

Abstract
Background
Limb-girdle muscular dystrophies (LGMD) are a heterogeneous group of genetically determined muscle disorders. TRAPPC11-related LGMD is an autosomal-recessive condition characterised by muscle weakness and intellectual disability.
Methods
A clinical and histopathological characterisation of 25 Roma individuals with LGMD R18 caused by the homozygous TRAPPC11 c.1287+5G>A variant is reported. Functional effects of the variant on mitochondrial function were investigated.
Results
The c.1287+5G>A variant leads to a phenotype characterised by early onset muscle weakness, movement disorder, intellectual disability and elevated serum creatine kinase, which is similar to other series. As novel clinical findings, we found that microcephaly is almost universal and that infections in the first years of life seem to act as triggers for a psychomotor regression and onset of seizures in several individuals with TRAPPC11 variants, who showed pseudometabolic crises triggered by infections. Our functional studies expanded the role of TRAPPC11 deficiency in mitochondrial function, as a decreased mitochondrial ATP production capacity and alterations in the mitochondrial network architecture were detected.
Conclusion
We provide a comprehensive phenotypic characterisation of the pathogenic variant TRAPPC11 c.1287+5G>A, which is founder in the Roma population. Our observations indicate that some typical features of golgipathies, such as microcephaly and clinical decompensation associated with infections, are prevalent in individuals with LGMD R18.
Keywords: neuromuscular diseases; genetics, population; movement disorders; epilepsy
What is already known on this topic
TRAPPC11-related limb-girdle muscular dystrophies (LGMD), which has been reported so far in no more than 20 individuals, is clinically characterised by muscle weakness, intellectual disability and hyperkinetic movements.
What this study adds
Our phenotypic characterisation of the pathogenic variant TRAPPC11 c.1287+5G>A, which is founder in the Roma population, indicates that some typical features of golgipathies, such as microcephaly and clinical decompensation associated with infections, are prevalent in individuals with TRAPPC11-related LGMD.
Our functional studies expanded the role of TRAPPC11 deficiency in mitochondrial function.
How this study might affect research, practice or policy
Our findings allow us to refine and expand the emerging concept of golgipathies through a better understanding of the phenotype associated with TRAPPC11-related LGMD.
Introduction
Limb-girdle muscular dystrophies (LGMD) are a heterogeneous group of genetically determined muscle disorders with predominant proximal weakness.1 The age of onset, severity and clinical course may vary among LGMD subtypes, ranging from early onset myopathy with rapid progression to adult-onset with long-time preserved ambulation and normal life span.2 3
TRAPPC11-related LGMD R18, previously known as LGMD2S,4 is an autosomal-recessive congenital disorder of glycosylation that is clinically characterised by muscle weakness, intellectual disability and hyperkinetic movements.5–7 TRAPPC11 encodes subunit 11 of the multiprotein TRAPP complex, a regulator of membrane trafficking and autophagy whose best characterised biochemical function is to act as a guanine nucleotide exchange factor to activate Ypt/Rab GTPases.8–10 Pathogenic variants or deletions of TRAPP subunits are involved in several neurodevelopmental disorders, known as TRAPPopathies.11 12 TRAPPopathies belong to the rapidly growing category of disorders of cell trafficking and in particular to membrane trafficking defects. The recent report of the cryo-EM structure of the Drosophila TRAPPIII complex has revealed how the TRAPP subunits assemble and the TRAPPC11 structure, shedding light on how disease-causing variants affect the structure and function of TRAPPC11.13
No more than 20 individuals and 12 variants in TRAPPC11 have been reported so far.5–7 14–19 Here, we report a founder pathogenic variant (TRAPPC11 c.1287+5G>A; p.Ala372_Ser429del) in the Roma population and provide a comprehensive phenotypic characterisation based on the description of 25 previously unreported Spanish Roma individuals who harbour the variant in homozygosis. There are no previous reports of a direct link between TRAPPC11 and mitochondrial function, but it is not uncommon to find associated mitochondrial defects in membrane trafficking disorders.20 21 We have therefore studied mitochondrial energy metabolism in individuals with the variant TRAPPC11 c.1287+5G>A to better understand the possible involvement of mitochondria in the pathophysiology of TRAPPC11-related muscular dystrophy.
Methods
Recruitment of patients, clinical examinations and molecular genetic analyses
Data of individuals with a genetically confirmed LGMD R18 were collected at 12 Spanish hospitals after sending a request for collaboration to all the members of the Spanish Paediatric Neurology Society. Demographic, clinical, genetic, electroencephalographic, neuroradiological and muscle biopsy data were collected in accordance with the ethics guidelines of each of the institutions involved. The degree of intellectual disability was classified according to the impression of the neurologist who followed each individual, but a neuropsychological assessment was not carried out systematically in all individuals.
Genomic DNA samples were extracted from peripheral blood of affected individuals. Different next-generation sequencing methodologies were used to prepare and capture genomic DNA libraries, from customised panels for selected genes to exome sequencing. The same single nucleotide variant c.1287+5G>A in TRAPPC11, classified as pathogenic according to the American College of Medical Genetics and Genomics guidelines for germline variant classification,22 was found in homozygosis in all the individuals. Segregation was performed on the asymptomatic parents of 13 individuals.
In silico analysis of variant TRAPPC11 c.1287+5G>A
A homology molecular model for human TRAPPC11 (positions 1–574) and TRAPPC2L (positions 1–138) was constructed based on Drosophila TRAPPIII complex structures (7B6H and 7B6R),13 using Modeller.23 Positions 61–75 and 378–405, with no determined structure in the template structure, were also modelled. Sequence identity was 42.1% and 52.9% for TRAPPC11 and TRAPPC2L, respectively. Human TRAPPC11 structural model was visualised using Pymol24 (http://www.pymol.org/pymol).
Muscle and skin biopsies, histological, immunohistochemical, immunofluorescence, ultrastructural studies and western blot analysis
Muscle biopsy samples were obtained for diagnostic purposes and processed following standard histological, histochemical and immunohistochemical protocols as previously described.25 Skin biopsy samples were obtained and fibroblasts were cultered according to Hospital Sant Joan de Déu standard clinical procedures. Specific antibodies used for immunohistochemical or immunofluorescence studies are listed in online supplemental table 1. Electron microscopy studies were performed on muscle samples from individual 5. Semi-quantitative western blot analysis on fibroblasts and muscle lysates using antibodies against TRAPPC11 and alpha-dystroglycan (IIH6 and VIA4) was performed as previously described.26 All the muscle biopsies and western blot analysis studies were expressly reviewed for this study by a pathologist (CJ), a clinical scientist (ACo) and a paediatric neurologist (DN-dB), all three specialists in neuromuscular disorders. Control muscle samples were taken from two children aged 5 and 10 years who did not have neuromuscular disease, from quadriceps and from an unknown muscle. Control fibroblasts were obtained from a skin biopsy of a child aged 10 years with no neuromuscular disease.
jmg-2022-109132supp001.pdf (47.4KB, pdf)
Characterisation of epilepsy
Classification of seizures, epilepsy course and available EEGs were reviewed by an epileptologist specialised in children (JD-C). Seizures and epilepsy were described and classified according to the International League Against Epilepsy classification.27 28 Bilateral tonic-clonic seizures in which the onset of the seizure was not observed and which could be either ‘generalised seizures’ or ‘focal seizures that evolve into bilateral tonic-clonic seizures’ were referred to as ‘bilateral tonic-clonic seizures’. Epilepsy course was classified as (a) ‘early seizure freedom’ (seizure-free within 6 months of starting treatment), (b) ‘delayed seizure freedom’ (seizures not immediately controlled by medication, but became seizure-free at some point after 6 months), (c) ‘fluctuating course’ (periods of seizure freedom of >12 months, interspersed with relapses) or (d) ‘refractory’ (never seizure-free for a continuous 12-month period) following the method defined by Brodie et al.29
Functional analysis of the variant on mitochondrial function
Functional studies have included the analysis of non-glycolytic ATP production and mitochondrial network visualisation. Mitochondrial (as in non-glycolytic) ATP concentration was measured as explained in the study by Oyarzabal et al.30 Briefly, cells from individuals with TRAPPC11-related muscular dystrophy and cells from controls were incubated with 2-deoxy-D-glucose for 2 hours to discriminate between total and non-glycolytic ATP. Finally, ATP concentration was assayed by bioluminescence using a luciferin-luciferase system (ATP Bioluminescence Assay Kit CLS II, Roche) according to the manufacturer’s instructions, corrected by protein concentration and expressed as folds over controls.
The mitochondrial network was visualised through immunofluorescence against TOMM20 (1:250, ab186735) following standard protocols. Fluorescence was imaged using Zeiss LSM 880 microscope (Zeiss, Germany) with a 63× magnification. Images were analysed using Fiji software and parametrised by Mitochondrial Analyzer plugin.31 All experiments are done at least three times with triplicates.
At the time the fibroblasts were biopsied, one of the individuals with TRAPPC11-related muscular dystrophy was 4 years old and the other was 12 years old. All controls, which included both males and females, were within this age range. All cells used were between passages 7 and 12.
Results
Demographics and molecular genetics
A total of 25 individuals with TRAPPC11-related muscular dystrophy (13 males, 12 females) from 18 families were collected. All of them were Spanish with a Roma origin and 22 of 23 cases (96%) were born from consanguineous parents. The mean age at the last examination was 11.8 years old (range 2–23). The previously reported homozygous splice-site variant TRAPPC11 (NM_021942.6) c.1287+5G>A was found in all the individuals. Segregation studies were performed on the parents of 13 individuals, confirming that the pathogenic variants were inherited each from one of the unaffected parents.
Structure consequence of TRAPPC11 c.1287+5G>A variant
The splice-site variant TRAPPC11 c.1287+5G>A results in an in-frame deletion of 58 amino acids (p.Ala372_Ser429del).5 Figure 1 shows human TRAPPC11 and TRAPPC2L structural model interaction. The deletion of residues 372–429 in TRAPPC11 is located within the conserved foie gras domain (residues 181–566), which is responsible for the surface interaction with TRAPPC2L.13 The lacking of this region has been recently described as resulting in defects in membrane trafficking,5 autophagy and N-linked glycosylation.17
Figure 1.
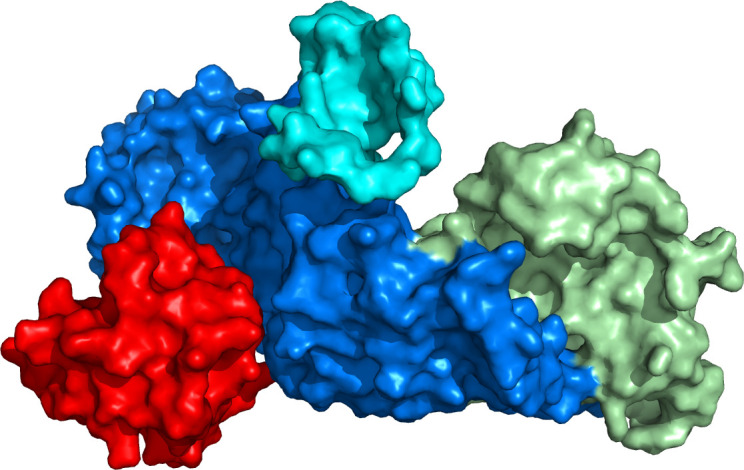
Molecular model of human TRAPPC11 (residues 1–574, in blue and green) interacting with human TRAPPC2L (residues 1–136, in red), in the TRAPPIII complex. TRAPPC11 foie gras domain (residues 181–566, in cyan blue) is essential for TRAPPC2L interaction. This domain contains residues 372–429 (in dark blue), which are lacking in TRAPPC11 c.1287+5G>A splice-site variant. The N-terminal domain of TRAPPC11 is shown in green.
Clinical features
A history of late premature birth was reported in 3/24 individuals (12.5%) and low birth weight in 3/23 (13%). Progressive and highly significant microcephaly was almost universally found (21/23; 91%), being detected at birth in 38% of individuals and acquired during the first years of life in 52%. The mean head circumference SD score was −3.45, ranging from −1.55 to −7.2. Individuals tended to be short, with a mean height percentile of p12 compared with the Spanish population (range p<1–p35).19 A height percentile below p2 was observed in 27% (4/15). Three individuals had neonatal hypotonia and none of them had congenital cataracts.
The most common initial presentation was a global psychomotor delay (21/25; 84%), while the presenting symptom was seizures in the four individuals who had seizures before the age of 7 months. A global psychomotor delay was observed at some point in 96% of individuals (24/25), and a subsequent intellectual disability was described in all individuals who were older than 4 years at the last follow-up (23/23). The intellectual disability was severe in eight individuals, moderate-to-severe in five, moderate in seven, mild in one and of an unknown degree in two. It is noteworthy that four individuals (16%) experienced a psychomotor regression, with loss of developmental milestones previously attained, triggered by infections. These triggering infections were even initially classified as encephalitis in these individuals, as there was drowsiness, febrile seizures and a slowed EEG. No causative microorganism was identified in the cerebrospinal fluid. These infections occurred at a median age of 13 months old (range: 4–36).
A severe language impairment was detected in 21/25 individuals (84%). Movement disorders, including hyperkinesia and choreiform movements, were reported in 79% of cases (19/24). None of the individuals with TRAPPC11-related muscular dystrophy showed signs of spasticity. The main clinical features are summarised in online supplemental table 2.
jmg-2022-109132supp002.pdf (43.7KB, pdf)
All individuals had elevated creatine kinase (CK) in serum, ranging from 300 to 5000 IU/L (mean: 1440 IU/L, SD: 1009 IU/L). Independent ambulation was acquired by 23 from 24 individuals (96%) at a mean age of 22 months, with a delay in the acquisition of autonomous ambulation—understood as above 18 months of age—observed in 40% (8/20). An early onset proximal weakness was detected in 71% of individuals (17/24), associated with myalgia and cramps in six of them (6/23; 26%). Rhabdomyolysis was not identified in any of the individuals.
Epilepsy, defined as recurrent seizures, was diagnosed in 12/25 individuals (48%), and the mean age at first seizure was 21 months (range: 4 months–8 years; median 12 months). All 12 individuals with epilepsy were treated with antiepileptic drugs. The most common type of seizure was bilateral tonic-clonic seizures, either with focal onset or with unknown onset, being experienced by 92% of individuals with epilepsy (11/12). Focal-onset seizures with impaired awareness were observed in 33% of individuals with epilepsy (4/12). Remarkably, half of individuals with epilepsy (6/12; 50%) had a history of status epilepticus, four of them in the context of fever. Seizures triggered by infections were observed in 83% of individuals (10/12). Seizure characteristics, as well as main electroclinical and neuroradiological features, are summarised in online supplemental table 2.
EEG reports were available in all 11 individuals with epilepsy. Selected illustrative EEG studies are shown in figure 2. Eight individuals (67%) showed interictal focal or multifocal epileptiform abnormalities. A high degree of variability was observed in the origin of the epileptiform abnormalities (online supplemental table 2). Although the temporal origin was slightly more frequent than in other locations, the difference was not significant and is not different from that observed in other individuals with epilepsy, given that temporal lobe epilepsy is the most common form of focal epilepsy.32 33 Background activity was slow in 42% of individuals with epilepsy (5/12). Photosensitivity was not present in any of the individuals.
Figure 2.

Awake EEG studies of individuals 5 and 8. Longitudinal bipolar montage at sensitivity 70 μV/cm is shown in all the cases. Individual 5: while EEG at 14 months old during his first seizures showed a slow and low voltage background activity without epileptic abnormalities (A1), the EEG at the age of 2 years, during wakefulness with eyes closed, showed a slow background activity without epileptiform abnormalities (A2). Individual 8: EEGs at the age of 13 years, during wakefulness with eyes closed (B1) and eyes open (B2) showed normal background activity, with epileptic abnormalities (sharp waves and spikes) in bilateral frontocentrotemporal region.
Long-term seizure control was assessed in the 12 individuals with epilepsy based on the Brodie classification system.29 Six individuals (50%) were seizure-free within 6 months of starting treatment (a), two individuals (17%) were seizure-free after 6 months of starting treatment (b) and 4 (33%) had a fluctuating course (c; periods of at least 12 months seizure-free, interspersed with relapses). A favourable response to monotherapy treatment was observed in 7/12 individuals (58%). A summary of medications used in each individual at the last follow-up is provided in online supplemental table 2. At the last visit, four individuals were seizure-free with levetiracetam monotherapy and three remained seizure-free after antiepileptic drug withdrawal. Non-pharmacological treatments, including the ketogenic diet, vagus nerve stimulation and epilepsy surgery, were not tried on any individual.
Brain MRI was performed in 84% of individuals (21/25). A generalised decrease in cortico-subcortical volume, including white matter, was found in 29% of them (6/21), with no other associated structural abnormalities. No hypomyelination was observed in the brain MRI of any individual. A prominence of the cerebellar folia was observed in individual 10 but obvious cerebellar atrophy or hypoplasia was not observed in any individual (figure 3 and online supplemental table 2).
Figure 3.

A progressive cortical and subcortical atrophy is observed in individual 5. MRIs at the age of 14 months (A) and 5 years (B) are shown, axial T1.
Scoliosis was identified in 50% of individuals (5/10) over 13 years of age. No cardiac involvement was observed in the 15 individuals in whom cardiac evaluation with ECG and echocardiography was performed, except in one individual with mild dilatation of the aortic root and ascending aorta.
Morphological findings in muscle biopsies
Muscle biopsies were performed on seven individuals, taken from either the quadriceps (five individuals), the deltoids (one individual) or an unknown location (one individual), at a mean age of 5.5 years (SD: 4.5; median: 3). The main features of muscle biopsies are shown in figure 4 and in online supplemental table 3. Muscle histology showed myopathic changes with variability in fibre size and some fibres with internalised nuclei. Intermyofibrillar pattern disruptions, with areas devoid of staining for oxidative enzymes (Nicotinadmide adenine dinucleotide and Succinate dehydrogenase), were observed in all the individuals. Occasional myonecrosis and regeneration (displayed as neonatal myosin-positive fibres) were observed, while increased endomysial or perimysial connective tissue with fibrosis or fatty infiltration were not observed in any of the cases. Fibre-type distribution was normal, with a mosaic pattern, in five cases while atrophy of type II fibres was observed in two individuals.
Figure 4.

Myopathic muscle pathology from quadriceps and deltoid biopsies. Biopsy from Individual 4 (A–D, I–L and U–W) was taken from quadriceps at 3 years and biopsy from individual 8 (E–H and M–P) was taken from deltoid at 12 years. Variability in fibre size and fibres with internalised nuclei were observed with H&E stain in individuals 4 and 8 (A, E), as well as hypercontracted fibres (arrow A, F). Increased connective tissue with fibrosis or fatty infiltration were not observed (B, F). Blurred unstained areas in some fibres were noted with SDH (C, G). Selective atrophy of fast fibres is noted with double immunostaining of myosins. Slow-type fibres stained red and fast-type fibres stained green (D, H). Sarcolemmal beta-dystroglycan was normal (I, M) compared with control (Q), which implies that the integrity of the sarcolemma is not altered. A reduction of sarcolemmal alpha-dystroglycan with a mosaic pattern (asterisks in fibres with reduced alpha-dystroglycan) was noted in individuals 4 and 8 with IIH6 (J and N) and VIA 4 (K and O) antibodies, compared with control (R and S). Merosin immunolabelling was normal in individuals 4 (L) and 8 (P) compared with control (T). Electron microscopy (U, V and W) performed in individual 4 showed focal Z-band streamings (arrow in U), a necrotic fibre with a structural disruption of the sarcomere (V) and electron-dense degraded material (arrow in W). Scale bar=100 µm in A–T. Scale bar=5 µm in U and W. Scale bar=2 µm in V.
jmg-2022-109132supp003.pdf (76.4KB, pdf)
Immunofluorescence labelling at the sarcolemma with two antibodies to alpha-dystroglycan (IIH6 and VIA4) showed a patchy or mosaic reduction of alpha-dystroglycan in 5/6 individuals, similar to four individuals with TRAPPC11-related muscular dystrophy recently described by Munot et al.18 Immunohistochemical studies for spectrin, dystrophin, sarcoglycans, beta-dystroglycan and laminin were normal. Overexpression of utrophin was not noted, except in regenerative fibres, which is physiological (figure 4).
Electron microscopy performed in individual 4 showed focal Z-band streamings, a necrotic fibre with an almost completely absent myofibrillar structure that was replaced by granular amorphous material as well as a fibre electron-dense degraded material (figure 4).
Variability in fibre size and the presence of atrophic fibres were the most prominent ultrastructural findings.
Western blot analysis studies
To study TRAPPC11 expression levels, a western blot analysis against TRAPPC11 was performed on fibroblast lysates from two individuals (individuals 5 and 6). No differences were observed compared with the control (figure 5A). An alpha-dystroglycan western blot analysis (VIA4 and IIH6 antibodies) was performed on five individuals and compared with controls to study whether glycosylation in muscle was altered. A reduction of alpha-dystroglycan expression was observed in two of the five individuals compared with the healthy control. The reduction in band intensity in individuals 8 and 11 was subtle-to-moderate. The intensity of the band in individuals 4, 5 and 13 was not reduced (figure 5B). No clear alteration in their mean molecular weight was observed compared with the control, except for individual 11, where the molecular weight appears slightly lower, which could be related to protein instability.
Figure 5.

Western blot analyses of TRAPPC11 (in fibroblasts) and alpha-dystroglycan (αDG) glycosylation (in muscle) in some patients. TRAPPC11 expression (A) was not different in fibroblasts from individuals (Ind) 5 and 6 compared with control while αDG glycosylation in muscle was reduced in some individuals. A subtle-to-moderate reduction was noted in Ind 8 and 11 compared with controls with antibodies VIA4 and IIH6. No reductions were noted in Ind 4, 5 and 13.
Pathophysiology of TRAPPC11 deficiency
We investigated the effect of TRAPPC11 deficiency on mitochondrial function in the fibroblasts of two individuals with TRAPPC11-related muscular dystrophy. Analysis of non-glycolytic ATP production (assumed as mitochondrial ATP) revealed a significant decrease of ~30% in the fibroblasts of individuals with TRAPPC11-related muscular dystrophy. Together with this finding, we observed some mild alterations in the mitochondrial network architecture, as individuals with TRAPPC11-related muscular dystrophy showed more mitochondria but with fewer and shorter branches, resulting in a ‘less structured mesh’ (figure 6).
Figure 6.

Analysis of the mitochondrial impact of TRAPPC11 deficiency. We explored mitochondrial function in terms of non-glycolytic ATP production (A) through a 2-hour incubation of fibroblasts from individuals with TRAPPC11-related muscular dystrophy and controls with 2-deoxy-D-glucose (DG) and a subsequent luciferin/luciferase assay. Results are presented as folds over control. Mitochondrial network was assayed by immunofluorescence against the mitochondrial marker TOMM20: representative images are shown in B and quantification results in C, showing an increase in mitochondrial counts, whose networks are less branched and with shorter mitochondria. Although only one representative image is shown for both control and patients, several fields of independent preparations in triplicate were analysed. The mitochondrial networks were analysed with the ImageJ plugin ‘Mitochondrial Analyzer’. Nuclei are marked in blue and mitochondria in magenta. Scale bar marks 10 µm. Images have been parametrised through Mitochondrial Analyzer. Unpaired Welch’s t-test was performed. *P<0.05; **p<0.01; ****p<0.0001.
Discussion
We report a comprehensive description of the phenotype of 25 individuals with LGMD due to a homozygous c.1287+5G>A splice-site variant in TRAPPC11. Variant c.1287+5G>A has been previously reported in homozygosity in five individuals from two Hutterite families who had myopathy, infantile hyperkinetic movements, ataxia and intellectual disability.5 Subsequently, it was also described in compound heterozygosity along with a frameshift variant (c.3379_3380insT) in a Spanish individual who was not Roma and showed hypotonia, spasticity, choreiform movements and remarkable cerebral atrophy.17 To our knowledge, this is the first time such a large number of TRAPPC11-related LGMD R18 individuals with the same homozygous variant are compiled in a single study. Furthermore, all individuals in our sample were Roma, suggesting TRAPPC11 c.1287+5G>A is a founder pathogenic variant in Roma ethnicity.
Founder pathogenic variants are often responsible for the high prevalence of rare genetic disorders in specific populations. It is known that the Hutterite population, a communal ethnoreligious group currently settled in the USA and Canada, was first established in the 16th century in Austria and subsequently travelled through various regions of Europe.34 In the 17th century, both Roma and Hutterite populations were present in Romania, which could be a hypothesis for the presence of the same variant in two ethnicities that are so remote from each other.
The main phenotypic characteristics of the individuals in our cohort do not differ substantially from those already described in previously reported individuals with pathogenic variants in TRAPPC11. Early onset muscle weakness, movement disorders, intellectual disability, epilepsy and elevation of CK in serum were almost constant findings in the individuals presented here. However, in our cohort, we have identified two additional phenotypic characteristics that are relevant and have not been previously reported. The first is a progressive and highly significant microcephaly that was almost universally found, often associated with a generalised decrease in cortico-subcortical volume. Although microcephaly has not been recognised as a feature associated with TRAPPC11 until now, having been reported in only one individual so far,18 postnatal microcephaly is a typical finding in most of the called ‘golgipathies’, a term used to describe those diseases caused by defects in Golgi-related genes.35–40 The second novel finding is that, according to the information gathered in our cohort, infections in the first years of life seemed to act as triggers for a psychomotor regression and onset of seizures in some of the TRAPPC11 individuals: psychomotor regression triggered by infections was observed in 16% of the individuals (4/25), and seizures triggered by infections were observed in 10/25 individuals (40%), including four who had a history of febrile status epilepticus. Even in four individuals with seizures and psychomotor regression the first diagnostic orientation was encephalitis. Indeed, other TRAPPopathies have been related to these episodes such as two novel homozygous variants in TRAPPC2L that associate postinfectious encephalopathy, developmental arrest, tetraplegia and rhabdomyolysis.41 42
These febrile illness-triggered seizures, usually presenting in the first 2 years of life, and occasionally associated with psychomotor regression, are a recognisable finding in many TRAPPC11 individuals and are similar to the phenotype showed by the few TRAPPC2L individuals reported so far.42 In addition, individuals with pathogenic variants in TRAPPC11 and TRAPPC2L share other clinical signs such as neurodevelopmental delay with speech difficulties, microcephaly and extrapyramidal symptoms. This marked phenotypic similarity leads us to hypothesise that variant TRAPPC11 c.1287+5G>A, located in the foie gras domain, which is essential for interaction with TRAPPC2L, may alter the interaction between TRAPPC2L and TRAPPC11 (figure 1). In an analogous manner, it is possible that the variants described in TRAPPC2L weaken the interaction between the two proteins, thus explaining this unique phenotypic similarity. The reason why pathogenic variants of several subunits of the TRAPP complex result in microcephaly (TRAPPC2L, TRAPPC6, TRAPPC9, TRAPPC11) while others do not is unclear but may be related to the function of these proteins within the TRAPP complex and their role in membrane traffic.
On the other hand, the acute episodes observed in TRAPPC11 individuals partially resemble the acute pseudometabolic crises triggered by infections that are well-described in individuals with pathogenic variants in TANGO2 43 and other cell trafficking disorders. Cell trafficking disorders, which encompass 346 genes so far, are mostly progressive diseases due to the homeostatic loss of crucial molecular functions involved in neurodegeneration such as lipid transfer and membrane remodelling.44 Although rhabdomyolysis and sustained CK elevation are hallmarks of some TRAPPopathies, they can also be observed in other anterograde trafficking disorders such as TANGO2, in which rhabdomyolysis during acute crises is associated with hypoglycaemia, hyperlactataemia and hyperammonaemia, or LPIN1, in which recurrent hyperCKaemia occurs.40 All these features point towards a major metabolic involvement and make it advisable that cell trafficking disorders are included in the differential diagnoses of inborn errors of metabolism of small molecules (in particular intoxication disorders) and energy defects. These findings suggest that stressful situations such as infections impact the normal functioning of the trafficking machinery between the endoplasmic reticulum (ER) and the Golgi apparatus in individuals with TRAPPC11 pathogenic variants and other disorders. The precise mechanisms leading to these manifestations need to be further described.
Although epilepsy represents a significant symptom of LGMD R18 with a serious impact on quality of life, to date neither the types of seizures nor the electroencephalographic abnormalities had been studied systematically in a well-described cohort of individuals. In our cohort, epilepsy occurred in more than half of individuals with TRAPPC11-related muscular dystrophy and was typically presented with early childhood-onset long-lasting bilateral tonic-clonic seizures. EEG studies showed focal or multifocal interictal epileptiform abnormalities in most of the cases. The response to antiepileptic drugs was good in a significant proportion of individuals but not in all.
Our pathological studies on muscle biopsy from seven individuals revealed a myopathic pattern with degenerative-regenerative fibres, isolated hypercontracted fibres and alterations in the intermyofibrillar pattern. Although the fibre-type distribution pattern was normal in most individuals, selective atrophy of fast fibres was identified in two cases. We detected a patchy reduction of alpha-dystroglycan of variable intensity, both with immunofluorescence and western blot analysis, which validates similar findings recently reported by Munot et al.18
TRAPPC11 is a protein involved in membrane trafficking10 44 and had been previously related to autophagy defects, with a role in membrane recruitment for the autophagosome formation.45 Our results expand the role of TRAPPC11 in mitochondrial function, as we observe a decreased mitochondrial ATP production capacity and network architecture in TRAPPC11-deficient fibroblasts. This sets the base for further research on the potential role of TRAPPC11 within mitochondria-Golgi-ER triad and its specific role on mitochondrial functions, which should be delimited with further experiments that are beyond the scope of this work, analysing the involvement of other mitochondrial functions in TRAPPC11-related muscular dystrophies such as the activity and expression of electron transport chain complexes, mitochondrial ultrastructure, reactive oxygen species production and metabolism, mitophagy and mitochondrial signalling. These findings may also help to develop targeted therapies.
In summary, we provide a comprehensive phenotypic characterisation of the pathogenic variant TRAPPC11 c.1287+5G>A, which is founder in the Roma population. Our observations indicate that glycosylation of alpha-dystroglycan is reduced in the skeletal muscle of individuals with LGMD R18 and we emphasise some features that are typical of golgipathies, such as microcephaly and clinical decompensation associated with infections, are probably more prevalent than previously thought.
Acknowledgments
We are indebted to the ‘Biobanc de l’HospitalInfantil Sant Joan de Déu per a la Investigació’, part of the Spanish Biobank Network of ISCIII for the sample and data procurement.
Footnotes
Twitter: @PochMarisa
ÀG-C and DN-dB contributed equally.
Contributors: MJ had a major role in the acquisition of the data, analysed the data, interpreted the data and drafted the manuscript for intellectual content. ÀG-C and DN-dB designed and conceptualised the study, had major role in the acquisition of the data, analysed the data, interpreted the data and drafted the manuscript for intellectual content. DN-dB is responsible for the overall content as the guarantor. The rest of the authors collected the data, performed the analysis and revised the manuscript for intellectual content.
Funding: DN-dB was supported by the Miguel Servet programme from Instituto de Salud Carlos III, Spain (CP22/00141). ÀG-C and NAJ-P are supportedby FIS P118/00111 and FI21/0073 ‘Instituto de Salud Carlos III’ and ‘Fondo Europeo de desarrollo regional (FEDER)’. CJ-M is partially supported by Instituto de Salud Carlos III and co-funded with European Regional Development Fund (ERDF) ‘A way to achieve Europe’ grant PI19/00122.
Competing interests: Authors report no disclosures. ÀG-C has received honoraria for lectures from PTC Therapeutics International GT, Immedica, Biomarin and Recordati Rare Diseases Foundation; she has also received a research grant from PTC Therapeutics and is a co-founder of the Sant Joan de Déu start up ‘Neuroprotect Life Sciences’.
Provenance and peer review: Not commissioned; externally peer reviewed.
Biobank: The HSJD Biobank is officially registered in the Spanish Institute of Health (Instituto de Salud Carlos III) under the number B.0000599. It is also authorised by the autonomic health system of Catalonia. All the samples of HSJD Biobank used in research are collected, managed and stored under strict quality measures (the Biobank is accredited by ISO 9001:2015 standard) and all donors or their relatives have signed an informed consent form. The HSJD Biobank website is: https://www.irsjd.org/es/servicios-cientificotecnicos/biobanco-pediatrico-para-la-investigacion/.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request.
Ethics statements
Patient consent for publication
Consent obtained from parent(s)/guardian(s).
Ethics approval
This study was approved by Hospital Sant Joan de Déu Clinical Research Ethics Committee (reference PIC-131-18). Participants gave informed consent to participate in the study before taking part.
References
- 1. Wicklund MP. The limb-girdle muscular dystrophies. CONTINUUM (Minneap Minn) 2019;25:1599–618. 10.1212/CON.0000000000000809 [DOI] [PubMed] [Google Scholar]
- 2. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 2018;5:1574–87. 10.1002/acn3.649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosales XQ, al-Dahhak R, Tsao C-Y. Childhood onset of limb-girdle muscular dystrophy. Pediatr Neurol 2012;46:13–23. 10.1016/j.pediatrneurol.2011.08.014 [DOI] [PubMed] [Google Scholar]
- 4. Straub V, Murphy A, Udd B, et al. 229th ENMC International workshop: limb girdle muscular dystrophies – Nomenclature and reformed classification naarden, the Netherlands, 17–19 March 2017. Neuromuscular Disorders 2018;28:702–10. 10.1016/j.nmd.2018.05.007 [DOI] [PubMed] [Google Scholar]
- 5. Bögershausen N, Shahrzad N, Chong JX, et al. Recessive TRAPPC11 mutations cause a disease spectrum of limb girdle muscular dystrophy and myopathy with movement disorder and intellectual disability. Am J Hum Genet 2013;93:181–90. 10.1016/j.ajhg.2013.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liang W-C, Zhu W, Mitsuhashi S, et al. Congenital muscular dystrophy with fatty liver and infantile-onset cataract caused by TRAPPC11 mutations: broadening of the phenotype. Skelet Muscle 2015;5:29. 10.1186/s13395-015-0056-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Matalonga L, Bravo M, Serra-Peinado C, et al. Mutations in TRAPPC11 are associated with a congenital disorder of glycosylation. Hum Mutat 2017;38:148–51. 10.1002/humu.23145 [DOI] [PubMed] [Google Scholar]
- 8. Lipatova Z, Segev N. Ypt/Rab GTPases and their TRAPP GEFs at the Golgi. FEBS Lett 2019;593:2488–500. 10.1002/1873-3468.13574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jones S, Newman C, Liu F, et al. The TRAPP complex is a nucleotide exchanger for Ypt1 and ypt31/32. Mol Biol Cell 2000;11:4403–11. 10.1091/mbc.11.12.4403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scrivens PJ, Noueihed B, Shahrzad N, et al. C4orf41 and TTC-15 are mammalian TRAPP components with a role at an early stage in ER-to-Golgi trafficking. Mol Biol Cell 2011;22:2083–93. 10.1091/mbc.E10-11-0873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brunet S, Sacher M. In sickness and in health: the role of TRAPP and associated proteins in disease. Traffic 2014;15:803–18. 10.1111/tra.12183 [DOI] [PubMed] [Google Scholar]
- 12. Sacher M, Shahrzad N, Kamel H, et al. TRAPPopathies: an emerging set of disorders linked to variations in the genes encoding transport protein particle (TRAPP)-associated proteins. Traffic 2019;20:5–26. 10.1111/tra.12615 [DOI] [PubMed] [Google Scholar]
- 13. Galindo A, Planelles-Herrero VJ, Degliesposti G, et al. Cryo-EM structure of metazoan TRAPPIII, the multi-subunit complex that activates the gtpase rab1. EMBO J 2021;40:e107608. 10.15252/embj.2020107608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Koehler K, Milev MP, Prematilake K, et al. A novel trappc11 mutation in two turkish families associated with cerebral atrophy, global retardation, scoliosis, achalasia and alacrima. J Med Genet 2017;54:176–85. 10.1136/jmedgenet-2016-104108 [DOI] [PubMed] [Google Scholar]
- 15. Fee DB, Harmelink M, Monrad P, et al. Siblings with mutations in TRAPPC11 presenting with limb-girdle muscular dystrophy 2S. J Clin Neuromuscul Dis 2017;19:27–30. 10.1097/CND.0000000000000173 [DOI] [PubMed] [Google Scholar]
- 16. Larson AA, Baker PR, Milev MP, et al. TRAPPC11 and gosr2 mutations associate with hypoglycosylation of α-dystroglycan and muscular dystrophy. Skelet Muscle 2018;8:17. 10.1186/s13395-018-0163-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Milev MP, Stanga D, Schänzer A, et al. Characterization of three trappc11 variants suggests a critical role for the extreme carboxy terminus of the protein. Sci Rep 2019;9:14036. 10.1038/s41598-019-50415-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Munot P, McCrea N, Torelli S, et al. TRAPPC11-related muscular dystrophy with hypoglycosylation of alpha-dystroglycan in skeletal muscle and brain. Neuropathol Appl Neurobiol 2022;48:e12771. 10.1111/nan.12771 [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Wu Y, Cui Y, et al. Novel trappc11 mutations in a chinese pedigree of limb girdle muscular dystrophy. Case Rep Genet 2018;2018:8090797. 10.1155/2018/8090797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Szymański J, Janikiewicz J, Michalska B, et al. Interaction of mitochondria with the endoplasmic reticulum and plasma membrane in calcium homeostasis, lipid trafficking and mitochondrial structure. Int J Mol Sci 2017;18:1576. 10.3390/ijms18071576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cohen S, Valm AM, Lippincott-Schwartz J. Interacting organelles. Curr Opin Cell Biol 2018;53:84–91. 10.1016/j.ceb.2018.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genetics in Medicine 2015;17:405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993;234:779–815. 10.1006/jmbi.1993.1626 [DOI] [PubMed] [Google Scholar]
- 24. Schrödinger. DeLano WL. PyMOL. 2020. Available: http://www.pymol.org/pymol
- 25. Healy EG. Muscle biopsy a practical approach 5th edition. Neuromuscular Disorders 2022;32:98. 10.1016/j.nmd.2021.10.004 [DOI] [Google Scholar]
- 26. Brockington M, Torelli S, Sharp PS, et al. Transgenic overexpression of large induces α-dystroglycan hyperglycosylation in skeletal and cardiac muscle. PLoS ONE 2010;5:e14434. 10.1371/journal.pone.0014434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fisher RS, van Emde Boas W, Blume W, et al. Epileptic seizures and epilepsy: definitions proposed by the international league against epilepsy (ILAE) and the international bureau for epilepsy (IBE). Epilepsia 2005;46:470–2. 10.1111/j.0013-9580.2005.66104.x [DOI] [PubMed] [Google Scholar]
- 28. Berg AT, Berkovic SF, Brodie MJ, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE commission on classification and terminology, 2005-2009. Epilepsia 2010;51:676–85. 10.1111/j.1528-1167.2010.02522.x [DOI] [PubMed] [Google Scholar]
- 29. Brodie MJ, Barry SJE, Bamagous GA, et al. Patterns of treatment response in newly diagnosed epilepsy. Neurology 2012;78:1548–54. 10.1212/WNL.0b013e3182563b19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oyarzabal A, Bravo-Alonso I, Sánchez-Aragó M, et al. Mitochondrial response to the BCKDK-deficiency: some clues to understand the positive dietary response in this form of autism. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2016;1862:592–600. 10.1016/j.bbadis.2016.01.016 [DOI] [PubMed] [Google Scholar]
- 31. Chaudhry A, Shi R, Luciani DS. A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cells. Am J Physiol Endocrinol Metab 2020;318:E87–101. 10.1152/ajpendo.00457.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gastaut H, Gastaut JL, Gonçalves e Silva GE, et al. Relative frequency of different types of epilepsy: a study employing the classification of the International League against epilepsy. Epilepsia 1975;16:457–61. 10.1111/j.1528-1157.1975.tb06073.x [DOI] [PubMed] [Google Scholar]
- 33. Ünver O, Keskin SP, Uysal S, et al. The epidemiology of epilepsy in children: a report from a Turkish pediatric neurology clinic. J Child Neurol 2015;30:698–702. 10.1177/0883073814539559 [DOI] [PubMed] [Google Scholar]
- 34. Hostetler JA. History and relevance of the Hutterite population for genetic studies. Am J Med Genet 1985;22:453–62. 10.1002/ajmg.1320220303 [DOI] [PubMed] [Google Scholar]
- 35. Rasika S, Passemard S, Verloes A, et al. Golgipathies in neurodevelopment: a new view of old defects. Dev Neurosci 2019;40:396–416. 10.1159/000497035 [DOI] [PubMed] [Google Scholar]
- 36. Passemard S, Perez F, Gressens P, et al. Endoplasmic reticulum and Golgi stress in microcephaly. Cell Stress 2019;3:369–84. 10.15698/cst2019.12.206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Passemard S, Perez F, Colin-Lemesre E, et al. Golgi trafficking defects in postnatal microcephaly: the evidence for “golgipathies.” Prog Neurobiol 2017;153:46–63. 10.1016/j.pneurobio.2017.03.007 [DOI] [PubMed] [Google Scholar]
- 38. Rawlins LE, Almousa H, Khan S, et al. Biallelic variants in TRAPPC10 cause a microcephalic trappopathy disorder in humans and mice. PLOS Genet 2022;18:e1010114. 10.1371/journal.pgen.1010114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaur P, Kadavigere R, Girisha KM, et al. Recurrent bi-allelic splicing variant c.454+3a > G in TRAPPC4 is associated with progressive encephalopathy and muscle involvement. Brain 2020;143:e29. 10.1093/brain/awaa046 [DOI] [PubMed] [Google Scholar]
- 40. Majethia P, Do Rosario MC, Kaur P, et al. Further evidence of muscle involvement in neurodevelopmental disorder with epilepsy, spasticity, and brain atrophy. Ann Hum Genet 2022;86:94–101. 10.1111/ahg.12452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Al-Deri N, Okur V, Ahimaz P, et al. A novel homozygous variant in TRAPPC2L results in a neurodevelopmental disorder and disrupts TRAPP complex function. J Med Genet 2021;58:592–601. 10.1136/jmedgenet-2020-107016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Milev MP, Graziano C, Karall D, et al. Bi-Allelic mutations in TRAPPC2L result in a neurodevelopmental disorder and have an impact on Rab11 in fibroblasts. J Med Genet 2018;55:753–64. 10.1136/jmedgenet-2018-105441 [DOI] [PubMed] [Google Scholar]
- 43. Bérat C-M, Montealegre S, Wiedemann A, et al. Clinical and biological characterization of 20 patients with TANGO2 deficiency indicates novel triggers of metabolic crises and NO primary energetic defect. J Inherit Metab Dis 2021;44:415–25. 10.1002/jimd.12314 [DOI] [PubMed] [Google Scholar]
- 44. García-Cazorla A, Oyarzábal A, Saudubray J-M, et al. Genetic disorders of cellular trafficking. Trends Genet 2022;38:724–51. 10.1016/j.tig.2022.02.012 [DOI] [PubMed] [Google Scholar]
- 45. Stanga D, Zhao Q, Milev MP, et al. TRAPPC11 functions in autophagy by recruiting ATG2B-WIPI4/WDR45 to preautophagosomal membranes. Traffic 2019;20:325–45. 10.1111/tra.12640 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmg-2022-109132supp001.pdf (47.4KB, pdf)
jmg-2022-109132supp002.pdf (43.7KB, pdf)
jmg-2022-109132supp003.pdf (76.4KB, pdf)
Data Availability Statement
Data are available on reasonable request.