

Abstract
The lung microenvironment plays a crucial role in maintaining lung homeostasis as well as the initiation and resolution of both acute and chronic lung injury. Acute chest syndrome (ACS) is a complication of sickle cell disease (SCD) like acute lung injury. Both the endothelial cells and peripheral blood mononuclear cells are known to secrete proinflammatory cytokines elevated during ACS episodes. However, in SCD, the lung microenvironment that may favor excessive production of proinflammatory cytokines and the contribution of other lung resident cells, such as alveolar macrophages and alveolar type 2 epithelial (AT-2) cells, to ACS pathogenesis is not completely understood. Here, we sought to understand the pulmonary microenvironment and the proinflammatory profile of lung alveolar macrophages (LAMs) and AT-2 cells at steady state in Townes sickle cell (SS) mice compared to control mice (AA). In addition, we examined lung function and micromechanics molecules essential for pulmonary epithelial barrier function in these mice. Our results showed that bronchoalveolar lavage (BAL) fluid in SS mice had elevated protein levels of pro-inflammatory cytokines interleukin (IL)-1β and IL-12 (p ⩽ 0.05) compared to AA controls. We showed for the first time, significantly increased protein levels of inflammatory mediators (Human antigen R (HuR), Toll-like receptor 4 (TLR4), MyD88, and PU.1) in AT-2 cells (1.4 to 2.2-fold) and LAM (17-21%) isolated from SS mice compared to AA control mice at steady state. There were also low levels of anti-inflammatory transcription factors (Nrf2 and PPARy) in SS mice compared to AA controls (p ⩽ 0.05). Finally, we found impaired lung function and a dysregulated composition of surfactant proteins (B and C). Our results demonstrate that SS mice at steady state had a compromised lung microenvironment with elevated expression of proinflammatory cytokines by AT-2 cells and LAM, as well as dysregulated expression of surfactant proteins necessary for maintaining the alveolar barrier integrity and lung function.
Keywords: Sickle cell disease, acute chest syndrome, alveolar type 2 epithelial cells, surfactant proteins, lung alveolar macrophages, inflammation
Impact statement
Inflammation can trigger acute chest syndrome (ACS), a leading cause of mortality in sickle cell disease (SCD). Existing studies on mechanism of ACS and attendant lung inflammation in SCD have not examined the alveolar epithelium and lung resident macrophages. In this study, we show evidence that lung alveolar macrophages (LAMs) and alveolar type 2 epithelial (AT-2) cells have a proinflammatory profile at “steady state” in Townes sickle cell (SS) mice, concomitant with low levels of anti-inflammatory and antioxidant transcription factors. Levels of surfactant proteins that protect the alveolar surfaces and decrease surface tension were also significantly lower in SS compared to AA (control) mice. These molecular changes in SS mice were also associated with an impairment in lung function. We conclude that the lung microenvironment in the SS mouse seemed primed for inflammation, thus tipping the balance toward a hyperinflammatory response and thus ACS when exposed to ACS triggers such as heme from hemolysis.
Introduction
Lung alveolar homeostasis is maintained by multiple resident cell types including the epithelial, endothelial, mesenchymal, and immune cells.1,2 Recent data from single-cell RNA sequencing, indicates the presence of robust intercellular communication between these cell types during homeostasis as well as in disease conditions.2,3 In uninjured lungs, the alveolar compartment is mostly quiescent, with slow turnover.1,4 On the contrary, injury activates multiple alveolar cell types to proliferate and differentiate into more mature cells that can affect repair by secreting surfactants or cytokines.1,5 The alveolar epithelium consists primarily of the alveolar type 1 epithelial (AT-1) and type 2 cells (AT-2).1,2,6 AT-1 cells are thin and flat, cover 95% of the alveolar surface area, and specialized mainly in gas exchange.1,4 AT-2 cells are cuboidal, and though they cover about 5% of the alveolar surface area, they represent 60% of alveolar epithelial cells by number. 6 AT-2 cells also self-renew to generate new AT-1 cells to repair acute injury in the postnatal lung tissue.1,7 They also secrete pulmonary surfactant that reduces the surface tension of the alveolar air–liquid interface, thereby preventing alveolar collapse.1,7 Pulmonary surfactants also have antimicrobial and anti-inflammatory properties which are important for protecting the lungs from inhaled microorganisms and potentially damaging inflammatory stimuli. 8 Pulmonary surfactants are composed mainly of phospholipids, neutral lipids, and four associated proteins categorized as SP-A, SP-B, SP-C, and SP-D.8,9 SP-A and SP-D are hydrophilic, collectively referred to as collectins (collagen-lectin) while SP-B and SP-C are hydrophobic.8,10,11 SP-A and SP-D interact with immune cells such as macrophages by associating with cell surface pattern-recognition receptors including Toll-like receptor (TLR) and CD14, thereby modulating phagocytosis and inflammatory cellular responses.11,12 Likewise, studies in humans have shown that the bronchoalveolar lavage (BAL) fluid concentrations of SP-A and SP-B were low in patients who are at risk for developing acute respiratory distress syndrome/acute lung injury and those with clinical lung injury, while the concentration of SP-D was normal.13,14 Also, studies of acute lung injury in mice have shown an abnormal surfactant function with reduced SP-B expression. 15
Macrophages are another cell type in the lung vital for maintaining homeostasis, initiation and resolution of inflammation, and tissue repair. There are two distinct populations of macrophages in the lungs: LAMs that are in contact with the AT-1 and AT-2 cells and the interstitial macrophages resident in the parenchyma space adjacent to the microvascular endothelium and alveolar epithelium.16,17 LAM acts as the first line of cellular defense against foreign invaders in the lower airways by scavenging and phagocytosing pathogens and recognizing microbial patterns via TLRs, NOD-like receptors (NODs) and other pattern-recognition pathways.18,19 Activated LAM release cytokines including type 1 interferon, tissue necrosis factor-α (TNF-α), interleukin (IL)-1β, and transforming growth factor (TGF) β family in either an NF-κB-dependent or independent pathway, stimulating complex crosstalk with neighboring epithelial cells in order to initiate or resolve lung inflammation and tissue repair via various processes.17 –19 The LAM is equipped with receptors to function as either pro- or anti-inflammatory, and the functional immune phenotype exhibited depends solely on the lung microenvironment.1,2
Maintaining the balance between pro- and anti-inflammatory states by alveolar epithelial cells and LAM is vital to pulmonary function and to preserving epithelial barrier integrity. This is evident in the damage to both endothelial and epithelial cell barriers leading to increased vascular permeability reported during acute lung injury, a disorder with a phenotype similar to acute chest syndrome (ACS) in SCD. 20 Notably, exaggerated vascular permeability has been shown to contribute to pulmonary edema, development of ACS, and pulmonary endothelial barrier dysfunction in SCD.21,22 In addition, pulmonary endothelial cells in contact with alveolar epithelial cells form a tight barrier characterized by adherens or tight junction proteins. This controls immune cell trafficking and the flux of liquid and solutes between the blood and surrounding tissue to maintain fluid homeostasis.23,24 Furthermore, serum from SCD patients and exosomes isolated from SCD patients who experienced one or more episodes of ACS contain biomarkers that reflect endothelial dysfunction with disrupted gap junction structures in vitro.25 –27
Still, the effect of alveolar epithelial cells, LAM, and the chronic proinflammatory cytokine status on maintaining optimal vascular barrier integrity during ACS in SCD are not clear. Understanding the steady-state profiles of pulmonary alveolar macrophages and AT-2 cells in SCD could help in elucidating the pathogenesis of ACS. Since most studies on mechanisms involved in the pathogenesis of ACS have been mostly on endothelial cells and monocytes,28 –31 in this study, we sought to gain better insights into the profiles of AT-2 and LAM by analyzing them at “steady-state” in SCD, allowing us to better delineate the impact of a perturbation in future experiments/studies. Broadening our understanding of the role of these cells may provide opportunities for identifying new therapeutic targets and/or intervention in acute and chronic lung injury that can restore the alveolar barrier integrity.
Materials and methods
Mice
All experiments with mice were conducted according to the protocol approved by Emory University’s Institutional Animal Care and Use Committee. Male and female Townes’ knock-in (B6;129-Hbbtm2(HBG1,HBB*)Tow/Hbbtm3(HBG1, HBB)Tow Hbatm1(HBA)Tow/J) humanized SS mouse and strain controls expressing normal human Hb (AA mice) were obtained from a colony maintained by our laboratory using the Emory University managed breeding service. All mice strains were 24 weeks old at the start of experimentation. Mouse genotypes were confirmed by polymerase chain reaction (PCR).
BAL fluid collection and processing
Mice were euthanized using an overdose of pentobarbital. An incision was made in the muscle around the trachea to expose the trachea, then a catheter was inserted into the trachea. Using a 1-ml syringe, phosphate-buffered saline (PBS, 0.9 ml) was slowly injected into the lung via the catheter and aspirated to recover the lavage fluid for a total of three times. Recovered BAL fluid placed in Eppendorf tubes was kept on ice followed by centrifugation at 400 × g for 8 min at 4°C to separate cellular content and BAL fluid. Recovered BAL fluid was either used immediately for surfactant or cytokines quantification or kept at −80°C for further analysis.
Isolation of AT-2 cells
Isolated lungs were perfused with PBS via catheterization of the pulmonary artery. 15 Digestion solution of Dispase was delivered via trachea catheter followed by 0.45 ml of low melt agarose (1%) to block the trachea and the airways. Lungs were transferred into a tube containing 2 ml Dispase and incubated for 45 min at room temperature. Lungs were then placed in a petri dish containing 0.01% DNase 1 in DMEM. Digested tissue was disrupted with fine-tipped forceps with gentle swirling for 5–10 min. Lung cells were suspended in newborn calf serum (NCS) and filtered through a 100 µm nylon cell strainer, then through a 20 µm nylon mesh. The filtered cells were pelleted by centrifugation at 300 × g at 4°C for 8 minutes. The supernatant was discarded, and cells were resuspended in DMEM without fetal bovine serum (FBS). Resuspended cells were plated onto IgG coated plates and incubated for 1 h at 37°C, to remove macrophages. Non-adherent cells (AT-2 cells) were removed from plates and centrifuged at 300 × g for 8 min. Recovered cell pellet was resuspended in complete culture media (DMEM, 1% Penicillin/Streptomycin, 20 mg Gentamicin Sulfate, 0.6 g Sodium Bicarbonate, and 0.18 g L-Glutamine) and used for subsequent assays.
Isolation of LAMs
LAM was isolated by a modified method previously described by Joshi et al. 32 Briefly, isolated lungs were lavaged twice with 1 ml of sterile cold PBS (pH 7.4). The recovered lavage fluid was centrifuged at 300 × g for 7 min. Cell pellet was resuspended in 400 µl of Ammonium-chloride-potassium red blood cell lysis buffer and incubated for 2 min at room temperature. Cold PBS (1 ml) was added to stop the lysis reaction and dilute the lysing buffer followed by centrifugation at 800 × g for 5 min at 4°C. Cell pellet was resuspended in PBS for downstream analysis. This procedure consistently produces cells that are greater than 98% viable by the Trypan blue exclusion test (DiffQuick, IMEB, Inc., San Marcos, CA). As previously published by others, CD32 staining showed that over 95% of cells recovered were alveolar macrophages.32,33
Cytokine and pulmonary surfactant quantification
BAL fluid cytokine concentrations were quantified using the Milliplex MAP magnetic bead panel kit (EMD Millipore, #MCYTOMAG-70K) following the manufacturer’s instructions. Total protein was quantified in the BAL fluid using the Pierce™ Coomassie (Bradford) Protein Assay Kit (ThermoFisher Scientific, #23200). Measured BAL cytokine levels were normalized to total BAL protein levels. Similarly, BAL fluid surfactant protein concentration was determined using enzyme-linked immunosorbent assay (ELISA) kit (Antibodies-online, Limerick, PA), according to the manufacturer’s protocols. The BAL fluid used for this part of the assay, was divided into four aliquots, each was used for the quantification of one surfactant protein. Measured BAL surfactant protein levels were also normalized to total BAL protein levels. Due to an unexpected assay contamination, we were not able to complete the assay for surfactant protein A and thus did not present the result of that assay as part of this report.
Measurement of lung mechanics
Lung function and mechanical properties such as lung elastance and inspiratory capacity (IC) were determined using the Flexivent system and FlexiWare software (SCIREQ, Montreal, Canada) in anesthetized and tracheotomized mice as described here.34 –36 Both open- and closed-chest conditions were evaluated.
Immunohistochemistry
Paraffin-embedded lung sections (4-5μm thickness) were de-waxed and rehydrated. Antigen retrieval was performed using a microwave, by heating the sections in 10mM citrate buffer (pH=6.0). We blocked endogenous peroxidase activity by incubating the sections in PBS buffer containing 0.3% hydrogen peroxidase for 30min. To block nonspecific binding, we incubated the tissue sections in 5% normal horse serum for one hour at room temperature. The tissue sections were sequentially incubated with primary antibodies at optimal dilutions (anti-HuR at 1:100, anti-TLR4 at 1:100) at 4°C overnight, and then proper secondary antibodies for 30min at room temperature (ImmPRESS REAGENT, VECTOR Lab, USA). The color was developed with the ImmPACT DAB kit (VECTOR Lab, USA). The tissue sections were then counterstained with hematoxylin and mounted using Permount medium (Fisher Scientific, #SP15-100). Two negative controls were included: (1) control slides stained without primary antibodies; (2) control slides incubated with specific blocking peptides (Santa Cruz, USA) prior to the primary antibody incubations. Images were captured using a 40X water dipping objective on an upright Olympus fluorescent microscope.
Immunocytochemistry
AT-2 cells plated in dishes were treated as described above under immunohistochemistry. HuR, TLR4, MyD88, PU.1, NRF2, and PPAR-γ, all purchased from Abcam were used as primary antibodies. Protein levels were measured as relative fluorescence units (RFUs) per cell.
Statistical analysis
GraphPad Prism 9 software was used for all statistical analyses. Results are reported as mean ± SEM. Group means were compared using parametric tests, such as the Mann–Whitney test or unpaired Student’s t-test. Statistical significance was set at p values of <0.05.
Results
Lung function and baseline surfactant proteins essential for pulmonary epithelial barrier function are dysregulated in SS mice
Lung function and mechanical properties such as lung elastance and IC were determined using the Flexivent system in tracheotomized mice. Both tissue elastance and change in IC have been used to measure pulmonary compliance; the ability of the lungs to stretch and expand during inflation. 37 Pulmonary compliance is altered by pathological changes in lung elastin fibers and the absence of surfactant that lowers alveolar surface tension.38,39 In our study, lung function and mechanics testing showed significant differences between SS and AA control mice. The change in IC was higher in AA control mice compared with SS mice (Figure 1(A)) Correspondingly, tissue elastance was significantly lower in SS compared to AA control mice (Figure 1(B), p ⩽ 0.05). Pulmonary surfactants are secreted into the alveolar space by epithelial (AT-2) cells to reduce surface tension and act as host defense against inhaled pathogens. 40 Furthermore, abnormal surfactant function caused by inflammatory cytokines plays a role in the pathogenesis of acute lung injury, 41 with alterations in lung function closely associated with the development of surfactant dysfunction and elevated expression of inflammatory cytokines including IL-1β. 15 Surfactant proteins B and C were significantly decreased in BAL fluid obtained from SS mice compared to AA controls (Figure 1(C) and (D)) while no differences were observed in the levels of surfactant protein D (Figure 1(E)). We also measured the levels of paraoxonase 1 (PON1), a hydrolytic antioxidant enzyme with the capability to protect against lipid oxidation. 42 The levels of PON1 were significantly reduced by about 66% in SS mice compared to AA controls (Figure 1(F)), suggesting that decreased protection against lipid oxidation by high circulating inflammatory cytokines in the lung of SS mice may contribute to lower surfactant B level seen in these mice at steady state. Overall, these results showed a decrease in lung function and mechanical properties implying a decreased ability to stretch during inflation while low levels of surfactant proteins in SS mice compared to AA mice controls may imply an abnormal alveolar surface tension.
Figure 1.

Lung function and surfactant protein expression in AA and SS mice at steady state. Lung function measured as (A) Change in IC (B) Tissue elastance. Quantification of relative protein content of surfactant proteins in BAL fluid collected from 24 weeks old mice. (C) SP-B, (D) SP-C and (E) SP-D. (F) Protein content of paraoxonase1 BAL fluid. Levels of surfactant proteins and paraoxonase1 were normalized to total protein in BAL fluid. Unpaired Student’s t-test (n = 6–15). Error bars indicate SEM. *p ⩽ 0.05, **p ⩽ 0.01. IC- inspiratory capacity.
Steady state profile of cytokines in BAL from SS mice
BAL fluid was collected from SS mice and AA control mice as described above. We first determined the total concentrations of markers of inflammation in BAL fluid using a multiplex bead assay. Compared with AA mice, the BAL fluid from SS mice showed significantly increased levels of pro-inflammatory cytokines IL-1β and IL-12 (Figure 2(A) and (B), p ⩽ 0.05). In contrast, levels of IL-9 and CXCL1 were decreased (Figure 2(D) and (C), p ⩽ 0.05). Similarly, we observed a significantly higher levels of HuR and TLR4 levels in AT-2 cells in SS mice (1.4- and 2.2-fold, respectively) compared to AA controls (Figure 3(A) and (B)). LAM from SS mice also have significantly higher expression of HuR and TLR4 compared to AA controls (Figure 3(E) and (F), p ⩽ 0.05). In addition, we quantified the expression of other associated markers of inflammation in isolated AT-2 cells and LAM. Myeloid differentiation primary response protein 88 (MyD88) is an important Toll-like and IL-1 receptor family signaling adaptor protein involved in innate immune response, chronic lung inflammation and fibrosis.43,44 PU.1 is a transcription factor required for NF-κB activation, neutrophilic lung inflammation, and pathological macrophage polarization in asthmatic airway inflammation.45,46 MyD88 and PU.1 level were unchanged in AT-2 cells (Figure 3(C) and (D)) while their levels were about 17% and 21% higher, respectively (p < 0.05 and 0.001) in LAM isolated from SS mice compared to AA controls (Figure 3(G) and (h)). These results were also confirmed with immunohistochemistry of the whole lung tissue where TLR4 and HuR were significantly elevated (Figure 4). These results are consistent with the observed elevated levels of proinflammatory IL-1β and IL-12 and decreased levels of pleiotropic cytokines (IL-9 and CXCL1) that regulate airway inflammation and hyper-responsiveness at steady state, in BAL from SS mice compared to AA controls (Figure 2). Taken together, these finding suggest that in the “steady state,” the lung microenvironment in SCD is already pro-inflammatory, and this might account for the hyper-responsiveness to heme that have been documented in prior studies.
Figure 2.
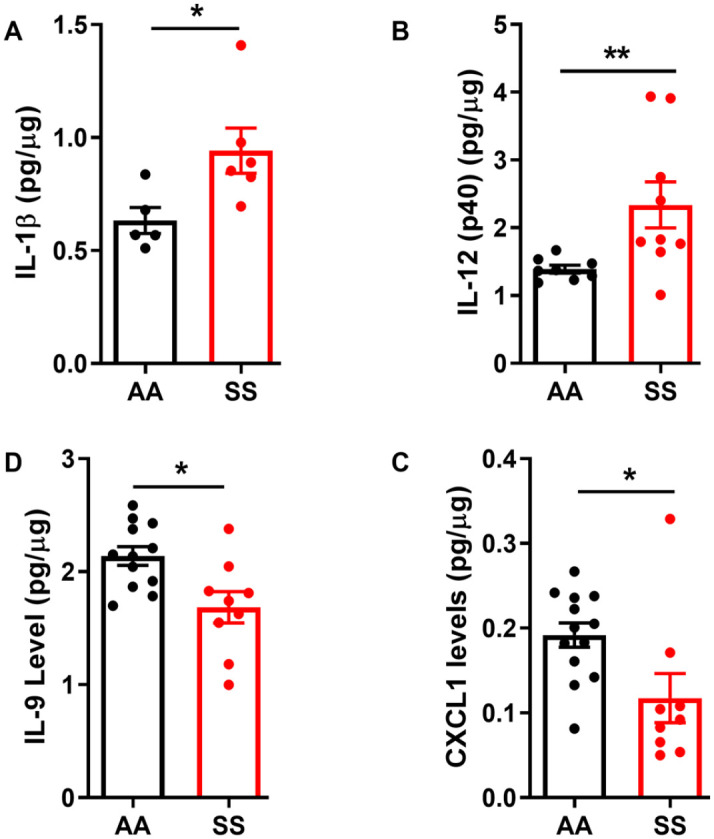
Lung BAL fluid expression of pro-inflammatory cytokines in SS and AA mice. BAL fluid protein levels of (A) IL-1β, (B) IL-9, (C) IL-12(p40), and (E) CXCL1 in naïve in 24-week-old SS mice compared to age-matched AA mice (n = 5–13). Individual cytokine levels measured were normalized to total protein in lavage fluid. Unpaired Student’s t-test. Error bars indicate SEM. *p ⩽ 0.05; **p ⩽ 0.01.
Figure 3.

Inflammatory mediators’ protein levels in lung epithelial cells and macrophages are elevated in SS mice compared to AA controls. AT-2 cells protein levels of (A) HuR, (B) TLR4, (C) MyD88, and (E) PU.1. LAM protein levels of (D) HuR, (E) TLR4, (F) MyD88, and (G) PU.1. Protein levels were detected using immunofluorescence and then quantified using image J and expressed in relative fluorescence units (RFU) per cell. Mann–Whitney test or Unpaired Student’s t-test. Error bars indicate SEM. *p ⩽ 0.05, ****p ⩽ 0.0001. AT-2: alveolar type 2 cells, LAM: lung alveolar macrophage.
Figure 4.

Immunohistochemical detection of HuR and TLR4 in the lung. Fluorescent (Olympus) microscopy images (magnification ×40) of lung sections stained with an antibody against HuR and TLR4. (A and C) AA mice and (B and D) SS mice. Staining was carried out in different lung sections of untreated 24-week-old mice (n = 4 per group). Scale bars = 30 µm. Blue staining represents nuclei with hematoxylin.
Expression of PPAR-γ and Nrf2 was significantly lower in AT-2 cells and LAM from SS mice
Our results above showed elevated expression of inflammatory markers in BAL fluid, AT-2 cells, LAM, and whole lung tissue from SS mice at steady-state. The major cellular defense mechanisms against inflammatory cytokines are the transcriptional regulators, nuclear factor (erythroid-derived 2)-like 2 (Nfe2L2 or Nrf2) and Peroxisome proliferator activated receptor gamma (PPAR-γ). Nrf2 is the master regulator of the cellular oxidative defense system.47,48 PPAR-γ is a ligand-dependent transcription factor that exerts anti-inflammatory properties by inhibiting the expression of inflammatory cytokines and modulates the immune cells’ differentiation toward anti-inflammatory phenotypes.49,50 Both antioxidant transcription factors Nrf2 and PPAR-γ proteins were significantly reduced in AT-2 cells and LAM from SS mice compared to AA controls (Figure 5, p ⩽ 0.05 to 0.0001). This suggests that dysregulation of the antioxidant pathway in non-endothelial cells in the lungs of SS mice may have a role in the pathobiology of the acute lung injury described by other investigators.
Figure 5.
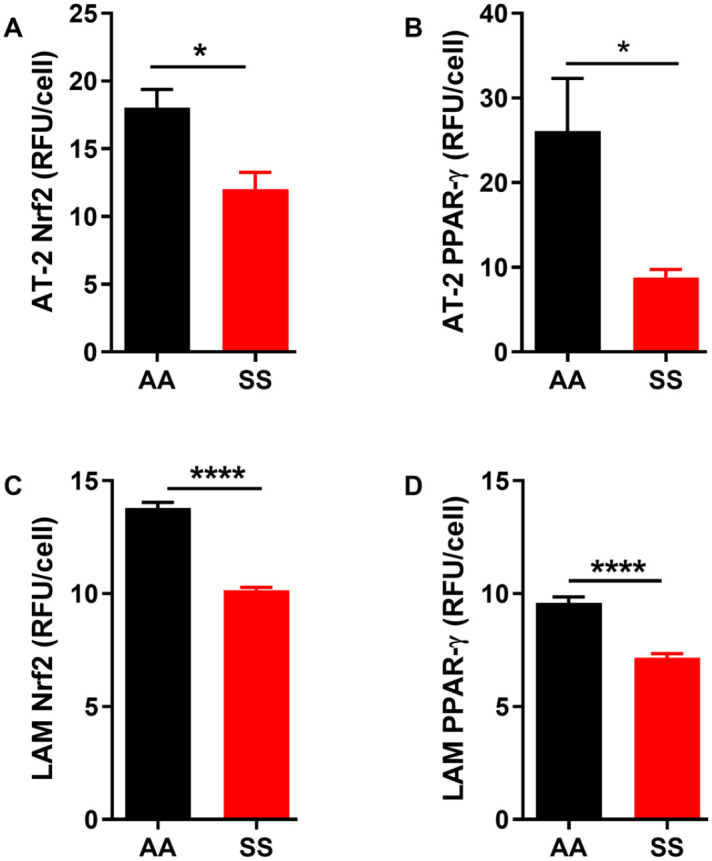
Protein levels of transcription factors involved in the regulation of antioxidant gene expression in the lungs are low in SS mice at steady state. Protein levels of AT-2 cells (A) Nrf2 and (B) PPAR-γ. Protein levels of LAM (C) Nrf2 and (D) PPAR-γ. Protein levels were detected using immunofluorescence and then quantifies using image J and expressed in relative fluorescence units (RFU) per cell. Unpaired Student’s t-test. Error bars indicate SEM. *p ⩽ 0.05, ****p < 0.0001. AT-2: alveolar type 2 cells, LAM: lung alveolar macrophage.
Discussion
Our study examines the profile of AT-2, LAM, and the composition of surfactant proteins at steady-state in SS mice compared to AA controls. First, we found impaired lung function and dysregulated composition of surfactant proteins necessary for maintaining alveolar barrier integrity in SS mice. Second, we showed that BAL fluid from SS mice has elevated levels of inflammatory cytokines. Finally, we showed for the first time that lung epithelial cells from SS mice at steady state have high protein levels of inflammatory mediators (HuR, TLR4, MyD88, and PU.1) with low levels of transcription factors that mediate antioxidant responses (Nrf2 and PPARy). Thus, in the “steady state” in SCD mice, the delicate balance between pro- and anti-inflammatory states in the lung microenvironment appears to be tipped toward inflammation and an impaired ability to respond to further oxidant stress.
Lung function measured as change in IC and tissue elastance were significantly diminished in SS mice lungs compared to AA controls. This agrees with previous studies showing reduced lung function in SS mice at steady-state which was exacerbated during heme-induced ACS. 29 Elevated levels of inflammatory mediators and low antioxidant levels in LAM and AT-2 cells reported in our study may contribute to the reduced lung function seen here. Reduced lung elastance has been shown to be mediated via TLR4 activation through MyD88-dependent or independent pathway in LPS and acid-induced acute lung injury model in mice. 51 Also, heme is a ligand for TLR4 expression in pulmonary endothelial cells during ACS and vaso-occlusive events resulting in reduced lung function.28,29 This reduction in lung function in the setting of elevated levels of inflammatory mediators at baseline reported in our study provides a unique insight into the rich source of cytokines in the alveolar epithelium in sickle mice that may significantly contribute to leakage of the alveolar barrier during ACS. In future studies, we will explore the interplay between elevated inflammatory cytokines and low levels of antioxidants in LAM and AT-2 cells and their effect on alveolar barrier integrity during ACS in SCD.
Furthermore, our study showed decreased levels of SP-B and C, while SP-D remained unchanged. We did not quantify SP-A in BAL fluid due to unexpected assay contamination. Several factors may be responsible for the surfactant dysregulation in SS mice reported here. The low levels of SP-B and C could be a direct result of the oxidative microenvironment in SS mice leading to degradation of the surfactant lipid and protein components by the abundance of free radicals generated by heme-bound iron, reactive oxygen species (ROS) released by activated immune cells, and chronic inflammation in SCD.52 –56 In addition to low levels of surfactants, our analysis of BAL fluid revealed low levels of PON1, an enzyme that protects against lipid oxidation. Although this significance was driven by an outlier, we will investigate this further in our next study. This strengthens our conclusion that decreased lung function in the lungs of SS mice is a feature of degradation of critical surfactant components in addition to oxidative modification. In addition, dysregulation in surfactants in SS mice reported in our study may result in increased exposure to pathogens and allergens.
Our results on elevated inflammatory immune cell profile and impaired lung function confirm and extend the findings of other groups showing that inflammatory mechanisms via increased circulating oxidative mediators or deficiency in antioxidants contribute to ACS and vaso-occlusion in SCD.28,29,31,57 –66 This dysregulated expression of inflammatory and antioxidant proteins in AT-2 and LAM at steady-state may contribute to the exaggerated response documented in SS mice when exposed to inflammatory stimuli. The elevated IL-1β and IL-12 proteins in BAL fluid, with low levels of IL-9 and CXCL1 we found in SS mice compared to AA controls could pose a supercharged microenvironment that may interfere with immune cells’ phenotype and response during initiation and resolution of ACS, alter the composition of the microbial flora, and affect other mechanisms that maintain homeostasis in healthy lungs. It is also possible that the highly inflammatory BAL fluid is not totally due to cytokines secreted in the alveolar cells, but also reflects, in part the systemic chronic inflammation found in SCD.63,67
Mechanistically, PU.1 and NLRP3 inflammasome pathways have been shown to regulate IL-9 production in T-cells and allergic airway inflammation in humans, 68 and lung injury in mice.68,69 In our study, IL-9 protein was significantly decreased in BAL fluid from SS mice compared to AA controls. Concurrently, PU.1 protein was significantly elevated in LAM isolated from these mice. Our results are consistent with the study by Chang et al. 68 where modulation of IL-9 levels resulted in high PU.1 expression in wild-type mice. The precise contribution of low protein levels of IL-9 and high PU.1 to the development of lung inflammation/injury and ACS in SS mice warrants further study.
Several cell types in the lung including LAM, endothelial, and epithelial cells are known to produce CXCL1, a chemokine that mediates neutrophil recruitment in acute lung inflammation.70 –73 Previous publications have shown that neutrophils are activated in SCD and their interaction with other blood cells and the endothelium initiate and propagate vaso-occlusion and inflammatory pathways.74 –76 Surprisingly, our data showed reduced protein levels of both IL-9 and CXCL1 in BAL fluid from SS mice compared to AA control mice. This reduction in CXCL1 and IL-9 protein in the BAL fluid of SS mice compared to AA controls may not be enough to protect the lung against injury in SCD since neutrophils interaction with the endothelium and other blood cells has been shown to induce vaso-occlusion in SCD.65,77,78 This suggests that lung dysfunction in SCD is due to multicellular pathological processes. Besides, proinflammatory cytokines and chemokines cellular signaling transduction networks are complex with the possibility of crosstalk and/or convergence of individual pathways through common stimuli or downstream receptors further complicated by the inflamed and deranged vascular bed in SCD.
Our study showed for the first time that other cell types in the lung of SS mice, specifically the AT-2 cells and LAM, express significantly higher levels of TLR4 and MyD88 compared to controls at steady-state. This was in agreement with other studies outside of SCD that reported the expression of TLR4 in lung alveolar and bronchial epithelial cells.79,80 TLR4 signaling in endothelial cells and monocytes has been linked to heme-induced ACS and vaso-occlusive crisis in past publications on SCD.28,29,31 This was reported to occur via the interaction of TLR4 with heme, a damage-associated molecular pattern molecule (DAMP), resulting in activation of endothelial Nuclear factor kappaB (NF-κB). 28 Furthermore, the molecular mechanisms that initiate inflammatory responses in acute lung injury require both TLR4 signaling and its adaptor protein, MyD88 for effective transepithelial migration of immune cells to the injury site for the repair of damaged tissues. 81 TLR4 activation transduced transmembrane signaling in a MyD88-dependent manner, thereby activating NF-κB and triggering the subsequent release of downstream inflammatory cytokines during inflammation.82,83 Although we did not verify the specific activation of NF-κB in AT-2 cells and LAM in our study, it has been previously shown that NF-κB is activated in the lungs of SS mice. 84 Nevertheless, the mechanisms involved in ACS and dysfunctional vascular permeability in SCD are not totally understood. It is possible that TLR4-MyD88 signaling pathway in AT-2 cells and LAM may contribute to the pathology of ACS in SCD, possibly via the activation of NF-κB, secretion of pro-inflammatory cytokines, and disruption of the pulmonary epithelial barrier.
Antioxidant levels are low in individuals with SCD and in SS mice due to the imbalance between excessive ROS generation and antioxidant levels. 85 Our results showing low levels of Nrf2 expression in LAM and AT-2 cells isolated from SS mice support and extend the works of Ghosh et al., 2016 and 2018 who reported that nonhematopoietic Nrf2 protects against tissue damage in SCD mouse model. Also, it has been shown that activation of Nrf2 by 3H-1,2-dithiole-3-thione (D3T) leads to increased heme-oxygenase 1 expression, resulting in less-severe lung damage and improved survival upon exposure to heme.58,59 Our results provide evidence that other alveolar cell types apart from the extensively studied endothelial cells may play a significant role in pulmonary inflammation and ACS pathology in SCD.
Our results showing a reduction in LAM and AT-2 cells PPAR-γ at baseline in SS mice support and extend previous publications on PPAR-γ expression in SCD.86 –90 However, it differs from these publications by documenting PPAR-γ expression in non-endothelial cells in the lung, suggesting that derangement of PPAR-γ in other alveolar cell types may contribute to pulmonary dysfunction in SCD. Peroxisome Proliferator Activated Receptors (PPARs) belong to the nuclear hormone receptor superfamily with three isoforms encoded by separate genes: PPAR-γ, PPARα, and PPARδ. 91 PPARs are ligand-activated transcription factors that regulate important genes involved in cell differentiation, apoptosis, and metabolic processes including lipid and glucose homeostasis.91,92 The low levels of PPARy protein that we found in both LAM and AT-2 cells may be due to heme released during hemolysis, a chronic feature of SCD, because heme has been shown to upregulate the expression of miRNAs (such as miR-27a) that negatively regulate PPARγ expression. 90 This low PPARy protein could predispose the alveolar milieu to be more sensitive to oxidative stress. Still, there is the possibility that low levels of antioxidant transcription factors such as PPARy in alveolar cells such as LAM and AT-2 may either prolong the initiation phase of the inflammatory process during acute lung injury on exposure to DAMPs such as heme or delay the resolution of the initial inflammatory reaction initiated by the presence of DAMPs.
In conclusion, our study suggests a role for LAM and AT-2 cells in alveolar epithelium pathology and possibly ACS in SCD. Such a role was previously undescribed in SCD. The LAM-epithelial cells-cytokines interaction is an important area that requires well-designed and detailed research and could have applications in developing new therapies or repurposing of existing therapies for ACS in SCD. In addition, our data showed a baseline reduction in transcription factors that regulate antioxidant molecules in LAMs and AT-2 cells in the lungs of SS mice compared with AA controls. Our future work will examine whether these findings correspond with a higher level of oxidative stress in the lungs or lower levels of antioxidant mediators such as glutathione in the lungs’ tissue and/or cells.
Footnotes
Authors’ Contributions: HIH designed the study. HIH executed the experiment with WL assisting with some of the experiments; OTG and HIH interpreted data and drafted the manuscript. HIH, OTG, L-ASB, and CHJ provided critical review of the manuscript. All authors reviewed and approved the final version.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health and National Heart Lung and Blood Institute (grant nos. U01HL117721 and R01HL138423). OTG is supported by the American Society of Hematology Scholar Award and the Parker B. Francis Fellowship in Pulmonary Research Award.
ORCID iDs: Oluwabukola T. Gbotosho  https://orcid.org/0000-0002-9176-7025
https://orcid.org/0000-0002-9176-7025
Hyacinth I Hyacinth
https://orcid.org/0000-0002-1991-7463
References
- 1. Basil MC, Katzen J, Engler AE, Guo M, Herriges MJ, Kathiriya JJ, Windmueller R, Ysasi AB, Zacharias WJ, Chapman HA, Kotton DN, Rock JR, Snoeck HW, Vunjak-Novakovic G, Whitsett JA, Morrisey EE. The cellular and physiological basis for lung repair and regeneration: past, present, and future. Cell Stem Cell 2020;26:482–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, Chang S, Conley SD, Mori Y, Seita J, Berry GJ, Shrager JB, Metzger RJ, Kuo CS, Neff N, Weissman IL, Quake SR, Krasnow MA. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature 2020;587:619–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, Brouwer S, Gomes T, Hesse L, Jiang J, Fasouli ES, Efremova M, Vento-Tormo R, Talavera-López C, Jonker MR, Affleck K, Palit S, Strzelecka PM, Firth HV, Mahbubani KT, Cvejic A, Meyer KB, Saeb-Parsy K, Luinge M, Brandsma CA, Timens W, Angelidis I, Strunz M, Koppelman GH, van Oosterhout AJ, Schiller HB, Theis FJ, van den Berge M, Nawijn MC, Teichmann SA. A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med 2019;25:1153–63 [DOI] [PubMed] [Google Scholar]
- 4. Li J, Wang Z, Chu Q, Jiang K, Li J, Tang N. The strength of mechanical forces determines the differentiation of alveolar epithelial cells. Dev Cell 2018;44:297–312 [DOI] [PubMed] [Google Scholar]
- 5. Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, Elias JA, Sheppard D, Erle DJ. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med 2002;8:885–9 [DOI] [PubMed] [Google Scholar]
- 6. Castranova V, Rabovsky J, Tucker JH, Miles PR. The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicol Appl Pharmacol 1988;93:472–83 [DOI] [PubMed] [Google Scholar]
- 7. Desai TJ, Brownfield DG, Krasnow MA. Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature 2014;507: 190–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mason RJ. Biology of alveolar type II cells. Respirology 2006;11:S12–5 [DOI] [PubMed] [Google Scholar]
- 9. Spragg R. Surfactant for acute lung injury. Am J Respir Cell Mol Biol 2007;37:377–8 [DOI] [PubMed] [Google Scholar]
- 10. Kuroki Y, Voelker DR. Pulmonary surfactant proteins. J Biol Chem 1994;269:25943–6 [PubMed] [Google Scholar]
- 11. Sano H, Kuroki Y. The lung collectins, SP-A and SP-D, modulate pulmonary innate immunity. Mol Immunol 2005;42:279–87 [DOI] [PubMed] [Google Scholar]
- 12. Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol 2001;63:521–54 [DOI] [PubMed] [Google Scholar]
- 13. Greene KE, Wright JR, Steinberg KP, Ruzinski JT, Caldwell E, Wong WB, Hull W, Whitsett JA, Akino T, Kuroki Y, Nagae H, Hudson LD, Martin TR. Serial changes in surfactant-associated proteins in lung and serum before and after onset of ARDS. Am J Respir Crit Care Med 1999;160:1843–50 [DOI] [PubMed] [Google Scholar]
- 14. Günther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, Yabut M, Temmesfeld B, Walmrath D, Morr H, Seeger W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med 1996;153:176–84 [DOI] [PubMed] [Google Scholar]
- 15. Ingenito EP, Mora R, Cullivan M, Marzan Y, Haley K, Mark L, Sonna LA. Decreased surfactant protein-B expression and surfactant dysfunction in a murine model of acute lung injury. Am J Respir Cell Mol Biol 2001;25:35–44 [DOI] [PubMed] [Google Scholar]
- 16. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015;518:547–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu G, Christman JW. Editorial: alveolar macrophages in lung inflammation and resolution. Front Immunol 2019;10:2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol 2011;2:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kopf M, Schneider C, Nobs SP. The development and function of lung-resident macrophages and dendritic cells. Nat Immunol 2015;16:36–44 [DOI] [PubMed] [Google Scholar]
- 20. Kawkitinarong K, Linz-McGillem L, Birukov KG, Garcia JG. Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am J Respir Cell Mol Biol 2004;31:517–27 [DOI] [PubMed] [Google Scholar]
- 21. Umapathy NS, Gonzales J, Makala LH, Xu H, Biddinger P, Pace BS. Impaired pulmonary endothelial barrier function in sickle cell mice. Haematologica 2017;102:e26–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ghosh S, Tan F, Ofori-Acquah SF. Spatiotemporal dysfunction of the vascular permeability barrier in transgenic mice with sickle cell disease. Anemia 2012;2012:582018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vandenbroucke E, Mehta D, Minshall R, Malik AB. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008;1123:134–45 [DOI] [PubMed] [Google Scholar]
- 24. Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am J Physiol Lung Cell Mol Physiol 2013;305:L665–181 [DOI] [PubMed] [Google Scholar]
- 25. Lapping-Carr G, Khalyfa A, Rangel S, Darlington W, Beyer EC, Peddinti R, Cunningham JM, Gozal D. Exosomes contribute to endothelial integrity and acute chest syndrome risk: preliminary findings. Pediatr Pulmonol 2017;52:1478–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gemel J, Mao Y, Lapping-Carr G, Beyer EC. Gap junctions between endothelial cells are disrupted by circulating extracellular vesicles from sickle cell patients with acute chest syndrome. Int J Mol Sci 2020;21:8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Santaterra VAG, Fiusa MML, Hounkpe BW, Chenou F, Tonasse WV, da Costa LNG, Garcia-Weber D, Domingos IF, de Lima F, Borba-Junior IT, Araújo ADS, Lucena-Araújo AR, Bezerra MAC, Dos Santos MNN, Costa FF, Millán J, De Paula EV. Endothelial barrier integrity is disrupted in vitro by heme and by serum from sickle cell disease patients. Front Immunol 2020;11:535147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, Smith A, Nath KA, Hebbel RP, Vercellotti GM. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 2014;123:377–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, Ofori-Acquah SF. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest 2013;123:4809–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gladwin MT, Ofori-Acquah SF. Erythroid DAMPs drive inflammation in SCD. Blood 2014;123:3689–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood 2000;96:2451–9 [PubMed] [Google Scholar]
- 32. Joshi PC, Mehta A, Jabber WS, Fan X, Guidot DM. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am J Respir Cell Mol Biol 2009;41:207–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Franke-Ullmann G, Pfortner C, Walter P, Steinmuller C, Lohmann-Matthes ML, Kobzik L. Characterization of murine lung interstitial macrophages in comparison with alveolar macrophages in vitro. J Immunol 1996;157:3097–104 [PubMed] [Google Scholar]
- 34. Zhou J, Wu Y, Henderson F, McCoy DM, Salome RG, McGowan SE, Mallampalli RK. Adenoviral gene transfer of a mutant surfactant enzyme ameliorates pseudomonas-induced lung injury. Gene Ther 2006;13:974–85 [DOI] [PubMed] [Google Scholar]
- 35. Chambers ED, White A, Vang A, Wang Z, Ayala A, Weng T, Blackburn M, Choudhary G, Rounds S, Lu Q. Blockade of equilibrative nucleoside transporter 1/2 protects against Pseudomonas aeruginosa-induced acute lung injury and NLRP3 inflammasome activation. FASEB J 2020;34:1516–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bonnardel E, Prevel R, Campagnac M, Dubreuil M, Marthan R, Berger P, Dupin I. Determination of reliable lung function parameters in intubated mice. Respir Res 2019;20:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schulte H, Mühlfeld C, Brandenberger C. Age-related structural and functional changes in the mouse lung. Front Physiol 2019;10:1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lopez-Rodriguez E, Boden C, Echaide M, Perez-Gil J, Kolb M, Gauldie J, Maus UA, Ochs M, Knudsen L. Surfactant dysfunction during overexpression of TGF-beta1 precedes profibrotic lung remodeling in vivo. Am J Physiol Lung Cell Mol Physiol 2016;310:L1260–171 [DOI] [PubMed] [Google Scholar]
- 39. Devos FC, Maaske A, Robichaud A, Pollaris L, Seys S, Lopez CA, Verbeken E, Tenbusch M, Lories R, Nemery B, Hoet PH, Vanoirbeek JA. Forced expiration measurements in mouse models of obstructive and restrictive lung diseases. Respir Res 2017;18:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Glasser SW, Witt TL, Senft AP, Baatz JE, Folger D, Maxfield MD, Akinbi HT, Newton DA, Prows DR, Korfhagen TR. Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am J Physiol Lung Cell Mol Physiol 2009;297:L64–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hallman M, Spragg R, Harrell JH, Moser KM, Gluck L. Evidence of lung surfactant abnormality in respiratory failure. Study of bronchoalveolar lavage phospholipids, surface activity, phospholipase activity, and plasma myoinositol. J Clin Invest 1982;70:673–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Litvinov D, Mahini H, Garelnabi M. Antioxidant and anti-inflammatory role of paraoxonase 1: implication in arteriosclerosis diseases. N Am J Med Sci 2012;4:523–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, Ryffel B, Couillin I. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest 2007;117:3786–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Couillin I, Vasseur V, Charron S, Gasse P, Tavernier M, Guillet J, Lagente V, Fick L, Jacobs M, Coelho FR, Moser R, Ryffel B. IL-1R1/MyD88 signaling is critical for elastase-induced lung inflammation and emphysema. J Immunol 2009;183:8195–202 [DOI] [PubMed] [Google Scholar]
- 45. Qian F, Deng J, Lee YG, Zhu J, Karpurapu M, Chung S, Zheng JN, Xiao L, Park GY, Christman JW. The transcription factor PU.1 promotes alternative macrophage polarization and asthmatic airway inflammation. J Mol Cell Biol 2015;7:557–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Karpurapu M, Wang X, Deng J, Park H, Xiao L, Sadikot RT, Frey RS, Maus UA, Park GY, Scott EW, Christman JW. Functional PU.1 in macrophages has a pivotal role in NF-kappaB activation and neutrophilic lung inflammation during endotoxemia. Blood 2011;118:5255–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kobayashi A, Ohta T, Yamamoto M. Unique function of the Nrf2-Keap1 pathway in the inducible expression of antioxidant and detoxifying enzymes. Methods Enzymol 2004;378:273–86 [DOI] [PubMed] [Google Scholar]
- 48. Motohashi H, Yamamoto M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol Med 2004;10:549–57 [DOI] [PubMed] [Google Scholar]
- 49. Heming M, Gran S, Jauch SL, Fischer-Riepe L, Russo A, Klotz L, Hermann S, Schafers M, Roth J, Barczyk-Kahlert K. Peroxisome proliferator-activated receptor-gamma modulates the response of macrophages to lipopolysaccharide and glucocorticoids. Front Immunol 2018;9:893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Martin H. Roles of peroxisome proliferator-activated receptor-gamma on brain and peripheral inflammation. Mutat Res 2010;690:57–63 [Google Scholar]
- 51. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, Ermolaeva M, Veldhuizen R, Leung YH, Wang H, Liu H, Sun Y, Pasparakis M, Kopf M, Mech C, Bavari S, Peiris JS, Slutsky AS, Akira S, Hultqvist M, Holmdahl R, Nicholls J, Jiang C, Binder CJ, Penninger JM. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008;133:235–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wang X, Mendelsohn L, Rogers H, Leitman S, Raghavachari N, Yang Y, Yau YY, Tallack M, Perkins A, Taylor JG, Noguchi CT, Kato GJ. Heme-bound iron activates placenta growth factor in erythroid cells via erythroid Krüppel-like factor. Blood 2014;124:946–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Francis RB, Jr, Haywood LJ. Elevated immunoreactive tumor necrosis factor and interleukin-1 in sickle cell disease. J Natl Med Assoc 1992;84:611–5 [PMC free article] [PubMed] [Google Scholar]
- 54. Perelman N, Selvaraj SK, Batra S, Luck LR, Erdreich-Epstein A, Coates TD, Kalra VK, Malik P. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood 2003;102:1506–14 [DOI] [PubMed] [Google Scholar]
- 55. Aft RL, Mueller GC. Hemin-mediated oxidative degradation of proteins. J Biol Chem 1984;259:301–5 [PubMed] [Google Scholar]
- 56. Mark L, Ingenito EP. Surfactant function and composition after free radical exposure generated by transition metals. Am J Physiol 1999;276: L491–500 [DOI] [PubMed] [Google Scholar]
- 57. Ghosh S, Flage B, Weidert F, Ofori-Acquah SF. P-selectin plays a role in haem-induced acute lung injury in sickle mice. Br J Haematol 2019;186: 329–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ghosh S, Hazra R, Ihunnah CA, Weidert F, Flage B, Ofori-Acquah SF. Augmented NRF2 activation protects adult sickle mice from lethal acute chest syndrome. Br J Haematol 2018;182:271–5 [DOI] [PubMed] [Google Scholar]
- 59. Ghosh S, Ihunnah CA, Hazra R, Walker AL, Hansen JM, Archer DR, Owusu-Ansah AT, Ofori-Acquah SF. Nonhematopoietic Nrf2 dominantly impedes adult progression of sickle cell anemia in mice. JCI Insight 2016;1:e81090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Belcher JD, Chen C, Nguyen J, Zhang P, Abdulla F, Nguyen P, Killeen T, Xu P, O’Sullivan G, Nath KA, Vercellotti GM. Control of oxidative stress and inflammation in sickle cell disease with the Nrf2 activator dimethyl fumarate. Antioxid Redox Signal 2017;26:748–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Belcher JD, Mahaseth H, Welch TE, Otterbein LE, Hebbel RP, Vercellotti GM. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. J Clin Invest 2006;116:808–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Belcher JD, Vineyard JV, Bruzzone CM, Chen C, Beckman JD, Nguyen J, Steer CJ, Vercellotti GM. Heme oxygenase-1 gene delivery by Sleeping Beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med 2010;88:665–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Conran N, Belcher JD. Inflammation in sickle cell disease. Clin Hemorheol Microcirc 2018;68:263–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vercellotti GM, Zhang P, Nguyen J, Abdulla F, Chen C, Nguyen P, Nowotny C, Steer CJ, Smith A, Belcher JD. Hepatic Overexpression of hemopexin inhibits inflammation and vascular stasis in murine models of sickle cell disease. Mol Med 2016;22:437–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Bennewitz MF, Jimenez MA, Vats R, Tutuncuoglu E, Jonassaint J, Kato GJ, Gladwin MT, Sundd P. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2017;2:e89761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Andemariam B, Adami AJ, Singh A, McNamara JT, Secor ER, Guernsey LA, Thrall RS. The sickle cell mouse lung: proinflammatory and primed for allergic inflammation. Transl Res 2015;166:254–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Elmariah H, Garrett ME, De Castro LM, Jonassaint JC, Ataga KI, Eckman JR, Ashley-Koch AE, Telen MJ. Factors associated with survival in a contemporary adult sickle cell disease cohort. Am J Hematol 2014;89:530–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, Jabeen R, McKinley C, Ahyi AN, Han L, Nguyen ET, Robertson MJ, Perumal NB, Tepper RS, Nutt SL, Kaplan MH. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol 2010;11:527–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wang T, Chai Z, Wang L, Liu B, Zhao J, Ren J, Yang B, Wei X, Jiang L, Liu F. IL-9 blockade attenuates inflammation in a murine model of mechanical ventilation-induced lung injury by inhibiting the NLRP3 inflammasome pathway. Inflammopharmacology 2022;30:1395–406. [DOI] [PubMed] [Google Scholar]
- 70. Bozic CR, Kolakowski LF, Jr, Gerard NP, Garcia-Rodriguez C, von Uexkull-Guldenband C, Conklyn MJ, Breslow R, Showell HJ, Gerard C. Expression and biologic characterization of the murine chemokine KC. J Immunol 1995;154:6048–57 [PubMed] [Google Scholar]
- 71. Frevert CW, Huang S, Danaee H, Paulauskis JD, Kobzik L. Functional characterization of the rat chemokine KC and its importance in neutrophil recruitment in a rat model of pulmonary inflammation. J Immunol 1995;154:335–44 [PubMed] [Google Scholar]
- 72. Ahuja N, Andres-Hernando A, Altmann C, Bhargava R, Bacalja J, Webb RG, He Z, Edelstein CL, Faubel S. Circulating IL-6 mediates lung injury via CXCL1 production after acute kidney injury in mice. Am J Physiol Renal Physiol 2012;303:F864–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bhatia M, Zemans RL, Jeyaseelan S. Role of chemokines in the pathogenesis of acute lung injury. Am J Respir Cell Mol Biol 2012;46:566–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol 1999;66:411–5 [DOI] [PubMed] [Google Scholar]
- 75. Barbu EA, Dominical VM, Mendelsohn L, Thein SL. Neutrophils remain detrimentally active in hydroxyurea-treated patients with sickle cell disease. PLoS ONE 2019;14:e0226583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhang D, Xu C, Manwani D, Frenette PS. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 2016;127:801–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A 2002;99:3047–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med 2009;15:384–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Guillot L, Medjane S, Le-Barillec K, Balloy V, Danel C, Chignard M, Si-Tahar M. Response of human pulmonary epithelial cells to lipopolysaccharide involves Toll-like receptor 4 (TLR4)-dependent signaling pathways: evidence for an intracellular compartmentalization of TLR4. J Biol Chem 2004;279:2712–8 [DOI] [PubMed] [Google Scholar]
- 80. Sha Q, Truong-Tran AQ, Plitt JR, Beck LA, Schleimer RP. Activation of airway epithelial cells by toll-like receptor agonists. Am J Respir Cell Mol Biol 2004;31:358–64 [DOI] [PubMed] [Google Scholar]
- 81. Jiang D, Liang J, Fan J, Yu S, Chen S, Luo Y, Prestwich GD, Mascarenhas MM, Garg HG, Quinn DA, Homer RJ, Goldstein DR, Bucala R, Lee PJ, Medzhitov R, Noble PW. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat Med 2005;11:1173–9 [DOI] [PubMed] [Google Scholar]
- 82. Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 1999;11:115–22 [DOI] [PubMed] [Google Scholar]
- 83. Lu M, Zhang Q, Chen K, Xu W, Xiang X, Xia S. The regulatory effect of oxymatrine on the TLR4/MyD88/NF-kappaB signaling pathway in lipopolysaccharide-induced MS1 cells. Phytomedicine 2017;36:153–9 [DOI] [PubMed] [Google Scholar]
- 84. Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, Bischof JC, Hebbel RP, Vercellotti GM. Transgenic sickle mice have vascular inflammation. Blood 2003;101:3953–9 [DOI] [PubMed] [Google Scholar]
- 85. Gbotosho OT, Kapetanaki MG, Kato GJ. The worst things in life are free: the role of free heme in sickle cell disease. Front Immunol 2020;11:561917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Gonsalves CS, Li C, Malik P, Tahara SM, Kalra VK. Peroxisome proliferator-activated receptor-alpha-mediated transcription of miR-301a and miR-454 and their host gene SKA2 regulates endothelin-1 and PAI-1 expression in sickle cell disease. Biosci Rep 2015;35:e00275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Li C, Mpollo MS, Gonsalves CS, Tahara SM, Malik P, Kalra VK. Peroxisome proliferator-activated receptor-alpha-mediated transcription of miR-199a2 attenuates endothelin-1 expression via hypoxia-inducible factor-1alpha. J Biol Chem 2014;289:36031–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Kalra VK, Zhang S, Malik P, Tahara SM. Placenta growth factor mediated gene regulation in sickle cell disease. Blood Rev 2018;32:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jang AJ, Chang SS, Park C, Lee CM, Benza RL, Passineau MJ, Ma J, Archer DR, Sutliff RL, Hart CM, Kang BY. PPARgamma increases HUWE1 to attenuate NF-kappaB/p65 and sickle cell disease with pulmonary hypertension. Blood Adv 2021;5:399–413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kang BY, Park K, Kleinhenz JM, Murphy TC, Sutliff RL, Archer D, Hart CM. Peroxisome proliferator-activated receptor gamma regulates the V-Ets avian erythroblastosis virus E26 oncogene homolog 1/microRNA-27a axis to reduce endothelin-1 and endothelial dysfunction in the sickle cell mouse lung. Am J Respir Cell Mol Biol 2017;56:131–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med 2002;53:409–35 [DOI] [PubMed] [Google Scholar]
- 92. Grygiel-Gorniak B. Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications–a review. Nutr J 2014;13:17. [DOI] [PMC free article] [PubMed] [Google Scholar]