

Abstract
The mitochondrial (m.) 3243A>G mutation is known to be associated with various mitochondrial diseases including mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS). Their clinical symptoms have been estimated to occur with an increased mitochondrial DNA (mtDNA) heteroplasmy and reduced activity of oxidative phosphorylation (OXPHOS) complexes, but their trends in the central nervous system remain unknown. Six autopsied mutant cases and three disease control cases without the mutation were enrolled in this study. The mutant cases had a disease duration of 1–27 years. Five of six mutant cases were compatible with MELAS. In the mutant cases, cortical lesions including a laminar necrosis were frequently observed in the parietal, lateral temporal, and occipital lobes; less frequently in the frontal lobe including precentral gyrus; and not at all in the medial temporal lobe. The mtDNA heteroplasmy in brain tissue samples of the mutant cases was strikingly high, ranging from 53.8% to 85.2%. The medial temporal lobe was preserved despite an inhospitable environment having high levels of mtDNA heteroplasmy and lactic acid. OXPHOS complex I was widely decreased in the mutant cases. The swelling of smooth muscle cells in the vessels on the leptomeninges, with immunoreactivity (IR) against mitochondria antibody, and a decreased nuclear/cytoplasmic ratio of choroidal epithelial cells were observed in all mutant cases but in none without the mutation. Common neuropathological findings such as cortical laminar necrosis and basal ganglia calcification were not always observed in the mutant cases. A high level of mtDNA heteroplasmy was observed throughout the brain in spite of heterogeneous cortical lesions. A lack of medial temporal lesion, mitochondrial vasculopathy in vessels on the leptomeninges, and an increased cytoplasmic size of epithelial cells in the choroid plexus could be neuropathological hallmarks helpful in the diagnosis of mitochondrial diseases.
Keywords: choroidal epithelial cell swelling, MELAS, mitochondrial 3243A>G mutation, mitochondrial vasculopathy, mtDNA heteroplasmy, stroke‐like episodes
1. BACKGROUND
Patients with the mitochondrial (m.) 3243A>G mutation in the mitochondrial tRNALeu(UUR) gene can show mitochondrial myopathy, encephalopathy, lactic acidosis, stroke‐like episodes (MELAS, OMIM 540000); Leigh syndrome (OMIM 256000); myoclonic epilepsy with ragged‐red fibers (MERRF, OMIM 545000); chronic progressive external ophthalmoplegia (CPEO, OMIM 157640); and maternally inherited diabetes‐deafness syndrome (MIDD, OMIM 520000) [1]. MELAS belongs to a heterogeneous group of mitochondrial disorders and is usually accompanied by neurosensory hearing loss, diabetes mellitus (DM), and mitochondrial myopathy with ragged‐red fibers (RRFs) [2]. Approximately 80% or 5% of MELAS cases involve a heteroplasmic point mutation of m.3243A>G or m.1351G>A, respectively, and approximately 80% of individuals with the m.3243A>G mutation are reported to show clinical symptoms compatible with MELAS [2]. The severity of mitochondrial disease is thought to be associated with the mitochondrial DNA (mtDNA) heteroplasmy, which is the ratio of mutant to wild‐type mtDNA, and the threshold value is thought to be in the range of 60%–90% [3]. The mtDNA heteroplasmy in peripheral blood derived from patients with juvenile‐form and adult‐form MELAS is reported to be 75.1% ± 11.2% and 53.2 ± 12.7%, respectively [4]. A study using a cytoplasmic hybrid technique revealed that >90% mtDNA heteroplasmy is needed to decrease oxidative phosphorylation (OXPHOS) [5], which is responsible for the bulk of cellular ATP. Recently, it was reported that serum fibroblast‐growth factor 21 (FGF21) [6, 7, 8] and growth and differentiation factor 15 (GDF15) [9, 10] are elevated in patients with mitochondrial disease, but the roles of FGF21 and GDF15 in the brain tissue remain unclear. Thus, we investigated the mtDNA heteroplasmy, mitochondrial content, and OXPHOS complexes using brain tissue samples in the autopsied mutant cases and compared the results to their neuropathological findings.
2. MATERIALS AND METHODS
2.1. Enrolled autopsied cases and their clinical information
Six mutant cases and three disease control cases derived from Aichi Medical University Karei Ikagaku Brain Resource Center (AKBRC) between 2003 and 2019 were enrolled in this study. All cases were Japanese. The disease control cases included patients with acute respiratory failure (C1), arrhythmia (C2), and sudden death (C3), and were confirmed not to have the m.3243A>G mutation. Two mutant cases had been excluded due to massive thalamic bleeding and a lack of brain samples. These cases were autopsied within 16 h after death and confirmed no massive brain edema to avoid superficial vacuolation secondary to autolysis or brain edema. Precise clinical information regarding their symptoms and treatments is shown in Table 1. Postmortem tissue samples of the brain (right frontal lobe, right precentral lobe, right parietal lobe, right occipital lobe, right lateral temporal lobe, right medial temporal lobe, right basal ganglia, right thalamus, right cerebellum, and thoracic spinal cord), sympathetic ganglia (SG), dorsal root ganglia (DRG), iliopsoas muscle, and cardiac muscle were collected at autopsies, and then snap‐frozen brain slices and tissue blocks were made using liquid nitrogen. We paid attention to avoid gross brain lesions such as those resulting from stroke‐like episodes when taking brain samples. One hundred and four tissue samples derived from 9 cases (including 93 brain tissue samples) were used for polymerase chain reaction (PCR), western blot (WB), and metabolomic analyses.
TABLE 1.
Clinical and autopsy information of cases with the mt‐3243A>G mutation and disease controls.
| M1 | M2 | M3 | M4 | M5 | M6 | C1 | C2 | C3 | |
|---|---|---|---|---|---|---|---|---|---|
| mt‐3243G>A mutation | + | + | + | + | + | + | − | − | − |
| Sex | F | M | M | F | M | M | M | F | M |
| Age at onset (years old) | 19 | 53 | 35 | 13 | 24 | 32 | N/A | N/A | N/A |
| Age at death (years old) | 29 | 54 | 46 | 29 | 37 | 59 | 79 | 37 | 60 |
| Disease duration (years) | 10 | 1 | 11 | 16 | 13 | 27 | N/A | N/A | N/A |
| Family history | − | + | + | − | + | N/A | − | − | − |
| Height (cm) | 150 | 142 | 142 | 143 | 156 | 167 | N/A | 163 | 175 |
| Stroke‐like episodes | + | + | + | + | + | − | − | − | − |
| Lactic acid elevation | + | + | + | + | + | − | − | − | − |
| Diabetes mellitus | + | − | + | − | − | + | − | − | − |
| Hearing loss | + | + | + | + | + | + | − | − | − |
| Muscle weakness | + | + | − | + | + | + | − | − | − |
| Ragged‐red muscle fibers | N/A | + | N/A | N/A | N/A | N/A | − | − | − |
| Cognitive disturbance | + | − | + | − | − | + | − | − | − |
| Headache | N/A | N/A | + | + | + | − | − | − | − |
| Epilepsy | + | − | + | + | − | − | − | − | − |
| Myoclonus | + | N/A | − | − | + | N/A | − | − | − |
| Cerebellar ataxia | + | − | − | − | N/A | − | − | − | − |
| Sensory neuropathy | + | − | N/A | − | N/A | + | − | − | − |
| Autonomic failure | N/A | + | N/A | − | N/A | − | − | − | − |
| Cardiopathy | − | − | − | + | N/A | − | − | − | − |
| Complicating disease | − | SMAS | − | − | − | IPUF | − | SZ | − |
| Treatment for mitochondrial disease | LA | − | LA | N/A | N/A | N/A | − | − | − |
| Brain weight (grams) | 700 | 890 | 915 | 940 | 1200 | 1220 | 1190 | 1270 | 1460 |
Abbreviations: F, female; IPUF, idiopathic pulmonary upper lobe fibrosis; LA, L‐arginine; M, male; N/A, not applicable; SMAS, superior mesenteric artery syndrome; SZ, schizophrenia.
2.2. Histological and immunohistochemical analyses
The left cerebrum, left cerebellum, brainstem, and spinal cord were fixed with 20% buffered formalin and embedded in paraffin. Histological examinations were performed using formalin‐fixed paraffin‐embedded (FFPE) sections stained with hematoxylin–eosin (HE), Klüver‐Barrera, and Gallyas‐Braak staining. Immunohistochemical examinations were performed as previously described [11]. The sections were incubated overnight with the following primary antibodies: Human Mitochondria antibody (M117, cline AF‐1; Leinco Technologies Inc., MO, USA; 1:200, pretreated by heat antigen retrieval), human PHF‐Tau (monoclonal, clone AT8; Thermo Scientific, IL, USA; 1:5000), amyloid β [4, 5, 6, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24] (monoclonal; Dako, Glostrup, Denmark; 1:1000, pretreated by heat antigen retrieval and formic acid), α‐synuclein (polyclonal; Santa Cruz Biotechnology, Santa Cruz, CA, USA; 1:20,000, pretreated by heat antigen retrieval and formic acid), and phosphorylated trans‐activation response DNA‐binding protein of 43 kDa (p‐TDP‐43, polyclonal; CosmoBio, Tokyo, Japan; 1:4000, pretreated by heat antigen retrieval and formic acid). Then, these sections were washed with phosphate‐buffered saline (PBS) and incubated with a secondary antibody (Histofine Simple Stain MAX PO (MULTI); Nichirei Bioscience Inc., Tokyo, Japan) for 1 h. The sections were visualized using 3,3′‐diaminobenzidine (DAB Tablet; FUJIFILM, Osaka, Japan), and Mayer's hematoxylin solution was used as a counterstain.
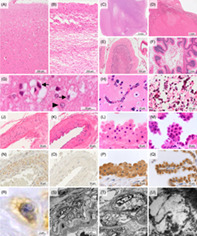
Based on the percent of occupied area against sampled area and severity of cortical lesion, cerebral cortex lesions in each brain region were graded as “absent,” “1–50% superficial vacuolations,” “>50% superficial vacuolations,” “1–50% laminar necrosis,” or “>50% laminar necrosis” (Figure 1A–D); cerebellar cortex lesions were also graded as “absent,” “1–50% ischemic lesion,” “>50% ischemic lesion,” “1–50% necrosis,” or “>50% necrosis” (Figure 1E, F). Cerebellar ischemic lesion was defined by loss of granule cells and Purkinje cells with Bergmann gliosis. Mineralization in the globus pallidus, putamen, caudate nucleus, cerebral peduncle, and cerebellar dentate nucleus was graded as “absent,” “capillary” (Figure 1H), or “coarse” (Figure 1I). Mitochondrial angiopathy of the left lateral temporal lobe assessed using immunohistochemistry (IHC) against mitochondria was graded as “absent” (Figure 1K, O) or “present” (Figure 1J, N). The mean total size, nuclear size, cytoplasmic size, and nuclear/cytoplasmic ratio of 20 epithelial cells in the choroid plexus were measured using ImageJ Ver. 1.53 t software (https://imagej.nih.gov/ij/) and HE images taken with a ×100 objective lens (Figure 1L, M, P, Q). Based on each diagnostic criteria, Alzheimer's disease was assessed using PHF‐Tau and amyloid β [4, 5, 6, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24] IHC of the temporal lobe [20, 25], Lewy body disease was assessed using α‐synuclein IHC of the substantia nigra, locus coeruleus, and dorsal nucleus of the vagus nerve [19], cerebral amyloid angiopathy was assessed using amyloid β [4, 5, 6, 9, 10, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24] IHC of the temporal and occipital lobes [18], and argyrophilic grains was assessed using PHF‐Tau IHC and Gallyas‐Braak staining of the amygdala and medial temporal lobe [26].
FIGURE 1.
Common neuropathological findings of cases with the m.3243A>G mutation. (A, G) Superficial vacuolation of the cerebral cortex in case M5. A high magnification view demonstrated type II astrocytes (arrowhead) and various sizes of vacuolations surrounding neurons (arrows). (B) Laminar necrosis of the cerebral cortex in case M3. (C, D) Necrotic lesions of the basal ganglia in case M1 (C) and thalamus in case M4 (D). (E) Ischemic lesion of the cerebellum in case M4, showing loss of granule cells and Purkinje cells with Bergmann gliosis. (F) Necrosis of the cerebellum in case M4. (H) Capillary mineralization in the putamen of case M3. (I) Coarse mineralization in the globus pallidus of case M3. (J, N, R) Mitochondrial vasculopathy showing swollen smooth muscle cells of the tunica media with small cytoplasmic vacuolations and mitochondria immunoreactivity in case M4. (K, O) Normal artery in case C1. (L, P) Swollen epithelial cells of the choroid plexus in case M4. (M, Q) Normal epithelial cells of the choroid plexus in case C1. (S–U) Electron microscopic images. Smooth muscle cells of the tunica media had many cytoplasmic vacuolations, presumably compatible with the ballooned mitochondria in case M1. (A–M): HE staining; (N‐Q): immunohistochemistry against mitochondria. Bar: 5 mm for (C), 1 mm for (D, F), 200 μm for (A, B, E), 100 μm for (I), 50 μm for (J, K, N, O), 20 μm for (G, H, L, M, P, Q), 5 μm for (S), 2 μm for (R, T), and 200 nm for (U). TI, tunica intima; TM, tunica media.
2.3. DNA sample preparation and m.3243A>G mtDNA heteroplasmy analysis
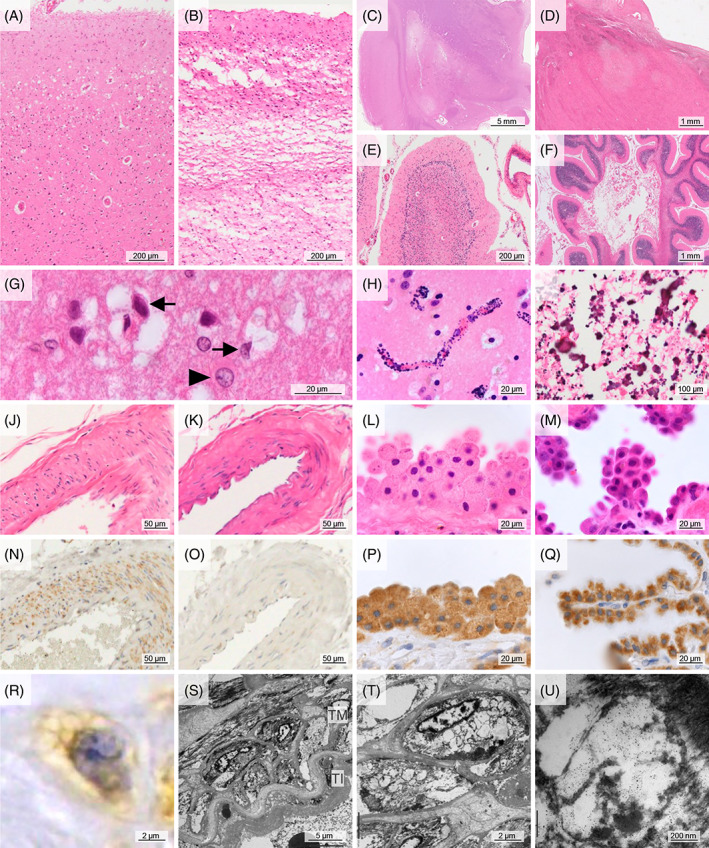
Total DNA was extracted from postmortem frozen samples of the brain, sympathetic ganglia (SG), dorsal root ganglia (DRG), iliopsoas muscle, and cardiac muscle using a QIAamp DNA Mini Kit (51304; QIAGEN, Venlo, Netherlands). A PCR‐based assay was used to determine the proportion of mutant and wild‐type mtDNA. The primers used were as follows: forward primer 5′‐CCTCCCTGTACGAAAGGA‐3′ and reverse primer 5’‐GCGATTAGAATGGGTACAATG‐3′. The dinucleotide substitution in the forward primer creates a restriction site for the restriction enzyme ApaI (Nippon Gene, Tokyo, Japan), which can cleave the mutant sequence (GGGCCC) when the m.3243A>G mutation is present. PCR amplification was carried out on a TaKaRa PCR Thermal Cycler Dice Gradient (TP600; TaKaRa, Shiga, Japan) using the following protocol: 1 min denaturation at 94°C, 1 min annealing at 55°C, and 45‐s extension at 72°C for 25 cycles. To quantitate the relative proportions of mutant and wild‐type mtDNA, two 237‐bp PCR samples were made, and the unilateral sample was digested with ApaI at 37°C for 60 min. These samples were run on a Novex 6% TBE Gel (EC62652BOX; Thermo Fisher Scientific, Aichi, Japan) and visualized using SYBR Green I Nucleic Acid Gel Stain (5760A, TaKaRa). ApaI digestion produces two units, one of 127 bp and one of 110 bp, which can easily be differentiated from the uncut wild‐type unit by gel electrophoresis. Iliopsoas muscle samples from cases M3 and C1 were shown as the representative PCR gel electrophoresis image (Figure 2A).
FIGURE 2.
Representative mitochondrial mutation analysis, and western blot in cases with the m.3243A>G mutation. (A) ApaI digestion products were seen in the mutant case (M3), in which the mtDNA heteroplasmy was calculated as 73.2%. (B) Fragments of OXPHOS complex I in case M1 were decreased compared to those in C1.
2.4. Protein extraction and WB analysis
The frozen tissue samples were homogenized in a 10‐fold volume of radioimmunoprecipitation (RIPA) lysis buffer containing a protease inhibitor and phosphatase inhibitors (SC‐24948; Santa Cruz, Dallas, TX, USA). Samples were separated on a 12% Bis‐Tris gel (NP0342BOX, Thermo Fisher Scientific) using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred to a nitrocellulose membrane (LC2000, Thermo Fisher Scientific). The transferred membrane was incubated with a primary antibody such as Total OXPHOS Human WB Antibody Cocktail (ab110411; Abcam, Kenbridge, MA, USA; 1:2000), Human Mitochondria antibody (M117; Leinco Technologies Inc; 1:100), Anti‐FGF21 antibody (ac17141; Abcam; 1:1000), GDF‐15 (sc‐377195; Santa Cruz; 1:50), and Beta Actin Mouse Monoclonal antibody (66009‐1‐Ig, NM_001101, Proteintech, IL, USA, 1:2000), and then visualized using Western Lightning Plus ECL (NEL104001EA, Perkin Elmer, Massachusetts, USA). Lysates taken from the control cases were also assessed for comparison. Parietal lobe samples from cases M1 and C1 were shown as the representative WB images (Figure 2B).
2.5. Electron microscopic analysis of mitochondrial vasculopathy
Several pieces cut from a glutaraldehyde‐fixed cerebral cortex sample containing leptomeningeal vessels in case M1 were postfixed with 1% osmium tetroxide, dehydrated using a graded ethanol series, and embedded in Epon 812 (TAAB Epon 812, Nisshin‐EM, Tokyo, Japan). Ultrathin sections were cut, stained with uranyl acetate and lead citrate, and examined with a JEM‐1400 electron microscope (JEOL Ltd., Tokyo, Japan) at 80 kV.
2.6. Metabolite extraction, metabolic derivatization, and metabolomic analysis
Metabolites were extracted using a modification of the Bligh‐Dyer method and then derivatized as described previously [27, 28]. The analysis of metabolites was performed by gas chromatography‐tandem mass spectrometry (GC–MS/MS). GC–MS/MS analysis was performed on a GCMS‐TQ8040 system (Shimadzu Corporation, Kyoto, Japan) equipped with a DB‐5 capillary column (30 m x 0.25 mm inner diameter, film thickness 1 μm; Agilent, Santa Clara, CA, USA). Metabolite detection was performed using the Smart Metabolites Database Ver. 3 software program (Shimadzu Corporation) using the method described in a previous study with some modifications [22]. Peak identification was performed automatically and then confirmed manually based on the specific precursor and product ions as well as the retention time using the method described in previous studies [14, 21]. Among 469 metabolites, lactic acid and pyruvic acid were assessed in this study.
2.7. Semi‐quantitative analysis of the PCR and WB results and heatmap visualization
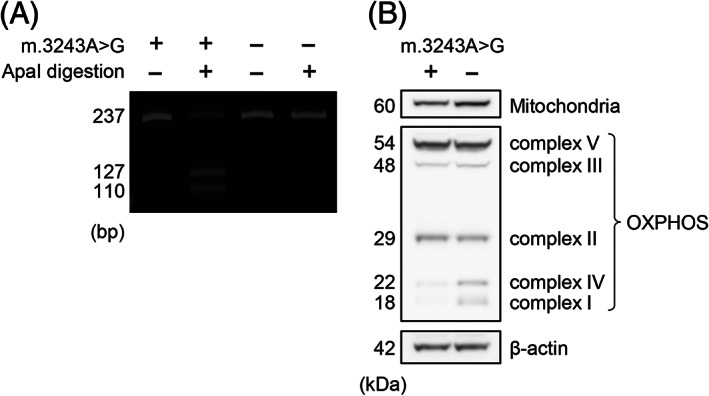
The density of the units derived from the PCR and WB methods was semi‐quantified using the functions “Plot Lanes” and “Measure” in ImageJ. The mtDNA heteroplasmy was calculated by dividing the two digested units by the total units. The total amount of mitochondria, FGF21, and GDF15 was standardized by dividing by the loading control, and OXPHOS complexes I to V were standardized by dividing by the standardized mitochondria. The semi‐quantitative values and the metabolite data were visualized as heatmaps using a Microsoft Excel 2019 MSD 64‐bit (RStudio, Boston, MA, USA). These semi‐quantitative data in cases with or without the mutation were averaged and projected onto brain images, as shown in Figure 4.
FIGURE 4.
Projected brain images in cases with or without the m.3243A>G mutation. Mean values in each brain region were projected into the brain images. The medial temporal lobe in the mutant cases was preserved despite an inhospitable environment with high mtDNA heteroplasmy and lactic acid. OXPHOS complex I was widely decreased in the mutant cases.
3. RESULTS
3.1. Clinical and autopsy information of cases with the m.3243A>G mutation
As shown in Table 1, the mutant cases developed mitochondrial disease at 13–53 years of age, died at 29–59 years of age, and had a disease duration of 1–27 years. Stroke‐like episodes and lactic acid elevation were confirmed in cases M1‐M5; DM in cases M1, M3, and M6; and hearing loss in all mutant cases. Cases M1‐M5 had neuropathological findings compatible with MELAS in this study, and were subdivided into those with the adult form of MELAS (cases M1‐3, M5) or the juvenile form of MELAS (case M4) based on their onset age. Case M6 had a pathological finding of muscle biopsy compatible with mitochondrial disorders but no stroke‐like episode nor an episode of lactic acid elevation; hence, case M6 was diagnosed as a mitochondrial disease, not MELAS. Cases M1‐M6 did not show clinical symptoms compatible with Leigh syndrome, MERRF, or CPEO. The efficacy of L‐arginine therapy was imponderable in this study because of the paucity of treatment records. The brain weight in 5 cases with stroke‐like episodes was decreased (700–1200 grams) compared to that in 4 cases without stroke‐like episodes (1190–1440 grams).
3.2. Neuropathological findings of cases with the m.3243A>G mutation
All mutant cases showed neuropathological findings such as laminar necrosis of the cerebral cortex or mineralization of the basal ganglia (Figure 3), and cases M1‐M5 showed neuropathological findings compatible with MELAS. Cerebral cortex lesions such as superficial vacuolations and laminar necrosis were seen frequently in the lateral temporal, parietal, and occipital lobes; less frequently in the frontal lobe including precentral gyrus; and not at all in the medial temporal lobe. Superficial vacuolations composed of increased Alzheimer type II astrocytes, indicating a metabolic brain disorder, and various sizes of vacuolations surrounding cortical neurons (Figure 1G), and distributed extensively compared to laminar necrosis. Ischemic lesions of the basal ganglia or thalamus were observed in one‐third of the mutant cases (Figure 1C, D), and cerebellar ischemic lesions were observed in five of six mutant cases. Brainstem ischemic lesions were observed only in case M5. Every ischemic lesion observed in our mutant cases was old and did not have inflammatory cell infiltration indicating fresh ischemic lesions. Moreover, no evidence of occlusion or thrombosis of the main cerebral arteries was found in our mutant cases. Capillary mineralization was confirmed in the globus pallidus, putamen, caudate nucleus, cerebellar dentate nucleus, or cerebral peduncle of cases M2–M6. Coarse mineralization was noted in the globus pallidus of cases M3, M5, and M6. Thicken vascular walls with swollen smooth muscle cells of the tunica media, which had small cytoplasmic vacuolations and immunoreactivity (IR) against mitochondria antibody, were detected only in the mutant cases, and were seen frequently on the leptomeninges but rarely in the brain parenchyma. The small cytoplasmic vacuolations of the smooth muscles were rimmed by the IR against mitochondria antibody (Figure 1R), presumably compatible with those seen in microscopic examinations (Figure 1S–U). The total epithelial cell size of the choroid plexus in cases M1‐M5 was enlarged (106.5–265.4 μm2) compared to that of the control cases (81.8–94.9 μm2). In case M6, the total epithelial cell size was not enlarged (92.6 μm2), but the nuclear/cytoplasmic ratio was reduced (0.178), similar to that in cases M1‐M5 (0.078–0.198), compared to that in the control cases (0.232–0.253). No definitive aging pathology such as neurodegenerative alterations caused by pathologic protein deposits was noted in any of the cases enrolled in this study.
FIGURE 3.
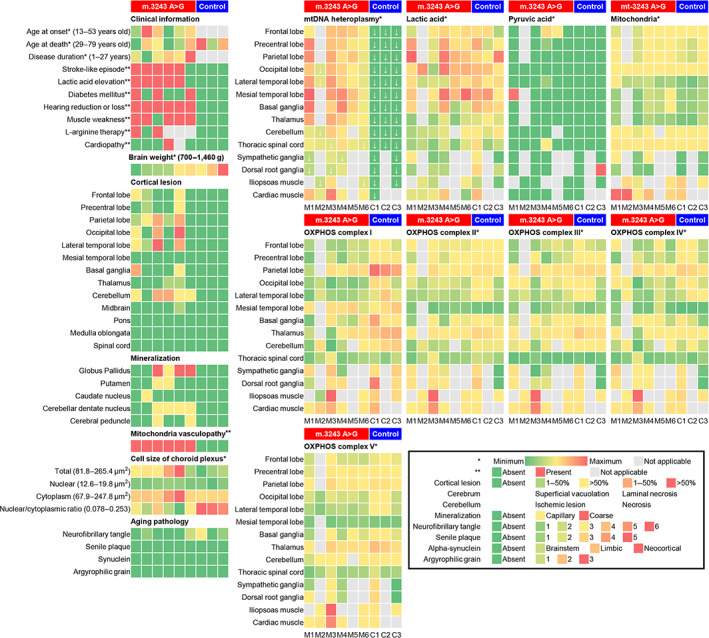
Heatmap visualization in cases with or without the m.3243A>G mutation. Each case was placed in ascending order of brain weight. In the mutant cases, cortical lesions were frequently observed in the lateral temporal, parietal, and occipital lobes, and the mtDNA heteroplasmy was entirely high. White down arrow symbols showed less than 60% of mtDNA heteroplasmy. OXPHOS complex I was decreased in the mutant cases compared to the control cases.
3.3. Heatmap visualization and projected brain images of the cortical lesions, mtDNA heteroplasmy, mitochondria, and OXPHOS complexes
Control cases had no cortical lesions and seemed to show slightly higher levels of lactic acid and OXPHOS complex I in the parietal lobe (Figure 3). The mtDNA heteroplasmy in 67 tissue samples of the mutant cases ranged from 39.9 to 85.2%, and that in 37 tissue samples of the control cases was 0%. In 53 brain samples of the mutant cases, the mtDNA heteroplasmy was strikingly high, ranging from 53.8% to 85.2%. Case M6, which had no clinical stroke‐like episode nor pathological cortical lesion, seemed to have lower mtDNA heteroplasmy (mean 64.8%) than the other mutant cases. Twelve of 67 tissue samples of the mutant cases had less than 60% mtDNA heteroplasmy (Figure 3, down arrow symbols). The medial temporal lobe, basal ganglia, and thalamus tended to show high mtDNA heteroplasmy (65.3%–85.2%), whereas the SG and DRG showed low mtDNA heteroplasmy (39.9%–60.2%). Cardiac muscles tended to show higher mtDNA heteroplasmy (61.2%–82.5%) than iliopsoas muscles (45.7%–73.2%). There was no cortical lesion in the medial temporal lobe despite an inhospitable environment having high mtDNA heteroplasmy and lactic acid. Pyruvic acid was detected sporadically in some tissue samples. OXPHOS complex I in the mutant cases was widely decreased compared to that in the control cases, but there was no distinctive relationship between OXPHOS complex and mtDNA heteroplasmy. OXPHOS complexes II‐V did not show definitive differences between cases with and without the mutation. Neither FGF21 nor GDF15 showed any type of tendency between cases or regions (data not shown).
4. DISCUSSION
In the mutant cases, cortical lesions such as ischemic lesions or laminar necrosis were frequently observed in the parietal, lateral temporal, and occipital lobes; less frequently in the frontal lobe including precentral gyrus; and not at all in the medial temporal lobe. The mtDNA heteroplasmy in 53 brain tissue samples of the mutant cases was elevated overall. The medial temporal lobe was preserved despite an inhospitable environment with high levels of mtDNA heteroplasmy and lactic acid. OXPHOS complex I was widely decreased in the mutant cases compared to the control cases. The swelling of vascular smooth muscle cells on the leptomeninges, with IR against mitochondria antibody, and a decreased nuclear/cytoplasmic ratio of choroidal epithelial cells were observed in all mutant cases but in none without the mutation.
Cortical lesions such as superficial vacuolations or laminar necrosis in the mutant cases were seen frequently in the parietal, lateral temporal, and occipital lobes, but the medial temporal lobe in the mutant cases had no cortical lesions despite the high levels of mtDNA heteroplasmy and lactic acid. Frequent temporal and occipital lesions or less frequent precentral gyrus lesion were compatible with published clinical characteristics such as frequent cortical blindness and homonymous hemianopia or less frequent hemiparesis and dysarthria [12], respectively. In a previous study using seven autopsied cases diagnosed with MELAS, cerebral necrotic lesions were reported to be dominant in the parietal and occipital lobes, which is basically compatible with this study, and the medial temporal lobe was also preserved in six of seven cases with MELAS [29]. Given that the one case having the medial temporal lobe lesion had not been evaluated the mutation in that study, it might be safe to say that the medial temporal lobe is relatively preserved in mutant cases with MELAS. It is well known that the medial temporal lobe is vulnerable to common brain ischemia, Alzheimer's disease, and medial temporal lobe epilepsy, but the reason why the medial temporal lobe in cases with MELAS was relatively preserved remains unclear. The medial temporal lobe might have undiscovered tolerance to decreased mitochondrial function or elevated lactate environment.
Twelve of 67 tissue samples derived from the mutant cases had a mtDNA heteroplasmy of less than 60%, and only 1 of the 12 tissue samples with low mtDNA heteroplasmy contained a mild ischemic lesion. Considering >60% of mtDNA heteroplasmy as the threshold value as previously reported [3], our cases with widespread high mtDNA heteroplasmy, regardless of ischemic lesions, were inferred to have a high risk of brain lesions. Tranah, et al. reported that mtDNA heteroplasmy of the m.3243A≥G mutation was elevated ranging 0%–19% even in healthy carriers and was associated with an increased risk of dementia, stroke, and mortality [30]. In that study, mtDNA was extracted from peripheral blood cells; hence, the mtDNA heteroplasmy might be underestimated compared to that extracted from brain and muscle tissue samples [31].
OXPHOS complex I in the mutant cases was widely decreased compared to the control cases, despite no definitive distributional difference in the total amount of mitochondria between the cases with or without the mutation. Furthermore, there was no distinctive relationship between OXPHOS complex I and mtDNA heteroplasmy. These results were consistent with those of previous reports using muscle biopsy samples or cell biological methods [15, 32].
Coarse mineralization in the basal ganglia and capillary mineralization resembled those of nonspecific aging. Recently, vascular mineralization in mitochondrial disease was thought to be associated with mineral dysregulation and accelerated cellular senescence secondary to mitochondrial dysfunction [24]; hence, the mineralized lesions of mitochondrial disease might resemble those of aging.
Moreover, the cytoplasm of epithelial cells was swollen in the choroid plexus of cases M1‐M5, and the nuclear/cytoplasmic ratio was decreased in all mutant cases but not in those without the mutation. Mitochondrial angiopathy, which was characterized by swollen vascular smooth muscle cells with both small cytoplasmic vacuolations and IR against mitochondria antibody, was observed in all mutant cases but in none without. The neuropathological hallmarks of mitochondrial diseases, including MELAS, have not been conclusive in the past; hence, a lack of medial temporal lesions, mitochondrial vasculopathy in vessels on the leptomeninges, an increased cytoplasmic size in choroidal epithelial cells could be the neuropathological hallmarks of mitochondrial diseases.
Serum FGF21 and GDF15 are thought to be promising diagnostic biomarkers of mitochondrial diseases [11, 16, 23, 25]. However, serum GDF15 levels are also known to be increased in acute respiratory distress syndrome [33], pulmonary hypertension [34], cardiac failure [35], cancer [36], and aging [37], and serum FGF21 levels are increased in obesity, type 2 DM [38, 39], and nonalcoholic fatty liver disease [17]. Since autopsied cases with some effects of the agonal stage were included and FGF21 and GDF15 were evaluated using tissue lysates, not serum, in this study, no reliable data on FGF21 and GDF15 are thought to have been acquired. Clinicians should consider other possible causes when assessing FGF21 or GDF15 in patients suspected of having mitochondrial disease.
Whether the cause of stroke‐like episodes is mitochondrial vasculopathy or cytopathy is still being debated. Koga Y, et al. suggested that mitochondrial vasculopathy played an important role in developing stroke‐like episodes via vasodilatation and vasogenic edema [16]. The distribution of stroke‐like episodes in our cases does not correspond to brain vascular territories, as previously described [23]; therefore, the conclusion that stroke‐like episodes are caused only by mitochondrial vasculopathy should be made with caution. Cytotoxic edema and hyperexcitability as the early mechanism of mitochondrial cytopathy were thought to be observed in stroke‐like episodes [16], but no evidence of mitochondrial cytopathy was observed because stroke‐like episodes in our cases were not acute lesions. Some researchers have proposed that both mitochondrial vasculopathy and cytopathy are related to the pathogenesis of stroke‐like episodes [13]. To clarify the positioning of mitochondrial vasculopathy and cytopathy in MELAS, cell biological or radiological research methods are thought to be more suitable than our research methods using human autopsied specimens. In conclusion, our findings are not sufficient to clarify the major cause of stroke‐like episodes, and continued debates across disciplines are needed.
This study assessing the mtDNA heteroplasmy, OXPHOS complexes, and neuropathological findings in autopsied cases with m.3243A>G mutation is thought to have certain worth and novelty. However, the limitations of our study were the paucity of clinical information, the use of autopsied human brain samples affected by the agonal stage, and the use of FFPE samples and frozen tissue samples harvested from contralateral sites. Regarding clinical information, the paucity of information regarding treatments, family history, and muscle biopsy was a considerable impediment to assessing the efficacy of treatments and the accuracy of the antemortem diagnosis. Autopsied brain samples were somewhat affected by the effects of the agonal stage, and fresh stroke‐like episodes with inflammatory cell infiltration were rarely observed. Considering that the distribution of brain lesions was not always symmetrical, our results derived from tissue samples taken from the contralateral side would have to be said to have a degree of bias.
Common neuropathological findings such as cortical laminar necrosis and basal ganglia calcification were not always observed in autopsied cases of the m.3243A>G mutation. Heterogeneous cortical lesions occurring frequently in the parietal, temporal, and occipital lobes were observed despite the extensive and high mtDNA heteroplasmy. A lack of medial temporal lesions, mitochondrial vasculopathy in the vessels on the leptomeninges, and an increased cytoplasmic size of epithelial cells in the choroid plexus could be neuropathological hallmarks helpful in diagnosis of mitochondrial diseases.
AUTHOR CONTRIBUTIONS
Hiroaki Miyahara conceptualized and made the original draft of the manuscript; Daisuke Tahara, Nao Tahara, Kazuhiro Takeda, Shusei Arafuka, and Hideyuki Moriyoshi assembled the clinical information; Chisato Tamai and Hiroaki Miyahara performed the western blot and mitochondrial DNA analyses; Masanori Inoue, Kazuhito Sekiguchi, and Kenji Ihara performed the GC–MS/MS analysis; Akio Akagi performed the electron microscopic procedures; Ryuichi Koizumi, Yuichi Riku, Jun Sone, Mari Yoshida, and Yasushi Iwasaki performed the histopathological analysis; Hiroaki Miyahara conducted data visualization; and all authors reviewed, edited, and approved the manuscript.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest.
FUNDING STATEMENTS
This research was supported by AMED under Grant Numbers JP18dm0107105 (M. Yoshida) and JP16kk0205009 (M. Yoshida). This work was also supported by Grants‐in‐Aid from the Research Committee of CNS Degenerative Diseases, Research on Policy Planning and Evaluation for Rare and Intractable Diseases, Health, Labour and Welfare Sciences Research Grants, the Ministry of Health, Labour and Welfare, Japan (K. Nakashima), the HORI Science and Arts Foundation (Y. Riku), and by JSPS KAKENHI Grant Number JP20K16586 (Y. Riku), 22K07359 (Y. Riku), and 20K08196 (H. Miyahara).
ETHICS STATEMENT
This study was performed in compliance with the principles of the Declaration of Helsinki. Approval was obtained from the institutional review boards of Aichi Medical University. Signed informed consent for autopsy, the archiving of tissue for research purposes, and genetic analysis were obtained from the family members of all cases in compliance with the Ethical Committee for Medical Researches of Aichi Medical University.
ACKNOWLEDGMENTS
We would like to thank Mami Sakurai for valuable help regarding a gas chromatography‐tandem mass spectrometry analysis.
Miyahara H, Tamai C, Inoue M, Sekiguchi K, Tahara D, Tahara N, et al. Neuropathological hallmarks in autopsied cases with mitochondrial diseases caused by the mitochondrial 3243A>G mutation. Brain Pathology. 2023;33(6):e13199. 10.1111/bpa.13199
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Goto Y, Nonaka I, Horai S. A mutation in the tRNA(Leu)(UUR) gene associated with the MELAS subgroup of mitochondrial encephalomyopathies. Nature. 1990;348(6302):651–653. [DOI] [PubMed] [Google Scholar]
- 2. Feigenbaum A, Chitayat D, Robinson B, MacGregor D, Myint T, Arbus G, et al. The expanding clinical phenotype of the tRNA(Leu(UUR)) A‐‐>G mutation at np 3243 of mitochondrial DNA: diabetic embryopathy associated with mitochondrial cytopathy. Am J Med Genet. 1996;62(4):404–409. [DOI] [PubMed] [Google Scholar]
- 3. Tuppen HA, Blakely EL, Turnbull DM, Taylor RW. Mitochondrial DNA mutations and human disease. Biochim Biophys Acta. 2010;1797(2):113–128. [DOI] [PubMed] [Google Scholar]
- 4. Koga Y. L‐arginine therapy on MELAS. Rinsho Shinkeigaku. 2008;48(11):1010–1012. [DOI] [PubMed] [Google Scholar]
- 5. Pang CY, Lee HC, Wei YH. Enhanced oxidative damage in human cells harboring A3243G mutation of mitochondrial DNA: implication of oxidative stress in the pathogenesis of mitochondrial diabetes. Diabetes Res Clin Pract. 2001;54(2):S45–S56. [DOI] [PubMed] [Google Scholar]
- 6. Maresca A, Del Dotto V, Romagnoli M, La Morgia C, Di Vito L, Capristo M, et al. Expanding and validating the biomarkers for mitochondrial diseases. J Mol Med (Berl). 2020;98(10):1467–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Riley LG, Nafisinia M, Menezes MJ, Nambiar R, Williams A, Barnes EH, et al. FGF21 outperforms GDF15 as a diagnostic biomarker of mitochondrial disease in children. Mol Genet Metab. 2022;135(1):63–71. [DOI] [PubMed] [Google Scholar]
- 8. Scholle LM, Lehmann D, Deschauer M, Kraya T, Zierz S. FGF‐21 as a potential biomarker for mitochondrial diseases. Curr Med Chem. 2018;25(18):2070–2081. [DOI] [PubMed] [Google Scholar]
- 9. Koga Y, Povalko N, Inoue E, Ishii A, Fujii K, Fujii T, et al. A new diagnostic indication device of a biomarker growth differentiation factor 15 for mitochondrial diseases: from laboratory to automated inspection. J Inherit Metab Dis. 2021;44(2):358–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Montero R, Yubero D, Villarroya J, Henares D, Jou C, Rodriguez MA, et al. GDF‐15 is elevated in children with mitochondrial diseases and is induced by mitochondrial dysfunction. PloS One. 2016;11(2):e0148709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hirano M, Iritani S, Fujishiro H, Torii Y, Kawashima K, Sekiguchi H, et al. Globular glial tauopathy type I presenting with behavioral variant frontotemporal dementia. Neuropathology. 2020;40(5):515–525. [DOI] [PubMed] [Google Scholar]
- 12. Iizuka T, Sakai F. Pathogenesis of stroke‐like episodes in MELAS: analysis of neurovascular cellular mechanisms. Curr Neurovasc Res. 2005;2(1):29–45. [DOI] [PubMed] [Google Scholar]
- 13. Ikawa M, Okazawa H, Arakawa K, Kudo T, Kimura H, Fujibayashi Y, et al. PET imaging of redox and energy states in stroke‐like episodes of MELAS. Mitochondrion. 2009;9(2):144–148. [DOI] [PubMed] [Google Scholar]
- 14. Kawahara Y, Hirashita Y, Tamura C, Kudo Y, Sakai K, Togo K, et al. Helicobacter pylori infection modulates endogenous hydrogen sulfide production in gastric cancer AGS cells. Helicobacter. 2020;25(5):e12732. [DOI] [PubMed] [Google Scholar]
- 15. Koga Y, Nonaka I, Kobayashi M, Tojyo M, Nihei K. Findings in muscle in complex I (NADH coenzyme Q reductase) deficiency. Ann Neurol. 1988;24(6):749–756. [DOI] [PubMed] [Google Scholar]
- 16. Koga Y, Povalko N, Nishioka J, Katayama K, Kakimoto N, Matsuishi T. MELAS and L‐arginine therapy: pathophysiology of stroke‐like episodes. Ann N Y Acad Sci. 2010;1201:104–110. [DOI] [PubMed] [Google Scholar]
- 17. Kralisch S, Fasshauer M. Fibroblast growth factor 21: effects on carbohydrate and lipid metabolism in health and disease. Curr Opin Clin Nutr Metab Care. 2011;14(4):354–359. [DOI] [PubMed] [Google Scholar]
- 18. Love S, Chalmers K, Ince P, Esiri M, Attems J, Jellinger K, et al. Development, appraisal, validation and implementation of a consensus protocol for the assessment of cerebral amyloid angiopathy in post‐mortem brain tissue. Am J Neurodegener Dis. 2014;3(1):19–32. [PMC free article] [PubMed] [Google Scholar]
- 19. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65(12):1863–1872. [DOI] [PubMed] [Google Scholar]
- 20. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, et al. National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012;123(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nishiuchi M, Sakai K, Tajima H, Katayama K, Kimura F, Hoshi S, et al. Orexigenic action of oral zinc: metabolomic analysis in the rat hypothalamus. Biosci Biotechnol Biochem. 2018;82(12):2168–2175. [DOI] [PubMed] [Google Scholar]
- 22. Nishiumi S, Kobayashi T, Kawana S, Unno Y, Sakai T, Okamoto K, et al. Investigations in the possibility of early detection of colorectal cancer by gas chromatography/triple‐quadrupole mass spectrometry. Oncotarget. 2017;8(10):17115–17126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ohama E, Ohara S, Ikuta F, Tanaka K, Nishizawa M, Miyatake T. Mitochondrial angiopathy in cerebral blood vessels of mitochondrial encephalomyopathy. Acta Neuropathol. 1987;74(3):226–233. [DOI] [PubMed] [Google Scholar]
- 24. Phadwal K, Vrahnas C, Ganley IG, MacRae VE. Mitochondrial dysfunction: cause or consequence of vascular calcification? Front Cell Dev Biol. 2021;9:611922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thal DR, Rub U, Orantes M, Braak H. Phases of a beta‐deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58(12):1791–1800. [DOI] [PubMed] [Google Scholar]
- 26. Saito Y, Ruberu NN, Sawabe M, Arai T, Tanaka N, Kakuta Y, et al. Staging of argyrophilic grains: an age‐associated tauopathy. J Neuropathol Exp Neurol. 2004;63(9):911–918. [DOI] [PubMed] [Google Scholar]
- 27. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959;37(8):911–917. [DOI] [PubMed] [Google Scholar]
- 28. Sekiguchi K, Miyahara H, Inoue M, Kiyota K, Sakai K, Hanada T, et al. Metabolome characteristics of liver autophagy deficiency under starvation conditions in infancy. Nutrients. 2021;13(9):3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tanahashi C, Nakayama A, Yoshida M, Ito M, Mori N, Hashizume Y. MELAS with the mitochondrial DNA 3243 point mutation: a neuropathological study. Acta Neuropathol. 2000;99(1):31–38. [DOI] [PubMed] [Google Scholar]
- 30. Tranah GJ, Katzman SM, Lauterjung K, Yaffe K, Manini TM, Kritchevsky S, et al. Mitochondrial DNA m.3243A>G heteroplasmy affects multiple aging phenotypes and risk of mortality. Sci Rep. 2018;8(1):11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hammans SR, Sweeney MG, Hanna MG, Brockington M, Morgan‐Hughes JA, Harding AE. The mitochondrial DNA transfer RNALeu(UUR) A‐‐>G(3243) mutation. A clinical and genetic study. Brain. 1995;118(Pt 3):721–734. [DOI] [PubMed] [Google Scholar]
- 32. Goto Y, Horai S, Matsuoka T, Koga Y, Nihei K, Kobayashi M, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke‐like episodes (MELAS): a correlative study of the clinical features and mitochondrial DNA mutation. Neurology. 1992;42(3 Pt 1):545–550. [DOI] [PubMed] [Google Scholar]
- 33. Clark BJ, Bull TM, Benson AB, Stream AR, Macht M, Gaydos J, et al. Growth differentiation factor‐15 and prognosis in acute respiratory distress syndrome: a retrospective cohort study. Crit Care. 2013;17(3):R92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Almudares F, Hagan J, Chen X, Devaraj S, Moorthy B, Lingappan K. Growth and differentiation factor 15 (GDF15) levels predict adverse respiratory outcomes in premature neonates. Pediatr Pulmonol. 2023;58(1):271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rochette L, Dogon G, Zeller M, Cottin Y, Vergely C. GDF15 and cardiac cells: current concepts and new insights. Int J Mol Sci. 2021;22(16):8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rochette L, Meloux A, Zeller M, Cottin Y. Vergely C (2020) functional roles of GDF15 in modulating microenvironment to promote carcinogenesis. Biochim Biophys Acta Mol Basis Dis. 1866;8:165798. [DOI] [PubMed] [Google Scholar]
- 37. Conte M, Giuliani C, Chiariello A, Iannuzzi V, Franceschi C, Salvioli S. GDF15, an emerging key player in human aging. Ageing Res Rev. 2022;75:101569. [DOI] [PubMed] [Google Scholar]
- 38. Cheng X, Zhu B, Jiang F, Fan H. Serum FGF‐21 levels in type 2 diabetic patients. Endocr Res. 2011;36(4):142–148. [DOI] [PubMed] [Google Scholar]
- 39. Xie T, Leung PS. Fibroblast growth factor 21: a regulator of metabolic disease and health span. Am J Physiol Endocrinol Metab. 2017;313(3):E292–E302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.