

Abstract
Neurons, astrocytes, and oligodendrocytes locally regulate protein translation within distal processes. Here, we tested whether there is regulated local translation within peripheral microglia processes (PeMPs) from mouse brain. We show that PeMPs contain ribosomes that engage in de novo protein synthesis, and these are associated with transcripts involved in pathogen defense, motility, and phagocytosis. Using a live slice preparation, we further show that acute translation blockade impairs the formation of PeMP phagocytic cups, the localization of lysosomal proteins within them, and phagocytosis of apoptotic cells and pathogen-like particles. Finally, PeMPs severed from their somata exhibit and require de novo local protein synthesis to effectively surround pathogen-like particles. Collectively, these data argue for regulated local translation in PeMPs and indicate a need for new translation to support dynamic microglial functions.
Introduction
Microglia serve crucial functions within the brain including phagocytic “pruning” of synapses1,2,3, clearance of apoptotic cells4, immune surveillance, pathogen defense, and providing trophic factors5. They achieve this via peripheral microglial processes (PeMPs) which dynamically extend, retract, and transiently contact nearby synapses, glial processes, and somata6,7,8. During surveillance, microglia are simultaneously sending and receiving signals via PeMPs, yet it is unclear how they individualize the interactions with different cellular partners at each PeMP.
Neurons similarly have thousands of inputs that need individualized responses to local activity to enable specific synaptic strengthening. Thus, they utilize ribosomes within processes to generate proteins in response to local signals: aka “local translation.” In neurons, local translation is required for chemically induced long-term potentiation in hippocampal slices9,10. Local translation is dynamically regulated by activity, influenced by untranslated region (UTR) sequences, and varied across neuronal cell types11,12. Similarly, astrocytes also locally translate proteins within their distal perisynaptic and endfeet processes13,14. Overall, local translation could rapidly direct specific proteins in specific spatiotemporal loci and conserve the energy required to transport proteins great distances.
Of particular interest is the rapidity by which microglia phagocytose apoptotic cells, supernumerary synapses, and pathogens. In live slices, microglia engulf apoptotic cells within about 30 minutes15. During phagocytosis, actin polymerization rearranges cytoskeleton and plasma membrane, forming a “phagocytic cup” (PC) around the target. Distal PeMPs can form PCs at > 50μm from the soma15. After engulfment, endocytosed phagosomes fuse with lysosomes to digest phagocytized material. Interestingly, phagocytosis of zymosan particles by macrophages (relatives of microglia), is blocked by the protein synthesis inhibitor cycloheximide16. However, it is unknown if protein synthesis occurs locally within PeMPs, or if it is necessary for phagocytosis.
In this study we first tested whether translation indeed occurs in PeMPs. We then developed and applied methods to identify the ribosome-bound mRNAs found in PeMPs, which included those involved in phagocytosis. Finally, we show functionally that inhibiting translation decreases the efficiency of phagocytosis, a key microglial process, and that even PeMPs severed from the soma surround pathogen-like particles in a translation-dependent manner.
Results
Translation occurs in peripheral microglial processes.
We first sought to determine if microglia exhibit local translation within PeMPs, by testing the prediction that ribosomes and new protein synthesis can be detected within PeMPs. To detect PeMP ribosomes, we generated mice with labeled ribosomes in microglia, as reported17,18, by crossing tamoxifen-inducible microglia-targeting Cre mice(CX3CR1-Cre(ERt2)), with floxed ‘TRAP’ mice (ribosomal protein L10a fused to eGFP)19; referred to hereafter as MG-TRAP. We hypothesized that local microglial protein synthesis could play a role in synaptic pruning, and thus focused on ages this occurs(p28-p35), yet limiting CX3CR1-creER-mediated developmental toxicity20 by delaying tamoxifen administration until p15.
We first visualized ribosomal location within cortical IBA1+ cells. A majority of which are microglia, plus a handful of CNS-resident macrophages sharing CX3CR1 and IBA1 expression. As noted in other reporters of CX3CR1-Cre(ERt2)21, all RPL10a-GFP+ cells colabeled with IBA1. Importantly for our hypothesis, we observed RPL10a-GFP signal within both somata and distal processes near synapses marked by presynaptic Synaptophysin (Fig. 1a). We also confirmed the presence of small ribosomal subunit RPS16 within PeMPs by immunolabeling (Fig. 1b–c, Supplementary Video 1). Since Synaptophysin alone cannot define synapses, we verified RPL10a-GFP in PeMPs through electron microscopy with immuno-DAB peroxidase staining against GFP, revealing signal within fine microglial processes located in hippocampal neuropil with adjacent synaptic structures (Fig. 1d–e).
Figure 1: Peripheral Microglial Processes (PeMPs) contain translating ribosomes.
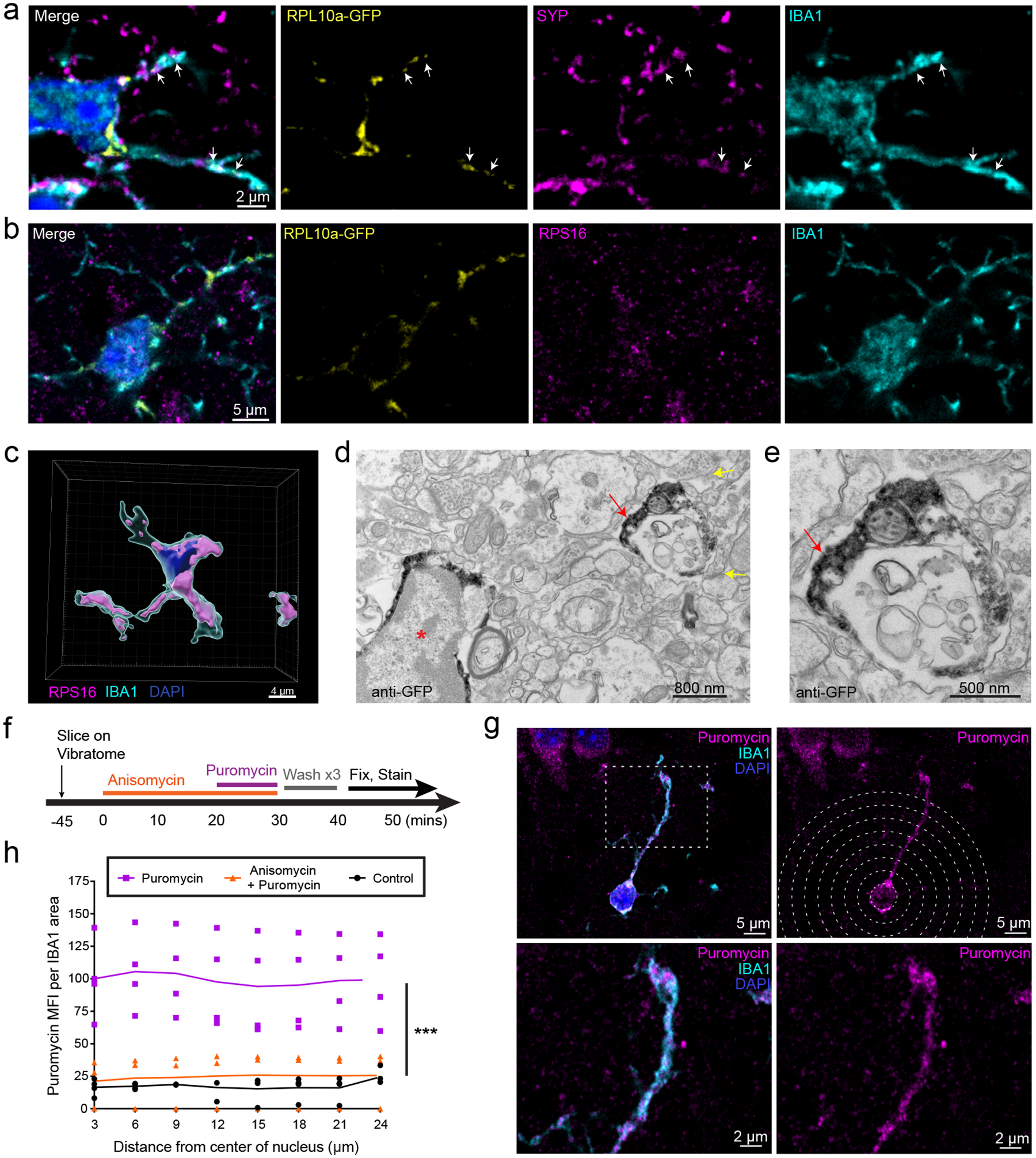
a-e, Brains were harvested from 4–5 week old CX3CR1-Cre(ERT2) x RPL10a-GFP fl/fl mice following tamoxifen induction from p15-p19. a-b, Immunostaining of somatosensory cortex shows localization of ribosomal subunits RPL10a-GFP (a,b) and RPS16 (b) within distal IBA1+ processes and adjacent to synaptophysin positive (a) presynaptic terminals. Images representative of 2 independent experiments from n = 3 mice. c, 3D rendering of RPS16 masked on an IBA1+ cell within somatosensory cortex. Image representative of 1 experiment from n=3 mice. (d,e) Immuno electron micrographs of mouse hippocampus stained with anti-GFP shows RPL10a-GFP present within a microglial soma (red asterisk) and PeMP (red arrow) with nearby synapses (yellow arrows).Images representative of 1 experiment from n = 4 mice. e, PeMP shown at high magnification. f-h, Acute live coronal slices were made from 4–5 week old C57bl6 mice and incubated as shown in the timeline in f. g, Immunostaining for the tRNA analog, puromycin, and IBA1 was performed to quantify local microglial protein synthesis by mean fluorescence intensity of puromycin staining at IBA1-colocalized sites (h), within 3 micron radial bins from the center of the nucleus out to distal PeMPs at increasing distances from the soma (n = 3 anisomycin + puromycin, n = 4 puromycin, n = 4 control brain slices across 2 non-littermate mice with each mouse generating at least 1 slice from each condition). For each slice between 12 and 17 microglia were analyzed and data points represent the mean value of all microglia within the slice while lines represent the means of each treatment group. Treatment is highly significant (p < 0.0001) by two-way ANOVA, while distance (p=0.9996) and interaction of treatment x distance (p=0.9999) are non-significant.
Next to demonstrate that these are functional ribosomes, we tested if active protein synthesis occurs in distal microglial processes. To localize de novo peptide synthesis, we generated live 300μm brain slices and incubated them with the tRNA analog puromycin, which labels newly synthesized peptides22. As a negative control, we preincubated slices with the protein synthesis inhibitor anisomycin (Fig. 1f). Slices were then fixed and immunostained to quantify mean fluorescence intensity (MFI) of anti-puromycin immunostaining within the IBA1+ area using concentric circles of increasing diameter from each microglia’s nucleus. We determined that protein synthesis occurs in the distal processes, which was blocked by anisomycin (Fig. 1g–h). Furthermore, the MFI of puromycin signal didn’t diminish away from the nucleus, suggesting substantial protein synthesis occurs within PeMPs even >20 microns away. Thus, PeMPs are actively engaged in de novo protein synthesis.
A subset of microglial transcripts is detectable in PEMPs
We next tested whether some microglial mRNA transcripts are enriched in PeMPs, or if all microglial transcripts were distributed evenly. We isolated dorsal cortex and hippocampus from MG-TRAP mice to generate synaptosomes using sucrose-density centrifugation23 in combination with translating ribosome affinity purification (TRAP) to enrich microglial ribosomes (Fig. 2a), similar to prior studies of perisynaptic neuronal12,24 and astrocytic transcripts13. This protocol leverages a ‘synaptosomal’ enrichment procedure that also contains fragments, presumably of fine processes, from non-neuronal cells as well13,24. We confirmed this synaptosomal enrichment protocol depletes nuclear proteins MeCP2 and LaminB1, enriches for synaptic protein Synaptophysin, and includes RPL10a-GFP, derived from CX3CR1-expressing microglia and macrophages (Extended Data Fig. 1). This strategy yielded four RNA isolates per replicate for sequencing: 1) Input: cortex and hippocampus homogenate RNA, 2) Microglial-TRAP (MG-TRAP): Anti-GFP ribosome affinity purification from the Input, 3) Synaptoneurosome-Input (SN-Input): Synaptoneurosome purification of the Input sample, 4) Peripheral Microglial Process-TRAP (PeMP-TRAP): Anti-GFP ribosome affinity purification from the SN-Input sample (Fig. 2a).
Figure 2: PeMP-TRAP defines transcripts enriched on peripheral microglial ribosomes.
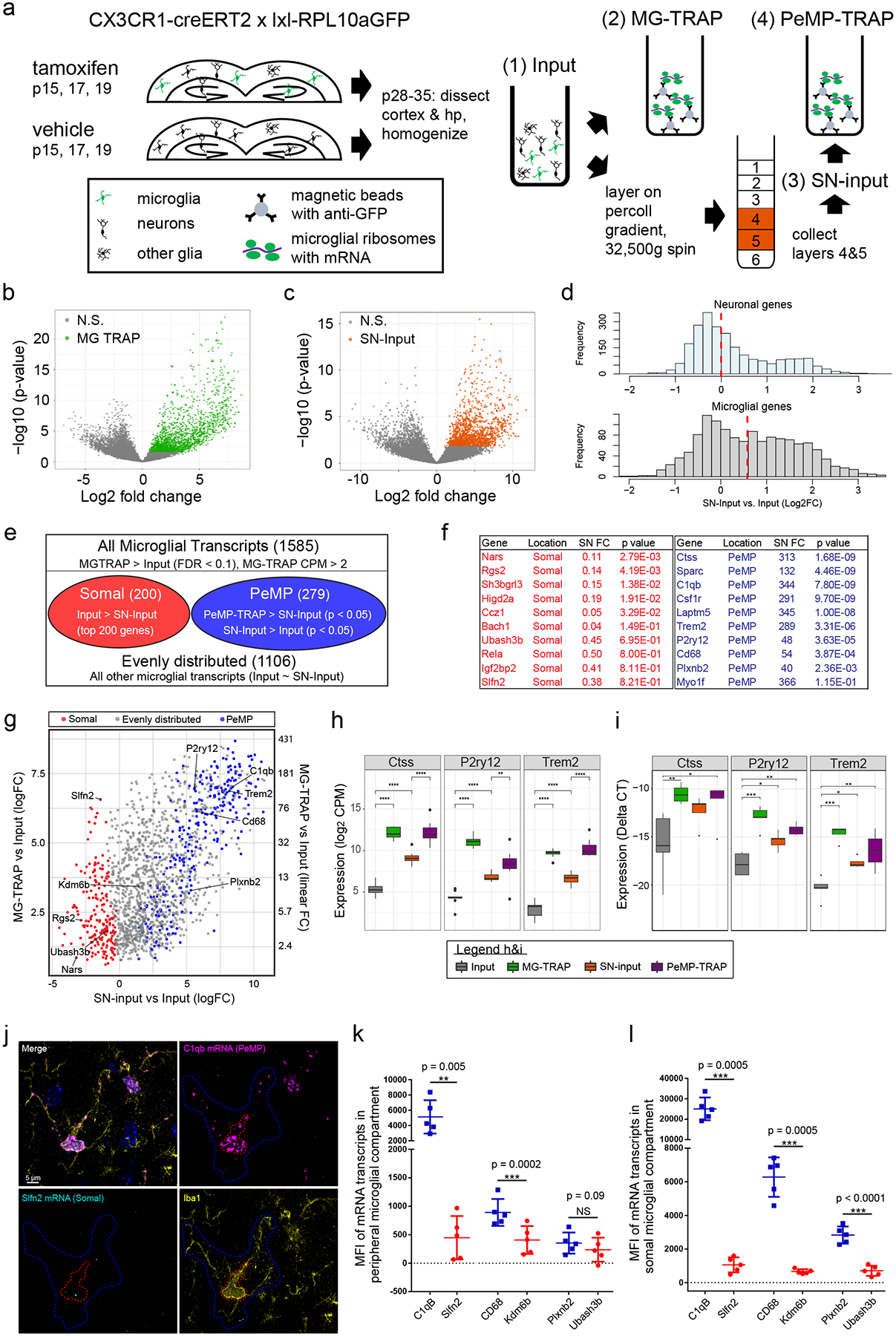
a-h, 44 p28–29 mice (pooled to n=8 replicates that received tamoxifen and n=3 that did not, from cortex and hippocampus) were harvested into four fractions(Input = tissue homogenate, MG-TRAP = Input subject to TRAP, SN-Input = Synaptosomes from Input, PeMP-TRAP = TRAP on SN-input. b-c, Volcano plots of MG-TRAP vs. Input (b) and SN-Input vs. Input (c), colored genes passed FDR correction by likelihood ratio test. d, Histograms of log2FC of SN-Input vs. Input for microglia(top), and neurons(row), red line: median. e, Definitions for PeMP-detectable (PeMP) and Somal transcripts using likelihood ratio tests for comparisons. f, SN input vs. Input table of example genes within PeMP and Somal microglial lists(FC: linear fold-change, p-values: FDR corrected likelihood ratio test). g, Scatter plot of Log2 Fold Changes (logFC) of SN-input vs. Input (x-axis) and MG-TRAP vs. Input (y-axis, linear and log2 FC both shown) highlighting example PeMP and somal genes. h, RNAseq expression levels for example PeMP transcripts showing enrichment in MG-TRAP, SN-input, and PeMP-TRAP (*p<0.05; **p<0.01; ***p<0.001 vs Input by Wilcoxon rank-sum test). Box plot elements, here and further: center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; points, data points beyond the 1.5x interquartile range. i, qRT-PCR validation of perisynaptic candidates in microglial TRAP and PeMP-TRAP vs. input (deltaCT to 18sRNA). One-way ANOVA (n=5 replicates from p28-p35 mice, 4 brains each replicate; **p<0.01, ***p<0.001): CTSS: F=5.879, p=0.00664; P2ry12: F=9.014, p=0.000995; TREM2: F=18.62, p=3.7e-5; Tukey’s HSD post hoc(*p<0.05; **p<0.01; ***p<0.001 vs Input). j, In situ hybridization (ISH) with IBA1 IF shows localization of transcripts within either peripheral (blue outline, quantified in k) or somal (red outline, quantified in l) cellular compartments. Images representative of 3 experiments, n = 5 mice and > 12 images per mouse. k-l, Mean fluorescent intensity (MFI) quantification of ISH signal (paired two-tailed T-test results shown on graph, n = 5 mice, > 14 microglia per animal, mean ± s.e.m.).
We first confirmed tamoxifen-dependent enrichment of microglial transcripts in MG-TRAP isolates vs. Cortex-Input isolates, consistent with prior ribotag (Ribosomal subunit L22-HA) studies25,26. MG-TRAP yielded a robust, tamoxifen-dependent enrichment for microglial transcripts among a panel of microglial, astrocyte, oligodendrocyte, endothelial, and neuronal markers (Extended Data Fig. 2, Extended Data Table 1)27 by RNASeq. Of note, MG-TRAP produced a slight enrichment for microglial transcripts even without tamoxifen, corroborating other studies using the CX3CR1-Cre(ERt2) line wherein ~2% of IBA1+ cells exhibit “leaky” cre activity even without tamoxifen21,25.
Comparing MG-TRAP vs. Cortex-Input, we identified 1585 transcripts significantly enriched within microglia (FDR < 0.05, MG-TRAP log2FC > 0 and log2CPM > 0) (Fig. 2b and Supplementary Table 1). Our list agrees well with published reports of microglia transcriptomes and translatomes (i.e, all 24 transcripts from Kang et al.26, all 49 from Hickman et al.28, 117 of 119 described from Haimon (Odds Ratio (OR) = 716, Fisher’s Exact Test p < 10−125)25, 487 of 885 from Ayata (OR = 26, Fisher’s Exact Test p < 10−300)17 and 342 of 465 from Zhang (OR=39, p < 10−273))27.
Additionally, to confirm SN fractionation enriched a separate synaptic transcriptome, we compared SN-Input to Input as desecribed12,13. We previously found these fractions contain fragments of a variety of cell types, including synaptoneurosomes from neurons. Consistent with this, we found significant enrichment for previously characterized synaptic-localized mRNAs such as Slc1a2, Shank3, and Gsn (Fig. 2c). Further, microglial transcripts showed a range enrichment in SN-Input over Input, consistent with a gradient of microglial RNA localization. We compared this to neuronal transcripts previously defined in a series of neuronal TRAP experiments12. Interestingly, the median SN enrichment was ~0 for neuronal transcripts, with bimodal data, consistent with a subset of mRNAs showing more localization to processes (Fig 2d). In contrast, MG transcripts are unimodal with a median of 0.67. This suggests that a larger proportion of microglial RNA overall exists in fine processes, perhaps reflecting the scant somata of microglia, especially when compared to larger neuronal somata.
Though there is a continuum of enrichment, from the microglial transcripts (1585) we identified the ~200 most “PeMP-detectable” and ~200 most somal microglial transcripts. “PeMP-detectable” transcripts (hereafter abbreviated to “PeMP transcripts”) were defined by the following two criteria 1) PeMP-TRAP > SN-Input and 2) SN-Input > Input. As a control comparison, the somal transcripts were defined by the two criteria: 1) MG-TRAP > Input and 2) Input > SN-Input (Fig. 2e–g). The PeMP transcripts (Supplementary Table 1) include several genes encoding proteins involved in microglia-neuron signaling and phagocytosis such as Ctss, CD68, C1qb, Trem2, and P2ry12 (Fig. 2f). We then validated these findings by quantitative PCR from additional mice (Fig. 2h, i), also extending these findings to slightly older ages (RNAseq p28-p29, QPCR p29-p35).
To compare our transcriptomics with spatial RNA localization, we conducted in situ hybridizations (ISH) on p28 somatosensory cortex, corpus callosum, and hippocampus tissue for PeMP and Somal transcripts in combination with IF for IBA1 (Fig. 2j–l). Each PeMP probe was paired with a Somal probe with similar RNA expression levels. All three PeMP probes (C1qB, CD68, Plxnb2) exhibited robust ISH signal within the peripheral microglia compartment (Fig. 2k). And two of these, C1qB and CD68, exhibited significantly greater peripheral MFI signal compared to paired Somal probes, Slfn2 and Kdm6b (Fig. 2k). Unexpectedly, we found that the PeMP-detectable transcripts also exhibited greater Somal ISH signal (Fig. 2l) than their paired somal transcripts. Notably, the somal compartment here includes the nucleus, while in our RNAseq protocol the nuclei were spun out, thus it can be difficult to compare the two methods directly. Likewise, two non-PeMP transcripts (Kdm6b and Ubash3b) and one PeMP transcript (Plxnb2) had considerable ISH signal also in non-IBA1+ cells (Extended Data Fig. 3), perhaps further explaining some of the disagreement between ISH and RNAseq data. Nonetheless, the higher overall levels of PeMP transcripts in all cellular compartments suggests one manner to achieve high RNA levels in the PeMP is via high levels of transcription and/or mRNA stability.
Analysis using Gene Ontologies (GO, Supplementary Table 2) of the PeMP transcript list showed that they are relatively enriched for immune response, cell motility, chemotaxis, and phagocytosis when compared against the full MG-TRAP transcript list (Fig. 3a). In contrast, the somal transcript list is enriched for transcriptional regulation, nucleoside metabolism, and RNA modification (Fig. 3b) when compared against the full MG-TRAP transcript list. Examples of microglial somal transcripts include Irf4, Rbm47, and Apobec2, which encodes an RNA-modification enzyme (Fig. 2f).
Figure 3: Distinguishing features of PeMP and somal microglial transcripts.
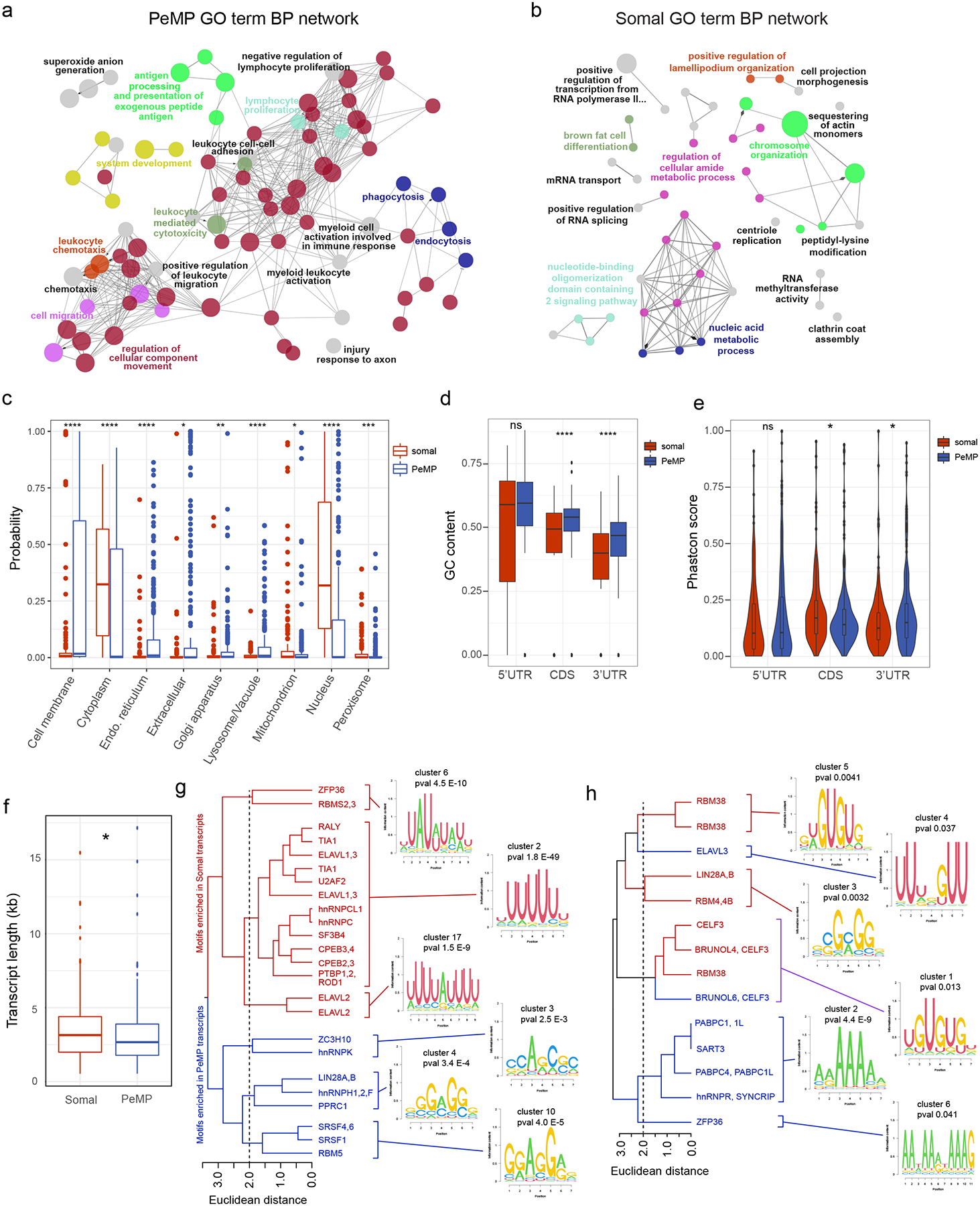
Network of connected and grouped Gene Ontology (GO) biological process terms generated using Cytoscape and Cluego plugin identified as enriched (a, p < 0.05 and b, p < 0.2 by two-sided hypergeometric test with Benjamini–Hochberg correction, n=279 PeMP and 200 somal transcripts) in PeMP (a) and somal (b) candidate lists against the full microglial background list (Supplementary Table 1), highlighting distinct functions of these transcripts. Similar GO terms within the same “tree” were grouped and share a color, but only the most significant GO term from each group is labeled (for full data, see Supplementary Table 2). c, DeepLoc analysis reveals the PeMP list is enriched in transcripts predicted to localize to membranes, endoplasmic reticulum, extracellular, golgi apparatus, and lysosomes, while somal transcripts localize disproportionately to the cytoplasm, mitochondrion, peroxisome and nucleus. c-f, p-values were generated with two-tailed Wilcoxon ranked sum tests, ns, not significant, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001, n = 279 PeMP and 200 somal transcripts). d, PeMP coding sequences (CDS, p=2.7e-05) and 3’ UTRs (p=8.6e-08) have higher GC content than Somal counterparts. e, Phastcon conservation scores were found to be higher in the somal transcripts’ CDS (p=0.039), but in PeMP transcripts’ 3’UTR (p=0.028). f, Transcript length was found to be significantly longer in Somal lists (p=0.019). g - h, Hierarchical clustering of RNA binding protein motifs discovered by Transite as significantly different (FDR-corrected Fischer’s exact test) in 3’ (g) and 5’ (h) UTRs illustrate average motif logos for the most significant (see Supplementary Table 3 for complete data and statistics) clusters found in PeMP and somal lists. Box and whisker plots (c,d,f) and violin plots (e) are defined by: center line, median; box limits, upper and lower quartiles; whiskers, 1.5* interquartile range, points, any data points outlying the box and whisker limits. Violin plots additionally include a rotated density plot on each side
Local translation in other cells supports subcellular localization of proteins to correct domains29. Therefore, if our approach was effective in microglia, we reasoned that the PeMP transcript list should encode proteins targeted for subcellular locations near the cell’s periphery such as the plasma membrane. Conversely, the somal transcript list should encode cytoplasmic and nuclear proteins. We used the Deeploc tool30, a deep learning algorithm to predict subcellular localization of proteins, to compare the PeMP to the somal list (Fig. 3c). Indeed, the PeMP transcript list had significantly more transcripts destined for the cell membrane, endoplasmic reticulum, extracellular, golgi apparatus, and of particular interest, the lysosome. Finally, we used the SignalP analysis tool31 and found that PeMP transcripts were more likely to encode secreted proteins based on greater signal peptide usage than somal transcripts (Extended Data Fig. 4).
To test the hypothesis that transcript localization may be a regulated sequence-specific process, we examined the transcripts’ 5’ untranslated region (UTR), coding, and 3’ UTR sequences to determine if sequence features differentiated PeMP from somal transcripts. Somal transcripts were slightly longer than PeMP transcripts (Fig. 3f). PeMP transcripts had higher GC content within their coding sequence and 3’UTR than somal transcripts, while GC content in the 5’UTR was not significantly different (Fig. 3d). While the coding sequence of somal transcripts was more highly conserved, 3’UTR conservation was higher in the PeMP transcript list (Fig. 3e). This suggests that preferential localization could involve sequences within the 3’ UTRs.
We next tested whether specific sequences underlie this spatial regulation. In neurons and other cells, RNA binding proteins (RBPs) bind to defined sequence motifs within UTRs to regulate mRNA localization, stability, and/or degradation. We therefore utilized the Transite set motif analysis tool32 to test for differential RBP motif usage in the 3’ and 5’ UTRs of PeMP and somal localized microglial transcripts (Supplementary Table 3). We found 46 differentially represented RBP motifs within the 3’ UTRs: 15 were more common in PeMP transcripts and 31 were more common in Somal transcripts. As motifs are often similar across RBPs, we used hierarchical clustering to group similar motifs (Fig. 3g–h). PeMP and somal RBP motifs tended to cluster with other PeMP and somal motifs, respectively.
Of particular interest, somal 3’UTRs were enriched for several AU-rich element motifs, including a UUAUUUAUU motif where ZFP36 (Tristetraproline) can bind. ZFP36 is highly expressed in microglia, suggesting it may regulate somal transcript retention or translation. RBP motifs significantly enriched within PeMP transcripts’ 3’ UTR include SRSF1 and hnRNPK. While there are too many different RBPs binding the same motifs to motivate RBP-knockout studies currently, the presence of such motifs strongly supports the conclusion that these gene sets contain distinct mechanisms of biological regulation.
Translation blockade impedes microglial phagocytosis
Since transcripts important for phagocytosis were enriched within PeMPs, we wondered if protein synthesis was necessary for PC assembly, and if key proteins of the phagolysosome are locally synthesized rather than transported from the soma.
We first examined whether translation occurs in PCs using an acute tissue injury model. Specifically, we made live 300 μm brain slices from 4–5-week-old mice. Near the surface of each slice, microglia display an injury response, assume an amoeboid morphology and phagocytize apoptotic cells and debris15. We briefly incubated the live slices with puromycin to measure protein synthesis within the first 25 μm of the surface using whole-mount immunolabeling (Fig. 4a) as described33. Microglia near the surface developed bulbous, phagocytic PeMPs which fully or partially surrounded nuclei of dying neighboring cells at 80 minutes post-slicing (Fig. 4b). We found that relative puromycin levels were higher within PeMPs with PC-like morphology (bulbous, spherical, often appearing as an IBA1+ ring) compared to PeMPs lacking these cup-like features (Fig. 4b–c). Furthermore, both puromycin MFI and ‘puromycin MFI colocalized with RPL10a-GFP’ were higher at PeMP contacts with neighboring nuclei (Supplementary Video 2) than at PeMPs with no visible contacts (Fig. 4d–e, Supplementary Video 3).
Figure 4: Translation is required for efficient formation of phagocytic structures in slice injury model.
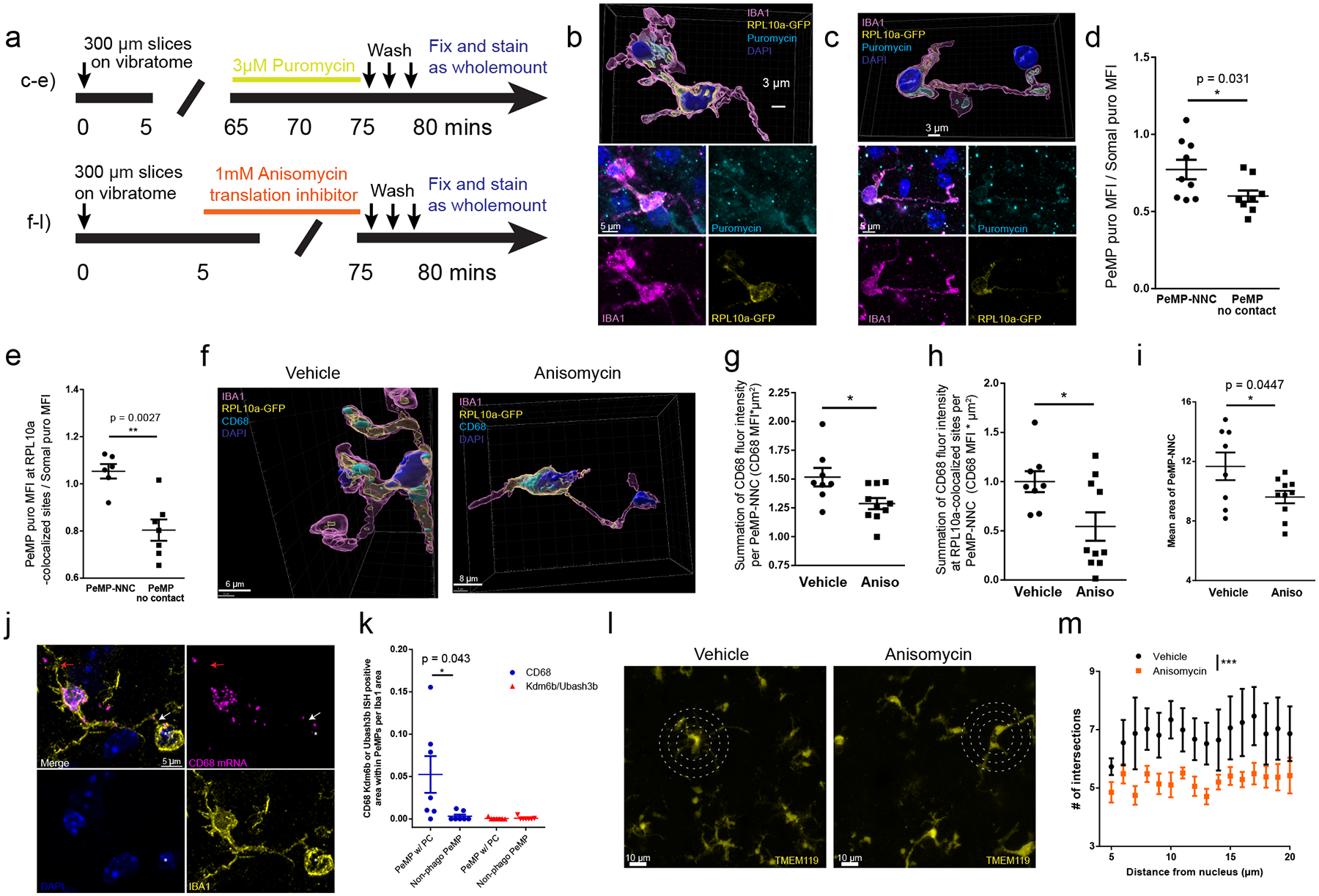
a-i and m-n All slices, with quantification of somatosensory cortex, from 4–5 week old CX3CR1-CreERt2 x RPL10a-GFP fl/fl, (b-i) or C57bl6 mice (l-m). a, Timelines for puromycin or translation inhibitor, anisomycin, for slice experiments (top-left: panels b-e, bottom-left: panels f-l). b-c, 3D (top) and maximum intensity projection (bottom) of IBA1, puromycin, GFP, and DAPI showing PeMP-neighboring nucleus contact (PeMP-NNC). b, A puromycin+ PeMP with phagocytic cup (PC) surrounding an apoptotic nucleus, (c) a non-phagocytic PeMP-NNC. d, Quantification of puromycin MFI at IBA1+ pixels (d) and IBA1+/RPL10+ pixels (e), in PEMP (1 uM radius), compared to whole image puromycin MFI. There is greater puromycin in PeMP-NNCs than in PeMPs without contacts. N = 8 slices from 3 mice, unpaired two-tailed T-test with Welch’s correction, * p = 0.031, ** p = 0.0027. f, 3D renderings from acute-injury slices with vehicle or anisomycin immunostained for CD68, RPL10a-GFP, IBA1, and DAPI. g-h, Quantification of PeMP-NNC CD68 pixel intensity (g) and CD68 pixel intensity at RPL10a-GFP colocalized sites (h) shows decreased PEMP CD68 upon translation blockade. Data were log-transformed to achieve normality and n=8 vehicle, n=10 anisomycin-treated slices from 3 animals (unpaired two-tailed T-tests, (g) p=0.027 and (h) p=0.021). i, Translation blockade impairs the mean area of PeMP-NNCs. n = 8 vehicle, n = 10 anisomycin-treated slices from 3 animals (unpaired two-tailed T-test). j, ISH from P28 WT mice brain tissue for CD68 and two non-phagocytosis associated mRNAs (Kdm6b and Ubash3b) comparing PC-shaped (white arrow) PeMPs surrounding pyknotic nuclei (asterisk) and non-phagocytic PeMPs (red arrow) quantified in (k) (7 PC-PeMPs and 7 non-phagocytic PeMPs from n = 7 microglia by paired two-tailed T-test). l, Sholl analysis of TMEM119-stained slices assessed process branching in vehicle and anisomycin-treated slices. Circles at 10, 15, and 20 μm radii. m, Quantification of (l) intersections with TMEM119 at concentric rings (5–25 μm) shows translation blockade decreases the process complexity. n = 6 slices from 2 animals per treatment group, imaging >7 microglia per slice (linear mixed model, *** p=7.98e-07). All data shown as mean ± s.e.m.
Given the elevated protein synthesis at PeMP phagocytic sites combined with our finding that transcripts involved in phagocytosis localize to PeMPs (Fig. 3a), we next tested whether a translation inhibitor would block the local accumulation of a PeMP-localized protein involved in phagocytosis. Specifically, we utilized MG-TRAP mouse acute live slices, blocked translation with anisomycin 5 minutes after sectioning, and then allowed the slices to incubate for an additional 70 minutes prior to fixation (Fig. 4a). We then performed whole-mount immunofluorescence staining for IBA1, the ribosomal subunit RPL10a-GFP, and CD68 molecule (hereafter abbreviated CD68), a microglia/macrophage specific phagolysosomal protein and PeMP transcript (Fig. 2). We outlined regions where a PeMP contacted an adjacent cell’s DAPI-stained nucleus (PeMP-NNCs) within 25μm of the slice surface. To be unbiased, we quantified every contact between a PeMP and a nucleus, regardless of morphology, in a blinded manner. Thus, we identified contacts ranging in area from 2.4 – 49 μm2 and likely includes both PeMPs contacting, but not phagocytosing neighboring somata (as described by Cserép et al.)34, in addition to PeMPs phagocytosing neighboring cells. At these PeMP-NNC sites, translation blockade significantly decreased the total CD68+ intensity (Fig. 4f, g). Translation blockade also decreased the total intensities of CD68+ at CD68 and RPL10a-GFP colocalized sites, which we measured as a proxy for de novo CD68 translation (Fig. 4f, h). Importantly, translation blockade did not affect microglial somal size, somal CD68 MFI, somal total CD68 intensity, somal RPL10a-GFP MFI, or somal sum of colocalized CD68 and RPL10a-GFP intensities (Extended Data Fig. 5 a–e). Interestingly, microglial somata supporting 2 or more larger PeMP-NNCs (> 10 μm2) did not have elevated somal CD68 compared to somata supporting either 0 or 1 PeMP-NNCs (Extended Data Fig. 5f) suggesting that somal synthesis and protein transport is not a major supplier of CD68 to PeMPs.
To determine whether new translation is required for PeMP-NNC formation, we blocked translation with anisomycin at 5 minutes post-sectioning in the live slice system. Anisomycin treatment significantly reduced the area of contact between PeMPs and neighboring nuclei (Fig 4i). Next, we sought to see if CD68 mRNA was also found near cups in uninjured tissue. We conducted ISH for CD68 on p28 WT brain tissue and were able to find 7 microglia (out of ~120 microglia) which exhibited a PeMP with a PC-like morphology surrounding a pyknotic nucleus (asterisk in Fig. 4j). CD68 mRNA signal within these phagocytic PeMPs (white arrow Fig. 4j) was higher than in non-phagocytic PeMPs (red arrow, Fig. 4j) from those same microglia (Fig. 4k). Furthermore in the same microglia, other mRNAs probed (Ubash3b or Kdm6b) were not enriched at these phagocytic PeMPs. Overall, this suggests that localization of CD68 mRNA is enhanced and production of CD68 protein is specifically upregulated within phagocytic PeMPs.
We then considered whether translational blockade influenced only PC-forming processes, or whether it also influences other morphological features. Indeed, anisomycin decreased the complexity of microglial processes (Fig 4l,m), without influencing soma size (Extended Data Fig. 5a), suggesting new translation supports process extension generally, including for phagocytosis. We also assessed microglial apoptosis during anisomycin treatment in live slices and found that expression of cleaved caspase 3 was unchanged (Extended Data Fig. 6)
To determine if these morphological changes corresponded to increased functional phagocytosis we incubated live slices from tamoxifen-injected CX3CR1-CreERT2 x TdTomato fl/fl mice with phRODO-tagged E.coli bioparticles, which fluoresce in acidic environments (e.g. phagolysosomes) enabling measurement of engulfment. We found anisomycin pretreatment impaired phagocytosis as measured by colocalization of phRODO and TdTomato (Fig 5a,b). Anisomycin treatment again did not, however, change the total area of the cells.
Figure 5: Local translation is required for efficient microglial phagocytosis of pathogen-like particles.
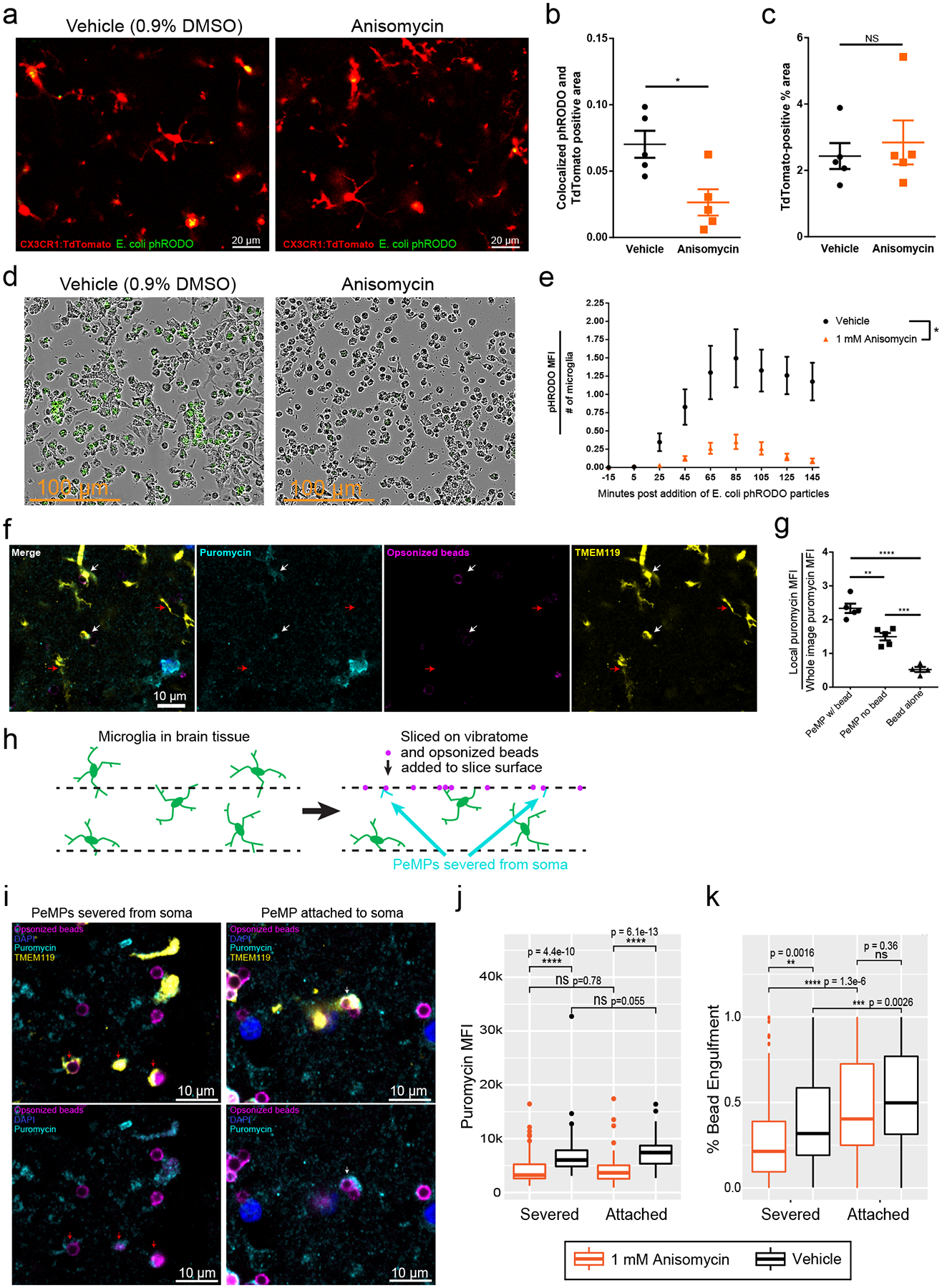
a, Representative images from tdTomato-labeled microglia phagocytosing phRODO-GFP E. coli bioparticles (green upon phagocytosis), with/without anisomycin. b, Phagocytosis of E. coli beads (phRODO-GFP&tdTomato colocalized area/tdTomato-area), is decreased by anisomycin (linear mixed model, * p=0.015, n=5 images from n=2 slices and n=2 mice/group). c, Microglial area was unchanged (N.S. by unpaired two-tailed t-test p=0.61). d, Microglial cultures pretreated with vehicle/anisomycin, then phRODO-green E. coli bioparticles, imaged every 10 minutes, (minute 85 shown - representative of 1 experiment with n=4 wells/treatment). e, Quantification of (d), phRODO-green MFI across time shows less phagocytosis with anisomycin. 3 images from each, n=4 wells (two-way ANOVA, effect of anisomycin p = 0.015, effect of time p<0.0001, interaction p<0.0001). Data are mean ± s.e.m. f, Rabbit-IgG opsonized beads added to slices (90 minutes, w/5-minute terminal puromycin), immunostained for TMEM119, puromycin, and rabbit-IgG. PeMPs contacting beads (white arrows) have greater puromycin MFI than PeMPs without contacts (red arrows), representative from 2 experiments from n = 5 mice. g, Quantification of normalized local puromycin MFI shows PeMPs contacting beads have greater puromycin than PeMPs without contacts (unpaired two-tailed T-test, n=5 slices/group **p = 0.0014, ***p=0.0002. ****p<0.0001). Data are mean ± s.e.m. h, Schematics of live slices, wherein some PeMPs are severed from somata. i, Immunostain of slices treated as in (f) showing soma-attached (white arrows) and severed PeMPs (red arrows), both containing puromycin and contacting opsonized beads. j, Quantification (puromycin MFI) shows anisomycin reduced puromycin levels in both severed and unsevered PeMPs (linear mixed model, effect of anisomycin p<2.2e-16, effect of attachment p=0.867, n=171 Anisomycin-treated PeMPs and 143 vehicle-treated PeMPs from 6 slices/group derived from 2 mice, post-hoc Wilcoxon tests shown on graph). k, Quantification of % bead area colocalized with TMEM119 shows that soma-attached status and anisomycin significantly affect % bead engulfment by microglia (linear mixed model, effect of attachment p=7.26e-10, effect of anisomycin p=0.0025, n=196 Anisomycin-treated PeMPs and 159 vehicle-treated PeMPs from 6 slices/group from 2 mice) and among severed PeMPs, anisomycin-treated PeMPs showed lower % engulfment (p = 0.0016 by post-hoc Wilcoxon test, other comparisons noted on graph).
We next tested if microglial phagocytosis’ dependence on new translation is cell-autonomous. Thus, we utilized CD11b+ bead selection to culture primary microglia from p15 rats, isolated from all other cell-types. Cultures were pre-treated with anisomycin (or vehicle), then phRODO-tagged E.coli bioparticles were added, and imaged over time. Anisomycin-treated cultures exhibited both a lower rate and lower peak of bioparticle engulfment (phRODO MFI) per microglia (Fig 5d,e, Supplementary Videos 4–5).
We also confirmed that the elevated protein synthesis at phagocytic contacts (e.g., Fig. 4b–c) was occurring within microglia rather than within their targets. We prepared live slices from WT mice and incubated them with rabbit-IgG-opsonized beads (60 minutes), followed by puromycin (5 minutes). PeMPs contacting/engulfing beads had greater levels of puromycin than PeMPs without contacts (Fig. 5f–g). Since beads have no translation, and background staining of puromycin at non-contacted beads is low (Fig. 5g), this indicates the increased puromycin in the phagocytic PeMP is not coming from the phagocytic target.
Finally, upon examination of slices’ surfaces, we made an intriguing observation: there were PeMPs surrounding opsonized beads that were not connected to microglial somata, presumably severed during the slicing process (Fig. 5h, Supplementary Video 6) thus providing a clear opportunity to understand the requirement for local translation in PeMP-autonomous behavior. Separately analyzing these severed PeMPs revealed those contacting opsonized beads had higher puromycin signal by immunofluorescence than severed PeMPs lacking bead contacts (Fig 5i,j), consistent with PeMP autonomous local translation. Finally, examining only the severed PeMPs and comparing anisomycin-treated vs. vehicle-treated slices revealed that translation blockade resulted in lower percent engulfment of opsonized beads (Fig. 5k, Supplementary Videos 7–8). Thus, we conclude that there is a local upregulation of protein synthesis within PeMPs near contacts with phagocytic targets, and that microglial protein synthesis is necessary for efficient phagocytosis to occur. Furthermore, local protein synthesis-dependent phagocytosis occurs even within PeMPs severed from the soma.
Discussion
Here, we show that distal microglial processes contain ribosomal subunits, mRNA transcripts, and synthesize new proteins. These PeMP-associated ribosomes are enriched with particular microglial mRNAs, biased towards pathogen defense, phagocytosis, and cell motility. Furthermore, PeMP-detectable transcripts are shorter, have higher GC content within coding and 3’ UTRs, and encode more membrane, lysosomal, endoplasmic reticulum, and secreted proteins. Using an acute tissue injury model, we further show that protein synthesis is abundant within microglial phagocytic structures. Translation blockade impairs phagocytic structure formation, particle engulfment, and localization of lysosomal protein CD68. Finally, severed PeMPs can surround opsonized beads in a process dependent upon protein synthesis. Taken together, we describe a mechanism by which microglia may translate distinct sub-pools of protein transcripts in specific processes to carry out specific functions, including the promotion of phagocytosis.
While individual transcripts may have distinct localization mechanisms, our study offers some insight into how PeMP transcripts may enrich in distal processes. First, compared to neurons, resting microglia have a much smaller somal volume, which may explain the greater proportion of microglial transcripts that are PeMP-expressed and enriched by SN-fractionation. When examined by ISH, the PeMP-detectable transcripts were higher expressed overall, in both peripheral processes and somata. This argues against selective mRNA transport as the main mechanism for most mRNAs and instead suggests that greater expression and/or stability may be essential. Comparing ISH data to PeMP-TRAP data also suggests that translation of specific transcripts within PeMPs could be a more selective mechanism for microglia than mRNA localization.
Interestingly, the coding sequence of PeMP transcripts was less conserved than somal transcripts The lower coding sequence conservation could reflect the abudance of genes involved in pathogen defense, which include many of the most rapidly evolving loci of mammalian genomes (e.g. MHCs, Fc Receptor genes, which respond to evolving pathogens)35,36. In contrast, the 3’UTR of PeMP transcripts was more conserved than somal transcripts - suggesting selection for localization to the cell membrane, secretory pathway, or lysosome via certain 3’ UTR sequences. One limitation is that for transcripts containing with >1 isoform, we utilized the longest transcript available for PeMP vs. Somal transcript feature comparisons. In the future, long-read mRNA sequencing may better inform actual isoform usage. Another caveat is that we were conservative in our criteria for PeMP-ribosome enrichment, requiring enrichment well above the level of the known non-specific background in TRAP experiments37. Thus, there may be additional transcripts discovered as TRAP methods refine.
UTRs of PeMP and somal transcripts are enriched in specific motifs including potential binding sites for RBPs. RBP motifs containing AU-rich elements including ELAVL1 (HuR), TIA1, and ZFP36, RBMS2, and RALY, were more abundant in somal 3’ UTRs. These AU-rich elements, and their corresponding RBPs, play key roles in negatively regulating the stability and translation of transcripts involved in DNA damage, cell cycle, and pro-inflammatory pathways38. These include important gatekeepers of inflammation such as Rela, a component of the NF-kb transcription factor complex, and interferon regulatory factors IRF2, IRF4, and IRF9. This suggests many somal transcripts are retained close to the nucleus via such RBPs. Motifs enriched within 3’ UTRs of PeMP transcripts include those predicted to bind SRSF1 and hNRNPK. SRSF1, in concert with IRAK2, regulates the nuclear export of certain proinflammatory mRNA transcripts39. hNRNPK has been found to localize to macrophage podosomes40; small extrusions involved in extracellular matrix remodeling, chemotaxis, and process motility. The enrichment of these motifs suggests these RBPs play similar roles within microglia.
Prior proteomics and localization studies of purified phagosomes from macrophages, which share lineage with microglia, may help interpret how local translation might aid phagocytosis. An electron microscopy study showed that the maturing macrophage phagosomal membrane contacts endoplasmic reticulum41, and a radiolabeling study revealed that several newly-synthesized ER-associated proteins become incorporated into the phagosomal membrane42. Furthermore, a mass-spectrometry study identified 19 different ribosomal subunits, including RPL10a, enriched within purified phagosomes43. Several other proteins enriched within purified phagosomes are encoded by PeMP transcripts (Figure 2e–f, Supplementary Table 1) including CD68, the lysosomal hydrolases legumain (LGMN), and cathepsins D, S, and Z (CTS-D,S,Z)43. Another PeMP transcript Myosin 1f, is important for sealing the PC around its target44. Ideally, we would block translation of this transcript specifically in just one process to further test the essentiality of local translation of this protein for cup sealing, however such methods do not currently exist. Nonetheless, taken together our results suggest that local protein synthesis near PCs is necessary for their maturation and function within microglia.
Why might microglia utilize local protein synthesis for phagocytosis? We speculate that since phagocytosis is a location-specific process, local protein synthesis could allow a single microglial process to phagocytose a nearby apoptotic cell while simultaneously contacting dozens of other healthy cells. In addition, while many microglial phagocytic proteins are expressed at low levels within healthy adult brains, when needed, rapid protein production is essential. Thus, local translation could be beneficial during brief windows of development, infection, or disease. In our tissue injury slice model, CD68 is already localized to cups at 80 minutes post-injury, which would be rapid for transcription, capping, polyadenylation, splicing, translation, and protein transport to all occur. Thus, it is possible transcripts pause on ribosomes for transport, enabling regulated unpausing for on-demand protein production, as demonstrated in neurons during mGluR-mediated long term depression45. In support of this hypothesis, our ISH in healthy brains revealed CD68 mRNA within many non-phagocytic PeMPs, but levels were increased within phagocytic PeMPs.
Beyond phagocytosis, transcripts relatively enriched in PeMPs identified several additional biological processes, including cell motility, cell migration, immune response, and pathogen defense. Indeed, anisomycin also decreased process complexity in general, suggesting new translation is important for process-extension generally. Thus, it would be of interest to determine if motility of distal microglial processes during homeostatic surveillance7 is dependent upon local protein synthesis, and whether this modulates synapses and/or microglial-somal interactions34. Furthermore, while we focused on healthy young mice, changes may occur within the PeMP-translatome during CNS disease, aging, or infection where microglial activation, morphology, and/or phagocytosis are altered, such as Alzheimer’s Disease, Parkinson’s Disease, or the maternal immune activation model of Autism. An examination of local transcripts may identify key biological processes occurring at specific sites (e.g., at microglial contacts with misfolded protein deposits or synapses) despite little change to total cellular levels of transcript.
Methods
Animals
All procedures involving animals were approved by the Washington University Institutional Animal Care and Use Committee (Protocol #20–0097). Mice or rats were group-housed and maintained in the vivarium at Washington University in St. Louis. The colony room lighting was 12:12 hr light/dark cycle; room temperature (~20–22°C) and relative humidity (50%) controlled automatically. Standard laboratory diet and water were freely available. We utilized 4–5 week old male and female C57Bl6/J (WT) or CX3CR1-Cre(ERt2) mice (official name: B6.129P2(C)-Cx3cr1tm2.1(cre/ERT2)Jung/J, JAX strain 020940) crossed to cre-dependent TRAP mice (official name B6.129S4-Gt(ROSA)26Sortm1(CAG-EGFP/Rpl10a,-birA)Wtp/J, JAX strain 022367) or cre-dependent tdTomato “Ai14” mice (official name: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J, JAX strain 007914). P11 Sprague Dawley (Taconic) rat pups were utilized for the primary microglial in vitro culture experiment. Both male and female mice or rats were used for all experiments within this study, but the study design was not powered to detect effects of sex. For PeMP-TRAP RNAseq specifically, male and female brain tissue was pooled together in each replicate(4 brains each) to provide enough yield for downstream analyses. For all brain-IF, slice-IF, ISH, and TRAP experiments, sets of animals from at least two separate litters were used, with littermate controls whenever possible.
Tamoxifen administration
The experimental cohort of mice was administered 100 mg/kg tamoxifen (Sigma T5648) intraperitoneally on postnatal days 15, 17, and 19, while a control cohort was administered vehicle (9:1 Sunflower oil: ethanol).
Electron microscopy
Brains from mice perfused with 4% paraformaldehyde (15710, Electron Microscopy Sciences) in PBS were fixed overnight, cut coronally a Leica VT1200S vibratome at 100 μm, and washed in PBS (3 × 10 min), incubated in.3% hydrogen peroxide for 15 min, washed in PBS (3 × 10 min), and placed into 1.0% sodium borohydride (NaBH4) for 15 min. After washing in PBS (3 × 10 min), slices were incubated with agitation overnight in biotinylated anti-GFP (AB6658, Abcam) diluted 1:1000 in 2% bovine serum albumin (BSA) in PBS at 4° C, then washed in PBS (3 × 10 min), incubated shaking for 45 min in ABC Elite kit solution (PK-6100, Vector Laboratories), washed in PBS (3 × 10 min), 0.1 M sodium acetate buffer (2 × 5 min), then incubated in nickel DAB solution (5.0 % Nickel ammonium sulfate in 0.2 M acetate buffer, 4 ml double deionized water, 1.0 ml DAB (5mg/ml) and 0.5–1.0 mg glucose oxidase) for 2–5 min, then washed in PBS (3 × 10 min), and post fixed in reduced osmium (1.0% OsO4, 1.5% KCN in PBS). After washing with MQ water (3 × 10 min), they were stained with 2% uranyl acetate for 1 hr in the dark, washed again in water (2 × 5 min), and dehydrated in acetone series (50%, 70%, 90% 100% × 2 – 10 min each). Infiltration was microwave assisted (350W with vacuum) in three steps of 3 min duration (50%, 100% X2). Samples were embedded between two glass microscope slides previously treated with Teflon (MS-143XD, Miller-Stephenson) and cured at 60° C for 48 hrs. Samples were sectioned at 70nm on a Leica UC7 ultramicrotome, picked up on formvar/carbon coated grids (FCF-100-CU, Electron Microscopy Sciences), and viewed in a JEOL JEM 1400 plus TEM at 120 kV accelerating voltage.
Immunolabeling of PFA-fixed frozen brain sections
Additional mice were perfused as described above, dissected, and postfixed for 24 hours. Brains were transferred to 30% sucrose solution for 48 hours, frozen in OCT (ThermoFisher #23-730-571) and then cryosectioned to 10 μm and stored at −80°C until use. We incubated slides with blocking solution (5% donkey serum, 0.1% Triton-X 100 in PBS) for 1 hour in a humidified chamber at room temperature, then with primary antibodies overnight at 4°C. Primaries included goat anti-IBA1 (1:200), rat anti-CD68 (1:200), and mouse anti-Synaptophysin (1:50). Following this incubation, slides were washed 3x in PBS, and incubated with donkey anti-species specific alexa fluor-conjugated secondary antibodies (all at 1:400 dilution) at room temperature for one hour in blocking solution. Slides were washed 3x, then nuclei were counterstained with DAPI at 1:20,000 dilution for 5 minutes, washed once, then mounted with Prolong Gold anti-fade mounting media (ThermoFisher #P36934).
Acute, live coronal brain slice preparation and treatment with puromycin and/or translation inhibitors
A vibratome was used to cut 300 μm-thick coronal sections through the somatosensory cortex in ice-cold slicing buffer (125 mM NaCl, 25 mM glucose, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 3 mM MgCl2). Slices were then allowed to temporarily recover (time denoted within figures) in oxygenated, room temperature artificial cerebrospinal fluid (ACSF): 125 mM NaCl, 25 mM glucose, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 3 mM MgCl2, and equilibrated with 95% oxygen and 5% CO2.
After recovery, slices were transferred to freshly oxygenated ACSF at 37°C with a gentle ~20RPM shake. The translation inhibitor anisomycin (Sigma #A9789) was added to some slices at either 5 or 45 minutes post-slicing as noted in figures 1f and 4a. To label de novo protein synthesis, live slices were incubated at 37°C with 5 or 10 minute terminal pulse with 3 μM puromycin (Tocris #40-895-0) as noted in figures 1f and 4a.
Confocal microscopy
We performed confocal microscopy using an LSM 700 AxioImager Z2 (Zeiss) with either a 40x or 63x magnification oil lens and an average of 1–3 microglia captured per image. Where noted in the figure legends, Z-stacks were acquired.
Quantification of puromycin within concentric circles radiating from soma
Custom ImageJ macros were used to draw concentric circles around the middle of a phagocytic cup (PC) with radii increasing by 3 μm. We created a thresholded IBA1+ cell mask and within each concentric circle and measured the mean intensity of puromycin signal at sites where it colocalized with IBA1 signal, divided by the area of IBA1+ pixels within the region.
Immunolabeling of 300μm whole-mount coronal brain slices adapted from Dissing-Olesen and MacVicar33
Each live brain slice was washed three times (2 minutes per wash) with 1 mL of ACSF on a platform shaker, then fixed by a 30 minute incubation with 1 mL of pre-warmed 4% PFA at 37 °C, and then stained as described33. Briefly, fixed slices were rinsed twice in PBS, then permeabilized for 2 hours in (2% Triton X-100, 20% DMSO in PBS), and blocked with 5% donkey serum and 5% goat serum in the same solution, overnight at 60 RPM at room temperature. Each section was then placed individually in a sealed custom small plastic packet containing 100 μL of a primary antibody solution with mouse anti-puromycin, rabbit anti-IBA1, and rat anti-CD68 (Refer to antibodies table for dilutions) in permeabilization solution with 2% donkey serum and 2% goat serum, mixed for four days at 4°C on a 360° rotisserie wheel. Sections were then washed once per hour over 4 hours with permeabilization solution followed by a two-day incubation with secondary antibody staining solution (1:400 goat anti-rabbit-Alexa-555, 1:400 goat anti-mouse IgG1-Alexa488, and 1:400 donkey anti-rat-Alexa647) in the dark. Sections were then washed 2 additional times with permeabilization solution, stained for 20 minutes with a 1:20,000 solution of DAPI (Sigma D9542) in permeabilization solution, then washed in permeabilization solution again. Immunolabeled sections were then mounted on slides as previously published33 except 2–3 drops of a 90% glycerol/10% PBS solution was added on top of slices before cover-slipping and sealing with nail polish.
Antibodies
| Goat anti-IBA1 | Abcam | ab5076 | IF | 1 in 200 | GR268568–3 |
| Rabbit anti-IBA1 | ThermoFisher | NC9288364 | IF | 1 in 300 | |
| Rabbit anti-IBA1 | Synaptic Systems | 234 003 | ISH-IF | 1 in 200 | |
| Mouse anti-synaptophysin | Abcam | ab8049 | IF | 1 in 50 | GR305661–1 |
| Mouse anti-synaptophysin | Abcam | ab8049 | WB | 1 in 1000 | GR305661–1 |
| Chicken anti-GFP | Aves labs | GFP-1020 | IF | 1 in 1000 | GFP697986 |
| Rabbit anti-GFP | Cell Signaling | 2555S | WB | 1 in 1000 | |
| Mouse anti-GFP 19c8 | Memorial Sloan Kettering ab core | AB_2716736 | TRAP | 50 ug per TRAP replicate | |
| Mouse anti-GFP 19f7 | Memorial Sloan Kettering ab core | AB_2716736 | TRAP | 50 ug per TRAP replicate | |
| Goat anti-GFP (biotinylated) | Abcam | AB6658 | EM | 1:1000 | |
| Rabbit anti-Lamin B1 | Abcam | ab16048 | WB | 1 in 500 | |
| Rabbit anti-MeCP2 | Cell Signaling | 3456 | WB | 1 in 1000 | |
| Rat anti-CD68 | eBiosciences | 14-0681-82 | IF | 1 in 200 | |
| Mouse anti-puromycin | Kerafast | EQ0001 | IF | 1 in 500 | |
| Rabbit anti-RPS16 | Sigma | SAB1100505 | IF | 1 in 1000 | |
| Guinea Pig anti-TMEM119 | Synaptic Systems | 400 004 | IF | 1:400 | |
| Rabbit anti-Cleaved caspase 3 | Cell Signaling | 9664 | IF | 1:500 | |
| Donkey anti-mouse alexa546 | Thermofisher-Invitrogen | a10036 | IF | 1 in 400 | |
| Donkey anti-chicken alexa488 | Jackson Immuno | 703-545-155 | IF | 1 in 400 | |
| Donkey anti-rat alexa647 (cross adsorbed) | Jackson Immuno | 712-605-153 | IF | 1 in 400 | |
| Donkey anti-rabbit alexa546 | Invitrogen | a10040 | IF | 1 in 400 | 1504518 |
| Donkey anti-goat alexa647 | Jackson Immuno | 705-605-003 | IF | 1 in 400 | |
| Goat anti-mouse alexa488 (IgG1 specific) | Invitrogen | A21121 | IF | 1 in 400 | |
| Donkey anti-guinea pig alexa647 | Jackson Immuno | 706-605-148 | IF | 1 in 400 | |
| Donkey anti-rabbit alexa647 | Invitrogen | a-31573 | IF | 1 in 400 | |
| Goat anti-mouse alexa647 (IgG1 specific) | Invitrogen | A21240 | IF | 1 in 400 | |
| Goat anti-rabbit alexa555 | Invitrogen | A21428 | IF | 1 in 400 |
Quantification of puromycin, CD68 and RPL10a-GFP incorporation within microglial phagocytic structures
Following wholemount immunostaining for IBA1, CD68, and either RPL10a-GFP or puromycin, Z-stack images (every 0.66μm) were taken with an LSM 700 AxioImager 2 (Zeiss) with a 63x oil lens within the first 25μm from the slice surface. We used the polygon tool in ImageJ to demarcate the perimeter around a PC using the IBA1 channel. The mean fluorescent intensity (MFI) of puromycin in a PC was measured by dividing the mean puromycin signal within the PC by the fraction of the PC area containing IBA1 signal. We accounted for differences in background puromycin signal by dividing the local puromycin MFI of each PC to the mean puromycin signal throughout the entire image because puromycin signal inversely scales with depth from slice surface.
Quantification of CD68 and RPL10a-GFP incorporation at microglia - neighbor nuclei contacts
Following wholemount immunostaining for IBA1, CD68, RPL10a-GFP, and nuclear stain DAPI, we used the polygon tool in ImageJ to demarcate the perimeter of any IBA1+ process contact with a DAPI+ nucleus (excluding any contacts within 5μm of the microglial soma). The mean fluorescent intensity (MFI) of puromycin within the PeMP was measured by dividing the local mean puromycin signal by the fraction of the cup area containing IBA1 signal. We accounted for differences in background puromycin signal by dividing the local puromycin MFI of each PeMP by the puromycin MFI throughout the entire image.
Synaptoneurosome fractionation and Translating Ribosome Affinity Purification (TRAP)
A dissection containing the dorsal cortex and hippocampus from p28-p29 MG-TRAP mice with or without tamoxifen administration (as above) was quickly isolated and added to an ice-cold homogenization buffer as previously described13. All reagents and buffers used are previously described24. Each replicate contained tissue from 4 mixed sex mice and was homogenized in 3.5 ml ice-cold homogenization buffer using a 7mL glass homogenizer (10 strokes each pestle, Kontes), and spun at 1000 × g in a Sorval RT7 for 10 min at 4°C. The supernatant from each replicate was split into two samples: For the input and MG-TRAP samples, 500 μl of the supernatant was incubated with 50 μl of Salt lysis buffer for 15 min and spun at 20,000 × g for 15 min at 4°C to reduce cell debris. The remaining supernatant (2 ml), was layered over a discontinuous 3–23% sucrose-Percoll gradient and spun at 32,500 × g for 5 min as described23. The synaptoneurosome band was collected by puncturing the bottom of the tube, and collecting fractions 4–5. Then, 90% of the Input and SN-Input samples (reserving 10% to purify and sequence the inputs) underwent TRAP for affinity purification of ribosomes, as described previously12. Briefly, samples were incubated with anti-eGFP-coated biotinylated, streptavidin MyOne T1 magnetic beads (ThermoFisher #65602) using 30 μl beads and 50 μg each of two anti-GFP antibodies (clone 19f7, 19c8, Memorial Sloan Kettering Cancer Center antibody core) per sample. After a 4 h incubation at 4°C, samples were washed using a high salt wash and resuspended in 0.15M KCl IP wash buffer. RNA from all input and TRAP samples were then isolated and purified using a Qiagen RNEasy MinElute kit.
Western Blotting
Sample protein concentration was determined via BCA assay and equal protein concentrations were loaded per lane. 15μl sample was combined with 15μl aliquot Laemmli Sample Buffer (Bio-Rad) with 5% beta-mercaptoethanol and boiled for 5 minutes. The electrophoresis chamber was filled with 1X SDS-PAGE Running Buffer. 30uls were loaded per well onto Mini-PROTEAN TGX gel (Bio-Rad CAT# 4561023) and electrophoresed at 80V for 10 minutes, then 120V for 1 hour. PVDF membrane was activated in 100% methanol for 10 minutes then washed in DI water, then transfer buffer (1x SDS-PAGE Running Buffer without SDS, 20% methanol) for 10 minutes. Gel was washed 2x in transfer buffer and transferred onto a PVDF membrane with a Trans-blot SD Semi-Dry transfer cell (Bio-Rad) and ran at 15V for 20 minutes. Transfer was confirmed via Ponceau stain. Membrane was washed 2x for 5 minutes in 1X TBST, then blocked in 5% Milk in 1x TBST (Blotting-Grade Blocker, Bio-Rad) for 1 hour at room temp. Membrane was incubated in 15mL of primary antibody in block containing 1:1000 mouse anti-synaptophysin (Abcam AB8049), or 1:1000 rabbit anti-GFP (Cell Signaling 2555S), or 1:500 rabbit anti-Lamin-B1 (abcam ab16048), or 1:1000 rabbit anti-MeCP2 (Cell Signaling 3456) over night at 4°C. 3 washes were completed in 1X TBST, 5 minutes each. The membrane was then incubated in 15mL of TBST with 5% milk plus secondary antibody - 1:2000 goat anti-mouse IgG HRP (Bio-Rad 170–6516) or 1:2000 anti-rabbit IgG, HRP-linked (Cell Signaling 7074S), respectively, for one hour at room temp. 3 washes were completed in 1X TBST, 5 minutes each. Membrane was visualized using myECL™ Imager (Thermo Scientific 1862591). The synaptophysin membrane was incubated in 1:1 mixtures of Clarity™ Western ECL Substrate (Bio-Rad CAT# 170–5060) in the dark for 5 minutes before imaging and exposed for up to 5 minutes. The GFP membrane was incubated in 1:1 mixtures of Super Signal™ West Femto Maximum Sensitivity Substrate (Thermo Scientific 34095) in dark for 5 minutes before imaging and exposed for up to 30 minutes in imager.
Library Preparation and RNA-sequencing
First batch (n=3 replicates): RNA was assessed for quality and concentration using an Agilent RNA 6000 Pico Kit on an Agilent Bioanalyzer. RNA samples were reverse transcribed into cDNA and amplified using Smartseq v4 Ultra Low Input RNA Kit for Sequencing (Takara #634889), per the manufacturer’s instructions. The following numbers of amplification cycles were used based on the concentration of input RNA per sample: 15 cycles for 63pg RNA input, 14 cycles for 125pg RNA input, 13 cycles for 250 pg RNA input, and 12 cycles for 500 pg RNA input. cDNA was then fragmented using a Covaris E210 sonicator using duty cycle 10, intensity 5, cycles/burst 200, time 180s, and a target insert size distribution around 200 bp was verified using an Agilent Tapestation 4200 and High Sensitivity D1000 ScreenTape and buffer (Agilent #5067–5585). Sequencing adaptors were then added on using NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs #E7645S) following the manufacturer’s instructions for a 200 bp insert size distribution and using AMpure XP beads (Beckman Coulter, #A63881) for size selection and purification. Libraries were then normalized and sequenced on an Illumina HiSeq3000 as single reads extending 50 bases, by the Genome Technology Access Center at Washington University.
Second batch (n=5 replicates): Total RNA integrity was determined using Agilent Bioanalyzer. Library preparation was performed with 10ng of total RNA. ds-cDNA was prepared using the SMARTer Ultra Low RNA kit for Illumina Sequencing (catalog#634828, Takara-Clontech) per manufacturer’s protocol. cDNA was fragmented using a Covaris E220 sonicator using peak incident power 18, duty factor 20%, cycles per burst 50 for 120 seconds. cDNA was blunt ended, had an A base added to the 3’ ends, and then had Illumina sequencing adapters ligated to the ends. Ligated fragments were then amplified for 15 cycles using primers incorporating unique dual index tags. Fragments were sequenced on an Illumina NovaSeq-6000 using paired end reads extending 150 bases.
Sequencing results from both batches were quality checked using FastQC version 0.11.7. Illumina sequencing adaptors were removed using Trimmomatic version 0.3846. Reads aligned to the mouse rRNA were removed by bowtie2 version 2.3.547. The remaining RNA-seq reads were then aligned to the mouse assembly (Ensembl 93) with STAR version 2.5.1a48. The number of uniquely mapped reads to each feature were counted using htseq-count version 0.9.149. Differential expression analysis was done using edgeR version 3.32.050. Only genes with CPM > 1 in at least 3 out of 24 replicates were retained for further analysis. A negative binomial generalized log-linear model (GLM) was fit to the counts for each gene. Then the likelihood ratio tests (LRT) were conducted for each comparison.
Quantitative PCR
To validate RNA transcript enrichment by PeMP-TRAP, five additional independent biological replicates of Input, TRAP, SN-input, and PeMP-TRAP were collected from p28-p35 animals as described above. cDNA samples were synthesized from RNA samples using Quanta qScript Reverse Transcriptase kit (QuantaBio, cat. no. 84002). Three technical replicates of each of the samples were quantified using iTaq Universal Sybrgreen (Bio-Rad, cat. no. 1725120) on QuantStudio 6 Flex (Applied Biosystems) in a 10 μL volume. To quantify enrichment, delta CT was calculated using 18S as an endogenous control. Data was analyzed using one-way ANOVA with Tukey’s HSD post-hoc test in R statistical software. Samples with undetermined amplification values were excluded from statistical analyses. The following primer sequences were used:
18s: F- GGGAGGTAGTGACGAAAAATAACAAT, R-TTGCCCTCCAATGGATCCT
CTSS: F-CCATTGGGATCTCTGGAAGAAAA, R-TCATGCCCACTTGGTAGGTAT (primer bank ID 10946582a1)
P2Ry12: F-ATGGATATGCCTGGTGTCAACA, R-AGCAATGGGAAGAGAACCTGG
Trem2: F-CTGGAACCGTCACCATCACTC, R-CGAAACTCGATGACTCCTCGG
In situ hybridization (ISH)
Brains from p28 C57Bl/6 mice were perfused with PBS and 4% PFA, cryoprotected, frozen in OCT, and sliced to 10 μm thickness, slide-mounted, and stored at −80 degrees for up to 4 weeks. Manufacturer’s instructions were followed using the RNAscope multiplex fluorescent v2 assay (Advanced Cell Diagnostics-ACD, Intro kit for new users, catalog #323136) for sequential ISH then immunostaining (Technical note: 323100-TN/Rev A) with a few modifications. The following steps were omitted from the protocol: 60 degree incubation, ethanol dehydration series, and boiling in co-detection target retrieval reagent. Protease plus was applied for 12 minutes and ISH probes were visualized using either Opal 570 or 650 dyes (Akoya Biosciences, FP1488001KT and FP1488001KT) at 1:1200 in the multiplex TSA buffer from the RNAscope multiplex fluorescent v2 kit. Following completion of the multiplex fluorescent v2 assay, slide-mounted sections were incubated for 15 minutes in blocking buffer consisting of co-detection antibody diluent (ACD #323180) with 5% normal donkey serum, then incubated overnight in blocking buffer plus rabbit anti-IBA1 antibody (Synaptic Systems #234003), washed three times in PBS, incubated for 1 hour in blocking buffer with 1:400 donkey anti-rabbit alexafluor 488 (Invitrogen), washed three times in PBS, incubated for 5 minutes with DAPI, and mounted with prolong gold antifade mount (Thermo Fisher #P36930).
Mus musculus RNAscope ISH mRNA probes and 3-plex negative control probes (catalog# 320871) were purchased from Advanced Cell Diagnostics and paired as follows:
| Pair | PeMP probes | Channel | Catalog # | non-PeMP probes | Channel | Catalog # |
|---|---|---|---|---|---|---|
| A |
C1qb Complement C1q-B chain |
C2 | 438101-C2 |
Slfn2
schlafen 2 |
C1 | 466671 |
| B |
CD68 CD68 antigen |
C2 | 316611-C2 |
Kdm6b
lysine demethylase 6B |
C3 | 477971-C3 |
| C |
Plxnb2 Plexin B2 |
C1 | 459181 |
Ubash3b ubiquitin associated and SH3 domain containing B |
C3 | 1045631-C3 |
These were imaged using a Zeiss LSM700 Axioscan2 microscope and a 63x oil lens in a confocal Z-stack consisting of 5 Z-slices (0.66μm step size) centered on the midpoint of a microglial soma. Fpr analysis we outlined the periphery and soma of each IBA1+ cell as depicted in fig 2j, masked on IBA1+ area, and calculated the MFI of ISH signal within each microglial compartment comparing each “PeMP-detectable” probe with its paired “non-PeMP” probe. Negative control probes (Advanced Cell Diagnostics, catalog# 320871) were used to determine the background levels of non-specific signal within each channel and this background signal was subtracted from the measured MFI to obtain the background-corrected MFI.
Gene Ontology (GO) term analysis and nodal networks
Following RNA sequencing, microglial somal and PeMP gene lists (Supplementary Table 1) were analyzed for GO term biological process (updated 12/10/2021) using the cluego plugin (v2.5.8)51 within the cytoscape application (v3.7.1) and using the full microglial transcript list (Extended Data Fig. 3) as the custom background. We then looked for enriched GO terms within the Somal and PeMP lists compared to the full microglial gene list via the built in two-sided hypergeometric test and Benjamini–Hochberg post-hoc correction with a cutoff of p < 0.05. A kappa score threshold of 0.4 was used to determine network connectivity. GO term tree levels included were minimum of 4 and maximum of 8. Similar GO terms were grouped with the most significant term displayed within a tree of minimum 2 common and maximum 5 different parents. Groupings are indicated by a shared color on the force directed nodal networks (Figure 3a–b and Supplementary Table 2). The nodal network graphic for PeMP transcripts was restricted to GO terms at p < 0.005 to fit the terms within the graphic.
RBP motif discovery using Transite transcript set motif analysis
RBP motifs were identified by entering transcript lists into the Transite transcript set motif analysis online tool (transite.mit.edu)32. We separately analyzed the 5’ and 3’ UTRs of PeMP and Somal gene lists using a matrix-based analysis, with the full MG-TRAP list as the background. Settings used were 50 maximum binding sites per mRNA, the transite motif database, and p-value adjustment with Benjamini-Hochberg. Enrichment scores for RBP motifs in the PeMP and Somal lists were then compared with Fisher’s exact test and false discovery rate correction to determine RBP motifs which were differentially represented within the PeMP and somal lists. The differentially represented RBP motifs were hierarchically clustered using vectorized position probability matrices (PPMs) for each motif and the hclust function with the “complete” method in R. Motif logos for each cluster were generated by seqLogo52 using the average PPMs across all cluster members.
Transcript feature identification
To compare transcript features, we queried the ensembl database for available transcripts corresponding to the ensembl gene IDs used to identify the genes in the somal and PeMP gene lists. For each gene in the lists we selected the longest available transcript to represent each gene. We then converted the transcript list to fasta format and submitted them to available web tools such as SignalP 5.0 and Deeploc. Web results were then analyzed by wilcox test to test for significance.
Live slice imaging and slice phagocytosis assay using E. coli BioParticles
Acute cortical slices (300 μm) were prepared from CX3CR1CreERT2 x AI14 (floxed TdTomato reporter) mice as described above. After recovery, slices were transferred to a 12-well plate with 1 mL freshly oxygenated ACSF and incubated with either 1mM anisomycin (Sigma #A9789) or vehicle (0.9% DMSO) at 37 °C with a gentle shake (~20 RPM). After 15 minutes, pHrodo™ Green E. coli BioParticles™ (Thermofisher P35366, prepared according to manufacturer’s instructions and diluted in ACSF) were gently pipetted over the surface of the slices. Slices were allowed to incubate with the beads for 75 minutes at 37 °C with a gentle shake (~20 RPM) and gently pipetting liquid from the bottom of the well over the surface of the slice every 20 minutes. Slices were then washed three times (2 minutes per wash) with 1 mL of ACSF with gentle shaking. Following washes, slices were placed in a custom perfusion chamber consisting of a silicone ring on top of a 4.5” x 6.25” sheet of no. 2 borosilicate glass (Brain Research Laboratories #5260–2) and continuously perfused with room-temperature, oxygenated ACSF while imaging. Slices were visualized on an upright Zeiss 880 confocal platform using a Zeiss 20x/1.0 water immersion objective and visible light laser excitation. Images consisting of 5–10 μm thick z-stacks at 0.76 μm step size were acquired every 2 minutes for 10 minutes using a QUASAR PMT detector.
Primary rat microglia culture and in vitro phagocytosis assay
Primary microglia were isolated from p11 rat pups following “basic protocol 2” outlined in Bohlen et al.53 except the microglia were cultured in media composed of DMEM/F-12 phenol red free (Gibco, #21041–025), CSF-1 at 10ng/mL (Peprotech, #400–28), 10% fetal bovine serum (Bio-Techne #S11150), TGF-β2 at 2 ng/mL (Peprotech, #100–35B), Heparan sulfate at 1 ug/mL (Galen lab supply, #GAG-HS01), and Pen-strep glutamine (Gibco #10378016 at 1:100 dilution). Briefly, rats were perfused and whole brain was chopped, dissociated, and pushed through a 70-micron cell strainer (Corning #CLS431751) to obtain a single cell suspension. Excess myelin was removed using myelin removal beads (Miltenyi, cat. no. 130-096-733) and then CD11b/c+ cells were positively selected using magnetic beads (Miltenyi, cat. no. 130-105-643) and LS columns (Miltenyi, cat. no. 130-042-401) as described53. Microglia were spot-plated at 30,000 cells per well in 20–30 uL of PBS for 10 minutes onto 24-well plates coated with Collagen IV (Corning, cat. no. 354233), then after 10 minutes, 500 uL of the DMEM/F12 media described above was added to each well. Cells were cultured in an IncuCyte S3 under conditions of 10% CO2 at 37 degrees C and imaged once every 6 hours to monitor growth. 50% of the media was exchanged for fresh media every 48 hours. Microglia were then cultured in vitro for 7–8 days prior to beginning phagocytosis assays.
For in vitro phagocytosis assay, cells were continuously imaged (GFP and phase) using a 10x lens beginning 5 minutes prior to anisomycin/vehicle addition and every 10 minutes thereafter in an IncuCyte S3 under conditions of 10% CO2 at 37 degrees C. 1mM anisomycin (Sigma #A9789) or vehicle (0.9% DMSO) was added to each well at time=5 minutes followed by 0.05 mg/ml of pHrodo Green E. coli BioParticles™ (Thermofisher P35366, diluted to a stock of 1 mg/mL in PBS and vigorously vortexed) 30 minutes following. Plates were imaged for 3 hours post BioParticle addition and data was plotted as pHrodo-GFP mean fluorescent intensity divided by # of microglia per field (microglia manually counted at first time point prior to anisomycin or vehicle addition).
Slice phagocytosis assay using opsonized beads
Acute cortical slices (300 μm) were prepared from C57Bl6/J mice as described in “Acute, live coronal brain slice preparation and treatment with puromycin and/or translation inhibitors”. Slices were held in ice-cold slicing buffer (125 mM NaCl, 25 mM glucose, 25 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 0.5 mM CaCl2, 3 mM MgCl2) for up to 30 minutes post-slicing, then at time=0 buffer was replaced with warm oxygenated ACSF and slices were incubated at 37 degrees C at a 20 RPM shake. At time = 5 minutes, some slices were treated with 1mM anisomycin or 0.9% DMSO (vehicle), followed by 15uL of 1% w/v opsonized, magnetic, 2.0–2.9 μm beads at time = 35 minutes. To generate 1% w/v opsonized beads, 400uL of amino magnetic bead particles (Spherotech #AM-20–10) were washed once in PBS (all washes on magnetic stand), then resuspended in 1 mL PBS with 60 ug/mL rabbit-IgG (Jackson ImmunoReseach #011-000-003) and incubated at room temperature for 2 hours with gentle 30RPM rotation, then washed twice with PBS, resuspended in 1 mL of ACSF, and stored for up to 1 week at 4 degrees C. A 5-minute pulse of 3 μM puromycin (Tocris #40-895-0) was added to some slices at t = 105 minutes, followed by fixation with 4% PFA at t = 110 minutes. Slices were processed as wholemount immunostains as above, using donkey anti-rabbit alexa647 at 1:400 to detect the opsonized beads and TMEM119 to identify microglia and their processes. Z-stack images (every 0.66μm) were taken with a LSM700 Axioimager-2 microscope with 63x oil lens, beginning 0.66μm above the slice surface and down to a depth of approximately 10μm deep from the slice surface.
For analysis, all microglial processes that were at least 2μm from their soma and located within the top 5μm of the slice were identified. First, each PeMP was manually outlined using imageJ within the Z-slice where the greatest portion of that PeMP resided (including only sections at least 2μm from the soma) and it was noted if the PeMP had any contacts with one or more opsonized beads. Several beads in each slice with no microglial contacts were also outlined as a control. Within these regions of interest, the local puromycin MFI (normalized to the whole image puromycin MFI) was calculated.
After this, a second round of analyses was conducted on these images. If the PeMP could be traced in either X, Y, or Z dimensions to a microglial soma (defined as TMEM119+ and dapi+) it was designated as an “attached PeMP.” If the process could be traced to the slice surface and could not be traced to either a microglial soma or any of the other 5 edges of the z-stack image (min X, max X, min Y, max Y, min Z), it was designated as a “severed PeMP.” Any PeMPs that could not be traced to a microglial soma but could be traced to one or more of the other 5 edges of the z-stack were not included in this second analysis because their status as “severed” or “attached” could not be determined (they could be attached to a microglial soma outside of the 3d image). For both attached and severed PeMPs, each contact with an opsonized bead, was manually outlined (using the Z-axis slice where the bead was centered) to include the bead area plus 0.5μm on all sides. Within this area, two measures were calculated using an imageJ macro for that Z-slice plus the slice above (+0.66μm) and slice below (−0.66μm) and then data from the three slices were averaged. 1) a TMEM119+ mask was applied and the local MFI of puromycin signal within the PeMP was calculated. 2) The % of bead area (alexa-647+) which co-localized with TMEM119+ area was calculated.
Blinding and randomization
All data collection and analyses were performed with the experimenter blinded to experimental condition. Where possible (live slice experiments), tissue was randomly assigned to treatment groups.
Statistical analyses
All data met assumptions for tests applied. In cases where data was not normal, data was log-transformed to achieve normality, or rank-based (e.g, Wilcoxon) tests were applied. No animals or data points were excluded from any analyses. Where data represented multiple observations from the same slice and animals, linear mixed models were utilized that accounted for this information. Linear mixed models were created and evaluated using the lme, stats, and lmerTest packages in R. All linear mixed models included scientifically impactful features such as treatment, genotype, or treatment-by-genotype interaction as co-variants, with mouse ID or day treated as random variables. After the main effects were analyzed by a linear mixed model the specific direction of effect was interpreted using either a Wilcoxon or a T-Test (depending on normality) post hoc test in R using the ggpubr package. For power calculations, no a priori power calculations were done. Rather, sample size was chosen based on prior studies where available12,13.
Box and whisker plots
Box plot elements, unless otherwise specified: center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; points, data points beyond the 1.5x interquartile range.
Scatter plots
Scatter plot graphs generated with Graphpad Prism v6.01. Points represent each data point, Mean +/− standard error of the mean shown on error bars.
Extended Data
Extended Data Fig. 1. Synaptosomal samples show RPL10a-eGFP, enrichment of synaptophysin (SYP), and depletion of nuclear proteins.
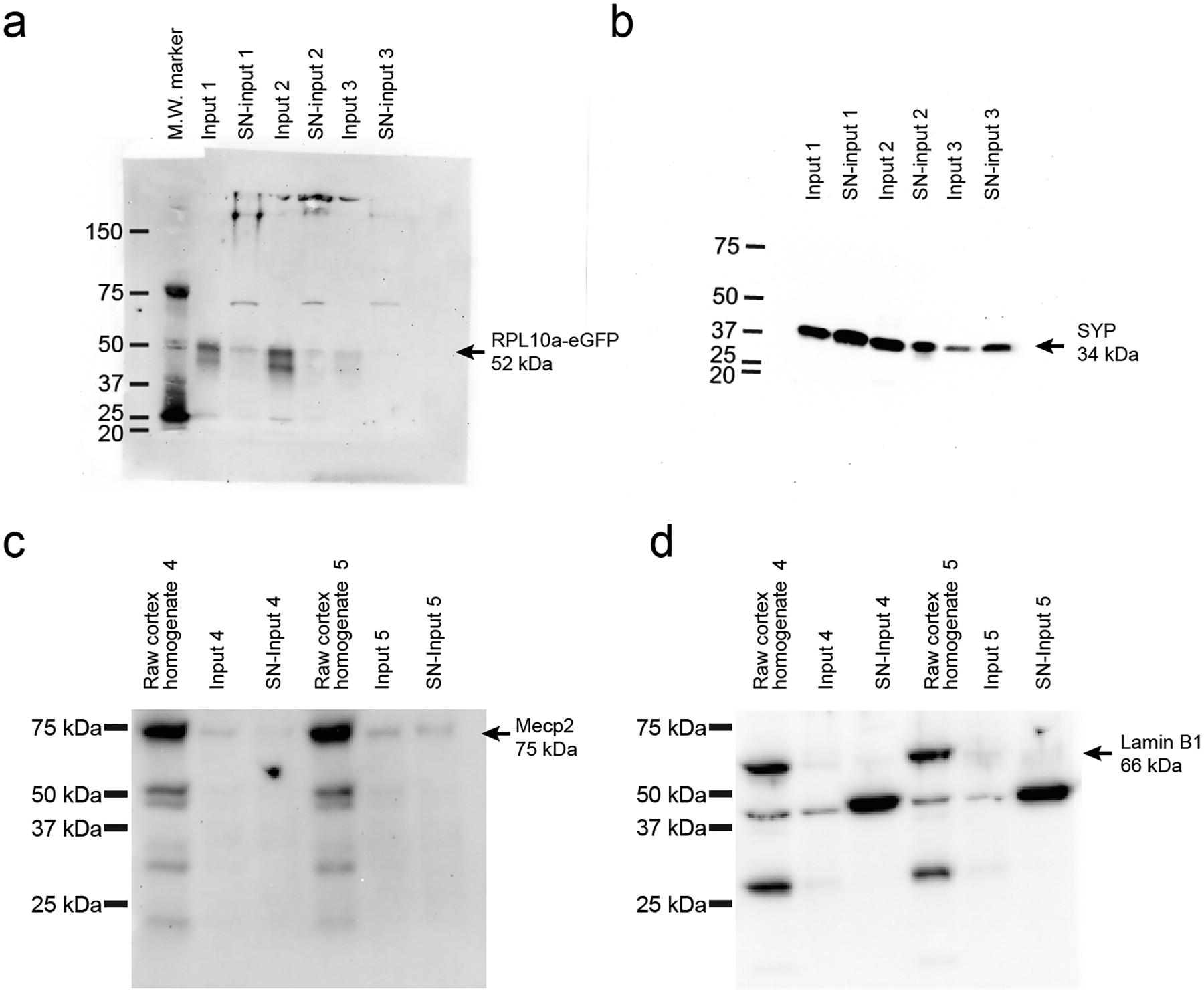
a, Western Blot using anti-GFP antibody detects the presence of the RPL10a-eGFP fusion protein (52 kDa) within 3 independent replicates of both Input and SN-Input fractions as described in Fig. 2 and methods. b, Western Blot using anti-SYP antibody detects the neuronal presynaptic protein, SYP (34 kDa), within three independent replicates’ SN-Input fractions. c-d, Western Blots show the detection of nuclear proteins, Mecp2 (c, 75 kDa with post-translational modifications) and Lamin B1 (d, 66 kDa) within the raw cortical homogenate (Isolated before the 1000g spin in the TRAP protocol), but shows their depletion within Input and SN-input fractions relative to the raw homogenate (two independent sets of replicates).
Extended Data Fig. 2. MG-TRAP yields a tamoxifen-dependent enrichment of microglial transcripts.
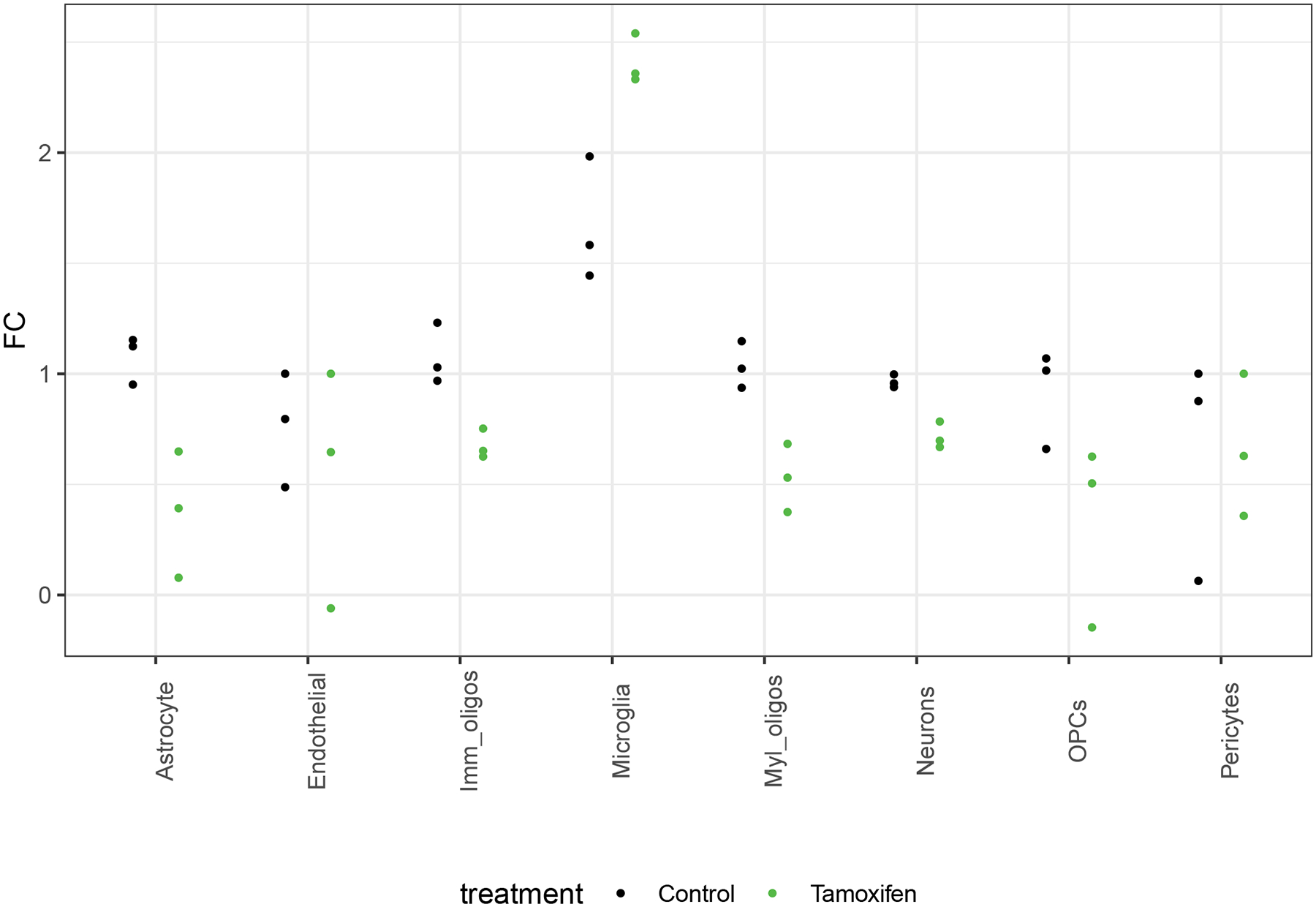
a) Median fold-change (FC) expression (MG-TRAP compared to Input) of the top 20–29 marker genes per cell type (as reported by Zhang et al., see Extended Data Table 1) from batch one replicate RNAseq experiments with (green dots) and without (black dots) tamoxifen injections reveals specific enrichment of microglial marker gene expression.
Extended Data Fig. 3. Multiplex in situ hybridization (ISH) shows transcript localization for PeMP and non-PeMP transcripts.
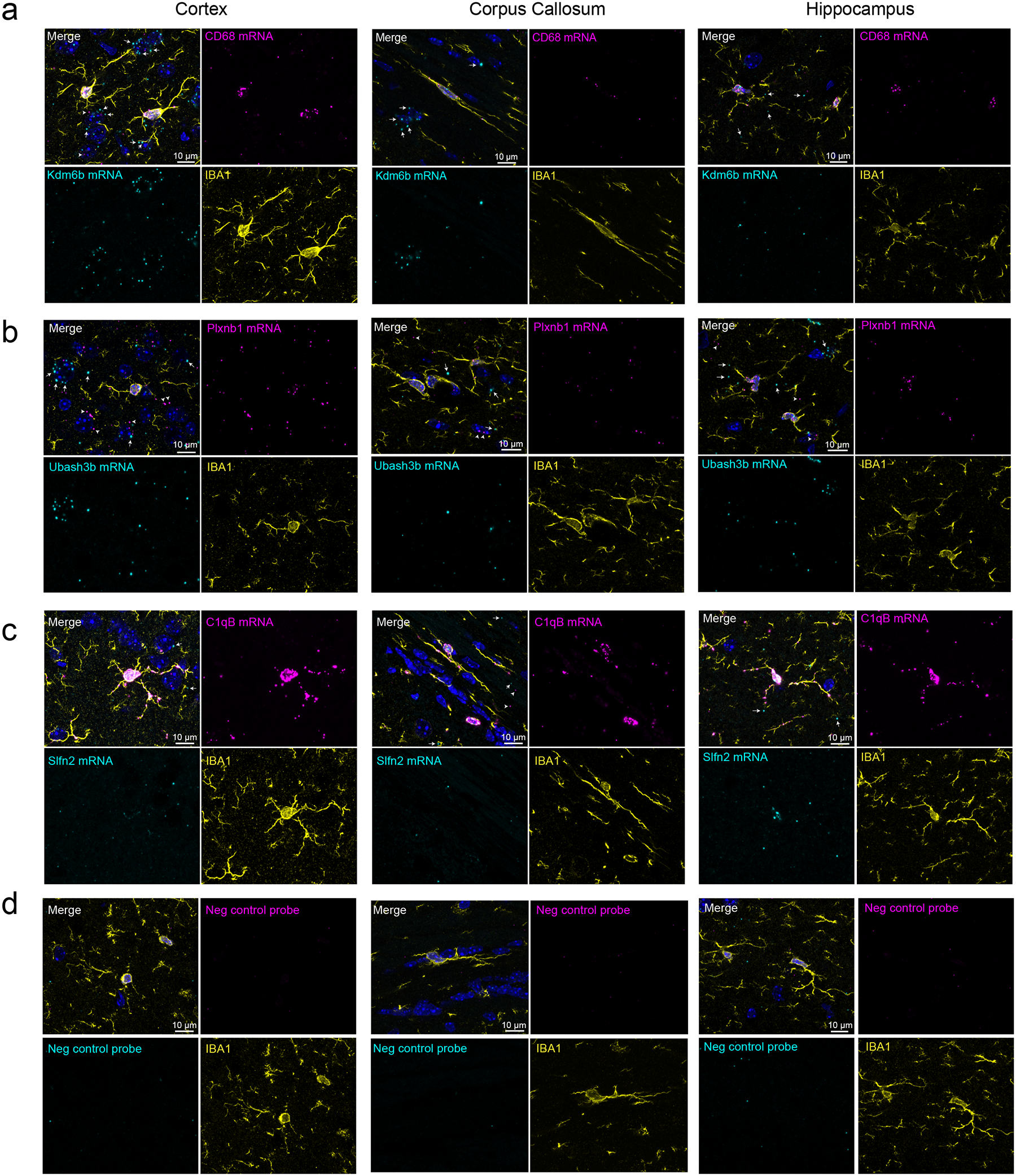
Maximum intensity projections of a 2 μm Z-stack image from ISH on p28 C57Bl/6 cortex, hippocampus, and corpus callosum tissue, for simultaneous PeMP-enriched vs. non-PeMP-enriched transcript detection in tandem with IBA1 immunofluorescence. Probes for PeMP-enriched transcripts shown in magenta and probes from non-PeMP-enriched transcripts shown in cyan. White arrows depict non-microglial signal emanating from somal-enriched probes, Kdm6b (a) and Ubash3b (b), while white arrowheads depict non-microglial signal emanating from PeMP-enriched probe, Plxnb1 (b). Images were also taken from negative control probes (d) to assess background signal levels. Quantifications are shown in Fig 2k–l. Representative images shown from n=5 mice and at least 4 z-stacks per brain region per mouse.
Extended Data Fig. 4. PeMP-detectable transcripts’ proteins have a higher probability of secretion using Signal-P algorithm.
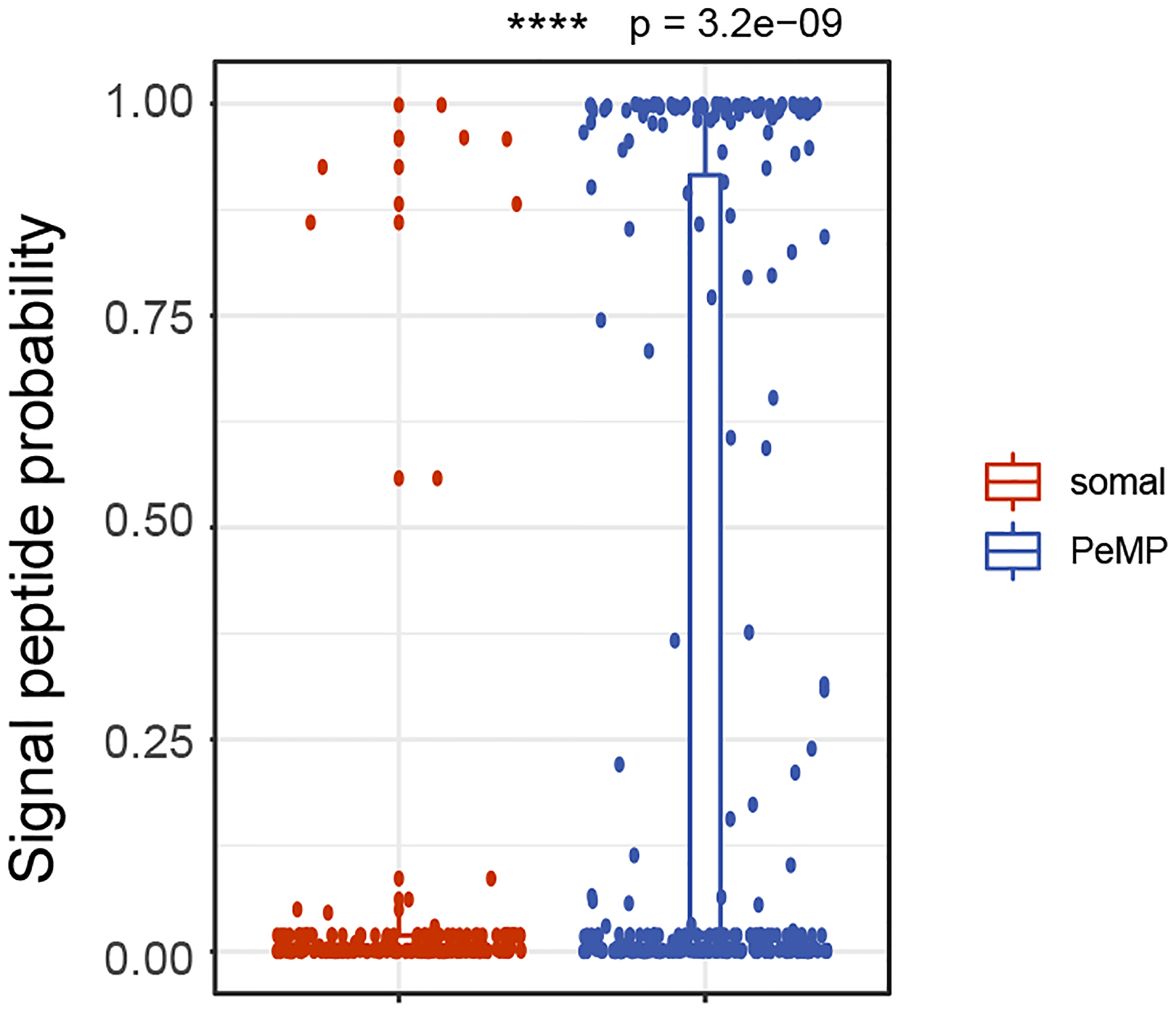
Predicted secretory signal peptide probablity is higher in PeMP than in Somal microglial transcripts. N = 200 somal and 279 PeMP transcripts. p = 3.2 x e-9 by two-tailed wilcoxon ranked sum test. Box plot elements: center line, median; box limits, upper and lower quartiles; whiskers, 1.5× interquartile range; points, data points beyond the 1.5x interquartile range.
Extended Data Fig. 5. Translation blockade does not affect microglial somal size, or somal CD68 and RPL10a expression.
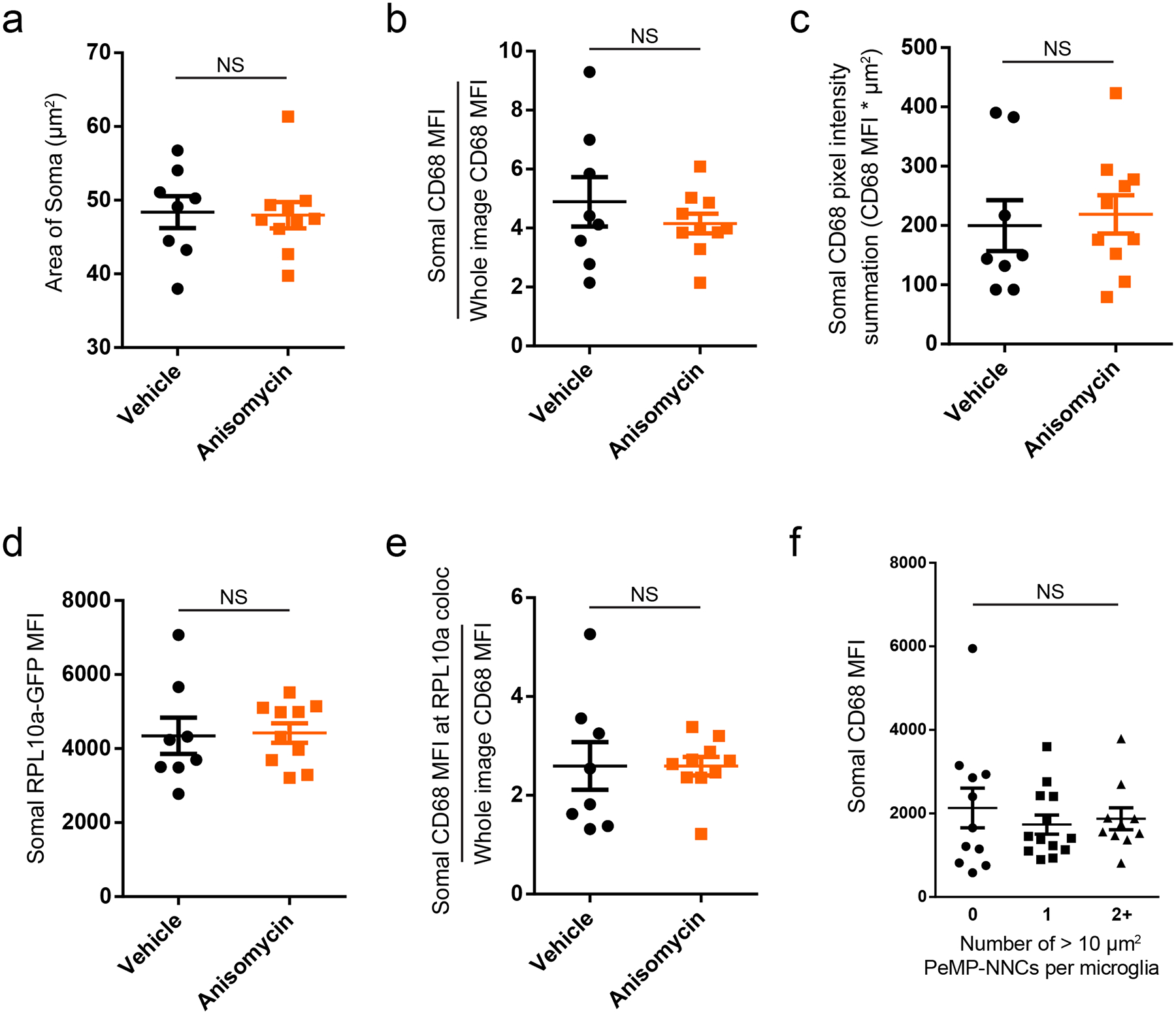
a-f) Whole mount immunostains were prepared from acute slices as in Figure 4d–i. a-e, No significant differences were detected between anisomycin and vehicle-treated slices in measurements of somal area (a, p = 0.89 by unpaired two-tailed t-test), somal CD68 mean fluorescence intensity (MFI) levels normalized to whole image CD68 MFI (b, p = 0.43 by unpaired two-tailed t-test with Welch’s correction), somal CD68 pixel intensity summation (c, p = 0.72 by unpaired two-tailed t-test), somal RPL10a-GFP MFI (d, p = 0.88 by unpaired two-tailed t-test), and somal CD68 MFI at RPL10a-GFP colocalized loci (e, p = 0.99 by unpaired two-tailed t-test with Welch’s correction). f, There was no statistical difference between vehicle-treated microglial somal CD68 MFI among those supporting either 0, 1, or 2 or more PeMP-NNCs > 10 um2 in area. p = 0.69 by one-way ANOVA. a-e, N = 8 vehicle and 10 anisomycin slices with at least 4 z-stack images per slice, derived from n = 3 mice. f, N = 34 microglial somata imaged from 4 independent vehicle-treated slices per group with at least 4 z-stack images per slice. Data are shown as mean ± s.e.m.
Extended data 6. Translation blockade does not affect cleaved caspase 3 staining in acute slice setting.
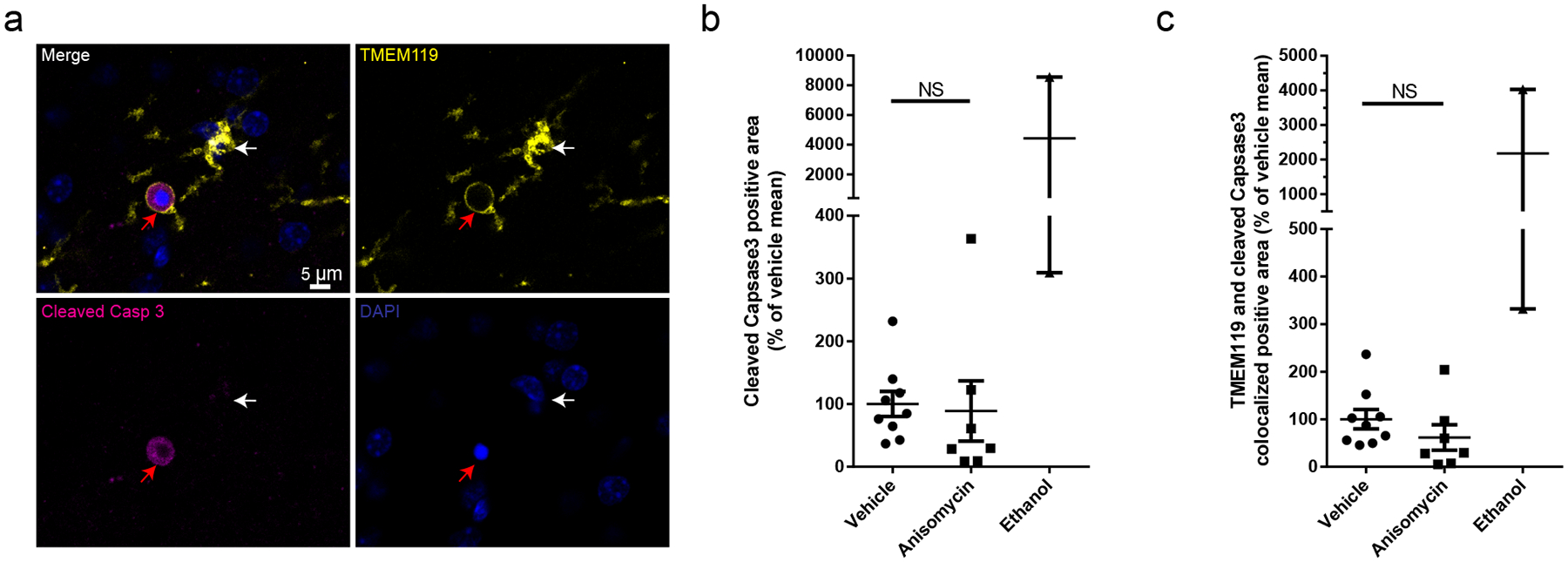
a) Whole mount immunostains were prepared from acute slices as in Figure 4d–i except immunostained with TMEM119, cleaved Caspase 3, and DAPI. Two slices were treated with 3% ethanol (30 minute treatment) as a positive control. A TMEM119+ microglial soma (white arrow) that has a PeMP encircling and forming a phagocytic cup around a cleaved caspase-3 positive apoptotic non-microglial cell (red arrow) within the ethanol-treated group. Images representative of 2 independent experiments. b-c, No significant differences were detected between anisomycin and vehicle-treated slices in measurements of cleaved caspase-3 postive area (b, p = 0.82 by unpaired two-tailed t-test), or cleaved caspase-3 colocalized with microglial marker, TMEM119 (c, p = 0.26 by unpaired two-tailed t-test). N = 9 vehicle, n = 7 anisomycin, and n=2 ethanol treated slices derived from n = 2 mice processed and stained on separate days. Data are shown as mean ± s.e.m.
Extended Data Table 1. Table of CNS cell type marker genes utilized for extended data fig. 2.
Most significant 20–29 marker genes per CNS cell type generated from the web tool (https://www.brainrnaseq.org/ - compare cell type of choice vs. all other CNS cell types) described in Zhang et al.
| Astrocytes | Endothelial | Microglia | Neurons | OPCs | Imm Oligos | Myl Oligos | Pericytes |
|---|---|---|---|---|---|---|---|
| Aqp4 | AU021092 | Ccl3 | Rein | Pdgfra | Cldn11 | Mog | fmod |
| Grm3 | Eltd1 | Tnf | Snhg11 | Lhfpl3 | Enpp6 | Cldn11 | rps2 |
| Fgfr3 | Cldn5 | Ccl4 | Sst | C1ql1 | Plp1 | Mobp | igf2 |
| Slc4a4 | Slco1a4 | Gdf15 | Npy | Cspg4 | Mog | Mbp | gpc3 |
| Aldh1l1 | Icam2 | Tmem119 | Islr2 | Nxph1 | Mobp | Opalin | ogn |
| Slc6a11 | Flt1 | Cd83 | Tmem130 | Cdo1 | Mag | Plp1 | Irrc32 |
| Slc15a2 | Slc38a5 | C1qa | A930038C07Rik | Mmp15 | Gjc2 | Tmem125 | flne |
| Slc7a10 | Tek | Selplg | Meg3 | Cacng4 | Mbp | Ermn | gjb2 |
| Mlc1 | Apold1 | Pld4 | Celf4 | Rlbp1 | Tmem88b | Gjc2 | itih2 |
| Slc1a3 | E030010A14Rik | C1qc | Stmn2 | Matn4 | Mai | Mag | rdh10 |
| Trim9 | Abcb1a | C1qb | Syt1 | Olfm2 | Lgi3 | Mai | bmp6 |
| Gja1 | Ctla2a | Trem2 | Tubb3 | Ncald | Gm98 | Ppp1r14a | aldh1a2 |
| Vcam1 | Prnd | Crybb1 | Crmp1 | Kcnip3 | Cnp | Tmem88b | postn |
| Daam2 | Esam | Csf1r | Ndrg4 | 3110035E14Rik | Nfasc | Gpr37 | sidt1 |
| Lrig1 | Cd93 | Laptm5 | Gap43 | Gpt2 | Fa2h | Lgi3 | Iamc3 |
| Cldn10 | Car4 | Ccrl2 | Ache | Cntn1 | Tspan2 | Cnp | slc22a6 |
| F3 | Tie1 | Fcrls | Atp1a3 | Sdc3 | Ugt8a | Lpar1 | clec3b |
| Fabp7 | Gpr116 | Cd68 | Gng2 | Lbh | Plxnb3 | Fa2h | slc6a13 |
| Acsbg1 | Kdr | Olfml3 | Syp | Vcan | Gsn | Gsn | bicc1 |
| Ncan | Slc16a4 | Tyrobp | Snap25 | Cspg5 | Tppp | Gjb1 | s100a10 |
| Igfbp2 | Ly6c1 | Fcer1g | Nsg2 | Epn2 | Bcas1 | Aspa | |
| Hes5 | Cdh5 | Hexb | Mllt11 | Olig1 | Tubb4 | Plekhb1 | |
| Slc1a2 | Lsr | Ctss | Syn2 | Sulf2 | Sirt2 | Gm98 | |
| Tlcd1 | Pglyrp1 | Ctsd | Ly6h | Tmeff2 | Pdlim2 | ||
| Gfap | Tm4sf1 | Itgb5 | Pcp4 | Plekhb1 | Ndrg1 | ||
| Ezr | Slc22a8 | Rcan1 | Uchl1 | Slc44a1 | Ugt8a | ||
| Ppap2b | Wfdcl | Ctsz | Dnm1 | Tspan2 | |||
| Fjx1 | Dll4 Itm2a |
Cst3 | Cryab |
Supplementary Material
Shown is a 3D reconstruction using Imaris software of Z-stack images from an immunostain for IBA1 (cyan), RPS16 (magenta - masked on IBA1 channel), and DAPI (blue) derived from 4 week old mouse cortex. The first half of the movie shows standard fluorescence view, while the second half shows a space-filled render on thresholded IBA1, RPS16, and DAPI channels.
Shown is a 3D reconstruction displayed with a space-filled rendering algorithm using Imaris software of Z-stack images from a whole-mount immunostain for IBA1 (magenta), puromycin (cyan - masked on IBA1 channel), RPL10a-eGFP (yellow) and DAPI (blue) derived from 4-week-old mouse cortex. The microglia’s soma is shown at center, whereas the DAPI+ nucleus in the corner is nearly 100% engulfed by a microglial process. The PeMP contacting and phagocytosing a neighboring nucleus (PeMP-NNC) shows high levels of puromycin and RPL10a-eGFP staining compared to a PeMP with no contacts (opposite corner)
Shown is a 3D reconstruction displayed with a space-filled rendering algorithm using Imaris software of Z-stack images from a whole-mount immunostain for IBA1 (magenta), puromycin (cyan - masked on IBA1 channel), RPL10a-eGFP (yellow) and DAPI (blue) derived from 4-week-old mouse cortex. The microglia can be seen with a long PeMP making contact with an IBA1-negative cell. The PeMP contacting this neighboring nucleus (PeMP-NNC) shows high levels of puromycin and RPL10a-eGFP staining compared to a PeMP with no contacts like the PeMP above the microglial soma.
Time lapse movie taken every 10 minutes for 4 hours with phase and green fluorescent overlay showing primary rat microglia at 7 days in vitro. Vehicle (0.9% DMSO) was added at t=5 minutes and pHRODO-tagged E.coli bioparticles (green) were added at t=35 minutes. Data are quantified in Fig. 5e.
Time lapse movie taken every 10 minutes for 4 hours with phase and green fluorescent overlay showing primary rat microglia at 7 days in vitro. Anisomycin (dissolved in 0.9% DMSO) was added at t=5 minutes and pHRODO-tagged E.coli bioparticles (green) were added at t=35 minutes. Data are quantified in Fig. 5e.
A descending series of 40 Z-stack images (shown at 2 images per second, 0.667 microns between steps) taken from 4 microns above the surface down to 22.7 microns below the surface of an acute-live slice that has been treated with opsonized beads and puromycin and then whole-mount immunostained for TMEM119 (yellow), puromycin (cyan), rabbit-IgG opsonized beads (magenta), and DAPI (dark blue). There are several TMEM119+ PeMPs surrounding opsonized beads at the slice surface which cannot be traced to somata deeper within the section indicating that these PeMPs have been severed from their somata in the slicing process.
Sheet names correspond to the two sample groups subject to the differential expression analysis performed. Table contains common gene symbol (external gene name), official Ensembl gene ID, and edgeR output comparing the two groups listed in the sheet title. logFC: log (base 2) fold change for enrichment when comparing the two groups in the sheet title. P-values by likelihood ratio tests and FDR (Benjamini-Hochberg) correction. For the “Microglial genes with location” sheet, only samples with log2 CPM > 1, FC > 0, and corrected p-value < 0.05 are included. Columns: Location: Somal, PeMP, or neither (NA) as defined in (Fig. 2 and methods). Unfiltered data is available at the Gene Expression Omnibus (GEO), accession no.GSE161460.
A 3D rendering of Z-stack images taken from surface and underlying 20 microns within the cortex of an acute-live slice that has been treated with opsonized beads and puromycin and then whole-mount immunostained for TMEM119 (yellow), puromycin (cyan), rabbit-IgG opsonized beads (magenta), and DAPI (dark blue).
GO term biological processes associated with PeMP and somal-enriched (separate tabs) microglial genes was calculated using the ClueGO plugin (Bindea et al 2009) for Cytoscape (see methods for settings) using the full microglial gene list (Supplementary Table 1) as a custom background and tested by FDR-corrected (Benjamini-Hochberg) two-sided hypergeometric test. Column headers are as defined by ClueGO. Only GO terms with corrected p < 0.05 are listed. Coloration/term groupings match Figure 3a–b. Within the PeMP-enriched list, GO term grouping was only carried out on terms with a corrected p value < 0.02 to match Figure 3a.
A 3D rendering of Z-stack images taken from surface and underlying 20 microns within the cortex of an acute-live slice that has been treated with opsonized beads and puromycin and then whole-mount immunostained for TMEM119 (yellow), puromycin (cyan), rabbit-IgG opsonized beads (magenta), and DAPI (dark blue).
Column headers are as defined by the output generated by Transite. Transite was run comparing PeMP list to all TRAP-enriched genes (columns D-F, J), and Somal list to all TRAP enriched genes (columns G-H, K). A Fisher’s Exact test (column K) was used to identify motifs found more in PeMP or Somal lists than expected by chance and corrected for multiple testing by Benjamin-Hochberg FDR (column P). Only FDR<.05 were retained. Ratio of motif enrichments was also calculated (column M). Results of hierarchical clustering (based on motif sequences Position Weight Matrices), are also included (columns R, S).
Acknowledgements
We would like to thank Peter Bayguinov, Gregory Strout, and the Washington University Center for Cellular Imaging for their expertise and training in live slice imaging and electron microscopy, Kristina Sakers for training and support, Claire Weichselbaum for comments on the manuscript, and the Genome Technology Access Center (GTAC@MGI) for sequencing and library preparation. Funding was provided by 5R01NS102272 (JD) and F32NS105363 (MV), and a microgrant from the Washington University Center for Cellular Imaging. GTAC is partially supported by P30 CA91842 and UL1TR002345. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Footnotes
Competing Interests Statement
JDD has previously received royalties related to TRAP. No other authors have competing interests.
Code Availability
All code is available at https://bitbucket.org/jdlabteam/vasek_microglia_local_translation_phagocytosis/src/master/
Data Availability
All data is available upon request. Raw and analyzed RNA-sequencing data are available at the Gene Expression Omnibus (GEO), accession no. GSE161460. Additional raw data is available at https://bitbucket.org/jdlabteam/vasek_microglia_local_translation_phagocytosis/src/master/
References
- 1.Schafer DP et al. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 74, 691–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hong S et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vasek MJ et al. A complement–microglial axis drives synapse loss during virus-induced memory impairment. Nature 534, 538–543 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marín-Teva JL et al. Microglia Promote the Death of Developing Purkinje Cells. Neuron 41, 535–547 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Butovsky O et al. Microglia activated by IL-4 or IFN-γ differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol. Cell. Neurosci 31, 149–160 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Davalos D et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci 8, 752–758 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Nimmerjahn A, Kirchhoff F & Helmchen F Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 308, 1314–1318 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Tremblay M-È, Lowery RL & Majewska AK Microglial Interactions with Synapses Are Modulated by Visual Experience. PLOS Biol. 8, e1000527 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller S et al. Disruption of Dendritic Translation of CaMKIIα Impairs Stabilization of Synaptic Plasticity and Memory Consolidation. Neuron 36, 507–519 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Kang H & Schuman EM A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273, 1402 (1996). [DOI] [PubMed] [Google Scholar]
- 11.Biever A, Donlin-Asp PG & Schuman EM Local translation in neuronal processes. Curr. Opin. Neurobiol 57, 141–148 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Ouwenga R, Lake AM, Aryal S, Lagunas T & Dougherty JD The Differences in Local Translatome across Distinct Neuron Types Is Mediated by Both Baseline Cellular Differences and Post-transcriptional Mechanisms. eNeuro 5, ENEURO.0320–18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakers K et al. Astrocytes locally translate transcripts in their peripheral processes. Proc. Natl. Acad. Sci 114, E3830–E3838 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boulay A-C et al. Translation in astrocyte distal processes sets molecular heterogeneity at the gliovascular interface. Cell Discov. 3, 17005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Petersen MA & Dailey ME Diverse microglial motility behaviors during clearance of dead cells in hippocampal slices. Glia 46, 195–206 (2004). [DOI] [PubMed] [Google Scholar]
- 16.D’Onofrio C, Paradisi F & Piccolo D The influence of some metabolic inhibitors on in vitro phagocytizing macrophages. I. The behaviour of human macrophages. Med. Microbiol. Immunol. (Berl.) 163, 195–207 (1977). [DOI] [PubMed] [Google Scholar]
- 17.Ayata P et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci 21, 1049–1060 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boutej H et al. Diverging mRNA and Protein Networks in Activated Microglia Reveal SRSF3 Suppresses Translation of Highly Upregulated Innate Immune Transcripts. Cell Rep. 21, 3220–3233 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Zhou P et al. Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc. Natl. Acad. Sci. U. S. A 110, 15395–15400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sahasrabuddhe V & Ghosh HS Cx3Cr1-Cre induction leads to microglial activation and IFN-1 signaling caused by DNA damage in early postnatal brain. Cell Rep. 38, (2022). [DOI] [PubMed] [Google Scholar]
- 21.Fonseca MI et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation 14, 48 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt EK, Clavarino G, Ceppi M & Pierre P SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Westmark PR, Westmark CJ, Jeevananthan A & Malter JS Preparation of synaptoneurosomes from mouse cortex using a discontinuous percoll-sucrose density gradient. J. Vis. Exp. JoVE (2011) doi: 10.3791/3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouwenga R et al. Transcriptomic Analysis of Ribosome-Bound mRNA in Cortical Neurites In Vivo. J. Neurosci 37, 8688–8705 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haimon Z et al. Re-evaluating microglia expression profiles using RiboTag and cell isolation strategies. Nat. Immunol 19, 636–644 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kang SS et al. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med 215, 2235–2245 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. Off. J. Soc. Neurosci 34, 11929–11947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hickman SE et al. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci 16, 1896–1905 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Condeelis J & Singer RH How and why does β-actin mRNA target? Biol. Cell 97, 97–110 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Almagro Armenteros JJ, Sønderby CK, Sønderby SK, Nielsen H & Winther O DeepLoc: prediction of protein subcellular localization using deep learning. Bioinformatics 33, 3387–3395 (2017). [DOI] [PubMed] [Google Scholar]
- 31.Almagro Armenteros JJ et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol 37, 420–423 (2019). [DOI] [PubMed] [Google Scholar]
- 32.Krismer K et al. Transite: A Computational Motif-Based Analysis Platform That Identifies RNA-Binding Proteins Modulating Changes in Gene Expression. Cell Rep. 32, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dissing-Olesen L & MacVicar BA Fixation and Immunolabeling of Brain Slices: SNAPSHOT Method. Curr. Protoc. Neurosci 71, 1.23.1–1.23.12 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Cserép C et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 367, 528–537 (2020). [DOI] [PubMed] [Google Scholar]
- 35.Sommer S The importance of immune gene variability (MHC) in evolutionary ecology and conservation. Front. Zool 2, 16 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lejeune J, Brachet G & Watier H Evolutionary Story of the Low/Medium-Affinity IgG Fc Receptor Gene Cluster. Front. Immunol 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dougherty JD, Schmidt EF, Nakajima M & Heintz N Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res. 38, 4218–4230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidali A et al. AU-Rich Element RNA Binding Proteins: At the Crossroads of Post-Transcriptional Regulation and Genome Integrity. Int. J. Mol. Sci 23, 96 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhou H et al. IRAK2 directs stimulus-dependent nuclear export of inflammatory mRNAs. eLife 6, e29630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cervero P, Himmel M, Krüger M & Linder S Proteomic analysis of podosome fractions from macrophages reveals similarities to spreading initiation centres. Eur. J. Cell Biol 91, 908–922 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Gagnon E et al. Endoplasmic Reticulum-Mediated Phagocytosis Is a Mechanism of Entry into Macrophages. Cell 110, 119–131 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Garin J et al. The phagosome proteome: insight into phagosome functions. J. Cell Biol 152, 165–180 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shui W et al. Membrane proteomics of phagosomes suggests a connection to autophagy. Proc. Natl. Acad. Sci 105, 16952–16957 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barger SR et al. Membrane-cytoskeletal crosstalk mediated by myosin-I regulates adhesion turnover during phagocytosis. Nat. Commun 10, 1249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Graber TE et al. Reactivation of stalled polyribosomes in synaptic plasticity. Proc. Natl. Acad. Sci. U. S. A 110, 16205–16210 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only References
- 46.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dobin A et al. STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Anders S, Pyl PT & Huber W HTSeq--a Python framework to work with high-throughput sequencing data. Bioinforma. Oxf. Engl 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robinson MD, McCarthy DJ & Smyth GK edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinforma. Oxf. Engl 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bindea G et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinforma. Oxf. Engl 25, 1091–1093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bembom Oliver & Ivanek Robert. seqLogo: Sequence logos for DNA sequence alignments. (2020).
- 53.Bohlen CJ, Bennett FC & Bennett ML Isolation and Culture of Microglia. Curr. Protoc. Immunol 125, e70 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Shown is a 3D reconstruction using Imaris software of Z-stack images from an immunostain for IBA1 (cyan), RPS16 (magenta - masked on IBA1 channel), and DAPI (blue) derived from 4 week old mouse cortex. The first half of the movie shows standard fluorescence view, while the second half shows a space-filled render on thresholded IBA1, RPS16, and DAPI channels.
Shown is a 3D reconstruction displayed with a space-filled rendering algorithm using Imaris software of Z-stack images from a whole-mount immunostain for IBA1 (magenta), puromycin (cyan - masked on IBA1 channel), RPL10a-eGFP (yellow) and DAPI (blue) derived from 4-week-old mouse cortex. The microglia’s soma is shown at center, whereas the DAPI+ nucleus in the corner is nearly 100% engulfed by a microglial process. The PeMP contacting and phagocytosing a neighboring nucleus (PeMP-NNC) shows high levels of puromycin and RPL10a-eGFP staining compared to a PeMP with no contacts (opposite corner)
Shown is a 3D reconstruction displayed with a space-filled rendering algorithm using Imaris software of Z-stack images from a whole-mount immunostain for IBA1 (magenta), puromycin (cyan - masked on IBA1 channel), RPL10a-eGFP (yellow) and DAPI (blue) derived from 4-week-old mouse cortex. The microglia can be seen with a long PeMP making contact with an IBA1-negative cell. The PeMP contacting this neighboring nucleus (PeMP-NNC) shows high levels of puromycin and RPL10a-eGFP staining compared to a PeMP with no contacts like the PeMP above the microglial soma.
Time lapse movie taken every 10 minutes for 4 hours with phase and green fluorescent overlay showing primary rat microglia at 7 days in vitro. Vehicle (0.9% DMSO) was added at t=5 minutes and pHRODO-tagged E.coli bioparticles (green) were added at t=35 minutes. Data are quantified in Fig. 5e.
Time lapse movie taken every 10 minutes for 4 hours with phase and green fluorescent overlay showing primary rat microglia at 7 days in vitro. Anisomycin (dissolved in 0.9% DMSO) was added at t=5 minutes and pHRODO-tagged E.coli bioparticles (green) were added at t=35 minutes. Data are quantified in Fig. 5e.
A descending series of 40 Z-stack images (shown at 2 images per second, 0.667 microns between steps) taken from 4 microns above the surface down to 22.7 microns below the surface of an acute-live slice that has been treated with opsonized beads and puromycin and then whole-mount immunostained for TMEM119 (yellow), puromycin (cyan), rabbit-IgG opsonized beads (magenta), and DAPI (dark blue). There are several TMEM119+ PeMPs surrounding opsonized beads at the slice surface which cannot be traced to somata deeper within the section indicating that these PeMPs have been severed from their somata in the slicing process.
Sheet names correspond to the two sample groups subject to the differential expression analysis performed. Table contains common gene symbol (external gene name), official Ensembl gene ID, and edgeR output comparing the two groups listed in the sheet title. logFC: log (base 2) fold change for enrichment when comparing the two groups in the sheet title. P-values by likelihood ratio tests and FDR (Benjamini-Hochberg) correction. For the “Microglial genes with location” sheet, only samples with log2 CPM > 1, FC > 0, and corrected p-value < 0.05 are included. Columns: Location: Somal, PeMP, or neither (NA) as defined in (Fig. 2 and methods). Unfiltered data is available at the Gene Expression Omnibus (GEO), accession no.GSE161460.
A 3D rendering of Z-stack images taken from surface and underlying 20 microns within the cortex of an acute-live slice that has been treated with opsonized beads and puromycin and then whole-mount immunostained for TMEM119 (yellow), puromycin (cyan), rabbit-IgG opsonized beads (magenta), and DAPI (dark blue).
GO term biological processes associated with PeMP and somal-enriched (separate tabs) microglial genes was calculated using the ClueGO plugin (Bindea et al 2009) for Cytoscape (see methods for settings) using the full microglial gene list (Supplementary Table 1) as a custom background and tested by FDR-corrected (Benjamini-Hochberg) two-sided hypergeometric test. Column headers are as defined by ClueGO. Only GO terms with corrected p < 0.05 are listed. Coloration/term groupings match Figure 3a–b. Within the PeMP-enriched list, GO term grouping was only carried out on terms with a corrected p value < 0.02 to match Figure 3a.
A 3D rendering of Z-stack images taken from surface and underlying 20 microns within the cortex of an acute-live slice that has been treated with opsonized beads and puromycin and then whole-mount immunostained for TMEM119 (yellow), puromycin (cyan), rabbit-IgG opsonized beads (magenta), and DAPI (dark blue).
Column headers are as defined by the output generated by Transite. Transite was run comparing PeMP list to all TRAP-enriched genes (columns D-F, J), and Somal list to all TRAP enriched genes (columns G-H, K). A Fisher’s Exact test (column K) was used to identify motifs found more in PeMP or Somal lists than expected by chance and corrected for multiple testing by Benjamin-Hochberg FDR (column P). Only FDR<.05 were retained. Ratio of motif enrichments was also calculated (column M). Results of hierarchical clustering (based on motif sequences Position Weight Matrices), are also included (columns R, S).
Data Availability Statement
All data is available upon request. Raw and analyzed RNA-sequencing data are available at the Gene Expression Omnibus (GEO), accession no. GSE161460. Additional raw data is available at https://bitbucket.org/jdlabteam/vasek_microglia_local_translation_phagocytosis/src/master/