated in regulating goal-directed and habitual behaviors, rodent dorsal striatum is devoid of D3Rs [33, 34]. Additionally, in humans and nonhuman primates, there is a larger distribution and higher density of D3R in the cerebral cortex when compared to rodents [35]. This brain region has been implicated in response inhibition and salience attribution, both of which are disrupted during substance misuse [36]. However, lower D3R densities in key brain regions in rodent models cannot be the primary reason for the discrepancy in (±)VK4-116 effects, since rodents appear more sensitive than nonhuman primates and humans. Similarly, different D3R distributions do not explain why (±)VK4-40 is efficacious in both species. It is important to note that (±)VK4-40 has ~20-fold higher affinity for human D3R as well as ~75-fold higher affinity for human D2R when compared with (±)VK4-116, suggesting higher affinity is advantageous in reducing oxycodone self-administration in nonhuman primates [13]. Further investigation of cell-based binding affinity, selectivity and functional data to in vivo potency and efficacy across species will be required. It is also possible that the differences in drug history, particularly total opioid use which is substantially higher in nonhuman primate studies, resulted in changes in D3R function and density that may account for these species differences. Determining the most effective pharmacological profile will inform future nonopioid medication development for successful treatment of patients with OUD.
The present study also examined whether there were sex differences in the behavioral effects of both D3R compounds. As noted earlier, epidemiological data suggest that women are at a greater risk for opioid misuse [37]. Cicero et al. found that opioids had reinforcing effects over a broader dose range in women compared with men [24]. When examining reinforcing effects under the FR scheduled of reinforcement, the dose of oxycodone that maintained peak rates was 0.01 mg/kg/injection in all four male monkeys and two of four female monkeys; for the other two females, higher doses were associated with peak response rates (0.03 and 0.056 mg/kg/injection). We found no evidence of sex differences in the potency or efficacy of both D3R compounds, which contrasts to another study from our laboratory showing the D2/D3R agonist quinpirole was more potent at eliciting yawning in male monkeys compared with female monkeys [38]. Whether sex differences are associated with D3R function (full agonist vs. partial agonist or antagonist) or unconditioned (e.g., drug-induced yawning) vs. conditioned (e.g., self-administration) behaviors remains to be determined.
Overall, the present findings further support the premise that D3R partial agonists may effectively reduce oxycodone reinforcement without affecting opioid-related antinociception, as determined in the warm water tail withdrawal assay. Future research should extend these findings to preclinical oxycodone-choice self-administration procedures and evaluate effectiveness of chronic, orally administered (±)VK4-40; administration of R-VK4-40 by that route has been shown to be behaviorally active in rodent models [14], but has not been done in nonhuman primates. Additionally, future research determining (±)VK4-40 and opioid interactions on pain-depressed behaviors (e.g., [39]) would also facilitate preclinical-to-clinical translatability.
Supplementary information
Acknowledgements
We would like to thank Michael Coller, Jillian H. Odom, Christopher Haggerty and Jianjing Cao for technical assistance.
Author contributions
KW, MLB, AHN and MAN designed the experiment. KW, JCC conducted experiments. MIA conducted data analyses. KW, MIA, MLB, AHN and MAN wrote the manuscript. All authors reviewed the manuscript.
Funding
This research was supported by DA017763, DA06634, DA037287, T32DA007027 and the NIDA Intramural Research Program Z1ADA000424.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kendall Woodlief, Mia I. Allen.
Supplementary information
The online version contains supplementary material available at 10.1038/s41386-023-01590-8.
References
- 1.Kelly RJ, Hearld LR. Service profiles in substance use disorder treatment: are treatment facilities consistent in service mix and service offerings? J Stud Alcohol Drugs. 2022;83:374–83. doi: 10.15288/jsad.2022.83.374. [DOI] [PubMed] [Google Scholar]
- 2.Lipari, RN and Van Horn, SL. Trends in substance use disorders among adults aged 18 or older. The CBHSQ Report: June 29, 2017. Center for Behavioral Health Statistics and Quality, Substance Abuse and Mental Health Services Administration, Rockville, MD. [PubMed]
- 3.Stephenson J. CDC warns of surge in drug overdose deaths during COVID-19. JAMA Health Forum. 2021;2:e210001–e210001. doi: 10.1001/jamahealthforum.2021.0001. [DOI] [PubMed] [Google Scholar]
- 4.Weiner SG, Baker O, Bernson D, Schuur JD. One-year mortality of patients after emergency department treatment for nonfatal opioid overdose. Ann Emerg Med. 2020;75:13–17. doi: 10.1016/j.annemergmed.2019.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stringfellow EJ, Lim TY, DiGennaro C, Zhang Z, Paramasivam P, Bearnot B et al. Long-term effects of increasing buprenorphine treatment seeking, duration, and capacity on opioid overdose fatalities: a model-based analysis. J Addict Med. 9900. 10.1097/ADM.0000000000001153. [DOI] [PMC free article] [PubMed]
- 6.Shipton EA, Shipton EE, Shipton AJ. A review of the opioid epidemic: what do we do about it? Pain Ther. 2018;7:23–36. doi: 10.1007/s40122-018-0096-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Volkow ND, Collins FS. The role of science in addressing the opioid crisis. N. Engl J Med. 2017;377:391–4. doi: 10.1056/NEJMsr1706626. [DOI] [PubMed] [Google Scholar]
- 8.You Z-B, Bi G-H, Galaj E, Kumar V, Cao J, Gadiano A, et al. Dopamine D3R antagonist VK4-116 attenuates oxycodone self-administration and reinstatement without compromising its antinociceptive effects. Neuropsychopharmacology. 2019;44:1415–24. doi: 10.1038/s41386-018-0284-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Newman AH, Xi Z-X and Heidbreder C. Current perspectives on selective dopamine D3 receptor antagonists/partial agonists as pharmacotherapeutics for opioid and psychostimulant use disorders. Curr Top Behav Neurosci. 2023;60:157–201. [DOI] [PMC free article] [PubMed]
- 10.Sokoloff P, Le B. Foll, The dopamine D3 receptor, a quarter century later. Eur J Neurosci. 2017;45:2–19. doi: 10.1111/ejn.13390. [DOI] [PubMed] [Google Scholar]
- 11.Appel NM, Li S-H, Holmes TH, Acri JB. Dopamine D3 receptor antagonist (GSK598809) potentiates the hypertensive effects of cocaine in conscious, freely-moving dogs. J Pharmacol Exp Ther. 2015;354:484–92. doi: 10.1124/jpet.115.224121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Keck TM, John WS, Czoty PW, Nader MA, Newman AH. Identifying medication targets for psychostimulant addiction: unraveling the dopamine D3 receptor hypothesis. J Med Chem. 2015;58:5361–80. doi: 10.1021/jm501512b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar V, Bonifazi A, Ellenberger MP, Keck TM, Pommier E, Rais R, et al. Highly selective dopamine D3 receptor (D3R) antagonists and partial agonists based on eticlopride and the D3R crystal structure: new leads for opioid dependence treatment. J Med Chem. 2016;59:7634–50. doi: 10.1021/acs.jmedchem.6b00860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Guglielmo G, Kallupi M, Sedighim S, Newman AH, George O. Dopamine D3 receptor antagonism reverses the escalation of oxycodone self-administration and decreases withdrawal-induced hyperalgesia and irritability-like behavior in oxycodone-dependent heterogeneous stock rats. Front Behav Neurosci. 2020;13:292. doi: 10.3389/fnbeh.2019.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jordan CJ, Humburg B, Rice M, Bi G-H, You Z-B, Shaik AB, et al. The highly selective dopamine D3R antagonist, R-VK4-40 attenuates oxycodone reward and augments analgesia in rodents. Neuropharmacology. 2019;158:107597. doi: 10.1016/j.neuropharm.2019.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jordan CJ, He Y, Bi GH, You ZB, Cao J, Xi ZX, et al. (±) VK4‐40, a novel dopamine D3 receptor partial agonist, attenuates cocaine reward and relapse in rodents. Br J Pharmacol. 2020;177:4796–807. doi: 10.1111/bph.15244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doyle MR, Peng LN, Cao J, Rice KC, Newman AH, Collins GT. 3, 4-methylenedioxypyrovalerone high-responder phenotype as a tool to evaluate candidate medications for stimulant use disorder. J Pharmacol Exp Ther. 2023;384:353–62. doi: 10.1124/jpet.122.001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galaj E, Bi G-H, Klein B, Hempel B, Shaik AB, Gogarnoiu ES, et al. A highly D3R-selective and efficacious partial agonist (S)-ABS01-113 compared to its D3R-selective antagonist enantiomer (R)-ABS01-113 as potential treatments for opioid use disorder. Neuropsychopharmacology. 2022;47:2309–18. doi: 10.1038/s41386-022-01379-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nunes EV, Gordon M, Friedmann PD, Fishman MJ, Lee JD, Chen DT, et al. Relapse to opioid use disorder after inpatient treatment: protective effect of injection naltrexone. J Subst Abus Treat. 2018;85:49–55. doi: 10.1016/j.jsat.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minhas M, Leri F. A multifaceted analysis of oxycodone addiction. Int J Ment Health Addiction. 2018;16:1016–32. doi: 10.1007/s11469-017-9827-y. [DOI] [Google Scholar]
- 21.Becker JB, McClellan ML, Reed BG. Sex differences, gender and addiction. J Neurosci Res. 2017;95:136–47. doi: 10.1002/jnr.23963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maria MM, Flanagan J, Brady K. Ovarian hormones and drug abuse. Curr psychiatry Rep. 2014;16:1–8. doi: 10.1007/s11920-014-0511-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Back SE, Payne RL, Wahlquist AH, Carter RE, Stroud Z, Haynes L, et al. Comparative profiles of men and women with opioid dependence: results from a national multisite effectiveness trial. Am J drug alcohol Abus. 2011;37:313–23. doi: 10.3109/00952990.2011.596982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cicero TJ, Aylward SC, Meyer ER. Gender differences in the intravenous self-administration of mu opiate agonists. Pharmacol Biochem Behav. 2003;74:541–9. doi: 10.1016/S0091-3057(02)01039-0. [DOI] [PubMed] [Google Scholar]
- 25.Townsend EA, Negus SS, Caine SB, Thomsen M, Banks ML. Sex differences in opioid reinforcement under a fentanyl vs. food choice procedure in rats. Neuropsychopharmacology. 2019;44:2022–9. doi: 10.1038/s41386-019-0356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Comer SD, Cooper ZD, Kowalczyk WJ, Sullivan MA, Evans SM, Bisaga AM, et al. Evaluation of potential sex differences in the subjective and analgesic effects of morphine in normal, healthy volunteers. Psychopharmacology. 2010;208:45–55. doi: 10.1007/s00213-009-1703-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cornelissen JC, Obeng S, Rice KC, Zhang Y, Negus SS, Banks ML. Application of receptor theory to the design and use of fixed-proportion mu-opioid agonist and antagonist mixtures in rhesus monkeys. J Pharmacol Exp Ther. 2018;365:37–47. doi: 10.1124/jpet.117.246439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richardson NR, Roberts DCS. Progressive ratio schedules in drug self-administration studies in rats: a method to evaluate reinforcing efficacy. J Neurosci Methods. 1996;66:1–11. doi: 10.1016/0165-0270(95)00153-0. [DOI] [PubMed] [Google Scholar]
- 29.Shaik AB, Kumar V, Bonifazi A, Guerrero AM, Cemaj SL, Gadiano A, et al. Investigation of novel primary and secondary pharmacophores and 3-substitution in the linking chain of a series of highly selective and bitopic dopamine D3 receptor antagonists and partial agonists. J Medicinal Chem. 2019;62:9061–77. doi: 10.1021/acs.jmedchem.9b00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woolverton WL and Nader MA. Experimental evaluation of the reinforcing effects of drugs. In M. W. Adler, A. Cowan (Eds.), Testing and evaluation of drugs of abuse. John Wiley & Sons; pp. 165–192, 1990.
- 31.Panlilio LV, Goldberg SR. Self‐administration of drugs in animals and humans as a model and an investigative tool. Addiction. 2007;102:1863–70. doi: 10.1111/j.1360-0443.2007.02011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levant B. Differential distribution of D3 dopamine receptors in the brains of several mammalian species. Brain Res. 1998;800:269–74. doi: 10.1016/S0006-8993(98)00529-0. [DOI] [PubMed] [Google Scholar]
- 33.Nakajima S, Gerretsen P, Takeuchi H, Caravaggio F, Chow T, Le Foll B, et al. The potential role of dopamine D3 receptor neurotransmission in cognition. Eur Neuropsychopharmacol. 2013;23:799–813. doi: 10.1016/j.euroneuro.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yager LM, Garcia AF, Wunsch AM, Ferguson SM. The ins and outs of the striatum: role in drug addiction. Neuroscience. 2015;301:529–41. doi: 10.1016/j.neuroscience.2015.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sokoloff P, Diaz J, Foll BL, Guillin O, Leriche L, Bezard E, et al. The dopamine D3 receptor: a therapeutic target for the treatment of neuropsychiatric disorders. CNS Neurol Disord-Drug Targets (Former Curr Drug Targets-CNS Neurol Disord) 2006;5:25–43. doi: 10.2174/187152706784111551. [DOI] [PubMed] [Google Scholar]
- 36.Goldstein RZ, Volkow ND. Dysfunction of the prefrontal cortex in addiction: neuroimaging findings and clinical implications. Nat Rev Neurosci. 2011;12:652–69. doi: 10.1038/nrn3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koons AL, Greenberg MR, Cannon RD, Beauchamp GA. Women and the experience of pain and opioid use disorder: a literature-based commentary. Clin Ther. 2018;40:190–6. doi: 10.1016/j.clinthera.2017.12.016. [DOI] [PubMed] [Google Scholar]
- 38.Martelle SE, Nader SH, Czoty PW, John WS, Duke AN, Garg PK, et al. Further characterization of quinpirole-elicited yawning as a model of dopamine D3 receptor activation in male and female monkeys. J Pharmacol Exp Ther. 2014;350:205–11. doi: 10.1124/jpet.114.214833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moerke MJ, Negus SS, Banks ML. Lack of effect of the nociceptin opioid peptide agonist Ro 64-6198 on pain-depressed behavior and heroin choice in rats. Drug Alcohol Depend. 2022;231:109255. doi: 10.1016/j.drugalcdep.2021.109255. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.