

Abstract
Coinhibition of TIGIT (T cell immunoreceptor with Ig and ITIM domains) and PD-1/PD-L1 (PD-1/L1) may improve response rates compared with monotherapy PD-1/L1 blockade in checkpoint naive non–small cell lung cancer with PD-L1 expression >50%. TIGIT mAbs with an effector-competent Fc can induce myeloid cell activation, and some have demonstrated effector T cell depletion, which carries a clinical liability of unknown significance. TIGIT Ab blockade translates to antitumor activity by enabling PVR signaling through CD226 (DNAM-1), which can be directly inhibited by PD-1. Furthermore, DNAM-1 is downregulated on tumor-infiltrating lymphocytes (TILs) in advanced and checkpoint inhibition–resistant cancers. Therefore, broadening clinical responses from TIGIT blockade into PD-L1low or checkpoint inhibition–resistant tumors, may be induced by immune costimulation that operates independently from PD-1/L1 inhibition. TNFSF14 (LIGHT) was identified through genomic screens, in vitro functional analysis, and immune profiling of TILs as a TNF ligand that could provide broad immune activation. Accordingly, murine and human bifunctional fusion proteins were engineered linking the extracellular domain of TIGIT to the extracellular domain of LIGHT, yielding TIGIT-Fc-LIGHT. TIGIT competitively inhibited binding to all PVR ligands. LIGHT directly activated myeloid cells through interactions with LTβR (lymphotoxin β receptor), without the requirement for a competent Fc domain to engage Fcγ receptors. LIGHT costimulated CD8+ T and NK cells through HVEM (herpes virus entry mediator A). Importantly, HVEM was more widely expressed than DNAM-1 on T memory stem cells and TILs across a range of tumor types. Taken together, the mechanisms of TIGIT-Fc-LIGHT promoted strong antitumor activity in preclinical tumor models of primary and acquired resistance to PD-1 blockade, suggesting that immune costimulation mediated by LIGHT may broaden the clinical utility of TIGIT blockade.
Key Points:
HVEM and LTβR are broadly expressed on tumor-infiltrating CD8+ T, NK, and myeloid cells.
LIGHT binding to HVEM/LTβR activates Fc-independent myeloid/T/NK cell activation.
TIGIT-Fc-LIGHT induces anti tumor responses in CPI primary/acquired resistant tumors.
Visual Abstract
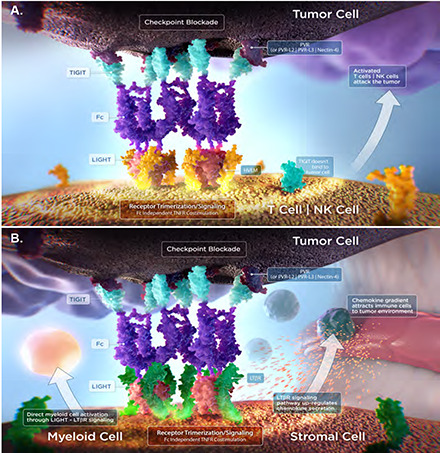
Introduction
Checkpoint inhibition (CPI) of PD-1/PD-L1 (PD-1/L1) or CTLA-4 has been transformative for patients with metastatic cancer across a wide range of tumor types. A subset of patients experience durable responses that translate into long-term survival benefit. However, even among checkpoint inhibitor–sensitive tumors there is heterogeneity of responses with most patients not responding to CPI (primary resistance) or relapsing shortly after initial benefit (acquired resistance) (1–3). Recent studies regarding T cell exhaustion in the context of antitumor immunity have shed light on the complex interplay of compensatory checkpoint pathways and the need for concurrent and efficient stimulation of immune cell populations to program immunological memory (4). Furthermore, immune responses are dynamic and continuously evolve in a given patient as a result of inherent genetic factors or in response to anticancer treatments such as surgery, radiotherapy, chemotherapy, or immunotherapy. Similar or overlapping mechanisms, which likely manifest at various times, enable tumor cells to evade antitumor immune responses, resulting in checkpoint-refractory disease (5). This results in a large unmet need population of patients who require innovative dosing/combination strategies designed to counteract established mechanisms of resistance to immunotherapy.
Targeting TIGIT (T cell immunoreceptor with Ig and ITIM domains, VSIG9) through Ab blockade is one such approach (6). While well tolerated, anti-TIGIT monotherapy has demonstrated minimal clinical activity in advanced solid tumors with objective response rates ranging from 0 to 3%. This lack of monotherapy activity may be due to the observation that TIGIT is expressed following upregulation of PD-1 or other coinhibitory receptors, including PVRIG, TACTILE, or LAG-3 (6–8). TIGIT is an inhibitory coreceptor that competes with an activating coreceptor, known as DNAM-1, for binding to PVR. Antitumor immunity following TIGIT Ab blockade is dependent on PVR-mediated costimulation of lymphocytes via DNAM-1 (6). PVR is overexpressed in a wide range of solid tumors, and high PVR expression has been associated with the proliferation of tumor cells, invasion, and a negative impact on survival (9). Clinical studies have shown that TIGIT blockade may enhance antitumor immunity when combined with PD-1/L1–blocking Abs (10, 11). The mechanism for this dependence is due to direct tyrosine dephosphorylation of the cytoplasmic tail of DNAM-1 by PD-1–mediated SHP-2 activation (6, 12–14). The convergent biology between PD-1–mediated lymphocyte inhibition and DNAM-1 inhibition may provide the mechanistic explanation for the relative lack of clinical activity in cancer patients with primary or acquired resistance to PD-1/L1 therapy.
Direct inhibition of DNAM-1 signaling by PD-1 activation does not address why combinations of anti-TIGIT and anti–PD-1/L1 Abs have not yet shown antitumor activity beyond what is expected for PD-1/L1 Ab monotherapy in PD-1/L1 acquired resistant cancer patients (11, 15, 16). A potential explanation for this result is the progressive downregulation of DNAM-1 among tumor-infiltrating lymphocytes (TILs) during tumor progression, which has been reported in multiple tumor types, including non–small cell lung cancer (NSCLC) and melanoma (17). Identification of additional costimulatory receptors with a more consistent expression pattern during tumor progression may provide opportunities for enhancing immune responses against advanced or checkpoint acquired resistant tumors.
An analysis of immune costimulatory molecule expression across The Cancer Genome Atlas (TCGA) identified high expression of lymphotoxin β receptor (LTβR) and herpes virus entry mediator A (HVEM), two receptors for TNFSF14 (LIGHT), across tumor types and at significantly higher transcript levels than DNAM-1. Analysis of single-cell RNA sequencing (scRNA-seq) and cell-surface protein analysis of primary human PBMCs confirmed these data and demonstrated increased expression of HVEM relative to DNAM-1 on both naive T (Tn) cells and T memory stem cells (Tscms). Furthermore, TILs from established syngeneic mouse tumors also demonstrated elevated protein expression of HVEM relative to DNAM-1.
A TIGIT-Fc-LIGHT bispecific fusion protein was generated and shown to bind all known TIGIT and LIGHT targets with high affinity. LIGHT signaling activated effector T cells through an overlapping pathway with DNAM-1, bypassing the need for DNAM-1 costimulation. A comparison of both Fc competent (IgG1) and silent (IgG4) versions of TIGIT-Fc-LIGHT demonstrated that LIGHT induced activation of myeloid cells irrespective of the type of Fc subclass present. The antitumor activity of TIGIT-Fc-LIGHT was dependent on both NK and CD8+ T cells, and was less dependent on concurrent inhibition of the PD-1/PD-L1 axis than TIGIT-blocking Abs. Furthermore, in a murine model of PD-1 acquired resistance, combinations of anti-TIGIT and anti–PD-1/L1 Abs did not improve response rates or provide survival benefit, consistent with current clinical data. In the same PD-1 acquired resistance models, TIGIT-Fc-LIGHT was observed to improve both response rates and overall survival alone or in combination with PD-1/L1 Abs. Finally, nonhuman primate toxicology studies demonstrated that TIGIT-Fc-LIGHT was well tolerated through doses of 40 mg/kg and stimulated rapid dose-dependent margination of total lymphocytes, including HVEM+ CD3+ T cells, following infusion. In addition, a dose-dependent increases in IL-2, CXCL10, MIP-1β, CCL17, and MCP-1 were observed in treated animals. Collectively, these data provide a rationale for clinical exploration of TIGIT-Fc-LIGHT in both checkpoint naive and PD-1/L1 acquired resistant cancer patients.
Materials and Methods
Construct generation, protein purification, and size-exclusion chromatography fractionation
The sequences of human and mouse TIGIT-Fc, Fc-LIGHT, and TIGIT-Fc-LIGHT were codon optimized and directionally cloned into mammalian expression vectors. Vectors were then either transiently transfected into Expi293 cells or stably transfected into CHO cells, and the resulting fusion protein was purified using affinity chromatography. Size-exclusion chromatography (SEC) was conducted on a Thermo Fisher Scientific Vanquish system with a TSKgel UltraSW aggregate column (Tosoh; 7.8 × 300 μm, 3 mm). The hexameric, tetrameric, and dimeric species of TIGIT-Fc-LIGHT were collected with an automated fraction collector. Molecular masses were estimated based on a gel filtration standard curve (Bio-Rad). Fractions were reinjected with the same conditions to confirm species purity, and the dual binding potency assay was used to assess fraction activity as described below.
Western blot
Human and mouse TIGIT-Fc-LIGHT proteins were treated with and without the deglycosylase PNGase F (NEB) for 1 h at 37°C according to the manufacturer’s recommendations, and then with and without the reducing agent 2-ME, and diluted in SDS loading buffer prior to separation by SDS-PAGE. Primary Abs used for probing human TIGIT-Fc-LIGHT and mouse (m)TIGIT-Fc-LIGHT were obtained from Cell Signaling Technology, Jackson ImmunoResearch Laboratories, and R&D Systems.
Meso Scale Discovery and ELISA detection
Dual binding potency assay
For TIGIT-Fc-LIGHT, Meso Scale Discover (MSD) multi-array plates were precoated overnight with recombinant human LTβR-Fc or HVEM-Fc (Acro Biosystems) at 4°C, and after incubation and washing analysis samples were added and serially diluted 3-fold in duplicate wells. Bound TIGIT-Fc-LIGHT was detected with recombinant biotinylated human PVR (Acro Biosystems), then streptavidin conjugated to ruthenium (MSD). The MSD MESO QuickPlex SQ 120 (model 1300) was used to generate relative light units. Analogous assays were performed using mTIGIT-Fc-LIGHT and murine recombinant PVR, HVEM, and LΤβR (Acro Biosystems).
Ab-based binding assay
The same methodology was performed as described for the dual potency assay above; however, TIGIT-Fc-LIGHT was captured with anti-human TIGIT and detected with anti-human LIGHT-biotin (all Abs from R&D Systems).
Decoy receptor 3 blocking assay
Human healthy donor (two distinct donors) and cancer patient (kidney, NSCLC, and prostate) serum samples were obtained from Innovative Research. MSD decoy receptor 3 (DcR3) detection reagents were used to profile the levels of soluble DcR3 in each sample. To assess whether serum levels of DcR3 were sufficient to inhibit TIGIT-Fc-LIGHT binding to PVR or HVEM, TIGIT-Fc-LIGHT was incubated in each serum sample for 20 min on ice, and then loaded onto MSD plates precoated with human recombinant HVEM. Plates were then processed according to the dual potency assay described above, with human recombinant PVR detection.
B and T lymphocyte attenuator/LIGHT competition assay
MSD plates coated with recombinant human HVEM-his were probed with an equimolar concentration of recombinant human BTLA (B and T lymphocyte attenuator)-his/biotin with and without 4.57 pM to 3 μM human TIGIT-Fc-LIGHT. Streptavidin-SULFO-TAG was used to detect bound BTLA and anti-human Fc-SULFO-TAG was used to detect bound TIGIT-Fc-LIGHT.
Biolayer interferometry–based affinity testing of receptor–ligand interactions
Biolayer interferometry was used to determine the on-rates (Ka), off-rates (Kd), and binding affinities (KD) of TIGIT-Fc-LIGHT to intended binding targets, using histidine- or biotin-tagged versions of the human recombinant proteins (PVR, HVEM, LTβR, PVRL2, PVRL3, and Nectin-4; purchased from Acro Biosystems or Sino Biological). Commercially available or internally produced single-sided fusion protein controls (TIGIT-Fc and Fc-LIGHT) were tested in parallel. Targets were immobilized at a concentration of 3 μg/ml in kinetics buffer (PBS/0.1% Tween 20/1% BSA; pH7.0) to anti-pentaHis– or streptavidin-coated biosensors. Direct binding of fusion proteins to recombinant target proteins was performed on an Octet-Red96 biolayer interferometry instrument, using association and dissociation times of 90 and 120 s, respectively.
Cell culture
CHO-K1, CT26/wild-type (WT), B16.F10, CT26/CPI-acquired resistance (AR), Jurkat, and A375 cells were obtained from American Type Culture Collection and cultured according to their guidelines (maintained at 37°C in 5% CO2). All parental cell lines in active culture were tested monthly using the Venor GeM Mycoplasma detection kit (Sigma-Aldrich). All transfected cell lines were tested an additional two times, separated by at least 2 wk, after transfection and confirmed to remain negative for mycoplasma.
In vitro cell line generation
Stable cell lines were generated to assess in vitro binding of human or mouse TIGIT-Fc-LIGHT, including CHO-K1/human (h)PVR, CHO-K1/hHVEM, and CHO-K1/mLTβR. cDNA vectors were obtained from R&D Systems or Origene and cloned into pcDNA3.1(−) (Thermo Fisher Scientific), and then parental CHO-K1 or Jurkat cells were nucleofected with the 4D-Nucleofector and Cell Line Nucleofector Kit SE (Lonza). After antibiotic selection and single-cell cloning using limiting dilution, receptor expression was verified using flow cytometry, and the resulting cell lines were used for in vitro binding assays.
In vitro functional assays
NF-κB signaling: noncanonical
U2OS/NIK/NF-κB reporter cells expressing LTβR were purchased from Eurofins/DiscoverX. On the day of the assay, 1 × 104 cells were plated into each well of a 96-well plate with either Fc-LIGHT or TIGIT-Fc-LIGHT. After 6 h, luminescence was assessed on a luminometer (Promega).
TIGIT/PVR/DNAM1 reporter assay
The CD155(PVR)/TIGIT blocking assay (Promega) was used according to the manufacturer’s instructions (see (Fig. 2G). Jurkat effector cells were confirmed to express human HVEM using flow cytometry. For the assay, Jurkat effector cells were plated into white 96-well plates (Costar) and incubated overnight at 37°C/5% CO2. The next day, cells were cocultured with CHO-K1 target cells and the following test articles: recombinant human IgG4 (negative control), anti-DNAM1 blocking Ab, Fc-LIGHT, or TIGIT-Fc-LIGHT. An anti-LIGHT blocking Ab was also used to block the function of the costimulatory domain of TIGIT-Fc-LIGHT. All Abs and reagents were purchased from Acro Biosystems, Sino Biologics, or R&D Systems. After 6 h of additional culture, luminescence was determined using a Promega Navigator luminometer.
FIGURE 2.
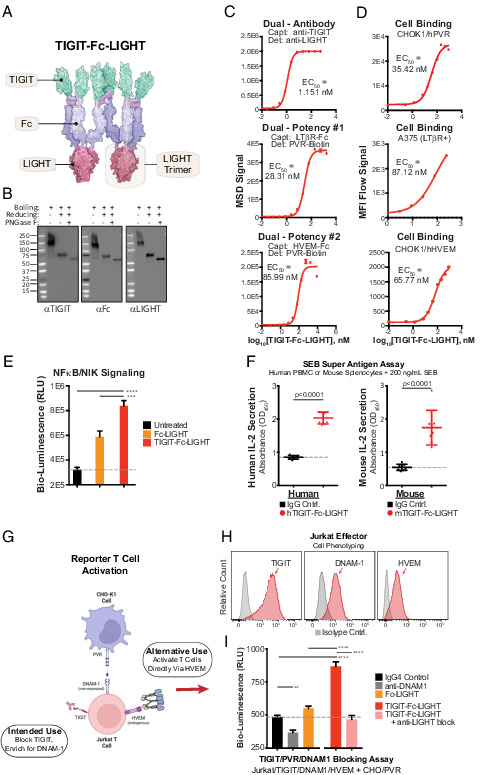
Hexameric TIGIT-Fc-LIGHT engages targets with high affinity and activates effector lymphocytes. (A) TIGIT-Fc-LIGHT was synthesized from a single expression vector in mammalian production cells. Shown is a depiction of the hexamer based on Protein Data Bank structures, with dimerization of the central IgG4 Fc domain and trimerization of the TNF-ligand domain. (B) SDS-PAGE Western blot was used to probe all three domains of TIGIT-Fc-LIGHT under nonreducing, 2-ME reduced, and reduced/deglycosylated (PNGaseF) conditions. (C) Individual MSD binding assays were developed to assess binding to each intended recombinant protein target. (D) Cell-based binding assays were developed to assess cell surface receptor binding (using CHOK1 cells engineered to express hPVR and hHVEM, and also A375 cells that express human LTβR). (E) NF-κB/NIK reporter cells were incubated with a recombinant Fc-LIGHT control or TIGIT-Fc-LIGHT (18 nM each), and signaling activity was assessed through luciferase detection. ***p < 0.001, ****p < 0.0001. (F) TIGIT-Fc-LIGHT (or the murine surrogate, both at 10 nM) activation of T cells through IL-2 induction was assessed in human PBMCs or mouse splenocytes cocultured with the superantigen SEB for 3 d. (G) Schematic of a two-cell reporter system in which PVR-expressing CHO-K1 cells are cocultured with Jurkat effector cells expressing TIGIT and DNAM-1. (H) Jurkat effector cells were phenotyped using flow cytometry for the expression of TIGIT, DNAM-1, and HVEM. (I) Jurkat effector and CHO/hPVR reporter cells were cocultured with (all at 150 nM) an IgG4 control, a DNAM-1–blocking Ab, TIGIT-Fc-LIGHT, or TIGIT-Fc-LIGHT preincubated with a LIGHT-blocking Ab for 6 h, and then luciferase signaling activity was assessed using a luminometer.
NK/T cell killing assay
CT26 tumor cells were plated into clear 96-well plates and cultured at 37°C/5% CO2 overnight. NK and total T cells were isolated from the spleens of BALB/c mice using magnetic cell isolation beads (STEMCELL Technologies). T cells were suboptimally stimulated with anti-mouse CD3/CD28 magnetic beads (STEMCELL Technologies; at 0.1 the recommended concentration) for 48 h. On the day of coculture, NK (2.5:1 E:T ratio) or T (5:1 E:T ratio) cells were added to plates containing CT26 tumors cells with and without mTIGIT-Fc-LIGHT, and a caspase-3/7 green reagent (Essen Bioscience). Images were taken on the Incucyte S3 platform, and fluorescent signal (increase in cell death) was quantitated over time.
Tn cell and Tscm differentiation and human CD3+ T cell activation
Healthy human donor PBMCs were obtained and CD8+ T cells were isolated using a naive CD8 magnetic isolation kit (STEMCELL Technology). Isolated cells were cultured in AIMV media (Life Technologies) in the presence of anti-human CD3/CD28 magnetic beads (Invitrogen) at a 1:3 cell/bead ratio, 20 IU/ml recombinant human IL-2 (R&D Systems), and 5 μM TWS119 (Selleck Chemicals) for 9 d (18). Following incubation, cells were stained with Abs to CD3, CD8, CCR7, CD45RO, CD62L, CD45RA, CD27, IL-7Rα, IL-2Rβ, and CD95 and assessed by flow cytometry (all Abs from BioLegend). Human T cells cultured for 48 h with ImmunoCult T cell activator beads and rhIL-2 (STEMCELL Technologies and R&D Systems) were assessed by flow cytometry for HVEM, 4-1BB, and CD69 expression pregated from forward/side scatter and then CD3+CD8+ populations.
AIMV proliferation assay and cytokine analysis
Healthy human donor PBMCs were plated at a density of 2 million cells per milliliter in AIM-V medium (Life Technologies) in 24-well plates with vehicle (PBS), TIGIT-Fc(IgG4)-LIGHT (150 nM), TIGIT-Fc(IgG1)-LIGHT (150 nM), anti-TIGIT (150 nM, IgG1 clone HuTIG1-IgG1.AA, Creative Biolabs), anti–PD-1 (150 nM, pembrolizumab), or the combination of anti-TIGIT and anti–PD-1. PBMC proliferative capacity with and without 150 nM TIGIT-Fc(IgG1)-LIGHT or TIGIT-Fc(IgG4)-LIGHT was assessed after 7 d in culture using the Promega MTS proliferation assay. On days 2 and 7, static images were taken to demonstrate differences in morphology between treatment groups. Replicate plates were seeded and were placed in a standard cell culture incubator at 37°C/5% CO2 for 2 d. Media were then removed and assessed for levels of IFN-γ, IL-8, IL-10, IL-12/p70, and SDF-1α (CXCL12) using an MSD multiplex array. PBMCs from a different human donor were used to assess single-sided fusion protein controls alone or in combination (TIGIT-Fc with and without Fc-LIGHT; 75 nM each), compared with TIGIT-Fc(IgG4)-LIGHT (150 nM) using the same protocol described above. MSD cytokine reagents were used to assess IFN-γ, IL-8, and IL-12/p70.
AIMV scRNA-seq
To assess the immune transcriptomic profile of PBMCs cultured for 2 d with 150 nM TIGIT-Fc(IgG4)-LIGHT or TIGIT-Fc(IgG1)-LIGHT, single cells from treated healthy donor human PBMCs were isolated using the 10x Genomics Chromium handler. Single-cell libraries were generated using 10x Genomics Chromium Next GEM Single Cell 3′ v3.1: dual index reagents. Libraries were sequenced on an Illumina NovaSeq 6000 and processed with the 10x Cell Ranger pipeline (v6.0.2) for demultiplexing, barcode, and unique molecular identifier counting and read alignments. Raw data were deposited at the National Center for Biotechnology Information Gene Expression Omnibus, accession number GSE201999 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE201999). Reads (>350 × 106) from >10,000 individual cells were mapped to the human hg38 reference genome (average 12,000 cells/sample). The filtered gene count matrices were then analyzed by Seurat (v4.0.2) for quality control, normalization, integration, dimension reduction, visualization, cell clustering, and differentially expressed gene (DEG) analysis. Cells with <200 genes, >2500 genes, or having >15% mitochondrial counts were filtered. Gene counts were normalized to total read counts and scaled to 10,000, then log transformed. All samples were integrated using 2000 anchor features to remove the batch effect. Integrated data were scaled and the top 30 principal components (PCs) were used for uniform manifold approximation and projection (UMAP) visualization. Shared nearest neighbor was identified using the top 30 PCs and resolution was set to 0.4 for cell clustering. SingleR (v 1.6.1) was used for cell type annotations. Three built-in references were used for training (19–21). Differential expression analysis for treated cells versus control within each cluster was performed using the Wilcox method. We limited tests to genes with log fold change threshold of 0.25 between groups.
Staphylococcal enterotoxin B superantigen assay
Primary PBMCs or mouse splenocytes were incubated with 200 ng/ml superantigen staphylococcal enterotoxin B (SEB) (List Biological Laboratories), in the presence of a human or mouse IgG control (10 μg/ml; Jackson ImmunoResearch Laboratories) or TIGIT-Fc-LIGHT. After 3 d, culture supernatants were collected and assessed by ELISA for levels of human or mouse IL-2 (BioLegend).
A375 cell stimulation
Human A375 cells expressing LTβR respond to costimulation via LIGHT (22). Cells were plated at 2 × 105 cells per well in 24-well plates overnight. The following morning, cells were either untreated or cultured with 100 nM of either human anti-LTβR (clone 71315, R&D Systems) or TIGIT-Fc-LIGHT. After 3 h, RNA was isolated from cells, reverse transcribed, and the resulting cDNA was used to assess gene expression of GAPDH, ACTB, CXCL8, and CCL2 (primers from OriGene). cDNA was amplified using SYBR Green reagents and the Bio-Rad CFX96 Touch real-time PCR detection system. Fold change in gene expression was determined using the ΔΔCt method, where target gene expression of the no treatment control versus the housekeeping gene ACTB was set at a value of 1. A second housekeeping gene (GAPDH) was also used as an example of a gene that did not change expression in response to treatment.
Flow cytometry
Cells were incubated with Fc Block (BioLegend) and then stained with fluorescent Abs for 30 min on ice in the dark (BioLegend and Abcam). The AH1-tetramer reagent (MBL International) was incubated with cells for 1 h on ice in the dark before adding the rest of the Ab mixture. Following incubation, stained cells were washed and resuspended in FACS buffer (1× PBS buffer containing 1% BSA, 0.02% sodium azide, and 2 mM EDTA). Flow cytometry was performed on a BD LSRFortessa.
Tumor model systems
For CT26/WT, CT26/AR, and B16.F10 studies, BALB/c or C57BL/6 mice, respectively, were s.c. implanted with 5 × 105 tumor cells into the rear flank. When tumor volume reached ∼80–115 mm3, indicating day 0, the mice were randomized by tumor volume and treatment was initiated. The mean starting tumor volume from each individual experiment is listed in the corresponding figure. On treatment days (days 0, 3, and 6), mice were treated by i.p. injection with vehicle (sterile PBS), anti–PD-1 (clone RMP1-14), anti–PD-L1 (clone 10F.9G2), anti-TIGIT (clone 1G9), anti-LTβR (clone 4H8 WH2), TIGIT-Fc, Fc-LIGHT, or mTIGIT-Fc-LIGHT (Bio X Cell, AdipoGen Life Sciences, and LSBio). Abs and single-sided fusion protein controls were given at doses of 100 μg and TIGIT-Fc-LIGHT at 200 μg. Tumor volume (mm3) and overall survival were assessed throughout the time course. Survival criteria included total tumor <1800 mm3 with no sign of tumor ulceration. Complete responders, in which tumors were established and were subsequently rejected, are listed in the appropriate figures along with group sizes. Cohorts of experimental mice were euthanized for immune profiling of tumor tissue using flow cytometry and Luminex cytokine analysis. Tumors were excised and dissociated using a tumor dissociation kit (Miltenyi Biotec) and homogenized through a 100-µm strainer to isolate tumor cells and infiltrating immune cells.
CD4, CD8, and NK depletion experiments
Some mice were i.p. injected with 100 μg of anti-CD4 (clone GK1.5), 100 μg of anti-CD8 (clone 2.43), or 500 μg of anti-NK (clone NK1.1) cells on days −1, 1, and 7. CD4, CD8, and NK cell populations in the peripheral blood were assessed by flow cytometry to verify depletion.
Generation of anti–PD-1-resistant CT26 tumors
CT26 tumor-bearing mice were given i.p. injections of anti–PD-1 (clone RMP1-14; Bio X Cell), consisting of 100 μg each on days 0, 3, and 6. Tumors were excised from mice that did not respond to anti–PD-1 therapy, dissociated using collagenase (STEMCELL Technologies), washed in 1× PBS, and plated in culture media. Cells were passaged two to four times and were then inoculated into new recipient mice. Again, when tumors reached 80–100 mm3, another treatment course of anti–PD-1 began. This process was repeated for a total of five rounds, at which point none of the treated mice responded to anti–PD-1 therapy. The cell lines generated after this in vivo pressure to develop anti–PD-1-acquired resistance are referred to as CT26/AR (previously characterized in https://doi.org/10.1101/2021.07.21.452854).
Human TIGIT-Fc-LIGHT administration in cynomolgus macaques
Study design
Purpose-bred, Asian-origin cynomolgus macaques (Macaca fascicularis) were housed at Charles River Laboratories (Mattawan, MI) and experiments with these animals were approved by the Institutional Animal Care and Use Committee for Charles River Laboratories. TIGIT-Fc-LIGHT or vehicle control was administered via i.v. infusion during 30 min. Vehicle (five males, five females), 0.1 mg/kg (three males, three females), 1 mg/kg (three males, three females), 10 mg/kg (three males, three females), or 40 mg/kg TIGIT-Fc-LIGHT (five males, five females) was infused every 7 d (days 1, 8, 15, and 22) for a total of four doses.
Clinical observations, hematology, and blood chemistry
Before and following test article administration, all animals were observed for potential clinical observations, body weight, and food consumption, and veterinary physical examinations, ophthalmic examinations, electrocardiology examinations, blood pressure assessment, and neurologic assessment were performed. Predose and postdose clinical pathology assessments, including hematology, coagulation, clinical chemistry, and urinalysis, were also performed. Reference ranges for the readouts presented in Supplemental Fig. 4F were provided by Charles River Laboratories.
Pharmacodynamic activity
Pharmacodynamic assessments included predose and postdose collection of peripheral blood using potassium EDTA anticoagulation for serum cytokines, and sodium heparin anticoagulation for peripheral blood flow cytometry studies. Ab panels used to stain peripheral immune cells included CD45, CD3, CD8, HVEM, DNAM-1, and PVR (BD Biosciences and BioLegend). HVEM, DNAM-1, and PVR expression levels were determined by establishing doublet exclusion gates from the forward and side scatter, and then gating on CD45+CD3+CD8+ cells. Serum cytokines were assessed using nonhuman primate serum cytokine multiplex reagents from MSD. On day 9, ∼24–36 h after the second infusion, surgical biopsies were obtained from the inguinal lymph node (two animals, one male and one female, from vehicle control, 10 and 40 mg/kg dose groups). Biopsy tissue was fixed in 10% neutral buffered formalin and paraffin embedded. Then, 5-μm sections were cut, H&E stained, and then scanned for analysis (Aperio). Five nonoverlapping randomly selected ×40 regions of interest were chosen, a minimum of 50 μm apart for each tissue (two animals per dose level). Images were imported into QuPath software for cell counting using default parameters. Measurement output was analyzed using SAS JMP software and plotted in GraphPad Prism software.
Experimental animal guidelines
All experimental mice used were female inbred BALB/C or C57BL/6 cohoused during the course of an experiment (The Jackson Laboratory). The numbers of animals used in each experiment are listed in the accompanying figure. All murine animal studies have been conducted in accordance with, and with the approval of, an Institutional Animal Care and Use Committee, and they were reviewed and approved by a licensed veterinarian. Experimental mice were monitored daily and euthanized by CO2 asphyxiation followed by cervical dislocation prior to any signs of distress.
Bioinformatics and statistical analysis
TCGA data were accessed through National Institutes of Health GDC (EBPlusPlusAdjustPANCAN_ IlluminaHiSeq_RNASeqV2.geneExp.tsv). Read counts were normalized to set the upper quartile count at 1000 for gene expression. Experimental replicates (n) are shown in figures and figure legends. Unless noted otherwise, values plotted represent the mean from a minimum of two distinct experiments, and error is SEM. Statistical significance (p value) was determined using unpaired t tests or one-way ANOVA with multiple comparisons. Significant p values are labeled as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Mantel–Cox statistical tests were used to determine the significance between the survival curves. The p values are noted in the legends to all figures.
Results
Identification of costimulatory receptor expression patterns in human tumors and lymphocytes
The expression of certain costimulatory receptors, such as DNAM-1, have been shown to be progressively downregulated in advanced tumors (17). Other costimulatory receptors may demonstrate more consistent expression patterns during tumor progression, and the identification of such receptors across a range of tumor types could provide the basis for targeting therapeutic immune stimulation in advanced cancers. Therefore, we assessed the expression of 53 immune costimulatory receptors across TCGA tumors (Fig. 1A). Target genes were rank ordered from high to low based on the mean expression of each gene across all TCGA tumors. Consistent with previously described downregulation of DNAM-1 expression in advanced tumors, we found that DNAM-1 ranked as one of the least abundant immune costimulators (Fig. 1A). Among the costimulatory receptors with more consistent expression patterns across tumor types, both HVEM (TNFRSF14) and LTβR (TNFRSF3), were ranked among the top 10 (Fig. 1A, 1B). HVEM and LTβR are both receptors for a TNF ligand known as LIGHT (TNFSF14, homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to HVEM, a receptor expressed on T lymphocytes) (23). Interestingly, intratumoral stimulation of HVEM via LIGHT has been shown to cause rejection of locally advanced and metastatic tumors in preclinical models (24).
FIGURE 1.

Immune costimulatory receptor expression across various peripheral blood cell subtypes and within human and mouse TILs. (A) Heatmap depicting the relative expression of 53 immune costimulatory genes across TCGA cancers ranked ordered based on high to low mean expression across all tumor types. The intensity of expression is depicted based on the minimum and maximum values in each column. Three genes of interest, that is, TNFRSF14 (HVEM), CD226 (DNAM-1), and LTBR (LTβR), are highlighted. (B) The normalized expression levels of TNFRSF14, CD226, and LTBR across all TCGA tumor types are plotted on the same scale to depict relative mRNA expression of these target genes in relationship to each other. (C) Human naive CD8+ T cells isolated from healthy donor PBMCs using magnetic separation were cultured in AIMV media with CD3/CD28 beads, human recombinant IL-2, and the GSK3β inhibitor TWS119 for 9 d. Following this time course, cells were isolated and assessed by flow cytometry according to a previously identified Ab panel that characterizes naive CD8+ T (Tn) cells and T stem cell memory CD8+ T cells (Tscms). The relative levels of HVEM and DNAM-1 are depicted and quantitated across both Tscms and Tn cells. (D) Human PBMCs cultured in AIMV media for 2 d were isolated and subjected to single-cell RNA sequencing (scRNA-seq). UMAP spatial organization was assessed using Seurat identified clusters (top) and SingleR to plot immune cell subtypes according to ImmuneExp (bottom). (E) Heatmap depicting the relative expression of 53 immune costimulatory genes across Seurat-identified clusters. Genes are ranked ordered based on high to low mean expression across all clusters. The intensity of expression is shown based on the minimum and maximum values in each column. Three genes of interest, that is, TNFRSF14 (HVEM), CD226 (DNAM-1), and LTBR (LTβR), are highlighted. (F) The normalized expression level of TNFRSF14, CD226, and LTBR across Seurat-identified clusters is plotted on the same scale to depict relative mRNA expression of these target genes in relationship to each other. (G) Murine CT26 wild-type colorectal (CT26/WT) tumors, CT26 tumors engineered to develop CPI-acquired resistance (CT26/AR), or B16.F10 melanoma tumors were inoculated into their respective recipient mice. Tumors were allowed to establish, and when they reached 80–110 mm3 (∼10–14 d after the initial inoculation), tumors were excised, dissociated, and the resulting tumor-infiltrating lymphocytes (TILs) were assessed by flow cytometry. Relative cell surface expression of HVEM and DNAM-1 was assessed in NK cells (gated on NKP46+ cells in CT26 tumors and NK1.1 in B16.F10 tumors) and CD8+ T cells (gated on CD3+CD8+ cells).
Tscms are a multipotent progenitor memory T cell that have been shown to have significant self-renewal and survival capacity (18, 25). Activation of Tscms was reported to be an important aspect of tumor control in the setting of PD-1 blockade and represents a potential reservoir for antitumor immunity for other checkpoints, including TIGIT (26). To evaluate the relative expression of HVEM and DNAM-1, a previously described in vitro differentiation protocol was used to isolate Tn cell and Tscm populations (18). Accordingly, we found that most Tn cells express HVEM (74.1% express HVEM alone and 21.8% coexpress HVEM and DNAM-1). Tscms expressed more DNAM-1 than did their Tn cell counterparts, but most DNAM-1+ cells also expressed HVEM (45.5% out of 54.25%). Furthermore, 86.7% of total Tscms expressed high levels of HVEM. These protein results are consistent with the mRNA expression of the same targets (Supplemental Fig. 1A) (18). In addition to HVEM and LTBR, the ligands for TIGIT were broadly expressed across tumor types, with PVR and PVRL2 mRNA being the most abundant, and much more highly expressed than CD274 (PD-L1) message (Supplemental Fig. 1B).
To extend this analysis to immune cells, we performed scRNA-seq on human PBMCs and bioinformatically defined cell populations through transcriptome clustering (Seurat) and immune cell type predictions (Fig. 1D). The expression of the same 53 immune costimulatory genes assessed above was also interrogated across the 16 Seurat-defined clusters (Fig. 1D). Genes were rank ordered from high to low based on average expression of each gene across all clusters. TNFRSF14 (HVEM) and CD226 (DNAM-1) ranked high in this heatmap, indicating that they were highly expressed in a number of immune cells, notably, in clusters 5 and 8, which corresponded to CD8+ T cells, and clusters 7, 10, and 11, which represented NK cells (Fig. 1E, 1F). Novershtern and Human Primary Cell Atlas annotations were also applied to the data and generated similar cell type predictions (Supplemental Fig. 1C). As expected, LTBR expression was low compared with other TNFRs in most PBMC populations, with the exception of cluster 6, which represents myeloid cells (Fig. 1F). It is noteworthy that transcriptomic-based ranking of costimulatory receptors/ligands provided initial directionality of expression, but protein analysis was used for confirmation, as message and protein expression of the same target can vary. The abundance of HVEM mRNA on human T cells, however, correlated with HVEM cell surface protein expression on CD3/CD28/IL-2–activated human T cells, and HVEM was shown to be expressed on the same cells as other markers of T cell activation that have been associated with tumor-reactive T cells, namely CD69 and 4-1BB (Supplemental Fig. 1D) (27).
To assess whether the preferential expression of HVEM over DNAM-1 on TILs translated preclinically to a range of murine tumor types, we inoculated mice with CT26/WT, CT26/AR (described in https://doi.org/10.1101/2021.07.21.452854), or B16.F10 tumors (Fig. 1G). Established tumors were isolated from mice, dissociated, and CD8+ T and NK cell infiltrates were assessed by flow cytometry. In all three tumor types, we found HVEM to be much more widely expressed on both CD8+ T and NK cells than DNAM-1 (Fig. 1G). Additionally, HVEM was found to be expressed at high levels on T and NK cells isolated from mouse splenocytes (Supplemental Fig. 1E). Taken together, these results provided a rationale for the exploration of a therapeutic candidate able to provide costimulatory signaling through HVEM and LTβR.
Production and characterization of TIGIT-Fc-LIGHT
TIGIT-Fc-LIGHT is a bifunctional Fc-linked fusion protein synthesized from a single expression vector in mammalian production cell lines and purified using affinity chromatography. Disulfide-induced dimerization of the central Fc domain, as well as charge-based trimerization of the TNF-ligand domain, yields a dimer of trimers in solution (28, 29). TIGIT-Fc-LIGHT was predicted to have a similar structure (Fig. 2A). The composition of TIGIT-Fc-LIGHT includes ∼80% hexamer, ∼15% tetramer, and ∼5% dimeric species, as shown by SEC (Supplemental Fig. 1F). Upon fractionation, the hexameric and tetrameric species produced indistinguishable potency (dual binding to LTβR and PVR or HVEM and PVR) from one another or to the unfractionated material, whereas the dimeric species demonstrated reduced potency (Supplemental Fig. 1G). Analysis of TIGIT-Fc-LIGHT under nonreduced SDS-PAGE confirmed the presence of a glycosylated disulfide-linked dimer of ∼140 kDa (SDS neutralizes the charge-based trimerization of TIGIT-Fc-LIGHT), which could be reduced to a deglycosylated monomer with an expected molecular mass of 59.3 kDa following incubation with 2-ME and PNGase-F (Fig. 2B). All three protein domains of both the human and mouse TIGIT-Fc-LIGHT constructs were recognized by commercial Abs via Western blot (Fig. 2B, Supplemental Fig. 1L).
Protein–protein interactions between the fusion proteins and their cognate binding partners were characterized using both recombinant protein and cell-based methods. First, biolayer interferometry–based affinity testing was used to assess the kinetics of receptor/ligand binding. TIGIT-Fc-LIGHT bound to recombinant human LTβR at 3.52 nM affinity (∼8-fold greater than a commercially available recombinant Fc-LIGHT control) and human PVR at 4.07 nM (consistent with human TIGIT-Fc). TIGIT-Fc-LIGHT bound to human HVEM at 6.49 nM, similar to Fc-LIGHT binding (2.12 nM) (Supplemental Fig. 1H). The increased affinity of TIGIT-Fc-LIGHT to LTβR may be at least partly explained by the increased avidity of the hexameric configuration of TIGIT-Fc-LIGHT. A combination of biolayer interferometry and MSD assays were developed to demonstrate binding to other PVR ligand family members, including PVRL2 (CD112), PVRL3 (CD113), and Nectin-4 (Supplemental Fig. 1H–I). MSD was also used to evaluate binding to individual targets, as well as the simultaneous binding of human TIGIT-Fc-LIGHT to both PVR and HVEM (EC50 of 28.31 nM) or PVR and LTβR (EC50 of 85.99 nM), indicating that the entire fusion protein was intact and able to engage its targets (Fig. 2C). LIGHT is also able to bind soluble DcR3, which can be elevated in certain autoinflammatory diseases (30). To assess whether soluble serum DcR3 in cancer patients would reach levels sufficient to interfere with the LIGHT domain of TIGIT-Fc-LIGHT, we quantitated the concentration of DcR3 in a range of healthy and cancerous human donor serum samples. We showed that in most serum samples, the DcR3 levels were 2000 pg/ml or less, with the exception of a prostate cancer sample that surpassed the upper limit of quantitation of the assay (>15,000 pg/ml)). In all cases when TIGIT-Fc-LIGHT was preincubated in each serum sample, these levels of DcR3 were not sufficient to interfere with TIGIT-Fc-LIGHT binding in the dual HVEM/PVR potency assay (Supplemental Fig. 1J). BTLA is expressed on some CD8+ T cell populations and serves as a weak immune checkpoint through both cis and trans interactions with HVEM (31, 32). Despite BTLA mRNA expression at low/undetectable levels on TILs (Supplemental Fig. 1B) and previous studies that have demonstrated that trimeric LIGHT preferentially binds to HVEM over BTLA, we developed an assay to measure TIGIT-Fc-LIGHT competition with recombinant BTLA for binding to HVEM (31, 32). When increasing concentrations of TIGIT-Fc-LIGHT were incubated with BTLA on a recombinant HVEM-coated MSD plate, a decrease in bound BTLA was detected that correlated with the increasing TIGIT-Fc-LIGHT signal, indicating that the LIGHT domain of TIGIT-Fc-LIGHT can preferentially bind to HVEM as compared with BTLA (Supplemental Fig. 1K).
Cell surface binding assays were developed to characterize the binding of both human and mouse fusion proteins to their targets expressed in cellular membranes using CHO-K1 cells engineered to stably express human PVR or HVEM. Flow cytometry analysis demonstrated that human TIGIT-Fc-LIGHT bound cell surface PVR (EC50 of 35.42 nM), cell surface LTβR (EC50 of 87.12 nM), and cell surface HVEM (EC50 of 65.77 nM) (Fig. 2D). Furthermore, mTIGIT-Fc-LIGHT was shown to bind in a dose-dependent manner to its targets using ELISA and to CT26/WT, CT26/AR, and B16.F10 tumor cells (Supplemental Fig. 1M, 1N). Thus, these three preclinical syngeneic mouse tumor models, when treated with mTIGIT-Fc-LIGHT, are relevant for the evaluation of antitumor efficacy.
Additional cell-based functional assays were used to inform on TIGIT-Fc-LIGHT activity. First, a NIK-dependent noncanonical NF-κB signaling assay, using the human bone osteosarcoma cell line, U2OS, which expresses high levels of LTβR (Fig. 2E), was employed to test the activity of the LIGHT domain of TIGIT-Fc-LIGHT. Upon treatment with TIGIT-Fc-LIGHT, bioluminescence was detected at levels significantly greater than what was generated with commercially available recombinant Fc-LIGHT (Fig. 2E). Second, TIGIT-Fc-LIGHT is expected to provide costimulation via HVEM to CD8+ T cells in an Ag-dependent manner. To evaluate this hypothesis, a SEB superantigen assay was used wherein human PBMCs or mouse splenocytes were cultured with SEB and either human or mouse TIGIT-Fc-LIGHT for 3 d, respectively. IL-2 secretion was then assessed in the culture media and indicated that human and mouse TIGIT-Fc-LIGHT were able to induce the production of adaptive cytokines (Fig. 2F). Third, ligation of LIGHT to HVEM on the surface of CD8+ T cells and NK cells is predicted to enhance killing of target tumor cells. Isolated murine T or NK effector cells were cocultured with CT26 tumor cells in the presence of a fluorescently activated cleaved caspase-3/7 reporter. The addition of mTIGIT-Fc-LIGHT stimulated an increase in caspase activity in target tumor cells, indicating that TIGIT-Fc-LIGHT actively increased the cytotoxic potential of the effector cells in culture (Supplemental Fig. 1O). Fourth, the human melanoma cell line A375 has been used as a direct functional readout for LIGHT/LTβR signaling via LTβR-dependent production of IL-8–associated genes following stimulation with LIGHT (22). When TIGIT-Fc-LIGHT or a commercially available LTβR agonist Ab was cultured with A375 cells, CXCL8 (gene encoding IL-8) and CCL2 were upregulated within 3 h, at a magnitude that was significantly greater with TIGIT-Fc-LIGHT as compared with the anti-LTβR control Ab (Supplemental Fig. 1P). Lastly, a commercially available TIGIT/DNAM-1 reporter system was used to determine whether HVEM costimulation of effector lymphocytes used redundant signaling pathways to DNAM-1. Because the TIGIT domain of TIGIT-Fc-LIGHT binds to PVR, this interaction will competitively inhibit the ability of PVR to interact with DNAM-1. Thus, DNAM-1–mediated costimulation is expected to be inhibited with recombinant constructs containing the extracellular domain of TIGIT. In this assay, TIGIT- and DNAM-1–expressing Jurkat T cells (effector) were cocultured with PVR-expressing CHO-K1 cells. The effector T cells also contain a luciferase reporter that is responsive to both TCR and DNAM-1 costimulation (Fig. 2G). Because the effector cells used in this assay were Jurkat T cells, we confirmed that these cells endogenously expressed human HVEM (Fig. 2H). In the CHO-K1 target cells, TIGIT-Fc-LIGHT was able to bypass DNAM-1 costimulation through direct HVEM costimulation by LIGHT (Fig. 2I). A DNAM-1–blocking Ab control was shown to decrease the fluorescence of the signal reporter in the assay, as expected. In contrast, incubation of these cells with TIGIT-Fc-LIGHT led to a significant increase in signal fluorescence, which was confirmed to be specific to the LIGHT domain of TIGIT-Fc-LIGHT, as it could be completely inhibited with a LIGHT-blocking Ab (Fig. 2I).
Myeloid cell activation and in vitro functional activity
The antitumor activity of TIGIT-blocking Abs has been linked to FcγR activation on myeloid cells (33). Whether these observations in preclinical systems translate to human clinical studies is an area of active debate, with some favoring a competing hypothesis that an effector-competent Fc domain could result in depletion of TIGIT-expressing effector T cells (34). The LIGHT/LTβR axis is important for lymphoid development, maturation of APCs including myeloid cells, and the initiation of immune responses both within lymph nodes and peripheral tissues (35). The observed expression of LTβR both within solid tumor tissues and specifically by myeloid cells (Fig. 1) raised the possibility that LIGHT could provide myeloid cell activation in a similar manner to what has been reported via engagement of Fcγ receptors.
To test this hypothesis, we generated native IgG1 and effector FcγR silent IgG4 variants of human TIGIT-Fc-LIGHT. The IgG1 variant was shown to have similar structural characteristics to the IgG4 variant, including the ability to engage effector Fcγ receptors (Supplemental Fig. 2A–C). The IgG1 and IgG4 variants were cultured with human PBMCs, and after 1 wk, both fusion proteins induced the adherence of cells to the cell culture plate, differentiation into a morphology consistent with myeloid cell activation, and stimulated significant proliferation (Fig. 3A, 3B). In fact, the clear morphological impact of both IgG1 and IgG4 TIGIT-Fc-LIGHT on myeloid differentiation of human PBMCs was evident after only 2 d in culture (Supplemental Fig. 2B). Both IgG1 and IgG4 variants of TIGIT-Fc-LIGHT also induced both effector (IFN-γ) and myeloid-specific (IL-8, IL12/p70, and CXCL12) cytokines (Fig. 3C). No significant impact on cellular morphology, magnitude of proliferation, or cytokine induction was found between the IgG1 and IgG4 fusion proteins (Fig. 3A–C). Cytokine response was assessed in PBMCs treated with a commercially available Fc-competent IgG1 TIGIT Ab both alone and in combination with the anti–PD-1 Ab pembrolizumab. Interestingly, in these studies only small inductions of IFN-γ and IL-8 were observed; however, they were not significantly different from control-treated cultures (Supplemental Fig. 2D). To better understand the contribution of components for TIGIT-Fc-LIGHT, single-sided TIGIT-Fc and Fc-LIGHT were generated and cytokine production was assessed with the AIMV assay. TIGIT-Fc(IgG4)-LIGHT again induced significant production of IFN-γ, IL-8, and IL-12/p70 from primary human PBMCs (Supplemental Fig. 2E). In comparison, Fc-LIGHT induced modest levels of cytokine production, and the combination with TIGIT-Fc did not appear to further increase activity, indicating that the induction of adaptive and innate immune cytokines from human PBMCs is predominantly driven by LIGHT (Supplemental Fig. 2E).
FIGURE 3.
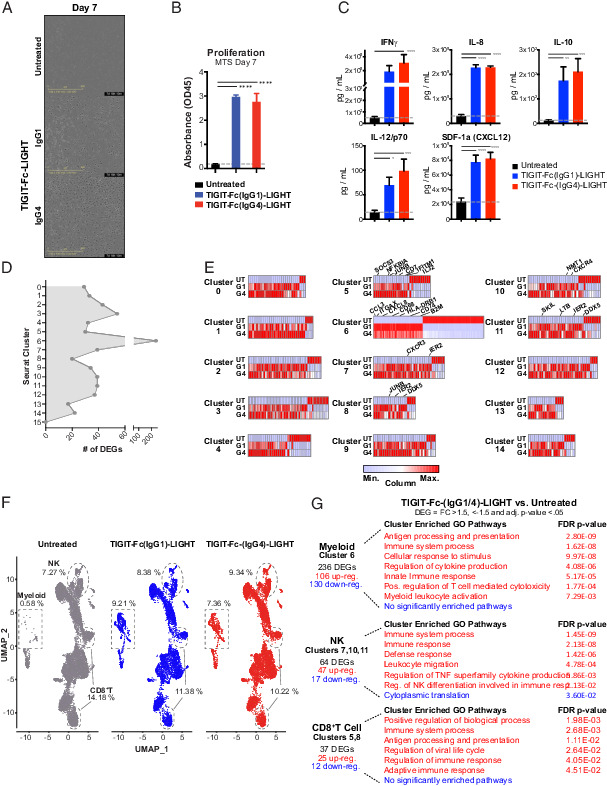
TIGIT-Fc-LIGHT directly activates myeloid, T, and NK cells regardless of Fc composition. Human donor PBMCs were either untreated (UT) or cultured in AIMV media with IgG1 or IgG4 variants of TIGIT-Fc-LIGHT, referred to as TIGIT-Fc(IgG1)-LIGHT or G1 and TIGIT-Fc(IgG4)-LIGHT or G4. (A) Phase contrast images of PBMCs cultured for 7 d with and without G1 or G4 TIGIT-Fc-LIGHT, depicting adherence of cells to the plate and the formation of a spindle-like myeloid morphology. (B) Proliferative capacity was assessed on PBMC cultures incubated for 7 d with and without 150 nM TIGIT-Fc(IgG1)-LIGHT or TIGIT-Fc(IgG4)-LIGHT using the Promega MTS proliferation assay. (C) After 2 d in culture, supernatant was removed and a series of cytokines were assessed using a MSD multiplex cytokine panel. (D) Day 2 PBMC (UT, G1, and G4 treated) single cells were isolated and subjected to single-cell RNA sequencing. Differentially expressed genes (DEGs) were identified between TIGIT-Fc-LIGHT G1/G4 and the untreated group across each of the 16 Seurat clusters identified in (Fig. 1. (E) Heatmaps depicting the relative differences in expression at DEGs identified across all 16 Seurat clusters. Genes of interest are labeled at clusters corresponding to myeloid, NK, and CD8+ T cell populations. (F) UMAP spatial distribution of untreated, TIGIT-Fc(IgG1)-LIGHT, and TIGIT-Fc(IgG4)-LIGHT datasets. The populations of cells that correspond to myeloid, NK, and CD8+ T cells (as identified using ImmuneExp annotations) are highlighted and the percentage of cells falling in those gates across treatment groups are shown. (G) The number and directionality of the DEGs are shown. Gene Ontology analysis (using PANTHER) was performed on the lists of upregulated and downregulated genes, and significantly enriched pathways (false discovery rate p value <0.05) are shown.
To further assess the immune stimulatory activity of TIGIT-Fc-LIGHT and potential functional differences between the IgG1 and IgG4 variants, scRNA-seq was performed 2 d after stimulation of human PBMC cultures. The distribution of cells expressing TIGIT, HVEM, LTBR, DNAM1, and all PVR ligands was visualized using UMAP (Supplemental Fig. 2F). DEGs were identified between both TIGIT-Fc-LIGHT variants and the untreated control across all 16 Seurat clusters identified (Fig. 3D). DEGs across clusters associated with myeloid, NK, and CD8+ T cells included CCL3, ITGAX (gene encoding CD11c), CXCL8, CD68, CD74 (gene encoding HLA-DR), B2M, IL-32, CD7, JUNB, IER2, and CXCR4 (Fig. 3E, Supplemental Fig. 2G). The upregulation of myeloid-activating genes was accompanied by an expansion in the cluster 6 population (the cluster with high expression of LTβR; see (Fig. 1F) following treatment with either the IgG1 or IgG4 versions of TIGIT-Fc-LIGHT (Fig. 3F). The NK cell population (found in clusters 7, 10, and 11 in (Fig. 3D, 3E) modestly increased, and despite a slight decrease in CD8+ T cells (clusters 5 and 8, note that T cell supportive cytokines were not added to the culture), genes associated with T cell and adaptive immune activation were upregulated (Fig. 3F). Gene Ontology pathway analysis on DEGs associated with the myeloid, NK, and CD8+ T cell clusters confirmed that both IgG1 and IgG4 versions of TIGIT-Fc-LIGHT stimulated broad immune cell activation across a range of immune cell fractions (Fig. 3G). Interestingly, out of all DEGs assessed across all Seurat clusters, only two genes in total were found to be differentially expressed between the IgG1 and IgG4 versions of TIGIT-Fc-LIGHT (Supplemental Fig. 2H). Collectively, these results provided evidence for direct myeloid cell activation by LIGHT, which was not further impacted by the selection of an FcγR-binding or nonbinding Fc domain.
In vivo antitumor efficacy
The in vivo antitumor activity of mTIGIT-Fc-LIGHT was evaluated in models of colorectal carcinoma (CT26/WT) and melanoma (B16.F10). Both are widely used as models that are sensitive (CT26/WT) or insensitive (B16.F10) to CPI. The B16.F10 model can be used to mimic primary resistance to anti–PD-1/L1 blockade.
BALB/c mice were inoculated on the hind flank with CT26/WT tumors, and when the mean starting tumor volume reached ∼84 mm3, the mice were randomized and treated with mTIGIT-Fc-LIGHT or benchmark Abs (Fig. 4A). mTIGIT-Fc-LIGHT significantly delayed tumor growth; following initial treatment, the average day in which the fusion protein–treated group reached tumor burden was day 28 (±6.87 d) following the initial treatment, compared with day 15 (±3.01 d) for the vehicle control group (Fig. 4B). TIGIT-Fc-LIGHT activity was compared with that of Abs targeting murine TIGIT, PD-1, PD-L1, LTβR, and Fc-LIGHT fusion protein, as well as the combination of these agents. The only commercially available anti-TIGIT (clone 1G9) is an effector silent mouse IgG1 and as a monotherapy demonstrates modest activity, increasing the time to tumor burden until day 18 (±4.38 d). Fc-LIGHT on its own delayed the time to tumor burden to day 20 (±1.92 d), but the combination with anti-TIGIT was unable to extend this further, suggesting that most antitumor activity was derived from LIGHT. mTIGIT-Fc-LIGHT significantly improved survival over the separate administration of anti-TIGIT with and without Fc-LIGHT or recombinant TIGIT-Fc with and without Fc-LIGHT, indicating that the colocalization of these inhibitory and costimulatory signals is critical for maximizing antitumor efficacy (Fig. 4C, Supplemental Fig. 3A–J). A commercially available LTβR Ab (clone 4H8 WH2) was also evaluated but did not show any activity in the CT26/WT model (Supplemental Fig. 3B). Encouragingly, mTIGIT-Fc-LIGHT monotherapy induced complete tumor regression in 12.9% of treated animals.
FIGURE 4.
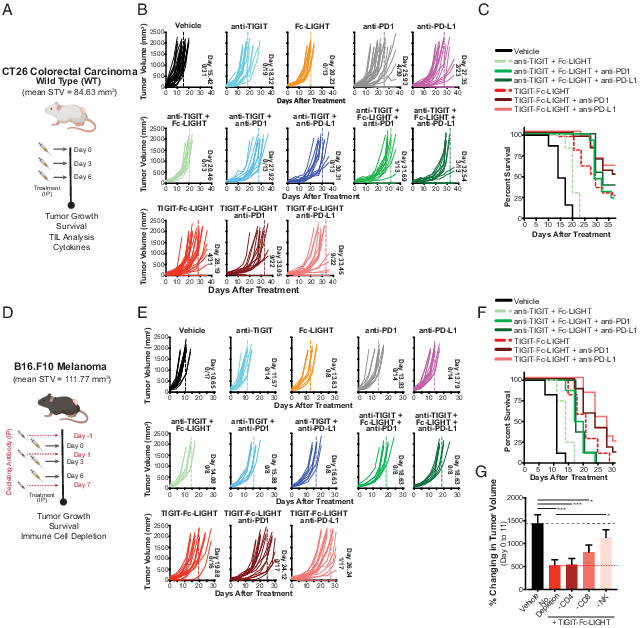
Mouse TIGIT-Fc-LIGHT induces antitumor activity in models of colorectal carcinoma and melanoma through the activation of T cells and NK cells. (A) When CT26 tumors reached an average starting tumor volume (STV) of 84.63 mm3 (indicating day 0), tumor-bearing mice were given three i.p. injections on days 0, 3, and 6, with each consisting of 200 μg of mouse (m)TIGIT-Fc-LIGHT, or 100 μg of anti-LTβR, anti-TIGIT, Fc-LIGHT, anti–PD-1, or anti–PD-L1, or the combinations shown above. (B) Each graph shows individual mouse tumor growth curves, the average day in which the entire group reached tumor burden (also indicated by dotted line), and the number of mice that completely rejected tumors/the total group size analyzed. ****p < 0.0001. (C) Kaplan–Meier survival plot through day 36 of the time course. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. (D) When B16.F10 tumors reached an average STV of 111.77 mm3 (indicating day 0), tumor-bearing mice were given three i.p. injections on days 0, 3, and 6, with each consisting of 200 μg of mTIGIT-Fc-LIGHT, or 100 μg of anti-TIGIT, Fc-LIGHT, anti–PD-1, or anti–PD-L1, or the combinations shown above. A cohort of mice were treated with a course of CD4, CD8, or NK depleting Abs on days −1, 1, and 7. (E) Each graph shows individual mouse tumor growth curves, the average day in which the entire group reached tumor burden (also indicated by dotted line), and the number of mice that completely rejected tumors/the total group size analyzed. (F) Kaplan–Meier survival plot through day 30 of the time course. (G) The percent change in tumor volume from day 0 to 11 is shown for the vehicle (PBS) only control (n = 9), the mTIGIT-Fc-LIGHT (n = 8 each), and each of the cell depletion groups (anti-CD4, anti-CD8, anti-NK1.1; n = 7 each).
Individual mice that rejected the primary tumor were rechallenged on day 29 with a second inoculation of CT26/WT tumor cells on the opposing hind flank without subsequent retreatment. Mice observed to have complete tumor rejection following an initial course of treatment with TIGIT-Fc-LIGHT were found to have protective immunity that prevented the progression of secondary tumors (Supplemental Fig. 3C). From these animals, which included six mice initially treated with mTIGIT-Fc-LIGHT and three mice treated with anti–PD-L1 monotherapy, peripheral blood was isolated on day 39 (10 d following the day 29 rechallenge). The comparative levels of double-positive effector memory cells (CD127+KLRG1+ out of total CD8+ PBMCs) were evaluated in these mice, and this population of cells was found to be significantly expanded in animals initially treated with mTIGIT-Fc-LIGHT (Supplemental Fig. 3D). The initial antitumor activity of mTIGIT-Fc-LIGHT was associated with increased tumor infiltration of IFN-γ+ NK cells, Ag-specific CD8+ T cells, and CD86+ dendritic cells, along with depletion of CD25+ and FoxP3+ CD4 regulatory T cells (Tregs) (Supplemental Fig. 3G). Interestingly, FOXP3 levels were also observed to be downregulated in human PBMCs following treatment with both IgG1 and IgG4 variants of human TIGIT-Fc-LIGHT, specifically in the ImmuneExp-defined annotation for Tregs (Supplemental Fig. 2G). In addition, in these murine studies, a series of effector and myeloid-specific cytokines were assessed in the tumor milieu, using supernatant generated from dissociated tumor tissue. Increased tumor levels of IL-2, TNF-α, CCL3, CCL4, IL-12(p70), and CCL2 were found within the tumor microenvironment of mTIGIT-Fc-LIGHT–treated animals (Supplemental Fig. 3H). The combination of anti-TIGIT with Fc-LIGHT and either anti–PD-1 or anti–PD-L1 yielded the greatest activity of the benchmark control groups, with tumor burden pushed to 31 d (±3.75 d) and 32 d (±3.82 d), and complete tumor rejection in 7.7 and 23.1% of treated animals, respectively (Fig. 4B). Combinations of mTIGIT-Fc-LIGHT with either anti–PD-1 or anti–PD-L1 also significantly improved efficacy, with tumor burden reached on day 33 (±4.94 or 4.64 d, respectively). Complete tumor rejection was observed in 40.9% of animals for both anti–PD-1 and anti–PD-L1 combinations.
mTIGIT-Fc-LIGHT also demonstrated monotherapy activity in B16.F10 melanoma, where the average time to reach tumor burden was seen at 19 d (±4.47 d), compared with 10 d (±2.00 d) in the vehicle control group, and 14 d (±2.27 d) in the anti-TIGIT + Fc-LIGHT combination group (Fig. 4D, 4E). The triplet combination of anti-TIGIT, Fc-LIGHT, and either anti–PD-1 or anti–PD-L1 provided intermediate benefit, delaying tumor growth to day 18 (±3.02 or 2.56 d, respectively). The combination of mTIGIT-Fc-LIGHT with anti–PD-1 or anti–PD-L1 improved efficacy and survival further (days 24 [±4.85] and 26 [±4.02 d], respectively), and the combination with anti–PD-L1 induced complete tumor rejection in one animal. These findings are significant given the aggressive tumor type and starting tumor volumes averaging >110 mm3 (Fig. 4E, 4F, Supplemental Fig. 3I, 3K). In the B16.F10 model, the contribution of immune cell subsets to antitumor activity was evaluated. A cohort of mice was treated with CD4-, CD8-, or NK cell–depleting Abs on days −1, 1, and 7 of the time course, and cell depletion was confirmed by flow cytometry (Fig. 4D, 4G, Supplemental Fig. 3L). The depletion of CD4 cells had no impact on mTIGIT-Fc-LIGHT activity; however, the depletion of CD8 cells or NK cells partially reduced tumor growth control, consistent with the expression pattern of TIGIT and HVEM (Fig. 4G). The fact that neither CD8 nor NK cell depletion completely abrogated antitumor responses suggested that both CD8+ T cells and NK cells contribute independently to antitumor activity, which was nonetheless enhanced when both cell populations were present. It is also interesting to note that in the CPI-responsive CT26/WT model, TIGIT-Fc-LIGHT+/− anti–PD-1/L1 performed similarly to the Ab combinations; however, in the CPI primary resistant B16.F10 tumor model, TIGIT-Fc-LIGHT monotherapy and anti–PD-1/L1 combination groups significantly outperformed the groups that combined the individual components (Supplemental Fig. 3I). These results indicate that TIGIT-Fc-LIGHT activity differentiates from Ab blockade of TIGIT and/or PD-1/L1, and may provide a unique benefit in settings in which checkpoint inhibitors do not work.
Preclinical model of CPI-acquired resistance
Previously, we generated and characterized a preclinical model of CPI-acquired resistance, which reflects many of the features observed in PD-1/L1–acquired resistant NSCLC patients (https://doi.org/10.1101/2021.07.21.452854). This model was developed by treating mice with established CT26/WT tumors with anti–PD-1 Abs, and excising tumors that were partially controlled but not rejected following treatment. The excised tumors were dissociated, expanded ex vivo, and reinoculated into new recipient mice for repeat treatment with anti–PD-1 Abs. This procedure was serially repeated five times in vivo to provide continuous, anti–PD-1 selective pressure, until none of the mice demonstrated benefit from PD-1 blockade. The resulting tumors were named CT26/AR. A comparison of the transcriptomes of CT26/AR tumors and tumors isolated from PD-1/L1 Ab–acquired resistance NSCLC patients were compared and found to share similar perturbations in JAK/STAT and IFN signaling pathways (https://doi.org/10.1101/2021.07.21.452854). Therefore, the CT26/AR model may be a useful tool for assessing the potential antitumor activity of novel therapeutics in the setting of PD-1/L1 resistance.
mTIGIT-Fc-LIGHT along with benchmark control treatment groups presented previously were evaluated in CT26/AR tumors after reaching a starting tumor volume of >100 mm3 in recipient mice (Fig. 5A). On average, CT26/AR tumors grew more quickly in vivo than their CT26/WT counterparts, with vehicle-treated animals reaching tumor burden at an average of 14 d (±3.02 d) following the initiation of treatment (Fig. 5B). Treatment with anti-TIGIT or anti–PD-1 did not appreciably delay tumor growth, whereas Fc-LIGHT and anti–PD-L1 delayed tumor burden to 17 d (±3.38 d) and 19 d (±5.07 d), respectively. The incremental delay in tumor growth for CT26/AR tumors treated with anti–PD-1 Abs is consistent with clinical data demonstrating that repeat treatment with anti–PD-1 Abs provides benefit in roughly 5% of patients (36). The modest monotherapy activity of Fc-LIGHT also suggested that LIGHT signaling through HVEM and/or LTβR functions independently from PD-1 and was distinct from monotherapy anti-TIGIT treatment (Fig. 5B, Supplemental Fig. 4A, 4B). Along these lines, the alignment of the cytoplasmic amino acid sequences between HVEM and DNAM-1 identified minimal homology, whereby HVEM lacked the tyrosine residues found in DNAM-1, which have been reported to become dephosphorylated by the SHP-2 domain of PD-1, involved in PD-1 inhibition of DNAM-1 costimulation (Supplemental Fig. 4C) (14, 37).
FIGURE 5.
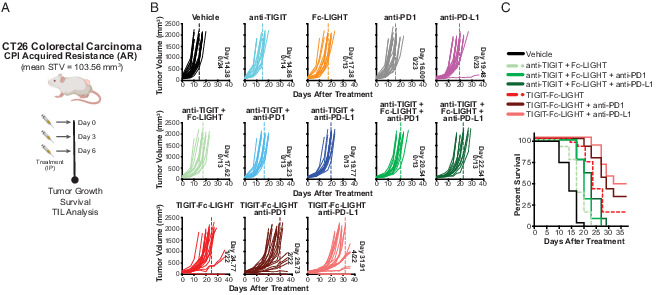
Mouse TIGIT-Fc-LIGHT extends antitumor responses to CPI-acquired resistance tumors through a broad activation of myeloid, CD8+ T, and NK cells. (A) When CT26/AR tumors reached an average starting tumor volume (STV) of 103.56 mm3 (indicating day 0), tumor-bearing mice were given three i.p. injections on days 0, 3, and 6, with each consisting of 200 μg of mTIGIT-Fc-LIGHT, or 100 μg of anti-TIGIT, Fc-LIGHT, anti–PD-1, or anti–PD-L1, or the combinations shown above. (B) Each graph shows individual mouse tumor growth curves, the average day in which the entire group reached tumor burden (also indicated by dotted line), and the number of mice that completely rejected tumors/the total group size analyzed. (C) Kaplan–Meier survival plot through day 36 of the time course.
mTIGIT-Fc-LIGHT monotherapy delayed tumor burden until day 24 (±6.05 d) and significantly improved survival as compared with the separate administration of anti-TIGIT and Fc-LIGHT (Mantel–Cox p value <0.0001; (Fig. 5B, 5C, Supplemental Fig. 4A, 4B). The combination of anti–PD-1 or anti–PD-L1 and TIGIT-Fc-LIGHT delayed tumor burden to 29 d (±5.95 d) and 31 d (±5.18 d) and induced complete tumor rejection in 9.1 and 18.2% of mice, respectively. Both TIGIT-Fc-LIGHT combinations significantly improved survival compared with triplet combinations of anti-TIGIT, Fc-LIGHT, and either anti–PD-1 or anti–PD-L1 (Fig. 5C, Supplemental Fig. 4A, 4B). Interestingly, CT26/AR tumors had slightly less CD8+ T cell infiltration than CT26/WT tumors, but similar levels of NK cells (Supplemental Fig. 4D, 4E). Whereas the ability to therapeutically enhance T cell infiltrate was constrained in CT26/AR tumors, TIGIT-Fc-LIGHT monotherapy still modestly enhanced Ag-specific CD8+ T cells into the tumor. Furthermore, in this tumor model, the combination of TIGIT-Fc-LIGHT with anti–PD-L1 led to a significant increase in effector CD8+ T cell infiltration that was comparable to the level observed in CT26/WT tumors (Supplemental Fig. 4D). NK cells appear to be less impacted in the CT26/AR model, and both TIGIT-Fc-LIGHT monotherapy and combinations with anti–PD-L1 led to a significant increase in NK cells (Supplemental Fig. 4E). These results are consistent with the immune cell contributions identified in the previously described cell depletion studies (Fig. 4G) and suggest that the broad immune stimulating activity of TIGIT-Fc-LIGHT may potentiate antitumor immunity in the setting of checkpoint-acquired resistance.
Safety and pharmacodynamic activity of TIGIT-Fc-LIGHT in nonhuman primates
Cynomolgus macaques were determined to be an appropriate species to examine the potential toxicity and immune properties of human TIGIT-Fc-LIGHT due to the cross-binding of human TIGIT and LIGHT to cynomolgus macaque targets (data not shown). Groups of treatment-naive cynomolgus macaques were repeatedly treated with i.v. TIGIT-Fc(IgG4)-LIGHT at doses of 0.1, 1.0, 10, and 40 mg/kg or vehicle control administered on days 1, 8, 15, and 22 of the study. TIGIT-Fc-LIGHT was well tolerated by all dose groups. Sporadic, mild, and transient increases in AST and ALT were noted in a few animals without a clear dose response and deemed potentially related to a skeletal muscle effect of TIGIT-Fc-LIGHT by the clinical pathologist. No associated microscopic changes were observed either in liver or muscle. All other organ function tests performed on animals treated with TIGIT-Fc-LIGHT remained in the normal reference range (Supplemental Fig. 4F). In prior studies, OX40L- and CD40L-containing fusion proteins were shown to stimulate selective margination of OX40- or CD40-expressing lymphocytes (28, 30). In this experiment, flow cytometry was performed to determine whether the observed changes in lymphocyte counts were driven by selective margination of HVEM-expressing lymphocytes out of the peripheral circulation. To begin, the levels of HVEM, DNAM-1, and PVR were assessed by flow cytometry on cynomolgus macaque CD8+ T cells (Supplemental Fig. 4G). Consistent with mice and humans, nonhuman primate CD8+ T cells expressed much greater levels of HVEM than DNAM-1. TIGIT-Fc-LIGHT stimulated a dose-dependent margination of peripheral lymphocytes that was evident in the complete blood count analysis immediately postdose (Fig. 6A). More specifically, TIGIT-Fc-LIGHT induced the margination of CD3+ T cells out of the periphery within 6 h after the initial infusion (Fig. 6B), and these cells were demonstrated to express high levels of HVEM (Supplemental Fig. 4G). The margination of immune cell populations bound to the TIGIT-Fc-LIGHT therapeutic take the drug with them and create a challenge when trying to assess pharmacokinetics; however, these analyses are underway. The changes in peripheral lymphocytes were restricted to the lymphoid compartment, and no significant changes were observed in the number of peripheral neutrophils, basophils, eosinophils, and platelets, or differences in the levels of hemoglobin or hematocrit (Supplemental Fig. 4F and data not shown). The observed decrease in peripheral lymphocytes correlated with an increase in lymphocyte counts in the infusion draining inguinal lymph node calculated from biopsy samples taken on day 9, ∼24–36 h after the second infusion (Supplemental Fig. 4H). Dose-dependent increases in the serum concentration of multiple adaptive immune and proinflammatory cytokines were noted, and unbiased principal component analysis (PCA) revealed a separation of dose treatment groups when all 30 cytokines from a multiplex array were included in the analysis (Fig. 6C). JMP software was used to generate a vector plot that identified cytokines that dominated the migration of individual animals across PCA quadrants based on a specific cytokine signature (Fig. 6D). The signature that led to animal responses migrating to quadrants 3 and 4 included proinflammatory cytokines such as CXCL10, CCL2, CCL4, CCL17, and IL-10. Adaptive immune cytokines were also observed to follow a dose response, and clustered in quadrant 2, including IL-2 and IL-17 (Fig. 6E, Supplemental Fig. 4I). Many of the serum cytokine responses were conserved with observations using murine TIGIT-Fc-LIGHT (Supplemental Fig. 4J). Human TIGIT-Fc-LIGHT shares ∼76% sequence identity with cynomolgus macaques, and anti-drug Ab responses were expected following the third infusion. The adaptive and innate immune cytokine signature observed with TIGIT-Fc-LIGHT occurred following each dose, including the first, and with similar kinetics in which the peak response was detected ∼2 h after infusion and largely returned to baseline within 24 h after infusion. Certain cytokines, including IL-2, escalated in peak concentration from the first dose to the second dose. Taken together, these results indicated that TIGIT-Fc-LIGHT was well tolerated through 40 mg/kg in cynomolgus macaques and exerted potent immunological effects based on the receptor/ligand interactions involved in the TIGIT and LIGHT pathways. Encouragingly, many of the serum cytokine changes observed in nonhuman primates overlapped with the cytokine changes observed in mouse in vivo and human in vitro assays (Supplemental Figs. 2–4).
FIGURE 6.
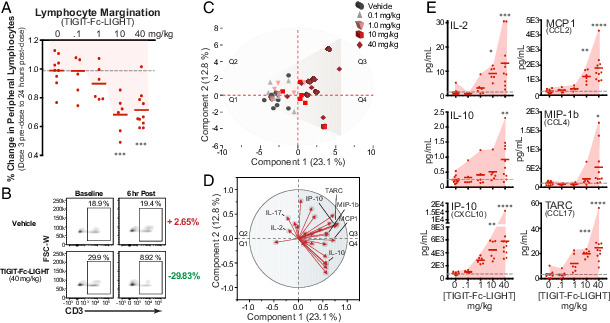
TIGIT-Fc-LIGHT on-target pharmacodynamic activity translates to immunological responses observed in nonhuman primates. (A) Cynomolgus macaques were given 4 weekly i.v. infusions of vehicle or TIGIT-Fc-LIGHT at 0 (10 animals, 0.1 (6 animals), 1.0 (6 animals), 10 (6 animals), and 40 (10 animals) mg/kg. Animals were monitored for various clinical hematology and chemistry parameters during the course of the study. Shown is the percent change in total lymphocyte count from dose 4 predose to 24 h after dose 4 at each individual animal, demonstrating a downward dose-dependent response as lymphocytes are marginating out of the periphery. One-way ANOVA was used for statistical analysis: ***p < 0.001. (B) Flow cytometry was used to assess the frequency of CD3+ T cells (pregated on CD45+ cells) in peripheral blood samples taken at baseline and 6 h after the day 1 infusion. Shown are example animals from the vehicle and TIGIT-Fc-LIGHT 40 mg/kg groups. (C) PCA was used to spatially visualize the distribution of 2-h postdose 2 for each vehicle- and TIGIT-Fc-LIGHT–treated animal, based on the detection levels of a 30-plex nonhuman primate MSD cytokine panel. (D) JMP software was used to generate a vector plot that ranks the influence of particular cytokines on the spatial distribution into each quadrant of the PCA plot in (C). The myeloid-associated cytokines IL-10, IP-10, MCP1, MIP-1β, and TARC, as well as the adaptive immune cytokine IL-2, were shown to be enriched in Q3. (E) The maximum postdose cytokine response is plotted across all dose groups, for each individual animal. Shading was used to highlight the dose response, and one-way ANOVA was used to assess significance: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Discussion
Ab-mediated blockade of TIGIT has translated to improved response rates for NSCLC patients in combination with anti–PD-1/L1 Abs. TIGIT blockade with an mAb leads to antitumor immunity by enabling PVR to bind and signal via DNAM-1. A mechanistic explanation for the dependence of TIGIT blockade upon concurrent PD-1/L1 blockade has been proposed, wherein PD-1 signaling directly inhibits costimulatory signaling via DNAM-1 (14, 37). This mechanism explains why concurrent PD-1/L1 blockade is necessary for clinical benefit of a TIGIT blocking Ab, but it does not explain why the combination of TIGIT and PD-1/L1 blockade has not provided clinical benefit for PD-1/L1–experienced cancer patients. A potential explanation for this observation includes the progressive downregulation of DNAM-1 during tumor progression, which was observed in NSCLC and melanoma (17).
Due to the progressive downregulation of DNAM-1 in advanced tumors, we undertook a systematic screen to identify other costimulatory receptors with a more consistent expression pattern on TILs that might serve as higher value targets for inducing antitumor immune responses. HVEM and LTβR, two receptors for the TNF ligand known as LIGHT, emerged as promising candidates from this screen. The overlapping expression pattern of DNAM-1 and HVEM on both CD8+ T and NK cells, and specifically including CD8+ Tscms, provided additional justification to further characterize LIGHT-mediated costimulation in the setting of TIGIT blockade.
There are potential benefits and liabilities to inhibiting the TIGIT/PVR axis using recombinant TIGIT proteins as opposed to TIGIT-binding mAbs. A possible benefit includes the potential for recombinant TIGIT to competitively inhibit all TIGIT ligands, including PVR, PVRL2, and nectins. A potential liability is that recombinant TIGIT will also block PVR interactions with DNAM-1 (34). Single-sided TIGIT-Fc fusion proteins have been reported both to induce tolerance and antitumor immunity, suggesting that the outcome of blocking all TIGIT ligands may be proinflammatory or anti-inflammatory depending on the experimental context (38–40). For many costimulatory receptors, redundant intracellular signaling pathways are used, and as a result homeostatic control is maintained through regulated expression of individual ligands and receptors. This was found to be the case with DNAM-1 and HVEM, wherein a recombinant TIGIT-Fc-LIGHT fusion protein stimulated downstream signaling pathways via HVEM in a reporter assay designed to assess costimulatory signaling via PVR/DNAM-1 interactions.
Another incompletely resolved question regarding the design of TIGIT-blocking Abs is whether FcR-competent Fc domains are an asset or liability. TIGIT is expressed by effector CD8+ T cells and NK cells, which have been shown to be important for the control of tumor growth in the setting of immune checkpoint blockade, including TIGIT blockade (10, 33, 41). If a TIGIT-blocking Ab has the ability to engage Fcγ receptors, then it is possible that Ab-dependent cellular cytotoxicity could lead to depletion of the TIGIT-expressing cells that are bound by the Fc-competent anti-TIGIT Ab. Indeed, depletion of CD8+ T cells was recently reported in human clinical studies with such an Ab(42, 43). In contrast, CD4+ Tregs also express TIGIT, which led to the hypothesis that perhaps anti-TIGIT Abs could lead to depletion of Tregs and enhance antitumor immunity; however, reasoning behind the selective depletion of CD4+ Tregs versus CD8+ effector T cells other than the surface expression of TIGIT on these cell types has not been clearly presented (44, 45). Others have reported that anti-TIGIT Abs with effector-competent Fc domains can engage Fc receptors on myeloid cells, enhancing expression of costimulatory molecules on their surface and priming of T cell immunity (33). Each of these hypotheses have merits, and consideration of these design issues is a necessary byproduct of Ab-based biologics. To date, however, there is no evidence for a physiological interaction between the TIGIT/PVR/DNAM-1 axis and Fc receptors, and this question is likely an artifact of utilizing Fc-containing therapeutics.
In consideration of this question, both FcγR-competent and silenced versions of TIGIT-Fc-LIGHT were constructed. Because many cell types that express Fc receptors also express LTβR, we evaluated whether Fc-mediated activation of myeloid cells could be independently detected in the presence of LIGHT. These studies demonstrated that LIGHT promoted proliferation, differentiation, and cytokine production of human peripheral blood leukocytes rapidly in vitro. The most prominently expanded leukocyte subset was found to correlate with the myeloid lineage and expressed high levels of LTβR. Single-cell gene analysis indicated that both IgG1 and IgG4 versions of TIGIT-Fc-LIGHT induced similar levels of gene activation associated with Ag processing and presentation, suggesting that the role of Fcγ receptor activation was either undetectable or superseded in the presence of LIGHT.
The combined observations that LIGHT could provide myeloid cell activation independent of Fc receptor engagement, while also activating CD8+ T cells, Tscms, and NK cells, suggested that the antitumor activity of TIGIT-Fc-LIGHT should be explored in vivo. Consistent with both the preclinical and emerging clinical data, treatment of established PD-1–sensitive tumors with the combination of PD-1/L1 and TIGIT blocking mAbs demonstrated antitumor activity that is consistent with prior reports (46). When established tumors (both CT26/WT and B16.F10 models) were treated with TIGIT-Fc-LIGHT, higher rates of tumor control and rejection were observed both as monotherapy and in combination with PD-1/L1 mAbs than what was observed for any of the Ab monotherapies or combinations. In the CT26/WT model, TIGIT-Fc-LIGHT monotherapy substantially altered the intratumoral microenvironment, with a higher proportion of activated APCs, NK cells, and Ag-specific CD8+ T cells infiltrating the tumor relative to vehicle-treated control animals. Furthermore, increased concentrations of multiple proinflammatory cytokines, including IL-2, TNF-α, CCL2, CCL3, CCL4, and IL-12, were observed. Most importantly, TIGIT-Fc-LIGHT demonstrated antitumor activity both alone and in combination with PD-1/L1 mAbs in the PD-1–acquired resistant CT26-AR tumor model, where head-to-head combinations of PD-1/L1 and TIGIT mAbs did not provide any discernible benefit. Consistent with the PD-1–sensitive CT26 model, TIGIT-Fc-LIGHT stimulated an increase in the proportion of both intratumoral NK cells and Ag-specific CD8+ T cells in the CT26-AR model, both as monotherapy and in combination with PD-1/L1 mAbs.
The potential for LIGHT to enable TIGIT blockade to control the growth of PD-1/L1–acquired resistant tumors prompted evaluation of TIGIT-Fc-LIGHT in nonhuman primate toxicology studies. The cynomolgus macaques used for these studies were tumor free; however, multiple indications of on-target activity were observed in the peripheral blood and secondary lymphoid tissue (lymph nodes) of the treated animals. As in humans, HVEM is widely expressed on PBLs in cynomolgus macaques, and rapid margination of HVEM-expressing lymphocytes was observed in a dose-dependent manner, in conjunction with increased concentrations of adaptive immune and proinflammatory cytokines, including IL-2. The similarity in the cytokine profile observed in mice treated with murine TIGIT-Fc-LIGHT as compared with monkeys treated with human TIGIT-Fc-LIGHT may indicate that overall function of LIGHT is well conserved between rodents and primates, and that some of the differences in mice could be related to the presence of an established tumor and the resulting antitumor immune response. Overall, TIGIT-Fc-LIGHT was well tolerated up to at least 40 mg/kg. Importantly, there was no evidence of cytokine release syndrome, despite the observed cytokine profile and relatively small increases in IL-6. The data from cynomolgus macaques indicate that monitoring peripheral lymphocyte counts and postdose cytokines could provide an indication of dose-dependent pharmacodynamic activity in humans.
In comparison with OX40L, CD40L, 4-1BBL, and GITRL, relatively little is known about the potential therapeutic role of LIGHT in cancer immunotherapy. Only a handful of reports have investigated this question, and they demonstrated that both systemic and local stimulation of HVEM and LTβR by LIGHT provides promising antitumor activity (24, 47–53). In this study, we have extended those findings and demonstrated that the LIGHT-mediated costimulation of CD8+ T cells and NK cells leads to durable antitumor immunity in the setting of TIGIT blockade. Importantly, these benefits extend to preclinical models of PD-1–acquired resistant tumors, both as monotherapy and in combination with anti–PD-1/L1 Abs. The observation that HVEM and LTβR are expressed at high levels even in advanced tumors, relative to DNAM-1, likely contributes to these observations. It is also likely that HVEM- and LTβR-mediated signaling is not directly inhibited by PD-1 due to the lack of homology between HVEM and DNAM-1 cytoplasmic domains and the presence of tyrosine phosphorylation sites, as was reported to be the case for DNAM-1 (Supplemental Fig. 4C) (14, 54). Overall, both human and mouse TIGIT-Fc-LIGHT fusion proteins were well tolerated and provided dose-dependent evidence supporting activation of CD8+ T cells, NK cells, and myeloid cells. The potential for therapeutic benefit of TIGIT-Fc-LIGHT in the setting of PD-1 inhibitor–acquired resistance will be further explored with future work.
Supplementary Material
This article is featured in Top Reads, p.
This work was supported by Shattuck Labs, Inc.
K.J.Y., K.J., L.E.G., A.P., C.W.S., Z.O., K.L., K.C., T.-A.N., J.M., A.M., and C.S. performed experiments. Y.-H.T. performed bioinformatics analysis. F.R. authored and edited text. S.d.S., T.H.S., and G.F. designed, executed, interpreted experiments, and edited the manuscript. T.H.S. and G.F. wrote the manuscript.
The raw data presented in this article have been submitted to the Gene Expression Omnibus under accession number GSE201999.
The online version of this article contains supplemental material.
- AR
- CPI-acquired resistance
- BTLA
- B and T lymphocyte attenuator
- CPI
- checkpoint inhibition
- DcR3
- decoy receptor 3
- DEG
- differentially expressed gene
- h
- human
- HVEM
- herpes virus entry mediator A
- LTβR
- lymphotoxin β receptor
- m
- mouse
- MSD
- Meso Scale Discovery
- NSCLC
- non–small cell lung cancer
- PCA
- principal component analysis
- PD-1/L1
- PD-1/PD-L1
- scRNA-seq
- single-cell RNA sequencing
- SEB
- staphylococcal enterotoxin B
- SEC
- size-exclusion chromatography
- TCGA
- The Cancer Genome Atlas
- TIGIT
- T cell immunoreceptor with Ig and ITIM domains
- TIL
- tumor-infiltrating lymphocyte
- Tn
- naive T
- Treg
- regulatory T cell
- Tscm
- T memory stem cell
- UMAP
- uniform manifold approximation and projection
- WT
- wild-type
Disclosures
All authors are employees of, or consultants for, Shattuck Labs and hold an equity interest in the company.
References
- 1. Zaretsky J. M., Garcia-Diaz A., Shin D. S., Escuin-Ordinas H., Hugo W., Hu-Lieskovan S., Torrejon D. Y., Abril-Rodriguez G., Sandoval S., Barthly L., et al. 2016. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375: 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shin D. S., Zaretsky J. M., Escuin-Ordinas H., Garcia-Diaz A., Hu-Lieskovan S., Kalbasi A., Grasso C. S., Hugo W., Sandoval S., Torrejon D. Y., et al. 2017. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 7: 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Donnell J. S., Long G. V., Scolyer R. A., Teng M. W., Smyth M. J.. 2017. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat. Rev. 52: 71–81. [DOI] [PubMed] [Google Scholar]
- 4. McLane L. M., Abdel-Hakeem M. S., Wherry E. J.. 2019. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 37: 457–495. [DOI] [PubMed] [Google Scholar]
- 5. Sharma P., Hu-Lieskovan S., Wargo J. A., Ribas A.. 2017. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Johnston R. J., Comps-Agrar L., Hackney J., Yu X., Huseni M., Yang Y., Park S., Javinal V., Chiu H., Irving B., et al. 2014. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell 26: 923–937. [DOI] [PubMed] [Google Scholar]
- 7. Khan O., Giles J. R., McDonald S., Manne S., Ngiow S. F., Patel K. P., Werner M. T., Huang A. C., Alexander K. A., Wu J. E., et al. 2019. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature 571: 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wherry E. J. 2011. T cell exhaustion. Nat. Immunol. 12: 492–499. [DOI] [PubMed] [Google Scholar]
- 9. Lee J. B., Hong M. H., Park S. Y., Chae S., Hwang D., Ha S. J., Shim H. S., Kim H. R.. 2021. Overexpression of PVR and PD-L1 and its association with prognosis in surgically resected squamous cell lung carcinoma. [Published erratum appears in 2021 Sci. Rep. 11: 15552.] Sci. Rep. 11: 8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yeo J., Ko M., Lee D. H., Park Y., Jin H. S.. 2021. TIGIT/CD226 axis regulates anti-tumor immunity. Pharmaceuticals (Basel) 14: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Niu J., Nagrial A., Voskoboynik M., Chung H. C., Lee D. H., Ahn M., Bauer T. M., Jimeno A., Chung V., Mileham K. F., et al. 2020. 1410P - Safety and efficacy of vibostolimab, an anti-TIGIT antibody, plus pembrolizumab in patients with anti-PD-1/PD-L1-naive NSCLC. Ann. Oncol. 31(Suppl. 4):S754–S840 (Abstr.). [Google Scholar]
- 12. Pauken K. E., Wherry E. J.. 2014. TIGIT and CD226: tipping the balance between costimulatory and coinhibitory molecules to augment the cancer immunotherapy toolkit. Cancer Cell 26: 785–787. [DOI] [PubMed] [Google Scholar]
- 13. Skokos D., Waite J. C., Haber L., Crawford A., Hermann A., Ullman E., Slim R., Godin S., Ajithdoss D., Ye X., et al. 2020. A class of costimulatory CD28-bispecific antibodies that enhance the antitumor activity of CD3-bispecific antibodies. Sci. Transl. Med. 12: eaaw7888. [DOI] [PubMed] [Google Scholar]
- 14. Wang B., Zhang W., Jankovic V., Golubov J., Poon P., Oswald E. M., Gurer C., Wei J., Ramos I., Wu Q., et al. 2018. Combination cancer immunotherapy targeting PD-1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci. Immunol. 3: eaat7061. [DOI] [PubMed] [Google Scholar]
- 15. Ahn M., Niu J., Kim D., Rasco D., Mileham K. F., Chung H. C., Vaishampayan U. N., Maurice-Dror C., Lo Russo P., Golan T., et al. 2020. 1400P - vibostolimab, an anti-TIGIT antibody, as monotherapy and in combination with pembrolizumab in anti-PD-1/PD-L1-refractory NSCLC. Ann. Oncol. 31 (Suppl. 4): S754–S840 (Abstr.). [Google Scholar]
- 16. Rodriguez-Abreu D., Johnson M. L., Hussein M. A., Cobo M., Patel A. J., Secen N. M., Lee K. H., Massuti B., Hiret S., Yang J. C.-H., et al. Primary analysis of a randomized, double-blind, phase II study of the anti-TIGIT antibody tiragolumab (tira) plus atezolizumab (atezo) versus placebo plus atezo as first-line (1L) treatment in patients with PD-L1-selected NSCLC (CITYSCAPE). J. Clin. Oncol. 38 (Suppl. 15): 9503. [Google Scholar]
- 17. Weulersse M., Asrir A., Pichler A. C., Lemaitre L., Braun M., Carrié N., Joubert M. V., Le Moine M., Do Souto L., Gaud G., et al. 2020. Eomes-dependent loss of the co-activating receptor CD226 restrains CD8+ T cell anti-tumor functions and limits the efficacy of cancer immunotherapy. Immunity 53: 824–839.e10. [DOI] [PubMed] [Google Scholar]
- 18. Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C. M., Quigley M. F., Almeida J. R., Gostick E., Yu Z., Carpenito C., et al. 2011. A human memory T cell subset with stem cell-like properties. Nat. Med. 17: 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmiedel B. J., Singh D., Madrigal A., Valdovino-Gonzalez A. G., White B. M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J. A., McVicker G., et al. 2018. Impact of genetic polymorphisms on human immune cell gene expression. Cell 175: 1701–1715.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Novershtern N., Regev A., Friedman N.. 2011. Physical module networks: an integrative approach for reconstructing transcription regulation. Bioinformatics 27: i177–i185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Monaco G., Lee B., Xu W., Mustafah S., Hwang Y. Y., Carre C., Burdin N., Visan L., Ceccarelli M., Poidinger M., et al. 2019. RNA-seq signatures normalized by mRNA abundance allow absolute deconvolution of human immune cell types. Cell Rep. 26: 1627–1640.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hehlgans T., Männel D. N.. 2001. Recombinant, soluble LIGHT (HVEM ligand) induces increased IL-8 secretion and growth arrest in A375 melanoma cells. J. Interferon Cytokine Res. 21: 333–338. [DOI] [PubMed] [Google Scholar]
- 23. Mauri D. N., Ebner R., Montgomery R. I., Kochel K. D., Cheung T. C., Yu G. L., Ruben S., Murphy M., Eisenberg R. J., Cohen G. H., et al. 1998. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8: 21–30. [DOI] [PubMed] [Google Scholar]
- 24. Yu P., Lee Y., Liu W., Chin R. K., Wang J., Wang Y., Schietinger A., Philip M., Schreiber H., Fu Y. X.. 2004. Priming of naive T cells inside tumors leads to eradication of established tumors. Nat. Immunol. 5: 141–149. [DOI] [PubMed] [Google Scholar]
- 25. Wang Y., Qiu F., Xu Y., Hou X., Zhang Z., Huang L., Wang H., Xing H., Wu S.. 2021. Stem cell-like memory T cells: the generation and application. J. Leukoc. Biol. 110: 1209–1223. [DOI] [PubMed] [Google Scholar]
- 26. Li Y., Cong Y., Jia M., He Q., Zhong H., Zhao Y., Li H., Yan M., You J., Liu J., et al. 2021. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat. Commun. 12: 951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Draghi A., Chamberlain C. A., Khan S., Papp K., Lauss M., Soraggi S., Radic H. D., Presti M., Harbst K., Gokuldass A., et al. 2021. Rapid identification of the tumor-specific reactive TIL repertoire via combined detection of CD137, TNF, and IFNγ, following recognition of autologous tumor-antigens. Front. Immunol. 12: 705422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. de Silva S., Fromm G., Shuptrine C. W., Johannes K., Patel A., Yoo K. J., Huang K., Schreiber T. H.. 2020. CD40 enhances type I interferon responses downstream of CD47 blockade, bridging innate and adaptive immunity. Cancer Immunol. Res. 8: 230–245. [DOI] [PubMed] [Google Scholar]
- 29. Fromm G., de Silva S., Johannes K., Patel A., Hornblower J. C., Schreiber T. H.. 2018. Agonist redirected checkpoint, PD1-Fc-OX40L, for cancer immunotherapy. J. Immunother. Cancer 6: 149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Siakavellas S. I., Sfikakis P. P., Bamias G.. 2015. The TL1A/DR3/DcR3 pathway in autoimmune rheumatic diseases. Semin. Arthritis Rheum. 45: 1–8. [DOI] [PubMed] [Google Scholar]
- 31. Cheung T. C., Oborne L. M., Steinberg M. W., Macauley M. G., Fukuyama S., Sanjo H., D’Souza C., Norris P. S., Pfeffer K., Murphy K. M., et al. 2009. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J. Immunol. 183: 7286–7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheung T. C., Steinberg M. W., Oborne L. M., Macauley M. G., Fukuyama S., Sanjo H., D’Souza C., Norris P. S., Pfeffer K., Murphy K. M., et al. 2009. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. [Published erratum appears in 2009 Proc. Natl. Acad. Sci. USA 106: 16535–16536.] Proc. Natl. Acad. Sci. USA 106: 6244–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han J. H., Cai M., Grein J., Perera S., Wang H., Bigler M., Ueda R., Rosahl T. W., Pinheiro E., LaFace D., et al. 2020. Effective anti-tumor response by TIGIT blockade associated with FcγR engagement and myeloid cell activation. [Published erratum appears in 2020 Front. Immunol. 11: 615755.] Front. Immunol. 11: 573405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dolgin E. 2020. Antibody engineers seek optimal drug targeting TIGIT checkpoint. [Published erratum appears in 2020 Nat. Biotechnol. 38: 1357.] Nat. Biotechnol. 38: 1007–1009. [DOI] [PubMed] [Google Scholar]
- 35. Gommerman J. L., Browning J. L.. 2003. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat. Rev. Immunol. 3: 642–655. [DOI] [PubMed] [Google Scholar]
- 36. Ahn M. N. J., Kim D., Rasco D., Mileham K. F., Chung H. C., Vaishampayan U. N., Maurice-Dror C., Lo Russo P., Golan T., Chartash E., et al. 2020. Vibostolimab, an anti-TIGIT antibody, as monotherapy and in combination with pembrolizumab in anti-PD-1/PD-L1-refractory NSCLC. Ann. Oncol. 31(Suppl 4): S887. [Google Scholar]
- 37. Banta K. L., Xu X., Chitre A. S., Au-Yeung A., Takahashi C., O’Gorman W. E., Wu T. D., Mittman S., Cubas R., Comps-Agrar L., et al. 2022. Mechanistic convergence of the TIGIT and PD-1 inhibitory pathways necessitates co-blockade to optimize anti-tumor CD8+ T cell responses. Immunity 55: 512–526.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shen X., Fu W., Wei Y., Zhu J., Yu Y., Lei C., Zhao J., Hu S.. 2021. TIGIT-Fc promotes antitumor immunity. Cancer Immunol. Res. 9: 1088–1097. [DOI] [PubMed] [Google Scholar]
- 39. Fu W., Cai R., Ma Z., Li T., Lei C., Zhao J., Hu S.. 2021. TIGIT-Fc as a potential therapeutic agent for fetomaternal tolerance. Front. Immunol. 12: 649135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang D., Hu W., Xie J., Zhang Y., Zhou B., Liu X., Zhang Y., Su Y., Jin B., Guo S., Zhuang R.. 2018. TIGIT-Fc alleviates acute graft-versus-host disease by suppressing CTL activation via promoting the generation of immunoregulatory dendritic cells. Biochim. Biophys. Acta Mol. Basis Dis. 1864(9 Pt B): 3085–3098. [DOI] [PubMed] [Google Scholar]
- 41. Chihara N., Madi A., Kondo T., Zhang H., Acharya N., Singer M., Nyman J., Marjanovic N. D., Kowalczyk M. S., Wang C., et al. 2018. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 558: 454–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Van den Mooter T. F. A., Migeotte A., Jungels C., Delafontaine B. R., Nguyen T. L.-A., Servane W., Truong C., De Henau O., Driessens G., Lager J., et al. 2021. Preliminary data from phase I first-in-human study of EOS884448, a novel potent anti-TIGIT antibody, monotherapy shows favorable tolerability profile and early signs of clinical activity in immune-resistant advanced cancers. Cancer Res. 81(Suppl. 13): CT118 (Abstr.). [Google Scholar]
- 43. Chen X., Xue L., Ding X., Liu Q., Zhang J., Jiang L., Liu S., Hou H., Zhu Q., Jiang B., et al. 2021. A Fc-competent anti-human TIGIT blocking antibody BGB-A1217 elicits strong immune responses and potent anti-tumor efficacy in pre-clinical models. 2021. Cancer Res. 81 (Suppl. 13): 1854 (Abstr.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chauvin J. M., Zarour H. M.. 2020. TIGIT in cancer immunotherapy. J. Immunother. Cancer 8: e000957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Preillon J., Cuende J., Rabolli V., Garnero L., Mercier M., Wald N., Pappalardo A., Denies S., Jamart D., Michaux A. C., et al. 2021. Restoration of T-cell effector function, depletion of Tregs, and direct killing of tumor cells: the multiple mechanisms of action of a-TIGIT antagonist antibodies. Mol. Cancer Ther. 20: 121–131. [DOI] [PubMed] [Google Scholar]
- 46. Hung A. L., Maxwell R., Theodros D., Belcaid Z., Mathios D., Luksik A. S., Kim E., Wu A., Xia Y., Garzon-Muvdi T., et al. 2018. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. OncoImmunology 7: e1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dai S., Lv Y., Xu W., Yang Y., Liu C., Dong X., Zhang H., Prabhakar B. S., Maker A. V., Seth P., Wang H.. 2020. Oncolytic adenovirus encoding LIGHT (TNFSF14) inhibits tumor growth via activating anti-tumor immune responses in 4T1 mouse mammary tumor model in immune competent syngeneic mice. Cancer Gene Ther. 27: 923–933. [DOI] [PubMed] [Google Scholar]
- 48. Fan Z., Yu P., Wang Y., Wang Y., Fu M. L., Liu W., Sun Y., Fu Y. X.. 2006. NK-cell activation by LIGHT triggers tumor-specific CD8+ T-cell immunity to reject established tumors. Blood 107: 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holmes T. D., Wilson E. B., Black E. V., Benest A. V., Vaz C., Tan B., Tanavde V. M., Cook G. P.. 2014. Licensed human natural killer cells aid dendritic cell maturation via TNFSF14/LIGHT. Proc. Natl. Acad. Sci. USA 111: E5688–E5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Qiao G., Qin J., Kunda N., Calata J. F., Mahmud D. L., Gann P., Fu Y. X., Rosenberg S. A., Prabhakar B. S., Maker A. V.. 2017. LIGHT elevation enhances immune eradication of colon cancer metastases. Cancer Res. 77: 1880–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Skeate J. G., Otsmaa M. E., Prins R., Fernandez D. J., Da Silva D. M., Kast W. M.. 2020. TNFSF14: LIGHTing the way for effective cancer immunotherapy. Front. Immunol. 11: 922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhai Y., Guo R., Hsu T. L., Yu G. L., Ni J., Kwon B. S., Jiang G. W., Lu J., Tan J., Ugustus M., et al. 1998. LIGHT, a novel ligand for lymphotoxin beta receptor and TR2/HVEM induces apoptosis and suppresses in vivo tumor formation via gene transfer. J. Clin. Invest. 102: 1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zou W., Zheng H., He T. C., Chang J., Fu Y. X., Fan W.. 2012. LIGHT delivery to tumors by mesenchymal stem cells mobilizes an effective antitumor immune response. Cancer Res. 72: 2980–2989. [DOI] [PubMed] [Google Scholar]
- 54. Stengel K. F., Harden-Bowles K., Yu X., Rouge L., Yin J., Comps-Agrar L., Wiesmann C., Bazan J. F., Eaton D. L., Grogan J. L.. 2012. Structure of TIGIT immunoreceptor bound to poliovirus receptor reveals a cell-cell adhesion and signaling mechanism that requires cis-trans receptor clustering. Proc. Natl. Acad. Sci. USA 109: 5399–5404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.