

Abstract
Allergic diseases arise in susceptible individuals in part because of decrements in protective pathways. The mechanism by which these anti-inflammatory molecules become repressed remains unclear. We have previously reported that epithelial dectin-1 prevents aberrant type 2 responses and is downregulated in the epithelium of allergic patients. Here we report that dectin-1 is constitutively expressed by the respiratory epithelium in humans, and that IL-33 specifically acts as a repressor of dectin-1. Mechanistically, this occurs via IL-33-dependent STAT3 activation and the subsequent repression of the dectin-1 gene, CLEC7A. We have identified a novel enhancer region upstream of the proximal promoter of CLEC7A that is only accessible in epithelial cells, but not in hematopoietic cells. Epigenetic repression of CLEC7A through this newly identified locus, downstream of an aberrant IL-33-STAT3 axis, occurs in the epithelium of allergic individuals. Collectively, our data identify a mechanism of epigenetic fine-tuning of dectin-1 expression in epithelial cells that may participate in allergenicity.
Keywords: allergy, allergic diseases, IL-33, dectin-1, epithelium
Introduction
Allergic diseases such as asthma, allergic rhinitis, chronic rhinosinusitis with nasal polyps (CRSwNP) are driven by dysregulated type 2 responses. There are several pathways that prevent overzealous activation of these responses and confer protection to the development of allergy. In susceptible individuals a decrement in these protective mechanisms can drive disease upon exposure to sources of allergens1–3. Several molecular pathways, including pattern recognition receptors like CLEC10A, TLR2, TLR7/8, and TLR9, as well as well-known anti-inflammatory intracellular mediators like A20, have been shown to be protective in asthma2–5. Some of these pathways are epithellialy-expressed and their activity can be decreased in allergic patients2,6. Consistent with this, we observed lower levels of dectin-1, a c-type lectin receptor (CLR) encoded by the CLEC7A gene, in epithelial cells from individuals with asthma and CRSwNP and found that it protects against the development of allergic asthma by recognizing invertebrate tropomyosin in house dust mite (HDM) extract and by preventing the aberrant release of IL-33, an epithelial-derived type 2-promoting cytokine1.
It is thought that in susceptible individuals, a combination of genetic and epigenetic mechanisms contributes to decrements in protective pathways. Single nucleotide polymorphisms (SNP) have been associated with the regulation of asthma-protective genes. For example, a coding SNP in the A20 gene, TNFAIP3, impairs protein function and increases the propensity of developing asthma2. Further, we found an intronic SNP in the CLEC7A locus associated with lower expression of CLEC7A in children with asthma1. However, in addition to genetic mechanisms of gene regulation, several epigenetic modifications in pro-inflammatory and anti-inflammatory genes have been identified in patients with allergic diseases7–10. These epigenetic mechanisms can, in combination with SNPs, control gene expression in disease, and have also been shown to regulate these transcripts in the asthmatic epithelium10–12. Epigenetic pathways involve not only DNA methylation and non-coding RNAs, but also histone modifications, which work in concert with repressors, enhancers and transcription factors to regulate gene expression7,10. The importance of these mechanisms in allergy is especially evident in studies done in T cells. For example, enhancer regions in the genes coding for type 2 cytokines in T cells are controlled by transcription factors like STAT6 and GATA3 and confer susceptibility to allergic diseases13,14. Moreover, a recent report shows differentially regulated enhancer regions are associated with upregulated and downregulated genes in epithelial cells from asthmatics12. Despite this, little remains known about the epigenetic regulation of gene expression in the epithelium and the identity of external mediators that can influence this biology.
Based on this, we set out to determine whether there are epigenetic mechanisms that repress epithelial dectin-1 expression in allergy. Understanding how these restorative mechanisms become dysregulated is an essential aspect in expanding our knowledge of allergic disease pathogenesis. Here we report that dectin-1 expression is inhibited by IL-33 in human and mouse respiratory epithelial cells, but not in immune cells. Our findings show the existence of a previously unknown enhancer region upstream of the dectin-1 gene that confers unique regulation of dectin-1 in the epithelium. We find that this region of chromatin can be bound by IL-33-induced STAT3 and acts to repress dectin-1 gene expression.
Materials and Methods
Mice
BALB/c mice were obtained from Jackson Labs (Bar Harbor, Maine) and bred in our facility. Il33cit/cit mice on the BALB/c background (gift from Dr. Andrew McKenzie) were bred to BALB/c to generate heterozygous animals. Il33cit/+ mice were used to generate homozygous progeny that was used for experiments. Mice were housed in a specific pathogen free animal facility in micro-isolator cages. Mice were provided autoclaved food (Lab diet 5010) and water ad libitum. All procedures were approved by the Animal Care and Use Committee of Johns Hopkins University.
House dust mite (HDM)
HDM was obtained from Stallergenes Greer. Vials used in experiments had Der p 1 content ranging from 114.21–245.95 microgram/vial, and endotoxin 42.25–2874 EU/vial.
HDM in vivo administration
Il33+/+ and Il33cit/cit mice were given PBS (40 ul) or 50 ug HDM intratracheally on days 0 and 14. Mice were sacrificed on day 17 for analysis of dectin-1 expression.
Flow cytometry
Mouse lung cells were obtained by digestion of lung tissue with 0.05 mg/ml Liberase TL (Roche) and 0.5 mg/ml DNaseI (Sigma) for 45 min at 37°C in 5% CO2. Digested tissue was filtered through a 70-um nylon mesh (BD Biosciences) and centrifuged. Pellet was resuspended in red blood cell lysis buffer (ACK lysis buffer). Recovered cells were counted (trypan blue exclusion), plated at 4–5×106 cells/ml. Cells were washed with PBS and labeled with Zombie Aqua live/dead dye (Biolegend) for 10 min at RT, and blocked with 10 ug/ml anti-CD16/32 (BioLegend or BioXCell) for an additional 20 min at RT. Cells were stained with fluorochrome-labeled antibodies as detailed in Table S1. Human sinonasal tissue was processed similarly to mouse lungs. Cells were labeled with live/dead dye (Zombie Aqua) for 10 min at RT and blocked with human TruStain FcX (BioLegend) for 20 min at RT. See Table S1 for a list of antibodies used. Data was acquired on an LSRII flow cytometer (BD Biosciences) and gated to exclude debris and to select single cells (SSC-W/SSC-A). Data was analyzed using FlowJo (BD Biosciences).
Human bronchial epithelial cell line culture
16HBE14o- (Sigma) and BEAS-2B (ATCC) were maintained in DMEM media (10% FBS). For transfections, 16HBE were seeded at 10,000 cells/well of a 96-well flat-bottom dish and transfected using FuGENE HD (Promega).
SNEC and NHBE cell culture
Human nasal epithelial cells and sinonasal mucosa were obtained from nasal brushings and tissue removed during endoscopic sinus surgery from control and patients with or without nasal polyps as previously described. The research protocol was approved through the Johns Hopkins Institutional Review process, and all subjects gave signed informed consent. Inclusion criteria included continuous symptoms of rhinosinusitis as defined by the AAO-HNS Chronic Rhinosinusitis Task Force for greater than 12 weeks, computed tomography of the sinuses revealing isolated or diffuse sinus mucosal thickening or air-fluid levels, and nasal polyps visible on diagnostic endoscopy. None of the subjects had a history of tobacco use, cystic fibrosis, ciliary dyskinesia, systemic inflammatory or autoimmune disease, or immunodeficiency. Characteristics of the subjects are summarized in Table S2. Isolated SNECs cells were grown in Bronchial Epithelial cell Growth Media (BEGM bullet kit, Lonza). At 80% confluency, cells were lifted using trypsin-EDTA (0.05%, Gibco) and seeded into a 96-well culture plate pre-coated with fibronectin-collagen. NHBE (Lonza) were cultured similarly to SNECs. Upon reaching 80–90% confluency, cells were starved overnight in BEGM media without the bovine pituitary extract (BEGMnoBP). Cells were treated with HDM (100 ug/ml) for 2h and cells were harvested for RNA. Where indicated, cells were pretreated for 2h with 5 uM stattic (Cayman) or DMSO, or 5 ug/ml isotype or neutralizing monoclonal anti-hIL-33 antibodies (R&D Systems, MAB36254), followed by stimulation with rIL-33 (Peprotech), HDM (100 ug/ml) for 2h. RNA was extracted (TRIzol), and reverse-transcribed to analyze CLEC7A expression. Primers are designed to span an intron to avoid co-amplification of genomic DNA (Table S3).
Phosphoflow
CRSwNP hSNECS were cultured in collagen/fibronectin-coated 6-well plates in BEGM media. Upon reaching 80–90% confluency, cells were starved overnight in BEGMnoBP. Cells were stimulated for 2h with media or recombinant human IL-33 (Peprotech). Cells were then placed on ice, washed with ice-cold PBS and incubated with ice-cold 0.25% trypsin on ice for 25–35 mins. Cells were detached with gentle pipetting and 32% paraformaldehyde (Electron Miscroscopy Sciences) was directly added to the media for a final concentration of 1.6%, and fixed for 10 min at RT. Cells were transferred to 15 ml conical tubes, spun, PFA was removed and cells were permeabilized with 500 μl ice-cold 90% methanol for 60 min at 4 °C. Cells were washed once in PBS+0.2% BSA and stained for 30 min with anti-human STAT3 pTyr705-Alexa Fluor 647 (BioLegend) in PBS+0.2% BSA at RT. Cells were washed twice in PBS+0.2% BSA.
Transfection of SNECs and NHBE
Transfection was done in BEGM media in 96-well dishes (65–85% confluent) with 0.098 ug CLEC7A.luc and 0.002 ug pGL4.PGK control vector per well (Promega) using Trans-IT X2 (Mirus). The following day, cells were starved in BEGMnoBP overnight. Media was removed, cells were rinsed once in PBS, and 150 ul/well BEGMnoBP was added, after 4–6h, 25 ul was collected for determination of secreted Nano-Glo, and cells lysed (25 ul/well, Passive Lysis Buffer, Promega) for detection of the firefly control (pGL4-PGK).
Mouse tracheal epithelial cells
Mouse tracheal epithelial cells were isolated from tracheas digested overnight at 4 °C in Ham’s F12 medium plus pronase (1 mg/ml; Roche). Cells were cultured for 3 h on Primaria plates (Falcon) for removal of fibroblasts. Non-adherent cells were resuspended in BEGM and plated at 25,000 cells per well of a 96-well dish. The following day, cells were harvested for RNA and determination of Clec7a expression (Table S3 for primers).
Dendritic cell culture
Human normal dendritic cells (Lonza) were cultured in 50 ng/ml GM-CSF and 50 ng/ml IL-4 for 5 days, after which they were stimulated with HDM (100 ug/ml) in combination with 10 ug/ml isotype control or neutralizing anti-IL-33 antibodies for 4h.
Plasmids
To generate CLEC7A.luc reporter vectors, a 3.5kb fragment of the human CLEC7A promoter region (chr12:10129421–10132905) was synthesized (GenScript) and cloned into the secreted Nano-Glo luciferase vector pNL1.3 (Promega). Mutation of the STAT3 binding site (wt: TTTCCAAGCAC, mut: TGGAAGGGCAC) in CENSER was performed commercially (GenScript). CENSER and proximal deletion constructs were done using the QuikChange mutagenesis kit (Agilent). Deletions were verified by gel electrophoresis.
Chromatin immunoprecipitation
0.5–1.0×106 epithelial cells (BEAS-2B) were trypisinized and fixed in 1% PFA (8 min) followed by quenching with 1 M glycine (10 min). Cells were washed twice in ice-cold PBS before storage at −80°C. Cells were lysed in 150 uL lysis buffer (1% SDS, 50 mM Tris-Cl (pH 8.1), 10 mM EDTA (pH 8.0)) and sonicated in TPX microtubes (Diagenode) in a 4°C-cooled water bath sonicator (Diagenode Bioruptor, HI setting, 30 sec ON/30 sec OFF) for 25–30 cycles to properly shear the DNA. Lysates were cleared by centrifugation (10 min, 18,000xg) and pre-cleared for non-specific binding with 40 ul BSA-blocked protein A/G magnetic Dynabeads (Invitrogen) for 4h at 4°C. Lysates were diluted 10-fold (1.1% Triton X-100, 0.01% SDS, 167 mM NaCl, 16.7 mM Tris-Cl pH 8.1, 1.2 mM EDTA) and incubated with 3.5 ug anti-STAT3 (clone ST3–5G7, Invitrogen) or anti-GFP (Santa-Cruz) antibodies overnight at 4°C. Antibodies were captured with 40 ul BSA-blocked protein A/G magnetic Dynabeads on a rocking platform for 4h at 4°C. Beads were washed in 1 mL low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.1, 150 mM NaCl), then, high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris pH 8.1, 500 mM NaCl), lithium chloride buffer (0.25 M LiCl, 1% Nonidet P-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris pH 8.1), followed by 2 washes in Tris-EDTA (pH 8.1). Chromatin was eluted from the beads using 200 ul elution buffer for 15 min at RT (1% SDS, 0.1 M NaHCO3), followed by de-crosslinking in 0.3 M NaCl at 65°C overnight. Chromatin was RNase-digested (30 min at 37°C), followed by proteinase K (1h at 56°C), and purified using PCR purification columns (Qiagen). PCR was performed using primers in the CLEC7A locus (see Table S3). All data was normalized to input and presented as enrichment over the IGX1A negative control region (primers from Qiagen).
Results
Respiratory epithelial cells express dectin-1
While the expression, regulation and function of prototypical PRRs such as TLRs have been extensively studied in the allergic epithelium, our understanding of the role of CLRs in allergic diseases is mostly unclear and has majorly been derived from their expression on hematopoietic cells (macrophages, dendritic cells, etc). Exploration of CLRs in human tracheal and bronchial epithelial cells (RNA-seq dataset GSE101993) reveals that several are constitutively expressed, including CLEC7A, the gene coding for dectin-1, suggesting that they may play an important role in regulating epithelial function (Fig. 1a). The majority of these CLRs are poorly understood, and many have unknown function, especially on the epithelium. Dectin-1 is probably the best studied CLR and its function is relatively well understood in myeloid cells in context of fungal infections. However, we1 and others15–20 have reported expression of dectin-1 in epithelial cells, and found that it has a key role in preventing aberrant IL-33 release in response to dust mite allergen. Further analysis of the CLEC7A locus using ENCODE data shows the transcribed exons across the human respiratory epithelium in nasal, tracheal, bronchial and airway epithelial cells (Fig. 1b). These data support the notion of epithelial cells as important non-hematopoietic dectin-1-expressing cells. In humans and mice, respiratory epithelial cells can be divided into subsets that can be deconvoluted based on EpCAM and CD104 or CD49f expression21–24. Where EpCAM+CD104− are luminal cells, EpCAM+CD104+ are luminal progenitors and EpCAMloCD104+ represent basal cells. We sought to characterize the expression of dectin-1 in these subsets. To this end, we used human sinonasal tissue, and we found that luminal progenitor and basal epithelial cells have the highest levels of dectin-1-expressing cells, with marginal expression in luminal epithelial cells (Fig. 1c). Our findings demonstrate that the epithelium maintains tonic levels of dectin-1 across the respiratory system.
Figure 1. Respiratory epithelial cells express dectin-1.
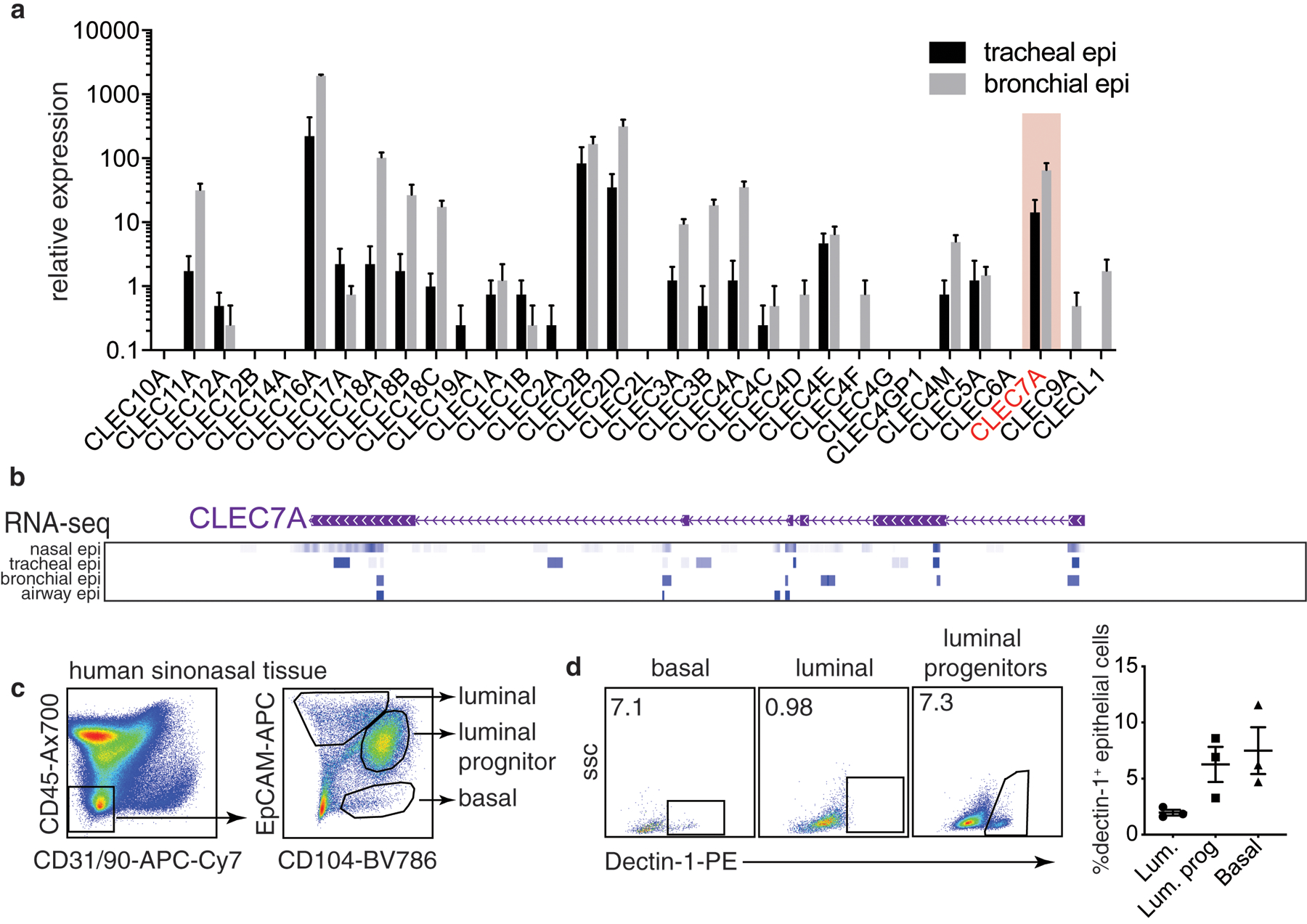
(a) Levels of CLR mRNAs in human bronchial and tracheal epithelial cells from healthy donors (RNA-seq, GSE101993). (b) RNA-seq from ENCODE highlighting CLEC7A exon transcription in nasal, tracheal, bronchial and airway epithelial cells. (c) gating scheme identifying various lung epithelial subsets by flow cytometry and (d) expression of dectin-1 in these populations. Data is means+SEM and pooled from 4 (a) or 3 (d) donors.
Allergen-induced IL-33 represses epithelial dectin-1
Our previous work has shown that dectin-1 is downregulated in the allergic epithelium, and that it functions to inhibits allergen-induced IL-331. Based on this, we postulated that dectin-1 is repressed by an autocrine pathway. As IL-33 is enriched in allergic airway tissue, we hypothesized that epithelial IL-33 may counter dectin-1 expression. To begin to explore this, we treated cultured human sinonasal epithelial cells (hSNECs) from nasal polyp patients with media or HDM and treated with isotype control or blocking anti-IL-33 mAbs. Interestingly, we observed that HDM reduced CLEC7A expression, and this repression was restored when IL-33 was neutralized (Fig. 2a). Interestingly, these culture conditions had no effect on expression of CLEC7A in human primary dendritic cells (Fig. 2b), suggesting a cell-type specific regulation of dectin-1. In line with this, treating hSNECs with rIL-33 (250 pg/ml) inhibited CLEC7A expression (Fig. 2c). Consistent with our human data, isolated mouse tracheal epithelial cells from IL-33 deficient (Il33cit/cit ) mice express more Clec7a than wildtype cells (Fig. 2d). Next, we explored the possibility that exposure to HDM could enhance responsiveness to autocrine IL-33 via upregulating its receptor, IL1RL1/ST2. We found that treating CRSwNP SNECs with HDM upregulated IL1RL1 (ST2) mRNA, but not in controls (supplementary Fig. 1) and suggests that allergen exposure in allergic epithelial cells enhances their production and responsiveness to IL-33. Overall, these data suggest a functional pathway in mice and human in which IL-33 represses epithelial dectin-1 expression.
Figure 2. Allergen-induced IL-33 represses epithelial dectin-1.
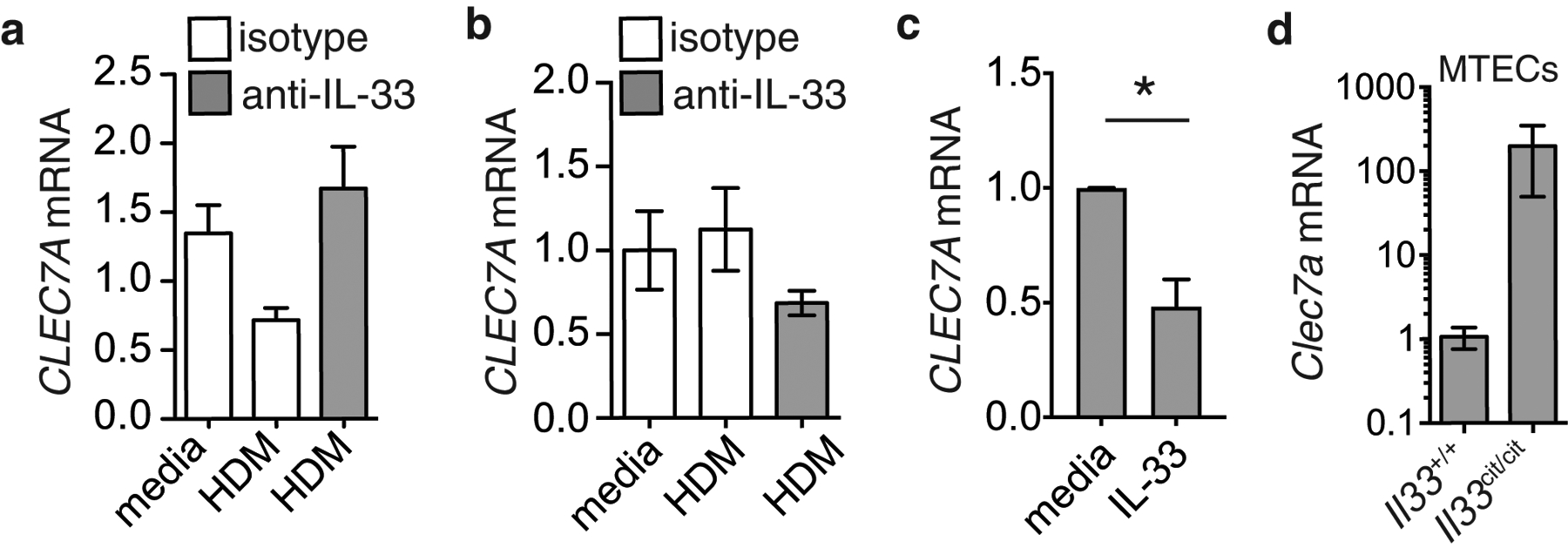
CLEC7A mRNA levels in (a) CRSwNP SNECs or (b) dendritic cells cultured in the presence of media, HDM (100 ug/ml) in combination with isotype or blocking anti-IL-33 antibodies (5 ug/ml) for 2h. (c) CLEC7A mRNA levels in CRSwNP SNECs treated with media or 0.25 ng/ml IL-33 for 2h. (d) Clec7a mRNA in cultured mouse tracheal epithelial cells from wildtype or Il33cit/cit mice. Data is means+SEM and represents representative data from 3 donors or 3 independent experiments.
IL-33 represses epithelial dectin-1 in vivo
To further examine the regulation by IL-33 of dectin-1 in vivo, we exposed, previously characterized IL-33 deficient (Il33cit/cit) mice25, to PBS or HDM. We find that lack of IL-33 results in an increase in the percentage of dectin-1+ basal epithelial cells in mice treated with HDM, when compared to control (Fig. 3a,b). Similarly, we find that the frequency of dectin-1+ luminal progenitor epithelial cells increases significantly in Il33-deficient animals after HDM exposure, as compared to IL33-sufficient mice (Fig. 3c,d). Surprisingly, we show that, while dectin-1 expression is abundant in myeloid cells, IL-33 does not appear to regulate it. Various myeloid populations were identified (see gating approach, supplementary Fig. 2), and we found that the frequency of dectin-1+ alveolar macrophages (Fig. 3e), interstitial macrophages (Fig. 3f), and CD11b+ dendritic cells (Fig. 3g) remains unaffected in Il33cit/cit animals. Overall, our data suggest that IL-33 represses the expression of dectin-1 specifically in epithelial cells and not hematopoietic cells, implying the involvement of cell-type specific epigenetic regulation of dectin-1. We did not observe that HDM caused a decrease in the frequency of mouse dectin-1+ epithelial cells, as it does in human nasal polyp-derived epithelial cells, suggesting that HDM may elicit non-epithelial factors that could promote survival or proliferation of respiratory epithelia. Nonetheless, these data overall demonstrate that allergen-induced IL-33 specifically represses epithelial dectin-1 in mouse and humans.
Figure 3. IL-33 represses epithelial dectin-1 in vivo.

Il33+/+ and Il33cit/cit mice were treated with PBS or HDM and lung dectin-1+ cells were analyzed. Dectin-1+ basal epithelial cells in (a) representative flow plots and (b) frequencies. Dectin-1+ luminal progenitors in (c) representative flow plots and (d) frequencies. Dectin-1+ (e) alveolar macrophages (f) interstitial macrophages and (g) CD11b+ dendritic cells. Data is means+SEM and representative of at least 2 independent experiments. Data was analyzed by one-way ANOVA followed by post-hoc test. *p<0.05; ***p<0.001.
Identification of a functional epithelial-specific enhancer region in the CLEC7A loci
To understand the epithelial-specific regulation of dectin-1, we investigated its regulatory landscape using ENCODE data where we analyzed DNaseI hypersensitive sites (DHS-seq) from primary human epithelial cells from various tissues, as well as from hematopoietic cells such as CD14+ monocytes and CD1c+ dendritic cells. This analysis revealed a previously reported proximal promoter26, which is active in both cell types, as demonstrated by an open chromatin configuration (Fig. 4a). In addition, we found a second, previously unreported, distal site of open chromatin, seen only in epithelial cells, we refer to this region as the ClEc7a Nonhematopoietic-SpEcific Region-CENSER. This suggests that CENSER acts as a unique regulatory region, differentially regulating dectin-1 expression between non-hematopoietic and hematopoietic cells. Moreover, we found this region to be enriched for the histone modifications H3K4me1 and H3K4me3 in epithelial cells (Supplementay Fig. 3), further supporting the role of CENSER as a regulator of CLEC7A expression in the epithelium. To evaluate the function of these regulatory regions of chromatin in controlling CLEC7A expression, we generated a 3.5kb promoter construct containing both the proximal and CENSER region upstream of secreted NanoLuc. We observed significant activity of this CLEC7A reporter construct in transiently transfected primary human sinonasal epithelial cells (Fig. 4b). Deletion of the proximal region attenuated CLEC7A promoter activity in primary human nasal epithelial cells, consistent with the important role of proximal promoters in regulating gene function (Figure 4b). Notably, removal of CENSER also reduced CLEC7A promoter activity in SNECs (Fig. 4b). Similarly, deletion of the CENSER and proximal regions also decreased CLEC7A promoter activity in human bronchial epithelial (NHBE) cells (Fig. 4c). Overall our data suggests a role for this CENSER region as an enhancer regulating dectin-1 expression in epithelial cells.
Figure 4. Identification of a functional epithelial-specific enhancer region in the CLEC7A loci.

(a) DHS-seq data from ENCODE for the CLEC7A locus for primary esophageal (EE), bronchial (BE), prostate (PE), and mammary epithelial (ME) cells, keratinocytes (KE), CD14+ monocytes and CD1c+ dendritic cells. CLEC7A promoter activity in (b) SNECs and (c) NHBE transfected with full-length or truncated promoter constructs. Levels of (d) TFAP2C, (e) MYC, (f) TFAP2A, (g) FOS, (h) BACH1, and (i) STAT3 mRNA in control and CRSwNP SNECs. Data is means+SEM and representative of 2–3 independent experiments. Data was analyzed by one-way ANOVA, Student’s T test, or Mann Whitney test (control vs. CRSwNP). *p<0.05, ***p<0.001, ****p<0.0001.
In order to elucidate the mechanism through which CENSER region controls dectin-1 promoter activity we analyzed ENCODE ChIP-seq data, and this revealed this region to be a target of the transcription factors TFAP2A, TFAP2C, MYC, FOS, BACH1 and STAT3 (Supplementary Fig. 4). We next assessed the levels of these CENSER-binding transcription factors (TFs) in cultured SNECs from control and CRSwNP patients (Fig. 4d–i). We found STAT3 mRNA to be significantly higher in the allergic epithelium (Fig. 4i). Consistently, exposure to HDM has been shown to activate STAT3 in mouse lung epithelial cells, leading to allergen-induced experimental asthma27,28. These data suggest a pro-allergic function for STAT3 where it could act as repressor of dectin-1 during aberrant Th2 responses.
STAT3 represses CLEC7A through CENSER.
IL-33 has been shown to phosphorylate STAT3 in skin, gut and breast epithelial cells29–32. Here we confirm that at low (250 pg/ml) or more standard (10 ng/ml) concentrations, IL-33 leads to an increase in the frequency of STAT3 pTyr705+ hSNECs (Fig. 5a,b). Next, to test the direct role of STAT3-CENSER interaction in controlling dectin-1 expression, we performed site-directed mutagenesis of the CENSER STAT3 binding site. We found that disrupting the STAT3 binding site significantly enhanced CLEC7A promoter activity in bronchial epithelial cell line (Fig. 5c), establishing the repressive function of STAT3 binding at the CLEC7A locus. Analysis of NCBI GEO data (GDS3106) also demonstrates that levels of Clec7a are higher in Stat3−/− mouse tracheal epithelial cells, as compared to controls (Fig. 5d). Next, we performed ChIP-PCR in bronchial epithelial cell line to assess STAT3 binding at the CLEC7A locus in response to IL-33. Using primers to amplify the regions around or at the proximal and CENSER, we found that, compared to control (anti-GFP), IL-33 promoted recruitment of STAT3 to the CENSER region, but not others (Fig. 5e). STAT3 is not only activated by IL-33, but can act as a transcriptional repressor33–35. Based on that and the fact that it can be enriched at CENSER, we hypothesized that an IL-33-STAT3 axis acts to repress CLEC7A transcription. We set out to test whether inhibition of STAT3 could upregulate CLEC7A in cultured CRSwNP SNECs. We found that HDM- (Fig. 5f) and IL-33-mediated (Fig. 5g) repression of CLEC7A in nasal polyp epithelial cells could be completely restored by the STAT3 inhibitor stattic. Taken together, these findings reveal that an aberrant IL-33-STAT3 axis represses CLEC7A through its interaction with a newly described enhancer region. These data uncover a potential novel epigenetic mechanism underlying the regulation of CLEC7A in the respiratory epithelium.
Figure 5. STAT3 represses CLEC7A through CENSER.

(a) representative flow plots and (b) frequencies of STAT3 pTyr705 phosphoflow in CRSwNP hSNECs treated with IL-33 for 2h. CLEC7A promoter activity in (c) bronchial epithelial cell line or (d) Clec7a mRNA in isolated mouse tracheal epithelial cells from Stat3+/+ and Stat3−/− mice. (e) STAT3 binding at the CLEC7A locus in bronchial epithelial cell line treated with media or IL-33 (10 ng/ml) for 2h. CLEC7A mRNA in CRSwNP SNECs treated with (f) HDM (100 ug/ml) or (g) IL-33 (0.25 ng/ml) and vehicle or 5 uM STAT3 inhibitor, stattic for 2h. Data is means+SEM and representative of 2–3 independent experiments, representative of 3 donors (a,b,f,g). Data was analyzed by one-way ANOVA followed by post-hoc test or two-way ANOVA followed by Bonferroni’s multiple comparisons test (panel e, where significances are displayed for IL-33 (anti-STAT3) vs. IL-33 (anti-GFP)). *p<0.05; **p<0.01; ***p<0.001, ****p<0.0001.
Discussion
In susceptible individuals, allergic airway diseases like asthma and CRS are thought to develop due to the aberrant expression of pathogenic pathways. However, we now know that insufficiencies in protective mechanisms also contribute to disease. Recent reports have uncovered molecular pathways, including PRRs like dectin-1 and CLEC10A, that mediate protection against aberrant type 2 responses1–4 and where decrement in these pathways, many of which in the epithelium1,2, result in a greater propensity to develop allergy. Thus, understanding the function and regulation of these protective epithelial sensors will be important to explain how individuals develop susceptibility to allergy.
Dectin-1, while traditionally associated with expression on macrophages and dendritic cells, is, as we found, constitutively expressed by human respiratory epithelial cells, and it is also expressed by epithelial cells of other barrier surfaces like the skin, eyes, and gut15–17,19,20,36–38. Nevertheless, little remains known about the regulation and function of epithelial dectin-1. Reports have shown that stimulation of human corneal epithelial cells with heat-killed Candida albicans results in the upregulation of dectin-139, and that it is required for the secretion of anti-fungal peptidoglycan recognition proteins (PGLYRP). Consistent with this, live, but not heat- killed, Aspergillus fumigatus conidia upregulated dectin-1 in primary human bronchial epithelial cells (HBE) in a TLR2-dependent manner40. Furthermore, exposure of HBE to A. fumigatus resulted in dectin-1-dependent cytokine release40. These findings are in line with the well-documented role of dectin-1 as a receptor for fungal β-glucan moieties, where this interaction triggers anti-fungal responses.
In addition to epithelial dectin-1 expression being triggered by fungus, some have shown that it can be induced by TLR2 ligand-containing bacteria. Specifically, while Mycobacterium tuberculosis (Mtb) and Staphylococcus aureus induced dectin-1 expression in the human alveolar epithelial A549 cell line36, TLR4-ligand containing bacteria, like Escherichia coli, could not activate dectin-136. Functionally, siRNA-mediated knockdown of dectin-1 resulted in impaired cytokine response to Mtb and elevated Mtb colony forming units (CFUs)36. Others have shown that A549 cells overexpressing dectin-1 and infected with non-typable Haemophilus influenzae are driven to produce cytokines but this is abolished in cells expressing a non-functional dectin-120. In the context of allergy, we have previously shown that in addition to these microbial ligands, the HDM protein allergen Der p 10, also known as invertebrate tropomyosin, is recognized by dectin-1 and this interaction prevent aberrant release of IL-33 by the epithelium1. Taken together, these data suggest that intact levels of epithelial dectin-1 promote anti-microbial responses and protect against allergic manifestations.
While some microbial interactions appear to drive dectin-1 expression in epithelial cells, the molecular mechanisms that regulate levels of epithelial dectin-1 are not understood. Although we have previously reported that an intronic SNP in CLEC7A is associated with lower gene expression in children with asthma1, epigenetic influences in response to inflammatory mediators also significantly contribute to gene expression. This supports the longstanding view that gene-environment interactions, especially in the epithelium, are regulators of susceptibility to allergy.
We found that exposure to dust-mite allergen led to a decrease in CLEC7A mRNA in epithelial cells from patients with nasal polyps (CRSwNP), which was restored by neutralizing IL-33. Moreover, mice lacking IL-33 also display significantly more dectin-1+ basal epithelial cells and dectin-1+ luminal progenitors. Consistent with this, IL-33 and other type 2-promoting innate epithelial cytokines like TSLP and IL-25 are known to downregulate molecules in human keratinocytes, such as filaggrin and claudin-1, that protect against atopic dermatitis30,31,41. Thus, IL-33, IL-25 and TLSP may promote disease by decreasing protective mucosal mechanisms in addition to directly driving type 2 inflammation.
We now know that many PRRs are expressed by epithelial cells, and reports have shown that they may function differently in the epithelium than they would on myeloid cells. For example, key studies have reported that epithelial TLR4 is a driver of type 2 responses to HDM42,43, whereas its expression on myeloid cells promotes neutrophilic responses to dust mite43. In addition to cell-type-specific function, PRR expression may also be differentially regulated in hematopoietic and non-hematopoietic compartments. Our data shows that while IL-33 represses epithelial dectin-1, blockade of IL-33 had no impact on haematopoietically expressed dectin-1.
We found that the cell-type specific (dys)regulation of CLEC7A in the human epithelium could be associated with CENSER, an upstream enhancer of the CLEC7A locus, that is in an open chromatin configuration only in epithelial cells, and not in human macrophages or DCs. Thus suggesting that this particular enhancer region confers an added level of regulation for dectin-1 expression in the epithelium. Regulatory regions that are designed to control gene expression in immune versus non-immune cells have been reported before. An epithelial-specific repressor in the TLR4 locus has been found to maintain low levels of TLR4 in gut epithelial cells, while having no effect in monocytes44. Perhaps there is an evolutionary advantage to epigenetically regulate the expression of PRRs differentially in mucosal epithelial cells versus immune cells. Maintaining expression of protective PRRs, like dectin-1, while keeping pro-Th2 PRRs like TLR4 at low levels at the epithelial interface would favor homeostatic responses to environmental exposures.
CENSER, acting as an enhancer, is bound by several experimentally verified transcription factors, including STAT3. Moreover, STAT3 is significantly upregulated in nasal polyp epithelial cells as compared to controls, suggesting that STAT3 may promote type 2 responses. This is supported by other reports showing that epithelial STAT3 promotes type 2 inflammation in the lungs in response to HDM in mice27, and that pharmacological blockade of STAT3 inhibits both IL-13 and IL-17A responses to HDM28 . As STAT3 has been reported to act as a transcriptional repressor33–35, in addition to several reports demonstrating that IL-33 activates STAT331,45,46, we postulated a role for an IL-33-STAT3 axis in inhibiting dectin-1 expression in epithelial cells. Our data shows that impairing STAT3 function enhances dectin-1 gene expression, and thus establishes STAT3 as repressor of dectin-1. Moreover, treating an epithelial cell line with IL-33 leads to the accumulation of STAT3 binding at CENSER, but not at the proximal promoter region. This is similar to other reports demonstrating that repression of filaggrin and claudin-1 in atopic dermatitis is driven by an IL-33-STAT3 axis30,31. This supports the notion that dysregulation of STAT3 in the allergic epithelium is a mechanism that disrupts homeostatic levels of protective molecules like dectin-1. Nevertheless, while we have identified STAT3 as inhibiting the CENSER enhancer, it remains to be determined which TF acts as a driver of CLEC7A transcription through this region.
Taken together, our findings suggest the existence of a pathophysiologically relevant pathway that regulates epithelial dectin-1 expression. We propose that aberrant production of IL-33 by respiratory epithelial cells from allergic individuals results in an autocrine-mediated increase in STAT3 activation. A dysregulated IL-33-STAT3 axis then targets the CENSER enhancer, only accessible in epithelial cells, to repress dectin-1 gene expression. Our results provide new insights into the mechanisms by which dysregulated innate epithelial cytokines may drive allergy through the reduction of protective pathways.
Supplementary Material
Supplementary Figure 1. IL1RL1 mRNA expression in hSNECs from control or CRSwNP following allergen exposure
Supplementary Figure 2. Gating scheme for lung immune cells.
Supplementary Figure 3. ENCODE ChIP-seq enrichment data for H3K4me1 and H3K4me3 at the human CLEC7A locus
Supplementary Figure 4. ENCODE DHS-seq and TF ChIP-seq data for the human CLEC7A locus.
Supplementary Table 1. List of flow antibodies
Supplementary Table 2. Summary of CRS patient characteristics used
Supplementary Table 3. Primer sequences
Acknowledgements:
Hwan Mee Yong has nothing to disclose, Dr. Gour has nothing to disclose, Dr. Sharma has nothing to disclose, Dr. Khalil has nothing to disclose, Dr. Lane reports grants from NIH (R01AI132590) as support during the conduct of the study. This work was funded by the National Institute of Allergy and Infectious Diseases (R01AI127644) to S.Lajoie
References
- 1.Gour N et al. Dysregulated invertebrate tropomyosin-dectin-1 interaction confers susceptibility to allergic diseases. Sci Immunol 3, doi: 10.1126/sciimmunol.aam9841 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuijs MJ et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 349, 1106–1110, doi: 10.1126/science.aac6623 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Kanemaru K et al. Clec10a regulates mite-induced dermatitis. Sci Immunol 4, doi: 10.1126/sciimmunol.aax6908 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Ventura S et al. A20-binding inhibitor of NF-kappaB (ABIN) 2 negatively regulates allergic airway inflammation. J. Exp. Med 215, 2737–2747, doi: 10.1084/jem.20170852 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zakeri A & Russo M Dual Role of Toll-like Receptors in Human and Experimental Asthma Models. Front. Immunol 9, 1027, doi: 10.3389/fimmu.2018.01027 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shikhagaie MM et al. Mapping of TLR5 and TLR7 in central and distal human airways and identification of reduced TLR expression in severe asthma. Clin. Exp. Allergy 44, 184–196, doi: 10.1111/cea.12176 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Yang IV & Schwartz DA Epigenetic mechanisms and the development of asthma. J. Allergy Clin. Immunol 130, 1243–1255, doi: 10.1016/j.jaci.2012.07.052 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reese SE et al. Epigenome-wide meta-analysis of DNA methylation and childhood asthma. J. Allergy Clin. Immunol 143, 2062–2074, doi: 10.1016/j.jaci.2018.11.043 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho SM Environmental epigenetics of asthma: an update. J. Allergy Clin. Immunol 126, 453–465, doi: 10.1016/j.jaci.2010.07.030 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moheimani F et al. The genetic and epigenetic landscapes of the epithelium in asthma. Respir. Res 17, 119, doi: 10.1186/s12931-016-0434-4 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li X et al. eQTL of bronchial epithelial cells and bronchial alveolar lavage deciphers GWAS-identified asthma genes. Allergy 70, 1309–1318, doi: 10.1111/all.12683 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McErlean P et al. Genome-wide profiling of an enhancer-associated histone modification reveals the influence of asthma on the epigenome of the airway epithelium. bioRxiv, 282889, doi: 10.1101/282889 (2018). [DOI] [Google Scholar]
- 13.Tanaka S et al. The enhancer HS2 critically regulates GATA-3-mediated Il4 transcription in T(H)2 cells. Nat. Immunol 12, 77–85, doi: 10.1038/ni.1966 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Koh BH et al. Th2 LCR is essential for regulation of Th2 cytokine genes and for pathogenesis of allergic asthma. Proc. Natl. Acad. Sci. U. S. A 107, 10614–10619, doi: 10.1073/pnas.1005383107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu W, Zhu N, Bai D, Miao J & Zou S The crosstalk between Dectin1 and TLR4 via NF-kappaB subunits p65/RelB in mammary epithelial cells. Int. Immunopharmacol 23, 417–425, doi: 10.1016/j.intimp.2014.09.004 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Zhu CC et al. Dectin-1 agonist curdlan modulates innate immunity to Aspergillus fumigatus in human corneal epithelial cells. Int J Ophthalmol 8, 690–696, doi: 10.3980/j.issn.2222-3959.2015.04.09 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Q et al. Role of Dectin-1 in the innate immune response of rat corneal epithelial cells to Aspergillus fumigatus. BMC Ophthalmol. 15, 126, doi: 10.1186/s12886-015-0112-1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu ZC et al. Up-regulation of Dectin-1 in airway epithelial cells promotes mice defense against invasive pulmonary aspergillosis. Int. J. Clin. Exp. Med 8, 17489–17497 (2015). [PMC free article] [PubMed] [Google Scholar]
- 19.Li C et al. Expression of dectin-1 during fungus infection in human corneal epithelial cells. Int J Ophthalmol 7, 34–37, doi: 10.3980/j.issn.2222-3959.2014.01.06 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heyl KA et al. Dectin-1 is expressed in human lung and mediates the proinflammatory immune response to nontypeable Haemophilus influenzae. MBio 5, e01492–01414, doi: 10.1128/mBio.01492-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li F, He J, Wei J, Cho WC & Liu X Diversity of epithelial stem cell types in adult lung. Stem Cells Int 2015, 728307, doi: 10.1155/2015/728307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rock JR et al. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. U. S. A 106, 12771–12775, doi: 10.1073/pnas.0906850106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McQualter JL, Yuen K, Williams B & Bertoncello I Evidence of an epithelial stem/progenitor cell hierarchy in the adult mouse lung. Proc. Natl. Acad. Sci. U. S. A 107, 1414–1419, doi: 10.1073/pnas.0909207107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holik AZ et al. The LIM-domain only protein 4 contributes to lung epithelial cell proliferation but is not essential for tumor progression. Respir. Res 16, 67, doi: 10.1186/s12931-015-0228-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardman CS, Panova V & McKenzie AN IL-33 citrine reporter mice reveal the temporal and spatial expression of IL-33 during allergic lung inflammation. Eur. J. Immunol 43, 488–498, doi: 10.1002/eji.201242863 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang C et al. Downregulation of PU.1 leads to decreased expression of Dectin-1 in alveolar macrophages during Pneumocystis pneumonia. Infect. Immun 78, 1058–1065, doi: 10.1128/IAI.01141-09 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Simeone-Penney MC et al. Airway epithelial STAT3 is required for allergic inflammation in a murine model of asthma. J. Immunol 178, 6191–6199 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Gavino AC, Nahmod K, Bharadwaj U, Makedonas G & Tweardy DJ STAT3 inhibition prevents lung inflammation, remodeling, and accumulation of Th2 and Th17 cells in a murine asthma model. Allergy 71, 1684–1692, doi: 10.1111/all.12937 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Xiao Y et al. Interleukin-33 Promotes REG3gamma Expression in Intestinal Epithelial Cells and Regulates Gut Microbiota. Cell Mol Gastroenterol Hepatol 8, 21–36, doi: 10.1016/j.jcmgh.2019.02.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryu WI et al. IL-33 down-regulates CLDN1 expression through the ERK/STAT3 pathway in keratinocytes. J. Dermatol. Sci 90, 313–322, doi: 10.1016/j.jdermsci.2018.02.017 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Ryu WI, Lee H, Bae HC, Ryu HJ & Son SW IL-33 down-regulates filaggrin expression by inducing STAT3 and ERK phosphorylation in human keratinocytes. J. Dermatol. Sci 82, 131–134, doi: 10.1016/j.jdermsci.2016.01.011 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Kim JY et al. Interleukin-33/ST2 axis promotes epithelial cell transformation and breast tumorigenesis via upregulation of COT activity. Oncogene 34, 4928–4938, doi: 10.1038/onc.2014.418 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Zhang H et al. STAT3 restrains RANK- and TLR4-mediated signalling by suppressing expression of the E2 ubiquitin-conjugating enzyme Ubc13. Nat Commun 5, 5798, doi: 10.1038/ncomms6798 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramadoss P, Unger-Smith NE, Lam FS & Hollenberg AN STAT3 targets the regulatory regions of gluconeogenic genes in vivo. Mol. Endocrinol 23, 827–837, doi: 10.1210/me.2008-0264 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Timofeeva OA et al. STAT3 suppresses transcription of proapoptotic genes in cancer cells with the involvement of its N-terminal domain. Proc. Natl. Acad. Sci. U. S. A 110, 1267–1272, doi: 10.1073/pnas.1211805110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee HM, Yuk JM, Shin DM & Jo EK Dectin-1 is inducible and plays an essential role for mycobacteria-induced innate immune responses in airway epithelial cells. J. Clin. Immunol 29, 795–805, doi: 10.1007/s10875-009-9319-3 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Peng XD et al. Fungus induces the release of IL-8 in human corneal epithelial cells, via Dectin-1-mediated protein kinase C pathways. Int J Ophthalmol 8, 441–447, doi: 10.3980/j.issn.2222-3959.2015.03.02 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cohen-Kedar S et al. Human intestinal epithelial cells respond to beta-glucans via Dectin-1 and Syk. Eur. J. Immunol 44, 3729–3740, doi: 10.1002/eji.201444876 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Hua X et al. A Novel Innate Response of Human Corneal Epithelium to Heat-killed Candida albicans by Producing Peptidoglycan Recognition Proteins. PLoS One 10, e0128039, doi: 10.1371/journal.pone.0128039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun WK et al. Dectin-1 is inducible and plays a crucial role in Aspergillus-induced innate immune responses in human bronchial epithelial cells. Eur. J. Clin. Microbiol. Infect. Dis 31, 2755–2764, doi: 10.1007/s10096-012-1624-8 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Kim BE, Bin L, Ye YM, Ramamoorthy P & Leung DYM IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J. Invest. Dermatol 133, 2678–2685, doi: 10.1038/jid.2013.223 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hammad H et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat. Med 15, 410–416, doi: 10.1038/nm.1946 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McAlees JW et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. 8, 863–873, doi: 10.1038/mi.2014.117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi K, Sugi Y, Hosono A & Kaminogawa S Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J. Immunol 183, 6522–6529, doi: 10.4049/jimmunol.0901271 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Kroeger KM, Sullivan BM & Locksley RM IL-18 and IL-33 elicit Th2 cytokines from basophils via a MyD88- and p38alpha-dependent pathway. J. Leukoc. Biol 86, 769–778, doi: 10.1189/jlb.0708452 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu F et al. STAT3 Inhibition Partly Abolishes IL-33-Induced Bone Marrow-Derived Monocyte Phenotypic Transition into Fibroblast Precursor and Alleviates Experimental Renal Interstitial Fibrosis. J. Immunol 203, 2644–2654, doi: 10.4049/jimmunol.1801273 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. IL1RL1 mRNA expression in hSNECs from control or CRSwNP following allergen exposure
Supplementary Figure 2. Gating scheme for lung immune cells.
Supplementary Figure 3. ENCODE ChIP-seq enrichment data for H3K4me1 and H3K4me3 at the human CLEC7A locus
Supplementary Figure 4. ENCODE DHS-seq and TF ChIP-seq data for the human CLEC7A locus.
Supplementary Table 1. List of flow antibodies
Supplementary Table 2. Summary of CRS patient characteristics used
Supplementary Table 3. Primer sequences