

Abstract
Key Clinical Message
Idiopathic multicentric Castleman disease (iMCD) is challenging to diagnose due to clinical similarities with other conditions, such as Still's disease. Once diagnosed, iMCD may be effectively managed with the anti‐interleukin‐6 antibody siltuximab.
Abstract
Here, we present the case of a 19‐year‐old Polish woman with persistent fever and enlarged lymph nodes and whose diagnosis remained inconclusive following initial clinical assessments and extensive laboratory analyses. The patient had subsequent complaints of joint pain and erythema which were suspicious of Still's disease and resolved with treatment with tocilizumab. Later, the progression of symptoms, such as lymphadenopathy, and elevated interleukin‐6 levels were consistent with Castleman disease, leading to the diagnosis of idiopathic multicentric Castleman disease seven years after the patient first reported symptoms. Treatment with the anti‐interleukin‐6 antibody siltuximab resulted in complete symptom resolution and normalization of inflammatory parameters. No adverse events were reported due to treatment with siltuximab.
Keywords: anti‐interleukin‐6 antibody, case report, diagnosis, idiopathic multicentric Castleman disease, siltuximab
1. INTRODUCTION
Castleman disease (CD) is a rare condition first described in 1956 by Castleman et al. as localized mediastinal lymphadenopathy. 1 , 2 , 3 CD encompasses a group of heterogenous lymphoproliferative disorders classified by the Castleman Disease Collaboration Network (CDCN) into unicentric CD (UCD) and multicentric CD (MCD), based on clinical and pathological features. 1 , 4 , 5 While UCD is a localized and reversible disease involving a single lymph node, MCD is a systemic and potentially life‐threatening disorder that affects multiple lymph nodes. 1 , 4 Approximately 50% of MCD cases are caused by uncontrolled human herpesvirus‐8 (HHV‐8) infections (HHV‐8‐associated MCD) which are most commonly observed in human immunodeficiency virus (HIV)‐positive patients, or by monoclonal plasma cells (polyneuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes [POEMS]‐associated MCD). 5 , 6 The etiology and pathogenesis of the remaining cases is less well understood, which have therefore been classified as idiopathic MCD (iMCD). 1 However, the cytokine interleukin‐6 (IL‐6) is thought to play an important role in the pathogenesis and symptom presentation in many patients with iMCD. 7 , 8 , 9 , 10 Clinical symptoms of iMCD include fever, night sweats, unintended weight loss, and anasarca, in addition to lymphadenopathy. 4 Some patients also exhibit hepatomegaly, splenomegaly, and/or liver and renal dysfunction. 4 Due to the clinical heterogeneity of the disease, the presence of nonspecific constitutional symptoms and the lack of biomarkers, diagnosis of iMCD can be challenging. 4 , 5 , 11 Clinical similarities between iMCD and other conditions, such as autoimmune diseases (e.g., Still's disease), cancer, and infections, often result in misdiagnosis and delays in initiating appropriate treatment. 5 To facilitate diagnosis of iMCD and guide treatment decisions, diagnostic consensus criteria were proposed by the CDCN in 2017, which include the presence of two major criteria (lymphadenopathy in ≥2 lymph node stations and histopathological confirmation of CD morphology) and ≥2 minor criteria, including ≥1 laboratory parameter (elevated C‐reactive protein [CRP] or erythrocyte sedimentation rate [ESR], anemia, thrombocytosis, polyclonal hypergammaglobulinemia, hypoalbuminemia, and constitutional symptoms). 5 , 6
Patients with iMCD are generally treated with anti‐IL‐6 therapies in combination with corticosteroids, regardless of symptom severity. 3 , 12 The monoclonal anti‐IL‐6 antibody siltuximab is the only therapy approved for iMCD in Europe and the United States, based on positive results from a randomized controlled Phase II trial. 7 , 13 , 14 Current treatment guidelines for iMCD recommend siltuximab in the first‐line setting, with the anti‐IL‐6 receptor antibody tocilizumab and the anti‐CD20 antibody rituximab generally recommended for later lines. 12 , 15 , 16
Here we report on a challenging diagnosis of iMCD in a patient who had complex systemic manifestations spanning 7 years that resolved following treatment with siltuximab.
2. CASE PRESENTATION
A 19‐year‐old Polish woman presented with fever and enlarged lymph nodes in October 2014. Lymphadenopathy was confirmed by ultrasound scan of peripheral nodes (cervical nodes: <10 mm diameter; axillary nodes: 10 mm diameter on the left side and 11 mm on the right side). Laboratory and imaging assessments were performed to rule out malignancy or infection. Laboratory analyses found elevated CRP levels (73 mg/L; normal <10 mg/L), increased ESR (56 mm/h; normal 0–20 mm/h), thrombocytosis, normocytic and microcytic anemia (mean corpuscular volume: 78 fl [normal 80–100 fl]; hemoglobin: 9.8 g/dL [normal 12.0–15.7 g/dL]), and positive antinuclear antibody titre (1:1000; negative <1:80). Procalcitonin levels were normal; however, ferritin levels were periodically elevated (1.5 times above normal range) and polyclonal hypergammaglobulinemia and hypoalbuminemia were present. Epstein–Barr virus, cytomegalovirus, and hepatitis B and C were all excluded. Immunoblot assays to detect autoimmune disease (including Sjógren's syndrome and lupus), assays for Borrelia burgdorferi, blood and urine cultures, and throat and anal swabs were all negative. Bone marrow biopsy was uncharacteristic, revealing granulocytic hyperplasia, and mild reticulin fibrosis. Chest and abdominal computed tomography (CT) performed in February 2015 demonstrated no abnormalities in these locations, including no lymphadenopathy or organomegaly. One month later, active neoplastic processes were identified in lymph nodes in the abdomen and pelvis using positron emission tomography (PET)–CT and a “watch and wait” approach was chosen. Six months after detection of these neoplastic processes, follow‐up imaging revealed complete spontaneous regression of lymphadenopathy. Results of the initial clinical examinations and laboratory analyses were inconclusive, and further investigations were required to confirm a diagnosis.
In April 2015, the patient reported pain in the right knee joint. Ultrasound and magnetic resonance imaging (MRI) detected a liquid–solid structure which was excised from the knee joint; histological examination confirmed a cavernous hemangioma which did not recur following removal. Seven months later, the patient reported headaches in addition to the persistent fever and was diagnosed with bacterial meningitis following examination of cerebrospinal fluid which revealed the presence of Acinetobacter lwoffii. The patient was subsequently treated with ceftriaxone, ampicillin, and acyclovir; however, no clinical improvements were observed. Methylprednisolone was started at an initial dose of 32 mg/day, which led to a reduction of inflammatory parameters by ~40% after 14 days but symptoms did not resolve.
In July 2017, histopathological examination of a supraclavicular lymph node identified an inflammatory process, with dense hyalinization of the paranodal connective tissue partially encasing the lymph node parenchyma. These findings were most consistent with the presence of an inflammatory pseudotumour. The patient also reported persistent pain in the joints of her hands, and transitory erythema was identified on the joint of her left ankle. Chest CT revealed a soft tissue mass in the superior‐anterior mediastinum which was suspicious of thymic hyperplasia, but MRI of the thymus demonstrated persistent residual thymus tissue without active neoplastic processes. The patient's leukocyte count was 8.33 × 103/μL. Treatment with methylprednisolone was initiated at a reduced dose of 4–8 mg/day. Subcutaneous methotrexate 15–25 mg/day once a week was added to the regimen for the treatment of suspected Still's disease, followed by azathioprine 150 mg/day and then ciclosporin 3 mg/kg/day; however, no clinical improvements were observed. The patient was subsequently diagnosed with adult‐onset Still's disease based on the elevated inflammatory markers in the blood, persistent pain in the hand joints, and erythema of the left ankle joint. Methylprednisolone, methotrexate, azathioprine, and ciclosporin were all discontinued, and the patient was started on tocilizumab at a dose of 8 mg/kg/day, once every 30 days for 24 cycles. Symptoms resolved after 2 cycles and CRP level and ESR normalized. However, following discontinuation of tocilizumab, the patient presented with fever, myalgia, sore throat, night sweats, and asthenia. Gradual elevations of CRP level and ESR were also observed. Tocilizumab was restarted but subsequently discontinued after 3 cycles due to unclear disease presentation.
Over the next few years, several additional examinations and analyses were carried out including next‐generation genetic testing to screen for congenital autoinflammatory syndromes. Pathogenic and potentially pathogenic mutations and human leukocyte antigen B27 were absent, and autoimmune lymphoproliferative syndrome and IgG4‐related disease were excluded. Laboratory analyses demonstrated markedly elevated concentrations of IL‐6 and serum amyloid A (SAA), which were 33.7 pg/mL (normal <7 pg/mL) and 571 mg/L (normal <6.4 mg/L), respectively. Adipose tissue samples were histologically examined due to elevated SAA but amyloidosis was not detected. CT scans of the chest and abdominal cavity revealed progression of lymphadenopathy in supraclavicular and mediastinal lymph nodes, hepatomegaly, and splenomegaly (Figure 1A). In addition, pleural effusion (<10 mm wide) was identified in the right pleural cavity with traces of fluid found in the interlobal fissures. Empiric treatment with colchicine was started in February 2021 due to elevated SAA levels and persistent fever consistent with an autoinflammatory disorder. Colchicine was administered at 1 mg/day for 4 weeks but was subsequently discontinued due to progression of lymphadenopathy and only a partial decrease in SAA concentration. Examination of the mediastinal lymph nodes with fine needle aspiration biopsy was inconclusive. Histological examination of a lymph node resected from the supraclavicular region revealed expansion of interfollicular areas with hypervascularity, hyalinization, and a marked increase in plasma cell count, but normal immunoglobulin light chain expression. The follicular zone was atrophic and demonstrated attenuated germinal centers with focal penetration by small blood vessels (Figure 2). No atypical CD30‐positive or CD15‐positive cells were detected and there were no significant changes in the topographic distribution of CD3‐positive T‐cells and CD20/PAX5‐positive B cells. Expression of HHV‐8 was negative, and the number of IgG4‐positive plasma cells was not elevated. Based on these findings, lymph node morphology was considered to be consistent with a mixed variant of CD. Our patient was diagnosed with iMCD in May 2021 according to both major CDCN criteria (histopathological lymph node features within the iMCD spectrum and lymphadenopathy in ≥2 lymph node regions) and several minor criteria (elevated CRP and ESR, anemia, thrombocytosis, hypalbuminemia, polyclonal hypergammaglobulinemia, constitutional symptoms, hepatomegaly and splenomegaly, and pleural effusion). 5
FIGURE 1.
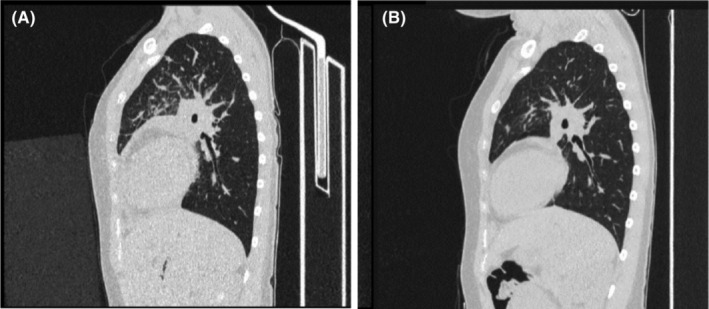
High‐resolution CT scans of the chest and abdomen demonstrating contrast‐enhanced lymph nodes at 3 months before the diagnosis of iMCD (A) and 9 months after initiation of siltuximab (B). (A) Demonstrates lymphadenopathy of the paratracheal mediastinal nodes, nodes in the hilum of the lungs, left subclavicular nodes, right supraclavicular nodes, left parasternal nodes, and retrosternal nodes in the anterior mediastinum. Hepatomegaly in the craniocaudal dimension of the right lobe and splenomegaly are also shown. (B) Illustrates partial regression of inflammatory infiltration and complete regression of fluid in the right pleural cavity on both sides of the lungs. Regression of lymphadenopathy of the mediastinal lymph nodes and lymph nodes located within the lungs is also shown. CT, computed tomography; iMCD, idiopathic multicentric Castleman disease.
FIGURE 2.
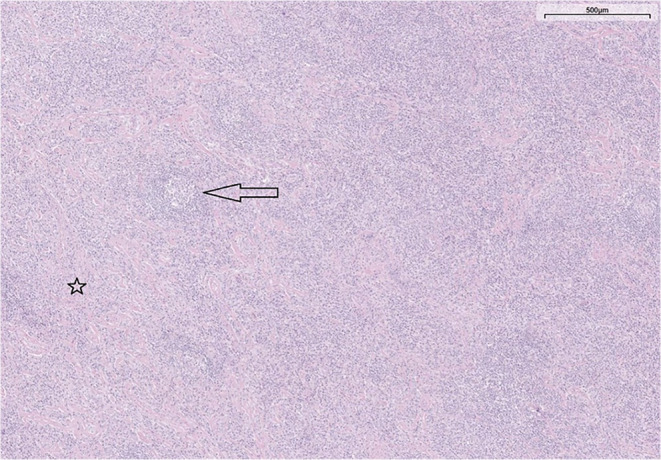
H&E staining of a lymph node from the supraclavicular region performed at the time of iMCD diagnosis, showing a disturbed histological pattern due to expansion of interfollicular areas with a dominant percentage of plasma cells, and attenuated germinal centers (arrow). Interfollicular hyalinization is also shown (star). H&E, hematoxylin and eosin; iMCD, idiopathic multicentric Castleman disease.
Following iMCD diagnosis, treatment with rituximab was initiated: the patient received 1 g on the first day followed by another 1 g 14 days later; however, no improvements in inflammatory parameters or symptoms were observed and rituximab was discontinued. The patient subsequently received treatment with intravenous siltuximab 500 mg once every 3 weeks, resulting in symptom resolution and normalization of inflammatory markers and SAA after 6 weeks. No siltuximab‐related adverse events were observed. Chest CT performed 9 months after initiation of siltuximab revealed regression of lymphadenopathy of the paratracheal mediastinal nodes, nodes in the hilum of the right and left lungs, left parasternal nodes, and retrosternal nodes in the anterior mediastinum (Figure 1B). As of August 2023, treatment with siltuximab was still ongoing.
3. DISCUSSION
iMCD is a rare and potentially life‐threatening systemic disorder which remains challenging to diagnose, despite the introduction of international, evidence‐based diagnostic criteria. 3 , 4 , 5 This is partly due to clinical and histological similarities between iMCD and several other conditions, such as some autoimmune diseases, infectious disorders, and malignancies. 5 Clinical confirmation of iMCD is complicated by patients often presenting with constitutional symptoms, which can lead to difficulties in making differential diagnoses and delays in treatment initiation. 4 , 5 Our patient initially presented with lymphadenopathy, fever, and laboratory features that were consistent with previous reports of iMCD; however, the initial absence of histopathological lymph node features of iMCD resulted in a significant delay in time to diagnosis. In addition, iMCD was not originally suspected due to the rarity of the condition. 3 , 5 , 17
Diagnosing our patient with iMCD involved extensive clinical, histological, and laboratory assessments at numerous follow‐up visits over a period of 7 years: physical examinations, CT–PET imaging, lymph node biopsies followed by histopathological analyses, various blood tests, bacterial cultures, and next‐generation sequencing tests were necessary to reach a differential diagnosis of iMCD.
The clinical examinations and laboratory analyses performed at initial presentation of our patient were inconclusive. Nearly a year later, the patient presented with headache in addition to persistent fever, which are both common symptoms of bacterial meningitis in adults. 18 The diagnosis was supported by examination of cerebrospinal fluid which revealed the presence of Acinetobacter lwoffii; however, treatment with antibiotics did not resolve the patient's symptoms. A year and a half after the diagnosis of bacterial meningitis, our patient was diagnosed with adult‐onset Still's disease, based on elevated inflammatory markers, persistent pain in the hand joints, and the development of erythema of the left ankle joint, which are all suggestive of this disorder. 19 Still's disease is a disorder that commonly poses challenges in clinical differentiation from iMCD. Clinical similarities between these diseases have been previously reported, with patients with MCD fulfilling several diagnostic criteria for Still's disease. 20 Our patient was treated with tocilizumab, which is an anti‐IL‐6 receptor antibody commonly used in the treatment of Still's disease. 16 , 21 Despite initial short‐term resolution of symptoms and normalization of inflammatory parameters, treatment with tocilizumab did not result in long‐term clinical improvement in our patient and was therefore discontinued.
Two and a half years later, new observations such as the progression of lymphadenopathy, the occurrence of hepatomegaly and splenomegaly, and a disturbed histological pattern in the right supraclavicular lymph node, in addition to our previous findings, were all suggestive of iMCD. Our patient now met the criteria for diagnosis of iMCD proposed by the CDCN: lymphadenopathy in ≥2 lymph node regions and several minor criteria (including elevated inflammatory markers, anemia, thrombocytosis, hypoalbuminemia, polyclonal hypergammaglobulinemia, constitutional symptoms, hepatomegaly and splenomegaly, and pleural effusion). 5 In addition, the absence of HHV‐8 ruled out HHV‐8‐related MCD, and markedly elevated levels of IL‐6 further supported the diagnosis of iMCD. 5 Patients with iMCD can be managed with targeted therapies. Current treatment guidelines recommend administration of anti‐IL‐6 monoclonal antibodies, such as siltuximab or tocilizumab, or the anti‐CD20 monoclonal antibody rituximab, with or without concomitant steroids. 5 , 7 , 12 , 15 , 16 Siltuximab is currently the only therapy indicated for the treatment of iMCD in Europe and is recommended in the first‐line setting by international guidelines. 7 , 12 Tocilizumab is recommended for subsequent treatment lines, or as first‐line therapy in regions where siltuximab is not approved or available. 12 Rituximab is often used as both first‐ or second‐line therapy in patients with iMCD in clinical practice and is formally recommended for subsequent treatment lines following siltuximab treatment, or as first‐line therapy in patients with mild symptoms. 12 , 22 At the time of treatment, siltuximab was not available for reimbursement by the national payer in Poland as first‐line treatment for iMCD. However, reimbursement was available providing our patient first received treatment with tocilizumab and rituximab, as stipulated by a special administrative procedure. Therefore, following the diagnosis of iMCD, treatment with rituximab was initially administered in our patient but was discontinued after one treatment course due to lack of clinical improvement. Following this, we started treatment with siltuximab, which has previously demonstrated superior progression‐free survival compared with rituximab. 23 In our patient, treatment with siltuximab over 6 weeks resulted in complete resolution of symptoms (such as lymphadenopathy) and normalization of inflammatory markers, including IL‐6 and SAA, which was not achieved with prior treatment with tocilizumab or colchicine. Siltuximab was also not associated with any adverse events. Interestingly, our patient's SAA levels corresponded with symptom severity throughout the duration of treatment, with normalization of SAA levels correlating with radiological remission. This suggests that SAA concentration could be a potential biomarker for iMCD. As of August 2023, treatment with siltuximab was still ongoing. The tolerability and efficacy of siltuximab observed in our patient is in line with previous evidence from both clinical practice and investigational trials of siltuximab in patients with iMCD. 14 , 24 , 25 , 26
4. CONCLUSION
This case report presents a complex presentation of iMCD resembling adult‐onset Still's disease and highlights the challenges commonly associated with the diagnosis of iMCD. Differentiating iMCD from Still's disease, in addition to malignancies and infection‐related disorders, is an essential aspect of diagnosing iMCD. While the introduction of diagnostic guidelines for iMCD in 2017 has helped to make diagnosis more efficient, reaching a timely and accurate diagnosis remains challenging. Our patient had undetected and progressive iMCD over a period of 7 years, which, once diagnosed, could be effectively managed with siltuximab, with symptom control being achieved soon after treatment initiation.
AUTHOR CONTRIBUTIONS
Magdalena Strach: Conceptualization; data curation; investigation; writing – original draft; writing – review and editing. Piotr Kuszmiersz: Data curation; investigation; writing – review and editing. Łukasz Chmura: Investigation; writing – review and editing. Mariusz Korkosz: Supervision; writing – review and editing.
FUNDING INFORMATION
Editorial support for the preparation of this manuscript was funded by EUSA Pharma.
CONFLICT OF INTEREST STATEMENT
The authors declare no competing interests.
CONSENT
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
ACKNOWLEDGMENTS
Editorial assistance was provided by Summer Tredgett of mXm Medical Communications and funded by EUSA Pharma.
Strach M, Kuszmiersz P, Chmura Ł, Korkosz M. A challenging diagnosis of idiopathic multicentric Castleman disease with complex systemic presentation: A case report. Clin Case Rep. 2023;11:e7981. doi: 10.1002/ccr3.7981
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author on reasonable request.
REFERENCES
- 1. Dispenzieri A, Fajgenbaum DC. Overview of Castleman disease. Blood. 2020;135(16):1353‐1364. doi: 10.1182/blood.2019000931 [DOI] [PubMed] [Google Scholar]
- 2. Castleman B, Iverson L, Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma. Cancer. 1956;9(4):822‐830. doi: [DOI] [PubMed] [Google Scholar]
- 3. Lomas OC, Streetly M, Pratt G, et al. The management of Castleman disease. Br J Haematol. 2021;195(3):328‐337. doi: 10.1111/bjh.17688 [DOI] [PubMed] [Google Scholar]
- 4. Carbone A, Borok M, Damania B, et al. Castleman disease. Nat Rev Dis Primers. 2021;7(1):84. doi: 10.1038/s41572-021-00317-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fajgenbaum DC, Uldrick TS, Bagg A, et al. International, evidence‐based consensus diagnostic criteria for HHV‐8‐negative/idiopathic multicentric Castleman disease. Blood. 2017;129(12):1646‐1657. doi: 10.1182/blood-2016-10-746933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Joshua D, Brandstadter DCF. How we manage idiopathic multicentric Castleman disease. Clin Adv Hematol Oncol. 2022;20(9):564‐571. [PMC free article] [PubMed] [Google Scholar]
- 7. European Medicines Agency . Sylvant Summary of Product Characteristics. 2019. Accessed 14 September, 2022. https://www.ema.europa.eu/en/documents/product‐information/sylvant‐epar‐product‐information_en.pdf
- 8. Fajgenbaum DC. Novel insights and therapeutic approaches in idiopathic multicentric Castleman disease. Blood. 2018;132(22):2323‐2330. doi: 10.1182/blood-2018-05-848671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Koga T, Sumiyoshi R, Kawakami A, Yoshizaki K. A benefit and the prospects of IL‐6 inhibitors in idiopathic multicentric Castleman's disease. Mod Rheumatol. 2019;29(2):302‐305. doi: 10.1080/14397595.2018.1532383 [DOI] [PubMed] [Google Scholar]
- 10. Sarosiek S, Shah R, Munshi NC. Review of siltuximab in the treatment of multicentric Castleman's disease. Ther Adv Hematol. 2016;7(6):360‐366. doi: 10.1177/2040620716653745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sumiyoshi R, Koga T, Kawakami A. Candidate biomarkers for idiopathic multicentric Castleman disease. J Clin Exp Hematop. 2022;62(2):85‐90. doi: 10.3960/jslrt.22010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. van Rhee F, Voorhees P, Dispenzieri A, et al. International, evidence‐based consensus treatment guidelines for idiopathic multicentric Castleman disease. Blood. 2018;132(20):2115‐2124. doi: 10.1182/blood-2018-07-862334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. United States Food and Drug Administration . Sylvant Prescribing Information, 2014. Accessed 14 September, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/125496s000lbl.pdf
- 14. van Rhee F, Wong RS, Munshi N, et al. Siltuximab for multicentric Castleman's disease: a randomised, double‐blind, placebo‐controlled trial. Lancet Oncol. 2014;15(9):966‐974. doi: 10.1016/S1470-2045(14)70319-5 [DOI] [PubMed] [Google Scholar]
- 15. European Medicines Agency . MabThera Summary of Product Characteristics. 2008. Accessed 14 September, 2022. https://www.ema.europa.eu/en/documents/product‐information/mabthera‐epar‐product‐information_en.pdf
- 16. European Medicines Agency . RoActemra Summary of Product Characteristics. 2013. Accessed 14 September, 2022. https://www.ema.europa.eu/en/documents/product‐information/roactemra‐epar‐product‐information_en.pdf
- 17. Yu L, Shi M, Cai Q, et al. A novel predictive model for idiopathic multicentric Castleman disease: the international Castleman disease consortium study. Oncologist. 2020;25(11):963‐973. doi: 10.1634/theoncologist.2019-0986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van de Beek D, Cabellos C, Dzupova O, et al. ESCMID guideline: diagnosis and treatment of acute bacterial meningitis. Clin Microbiol Infect. 2016;22(Suppl 3):S37‐S62. doi: 10.1016/j.cmi.2016.01.007 [DOI] [PubMed] [Google Scholar]
- 19. Gerfaud‐Valentin M, Jamilloux Y, Iwaz J, Sève P. Adult‐onset Still's disease. Autoimmun Rev. 2014;13(7):708‐722. doi: 10.1016/j.autrev.2014.01.058 [DOI] [PubMed] [Google Scholar]
- 20. González García A, Giraldo WAS, Hita JLM, de la Peña JLP. Comparison of the clinical and laboratory features at onset between multicentric Castleman's disease and adult‐onset Still's disease. Ann Rheum Dis. 2017;76:413 (abstract). [Google Scholar]
- 21. National Institute for Health and Care Excellence . Anakinra for treating Still's disease. 2021. Accessed 14 September, 2022. https://www.nice.org.uk/guidance/ta685/resources/anakinra‐for‐treating‐stills‐disease‐pdf‐82609381733317
- 22. Fajgenbaum DC, van Rhee F, Nabel CS. HHV‐8‐negative, idiopathic multicentric Castleman disease: novel insights into biology, pathogenesis, and therapy. Blood. 2014;123(19):2924‐2933. doi: 10.1182/blood-2013-12-545087 [DOI] [PubMed] [Google Scholar]
- 23. Yu L, Tu M, Cortes J, et al. Clinical and pathological characteristics of HIV‐ and HHV‐8‐negative Castleman disease. Blood. 2017;129(12):1658‐1668. doi: 10.1182/blood-2016-11-748855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lang E, Sande B, Brodkin S, van Rhee F. Idiopathic multicentric Castleman disease treated with siltuximab for 15 years: a case report. Ther Adv Hematol. 2022;13:20406207221082552. doi: 10.1177/20406207221082552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chrysochoou S, Kreft A, Schneider E. Siltuximab‐related favorable clinical outcome for a patient suffering from idiopathic multicentric Castleman disease. Case Rep Hematol. 2022;2022:1840589. doi: 10.1155/2022/1840589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. van Rhee F, Casper C, Voorhees PM, et al. A phase 2, open‐label, multicenter study of the long‐term safety of siltuximab (an anti‐interleukin‐6 monoclonal antibody) in patients with multicentric Castleman disease. Oncotarget. 2015;6(30):30408‐30419. doi: 10.18632/oncotarget.4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author on reasonable request.