

Summary
Neutrophil swarming is a complex coordinated process in which neutrophils sensing pathogen or damage signals are rapidly recruited to sites of infections or injuries. This process involves cooperation between neutrophils where autocrine and paracrine positive-feedback loops, mediated by receptor/ligand pairs including lipid chemoattractants and chemokines, amplify localized recruitment of neutrophils. This review will provide an overview of key pathways involved in neutrophil swarming and then discuss the cell intrinsic and systemic mechanisms by which NADPH oxidase 2 (NOX2) regulates swarming, including modulation of calcium signaling, inflammatory mediators, and the mobilization and production of neutrophils. We will also discuss mechanisms by which altered neutrophil swarming in disease may contribute to deficient control of infections and/or exuberant inflammation. Deeper understanding of underlying mechanisms controlling neutrophil swarming and how neutrophil cooperative behavior can be perturbed in the setting of disease may help to guide development of tools for diagnosis and precision medicine.
Subject areas: Immunology, Molecular biology
Graphical abstract
Immunology; Molecular biology
Introduction
Neutrophils are the most abundant white blood cells in humans and are in the first line of host defense. In addition to fighting infections and participating in tissue repair, neutrophils are also involved in inflammatory and autoimmune diseases and cancer.1,2,3 Neutrophils are produced and stored in the bone marrow and originate from hematopoietic stem and progenitor cells (HSCPs). Under the influence of both extracellular and intracellular molecules, such as cytokines and transcription factors, HSCPs develop into neutrophils following lineage determination and committed granulopoiesis.1,3,4 Neutrophils have a short half-life and are continuously generated and released into the peripheral blood at steady state. In the setting of acute infection or inflammation, the hematopoietic system switches from steady state to demand, or “emergency,” granulopoiesis to adapt to the increased demand for neutrophils.5 Neutrophils have lobulated nuclei, giving them flexibility to transmigrate through the endothelium and into the tissue interstitium. Neutrophils are recruited to the inflammatory sites via a multiple-step process mediated by their adhesion and migration guided by chemokines, lipid chemoattractants, complement factors, formyl-peptides and their relevant receptors.1,3,6,7 Once neutrophils are recruited to the site of inflammation, they clear pathogens through phagocytosis, degranulation, the release of reactive oxygen species (ROS) and neutrophil extracellular traps (NETs).1,3 Neutrophil activation and migration are enhanced through autocrine and paracrine feedback amplification.6 Neutrophils can communicate with other cells, including immune cells and non-immune cells, through the release of cytokines, signaling molecules or exosomes.1,2,6,8 Recent studies have illustrated a phenomenon called neutrophil swarming, a rapid focal accumulation of neutrophils mediated by intercellular communication that generates feedforward loops to coordinate neutrophil recruitment at local sites.9,10,11,12,13,14 Neutrophil swarming has emerged as an important neutrophil response with implications for microbial infection, inflammation, immune regulation, tissue destruction and repair. The mechanisms underlying swarming have been the subject of several recent reviews.6,9,10,11,13,14 Here, we first provide an overview of the current understanding of cellular and molecular mechanisms involved in initiation and modulation of neutrophil swarming. We then highlight emerging direct and indirect evidence that show how NADPH oxidase 2 (NOX2) and related signals regulate neutrophil swarming as well as disease states potentially associated with aberrant neutrophil swarming.
Neutrophil swarming signals
Neutrophil swarming is a conserved response among species and has been studied in mice and zebrafish in vivo using intravital imaging, and in human neutrophils in vitro using engineered platforms or other settings.12,15,16,17,18,19,20 Both Pathogen Associated Molecular Patterns (PAMPs) and Damage-Associated Molecular Patterns (DAMPs) can trigger neutrophil swarming (Figure 1). Neutrophil swarming occurs widely in different tissues, such as skin, ear, liver, lung, spleen, lymph node and brain,11 suggesting that swarming is a conserved protective mechanism in the host response to tissue injury and can also facilitate pathogen containment.
Figure 1.

Initial and stop signals of neutrophil swarming
Both Pathogen Associated Molecular Patterns (PAMPs) and Damage-Associated Molecular Patterns (DAMPs) can trigger neutrophil swarming. After tissue injury or infection, pioneer neutrophils are recruited to the site of inflammation or infection. These pioneer neutrophils produce chemoattractants such as LTB4 and CXCL2, which further coordinate a feedforward loop to amplify the intercellular signaling and swarm aggregation. Calcium, complement, ATP, connexin 43 and integrins positively regulate the chemoattractants' signals during the swarming process, whereas NOX2 is a negative regulator. Desensitization of the GPCRs by GPCR kinase 2 (GRK2) is essential to stop neutrophil swarming. Lipoxin A4 (LXA4), resolvin E3 (RvE3), and ω-OH-LTB4 also contribute to the termination of neutrophil swarming.
Although much remains to be learned regarding various mechanisms regulating neutrophil swarming, studies have begun to define the initiating signals and subsequent feedback loops that coordinate this process. In the initial steps, “pioneer” neutrophils that encounter pathogens, injured or dying cells initiate a signal relay to recruit additional neutrophils to the site.9,11,12,14,15,17,21 For example, calcium alarm signals in frontline neutrophils at wound sites initiate production of the lipid mediator leukotriene B4 (LTB4) to promote neutrophil activation and recruitment.16 Feedforward loops involving intercellular communication via neutrophil-produced chemoattractants such as LTB4 and chemokines attract additional neutrophils, leading to a rapid and exponential accumulation of neutrophils at local sites (Figure 1).9,10,11,12,13,14,15,22 Neutrophil delivery into the tissues also requires a continuous supply of neutrophils from the blood stream – and neutrophil accumulation can thus be blunted if neutrophils are not available. Thus, neutrophils and other cells produce not only mediators important locally for neutrophil accumulation, but also engage those that act systemically, most notably the IL-1-G-CSF axis. This sustains neutrophil recruitment into tissues by rapidly mobilizing neutrophils into the peripheral blood from the marrow storage pool and by increasing granulopoiesis.5,23
LTB4 is indispensable in neutrophil swarming
LTB4 plays a central role in initiating neutrophil swarming.12,15,16,17 LTB4 is primarily produced by neutrophils and macrophages and is synthesized in several rate-limiting steps.24 Free arachidonic acid (AA) is generated by phospholipase A (PLA) from membrane phospholipids. AA is then metabolized to Leukotriene A4 (LTA4) by arachidonate 5-lipoxygenase (5-LO) and 5-Lipoxygenase Activating Protein (FLAP) through a two-step reaction, and LTA4 is further converted to LTB4 through LTA4 hydrolase.24,25 LTB4 and related enzymes 5-LO, FLAP and LTA4 are packaged and released in exosomes, which then work through autocrine and paracrine routes to mediate the neutrophil chemotaxis.26,27 LTB4 signaling is mainly mediated through two GPCRs, a high-affinity receptor BLT1 (LTB4R1) and a low-affinity receptor BLT2 (LTB4R2). The importance of LTB4, its receptors, and components of the synthesis pathway have been well demonstrated across species, both in vitro and in vivo. Mouse neutrophils lacking BLT1 had severely impaired swarm formation to focal laser injury.12 The size of human neutrophil swarms triggered by zymosan was also reduced in the presence of BLT1 and BLT2 antagonist U75302 and LY255283.15 5-LO translocation in neutrophils within clusters was detected by spinning-disk microscopy following acute laser wounding in zebrafish.16 CRISPR/Cas9-mediated knockdown of Blt1 and Lta4h reduced the neutrophil recruitment at the wound sites of zebrafish.17 These experiments demonstrate that LTB4/LTB4R signaling is indispensable for neutrophil swarming in different species. Of note, LTB4 can also be synthesized transcellularly. 5-LO is primarily expressed in leukocytes, but LTA4H is expressed in many cells lacking significant 5-LO activity.25 Lta4hKO neutrophils and Alox5KO neutrophils mixed together can form swarms normally in response to C. albicans, but they cannot form swarms by themselves.28 This result supports the notion of neutrophil swarming as a highly coordinated process within a group of neutrophils, not simply a sum of individual neutrophil activities.9,28 Recently, a genetically encoded biosensor was developed for visualizing LTB4 levels.29 This technology might be a powerful tool for future studies analyzing the spatiotemporal dynamics of LTB4 activity during neutrophil swarming in vivo.
Signals regulating LTB4 production affect neutrophil swarming
Given the central role of LTB4 in driving neutrophil swarming, many pathways found to regulate swarming converge at LTB4 synthesis. In a zebrafish wound model, the damage signal ATP is released via connexin-43 (Cx43) hemichannels in clustered pioneer neutrophils contacting necrotic tissue. ATP signaling through P2X1 receptors induces calcium influx (“calcium alarm signal”), which further promotes local LTB4 production in neutrophils,16 suggesting that ATP-Cx43-calcium-LTB4 signaling is important in the initial phase of neutrophil swarming. Complement-regulated LTB4 production is also important in neutrophil swarming.15,30,31,32 Mouse neutrophils incubated with C. neoformans in the presence of C3−/− plasma were not able to form swarms and notably demonstrated impaired LTB4 production,31 suggesting a complement-LTB4 axis in regulating mouse neutrophil swarming in response to C. neoformans in vitro. Furthermore, human neutrophils in response to patterned zymosan particles also showed reduced swarming in heat-inactivated serum compared to regular serum, suggesting heat-labile factors such as complement contribute to neutrophil swarming.15 In vivo, C3 deficiency or C5aR antibody blockade reduced LTB4 production and pulmonary intravascular cluster formation in a C. albicans induced sepsis model.32 Taken together, complement-induced LTB4 production is vital in neutrophil swarming. Finally, the enzymatic activity of NOX2 and its effect on intracellular calcium can regulate LTB4 production and neutrophil swarming.16,23,33,34,35 We will discuss this in more detail in NOX2, swarming, and neutrophilic hyperinflammation.
Chemokines and cytokines that regulate neutrophil swarming
The chemokine CXCL2 works synergistically with LTB4 signaling in neutrophil swarming. Neutrophils lacking the CXCL2 receptor, CXCR2, showed impaired aggregation in laser-induced focal damage to mouse ear dermis.12 The presence of CXCL2 was also confirmed in mouse neutrophil clusters by immunofluorescence staining.12 Blockade of CXCR1 and CXCR2 alone did not affect human neutrophil swarming in response to patterned zymosan particle clusters; however, blockade CXCR1 and CXCR2 together with inhibition of BLT1 and BLT2 additionally reduced the chemotactic index compared to inhibition of BLT1 and BLT2 alone.15 These results suggest that CXCL8 (CXCR1/CXCR2 ligand) cooperates with LTB4 to promote neutrophil swarming.
Integrins and extracellular matrix in neutrophil swarming
Integrins and extracellular matrix are also involved in mediating neutrophil swarming. fMLF-primed human neutrophils formed clusters on immobilized fibrinogen (Fn) and β-glucan, but not Fn or β-glucan alone. While Fn and β-glucan are both recognized by Complement Receptor 3 (CR3, CD11b/CD18, αMβ2), the binding domains are distinct, suggesting that engagement of both the C3R I-domain (Fn) and lectin-like domain (β-glucan) may be required for clustering.36 Blockade of CR3 or VLA3 (α3β1) but not VLA5 (α5β1) or VLA6 (α6β1) by corresponding antibodies completely inhibited human neutrophil cluster formation on Fn+β-glucan,37 suggesting that human neutrophil swarming is regulated by multiple integrins. Further evidence for the role of integrins in neutrophil swarming comes from mouse studies. In vitro, CD11b-deficient neutrophils could not form stable clusters around C. neoformans.31 However, since CD11b is important for neutrophil recognition of Cryptococcus, it is unclear whether decreased swarming in this setting is due to lack of pathogen recognition, or a requirement for CD11b/β2 integrin in organizing the neutrophil swarm. In vivo, integrins have been demonstrated to assist in neutrophil localization in the central swarm. After focal laser injury, collagen fibers in the wound center are physically displaced by the dense neutrophil cluster and co-injection of WT and neutrophils deficient in CD11b, CD18, CD11a or the integrin-associated adaptor Talin demonstrated that integrins are required for neutrophil accumulation in the collagen-free zone.12 In contrast to the in vitro studies with human neutrophils, recruitment and clustering of Itgb1−/− neutrophils were similar to WT neutrophils in this model.12 Whether these conflicting results are due to differences in species or the in vitro versus in vivo model system is unclear. Taken together, the involvement of integrins in neutrophil swarming is complex and may depend on the cellular context, extracellular matrix, phases of neutrophil swarming etc. Furthermore, the underlying mechanism through which integrins regulate neutrophil swarming, including whether these involve integrin-dependent production of LTB4, still need further investigation. More studies are required to fully outline the spatiotemporal involvement of integrins and extracellular matrix.
Stop signals for neutrophil swarming
How neutrophil swarming is terminated and resolved is still largely unknown. Recently, GPCR desensitization has been proposed to be essential to both limit neutrophil swarm size, and to allow neutrophils to fully arrest within the developing swarm.22 GPCR kinases (GRKs) are crucial for GPCR desensitization and have been shown to induce arrest of neutrophil migration when they sense high concentrations of chemoattractants. Kienle et al. utilized intravital microscopy of lymph nodes during P. aeruginosa infection, combined with in vitro co-culture modeling, to demonstrate that GRK2 provides an essential “stop” signal in neutrophil swarms. Grk2−/− neutrophils formed larger clusters in vitro around P. aeruginosa aggregates, but notably lacked the arrest phase and moved out of neutrophil clusters again at high speed. Interestingly, this disorganization led to impaired phagocytosis and control of bacteria growth,22 suggesting that neutrophil arrest in the central swarm is necessary for efficient host defense. CXCR1 desensitization in neutrophils was also reported in an injury model of zebrafish,38 suggesting that GPCR desensitization might be a conserved mechanism for preventing excessive neutrophil aggregation. The lipid pro-resolving mediators lipoxin A4 (LXA4) and resolvin E3 (RvE3) have also been identified as stop signals for human neutrophil swarming in vitro.15 ω-OH-LTB4, an LTB4 metabolite synthesized by the enzyme omega-hydroxylase, has also been shown to be a natural inhibitor of LTB4-mediated responses by competition for BLT1 receptor binding.39 Human neutrophils showed strong LTB4 production with stimulation by the bacterial chemoattractant fMLF after preincubation with Salmonella typhimurium. When the bacteria:neutrophil ratio was increased, the transformation of LTB4 to ω-OH-LTB4 was suppressed.40 Though the underlying mechanism is unclear, it suggests neutrophils may favor LTB4 to ω-OH-LTB4 transformation when the microbe numbers are decreased and contribute to resolution of neutrophil swarming. However, whether LXA4, RvE3 and ω-OH-LTB4 participate in controlling the neutrophil swarming in vivo and how they coordinate the initiation, maintenance, and resolution signals still needs further investigation.
Communication with other cells during neutrophil swarming
Neutrophil swarm formation also initiates the recruitment of other cells, including monocytes and macrophages, to the swarm location. In laser-induced tissue injury in mice, monocytes and macrophages often assemble around the neutrophil cluster and may have a role in containing neutrophil swarm growth at the wound site.12 In studies analyzing the response to bioparticles in vitro, monocytes only accumulate when neutrophils are present, suggesting that the neutrophil swarm is a required first step for secondary recruitment of monocytes.41 In addition, tissue-resident macrophages can sense death signals from injured cells and extend processes to cloak the tissue microlesions to prevent neutrophil swarm initiation and neutrophil-driven tissue damage.20 Taken together, cross-talk between neutrophils, monocytes and macrophages likely plays an important role in regulating initiation and modulation of swarm growth.
Calcium signaling regulates neutrophil swarming
It is well known that calcium plays a vital role in neutrophil activation and function. Intracellular calcium levels are tightly controlled and in resting cells, calcium is sequestered within intracellular organelles including the endoplasmic reticulum (ER) and in the extracellular space. In non-excitable cells, including neutrophils, the primary mechanism of calcium entry into the cell is termed store-operated calcium entry (SOCE), whereby receptor signaling cascades lead to calcium release from the ER. Calcium sensing STIM proteins in the ER undergo conformational change and gate plasma membrane Ca2+ release-activated Ca2+ (CRAC) channels, comprised of ORAI family members, to permit sustained calcium entry from the extracellular space and activation of downstream calcium-dependent signaling.42,43,44 The role of calcium in regulating neutrophil swarming is emerging. Based on the current knowledge of mechanisms driving neutrophil swarming, there are several pathways that are anticipated to be calcium-regulated.
Calcium-dependent regulation of swarming mediators
LTB4 is a notable example of a mediator of neutrophil swarming that is exquisitely sensitive to intracellular calcium levels, where activation of key components of the LTB4 synthesis pathway, phospholipase A (PLA) and 5-LO, are dependent on calcium signaling.45 Though calcium is not responsible for cytosolic phospholipase A2 catalysis, it is needed for translocation of cPLA2 from cytosol to nuclear membrane and endoplasmic reticulum.46 Calcium can also bind to the N-terminal of 5-LO and is required for 5-LO translocation from cytosol to nuclear membrane and enzyme activation.47,48 Signaling though multiple receptor families including Gq-associated GPCRs (e.g., CXCR2, CXCR1, C5aR) and tyrosine kinase-associated receptors (TKARs) initiate SOCE. TKARs include key neutrophil receptors such as integrins, C-type lectin receptors, and Fc receptors; therefore, swarms that receive signals from these receptors may rely on calcium signaling for swarm amplification through LTB4 production. Although calcium-dependent LTB4 production is clearly an important mediator that influences neutrophil swarm formation, calcium signaling promotes many other neutrophil processes.44 SOCE via ORAI1 is also required for integrin outside-in signaling,49 and therefore may participate in neutrophil accumulation at the center swarm. Whether other calcium-dependent neutrophil processes such as degranulation and cytokine/chemokine production contribute to swarming remains to be studied.
Direct evidence for calcium signaling as a regulator of neutrophil swarms
Several studies have established a role for calcium signaling in the initiation and propagation of neutrophil swarms. To investigate whether calcium signal alone is sufficient in triggering neutrophil swarming, Khazen et al. performed an elegant experiment using an optogenetically activatable STIM1. Using this model, this group demonstrated that STIM1 activation and subsequent SOCE-initiated neutrophil clustering.35 The size and number of the clusters were reduced when STIM1 was photoactivated in the presence of BLT1/BLT2 antagonists, suggesting that calcium signaling in pioneer neutrophils attracts neighboring neutrophils via LTB4 dependent mechanisms.33,35,50 Spatiotemporal dynamics of calcium signals in neutrophil swarming around fungal particles zymosan were also defined using calcium dye Indo-1 by time-lapse imaging. The first pioneer neutrophils showed a strong calcium elevation upon contact with zymosan and cell arrest, followed by a transient calcium wave in neutrophils within a radius of 100–150 μm. These neutrophils then joined the cluster contacting zymosan and acquired sustained calcium elevation, suggesting that the strong calcium signal in the cluster center will amplify further waves of neutrophil recruitment in a calcium-dependent feedforward loop. Staining of propidium iodide did not show cell death in the neutrophil cluster center with zymosan, suggesting initial neutrophil swarming is not triggered by DAMPs in this setting.35 The zymosan receptor Dectin-1 triggers strong activation of SOCE dependent on STIM1 and ORAI1,50,51 however whether this signaling pathway is responsible for the intracellular calcium signal generated by the pioneer neutrophils remains to be seen. As discussed above, studies in zebrafish have demonstrated that sterile tissue injury can also initiate a calcium alarm signal via ATP release from connexin-43 (Cx43) hemichannels and subsequent ATP sensing by P2X1 receptor channels.16 Calcium influx through these P2X1 channels is required for neutrophil cluster formation via LTB4. However, pharmacological inhibition of P2X receptors or connexins did not affect mouse neutrophil cluster formation in response to zymosan,35 suggesting that stimulus-specific signal pathways are involved during neutrophil swarming in different species. Together these studies suggest that elevation in cytosolic calcium is crucial for both PAMP- and DAMP-triggered neutrophil swarming.
Priming modulates neutrophil calcium signaling
Primed neutrophils may have enhanced calcium influx following a second stimulus, which could augment neutrophil swarms. GM-CSF primed neutrophils showed more sustained elevation of intracellular calcium than unprimed neutrophils with the stimulation of soluble complexes or in response to chemotactic factors.52,53 However, there was no difference of the elevation of intracellular calcium between GM-CSF primed neutrophils and unprimed neutrophils in calcium-free media with the presence of EGTA,53 suggesting extra calcium elevation in primed neutrophils is primarily due to calcium influx. Intriguingly, Hopke et al. showed that the addition of GM-CSF or G-CSF enhanced the swarming of human neutrophils to C. albicans, although effects on intracellular calcium were not studied.34 Exposure to bacteria may also exert priming effects impacting calcium influx. For example, preincubation of human neutrophils with Salmonella typhimurium increased the LTB4 production triggered by the bacterial chemoattractant fMLF, which is associated with augmented calcium influx in primed neutrophils.40 This suggests that bacterial infection may boost neutrophil swarming in a calcium-LTB4 dependent manner.
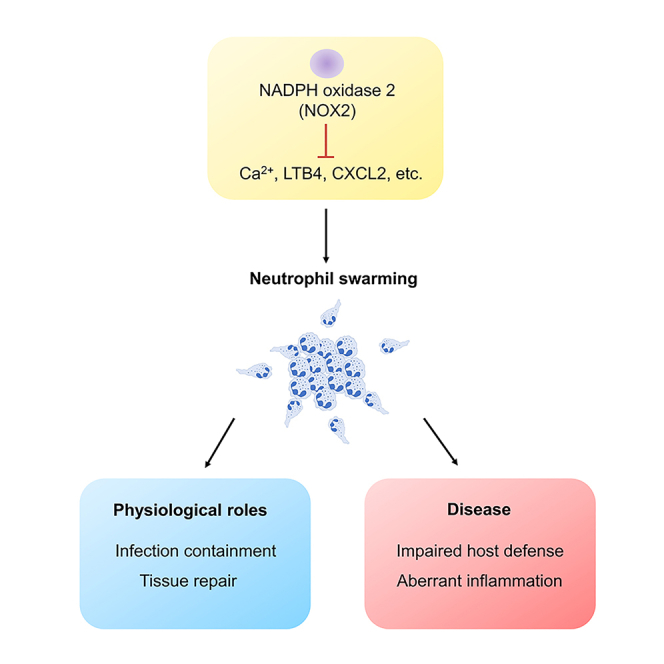
NOX2, swarming, and neutrophilic hyperinflammation
NADPH oxidase 2 (NOX2) is the major source of reactive oxygen species in activated neutrophils. NOX2 is an enzyme with multiple subunits, including two membrane-bound subunits CYBB and CYBA, cytosolic subunits NCF2, NCF1 and NCF4, and the small GTPase Rac. During neutrophil activation, cytosolic subunits and Rac are recruited to the phagosome and plasma membrane. The assembled oxidase then mediates electron transfer from NADPH to oxygen to generate superoxide, which is the precursor of other ROS (Figure 2).54,55 NOX2 assembly in neutrophils can be triggered through activation of signaling pathways downstream of several receptor families, including GPCRs (e.g., formyl peptide receptor, LTB4 receptor, other chemokine receptors) and tyrosine kinase-associated receptors (e.g., integrins, Fcγ receptors, C-type lectin receptors such as Dectin).56 Inactivating NOX2 mutations result in the inherited immunodeficiency, chronic granulomatous disease (CGD), which is associated with recurrent bacterial and fungal infections as well as aberrant inflammation, including neutrophilic hyperinflammation. Genetic variants in NOX2 subunits that partially reduce NOX2 activity, while not associated with infections, are linked to autoimmune and inflammatory disorders, such as systemic lupus erythematosis, rheumatoid arthritis, and inflammatory bowel disease.55 Thus, NOX2-derived ROS are not only important for host defense against microbes, but also participate in immune regulation.55
Figure 2.

NADPH oxidase limits LTB4 and cytokine production induced by fungal particles
NOX2 is a multi-subunit enzyme, including two membrane-bound subunits CYBB and CYBA, cytosolic subunits NCF2, NCF1 and NCF4, and a small GTPase Rac. Cytosolic subunits are recruited to the plasma membrane CYBB/CYBA heterodimer after neutrophil activation by soluble or particulate stimuli, such as the fungal cell wall particle zymosan. The assembled oxidase then catalyzes electron transfer from NADPH to oxygen to generate superoxide, which is the precursor of other ROS. The electron transfer across the plasma membrane leads to depolarization, which decreases the driving force of calcium entry. Intracellular calcium elevation activates cytosolic PLA2 (cPLA2), which leads to the release of arachidonic acid (AA) from membrane phospholipids. AA is then metabolized to Leukotriene A4 (LTA4) by arachidonate 5-lipoxygenase (5-LO) and 5-Lipoxygenase Activating Protein (FLAP), and LTA4 is further converted to LTB4 through LTA4 hydrolase. 5-LO is a rate-limiting enzyme for LTB4 synthesis and its activation is also dependent on calcium. LTB4 binds to its receptor BLT1, which further increases the intracellular calcium signal through an autocrine or paracrine route. Zymosan can activate TLR or Dectin-1 on neutrophils, which leads to pro-inflammatory cytokine production via TLR-MyD88-AP1/NF-κB and in parallel, Dectin-1-Card9- NF-κB. NOX2 can inhibit these signaling pathways or directly inhibit NF-κB, which limits the production of pro-inflammatory cytokines. In the absence of NOX2, the negative membrane potential in CGD neutrophils leads to overload of intracellular calcium, which triggers increased LTB4 production and a further enhanced LTB4-BLT1 feedback loop. Therefore, more pro-inflammatory cytokines are produced without the dampening of signal transduction pathways by NOX2.
Experimental studies support a role for increased acute recruitment of neutrophils as one component of the dysregulated inflammation associated with CGD. We demonstrated that CGD mice display neutrophilic hyperinflammation following inhalation of sterile fungal PAMPs (zymosan particles).23,33 Histologically, the lungs of CGD mice display significantly larger neutrophil foci in lung parenchyma compared to wild type (WT) mice at 8 h following zymosan challenge, which continued to increase in size over the next 16 h. Although the dynamics of neutrophil behavior have not yet been visualized using intravital microscopy, these foci are reminiscent of those formed by neutrophil swarms. Studies with mouse neutrophils demonstrated that CGD neutrophils spontaneously formed larger and increased numbers of neutrophil clusters in vitro within the first 60 min of exposure to zymosan.33 These findings are further supported by a recent study by Hopke et al., who used video microscopy to show that human CGD neutrophils formed significantly larger swarms against C. albicans in vitro.34
NOX2 activity limits neutrophil swarms via electrogenic effects on calcium signaling and LTB4
Insight into the mechanisms driving this exaggerated swarming response in the absence of NOX2 points back to the central role of LTB4 in neutrophil swarming.34 The 5-LO inhibitor Zileuton and BLT1 antagonist U75302 significantly inhibited CGD neutrophil cluster formation with zymosan in vitro and, importantly, reduced the size of neutrophil foci in CGD lung in the first 24 h following challenge with zymosan in vivo.33
Prior studies focused on the role of ROS in leukotriene degradation; however, the addition of ROS scavengers superoxide dismutase (SOD) and catalase only modestly increased the LTB4 production in WT mouse neutrophils.33 Myeloperoxidase (MPO) inhibitors 4-aminobenzoic acid hydrazide or exogenous H2O2 or H2O2 generation system by glucose/glucose oxidase, with or without MPO, also did not affect LTB4 levels in WT mouse neutrophils in response to zymosan, suggesting that increased production of LTB4 is not due to ROS-mediated degradation of LTB4.33 Though in cell-free systems, LTB4-mediated chemotaxis is decreased by hydroxyl radicals, there is still no direct evidence that LTB4 is degraded by NOX2-derived ROS in cells.57,58,59
A key feature of NOX2 is that this complex is an electron transport system, where the transfer of an electron across the membrane to oxygen in the extracellular (or phagosomal) space also generates an intracellular proton (Figure 2). The neutrophil resting membrane potential (Vm) is around −70mV and theoretical calculations estimate that unchecked, NOX2 activation could drive Vm to +200mV.60 However, compensatory mechanisms counter-balance this depolarizing force, including the voltage-gated proton channel (Hvcn1), which opens to allow H+ efflux, pH maintenance, and continued oxidase activity.61,62,63 The measured Vm during ROS production is thus ∼+30-40mV, still comparable in magnitude to a neuronal action potential.60 As discussed above, calcium entry in non-excitable cells occurs primarily via SOCE, which displays an inverse relationship of Ca2+/Vm where the influx of positively charged Ca2+ is enhanced by the negative Vm. Factors that result in Vm depolarization inhibit calcium influx. The effect of ROS-mediated depolarization was first demonstrated when the Ligeti lab observed enhanced calcium influx in CGD neutrophils, then connected this finding to hyperpolarization in the absence of NOX2 function.64,65,66,67,68 Calcium entry is also impaired in Hvcn1−/− neutrophils due to increased ROS-driven depolarization.63 Our study confirmed that LTB4 production is tightly correlated to calcium influx, and that calcium influx is enhanced in CGD neutrophils (Figure 2).33 This suggests that the electrogenic interaction between NOX2 and calcium signaling is reflected in calcium overload and excessive LTB4 production driving neutrophil swarming in CGD. Notably, intracellular calcium also activates signaling pathways that promote NOX2 activation, suggesting that electrogenic inhibition of calcium signaling may be a central feedback mechanism for regulating ROS production and other calcium-dependent neutrophil processes.
NOX2 activity may limit neutrophil swarms by dampening their production of chemokines and cytokines
In addition to regulation of LTB4, NOX2 also limits neutrophil pro-inflammatory cytokine and chemokine production.55,69,70,71,72,73 CXCL2 levels in CGD mouse neutrophils and CXCL8 levels in human neutrophils treated with an NOX2 inhibitor DPI were significantly increased after zymosan stimulation.72 Kobayashi et al. also demonstrated enhanced pro-inflammatory gene expression in human CGD neutrophils.74 The mechanisms underlying this hyperactivation in the absence of NOX2 are multifactorial and only partially understood. Most studies focus on the direct effects of ROS as a second messenger, such as oxidative modification of signaling pathways.73,75,76 For example, ROS-dependent NF-kB oxidation has been shown to modify neutrophil IL1β production.77 Absence of NOX2 also results in enhanced activation of tyrosine phosphatase SHP2-Syk and downstream Card9-dependent NFkB and Card9-independent JNK-c-Jun in neutrophils stimulated with fungal particles.72 Cytokine production may be directly related to the impact of ROS produced by NOX2 since the addition of exogenous oxidants hydrogen peroxide or xanthine oxidase/hypoxanthine significantly reduced CXCL2 production, and the ROS scavenger catalase significantly increased CXCL2 levels in mouse neutrophils stimulated with zymosan.72 However, whether excessive production of CXCL2/CXCL8 contributes to an exaggerated swarming in neutrophils with NOX2 deficiency still needs further investigation. ROS also negatively regulates the levels of other cytokines after fungal stimulation, including TNF-α,72 which may also contribute to limiting neutrophil swarming via direct or indirect effects on neutrophil functions.
NOX2 activity regulates neutrophil accumulation through the IL-1-G-CSF axis
Mediators produced locally are important for rapid neutrophil accumulation at infected or inflamed sites, but to accelerate and sustain this recruitment requires a continuous supply of neutrophils from the blood stream, including neutrophils newly released from the marrow storage pool in response to systemic signals. An important pathway involves the local production of the alarmins IL-1α or IL1-β, leading to upregulation of pro-inflammatory cytokine expression in the local environment. This includes G-CSF, a key cytokine that induces increased release of marrow neutrophils into the bloodstream for delivery into inflamed sites, as well as stimulates emergency granulopoiesis.
Recent studies have revealed the IL-1β-G-CSF axis is amplified systemically in the absence of NOX2, and synergizes with increased LTB4 to promote focal neutrophil accumulation in CGD mice during acute inflammation.23,33 Neutrophils themselves can be an important source of IL-1β in both infection and sterile inflammation.23,78,79,80,81 CGD neutrophils produce increased IL-1β in response to fungal cell walls in vitro,72 and many more IL-1β-expressing neutrophils, recruited via LTB4, accumulate in the lungs of CGD mice following inhalation of zymosan fungal particles, resulting in markedly elevated IL-1β levels in the airways.72 This was accompanied by elevated local and plasma G-CSF levels and increased marrow release of mature and immature neutrophils.23 Blocking either IL-1β or G-CSF limited the size of lung neutrophil foci at 24 h, showing that the IL-1β- G-CSF axis contributes to the increased neutrophil inflammation in CGD, acting sequentially but non-redundantly with LTB4 (Figure 3).23,33 These findings are also consistent with the importance of lipid mediator-cytokine cascades in the acute inflammatory response, and how their dysregulation in the absence of NOX2 may contribute to increased neutrophil swarming and neutrophilic inflammation.
Figure 3.

NADPH oxidase controls neutrophil swarming through limiting LTB4 and cytokine levels
Neutrophils produce the chemoattractants LTB4 and CXCL2 to mediate neutrophil swarming in order to combat infections or inflammation. Neutrophils also generate IL-1β, which is vital for neutrophil activation, and can locally regulate other cytokine production. This includes IL-1β-induced production of G-CSF, which then acts systemically to increase neutrophil mobilization from the marrow into the peripheral blood and to increase granulopoiesis. In the absence of NOX2, feedforward loops are greatly amplified. The increase in LTB4 and IL-1β levels, and indirectly, G-CSF, reflects not only increased production by individual neutrophils but the much larger numbers of neutrophils present in the airways via the amplified feedforward loop.
Heterogeneity in neutrophil NOX2 responses
Given the multiple mechanisms by which ROS production regulates inflammatory responses, heterogeneity in NOX2 activation may be an important modifier of neutrophil swarm dynamics. Numerous factors generate heterogeneity in oxidase activation. Neutrophils express abundant cell surface markers,42 and many are involved in ROS production via direct activation by ligand-receptor recognition or priming the cell for enhanced activation of NOX2.82 Different receptors and agonist concentration can initiate varying strength and kinetics of NOX2 activation.83 For example, GPCRs tend to activate rapid but transient bursts of NOX2 activation that are sustained for seconds to minutes, while Tyrosine Kinase-Associated Receptors (TKARs), at least in vitro, induce sustained ROS production for up to hours.50 The cellular context also dictates NOX2 activation where adhesion to the extracellular matrix, or concurrent priming signals from neighboring cells can augment ROS production.84 In addition, metabolic pathways and membrane phospholipids regulate NOX2 activation status.56,85 Diversity in neutrophil phenotype and NOX2 activity can also arise through neutrophil maturation, in response to microenvironmental and tissue-specific cues, and following infectious or inflammatory stimuli and with aging.86,87,88,89,90,91,92,93,94 Recent studies utilizing single cell RNAseq report the presence of transcriptionally distinct subsets in healthy mice and humans that could lead to variable NOX2 activation among the neutrophil populations.95,96 Other studies have reported heterogeneity in the onset of ROS production within neutrophil phagosomes in the first 30 min following stimulation with zymosan particles,97 which appears to reflect differences in timing of NOX2 assembly on individual phagosomes.98 Underlying mechanisms and how this influences the kinetics and heterogeneity of neutrophil NOX2 responses remain to be clarified. Intriguingly, the overall magnitude of an individual’s neutrophil NOX2 ROS response to immune complexes, bacterial ligands, or pathogens is a fixed phenotype and is heritable, suggesting that this may be at least in part genetically determined.99 Thus, there are multiple factors, both extrinsic as well as neutrophil-intrinsic differences that can impact neutrophil NOX2 responses.
Since NOX2 activity is important in the regulation of calcium signaling and LTB4 synthesis, it is interesting to consider how factors that impact NOX2 activity may affect the efficiency of LTB4 production and the neutrophil swarming response. Experimental support for this concept comes from a recent study where S100A8-Cre-mediated deletion of Ncf2 in neutrophils yields “Ncf2S100A8Cre” neutrophils with ∼20% NOX2 activity of WT neutrophils.100 LTB4 production by Ncf2S100A8Cre neutrophils stimulated with zymosan in vitro was significantly higher than Ncf2fl/fl neutrophils but not as elevated as Ncf2−/− neutrophils entirely lacking NOX2 activity,100 supporting the notion that NOX2 activity limits LTB4 production in a dose-dependent manner. Moreover, Ncf2S100A8Cre mice had increased neutrophilic lung inflammation at 24 h following zymosan inhalation. Notably, the size of focal neutrophil lung infiltrates, while larger than those seen in WT mice, were smaller than in Ncf2−/− mice,100 suggesting this dose-dependence also translates to neutrophil swarming triggered in vivo. Thus, heterogeneity in NOX2 activation driven by variation in cellular or disease context may serve as a spatiotemporal tuner of neutrophil swarm formation.
ROS, size matters?
It has been proposed that ROS are sensors for microbe size.77 In vitro, human neutrophils only formed swarms when the size of zymosan particle patterns was larger than 17.5 μm (more than three zymosan particles in a cluster), suggesting that neutrophil swarming may also be related to the size of target microbes.15 ROS localization could contribute to this size discrimination, since phagocytosis of small microbes triggers intracellular ROS production, whereas microbes that are too large to ingest, such as fungal hyphae, trigger extracellular ROS production.72,77 ROS regulate NF-κB activity through multiple mechanisms, such as direct oxidation of NF-κB (p50) to inhibit its DNA binding activity or indirect inhibition of upstream NF-κB activating pathways by oxidation, such as IκBα, IKK and Akt.101 Intracellularly produced ROS may have more access to these signaling pathways. This notion is supported by studies demonstrating that intracellularly produced neutrophil ROS limits their cytokine production, such as IL-1β, CXCL-2 and TNF-α,72,77 whereas extracellular ROS triggered by Candida hyphae did not dampen neutrophil IL-1β expression, associated with recruitment of more neutrophils to form clusters compared to phagocytosed Candida spores.77 How differences in the localization of NOX2-generated ROS regulates neutrophil swarming to combat microbes of different sizes and its impact on LTB4 production is unknown and will require further investigation.
Physiologic role of neutrophil swarms
Neutrophil swarming can be viewed as a distinct functional response by neutrophils involving a coordinated group behavior leading to a rapid influx of neutrophils at local sites. Despite the growing body of literature elucidating the molecular mechanisms that support neutrophil swarming, the biologic importance of swarming in controlling infections as well as responding appropriately to injury remains to be fully elucidated. Given the importance of neutrophils in host defense against bacterial and fungal pathogens, it is reasonable to hypothesize that neutrophil cooperative behavior is an important mechanism for organizing the host response to infection. Numerous studies utilizing in vivo imaging have demonstrated that neutrophil swarms form almost ubiquitously in response to a nidus of infection.102,103,104 In several studies, neutrophil depletion or blocking neutrophil receptors required for entry into the infection site results in increased pathogen growth or dissemination, including S. aureus.102,103 In addition, neutrophil swarms mediated by Cx43 can restrict opportunistic bacterial infection in a wound infection model in zebrafish.16 While supporting the notion that neutrophil swarms contribute to host defense, these experiments do not distinguish between a general requirement for neutrophil recruitment and neutrophil-dependent microbial killing versus specific features of swarm organization. Indeed, Kienle et al. recently demonstrated that swarm formation alone is not sufficient for bacterial containment. GRK2 mediates GPCR desensitization and Grk2−/− neutrophils formed larger clusters compared to WT in response to sterile or infectious stimuli.22 However, without GPCR desensitization these neutrophils also rapidly exit the clusters, resulting in a lack of “focus” and poor bacterial control when challenged with Pseudomonal infection. The development of micropatterning chips by Irimia’s group for in vitro studies has enabled more direct inquiry into the role of swarming in pathogen containment and killing. By printing reproducible clusters of fungal particles onto a slide, these investigators were able to demonstrate that neutrophil swarms delay germination and fungal growth, and identified factors, including LTB4 signaling, NOX2, and NET formation that were important for pathogen control.15,34 Although studies of other pathogens remain limited, this technology is a powerful tool to directly query the signaling pathways that are necessary for swarm organization and pathogen killing.
Although swarming is certainly a mechanism to recruit large numbers of neutrophils, the physical structure of neutrophil swarms in relation to the surrounding tissue may also be relevant for swarm function. Studies by Waite et al. showed that during Listeria infection, myeloid swarms in the spleen restricted blood flow to the infected region and supported bacterial containment103 In the skin, intravital imaging also demonstrates degradation of the collagen ECM matrix by the neutrophil swarm,12 however whether this process plays a physiological role in host defense, wound healing, or even hemostasis is unknown and will be an important area for further studies.
Neutrophil swarms in disease
As we learn in more depth the complex array of signals that allow the neutrophil cooperative behavior in swarming and the biological function of this behavior, this knowledge reveals an equally complex number of ways that swarming could become dysfunctional in disease states. Excessive swarming may also lead to collateral tissue damage. Neutrophils are inherently toxic cells, releasing abundant quantities of proteases and reactive oxygen species that not only kill pathogens but are also injurious to host tissues. As such, the feedforward amplification that drives swarm formation could quickly become a liability if inadequately regulated. CGD is an excellent example where neutrophil swarms may be deleterious and contribute to disease pathology. As also discussed above, CGD is notable not only for susceptibility to bacterial and fungal infection, but also hyperinflammation. The mechanisms driving inflammation in CGD are likely multifactorial, but the current evidence in mice and human cells suggests that dysregulated calcium signaling, LTB4-dependent, and LTB4-independent mechanisms that in part involve the IL-1-G-CSF axis contribute to enhanced swarm formation. The combination of poor pathogen control and exaggerated swarming likely compound the severity of the disease. Similarly, exaggerated local LTB4/BLT1 signaling was associated with excessive neutrophil infiltration and poor control of methicillin-resistant Staphylococcus aureus (MRSA) skin infection in diabetic mice.105 Aberrant LTB4 production is also thought to be detrimental in diseases including arthritis and atherosclerosis.106 Joint inflammation in K/BxN serum-transfer arthritis model involves LTB4, IL-1, and CCR1 and CXCR2 ligands and is increased in CGD mice.81,107,108 NOX2-deficient neutrophils also show enhanced proinflammatory genes expression and diminished anti-inflammatory functions including less suppressive activity to T cells and lower PD-L1 expression.109 Whether LTB4 drives pathological neutrophil swarming within the joint space remains to be seen. While further studies are required, this suggests that targeting swarming using LTB4 or calcium channel inhibitors might be a novel therapeutic approach for the inflammatory manifestations of CGD or other diseases manifested by excessive neutrophil swarming.
Tissue- and pathogen-specific factors are likely important in determining whether swarming is helpful or harmful to the host response. While swarm formation at the site of a skin lesion might be beneficial for pathogen containment and organization of the healing wound, robust swarming in vital organs such as heart or brain could lead to irrecoverable tissue injury. Similarly, in a C. albicans yeast sepsis mouse model, LTB4-mediated intravascular swarms caused a “traffic jam” within the small capillaries and resulted in pulmonary hemorrhage and hypoxemia.32 The clearance of C. albicans was comparable between Ltb4r−/− mice and WT mice at 24 h, which suggests that excessive neutrophil swarms and intravascular inflammation, rather than impaired pathogen control led to the worse outcomes. Pathogens are well-practiced at using virulence factors to escape innate immune recognition and killing; thus the swarming response also represents a prime target for bacterial and fungal virulence factors.
Systemic factors that alter neutrophil NOX2 activity, priming or differentiation state such as chronic illness may alter neutrophil swarming due to changes in cell surface receptors or intracellular signaling mediators that impact intra- and intercellular communication.86,87,88,89,90,91,92,93,94 Indeed, patients following solid organ or hematopoietic stem cell transplantation are known to be at high risk for bacterial and fungal infection, and are well-known to have functional defects in neutrophil chemotaxis, ROS generation, and intracellular killing. A recent study demonstrated that neutrophils from these patients often showed smaller swarms and regardless of swarm size, defective ability to control C. albicans growth and escape in vitro.110 Others report that the size of neutrophil swarms in response to zymosan was reduced in patients after major trauma (particularly in the early phase), acute generalized exanthematous pustulosis (AGEP) and sepsis.15 In cirrhosis, neutrophil swarms from patients showed reduced ability to control fungal growth, despite a similar swarm size compared to healthy controls, suggesting that in this setting global impairment of neutrophil function impairs neutrophil bactericidal responses with the swarm.111
Patients with cystic fibrosis, characterized by mutations in the CFTR chloride channel, develop recurrent pulmonary bacterial infections due to abnormal mucociliary clearance as well as primary neutrophil dysfunction due to loss of CFTR. Even in the absence of acute infection, this is a chronic systemic illness with marked changes in pulmonary bacterial colonization and often low grade chronic inflammation.112 In a microscale cluster neutrophil swarming assay, cystic fibrosis (CF) neutrophils formed larger swarms on C. albicans clusters compared to non-CF neutrophils. However, when taken from hospitalized CF patients, neutrophils formed smaller swarms compare to outpatient CF neutrophils,113 suggesting that neutrophil swarming is altered both by acute and chronic aspects of cystic fibrosis disease. Although larger, neutrophil swarms from CF-hospitalized patients had a reduced ability to restrict fungal growth as compared to neutrophils from non-CF or CF outpatients. Taken together, this suggests that altered swarm function under disease conditions may impair control of infections, and much work is required to better understand the mechanisms by which these systemic diseases alter swarm formation and function, which may provide new targets for diagnosis and drug development.
Conclusions and remaining questions
The cooperative behavior exhibited by swarming neutrophils is a remarkable example of intercellular communication between immune cells. Although the past decade has seen significant advances from the descriptive and mechanistic studies performed using animal models and new in vitro modeling systems, many questions remain. In particular, there is a great need to better understand the biological role of this group behavior in host defense, inflammation, and wound healing. Are swarms necessary for host defense? What are the necessary features of the swarm that enable bacterial control? What is the role of neutrophil swarms in acute vs. chronic inflammation? Is the organization of the neutrophil swarm instructive or destructive for the subsequent wound healing process? NOX2 is emerging as an important regulator of neutrophil function, and current evidence suggests that NOX2 is similarly important for the regulation of swarm formation through the electrogenic effects of the oxidase on calcium signaling and downstream mediators including LTB4, via direct effects of ROS on multiple signaling pathways, and perhaps additional components that regulate swarming but are not yet identified. Further investigation into how NOX2 regulates swarming and how swarming contributes to inflammatory pathology is anticipated to both deepen our understanding of the fundamental mechanisms driving neutrophil swarming, as well as provide insight into the mechanisms that drive inflammatory pathology in CGD. Moreover, many acquired disorders as well as aging are associated with impaired neutrophil chemotaxis and NOX2 activity, both of which could alter swarming behavior and have important implications for host defense and inflammation. Finally, this observation of intercellular communication that markedly impacts neutrophil migratory behavior also provokes the question of whether other neutrophil functions such as phagocytosis or degranulation, typically considered to be cell autonomous functions, may be similarly modulated by group behavior. For example, does phagocytosis of a particle or pathogen by one neutrophil release signals that alter the phagocytic capacity of neighboring cells? How are neutrophils otherwise communicating to coordinate the behavior of the group? The gaps in knowledge remain quite vast, however progress in these areas have great potential to offer more complete understanding of neutrophil behavior in disease and to identify new therapeutic targets to modulate inflammation and host defense.
Acknowledgments
The authors thank Tina McGrath for assistance with article preparation. This work was supported by a grant from the Children’s Discovery Institute of Washington University and St. Louis Children's Hospital (M.C.D.) and AI166793 (R.A.C.).
Author contributions
Z.S. and S.B. conceived and performed investigation. Z.S., S.B., R.A.C., and M.C.D. wrote the article. R.A.C and M.C.D. supervised the article. R.A.C and M.C.D reviewed and edited the article.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Contributor Information
Regina A. Clemens, Email: clemensra@wustl.edu.
Mary C. Dinauer, Email: mdinauer@wustl.edu.
References
- 1.Nauseef W.M., Borregaard N. Neutrophils at work. Nat. Immunol. 2014;15:602–611. doi: 10.1038/ni.2921. [DOI] [PubMed] [Google Scholar]
- 2.Burn G.L., Foti A., Marsman G., Patel D.F., Zychlinsky A. The Neutrophil. Immunity. 2021;54:1377–1391. doi: 10.1016/j.immuni.2021.06.006. [DOI] [PubMed] [Google Scholar]
- 3.Mayadas T.N., Cullere X., Lowell C.A. The multifaceted functions of neutrophils. Annu. Rev. Pathol. 2014;9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cowland J.B., Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol. Rev. 2016;273:11–28. doi: 10.1111/imr.12440. [DOI] [PubMed] [Google Scholar]
- 5.Manz M.G., Boettcher S. Emergency granulopoiesis. Nat. Rev. Immunol. 2014;14:302–314. doi: 10.1038/nri3660. [DOI] [PubMed] [Google Scholar]
- 6.Németh T., Mócsai A. Feedback Amplification of Neutrophil Function. Trends Immunol. 2016;37:412–424. doi: 10.1016/j.it.2016.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Futosi K., Fodor S., Mócsai A. Reprint of Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013;17:1185–1197. doi: 10.1016/j.intimp.2013.11.010. [DOI] [PubMed] [Google Scholar]
- 8.Tsai C.Y., Hsieh S.C., Liu C.W., Lu C.S., Wu C.H., Liao H.T., Chen M.H., Li K.J., Shen C.Y., Kuo Y.M., Yu C.L. Cross-Talk among Polymorphonuclear Neutrophils, Immune, and Non-Immune Cells via Released Cytokines, Granule Proteins, Microvesicles, and Neutrophil Extracellular Trap Formation: A Novel Concept of Biology and Pathobiology for Neutrophils. Int. J. Mol. Sci. 2021;22:3119. doi: 10.3390/ijms22063119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irimia D. Neutrophil Swarms Are More Than the Accumulation of Cells. Microbiol. Insights. 2020;13 doi: 10.1177/1178636120978272. 1178636120978272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kienle K., Lämmermann T. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol. Rev. 2016;273:76–93. doi: 10.1111/imr.12458. [DOI] [PubMed] [Google Scholar]
- 11.Lämmermann T. In the eye of the neutrophil swarm-navigation signals that bring neutrophils together in inflamed and infected tissues. J. Leukoc. Biol. 2016;100:55–63. doi: 10.1189/jlb.1MR0915-403. [DOI] [PubMed] [Google Scholar]
- 12.Lämmermann T., Afonso P.V., Angermann B.R., Wang J.M., Kastenmüller W., Parent C.A., Germain R.N. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–375. doi: 10.1038/nature12175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rocha-Gregg B.L., Huttenlocher A. Swarming motility in host defense. Science. 2021;372:1262–1263. doi: 10.1126/science.abj3065. [DOI] [PubMed] [Google Scholar]
- 14.Mihlan M., Glaser K.M., Epple M.W., Lämmermann T. Neutrophils: Amoeboid Migration and Swarming Dynamics in Tissues. Front. Cell Dev. Biol. 2022;10:871789. doi: 10.3389/fcell.2022.871789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reátegui E., Jalali F., Khankhel A.H., Wong E., Cho H., Lee J., Serhan C.N., Dalli J., Elliott H., Irimia D. Microscale arrays for the profiling of start and stop signals coordinating human-neutrophil swarming. Nat. Biomed. Eng. 2017;1:0094. doi: 10.1038/s41551-017-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poplimont H., Georgantzoglou A., Boulch M., Walker H.A., Coombs C., Papaleonidopoulou F., Sarris M. Neutrophil Swarming in Damaged Tissue Is Orchestrated by Connexins and Cooperative Calcium Alarm Signals. Curr. Biol. 2020;30:2761–2776.e7. doi: 10.1016/j.cub.2020.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isles H.M., Loynes C.A., Alasmari S., Kon F.C., Henry K.M., Kadochnikova A., Hales J., Muir C.F., Keightley M.C., Kadirkamanathan V., et al. Pioneer neutrophils release chromatin within in vivo swarms. Elife. 2021;10:e68755. doi: 10.7554/eLife.68755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chtanova T., Schaeffer M., Han S.J., van Dooren G.G., Nollmann M., Herzmark P., Chan S.W., Satija H., Camfield K., Aaron H., et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. 2008;29:487–496. doi: 10.1016/j.immuni.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng L.G., Qin J.S., Roediger B., Wang Y., Jain R., Cavanagh L.L., Smith A.L., Jones C.A., de Veer M., Grimbaldeston M.A., et al. Visualizing the neutrophil response to sterile tissue injury in mouse dermis reveals a three-phase cascade of events. J. Invest. Dermatol. 2011;131:2058–2068. doi: 10.1038/jid.2011.179. [DOI] [PubMed] [Google Scholar]
- 20.Uderhardt S., Martins A.J., Tsang J.S., Lämmermann T., Germain R.N. Resident Macrophages Cloak Tissue Microlesions to Prevent Neutrophil-Driven Inflammatory Damage. Cell. 2019;177:541–555.e17. doi: 10.1016/j.cell.2019.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Afonso P.V., Janka-Junttila M., Lee Y.J., McCann C.P., Oliver C.M., Aamer K.A., Losert W., Cicerone M.T., Parent C.A. LTB4 is a signal-relay molecule during neutrophil chemotaxis. Dev. Cell. 2012;22:1079–1091. doi: 10.1016/j.devcel.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kienle K., Glaser K.M., Eickhoff S., Mihlan M., Knöpper K., Reátegui E., Epple M.W., Gunzer M., Baumeister R., Tarrant T.K., et al. Neutrophils self-limit swarming to contain bacterial growth in vivo. Science. 2021;372:eabe7729. doi: 10.1126/science.abe7729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Song Z., Bhattacharya S., Huang G., Greenberg Z.J., Yang W., Bagaitkar J., Schuettpelz L.G., Dinauer M.C. NADPH oxidase 2 limits amplification of IL-1β-G-CSF axis and an immature neutrophil subset in murine lung inflammation. Blood Adv. 2023;7:1225–1240. doi: 10.1182/bloodadvances.2022007652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Werz O. 5-lipoxygenase: cellular biology and molecular pharmacology. Curr. Drug Targets - Inflamm. Allergy. 2002;1:23–44. doi: 10.2174/1568010023344959. [DOI] [PubMed] [Google Scholar]
- 25.Wan M., Tang X., Stsiapanava A., Haeggström J.Z. Biosynthesis of leukotriene B4. Semin. Immunol. 2017;33:3–15. doi: 10.1016/j.smim.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 26.Arya S.B., Chen S., Jordan-Javed F., Parent C.A. Ceramide-rich microdomains facilitate nuclear envelope budding for non-conventional exosome formation. Nat. Cell Biol. 2022;24:1019–1028. doi: 10.1038/s41556-022-00934-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Majumdar R., Tavakoli Tameh A., Arya S.B., Parent C.A. Exosomes mediate LTB4 release during neutrophil chemotaxis. PLoS Biol. 2021;19:e3001271. doi: 10.1371/journal.pbio.3001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hopke A., Lin T., Scherer A.K., Shay A.E., Timmer K.D., Wilson-Mifsud B., Mansour M.K., Serhan C.N., Irimia D., Hurley B.P. Transcellular biosynthesis of leukotriene B4 orchestrates neutrophil swarming to fungi. iScience. 2022;25:105226. doi: 10.1016/j.isci.2022.105226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tamás S.X., Roux B.T., Vámosi B., Dehne F.G., Török A., Fazekas L., Enyedi B. A genetically encoded sensor for visualizing leukotriene B4 gradients in vivo. Nat. Commun. 2023;14:4610. doi: 10.1038/s41467-023-40326-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muldur S., Vadysirisack D.D., Ragunathan S., Tang Y., Ricardo A., Sayegh C.E., Irimia D. Human Neutrophils Respond to Complement Activation and Inhibition in Microfluidic Devices. Front. Immunol. 2021;12:777932. doi: 10.3389/fimmu.2021.777932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun D., Shi M. Neutrophil swarming toward Cryptococcus neoformans is mediated by complement and leukotriene B4. Biochem. Biophys. Res. Commun. 2016;477:945–951. doi: 10.1016/j.bbrc.2016.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee E.K.S., Gillrie M.R., Li L., Arnason J.W., Kim J.H., Babes L., Lou Y., Sanati-Nezhad A., Kyei S.K., Kelly M.M., et al. Leukotriene B4-Mediated Neutrophil Recruitment Causes Pulmonary Capillaritis during Lethal Fungal Sepsis. Cell Host Microbe. 2018;23:121–133.e4. doi: 10.1016/j.chom.2017.11.009. [DOI] [PubMed] [Google Scholar]
- 33.Song Z., Huang G., Chiquetto Paracatu L., Grimes D., Gu J., Luke C.J., Clemens R.A., Dinauer M.C. NADPH oxidase controls pulmonary neutrophil infiltration in the response to fungal cell walls by limiting LTB4. Blood. 2020;135:891–903. doi: 10.1182/blood.2019003525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hopke A., Scherer A., Kreuzburg S., Abers M.S., Zerbe C.S., Dinauer M.C., Mansour M.K., Irimia D. Neutrophil swarming delays the growth of clusters of pathogenic fungi. Nat. Commun. 2020;11:2031. doi: 10.1038/s41467-020-15834-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khazen R., Corre B., Garcia Z., Lemaître F., Bachellier-Bassi S., d'Enfert C., Bousso P. Spatiotemporal dynamics of calcium signals during neutrophil cluster formation. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2203855119. e2203855119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Byrd A.S., O'Brien X.M., Johnson C.M., Lavigne L.M., Reichner J.S. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to Candida albicans. J. Immunol. 2013;190:4136–4148. doi: 10.4049/jimmunol.1202671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson C.M., O'Brien X.M., Byrd A.S., Parisi V.E., Loosely A.J., Li W., Witt H., Faridi M.H., LeFort C.T., Gupta V., et al. Integrin Cross-Talk Regulates the Human Neutrophil Response to Fungal beta-Glucan in the Context of the Extracellular Matrix: A Prominent Role for VLA3 in the Antifungal Response. J. Immunol. 2017;198:318–334. doi: 10.4049/jimmunol.1502381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coombs C., Georgantzoglou A., Walker H.A., Patt J., Merten N., Poplimont H., Busch-Nentwich E.M., Williams S., Kotsi C., Kostenis E., Sarris M. Chemokine receptor trafficking coordinates neutrophil clustering and dispersal at wounds in zebrafish. Nat. Commun. 2019;10:5166. doi: 10.1038/s41467-019-13107-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Archambault A.S., Poirier S., Lefebvre J.S., Robichaud P.P., Larose M.C., Turcotte C., Martin C., Provost V., Boudreau L.H., McDonald P.P., et al. 20-Hydroxy- and 20-carboxy-leukotriene (LT) B(4) downregulate LTB(4) -mediated responses of human neutrophils and eosinophils. J. Leukoc. Biol. 2019;105:1131–1142. doi: 10.1002/JLB.MA0718-306R. [DOI] [PubMed] [Google Scholar]
- 40.Golenkina E.A., Galkina S.I., Pletjushkina O., Chernyak B., Gaponova T.V., Romanova Y.M., Sud'ina G.F. Gram-Negative Bacteria Salmonella typhimurium Boost Leukotriene Synthesis Induced by Chemoattractant fMLP to Stimulate Neutrophil Swarming. Front. Pharmacol. 2021;12:814113. doi: 10.3389/fphar.2021.814113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walters N., Zhang J., Rima X.Y., Nguyen L.T.H., Germain R.N., Lämmermann T., Reátegui E. Analyzing Inter-Leukocyte Communication and Migration In Vitro: Neutrophils Play an Essential Role in Monocyte Activation During Swarming. Front. Immunol. 2021;12:671546. doi: 10.3389/fimmu.2021.671546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Futosi K., Fodor S., Mócsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013;17:638–650. doi: 10.1016/j.intimp.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Immler R., Simon S.I., Sperandio M. Calcium signalling and related ion channels in neutrophil recruitment and function. Eur. J. Clin. Invest. 2018;48:e12964. doi: 10.1111/eci.12964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Clemens R.A., Lowell C.A. CRAC channel regulation of innate immune cells in health and disease. Cell Calcium. 2019;78:56–65. doi: 10.1016/j.ceca.2019.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters-Golden M., Brock T.G. 5-lipoxygenase and FLAP. Prostaglandins Leukot. Essent. Fatty Acids. 2003;69:99–109. doi: 10.1016/s0952-3278(03)00070-x. [DOI] [PubMed] [Google Scholar]
- 46.Perisic O., Fong S., Lynch D.E., Bycroft M., Williams R.L. Crystal structure of a calcium-phospholipid binding domain from cytosolic phospholipase A2. J. Biol. Chem. 1998;273:1596–1604. doi: 10.1074/jbc.273.3.1596. [DOI] [PubMed] [Google Scholar]
- 47.Hammarberg T., Provost P., Persson B., Rådmark O. The N-terminal domain of 5-lipoxygenase binds calcium and mediates calcium stimulation of enzyme activity. J. Biol. Chem. 2000;275:38787–38793. doi: 10.1074/jbc.M006136200. [DOI] [PubMed] [Google Scholar]
- 48.Flamand N., Lefebvre J., Surette M.E., Picard S., Borgeat P. Arachidonic acid regulates the translocation of 5-lipoxygenase to the nuclear membranes in human neutrophils. J. Biol. Chem. 2006;281:129–136. doi: 10.1074/jbc.M506513200. [DOI] [PubMed] [Google Scholar]
- 49.Dixit N., Yamayoshi I., Nazarian A., Simon S.I. Migrational guidance of neutrophils is mechanotransduced via high-affinity LFA-1 and calcium flux. J. Immunol. 2011;187:472–481. doi: 10.4049/jimmunol.1004197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clemens R.A., Chong J., Grimes D., Hu Y., Lowell C.A. STIM1 and STIM2 cooperatively regulate mouse neutrophil store-operated calcium entry and cytokine production. Blood. 2017;130:1565–1577. doi: 10.1182/blood-2016-11-751230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Grimes D., Johnson R., Pashos M., Cummings C., Kang C., Sampedro G.R., Tycksen E., McBride H.J., Sah R., Lowell C.A., Clemens R.A. ORAI1 and ORAI2 modulate murine neutrophil calcium signaling, cellular activation, and host defense. Proc. Natl. Acad. Sci. USA. 2020;117:24403–24414. doi: 10.1073/pnas.2008032117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naccache P.H., Hamelin B., Gaudry M., Bourgoin S. Priming of calcium mobilization in human neutrophils by granulocyte-macrophage colony-stimulating factor: evidence for an involvement of phospholipase D-derived phosphatidic acid. Cell. Signal. 1991;3:635–644. doi: 10.1016/0898-6568(91)90040-2. [DOI] [PubMed] [Google Scholar]
- 53.Watson F., Edwards S.W. Stimulation of primed neutrophils by soluble immune complexes: priming leads to enhanced intracellular Ca2+ elevations, activation of phospholipase D, and activation of the NADPH oxidase. Biochem. Biophys. Res. Commun. 1998;247:819–826. doi: 10.1006/bbrc.1998.8524. [DOI] [PubMed] [Google Scholar]
- 54.Nunes P., Demaurex N., Dinauer M.C. Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis. Traffic. 2013;14:1118–1131. doi: 10.1111/tra.12115. [DOI] [PubMed] [Google Scholar]
- 55.Dinauer M.C. Inflammatory consequences of inherited disorders affecting neutrophil function. Blood. 2019;133:2130–2139. doi: 10.1182/blood-2018-11-844563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paclet M.H., Laurans S., Dupré-Crochet S. Regulation of Neutrophil NADPH Oxidase, NOX2: A Crucial Effector in Neutrophil Phenotype and Function. Front. Cell Dev. Biol. 2022;10:945749. doi: 10.3389/fcell.2022.945749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Henderson W.R., Klebanoff S.J. Leukotriene production and inactivation by normal, chronic granulomatous disease and myeloperoxidase-deficient neutrophils. J. Biol. Chem. 1983;258:13522–13527. [PubMed] [Google Scholar]
- 58.Henderson W.R., Klebanoff S.J. Leukotriene B4, C4, D4 and E4 inactivation by hydroxyl radicals. Biochem. Biophys. Res. Commun. 1983;110:266–272. doi: 10.1016/0006-291x(83)91290-1. [DOI] [PubMed] [Google Scholar]
- 59.Hamasaki T., Sakano T., Kobayashi M., Sakura N., Ueda K., Usui T. Leukotriene B4 metabolism in neutrophils of patients with chronic granulomatous disease: phorbol myristate acetate decreases endogenous leukotriene B4 via NADPH oxidase-dependent mechanism. Eur. J. Clin. Invest. 1989;19:404–411. doi: 10.1111/j.1365-2362.1989.tb00249.x. [DOI] [PubMed] [Google Scholar]
- 60.Murphy R., DeCoursey T.E. Charge compensation during the phagocyte respiratory burst. Biochim. Biophys. Acta. 2006;1757:996–1011. doi: 10.1016/j.bbabio.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 61.Morgan D., Cherny V.V., Finnegan A., Bollinger J., Gelb M.H., DeCoursey T.E. Sustained activation of proton channels and NADPH oxidase in human eosinophils and murine granulocytes requires PKC but not cPLA2 alpha activity. J. Physiol. 2007;579:327–344. doi: 10.1113/jphysiol.2006.124248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Femling J.K., Cherny V.V., Morgan D., Rada B., Davis A.P., Czirják G., Enyedi P., England S.K., Moreland J.G., Ligeti E., et al. The antibacterial activity of human neutrophils and eosinophils requires proton channels but not BK channels. J. Gen. Physiol. 2006;127:659–672. doi: 10.1085/jgp.200609504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.El Chemaly A., Okochi Y., Sasaki M., Arnaudeau S., Okamura Y., Demaurex N. VSOP/Hv1 proton channels sustain calcium entry, neutrophil migration, and superoxide production by limiting cell depolarization and acidification. J. Exp. Med. 2010;207:129–139. doi: 10.1084/jem.20091837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Geiszt M., Kapus A., Német K., Farkas L., Ligeti E. Regulation of capacitative Ca2+ influx in human neutrophil granulocytes. Alterations in chronic granulomatous disease. J. Biol. Chem. 1997;272:26471–26478. doi: 10.1074/jbc.272.42.26471. [DOI] [PubMed] [Google Scholar]
- 65.Rada B.K., Geiszt M., Van Bruggen R., Nemet K., Roos D., Ligeti E. Calcium signalling is altered in myeloid cells with a deficiency in NADPH oxidase activity. Clin. Exp. Immunol. 2003;132:53–60. doi: 10.1046/j.1365-2249.2003.02138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Geiszt M., Kapus A., Ligeti E. Chronic granulomatous disease: more than the lack of superoxide? J. Leukoc. Biol. 2001;69:191–196. [PubMed] [Google Scholar]
- 67.Tintinger G.R., Theron A.J., Steel H.C., Anderson R. Accelerated calcium influx and hyperactivation of neutrophils in chronic granulomatous disease. Clin. Exp. Immunol. 2001;123:254–263. doi: 10.1046/j.1365-2249.2001.01447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rada B.K., Geiszt M., Hably C., Ligeti E. Consequences of the electrogenic function of the phagocytic NADPH oxidase. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2005;360:2293–2300. doi: 10.1098/rstb.2005.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Morgenstern D.E., Gifford M.A., Li L.L., Doerschuk C.M., Dinauer M.C. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 1997;185:207–218. doi: 10.1084/jem.185.2.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Segal B.H., Han W., Bushey J.J., Joo M., Bhatti Z., Feminella J., Dennis C.G., Vethanayagam R.R., Yull F.E., Capitano M., et al. NADPH oxidase limits innate immune responses in the lungs in mice. PLoS One. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Endo D., Fujimoto K., Hirose R., Yamanaka H., Homme M., Ishibashi K.I., Miura N., Ohno N., Aratani Y. Genetic Phagocyte NADPH Oxidase Deficiency Enhances Nonviable Candida albicans-Induced Inflammation in Mouse Lungs. Inflammation. 2017;40:123–135. doi: 10.1007/s10753-016-0461-9. [DOI] [PubMed] [Google Scholar]
- 72.Yoo D.G., Paracatu L.C., Xu E., Lin X., Dinauer M.C. NADPH Oxidase Limits Collaborative Pattern-Recognition Receptor Signaling to Regulate Neutrophil Cytokine Production in Response to Fungal Pathogen-Associated Molecular Patterns. J. Immunol. 2021;207:923–937. doi: 10.4049/jimmunol.2001298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singel K.L., Segal B.H. NOX2-dependent regulation of inflammation. Clin. Sci. 2016;130:479–490. doi: 10.1042/CS20150660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kobayashi S.D., Voyich J.M., Braughton K.R., Whitney A.R., Nauseef W.M., Malech H.L., DeLeo F.R. Gene expression profiling provides insight into the pathophysiology of chronic granulomatous disease. J. Immunol. 2004;172:636–643. doi: 10.4049/jimmunol.172.1.636. [DOI] [PubMed] [Google Scholar]
- 75.Zeng M.Y., Miralda I., Armstrong C.L., Uriarte S.M., Bagaitkar J. The roles of NADPH oxidase in modulating neutrophil effector responses. Mol. Oral Microbiol. 2019;34:27–38. doi: 10.1111/omi.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuijpers T., Lutter R. Inflammation and repeated infections in CGD: two sides of a coin. Cell. Mol. Life Sci. 2012;69:7–15. doi: 10.1007/s00018-011-0834-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warnatsch A., Tsourouktsoglou T.D., Branzk N., Wang Q., Reincke S., Herbst S., Gutierrez M., Papayannopoulos V. Reactive Oxygen Species Localization Programs Inflammation to Clear Microbes of Different Size. Immunity. 2017;46:421–432. doi: 10.1016/j.immuni.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sun Y., Abbondante S., Karmakar M., de Jesus Carrion S., Che C., Hise A.G., Pearlman E. Neutrophil Caspase-11 Is Required for Cleavage of Caspase-1 and Secretion of IL-1β in Aspergillus fumigatus Infection. J. Immunol. 2018;201:2767–2775. doi: 10.4049/jimmunol.1701195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Karmakar M., Katsnelson M., Malak H.A., Greene N.G., Howell S.J., Hise A.G., Camilli A., Kadioglu A., Dubyak G.R., Pearlman E. Neutrophil IL-1β processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J. Immunol. 2015;194:1763–1775. doi: 10.4049/jimmunol.1401624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mohammadi N., Midiri A., Mancuso G., Patanè F., Venza M., Venza I., Passantino A., Galbo R., Teti G., Beninati C., Biondo C. Neutrophils Directly Recognize Group B Streptococci and Contribute to Interleukin-1β Production during Infection. PLoS One. 2016;11:e0160249. doi: 10.1371/journal.pone.0160249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chou R.C., Kim N.D., Sadik C.D., Seung E., Lan Y., Byrne M.H., Haribabu B., Iwakura Y., Luster A.D. Lipid-cytokine-chemokine cascade drives neutrophil recruitment in a murine model of inflammatory arthritis. Immunity. 2010;33:266–278. doi: 10.1016/j.immuni.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nguyen G.T., Green E.R., Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front. Cell. Infect. Microbiol. 2017;7:373. doi: 10.3389/fcimb.2017.00373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mol S., Hafkamp F.M.J., Varela L., Simkhada N., Taanman-Kueter E.W., Tas S.W., Wauben M.H.M., Groot Kormelink T., de Jong E.C. Efficient Neutrophil Activation Requires Two Simultaneous Activating Stimuli. Int. J. Mol. Sci. 2021;22:10106. doi: 10.3390/ijms221810106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fumagalli L., Campa C.C., Germena G., Lowell C.A., Hirsch E., Berton G. Class I phosphoinositide-3-kinases and SRC kinases play a nonredundant role in regulation of adhesion-independent and -dependent neutrophil reactive oxygen species generation. J. Immunol. 2013;190:3648–3660. doi: 10.4049/jimmunol.1201951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Miralda I., Uriarte S.M., McLeish K.R. Multiple Phenotypic Changes Define Neutrophil Priming. Front. Cell. Infect. Microbiol. 2017;7:217. doi: 10.3389/fcimb.2017.00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khoyratty T.E., Ai Z., Ballesteros I., Eames H.L., Mathie S., Martín-Salamanca S., Wang L., Hemmings A., Willemsen N., von Werz V., et al. Distinct transcription factor networks control neutrophil-driven inflammation. Nat. Immunol. 2021;22:1093–1106. doi: 10.1038/s41590-021-00968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hedrick C.C., Malanchi I. Neutrophils in cancer: heterogeneous and multifaceted. Nat. Rev. Immunol. 2022;22:173–187. doi: 10.1038/s41577-021-00571-6. [DOI] [PubMed] [Google Scholar]
- 88.Bongers S.H., Chen N., van Grinsven E., van Staveren S., Hassani M., Spijkerman R., Hesselink L., Lo Tam Loi A.T., van Aalst C., Leijte G.P., et al. Kinetics of Neutrophil Subsets in Acute, Subacute, and Chronic Inflammation. Front. Immunol. 2021;12:674079. doi: 10.3389/fimmu.2021.674079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Evrard M., Kwok I.W.H., Chong S.Z., Teng K.W.W., Becht E., Chen J., Sieow J.L., Penny H.L., Ching G.C., Devi S., et al. Developmental Analysis of Bone Marrow Neutrophils Reveals Populations Specialized in Expansion, Trafficking, and Effector Functions. Immunity. 2018;48:364–379.e8. doi: 10.1016/j.immuni.2018.02.002. [DOI] [PubMed] [Google Scholar]
- 90.Simmons S.R., Bhalla M., Herring S.E., Tchalla E.Y.I., Bou Ghanem E.N. Older but Not Wiser: the Age-Driven Changes in Neutrophil Responses during Pulmonary Infections. Infect. Immun. 2021;89:e00653-20. doi: 10.1128/IAI.00653-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sun K., Metzger D.W. Influenza infection suppresses NADPH oxidase-dependent phagocytic bacterial clearance and enhances susceptibility to secondary methicillin-resistant Staphylococcus aureus infection. J. Immunol. 2014;192:3301–3307. doi: 10.4049/jimmunol.1303049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jasper A.E., McIver W.J., Sapey E., Walton G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Res. 2019;8 doi: 10.12688/f1000research.18411.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Engelich G., Wright D.G., Hartshorn K.L. Acquired disorders of phagocyte function complicating medical and surgical illnesses. Clin. Infect. Dis. 2001;33:2040–2048. doi: 10.1086/324502. [DOI] [PubMed] [Google Scholar]
- 94.Dowey R., Iqbal A., Heller S.R., Sabroe I., Prince L.R. A Bittersweet Response to Infection in Diabetes; Targeting Neutrophils to Modify Inflammation and Improve Host Immunity. Front. Immunol. 2021;12:678771. doi: 10.3389/fimmu.2021.678771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wigerblad G., Cao Q., Brooks S., Naz F., Gadkari M., Jiang K., Gupta S., O'Neil L., Dell'Orso S., Kaplan M.J., Franco L.M. Single-Cell Analysis Reveals the Range of Transcriptional States of Circulating Human Neutrophils. J. Immunol. 2022;209:772–782. doi: 10.4049/jimmunol.2200154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Xie X., Shi Q., Wu P., Zhang X., Kambara H., Su J., Yu H., Park S.Y., Guo R., Ren Q., et al. Single-cell transcriptome profiling reveals neutrophil heterogeneity in homeostasis and infection. Nat. Immunol. 2020;21:1119–1133. doi: 10.1038/s41590-020-0736-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Albrett A.M., Ashby L.V., Dickerhof N., Kettle A.J., Winterbourn C.C. Heterogeneity of hypochlorous acid production in individual neutrophil phagosomes revealed by a rhodamine-based probe. J. Biol. Chem. 2018;293:15715–15724. doi: 10.1074/jbc.RA118.004789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li X.J., Tian W., Stull N.D., Grinstein S., Atkinson S., Dinauer M.C. A fluorescently tagged C-terminal fragment of p47phox detects NADPH oxidase dynamics during phagocytosis. Mol. Biol. Cell. 2009;20:1520–1532. doi: 10.1091/mbc.e08-06-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maskarinec S.A., McKelvy M., Boyle K., Hotchkiss H., Duarte M.E., Addison B., Amato N., Khandelwal S., Arepally G.M., Lee G.M. Neutrophil functional heterogeneity is a fixed phenotype and is associated with distinct gene expression profiles. J. Leukoc. Biol. 2022;112:1485–1495. doi: 10.1002/JLB.4A0322-164R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Idol R.A., Bhattacharya S., Huang G., Song Z., Huttenlocher A., Keller N.P., Dinauer M.C. Neutrophil and Macrophage NADPH Oxidase 2 Differentially Control Responses to Inflammation and to Aspergillus fumigatus in Mice. J. Immunol. 2022;209:1960–1972. doi: 10.4049/jimmunol.2200543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Morgan M.J., Liu Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011;21:103–115. doi: 10.1038/cr.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liese J., Rooijakkers S.H.M., van Strijp J.A.G., Novick R.P., Dustin M.L. Intravital two-photon microscopy of host-pathogen interactions in a mouse model of Staphylococcus aureus skin abscess formation. Cell Microbiol. 2013;15:891–909. doi: 10.1111/cmi.12085. [DOI] [PubMed] [Google Scholar]
- 103.Waite J.C., Leiner I., Lauer P., Rae C.S., Barbet G., Zheng H., Portnoy D.A., Pamer E.G., Dustin M.L. Dynamic imaging of the effector immune response to listeria infection in vivo. PLoS Pathog. 2011;7:e1001326. doi: 10.1371/journal.ppat.1001326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bogoslowski A., Butcher E.C., Kubes P. Neutrophils recruited through high endothelial venules of the lymph nodes via PNAd intercept disseminating Staphylococcus aureus. Proc. Natl. Acad. Sci. USA. 2018;115:2449–2454. doi: 10.1073/pnas.1715756115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Brandt S.L., Wang S., Dejani N.N., Klopfenstein N., Winfree S., Filgueiras L., McCarthy B.P., Territo P.R., Serezani C.H. Excessive localized leukotriene B4 levels dictate poor skin host defense in diabetic mice. JCI Insight. 2018;3:e120220. doi: 10.1172/jci.insight.120220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Brandt S.L., Serezani C.H. Too much of a good thing: How modulating LTB4 actions restore host defense in homeostasis or disease. Semin. Immunol. 2017;33:37–43. doi: 10.1016/j.smim.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sadik C.D., Kim N.D., Luster A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011;32:452–460. doi: 10.1016/j.it.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sadik C.D., Luster A.D. Lipid-cytokine-chemokine cascades orchestrate leukocyte recruitment in inflammation. J. Leukoc. Biol. 2012;91:207–215. doi: 10.1189/jlb.0811402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liao Y.C., Wu S.Y., Huang Y.F., Lo P.C., Chan T.Y., Chen C.A., Wu C.H., Hsu C.C., Yen C.L., Chen P.C., Shieh C.C. NOX2-Deficient Neutrophils Facilitate Joint Inflammation Through Higher Pro-Inflammatory and Weakened Immune Checkpoint Activities. Front. Immunol. 2021;12:743030. doi: 10.3389/fimmu.2021.743030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barros N., Alexander N., Viens A., Timmer K., Atallah N., Knooihuizen S.A.I., Hopke A., Scherer A., Dagher Z., Irimia D., Mansour M.K. Cytokine Augmentation Reverses Transplant Recipient Neutrophil Dysfunction Against the Human Fungal Pathogen Candida albicans. J. Infect. Dis. 2021;224:894–902. doi: 10.1093/infdis/jiab009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Knooihuizen S.A.I., Alexander N.J., Hopke A., Barros N., Viens A., Scherer A., Atallah N.J., Dagher Z., Irimia D., Chung R.T., Mansour M.K. Loss of Coordinated Neutrophil Responses to the Human Fungal Pathogen, Candida albicans, in Patients With Cirrhosis. Hepatol. Commun. 2021;5:502–515. doi: 10.1002/hep4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wang G., Nauseef W.M. Neutrophil dysfunction in the pathogenesis of cystic fibrosis. Blood. 2022;139:2622–2631. doi: 10.1182/blood.2021014699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yonker L.M., Marand A., Muldur S., Hopke A., Leung H.M., De La Flor D., Park G., Pinsky H., Guthrie L.B., Tearney G.J., et al. Neutrophil dysfunction in cystic fibrosis. J. Cyst. Fibros. 2021;20:1062–1071. doi: 10.1016/j.jcf.2021.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]