

Highlights
-
•
First report on the structural characterization of DIV1 MCP with repurposing drugs.
-
•
DIV1 repurposing potential of Chloroquine, Rimantadine, and CAP-1 were studied.
-
•
Findings enhance understanding of DIV MCP structure-function-dynamics relationships.
Keywords: Shrimp hemocyte iridescent virus, Antiviral drug, Decapod iridescent virus 1, Chloroquine, Rimantadine, CAP-1, Drug repurposing, Aquaculture
Abstract
Drug repurposing is a methodology of identifying new therapeutic use for existing drugs. It is a highly efficient, time and cost-saving strategy that offers an alternative approach to the traditional drug discovery process. Past in-silico studies involving molecular docking have been successful in identifying potential repurposed drugs for the various treatment of diseases including aquaculture diseases. The emerging shrimp hemocyte iridescent virus (SHIV) or Decapod iridescent virus 1 (DIV1) is a viral pathogen that causes severe disease and high mortality (80 %) in farmed shrimps caused serious economic losses and presents a new threat to the shrimp farming industry. Therefore, effective antiviral drugs are critically needed to control DIV1 infections. The aim of this study is to investigate the interaction of potential existing antiviral drugs, Chloroquine, Rimantadine, and CAP-1 with DIV1 major capsid protein (MCP) with the intention of exploring the potential of drug repurposing. The interaction of the DIV1 MCP and three antivirals were characterised and analysed using molecular docking and molecular dynamics simulation. The results showed that CAP-1 is a more promising candidate against DIV1 with the lowest binding energy of -8.46 kcal/mol and is more stable compared to others. We speculate that CAP-1 binding may induce the conformational changes in the DIV1 MCP structure by phosphorylating multiple residues (His123, Tyr162, and Thr395) and ultimately block the viral assembly and maturation of DIV1 MCP. To the best of our knowledge, this is the first report regarding the structural characterisation of DIV1 MCP docked with repurposing drugs.
1. Introduction
Shrimp aquaculture is the practice of commercial rearing and harvesting of shrimp species in man-made environments. Its development has decreased the pressure caused by overfishing in marine ecosystems and raised the commercial supply of marine-based food, necessitating the development of a sustainable and eco-friendly industry. Aquaculture practices create artificial environments that allow exposure to biotic (pathogens) and abiotic conditions (pH) not encountered in their natural habitat [1]. Poor water quality and dense population may allow the transmission of pathogens, especially when crossing species barriers. Shrimp aquaculture, found in 60 countries accounts for just over half the world's shrimp supply with over 80 % of production is based in Asia. Demand for shrimp is expected to stay robust and interest in increasing this value chain include the development of disease control technologies for emerging diseases [2].
The shrimp hemocyte iridescent virus (SHIV), also known as Decapod iridescent virus 1 (DIV1), is an iridescent virus with a double-stranded DNA genome, classified within the proposed genus Decapodiridovirus within the family Iridoviridae. First reported in 2014, pond reared Litopenaeus vannamei shrimp in Zhejiang Province, China experienced massive die-offs while exhibiting hepatopancreatic atrophy with fading colour, empty stomach and guts, and softshell [3]. Histological sections revealed viral aetiology but with pathogenic symptoms that did not match the histopathologic characteristics of any known viruses. Genome sequencing has revealed that SHIV and Cherax quadricarinatus iridovirus (CQIV), identified from freshwater red claw crayfish Cherax quadricarinatus, are likely to be different strains of the same virus species [4]. In March 2019, the Executive Committee of the International Committee on Taxonomy of Viruses (ICTV) approved a proposal for a new species of Decapod Iridescent Virus 1 (DIV1) in the new genus Decapodiridovirus, to include SHIV and CQIV [5]. Species that are known to be susceptible is the Pacific white shrimp Penaeus (Litopenaeus) vannamei with detection in many susceptible penaeid species such as the Giant freshwater prawn Macrobrachium rosenbergii [6], Louisiana swamp crayfish Procambarus clarkii [3], Oriental freshwater shrimp Macrobrachium nipponense, Oriental prawn Exopalaemon carinicauda [6] and Redclaw crayfish Cherax quadricarinatus [7] indicating that DIV1 poses a new threat to the shrimp farming industry.
To date, the search for preventive treatment for SHIV/DIV1 is still ongoing. Current measures being utilised are conventional biosecurity practices such as surveillance and movement restrictions for farms, quarantine and removal of moribund or dead individuals from affected farms and testing for DIV1 in brood stock and post larvae. Treatment of aquaculture diseases has been reliant on the implementation of best practices and chemical treatment (chlorine). Novel experimental treatments or prevention of shrimp disease have been reported using probiotics [8,9], sulfate polysaccharides in the algal crude extracts [10], bioactive secondary metabolites [11], silver nanoparticles (AgNPs) [12] and immunostimulants [13]. Molecular biology efforts that have been attempted include the use of double-stranded RNA (dsRNA) to induce the host RNA interference (RNAi) antiviral mechanism and the use of endogenous viral elements (EVE) to provide shrimp with heritable tolerance to viral infections [14]. In addition, the development of vaccines for shrimp disease is ineffective as shrimp lack the adaptive immune system and rely on the inherent immune system to fight pathogens. Currently, there is no reported effort to explore the use of antiviral drugs in the treatment of viral diseases in shrimp aquaculture.
Focusing the Major Capsid Protein (MCP) as the target protein for this study on drug repurposing for DIV1 is a well-founded decision. MCP is a crucial component of the viral capsid, which is the protective outer shell of the virus [15,16]. It is responsible for forming the structural framework of the viral particle in several viruses, including DIV1 [17], [18], [19]. By targeting structural proteins like MCP, it is possible to disrupt multiple stages of the viral lifecycle. In the case of DIV1, interfering with MCP can impair viral assembly, maturation, and, ultimately, the virus's ability to infect host cells. Furthermore, MCP is often a conserved protein within a viral species, making it a constant and reliable target for drug development [17,20]. This consistency minimizes the probability of resistance forming quickly, which is a concern in the development of antiviral drugs. This fact is supported by the availability of structural information about MCP and allows researchers to predict how potential drugs might interact with the protein, which is a key step in drug repurposing research. Moreover, if this study successfully recognizes existing drugs that interact with MCP and inhibit DIV1, it could mitigate economic losses in shrimp farming by controlling DIV1 using MCP-targeted drugs.
Well-known examples of drug repurposing are Sildenafil that was developed for hypertension treatment but commercialized for the treatment of erectile dysfunction, dimethyl fumarate that has been repurposed from the treatment of psoriasis to multiple sclerosis and the sedative thalidomide being repurposed for the treatment of multiple myeloma and leprosy. However, these successfully reported drug repositioning is often a product of serendipity [21]. To further increase the success of drug repurposing to overcome the financial and time constraints faced during the traditional drug discovery process, researchers have relied more on the utilisation of bioinformatics and computational tools. The establishment of the Drug Repurposing Hub database (https://clue.io/repurposing) has boosted the computational screening of drugs by providing an annotated collection of FDA-approved drugs, clinical trial drugs, and pre-clinical tool compounds with a companion information resource [22].
Computational approaches that include molecular modelling [23], transcriptomic data mining [24] and molecular docking [25] have contributed to the screening of drugs for repurposing. As of date, computational screening has produced promising results such as the use of malaria drug amodiaquine as a potential anti-cancer agent [26] and ciprofloxacin for the treatment of Chagas disease that is caused by the parasite Trypanosoma cruzi [25], Ethambutol for African sleeping sickness [27] and Comtan for the treatment of tuberculosis [28]. The use of big-data, -omics technologies, machine learning algorithms and artificial intelligence will all offer unprecedented knowledge and tools to aid drug repurposing [29]. The present paper is the first structural characterization of DIV1 MCP. The 3D model of DIV1 MCP was generated and used for comparative modelling. Molecular docking and molecular dynamics simulation is used to identify and characterise the inhibition mode of three potential antivirals, Chloroquine, Rimantadine and CAP-1 against DIV1.
2. Methodology
2.1. Physiochemical characterization and sequence alignment
The target sequence was retrieved from the GenBank Database under accession no. ATE87157.1 and the full amino acid sequence was included in the supplementary file (Table S1). BLAST was employed to analyse the major capsid protein (MCP) derived from Decapod Iridescent Virus 1 (DIV1 MCP), which was isolated from the white leg shrimp, Litopenaeus vannamei. We conducted a BLAST search using the Protein Data Bank (PDB) as the database to identify a suitable crystal structure to serve as a template for our model. The physiochemical of DIV1 MCP were computed using the Expasy's ProtParam tool [30]. SUPER-FAMILY HMM server was utilised to identify the conserved domain and the potential protein family [31]. The multiple sequence alignment then was developed using Clustal Omega server [32] and ESPript 3 [33].
2.2. Model development and evaluation
The potential template of MCP was screened by using different servers that were PSI- BLAST [34], Phyre 2 [35] and Swiss Model [36]. The sequence of MCP has been optimized and the three-dimensional (3D) structure was built by using Swiss Model server. The 3D-model has been refined using 3D refine server [37] and evaluated by utilising PROCHECK [38] and ERRAT [39] server. The model was superimposed with the best template structure and their structural differences were determined. The chosen model of MCP was analysed its topology using PDBsum [40]. The quality scores of alignments between root mean square deviations (RMSD) were determined using TM-align program [41,42].
2.3. Molecular docking
The binding site was predicted using the binding site analysis tool in FTSite server [43] and Conserved Domain Database (CDD). The structure of antiviral drugs, Chloroquine (PubChem CID: 2719), Rimantadine (PubChem CID: 5071) and CAP-1 (PubChem CID: 5,082,818) were extracted from the PubChem server [44]. Molecular docking was performed by using SwissDock and the protein-ligand with the lowest binding energy was selected as a good binding affinity. Interactions of a protein-ligand complex was analysed using UCSF Chimera [45] and shown in schematic diagrams in 3D and 2D using BIOVIA Discovery Studio version 2020 [46].
2.4. Molecular dynamics simulation
To study the stability and molecular dynamics of DIV1 MCP interaction with the antiviral drugs, all proteins were simulated with different complexes (Apo, Chloroquine, Rimantadine and CAP-1). All protein-ligand docked complexes were subjected to MD simulation using GROMACS 2021 software package [47] and CHARMM36 force field [48]. The topologies and force filed parameters of all ligands were built separately using CGenFF server due to limitation of GROMACS program to parameterize the heteroatom groups and small molecules. The structures were placed at the centre of the cubic box which at least 10 Å from the edges and solvated with Simple Point Charge (SPC) water molecules. The system was neutralized by adding in accordance sodium ions to create zero charged system. Periodic boundary conditions (PBC) were applied to the proteins, and electrostatic interactions were improved by utilising the Particle Mesh Ewald (PME) summation technique, which is the best method for calculating long-range electrostatics. The energy minimization was performed using the steepest descent minimization of 5000 steps followed by equilibration for 100 ps of solute-position-restrained MD. LINCS algorithm (LINear Constraint Solver) was applied to constraint all bonds in the system. The MD simulations of the equilibrated structures were conducted in triplicate for a duration of 100 ns (100,000 ps). To assess the stability and compactness of the protein-ligand structure, GROMACS utilities; root mean square deviation (RMSD) of the backbone and radius of gyration (Rg) were calculated.
3. Results and discussion
3.1. Physicochemical characterization and sequence alignment
The physiochemical of MCP were computed using the Expasy's ProtParam tool. The result shows the molecular weight and theoretical pI of the target protein was 53,857.50 kDa and pI 7.59. The total number of negatively (Asp + Glu) and positively (Arg + Lys) charged residues was 50 –R and 51 + R. This target protein is classified as stable since the instability index was lower than 40. Decapod iridescent virus 1 (DIV1) is a large double-stranded DNA (dsDNA) virus in the family Iridoviridae. The protein sequence of DIV1 MCP was subjected to several proteins functional database PFAM, Conserved Domain Database (CDD) and SUPERFAMILY HMM server DIV1 MCP was classified as Major capsid protein belong to family nucleocytoplasmic large DNA viruses (Table S1). The sequence alignment of major capsid protein from DIV1 showed high homology to MCP from Singapore Grouper Iridovirus (PDB ID: 6OJN) and Paramecium bursaria Chlorella virus 1 (PDB ID: 5TIP) with 38 % and 23 % sequence identity respectively.
3.2. Model development and evaluation
The potential template that best resembles the target protein was identified using three different servers, PSI-Blast, Phyre 2, and Swiss Model. As a result, two templates 6OJN and 5TIP appeared with different percentages of identity obtained from different servers, and amongst both templates, 6OJN was chosen as the best template as it showed the highest sequence identity with the target protein (DIV1 MCP) compared to 5TIP (Table 1). Thus, the three-dimensional (3D) structure of DIV1 MCP was constructed based on the crystal structure of Singapore grouper iridovirus (PDB ID:6OJN) as a template using the Swiss Model server. The initial 3D structures of DIV1 MCP were further optimized by 3D refine server to exclude poor molecular contacts and refine the sidechains. Two structure evaluation tools, PROCHECK and ERRAT were implemented to validate the quality of the created 3D models before and after structure refinement (Table 2). The Psi/Phi Ramachandran plot from the PROCHECK analysis was used to examine the backbone conformations of the protein. According to the Ramachandran plot, 99.70 % of residues are in allowed regions and only 0.3 % of amino acid residue are in disallowed regions. A model with a score of more than 90 % is considered to be of good stereochemical quality [38]. To calculate the overall quality score for non-bonded atomic interaction, model validation was also performed by ERRAT. The ERRAT score of the MCP model was significantly improved with a score 73.56 %. Based on [49], the normal range of ERRAT score to be accepted for a high-quality model must be greater than 50 % therefore, indicate the DIV1 MCP model as a good quality model. The superimposition of the model (DIV1 MCP) with the template (6OJN) revealed a good structure alignment with RMSD of 0.38 Å and a 98 % coverage of the backbone atoms. The result of TM-score for the alignments showed 0.98 indicating that this structure alignment represents a better template as a score above 0.4 is regarded as significant. Subsequently, we also performed a comparative analysis with AlphaFold. The structural alignment assessment demonstrated that the model generated by Swiss Model exhibited a lower RMSD value and achieved a higher Tm-align score in relation to the template 6OJN, as compared to the model produced by AlphaFold. Based on this evaluation, we opted to employ the model produced by the Swiss Model server (Fig. S1). Despite their low sequence similarity, MCP and the other members of the nucleocytoplasmic large DNA virus family have a high degree of structural similarity. In addition, the secondary structure of the DIV1 MCP trimer displayed eight α-helices and a signature double jelly-roll fold with 16 ß-strands forming two antiparallel sheets as depicted in Fig. 1. This prevalence of double jelly-roll fold is known as a core structural motif that appears amongst capsid proteins and might be related to the viral shell formation of varying sizes [50,51].
Table 1.
DIV1 MCP's top three proposed templates by different servers.
| Protein enzyme | Server name | Template | Organism | Family | Length | Identity | E-value |
|---|---|---|---|---|---|---|---|
| Major Capsid Protein (ATE87157.1) | PSI-Blast | 6OJN | Singapore grouper iridovirus |
Capsid_NCLDV | 537 | 38 % | 3e-107 |
| 5TIP |
Paramecium bursaria Chlorella virus 1 |
Capsid_NCLDV | 436 | 23 % | 4e-19 | ||
| Phyre 2 | 6OJN | Singapore grouper iridovirus |
Capsid_NCLDV | 537 | 38 % | 0 | |
| 5TIP |
Paramecium bursaria Chlorella virus 1 |
Capsid_NCLDV | 436 | 22 % | 0 | ||
| Swiss Model | 6OJN | Singapore grouper iridovirus |
Capsid_NCLDV | 537 | 39 % | N/A | |
| 5TIP |
Paramecium bursaria Chlorella virus 1 |
Capsid_NCLDV | 436 | 21 % | N/A |
Table 2.
Summary of model validation using different tools.
| Model evaluation tools | Normal range of the score | Obtained score | ATE87157.1 |
|---|---|---|---|
| PROCHECK | >90 % | Before refinement | 99.70 % |
| After refinement | 99.70 % | ||
| ERRAT | >50 % | Before refinement | 67.43 % |
| After refinement | 73.56 % |
Fig. 1.

Overall structure of DIV1 capsid protein. (a) Side view (left) and top view of DIV1 MCP trimer. (b) The secondary structure and (c) topology diagram of DIV1 MCP monomer. The β-strands are presented in light blue while α-helices in dark blue.
3.3. Molecular docking
Before starting a molecular docking, the target protein was submitted to FTSite server to find the binding site region. Binding site identification is important for structure-based prediction of function and drug design. With near-experimental accuracy, FTSite is capable of predicting the binding sites in over 94 % of all proteins [43,52]. Fig. 2 depicts the position of the structural target protein binding site predictions in trimer: pink, green, and blue. The amino acids such as Asn120, His123, Asp147, Asn150, Gln151, Tyr162, Asn163, Ile166, Gly167, and Arg323 were predicted as binding sites for the substrate-binding site of DIV1 MCP protein.
Fig. 2.

The predicted binding pockets for DIV1 MCP. (a) The binding pocket of the MCP trimer. The predicted residues are represented in the mesh surface (blue, green and pink mesh surfaces. (b) A close up of the substrate-binding site is shown in orange surface representation. The binding sites were predicted using FTsite server.
Chloroquine, Rimantadine and CAP-1 were chosen as potential antiviral drug due to these compounds have several abilities to block viral capsid. Different compounds are categorized based on the viral cycle stage with which they interact, and their mechanisms were summarized in Fig. 3. Rimantadine is an M2 ion channel inhibitor and is used to suppress influenza A infection. It prevents viruses from replicating by interfering with the virus's uncoating process [53,54]. As for CAP-1, according to [55] and [56], CAP-1 is an antiviral drug used to inhibit HIV replication by preventing the assembly of viral capsids. Recent studies reported that Chloroquine used as an antiviral drug and potently fights the SARS-CoV-2 (Covid-19) pandemic by blocking the entry stage of viral replication even at low concentrations were used [57], [58], [59]. However, these compounds have been tested in humans, and to the best of our knowledge, there is no reported data regarding those compounds being used as antiviral drugs to treat viral infections in shrimp aquaculture. Thus, this is the first report on the structural characterisation of DIV1 MCP docked with repurposing drugs.
Fig. 3.

Schematic illustration of antiviral drug mechanism against DIV1 virus replication. Modified from Refs. [60,61]. This illustration was created with BioRender.com.
The finding revealed that three drugs i.e. Chloroquine, Rimantadine, and CAP-1 showed significant binding energies with DIV1 MCP protein as presented in Table 3. Amongst those compounds, CAP-1 has shown such a promising repurpose drug candidate against DIV1 as it possesses the lowest binding energy of −8.46 kcal/mol. Besides, the respective compounds have shown various interactions including van der Waals, hydrogen bonds, and alkyl while their active sites lie in a deep pocket shaped by α helical barrel loops as presented in Fig. 4.
Table 3.
The binding energy of the potential drugs with the DIV1 MCP.
| Drug | PubChem CID | Structure | Binding energy (kcal/mol) |
|---|---|---|---|
| Chloroquine | 2719 | 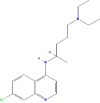 |
−7.25 |
| Rimantadine | 5071 | 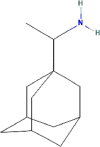 |
−6.62 |
| CAP-1 | 5,082,818 | 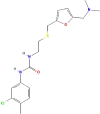 |
−8.46 |
Fig. 4.

The interactions of DIV1 MCP with (a) Chloroquine (b) Rimantadine and (c) CAP-1 compounds. The residues involved in the various interactions including van der Waals, hydrogen bond, and alkyl were indicated in light green, green, and lavender colours, respectively. Those interactions were created by using 2D schematic diagrams using Discovery Studio 4.0 (DS 4.0).
Docking results showed that the active centre pocket of the DIV1 MCP-ligands (Chloroquine and Rimantadine) complexes consists of ten binding residues which are similar to the prediction result of the substrate-binding site obtained by FTsite server. Meanwhile, the DIV1-CAP-1 complex showed nine binding residues (Gly167 is excluded) recognised as active sites. As can be seen from Fig. 4, most residues of DIV1 MCP interacted with three compounds via van der Waals forces. These interactions occur between hydrophobic side chains which are essential to protein stability and function, especially for macromolecular complex structures such as DIV1 MCP trimer [62].
3.4. Molecular dynamics simulation
Molecular dynamics simulations were conducted in triplicate, with replicate 1 (R1), replicate 2 (R2), and replicate 3 (R3), each running for 100 ns. These simulations were performed to investigate the stability of DIV1 capsid protein complexes. The RMSD of the apo structure exhibited the highest deviations compared to the complex structure of the DIV1 capsid protein with antivirals, with RMSD values of 0.77 nm (R1), 0.71 nm (R2) and 0.73 nm (R3). The DIV1 MCP-CAP-1 complex demonstrated the most stability with RMSD values of 0.59 nm (R1), 0.63 nm (R2) and 0.62 nm (R3) compared to other complexes. The DIV1 MCP- Chloroquine and DIV1 MCP-Rimantadine exhibited slightly lower stability, as indicated by their RMSD values ranging from 0.66 nm to 0.69 nm (Fig. 5). These findings indicate that the sustained stability of the complexes are implied by the binding of the antivirals to DIV1 MCP. By taking together the result of docking and molecular dynamics simulation, therefore, it has been proved that the DIV1 MCP-CAP-1 complex is the most stable compared to other complexes.
Fig. 5.

Molecular dynamics analysis of apo and DIV1 capsid protein-ligand complexes (Chloroquine, Rimantadine, and CAP-1) trajectories generated by GROMACS. The MD simulations were conducted in triplicate (referred to as R1, R2, and R3) for a duration of 100 ns each.
The data of structural alignment evaluation of the DIV1 MCP model with its template, Singapore grouper Iridovirus (6OJN) showed 98 % of coverage, describing that their phylogenetic relationship of iridovirus was related. Decapod iridescent virus 1 (DIV1) is the only species of the new genus Decapod iridovirus and is included in the family Iridoviridae. It is an emerging viral pathogen that critically threatens shrimp aquaculture [6,63,64]. Thus, the mechanism of control and prevention for this disease remains scarce and elusive [60] reported that proteomic analysis of infected cells by iridovirid type revealed that the iridoviral replication involved phosphorylation reactions to increase the expression of that viral genome. Therefore, to stop this type of viral replication, a selective inhibitor or drug should be derived from a group that could block the assembly of viral at the late stage because the phosphorylation reaction is one of the post-translational modifications that occur at the final stage (Fig. 3) [65]. This fact can be related to the mechanism of antiviral drug, CAP-1 which functions to prevent the assembly of viral capsids and lead to the inhibition of viral replication [61].
Intriguingly, our findings demonstrated that CAP-1 showed the highest binding affinity (−8.46 kcal/mol) to DIV1 MCP protein, and compared to other drugs, CAP-1 had phosphorylated multiple residues such as His123, Tyr162, and Thr395. This fact has been corroborated by previous studies where CAP-1 had decreased the multimerization of viral and affected the stability and assembly of viral capsids which occur at the late stage [56]. Moreover, CAP-1 binding to viral protein induced the conformational changes that cause displace of residues Phe32, His62, and Tyr145 within the pocket of binding site and shifted the loop between two alpha helices of the viral protein structure. Consequently, it causes a loss of mature capsid assembly [56,[66], [67], [68]]. Therefore, we speculated that CAP-1, a repurposing drug could inhibit the viral assembly and maturation of DIV1 MCP by interfering phosphorylation of the viral protein.
Regardless, this study is primarily focused on the prediction of potential antiviral drugs targeting DIV1 replication and viral infectivity. While the results of molecular docking and molecular dynamics simulations have revealed intriguing interactions between the selected drugs and the DIV1 MCP, it's important to note that the scope of the study is limited to computational predictions. The aim here is to identify candidate drugs for further investigation, and as such, the study does not include experimental validation or verification of these predictions. Future research and experimental studies would be essential to confirm the effectiveness of the identified drugs in inhibiting DIV1 replication and controlling viral infectivity in practical settings, particularly within the context of shrimp farming.
4. Conclusion
In conclusion, the present study determined the physicochemical features, model development of the DIV1 MCP, and investigated the interactions of DIV1 MCP with three antiviral drugs i.e. Chloroquine, Rimantadine, and CAP-1. Amongst those drugs, CAP-1 has shown such a promising repurpose drug candidate against DIV1 as it possesses the lowest binding energy and good stability. We speculate that CAP-1 binding may induce the conformational changes in DIV1 MCP structure by phosphorylating multiple residues such as His123, Tyr162, and Thr395 and ultimately block the viral assembly and maturation of DIV1 MCP. This fact indicates that viral infectivity is critically dependant on capsid formation and stability, making the capsid protein a potentially attractive antiviral target. To the best of our knowledge, this is the first report regarding the structural characterisation of DIV1 MCP docked with repurposing drugs. In the future, in vitro and in vivo studies are required to corroborate the ability of CAP-1, a repurposing drug to treat the infection of Decapod Iridescent Virus 1 (DIV1) in shrimp farming.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
This work was supported by Ministry of Higher Education (MoHE) Malaysia through Fundamental Research Grants Scheme (FRGS/1/2021/STG01/UMT/02/2) and Universiti Malaysia Terengganu through Talent and Publication Enhancement Research Grant (UMT/TAPE-RG/2020/55298).
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.fsirep.2023.100120.
Appendix. Supplementary materials
Data availability
Data will be made available on request.
References
- 1.Millard R.S., P.Ellis R., S.Bateman K., K.Bickley L., R.Tyler C., Aerle R., Santos E.M. How do abiotic environmental conditions influence shrimp susceptibility to disease? A critical analysis focussed on white spot disease. J. Invertebr. Pathol. 2021;186 doi: 10.1016/j.jip.2020.107369. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher, R. (2019). Fresh insights into the $26.7 billion shrimp sector. The Fish Site. https://thefishsite.com/articles/fresh-insights-into-the-26-7-billion-shrimp-sector.
- 3.Qiu L., Chen M.M., Wan X.Y., Li C., Zhang Q.L., Wang R.Y., Cheng D.Y., Dong X., Yang B., Wang X.H., Xiang J.H., Huang J. Characterization of a new member of Iridoviridae, Shrimp hemocyte iridescent virus (SHIV), found in white leg shrimp (Litopenaeus vannamei) Sci. Rep. 2017;7:11834. doi: 10.1038/s41598-017-10738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu L., Wang T., Li F., Yang F. Isolation and preliminary characterization of a new pathogenic iridovirus from redclaw crayfish Cherax quadricarinatus. Dis. Aquat. Org. 2016;120(1):17–26. doi: 10.3354/dao03007. [DOI] [PubMed] [Google Scholar]
- 5.Bunnajirakul S. Emerging disease in shrimp; decapod iridescent virus 1 (DIV1) infection. J. Mahanakorn Vet. Med. 2020;15(2):223–230. [Google Scholar]
- 6.Qiu L., Chen X., Zhao R., Li C., Gao W., Zhang Q., Huang J. Description of a natural infection with Decapod Iridescent Virus 1 in farmed giant freshwater prawn, Macrobrachium rosenbergii. Viruses. 2019;11(4):354. doi: 10.3390/v11040354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang H., Wei X., Wang R., Zeng L., Yang Y., Huang G., Shafique L., Ma H., Lv M., Ruan Z., Naz H., Lin Y., Huang L., Chen T. Transcriptomics of Cherax quadricarinatus hepatopancreas during infection with Decapod iridescent virus 1 (DIV1) Fish Shellfish Immunol. 2020;98:832–842. doi: 10.1016/j.fsi.2019.11.041. [DOI] [PubMed] [Google Scholar]
- 8.Jamal M.T., Abdulrahman I.A., Harbi M.Al, Chithambaran S. Probiotics as alternative control measures in shrimp aquaculture: a review. J. Appl. Biol. Biotechnol. 2019;7(3):69–77. doi: 10.7324/JABB.2019.70313. [DOI] [Google Scholar]
- 9.Knipe H., Temperton B., Lange A., Bass D., Tyler C.R. Probiotics and competitive exclusion of pathogens in shrimp aquaculture. Rev. Aquac. 2021;13(1):324–352. doi: 10.1111/raq.12477. [DOI] [Google Scholar]
- 10.Klongklaew N., Praiboon J., Tamtin M., Srisapoome P. Antibacterial and antiviral activities of local Thai green macroalgae crude extracts in pacific white shrimp (Litopenaeus vannamei) Mar. Drugs. 2020;18(3):140. doi: 10.3390/md18030140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Namitha R., Vasantharaja R., Radhakrishnan M., Govindaraju K. Actinobacteria and their bioactive molecules for anti-WSSV activity: a mini review. Aquac. Res. 2021;52(3):883–889. doi: 10.1111/are.14942. [DOI] [Google Scholar]
- 12.Camacho-Jiméneza L., RuthÁlvarez-Sánchez A., Mejía-Ruíz C.H. Silver nanoparticles (AgNPs) as antimicrobials in marine shrimp farming: a review. Aquac. Rep. 2020;18 [Google Scholar]
- 13.Mohan K., Ravichandran S., Muralisankar T., Uthayakumar V., Chandirasekar R., Seedevi P., Rajan D.K. Potential uses of fungal polysaccharides as immunostimulants in fish and shrimp aquaculture: a review. Aquaculture. 2019;500:250–263. doi: 10.1016/j.aquaculture.2018.10.023. [DOI] [PubMed] [Google Scholar]
- 14.Labreuche Y., Warr G.W. Insights into the antiviral functions of the RNAi machinery in penaeid shrimp. Fish Shellfish Immunol. 2013;34(4):1002–1010. doi: 10.1016/j.fsi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Krupovic M., Koonin E.V. Multiple origins of viral capsid proteins from cellular ancestors. Proc. Natl. Acad. Sci. U. S. A. 2017;114(12):E2401–E2410. doi: 10.1073/pnas.1621061114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Throngnumchai B., Jitrakorn S., Sangsuriya P., Unajak S., Khunrae P., Dong H.T., Saksmerprome V., Rattanarojpong T. Refolded recombinant major capsid protein (MCP) from infectious spleen and kidney necrosis virus (ISKNV) effectively stimulates serum specific antibody and immune related genes response in Nile tilapia (Oreochromis niloticus) Protein Expr. Purif. 2021;184 doi: 10.1016/j.pep.2021.105876. [DOI] [PubMed] [Google Scholar]
- 17.Qin P., Munang'andu H.M., Xu C., Xie J. Megalocytivirus and other members of the family iridoviridae in finfish: a review of the etiology, epidemiology, diagnosis, prevention and control. Viruses. 2023;15(6):1359. doi: 10.3390/v15061359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu L., Chen X., Gao W., Li C., Guo X.M., Zhang Q.L., Yang B., Huang J. Molecular epidemiology and histopathological study of a natural infection with decapod iridescent virus 1 in farmed white leg shrimp, Penaeus vannamei. Aquaculture. 2021;533 doi: 10.1016/J.AQUACULTURE.2020.736105. [DOI] [Google Scholar]
- 19.Xu Y., Wang Y., Hu J., Bao Z., Wang M. Development and visualization improvement for the rapid detection of decapod iridescent virus 1 (DIV1) in Penaeus vannamei based on an isothermal recombinase polymerase amplification assay. Viruses. 2022;14(12) doi: 10.3390/v14122752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang S.M., Tu C., Tseng C.H., Huang C.C., Chou C.C., Kuo H.C., Chang S.K. Genetic analysis of fish iridoviruses isolated in Taiwan during 2001-2009. Arch. Virol. 2011;156(9):1505–1515. doi: 10.1007/s00705-011-1017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parisi D., F.Adasme M., Sveshnikova A., Bolz S.N., Moreau Y., Schroeder M. Drug repositioning or target repositioning: a structural perspective of drug-target-indication relationship for available repurposed drugs. Comput. Struct. Biotechnol. J. 2020;18:1043–1055. doi: 10.1016/j.csbj.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Corsello S.M., Bittker J.A., Liu Z., Gould J., McCarren P., Hirschman J.E., Johnston S.E., Vrcic A., Wong B., Khan M., Asiedu J., Narayan R., Mader C.C., Subramanian A., Golub T.R. The drug repurposing hub: a next-generation drug library and information resource. Nat. Med. 2017;23(4):405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu X., Huang M., Zou X. Docking-based inverse virtual screening: methods, applications, and challenges. Biophys. Rep. 2018;4(1):1–16. doi: 10.1007/s41048-017-0045-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang Y., Yella J., Jegga A.G. Transcriptomic data mining and repurposing for computational drug discovery. Methods Mol. Biol. 2019;1903:73–95. doi: 10.1007/978-1-4939-8955-3_5. [DOI] [PubMed] [Google Scholar]
- 25.Adasme M.F., Bolz S.N., Adelmann L., Salentin S., Haupt V.J., Moreno-Rodríguez A., Nogueda-Torres B., Castillo-Campos V., Yepez-Mulia L., De Fuentes-Vicente J.A., Rivera G., Schroeder M. Repositioned drugs for chagas disease unveiled via structure-based drug repositioning. Int. J. Mol. Sci. 2020;21(22):8809. doi: 10.3390/ijms21228809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Salentin S., Adasme M.F., Heinrich J.C., Haupt V.J., Daminelli S., Zhang Y., Schroeder M. From malaria to cancer: computational drug repositioning of amodiaquine using PLIP interaction patterns. Sci. Rep. 2017;7:11401. doi: 10.1038/s41598-017-11924-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rashmi M., Swati D. In silico drug re-purposing against African sleeping sickness using GlcNAc-PI de-N-acetylase as an experimental target. Comput. Biol. Chem. 2015;59:87–94. doi: 10.1016/j.compbiolchem.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 28.Kinnings S.L., Liu N., Buchmeier N., Tonge P.J., Xie L., Bourne P.E. Drug discovery using chemical systems biology: repositioning the safe medicine Comtan to treat multi-drug and extensively drug resistant tuberculosis. PLoS Comput. Biol. 2009;5(7) doi: 10.1371/journal.pcbi.1000423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mottini C., Napolitano F., Li Z., Gao X., Cardone L. Computer-aided drug repurposing for cancer therapy: approaches and opportunities to challenge anticancer targets. Semin. Cancer Biol. 2021;68:59–74. doi: 10.1016/j.semcancer.2019.09.023. [DOI] [PubMed] [Google Scholar]
- 30.Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M.R., Appel R.D., Bairoch A. The Proteomics Protocols Handbook. Humana Press; 2005. Protein identification and analysis tools on the ExPASy server; pp. 571–607. J. M. Walker. [DOI] [Google Scholar]
- 31.Razali S.A., Sarah Diana P., Shamsir M.S., Mahadi N.M., Mohd Illias R. Substrate and cofactor binding interaction studies of galactitol -1- Phosphate 5- dehydrogenase from peptoclostridium difficile. J. Teknol. 2016;78(6):199–210. doi: 10.11113/jt.v78.7598. [DOI] [Google Scholar]
- 32.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J.D., Higgins D.G. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7(1):539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robert X., Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42(W1):W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li W., McWilliam H., Goujon M., Cowley A., Lopez R., Pearson W.R. PSI-Search: iterative HOE-reduced profile SSEARCH searching. Bioinformatics. 2012;28(12):1650–1651. doi: 10.1093/bioinformatics/bts240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelley L.A., Mezulis S., Yates C., Wass M., Sternberg M. The Phyre2 web portal for protein modelling, prediction, and analysis. Nat. Protoc. 2015;10(6):845–858. doi: 10.1038/nprot.2015-053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., De Beer T.A.P., Rempfer C., Bordoli L., Lepore R., Schwede T. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46(W1):W296–W303. doi: 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bhattacharya D., Nowotny J., Cao R., Cheng J. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Res. 2016;44(W1):W406–W409. doi: 10.1093/NAR/GKW336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laskowski R.A., Macarthur M.W., Thornton J.M. PROCHECK : validation of protein-structure coordinates. Int. Tables Crystallogr. 2012;F(Chapter 21.4):684–687. [Google Scholar]
- 39.Colovos C., Yeates T.O. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2(9):1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Laskowski R.A., Jabłońska J., Pravda L., Vařeková R.S., Thornton J.M. PDBsum: structural summaries of PDB entries. Protein Sci. 2018;27(1):129–134. doi: 10.1002/pro.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Razali S.A., Shamsir M.S. Characterisation of a catalytic triad and reaction selectivity in the dual mechanism of the catalyse hydride transfer in xylitol phosphate dehydrogenase. J. Mol. Graph. Model. 2020;97 doi: 10.1016/j.jmgm.2020.107548. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y., Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33(7):2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ngan C.H., Hall D.R., Zerbe B., Grove L.E., Kozakov D., Vajda S. FtSite: high accuracy detection of ligand binding sites on unbound protein structures. Bioinformatics. 2012;28(2):286–287. doi: 10.1093/bioinformatics/btr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y., Bryant S.H., Cheng T., Wang J., Gindulyte A., Shoemaker B.A., Thiessen P.A., He S., Zhang J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2017;45(D1):D955–D963. doi: 10.1093/nar/gkw1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C.C., Meng E.C., Morris J.H., Pettersen E.F., Ferrin T.E. Enhancing UCSF chimera through web services. Nucleic Acids Res. 2014;42(W1):W478–W484. doi: 10.1093/nar/gku377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Systèmes, D. (2020). BIOVIA discovery visualizer. [Version 2020]. San Diego: Dassault Systèmes. 10.1054/midw.2001.0283.
- 47.Páll S., Zhmurov A., Bauer P., Abraham M., Lundborg M., Gray A., Hess B., Lindahl E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020;153 doi: 10.1063/5.0018516. [DOI] [PubMed] [Google Scholar]
- 48.Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., Groot B.L., Grubmüller H., MacKerell A.D., Jr CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017;14(1):71–73. doi: 10.1038/nmeth.4067.CHARMM36m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chaitanya M., Babajan B., Anuradha C.M., Naveen M., Rajasekhar C., Madhusudana P., Kumar C.S. Exploring the molecular basis for selective binding of Mycobacterium tuberculosis Asp kinase toward its natural substrates and feedback inhibitors: a docking and molecular dynamics study. J. Mol. Model. 2010;16(8):1357–1367. doi: 10.1007/s00894-010-0653-4. [DOI] [PubMed] [Google Scholar]
- 50.Cheng S., Brooks C.L. Viral capsid proteins are segregated in structural fold space. PLoS Comput. Biol. 2013;9(2) doi: 10.1371/journal.pcbi.1002905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krupovic M., Makarova K.S., Koonin E.V. Cellular homologs of the double jelly-roll major capsid proteins clarify the origins of an ancient virus kingdom. Proc. Natl. Acad. Sci. U. S. A. 2022;119(5) doi: 10.1073/pnas.2120620119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kozakov D., Grove L.E., Hall D.R., Bohnuud T., Mottarella S.E., Luo L., Xia B., Beglov D., Vajda S. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat. Protoc. 2015;10(5):733–755. doi: 10.1038/nprot.2015.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kausar S., Said Khan F., Ishaq Mujeeb Ur Rehman M., Akram M., Riaz M., Rasool G., Hamid Khan A., Saleem I., Shamim S., Malik A. A review: mechanism of action of antiviral drugs. Int. J. Immunopathol. Pharmacol. 2021;35:1–12. doi: 10.1177/20587384211002621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koff W.C., Knight V. Inhibition of influenza virus uncoating by rimantadine hydrochloride. J. Virol. 1979;31(1):261–263. doi: 10.1128/jvi.31.1.261-263.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klumpp K., Crépin T. Capsid proteins of enveloped viruses as antiviral drug targets. Curr. Opin. Virol. 2014;5:63–71. doi: 10.1016/j.coviro.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 56.McFadden W.M., Snyder A.A., Kirby K.A., Tedbury P.R., Raj M., Wang Z., Sarafianos S.G. Rotten to the core: antivirals targeting the HIV-1 capsid core. Retrovirology. 2021;18:41. doi: 10.1186/s12977-021-00583-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends. 2020;14(1):72–73. doi: 10.5582/BST.2020.01047. [DOI] [PubMed] [Google Scholar]
- 58.Spinelli F.R., Ceccarelli F., Franco M.Di, Conti F. To consider or not antimalarials as a prophylactic intervention in the SARS- CoV-2 (Covid-19) pandemic. Ann. Rheum. Dis. 2020;79:666–667. doi: 10.1093/cid/ciaa237. [DOI] [PubMed] [Google Scholar]
- 59.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.İnce İ.A., Özcan O., Ilter-Akulke A.Z., Scully E.D., Özgen A. Invertebrate iridoviruses: a glance over the last decade. Viruses. 2018;10(4):161. doi: 10.3390/v10040161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pereiro P., Figueras A., Novoa B. Compilation of antiviral treatments and strategies to fight fish viruses. Rev. Aquac. 2021;13(3):1223–1254. doi: 10.1111/raq.12521. [DOI] [Google Scholar]
- 62.Li J., Wang Y., An L., Chen J., Yao L. Direct observation of CH/CH van der Waals interactions in proteins by NMR. J. Am. Chem. Soc. 2018;140(9):3194–3197. doi: 10.1021/jacs.7b13345. [DOI] [PubMed] [Google Scholar]
- 63.Liao X., He J., Li C. Decapod iridescent virus 1: an emerging viral pathogen in aquaculture. Rev. Aquac. 2022:1–2. doi: 10.1111/raq.12672. [DOI] [Google Scholar]
- 64.Qiu L., Chen X., Gao W., Li C., Guo X.M., Zhang Q.L., Bing Y., Huang J. Molecular epidemiology and histopathological study of a natural infection with decapod iridescent virus 1 in farmed white leg shrimp, Penaeus vannamei. Aquaculture. 2021;533 [Google Scholar]
- 65.Ramazi S., Zahiri J. Post-translational modifications in proteins: resources, tools and prediction methods. Database. 2021;2021:baab012. doi: 10.1093/database/baab012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelly B.N., Kyere S., Kinde I., Tang C., Howard B.R., Robinson H., Sundquist W.I., Summers M.F., Hill C.P. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J. Mol. Biol. 2007;373(2):355–366. doi: 10.1016/j.jmb.2007.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lemke C.T., Titolo S., von Schwedler U., Goudreau N., Mercier J.F., Wardrop E., Faucher A.M., Coulombe R., Banik S.S.R., Fader L., Gagnon A., Kawai S.H., Rancourt J., Tremblay M., Yoakim C., Simoneau B., Archambault J., Sundquist W.I., Mason S.W. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J. Virol. 2012;86(12):6643–6655. doi: 10.1128/jvi.00493-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pornillos O., Ganser-Pornillos B.K., Kelly B.N., Hua Y., Whitby F.G., David Stout C., Sundquist W.I., Hill C.P., Yeager M. X-ray structures of the hexameric building block of the HIV capsid. Cell. 2009;137(7):1282–1292. doi: 10.1016/j.cell.2009.04.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.