

Abstract
Given the key role of the IL-23/Th17 axis in the pathogenesis of moderate-to-severe plaque psoriasis, several specific inhibitors of the p19 subunit of IL-23 have been approved to treat this chronic inflammatory disease. Clinical data indicate that guselkumab, one such selective IL-23 inhibitor, achieves greater clinical efficacy compared with ustekinumab, which inhibits both IL-12 and IL-23 via binding their shared p40 subunit. To understand mechanisms underlying the enhanced efficacy observed with the p19 subunit of IL-23–specific inhibition, we explored cellular and molecular changes in skin of psoriasis patients treated with ustekinumab or guselkumab and in ustekinumab inadequate responders (Investigator’s Global Assessment of psoriasis score ≥ 2) subsequently treated with guselkumab (ustekinumab→guselkumab). Skin biopsies were collected pretreatment and posttreatment to assess histologic changes and molecular responses in ustekinumab- and guselkumab-treated patients. Serum cytokines and skin transcriptomics from the subset of ustekinumab→guselkumab-treated patients were also analyzed to characterize differential treatment effects. Ustekinumab and guselkumab demonstrated differential effects on secretion of pathogenic Th17-related cytokines induced by IL-23 in in vitro assays, which suggest guselkumab is a more potent therapeutic agent. Consistent with these findings, guselkumab elicited a significantly greater reduction in cellular and molecular psoriasis-related disease indicators than ustekinumab. In ustekinumab→guselkumab patients, suppression of serum IL-17A and IL-17F levels and neutralization of molecular scar and psoriasis-related gene markers in skin were significantly greater compared with patients continuing ustekinumab. This comparative study demonstrates that guselkumab inhibits psoriasis-associated pathology, suppresses Th17-related serum cytokines, and normalizes the psoriasis skin gene expression profile more effectively than ustekinumab.
Introduction
The IL-23/IL-17 axis has emerged as the key signaling pathway in the pathogenesis of chronic plaque psoriasis (1, 2), an inflammatory skin disorder that affects 2–3% of the population. Several biologic therapies for psoriasis that target the IL-23/IL-17 pathway and are approved by the Food and Drug Administration and European Medicines Agency demonstrate higher clinical benchmarks of efficacy, skin clearance, and sustained response when compared with more broadly acting biologics such as TNF inhibitors (3, 4).
Ustekinumab, which targets both IL-12 and IL-23 through blockade of their shared p40 protein subunit (and inhibits both the Th1 and Th17 pathways, respectively), was shown to be safe and effective for the treatment of moderate-to-severe plaque psoriasis in adults and adolescents (5–7). Targeted inhibition of IL-17A by ixekizumab or secukinumab, as well as antagonism of IL-17RA by brodalumab, have also demonstrated significant efficacy in plaque psoriasis. Furthermore, ixekizumab, brodalumab, and secukinumab were all shown to elicit greater levels of efficacy, resolution of psoriasis-related pain, itching, and scaling, and improvement in quality of life than ustekinumab in phase 3 trials (8, 9). As such, these studies affirmed that more robust efficacy can be achieved in psoriasis patients with selective blockade of pathogenic IL-17 activity than with dual blockade of IL-12 and IL-23.
More recent data indicate that inhibition of the p19 subunit of IL-23 (IL-23p19) can also afford greater efficacy compared with inhibition of IL-12/23p40 in patients with moderate-to-severe psoriasis. Although IL-23 is a member of the IL-12 family of cytokines (10), it comprises the IL-12/23p40 subunit paired with a unique p19 subunit and plays a role in the maintenance and proliferation of Th17 cells. Guselkumab, tildrakizumab, risankizumab, brazikumab, and mirikizumab are all mAb therapies that specifically bind to IL-23p19. In pivotal phase 3 clinical studies, ≥ 70% of guselkumab- and risankizumab-treated psoriasis patients achieved skin responses representing at least 90% improvement from baseline to week 16 (4). The promise and potential value of IL-23p19 subunit inhibitors may be best exemplified by the sustained duration of clinical response. Selective blockade of IL-23 with guselkumab through 5 y produced skin responses representing at least 90% improvement from baseline in > 80% of patients (11).
The cellular and molecular mechanisms of therapeutic Abs targeting the IL-12/23p40 subunit, IL-17 or its receptor, or IL-23p19 are beginning to be better understood. In this study, we explored cellular and molecular changes in the skin of psoriasis patients treated with ustekinumab or guselkumab to better delineate the mechanistic differences underlying selective IL-23 inhibition via binding to its p19 subunit versus inhibition of both IL-12 and IL-23 through p40 subunit binding.
Materials and Methods
Study populations and skin biopsies
To compare the cellular and molecular mechanisms of guselkumab (TREMFYA; Janssen Biotech, Horsham, PA) and ustekinumab (STELARA; Janssen Biotech, Horsham, PA) in the context of psoriasis, skin biopsy data were derived from subsets of 14 patients who received ustekinumab 90 mg in the phase 3 ACCEPT study (3) and 10 patients who received guselkumab 100 or 300 mg (1 patient had missing baseline and week 1 data; 9 had paired data) in a phase 1 first-in-human study in psoriasis patients (12). In both studies, biopsies were collected from lesional and nonlesional (i.e., macroscopically normal) skin at baseline and from lesional skin at weeks 1 and 12 of treatment. Biopsies from 24 healthy subjects served as controls for the 24 psoriasis patients. The subset of samples was selected because it represented the higher dose of each treatment, and the cohorts were comparable in sample size, body mass index, and baseline disease severity by Psoriasis Area and Severity Index (PASI) (Table I). To avoid technical bias, samples from the two cohorts were hybridized in the same microarray batch. There was > 80% overlap between the two cohorts in disease expression profile (i.e., lesional versus nonlesional at baseline), indicating that the two cohorts were also comparable at the disease expression profile level.
Table I. Baseline demographic and disease characteristics of psoriasis patient cohorts.
| Baseline Characteristics | Phase 1 FIH: Guselkumab | Phase 3 ACCEPT: Ustekinumab 90 mg | p Valuea |
|---|---|---|---|
| No. of patients | 24b | 14 | |
| Age (y), mean ± SD | 40.8 ± 10.2 | 46.6 ± 13.1 | 0.14 |
| Age of onset (y), mean ± SD | 23.6 ± 13.5 | 28.1 ± 14.4 | 0.34 |
| Male, n (%) | 15 (62.5) | 13 (92.9) | 0.06 |
| Race, n (%) | 1.0 | ||
| White | 20 (83.3) | 12 (85.7) | |
| Asian | 3 (12.5) | 2 (14.3) | |
| Other | 1 (4.2) | 0 (0) | |
| BMI (kg/m2), mean ± SD | 31.8 ± 4.2 | 33.4 ± 8.6 | 0.44 |
| Disease duration (y), mean ± SD | 16.4 ± 9.9 | 18.5 ± 10.9 | 0.57 |
| PASI score, mean ± SD | 17.1 ± 6.4 | 20.8 ± 13.2 | 0.25 |
Derived from t test.
Ten of these patients received either guselkumab 100 mg (n = 5) or 300 mg (n = 5).
BMI, body mass index; FIH, first-in-human.
Samples from an independent phase 3 study (NAVIGATE) (13) were used to assess serum cytokine and skin transcriptomic changes in psoriasis patients receiving ustekinumab and guselkumab treatment. In NAVIGATE, 871 patients received open-label treatment with ustekinumab (45 or 90 mg for patients ≤ 100 kg or > 100 kg, respectively) at weeks 0 and 4. At week 16, 268 patients with an inadequate response (Investigator’s Global Assessment of psoriasis [IGA] ≥ 2) were randomized to receive either guselkumab 100 mg or continued ustekinumab treatment, whereas 585 patients with IGA = 0/1 at week 16 continued open-label ustekinumab. Serum samples were collected from all patients at weeks 0, 16, 40, and 52, and skin biopsies were obtained from a subset of patients at weeks 0, 16, and 40. All patients provided written informed consent before participation in any study-related procedures.
Specificity and binding of ustekinumab and guselkumab to IL-12/23p40 and IL-23p19
Plates were coated with ustekinumab (specific to IL-12/23p40), guselkumab (specific to IL-23p19), and a negative control Ab. Recombinant human (rh) IL-23, IL-12, or p40 monomer was added, and detection was performed using a biotinylated anti-p40 Ab and streptavidin protein covalently conjugated to HRP enzyme (SA-HRP) with 3,3′, 5,5′-tetramethylbenzidine substrate. Data are presented as mean OD values of duplicate samples. The rh proteins and Abs were generated at Janssen Research & Development (Spring House, PA) or purchased from R&D Systems (Minneapolis, MN).
Receptor binding and STAT signaling
Chimeric IL-12Rβ1-Fc and IL-23R-Fc proteins were coated on ELISA plates. Ustekinumab, guselkumab, or an isotype control were then titrated against rhIL-23 or rhL-12. Cytokine binding was detected with SA-HRP development of a biotinylated secondary IL-12/23p40–specific mAb that does not compete for ustekinumab binding.
Detection of intracellular phospho–STAT-3 and phospho–STAT-4
Aliquots of rhIL-12 or rhIL-23 were preincubated with ustekinumab, guselkumab, or an isotype control Ab. Cells were stimulated with rhIL-12 or rhIL-23, alone or with preincubated mixtures of Ab. Detection of intracellular phospho–STAT-3 or phospho–STAT-4 was performed by flow cytometry using PE-conjugated phospho–STAT3- and phospho–STAT4-specific Abs from BD Biosciences (San Jose, CA). The rh proteins and Abs were generated at Janssen Research & Development (Spring House, PA).
Neutralization of human IL-23–induced Th17 cytokine production by mouse splenocytes
Single-cell suspensions were prepared from mouse spleens. Aliquots of rhIL-12 and rhIL-23 were added in the presence of ustekinumab, guselkumab, or a negative isotype control to cells for 1–3 d. Cell supernatants were collected and assayed by ELISA for IL-17A, IL-17F, IL-22, and IFN-γ. Data are summarized as mean values of quadruplicate samples for IL-17A, IL-17F, and IL-22 and as mean values of duplicate samples for IFN-γ.
Histologic features and photomicrograph visualization of treated psoriatic skin lesions
Skin biopsy specimens were collected at baseline and week 12 from psoriasis patients treated with guselkumab (100 and 300 mg doses combined to increase analytical power) or with ustekinumab (90 mg). Specimens were evaluated to assess histopathologic changes after treatment at week 12 relative to baseline. Epidermal thickness, CD3+ T cell counts, and CD11c+ myeloid dendritic cell counts were enumerated. Changes in histologic features of skin biopsies from guselkumab- versus ustekinumab-treated patients were visualized via photomicrographs.
Neutralization of psoriasis transcriptome by guselkumab and ustekinumab
The psoriasis transcriptome was determined by comparing lesional versus nonlesional skin biopsies collected at baseline from each study cohort. Molecular effects were evaluated at week 12 versus baseline in skin biopsies from patients achieving ≥ 50% improvement in the PASI (PASI50) score with guselkumab (phase 1 first-in-human) or ustekinumab (phase 3 ACCEPT). Probes with false discovery rate < 0.05 and fold change ≥ 2 were considered the “significant neutralization” set. The residual disease gene expression profile, or “molecular scar,” at week 12 was defined as genes with < 75% recovery after treatment with guselkumab or ustekinumab among probes identified in the psoriasis transcriptome that remained in resolved skin lesions. Also, for genes in the psoriasis transcriptome, percent improvement in top enriched gene expression signatures at weeks 1 and 12 was compared between ustekinumab- and guselkumab-treated patients via paired t tests.
Serum cytokine profiling in NAVIGATE
Blood samples were collected at weeks 0, 16, and 40 to quantify serum concentrations of IL-17A, IL-17F, and IL-22 (n = 40 per week 16 treatment group) using a high-sensitivity, single-molecule counting platform (Singulex, Alameda, CA). IFN-γ was measured using an electrochemiluminescence platform (Meso Scale Discovery, Rockville, MD). Healthy control sera (n = 25) were obtained from an independent cohort (Bioreclamation, Westbury, NY). Serum biomarker concentrations were compared between samples obtained at weeks 0 and 16, between patients with adequate versus patients with inadequate response at week 16, between weeks 16 and 40 and weeks 16 and 52 among patients randomized to guselkumab after inadequate response to open-label ustekinumab at week 16, and between patients randomized to switch to guselkumab versus continue ustekinumab treatment after inadequate response to open-label ustekinumab at week 16.
Transcriptional profiling
RNA samples were derived from skin biopsies obtained at weeks 0, 16, and 40 in the phase 1 first-in-human and phase 3 ACCEPT studies and preserved in RNAlater. Transcriptomic profiling was performed using the Affymetrix GeneChip HT HG-U1331 PM Array (Affymetrix, Santa Clara, CA), and expression measures were obtained by using the robust multiarray average algorithm. Raw data have been deposited in the National Center for Biotechnology Information’s Gene Expression Omnibus (accession number GSE51440 for the guselkumab phase 1 first-in-human study and accession number GSE106992 for the ustekinumab phase 3 ACCEPT study). Microarray hybridization was performed within the same batch, and analyses were conducted using the same methods and cutoffs.
Skin transcriptional profiles in the NAVIGATE study were defined by RNA sequencing (RNA-Seq) analysis. Molecular pathways with > 25 psoriasis transcriptome genes and ≥ 70% prevalence in psoriasis-relevant pathways were identified. Improvement was calculated only for psoriasis transcriptome genes (lesional versus nonlesional differentially expressed genes at baseline). Treatments were compared via t tests; averaged RNA-Seq expression data [in log2(fragments per kilobase of exon per million mapped fragments + 0.25)] were plotted for each treatment group at different time points.
Results
Psoriasis patient subsets used to compare cellular and molecular mechanisms
Patient data from a phase 1 first-in-human guselkumab trial (ClinicalTrials.gov identifier, NCT00925574) (14) and the phase 3 ACCEPT trial (ClinicalTrials.gov identifier, NCT00454584) (3) were employed in psoriasis transcriptome analyses. Baseline demographic and disease characteristics were generally comparable between the phase 1 first-in-human guselkumab study population (24 patients randomized to treatment in the second part of the study, including 5 who received guselkumab 100 mg and 5 who received guselkumab 300 mg) and the 14 patients who received ustekinumab 90 mg in the phase 3 ACCEPT study, although in the ACCEPT study cohort, patients were somewhat older with disease onset at an earlier age, and there was a numerically higher mean body mass index and numerically higher proportion of males (Table I). At the transcriptomic level, baseline disease profiles (identified via baseline lesional versus nonlesional skin false discovery rate < 0.05 and fold change ≥ 2) for the phase 1 first-in-human (guselkumab) and phase 3 ACCEPT (ustekinumab) patient subsets demonstrated > 70% concordance (Supplemental Table I).
In vitro inhibition of IL-12 and IL-23 bioactivity
Primary pharmacodynamic studies were performed to characterize the in vitro binding interactions, mechanism of action, and functional effects of ustekinumab and guselkumab with respect to neutralizing IL-12 and IL-23 bioactivity. By binding to human IL-12/23p40, ustekinumab prevents the interaction of both IL-12 and IL-23 with IL-12Rβ1, a type 1 transmembrane receptor that specifically associates with the p40 subunit common to both cytokines (15). Conversely, by binding specifically to human IL-23p19, but not to IL-12 or IL-12/23p40, guselkumab prevents the interaction of IL-23 with IL-23R, but not with IL-12Rβ1 (Supplemental Fig. 1, Fig. 1). These observations are consistent with ustekinumab’s cocrystal structure with IL-12, confirming that ustekinumab binds to the IL-12/23p40 subunit (16, 17), whereas guselkumab selectively inhibits IL-23 by binding its p19 subunit.
FIGURE 1.
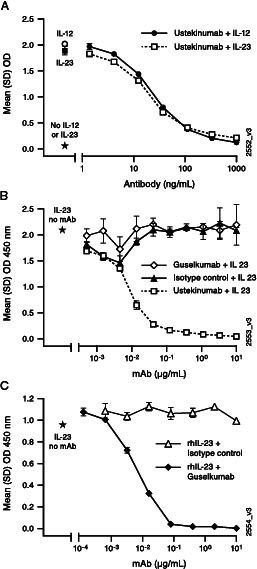
Neutralization of IL-12 and IL-23 by guselkumab and ustekinumab.
(A) Ustekinumab demonstrates equivalent neutralization of human IL-12 and IL-23 binding to human IL-12Rβ1. Ustekinumab was titrated against 10 ng/ml human IL-23 or IL-12 on IL-12Rβ1-Fc–coated plates. Cytokine binding was detected with SA-HRP development of a biotinylated secondary IL-12/23p40–specific mAb that does not compete for ustekinumab binding. (B and C) Guselkumab blocks human IL-23p19 binding to IL-23R, but not to IL-12Rβ1. Chimeric IL-12Rβ1-Fc (B) and IL-23R-Fc (C) were coated on ELISA plates. Preincubated mixtures of rhIL-23 with guselkumab or with a positive control (ustekinumab) or an isotype control were titrated. Binding of IL-23 was detected with a biotinylated IL-12/23p40–specific mAb followed by SA-HRP. Data are presented as mean OD values of duplicate wells. Stars indicate signal without mAb.
Functionally, ustekinumab inhibits both IL-12–mediated STAT-4– and IL-23–mediated STAT-3 phosphorylation, whereas guselkumab selectively inhibits only IL-23–mediated STAT-3 phosphorylation (Supplemental Fig. 2). Because human IL-23 can induce IL-17A, IL-17F, and IL-22 production by mouse splenocytes, we used this cell-based in vitro system to examine the effects of ustekinumab and guselkumab on IL-23–induced Th17 cytokine production. Both Abs inhibited IL-23–induced IL-17A, IL-17F, and IL-22 production; however, guselkumab demonstrated greater inhibitory potency in vitro than ustekinumab (Fig. 2A–C). As expected, only ustekinumab inhibited IL-12–induced IFN-γ production (Fig. 2D). The IC50 of ustekinumab and guselkumab on IL-17A, IL-17F, and IL-22 production in these in vitro conditions are provided in Table II.
FIGURE 2.
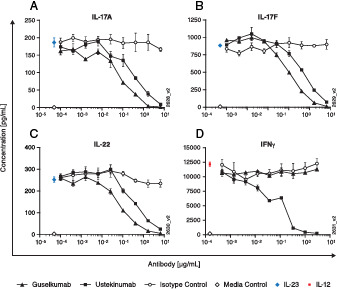
Ustekinumab and guselkumab inhibit IL-23–induced cytokine production in vitro.
(A–C) were harvested from female C57BL/6 mice, and single-cell suspensions were prepared. rhIL-23 was preincubated with guselkumab, ustekinumab, or an isotype control Ab and then added to cells. Cells were incubated for 3 d. Supernatants were assayed by ELISA for IL-23–induced (A) IL-17A, (B) IL-17F, and (C) IL-22 production. (D) Human NK92MI cells were plated, and rhIL-12 was preincubated with guselkumab, ustekinumab, or an isotype control Ab and added to cells. Cell supernatants were collected and assayed by cytokine-specific ELISA for IL-12–induced (D) IFN-γ production. Data shown are means of quadruplicate (A–C) or duplicate (D) samples.
Table II. In vitro potency of ustekinumab and guselkumab on cytokine production.
| IC50 (nM) | |||
|---|---|---|---|
| Cytokine | GUS | UST | Increased Potency of GUSa |
| IL-17A | 0.10 | 0.38 | 3.8 |
| IL-17F | 0.11 | 0.49 | 4.5 |
| IL-22 | 0.09 | 0.40 | 4.4 |
UST IC50:GUS IC50.
GUS, guselkumab; UST, ustekinumab.
Collectively, results of these experiments demonstrate that the mechanism of action for ustekinumab-mediated inhibition of human IL-12 and IL-23 bioactivity is blockade of these cytokines from binding to IL-12Rβ1, thereby preventing receptor interactions and signaling via STAT-4 and STAT-3, respectively. In contrast, the selective mechanism of action for guselkumab-mediated inhibition of human IL-23 bioactivity is prevention of IL-23p19 binding to IL-23R, thereby impeding downstream signaling via STAT-3. Thus, both ustekinumab and guselkumab inhibit IL-23–mediated cytokine production in vitro, with guselkumab demonstrating ∼4-fold (3.6- to 4.5-fold) greater activity under these testing conditions.
Greater normalization of histologic features of psoriasis by guselkumab than ustekinumab
Relative to psoriasis patients receiving ustekinumab 90 mg, IL-23p19 neutralization in those receiving guselkumab 100 or 300 mg, reductions in epidermal thickness, T cell counts, and myeloid dendritic cell counts were clearly evident by routine microscopy using H&E and immunohistochemistry staining (Fig. 3A). Significantly greater median percent reductions from baseline to week 12 were seen in lesional skin epidermal thickness (66.8% versus 58.7%; p = 0.0403; Fig. 3B), T cell counts (CD3; 92.5% versus 71.4%; p = 0.0257; Fig. 3C), and myeloid dendritic cell counts (CD11c; 100.0% versus 72.6%; p = 0.0160; Fig. 3D).
FIGURE 3.
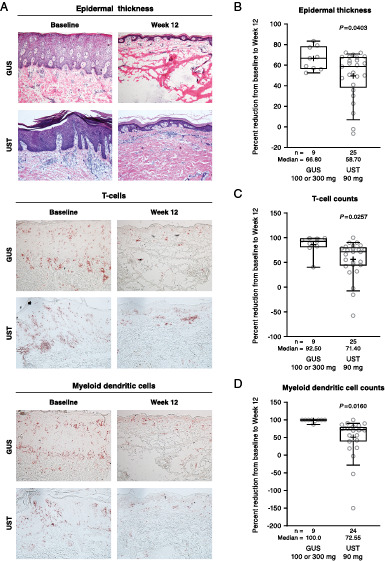
Histologic changes observed from baseline to week 12 in guselkumab- versus ustekinumab-treated psoriasis patients.
(A) Photomicrograph documentation (85-fold original magnification) of histologic changes from baseline to week 12 in epidermal thickness (top panels; H&E staining), T cell counts (middle panels; CD3 staining), and myeloid dendritic cell counts (bottom panels; CD11c staining). Percent reductions from baseline to week 12 for (B) epidermal thickness, (C) T cell counts, and (D) myeloid dendritic cell counts. GUS, guselkumab; UST, ustekinumab.
More efficacious neutralization of the psoriasis transcriptome by guselkumab than ustekinumab
A total of 1225 and 1375 probes were significantly neutralized (false discovery rate < 0.05 and fold change ≥ 2) in psoriasis lesional skin biopsies relative to baseline nonlesional skin in the phase 1 first-in-human (guselkumab) and phase 3 ACCEPT (ustekinumab) cohorts, respectively, representing a combined set of 1725 unique probes. Transcriptomic analyses were limited to patients achieving PASI50 because of the limited number of biopsies from patients achieving PASI75 or PASI90. Among patients achieving PASI50 at week 12, guselkumab significantly neutralized 76.7% of the psoriasis transcriptome in skin biopsies (1225 probes), whereas ustekinumab neutralized 45.2% of the psoriasis transcriptome (1375 probes) using the same cutoffs in skin biopsies (Fig. 4A). To evaluate the magnitude of neutralization, we further applied a 75% recovery cutoff to the set of probes with significant neutralization (false discovery rate < 0.05 and fold change > ±2× at week 12 versus baseline) by each treatment and observed that guselkumab resolved 75.4% of the psoriasis transcriptome, whereas ustekinumab resolved only 26.7% of the psoriasis transcriptome at week 12 (Fig. 4A). Taken together, these findings indicate that the degree of disease neutralization elicited by guselkumab is both more extensive and of larger magnitude than that afforded by ustekinumab.
FIGURE 4.
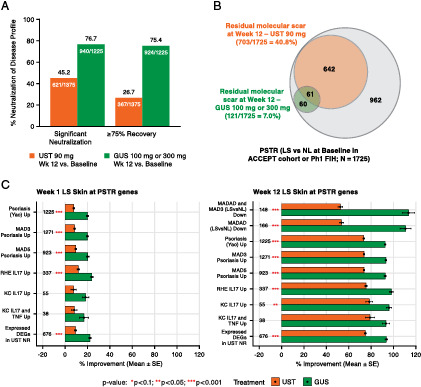
Psoriasis transcriptome neutralization in PASI50 responders at week 12 of guselkumab (100 or 300 mg; n = 10) versus ustekinumab (90 mg; n = 12) treatment.
(A) Psoriasis transcriptome % neutralization from baseline at week 12. Significant neutralization was defined as false discovery rate < 0.05 and fold change ≥ 2; ≥ 75% recovery defined as significant neutralization and ≥ 75% modulation toward baseline nonlesional levels. (B) Venn diagram illustrating “molecular scar” overlap at week 12. (C) % improvement (lesional versus nonlesional at baseline) of top enriched psoriasis transcriptome genes from baseline at weeks 1 and 12 (red asterisks represent significant difference between treatments via t test, with *p < 0.1, **p < 0.05, ***p < 0.001). DEG, differentially expressed gene; GUS, guselkumab; KC, keratinocyte; LS, lesional; MAD, meta-analysis derived; MADAD, meta-analysis–derived atopic dermatitis; NL, nonlesional; NR, nonresponder; Ph1, phase 1; PSTR, psoriasis transcriptome; RHE, reconstructed human epidermal; UST, ustekinumab.
We also identified a set of genes showing persistently dysregulated expression, considered the residual disease gene expression profile and often referred to as the “molecular scar,” after treatment with either ustekinumab or guselkumab. The molecular scar represents a group of genes with persistent expression after 12 wk of successful treatment, i.e., when visible signs of skin inflammation are absent and clinical symptoms have resolved. A set of genes representing the molecular scar was previously defined for both etanercept and ustekinumab (18, 19) and is similarly defined in this study. Consistent with guselkumab’s more robust and extensive resolution of genes associated with the psoriasis transcriptome, a larger residual molecular scar was observed at week 12 for ustekinumab (703/1725 probes; 40.8%) than for guselkumab (121/1725 probes; 7%) treatment (Fig. 4B). Notably, guselkumab treatment normalized >90% of the ustekinumab molecular scar, almost to the level of baseline nonlesional skin (Supplemental Table I).
When assessing the percent of psoriasis transcriptome improvement among the top enriched gene expression signatures, greater improvement in the psoriasis and Th17 signatures was observed with guselkumab than ustekinumab at week 1 (Fig. 4C). More extensive differences (p < 0.01) were observed at week 12, suggesting stronger suppression of disease- and mechanism-related signatures via targeting the IL-23p19 subunit compared with the IL-12/23p40 subunit (Fig. 4C). Furthermore, guselkumab fully downregulated many psoriasis core pathway–associated genes to the expression levels of nonlesional skin collected at baseline.
Switching from ustekinumab to guselkumab treatment enhances suppression of Th17-related cytokines
Cytokines associated with the IL-23/Th17 pathway were evaluated in an independent phase 3 clinical study (NAVIGATE; ClinicalTrials.gov identifier: NCT02203032), in which patients with inadequate response (defined by IGA score ≥ 2, i.e., mild-to-severe psoriasis) to open-label ustekinumab treatment at week 16 were randomized to either switch to guselkumab or continue treatment with ustekinumab through week 52 (13). Patients initially treated with ustekinumab who did not achieve clear (IGA = 0) or almost clear (IGA = 1) skin by week 16 derived significant clinical benefit from switching to guselkumab. A subset of NAVIGATE patients (n = 40 per week 16 treatment group) was selected for serum cytokine profiling. Baseline demographic and disease characteristics were consistent between patients included in the serum cytokine subset (n = 120) and the full study cohort (n = 853) (Supplemental Table II).
After 16 wk of open-label ustekinumab treatment, patients achieving clear or almost clear skin (IGA = 0/1) exhibited significantly lower serum IL-17A and IL-17F concentrations relative to patients with an inadequate response (IGA ≥ 2; p < 0.05) (Fig. 5A, 5B). No further suppression was observed beyond week 16 in either group continuing ustekinumab treatment. However, ustekinumab inadequate responders randomized to switch to guselkumab exhibited greater suppression of IL-17A, IL-17F, and IL-22 at weeks 40 and 52 versus week 16, with further suppression of IL-17A levels reaching significance at and beyond week 40 (Fig. 5A–C). Cytokine levels in the group randomized to continue ustekinumab were stable from week 16 through week 52. Overall, these data are consistent with the increased clinical benefit observed in NAVIGATE after a switch from ustekinumab→guselkumab treatment (13).
FIGURE 5.
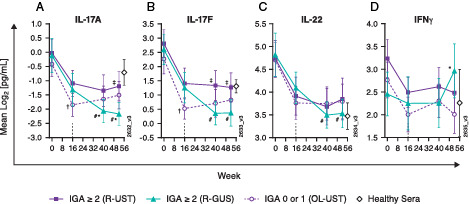
Switching from IL-12p40 to IL-23p19 blockade enhances suppression of Th17 cytokines.
Serum concentrations of (A) IL-17A, (B) IL-17F, and (C) IL-22 were measured using a high-sensitivity, single-molecule counting platform. (D) IFN-γ concentrations were measured using an electrochemiluminescence ELISA platform (n = 40 per week 16 treatment group; n = 25 for healthy control sera). †p < 0.05, week 16 IGA = 0/1 versus IGA ≥ 2 (OL ustekinumab); #p < 0.05 versus week 16; ‡p < 0.05, randomized to guselkumab versus randomized to ustekinumab; *p < 0.05, randomized to guselkumab versus OL ustekinumab. GUS, guselkumab; OL, open-label; R, randomized; UST, ustekinumab.
Conversely, although reduced during treatment, serum concentrations of IL-22 were not significantly different between ustekinumab responders (IGA = 0/1) and those with inadequate response (IGA ≥ 2). However, switching to selective IL-23 inhibition with guselkumab further suppressed IL-22 to levels that were significantly lower at weeks 40 and 52 relative to week 16. Consistent with in vitro observations that guselkumab does not inhibit IL-12–mediated IFN-γ production, serum IFN-γ concentrations increased by week 52 in patients who switched from ustekinumab→guselkumab at week 16 (Fig. 5D).
Resolution of the psoriasis transcriptome after switching from ustekinumab to guselkumab treatment
Transcriptomic profiling of skin from a subset of patients (n = 13) from NAVIGATE enabled comparison of the psoriasis transcriptome in ustekinumab patients with inadequate response (IGA ≥ 2) who were randomized at week 16 to either switch to guselkumab treatment or continue ustekinumab treatment through week 52. After 16 wk of open-label exposure to ustekinumab, minimal differences in the top enriched gene expression signatures were observed between the randomized inadequate response subgroups; the week 16 levels of gene expression then served as a new baseline for comparison after randomization to either guselkumab or continued ustekinumab treatment (13). With the switch to guselkumab, significant differences in the top enriched gene expression signatures emerged by week 40, including greater improvements in the psoriasis and Th17 signatures, compared with continuing ustekinumab treatment (Fig. 6A). Although further neutralization of the residual disease gene expression profile, or molecular scar, was observed with an additional 24 wk of ustekinumab treatment relative to week 16, only guselkumab treatment normalized the molecular scar to a degree approximating baseline nonlesional skin at week 40 (Fig. 6B).
FIGURE 6.
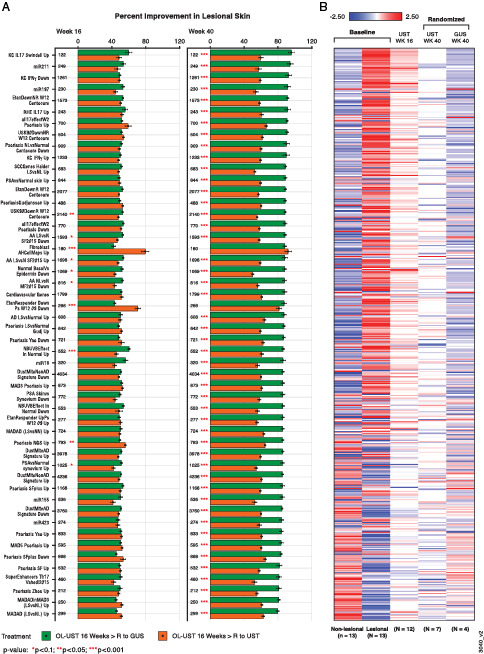
Resolution of the psoriasis transcriptome and residual molecular scar after switching from ustekinumab to guselkumab treatment.
(A) Percent improvement in the top enriched gene expression signatures with >25 psoriasis transcriptome genes and ≥70% prevalence in the signatures in lesional skin biopsies from psoriasis patients with inadequate response (IGA ≥ 2) to open-label ustekinumab treatment at week 16 who either continued treatment with ustekinumab (orange bars) or crossed over to guselkumab (green bars) treatment through week 40. Improvement is calculated only for genes in the psoriasis transcriptome (lesional versus nonlesional DEGs at baseline). Red asterisks represent significant difference between treatments (*p < 0.1, **p < 0.05, ***p < 0.001) derived from t tests. (B) Residual molecular scar over time defined by RNA-Seq analysis. Plotted are averaged RNA-Seq expression data [in log2(fragments per kilobase of exon per million mapped fragments + 0.25)] for each treatment group at different time points. AA, alopecia areata; AD; atopic dermatitis; DEG, differentially expressed gene; GUS, guselkumab; KC, keratinocyte; LS, lesional; MAD, meta-analysis derived; MADAD, meta-analysis–derived atopic dermatitis; MF, mycosis fungoides; miR, microRNA; NGS, next generation sequencing; NL, nonlesional; NN, normal healthy control; NVUVB, narrow-band UVB; OL, open-label; PSA, psoriasis; R, randomized; RHE, reconstructed human epidermal; SCC, squamous cell carcinoma; SF, Suárez-Fariñas; UST, ustekinumab.
Discussion
Guselkumab was the first in the class of selective IL-23 inhibitors to be approved in the United States (20) and European Union (21) to treat adults with moderate-to-severe psoriasis. Guselkumab demonstrated greater efficacy and durability of response in the treatment of plaque-type psoriasis compared with both placebo and adalimumab in the VOYAGE clinical trials and versus secukinumab in the ECLIPSE study (4, 22–25). In the latter, guselkumab was superior to secukinumab in long-term efficacy for treatment of moderate-to-severe psoriasis based on PASI90 response at week 48 (25). In the NAVIGATE study, patients with inadequate response to ustekinumab who switched to guselkumab experienced significantly improved therapeutic benefit (13).
These clinical studies confirm the critical role of IL-23 in the pathogenesis of psoriasis; however, only a few published reports have sought to elaborate the mechanism by which agents specifically targeting the IL-23p19 subunit correct disease pathophysiology and resolve clinical disease compared with those with different or overlapping mechanisms of action (14, 26). In our study, we compared molecular mechanisms of action, potencies of activity, and cellular and molecular changes mediated between ustekinumab and guselkumab in the skin of patients with moderate-to-severe psoriasis.
In this study, we confirmed guselkumab’s mechanism of action through selective inhibition of IL-23 via binding to its p19 subunit. Functionally, ustekinumab, by binding to the p40 subunit shared by IL-12 and IL-23, inhibits both IL-12–mediated STAT-4 and IL-23–mediated STAT-3 phosphorylation, whereas guselkumab selectively inhibits only IL-23–mediated STAT-3 phosphorylation. In vitro, guselkumab inhibited IL-23–induced IL-17A, IL-17F, and IL-22 production more potently than ustekinumab.
In a phase 1 first-in-human study, guselkumab was shown to improve pathophysiologic features of psoriasis, as assessed by epidermal thickness, T cell counts, and myeloid dendritic cell counts, in lesional skin as early as 1 wk after starting treatment (14). In this study, we demonstrate that guselkumab (100 or 300 mg) significantly normalizes histologic features characteristic of psoriatic skin lesions to a much greater extent than the 90 mg dose of ustekinumab. Relative to baseline, epidermal thickness was reduced by 67% for guselkumab versus 58% for ustekinumab, and T cell and myeloid dendritic cell counts were normalized to nonlesional or normal levels in guselkumab-treated patients in week 12 biopsies. Together, these findings demonstrate the greater impact of selectively inhibiting IL-23 by binding its p19 subunit, when compared with blocking both IL-12 and IL-23 via their shared IL-12/23p40 subunit, in abating inflammatory processes associated with psoriasis.
Consistent with the dramatic changes in skin histopathology, guselkumab also neutralized the psoriasis transcriptome to a greater extent than ustekinumab. Measured 3 mo after starting treatment, guselkumab-treated patient lesional skin had a greater total number of psoriasis transcriptome genes normalized and a larger magnitude of gene expression changes, when compared with biopsies from ustekinumab-treated patients. Furthermore, the set of residual molecular scar genes expressed after guselkumab treatment was considerably smaller than that after ustekinumab treatment (121 or 7% of probes versus 703 or 41% of probes, respectively).
The differences in clinical efficacy between ustekinumab and guselkumab are clearly demonstrated through comparison of pivotal clinical trial results for each drug. This point is further exemplified in the phase 3 NAVIGATE study, in which psoriasis patients with an inadequate response to ustekinumab (i.e., IGA ≥ 2 at week 16) randomized to switch to guselkumab at week 16 achieved significantly better IGA and PASI75/90/100 response rates at week 52 than those who continued ustekinumab treatment (13). In this study, among patients who achieved clear or almost clear skin after switching to guselkumab, serum levels of Th17-related cytokines (IL-17A, IL-17F, IL-22) were further reduced compared with week 16 levels observed with inadequate response to ustekinumab. Similarly, transcriptomic profiling of skin biopsies showed that modulation of psoriasis and Th17 gene signatures at week 40 was much stronger in patients randomized to guselkumab than in those who continued ustekinumab treatment. Likewise, the residual disease gene expression profile, or molecular scar, was more closely normalized to baseline nonlesional levels in patients randomized to guselkumab. Taken together, these mechanistic observations are consistent with the improved clinical responses seen with switching to guselkumab in moderate-to-severe psoriasis patients who did not achieve an optimal response with ustekinumab.
These clinical response results are comparable with those from a recent report of head-to-head phase 3 trials (UltIMMa-1 and UltIMMa-2) in which another selective IL-23 inhibitor, risankizumab, demonstrated superior efficacy in the treatment of moderate-to-severe plaque psoriasis compared with ustekinumab (26). On the cellular and molecular levels, lesional skin biopsies from risankizumab-treated psoriasis patients have also shown greater normalization of psoriasis-associated gene expression than achieved by ustekinumab at week 12 of a phase 2 study (27). That study, however, did not include nonlesional baseline biopsies, and it was not possible to assess improvement in the residual disease gene expression profile, or molecular scar, associated with risankizumab treatment of psoriasis. Our study allowed for such assessments through up to 1 y, which is important in psoriasis, because it is a chronic inflammatory condition prone to recurring at the same body sites (28).
When considering the clinical, histologic, and molecular differences between broader targeting of IL-12 and IL-23 via their shared p40 subunit and selectively inhibiting IL-23 through p19 subunit binding, it is important to acknowledge the apparent divergence in roles of IL-12 and IL-23 in inflammatory pathways (29). In preclinical murine models, the IL-12 pathway has been shown to play a protective or regulatory role regarding inflammation in psoriasis, in part by limiting the accumulation of IL-17–producing cells in the skin. Moreover, IL-12 may have a protective effect on keratinocytes, as demonstrated by the modulation of psoriasis-related genes in human keratinocytes treated with IL-12 in vitro (30). Together, these observations suggest that also targeting IL-12 with ustekinumab might have a somewhat limiting effect in the treatment of psoriasis compared with directly targeting only IL-23.
Although clinical, histopathologic, proteomic, and transcriptomic differences between guselkumab and ustekinumab may be explained based on their relative effects on cytokines and downstream pathways, we cannot rule out the possibility that such differences may also relate to potency. Our in vitro studies show that guselkumab more potently neutralizes production of IL-17A, IL-17F, and IL-22 cytokines than ustekinumab while preserving IFN-γ production. Exposure-response modeling also suggests that guselkumab may be more potent than ustekinumab. Models based on a large database of ustekinumab-treated psoriasis patients and guselkumab phase 2 and 3 clinical trial data suggest that higher (∼3× those of guselkumab) circulating levels of ustekinumab would be needed to achieve levels of efficacy comparable with those achieved with guselkumab (data not shown). In turn, it must also be considered that disparities in dosing may contribute to observed differences.
From this and other studies, it is becoming clear that selectively targeting IL-23p19 delivers greater clinical efficacy in the treatment of moderate-to-severe psoriasis than TNF (4, 22) or IL-12/23p40 blockade (with the exception of tildrakizumab, which has shown efficacy generally equivalent to ustekinumab (26, 31). Clinical improvement of psoriasis with guselkumab treatment is associated with strong reductions in the expression of genes associated with the IL-17 axis. Therefore, similar studies comparing the cellular and molecular effects of IL-23–specific inhibition with those mediated by targeting IL-17 would be of great interest. More importantly, this study provides insight into long-term effects of guselkumab, specifically after switching from ustekinumab. Analysis of skin biopsies from the NAVIGATE study demonstrated that the improved clinical response associated with guselkumab (through week 40) is associated with normalization of the residual disease gene expression profile or molecular scar nearly equivalent to baseline nonlesional levels. This suggests that long-term control of psoriasis correlates with controlling overactivity of the IL-23 pathway and suppressing expression of multiple downstream cytokines, including IL-17A, IL17-F, and IL-22 (13). This, in turn, may help explain the durability of response of guselkumab demonstrated clinically (25).
Some limitations of this study should be noted, including comparison of patients from two independent studies and small sample size. Clinical and disease characteristics were generally comparable between ustekinumab- and guselkumab-treated patients; however, skin biopsies for these analyses were obtained from two independent phase 1 and phase 3 studies and not a head-to-head comparative trial. The small sample size of the phase 1 first-in-human and phase 3 ACCEPT cohorts gave rise to a less extensive psoriasis transcriptome relative to previous reports (19), resulting in reduced power to delineate gene expression profiles representing residual molecular scars and overall impact of either agent on psoriasis. Although transcriptomic analyses from the NAVIGATE study allowed for direct assessment of the effect of guselkumab treatment on the ustekinumab residual molecular scar, the sample size was also small. Validation and confirmation of these findings in independent cohorts of larger size will be important to solidify understanding of requirements for fully controlling pathogenic pathways and potentially achieving remission in a life-altering disease such as psoriasis.
In summary, this study demonstrates that guselkumab, which specifically binds to IL-23p19, significantly normalizes histologic features characteristic of psoriatic skin lesions to a much greater extent than ustekinumab, which targets both IL-12 and IL-23 through binding to their shared p40 protein subunit. In addition, guselkumab neutralizes the psoriasis transcriptome to a greater extent than ustekinumab, and the set of residual molecular scar genes expressed after guselkumab treatment was considerably smaller than after ustekinumab treatment.
Supplementary Material
Acknowledgments
Carrie Brodmerkel, Keying Ma, Karen Leander, Carol Franks, and Brittney Scott contributed serum protein data from the guselkumab NAVIGATE study (ClinicalTrials.gov identifier: NCT02203032). Medical writing support was provided by Michelle L. Perate, a professional medical writer, and Amy C. Porter of Certara Synchrogenix under the direction of the authors in accordance with Good Publication Practice guidelines (32) and was supported by Janssen Scientific Affairs, LLC.
Footnotes
This work was supported by the Janssen Research & Development, LLC, which funded the studies contributing data to these analyses.
The online version of this article contains supplemental material.
The raw data presented in this article have been submitted to the National Center for Biotechnology Information’s Gene Expression Omnibus under accession number GSE51440 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE51440) for the guselkumab phase 1 first-in-human study and accession number GSE106992 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE106992) for the phase 3 ACCEPT study.
- IGA
- Investigator’s Global Assessment of psoriasis
- IL-23p19
- p19 subunit of IL-23
- PASI
- Psoriasis Area and Severity Index
- PASI50
- ≥50% improvement in the Psoriasis Area and Severity Index
- rh
- recombinant human
- RNA-Seq
- RNA sequencing
- SA-HRP
- streptavidin protein covalently conjugated to HRP enzyme
Disclosures
K.C., K.L., P.B., Y.O., and Y.C. are employed by Janssen Research & Development, LLC, a wholly owned subsidiary of Johnson & Johnson (J&J), and own J&J stock/stock options. F.Y., M.M.E., and J.B. were employed by Janssen Research & Development, LLC, a wholly owned subsidiary of Johnson & Johnson (J&J), at the time of this work and own J&J stock/stock options. S.G. declares no relevant conflicts of interest. J.G.K. has received research support (grants paid to his institution) from AbbVie, Akros, Allergen, Amgen, Avillion, BMS, Biogen MA, Boehringer, Botanix, Celgene, Eli Lilly, Incyte, Innovaderm, Janssen, Leo Pharma, Novan, Novartis, Paraxel, Pfizer, Regeneron, Sienna, UCB, and Vitae and personal fees from AbbVie, Acros, Allergan, Almirall, Amgen, Arena, Aristea, Asana, Aurigne, BiogenIdec, Boehringer, Celgene, Eli Lilly, Escalier, Janssen, Leo Pharma, Menlo, Nimbus, Novartis, Pfizer, Sanofi, Sienna, Sun Pharma, UCB, and Valeant.
References
- 1. Kim, J., Krueger J. G.. 2017. Highly effective new treatments for psoriasis target the IL-23/Type 17 T cell autoimmune axis. Annu. Rev. Med. 68: 255–269. [DOI] [PubMed] [Google Scholar]
- 2. Puig, L. 2017. The role of IL 23 in the treatment of psoriasis. [Published erratum appears in 2017 Exp. Rev. Clin. Immunol. 13: 6, ix.] Expert Rev. Clin. Immunol. 13: 525–534. [DOI] [PubMed] [Google Scholar]
- 3. Griffiths, C. E., Strober B. E., van de Kerkhof P., Ho V., Fidelus-Gort R., Yeilding N., Guzzo C., Xia Y., Zhou B., Li S., et al. ACCEPT Study Group . 2010. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N. Engl. J. Med. 362: 118–128. [DOI] [PubMed] [Google Scholar]
- 4. Blauvelt, A., Papp K. A., Griffiths C. E., Randazzo B., Wasfi Y., Shen Y. K., Li S., Kimball A. B.. 2017. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. [Published erratum appears in 2017 J. Am. Acad. Dermatol. 76: 1226.] J. Am. Acad. Dermatol. 76: 405–417. [DOI] [PubMed] [Google Scholar]
- 5. Leonardi, C. L., Kimball A. B., Papp K. A., Yeilding N., Guzzo C., Wang Y., Li S., Dooley L. T., Gordon K. B.; PHOENIX 1 study investigators . 2008. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 76-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 1). Lancet 371: 1665–1674. [DOI] [PubMed] [Google Scholar]
- 6. Papp, K. A., Langley R. G., Lebwohl M., Krueger G. G., Szapary P., Yeilding N., Guzzo C., Hsu M. C., Wang Y., Li S., et al. PHOENIX 2 study investigators . 2008. Efficacy and safety of ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: 52-week results from a randomised, double-blind, placebo-controlled trial (PHOENIX 2). Lancet 371: 1675–1684. [DOI] [PubMed] [Google Scholar]
- 7. Landells, I., Marano C., Hsu M. C., Li S., Zhu Y., Eichenfield L. F., Hoeger P. H., Menter A., Paller A. S., Taieb A., et al. 2015. Ustekinumab in adolescent patients age 12 to 17 years with moderate-to-severe plaque psoriasis: results of the randomized phase 3 CADMUS study. J. Am. Acad. Dermatol. 73: 594–603. [DOI] [PubMed] [Google Scholar]
- 8. Lebwohl, M., Strober B., Menter A., Gordon K., Weglowska J., Puig L., Papp K., Spelman L., Toth D., Kerdel F., et al. 2015. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N. Engl. J. Med. 373: 1318–1328. [DOI] [PubMed] [Google Scholar]
- 9. Bagel, J., Nia J., Hashim P. W., Patekar M., de Vera A., Hugot S., Sheng K., Xia S., Gilloteau I., Muscianisi E., et al. 2018. Secukinumab is superior to ustekinumab in clearing skin in patients with moderate to severe plaque psoriasis (16-week CLARITY results). Dermatol. Ther. (Heidelb.) 8: 571–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oppmann, B., Lesley R., Blom B., Timans J. C., Xu Y., Hunte B., Vega F., Yu N., Wang J., Singh K., et al. 2000. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13: 715–725. [DOI] [PubMed] [Google Scholar]
- 11. Reich, K., Gordon K. B., Strober B. E., Armstrong A. W., Miller M., Shen Y. K., You Y., Han C., Yang Y. W., Foley P., Griffiths C. E. M.. 2021. Five-year maintenance of clinical response and health-related quality of life improvements in patients with moderate-to-severe psoriasis treated with guselkumab: results from VOYAGE 1 and VOYAGE 2. Br. J. Dermatol. 185: 1146–1159. [DOI] [PubMed] [Google Scholar]
- 12. Zhuang, Y., Calderon C., S. J. Marciniak, Jr., Bouman-Thio E., Szapary P., Yang T. Y., Schantz A., Davis H. M., Zhou H., Xu Z.. 2016. First-in-human study to assess guselkumab (anti-IL-23 mAb) pharmacokinetics/safety in healthy subjects and patients with moderate-to-severe psoriasis. Eur. J. Clin. Pharmacol. 72: 1303–1310. [DOI] [PubMed] [Google Scholar]
- 13. Langley, R. G., Tsai T. F., Flavin S., Song M., Randazzo B., Wasfi Y., Jiang J., Li S., Puig L.. 2018. Efficacy and safety of guselkumab in patients with psoriasis who have an inadequate response to ustekinumab: results of the randomized, double-blind, phase III NAVIGATE trial. Br. J. Dermatol. 178: 114–123. [DOI] [PubMed] [Google Scholar]
- 14. Sofen, H., Smith S., Matheson R. T., Leonardi C. L., Calderon C., Brodmerkel C., Li K., Campbell K., S. J. Marciniak, Jr., Wasfi Y., et al. 2014. Guselkumab (an IL-23-specific mAb) demonstrates clinical and molecular response in patients with moderate-to-severe psoriasis. J. Allergy Clin. Immunol. 133: 1032–1040. [DOI] [PubMed] [Google Scholar]
- 15. Robinson, R. T. 2015. IL12Rβ1: the cytokine receptor that we used to know. Cytokine 71: 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luo, J., Wu S. J., Lacy E. R., Orlovsky Y., Baker A., Teplyakov A., Obmolova G., Heavner G. A., Richter H. T., Benson J.. 2010. Structural basis for the dual recognition of IL-12 and IL-23 by ustekinumab. J. Mol. Biol. 402: 797–812. [DOI] [PubMed] [Google Scholar]
- 17. Benson, J. M., Sachs C. W., Treacy G., Zhou H., Pendley C. E., Brodmerkel C. M., Shankar G., Mascelli M. A.. 2011. Therapeutic targeting of the IL-12/23 pathways: generation and characterization of ustekinumab. Nat. Biotechnol. 29: 615–624. [DOI] [PubMed] [Google Scholar]
- 18. Tian, S., Krueger J. G., Li K., Jabbari A., Brodmerkel C., Lowes M. A., Suárez-Fariñas M.. 2012. Meta-analysis derived (MAD) transcriptome of psoriasis defines the “core” pathogenesis of disease. PLoS One 7: e44274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brodmerkel, C., Li K., Garcet S., Hayden K., Chiricozzi A., Novitskaya I., Fuentes-Duculan J., Suarez-Farinas M., Campbell K., Krueger J. G.. 2019. Modulation of inflammatory gene transcripts in psoriasis vulgaris: differences between ustekinumab and etanercept. J. Allergy Clin. Immunol. 143: 1965–1969. [DOI] [PubMed] [Google Scholar]
- 20. U.S. Food and Drug Administration . 2017. Tremfya (guselkumab): U.S. prescribing information. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761061s000lbl.pdf. Accessed: April 10, 2023.
- 21. U.S. Food and Drug Administration . 2017. Summary of product characteristics: Tremfya (guselkumab) 100 mg solution for injection. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761061s000lbl.pdf. Accessed: March 9, 2023.
- 22. Reich, K., Armstrong A. W., Foley P., Song M., Wasfi Y., Randazzo B., Li S., Shen Y. K., Gordon K. B.. 2017. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double-blind, placebo- and active comparator-controlled VOYAGE 2 trial. [Published erratum appears in 2017 J. Am. Acad. Dermatol. 76: 1226.] J. Am. Acad. Dermatol. 76: 418–431. [DOI] [PubMed] [Google Scholar]
- 23. Gordon, K. B., Blauvelt A., Foley P., Song M., Wasfi Y., Randazzo B., Shen Y. K., You Y., Griffiths C. E. M.. 2018. Efficacy of guselkumab in subpopulations of patients with moderate-to-severe plaque psoriasis: a pooled analysis of the phase III VOYAGE 1 and VOYAGE 2 studies. Br. J. Dermatol. 178: 132–139. [DOI] [PubMed] [Google Scholar]
- 24. Foley, P., Gordon K., Griffiths C. E. M., Wasfi Y., Randazzo B., Song M., Li S., Shen Y. K., Blauvelt A.. 2018. Efficacy of guselkumab compared with adalimumab and placebo for psoriasis in specific body regions: a secondary analysis of 2 randomized clinical trials. JAMA Dermatol. 154: 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Reich, K., Armstrong A. W., Langley R. G., Flavin S., Randazzo B., Li S., Hsu M. C., Branigan P., Blauvelt A.. 2019. Guselkumab versus secukinumab for the treatment of moderate-to-severe psoriasis (ECLIPSE): results from a phase 3, randomised controlled trial. Lancet 394: 831–839. [DOI] [PubMed] [Google Scholar]
- 26. Gordon, K. B., Strober B., Lebwohl M., Augustin M., Blauvelt A., Poulin Y., Papp K. A., Sofen H., Puig L., Foley P., et al. 2018. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet 392: 650–661. [DOI] [PubMed] [Google Scholar]
- 27. Visvanathan, S., Baum P., Vinisko R., Schmid R., Flack M., Lalovic B., Kleiner O., Fuentes-Duculan J., Garcet S., Davis J. W., et al. 2019. Psoriatic skin molecular and histopathologic profiles after treatment with risankizumab versus ustekinumab. J. Allergy Clin. Immunol. 143: 2158–2169. [DOI] [PubMed] [Google Scholar]
- 28. Benezeder, T., Wolf P.. 2019. Resolution of plaque-type psoriasis: what is left behind (and reinitiates the disease). Semin. Immunopathol. 41: 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cua, D. J., Sherlock J., Chen Y., Murphy C. A., Joyce B., Seymour B., Lucian L., To W., Kwan S., Churakova T., et al. 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421: 744–748. [DOI] [PubMed] [Google Scholar]
- 30. Kulig, P., Musiol S., Freiberger S. N., Schreiner B., Gyülveszi G., Russo G., Pantelyushin S., Kishihara K., Alessandrini F., Kündig T., et al. 2016. IL-12 protects from psoriasiform skin inflammation. Nat. Commun. 7: 13466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sbidian, E., Chaimani A., Garcia-Doval I., Doney L., Dressler C., Hua C., Hughes C., Naldi L., Afach S., Le Cleach L.. 2021. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst. Rev. 4: CD011535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeTora, L. M., Toroser D., Sykes A., Vanderlinden C., Plunkett F. J., Lane T., Hanekamp E., Dormer L., DiBiasi F., Bridges D., et al. 2022. Good publication practice (GPP) guidelines for company-sponsored biomedical research: 2022 update. Ann Intern Med 175: 1298–1304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.