

Abstract
DNA methylation is a conserved modification that must be precisely regulated during development to facilitate its roles in silencing transposable elements and regulating gene expression. In plants, DNA methylation changes during reproduction are widely documented and, in many cases, the underlying mechanisms are well understood. In somatic tissues, the diversity of methylation patterns are only recently emerging but they are often associated with the RNA-directed DNA methylation (RdDM) pathway. Here, we discuss advances in our understanding of how the locus-specific targeting and tissue-specific expression of RdDM proteins regulate methylation patterns, how the targeting of methylation at loci with imperfect homology expands the purview of RdDM, and how natural variation within RdDM factors impacts DNA methylation patterns.
Introduction
As DNA methylation is associated with the silencing of transposable elements (TEs) throughout the genome, and with the regulation of some protein-coding genes, it is critical to establish and maintain the appropriate methylation patterns throughout plant development. Such patterns reflect a balance of largely distinct pathways controlling the establishment, maintenance, and removal of DNA methylation. Briefly, methylation is initially established in all sequence contexts (CG, CHG, and CHH, where H=A, T, or C) by the RNA-directed DNA methylation (RdDM) pathway, which uses the sequence information provided by non-coding RNAs to specify where DNA methylation should be deposited by DOMAINS REARRANGED METHYLTRANSFERASE 2 (DRM2) (Fig. 1)[1]. Once established, DNA methylation is maintained via the activity of several additional DNA methyltransferases to generate heritable methylation patterns[2,3]. However, DNA methylation can also be actively removed by demethylases or passively lost without a functional maintenance system[4]. Depending on the cell type and developmental stage, DNA methylation changes during plant reproduction have been attributed to all the aforementioned processes, revealing diverse mechanisms that enable DNA methylation dynamics (reviewed in[5]). In somatic tissues, herein defined as those comprised of diploid as well as sporophytic cells, how these pathways generate methylation diversity remains less clear, but factors controlling the locus-specific targeting of both the demethylases (reviewed in[4]) and the RdDM machinery (covered below) are providing key mechanistic insights into the control of DNA methylation patterns during plant development.
Figure 1.
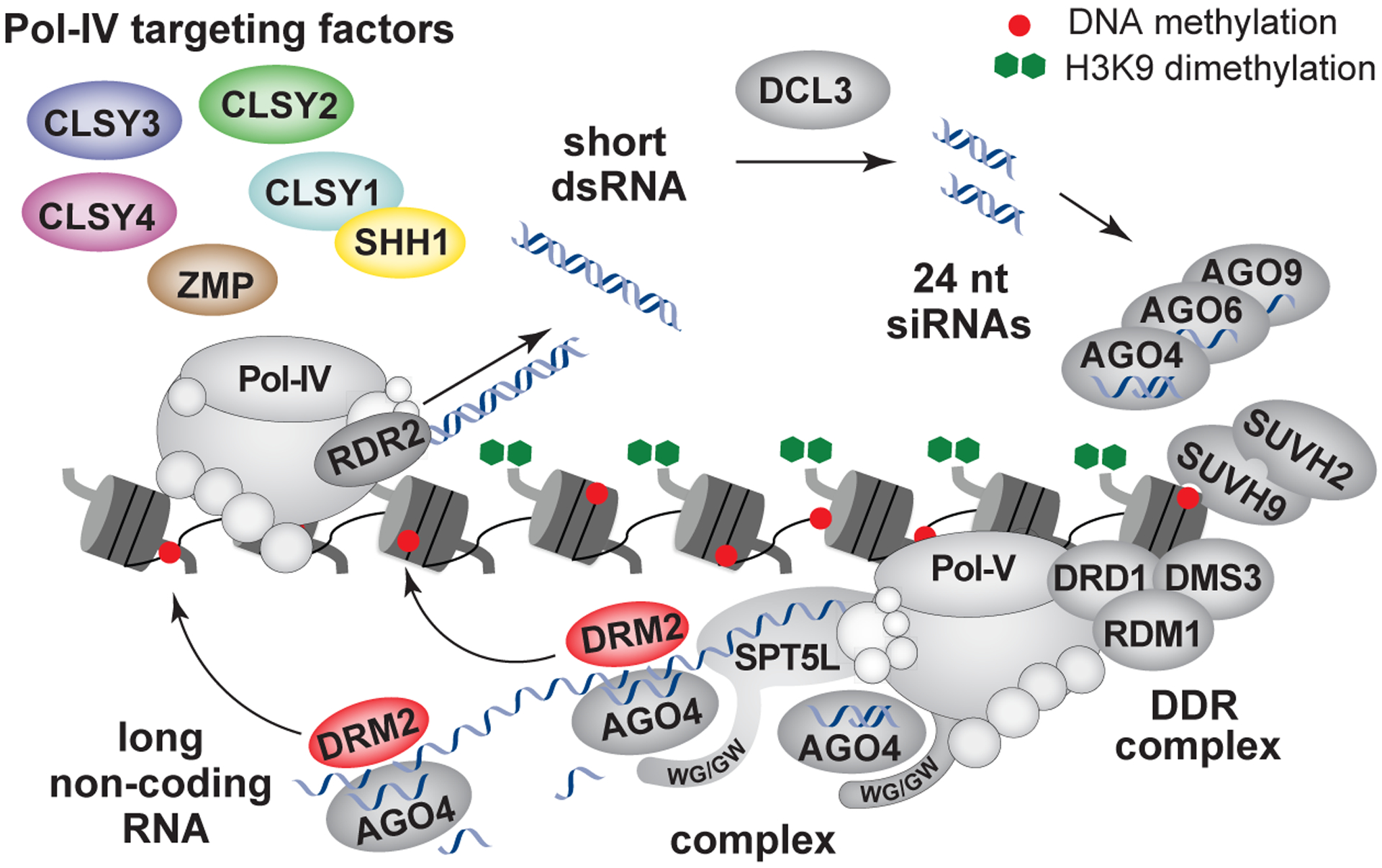
Detailed view of the canonical RdDM pathway, which utilizes two different types of non-coding RNAs to target DNA methylation and silence genes and transposons. Briefly, RdDM is initiated by the Pol-IV polymerase, which interacts with, and is targeted to chromatin by, a number of proteins, including SHH1, all four CLSYs, and ZMP. Pol-IV transcripts are processed co-transcriptionally by RDR2 to generate short double-stranded RNAs (dsRNAs). These RNAs are chopped by DCL3 into 24nt siRNAs, which are loaded into various AGO effector complexes. Pol-V, which is targeted by the DDR complex and two SUVH proteins, generates long non-coding RNAs. AGO effector complexes are recruited to RdDM loci via interactions with the WG/GW motifs present in Pol-V and SPT5L and interactions with the nascent Pol-V transcript. Sliced Pol-V transcripts bound by AGO4 are proposed to promote the association with DRM2 to enable efficient deposition of DNA methylation. Non-canonical RdDM pathways, which generate siRNAs in a Pol-IV independent manner, are reviewed in[43]. The red dots and green octagons represent DNA and histone methylation, respectively.
The RdDM pathway uses two plant-specific RNA polymerases, Pol-IV and Pol-V, to generate non-coding RNAs that enable the targeting of methylation at transposons and repeats (Fig. 1). Pol-IV associates with multiple accessory proteins, including SAWADEE HOMEODOMAIN HOMOLOG 1 (SHH1)[6], a family of four related putative chromatin remodeling factors, CLASSY (CLSY) 1–4[6], and ZINC FINGER, MOUSE DOUBLE-MINUTE/SWITCHING COMPLEX B, PLUS-3 Protein (ZMP)[7], which facilitate its localization to chromatin[6–8]. Once targeted, Pol-IV produces short (~26–45 nt) single-stranded RNAs that are rapidly converted into double-stranded RNA via a tight physical and biochemical connection with RNA-DEPENDENT RNA POLYMERASE 2 (RDR2)[1,9–11]. These double-stranded RNAs are then cleaved into 24nt small interfering RNAs (siRNAs) by DICER-LIKE 3 (DCL3) and loaded into either ARGONAUTE 4 (AGO4), AGO6, or AGO9 effector proteins[1,12]. The siRNA-loaded AGO complexes are then recruited to loci transcribed by Pol-V. This polymerase is targeted via interactions with its own unique set of factors, including the DDR complex (DEFECTIVE IN MERISTEM SILENCING 3 (DMS3), DEFECTIVE IN RNA-DIRECTED DNA METHYLATION 1 (DRD1), and RNA-DIRECTED DNA METHYLATION 1 (RDM1)) and two related methyl-DNA-binding factors, SU(VAR)3–9 HOMOLOG 2 (SUVH2) and SUVH9[13]. Pol-V also interacts with SUPPRESSOR OF TY INSERTION 5-LIKE (SPT5L) which, like the C-terminal domain of the largest Pol-V subunit, possesses WG/GW motifs that interact with AGO proteins[1]. At least in the case of AGO4, this recruitment results in the cleavage of nascent Pol-V transcripts[1], which is important for efficient targeting of DNA methylation by DRM2 at transposons and repeats throughout the genome[14].
While the core components and general process of RdDM are well established, the mechanisms through which this pathway is modulated to generate diverse methylation patterns are only now emerging. Here we cover three different aspects of RdDM regulation that contribute to DNA methylation diversity, focusing mainly on somatic tissues. First, we feature literature revealing the mechanisms employed by Pol-IV targeting factors to control locus- and tissue-specific DNA methylation patterns. We then highlight recent findings demonstrating that siRNAs can target methylation at loci with imperfect homology, either within or between tissues, to regulate additional protein-coding genes. Finally, we discuss the role of natural variants in shaping somatic methylation patterns. Throughout, we highlight examples where altered methylation at RdDM targets display developmental consequences.
Controlling Pol-IV targeting to regulate DNA methylation patterns in a locus-specific manner
With its early role in the RdDM pathway, regulating Pol-IV targeting has proven an effective way to modulate the expression levels of siRNAs, resulting in the generation of diverse methylation profiles. In this section, we detail advances in our understanding of Pol-IV targeting stemming from the identification and characterization of Pol-IV-interacting proteins, including SHH1[6], the CLSYs[6], and ZMP[7]. Together, these studies reveal how combining locus-specific targeting with tissue-specific expression allows the generation of diverse DNA methylation patterns during plant development.
Although initially only CLSY1 was known to play a role in RdDM[15], purification of all four CLSYs with Pol-IV[6] suggested roles for the whole family—making them excellent candidates for imparting distinct capabilities to the Pol-IV complex. Indeed, profiling siRNAs, DNA methylation, and Pol-IV chromatin association in various clsy mutant backgrounds resulted in the following key discoveries (Fig. 2A, B). First, clsy quadruple mutants show global losses of siRNAs, phenocopying pol-iv mutants and demonstrating that together, the 4 CLSY proteins are master regulators of Pol-IV function[8,16]. Second, each clsy single mutant affects siRNA production and DNA methylation levels at largely nonoverlapping RdDM targets, establishing them as locus-specific regulators of Pol-IV function[8,16]. Third, there is redundancy within the CLSY family as clsy1,2 and clsy3,4 double mutants affect more loci than any of their single mutant counterparts[8]. Interestingly, this redundancy revealed that loci controlled by CLSY1 and CLSY2 versus CLSY3 and CLSY4 are not randomly distributed in the genome, but rather enriched in the chromosome arms or pericentromeric heterochromatin, respectively[8], suggesting a division of labor depending on different chromatin states. Consistent with this hypothesis, targeting of Pol-IV by the CLSYs relies on different chromatin modifications: CLSY1 and CLSY2 control siRNA production in an H3K9 methylation-dependent manner by mediating the interaction between Pol-IV and SHH1[8], a chromatin reader that specifically recognizes unmodified H3K4 and methylated H3K9 residues to facilitate Pol-IV targeting (Fig. 3A)[17,18]. On the contrary, siRNA production at loci controlled by CLSY3 and CLSY4 is independent of H3K9 methylation[8] and instead, shows a partial dependence on CG methylation via mechanisms that remain to be determined (Fig. 3B)[8]. Taken together, these findings demonstrate that the CLSYs are crucial for mediating key associations within the Pol-IV complex and for leveraging distinct chromatin features to enable Pol-IV targeting in a highly locus-specific manner.
Figure 2.
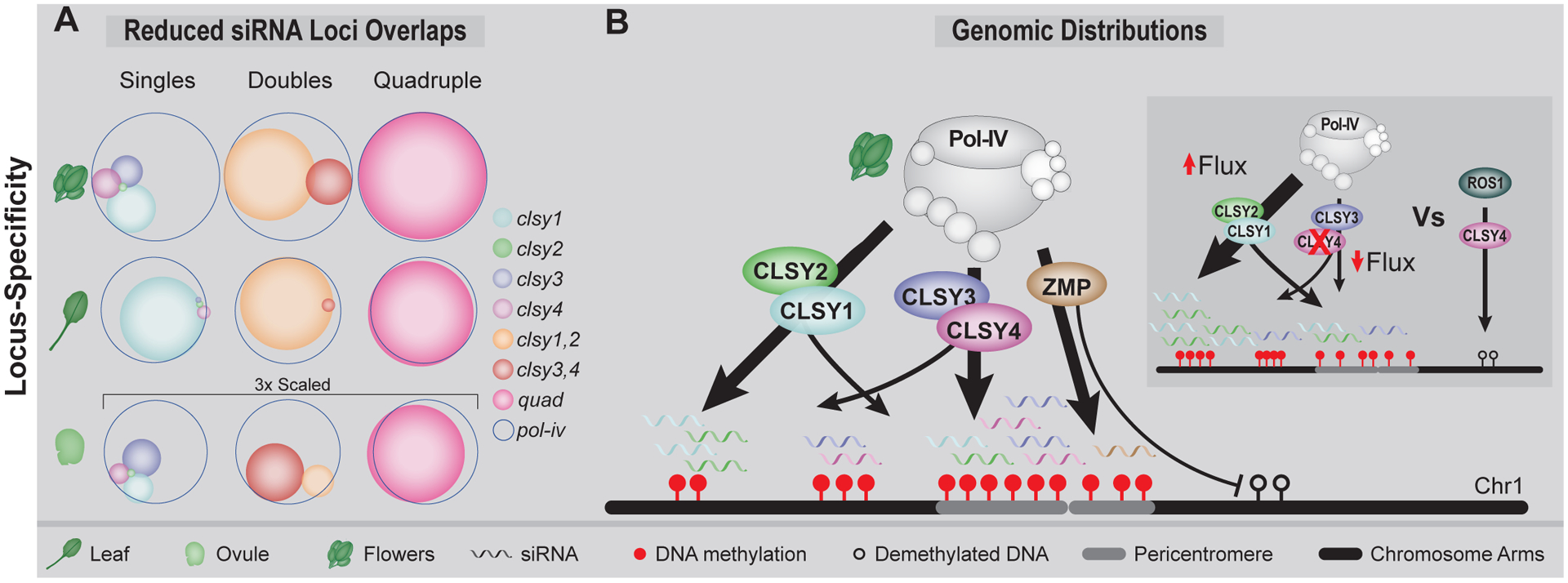
Locus- and tissue-specificity of Pol-IV targeting factors. (A) Scaled Venn diagrams adapted from[20] showing the overlaps of reduced siRNA clusters in the indicated genotypes and tissues. Overlaps in ovules are shown on a 3x scale. (B) Cartoon depicting the genomic distributions of loci regulated by the CLSYs and ZMP in flowers, where arrow thicknesses correspond to the relative numbers of siRNA clusters regulated by these factors. An inlay showing two alternative models for the hypermethylation phenotypes observed in clsy mutants, using clsy4 as an example: (Left) Redistribution of Pol-IV in the absence of CLSY4 vs (Right) Direct role for CLSY4 in targeting the DNA demethylase, ROS1.
Figure 3.
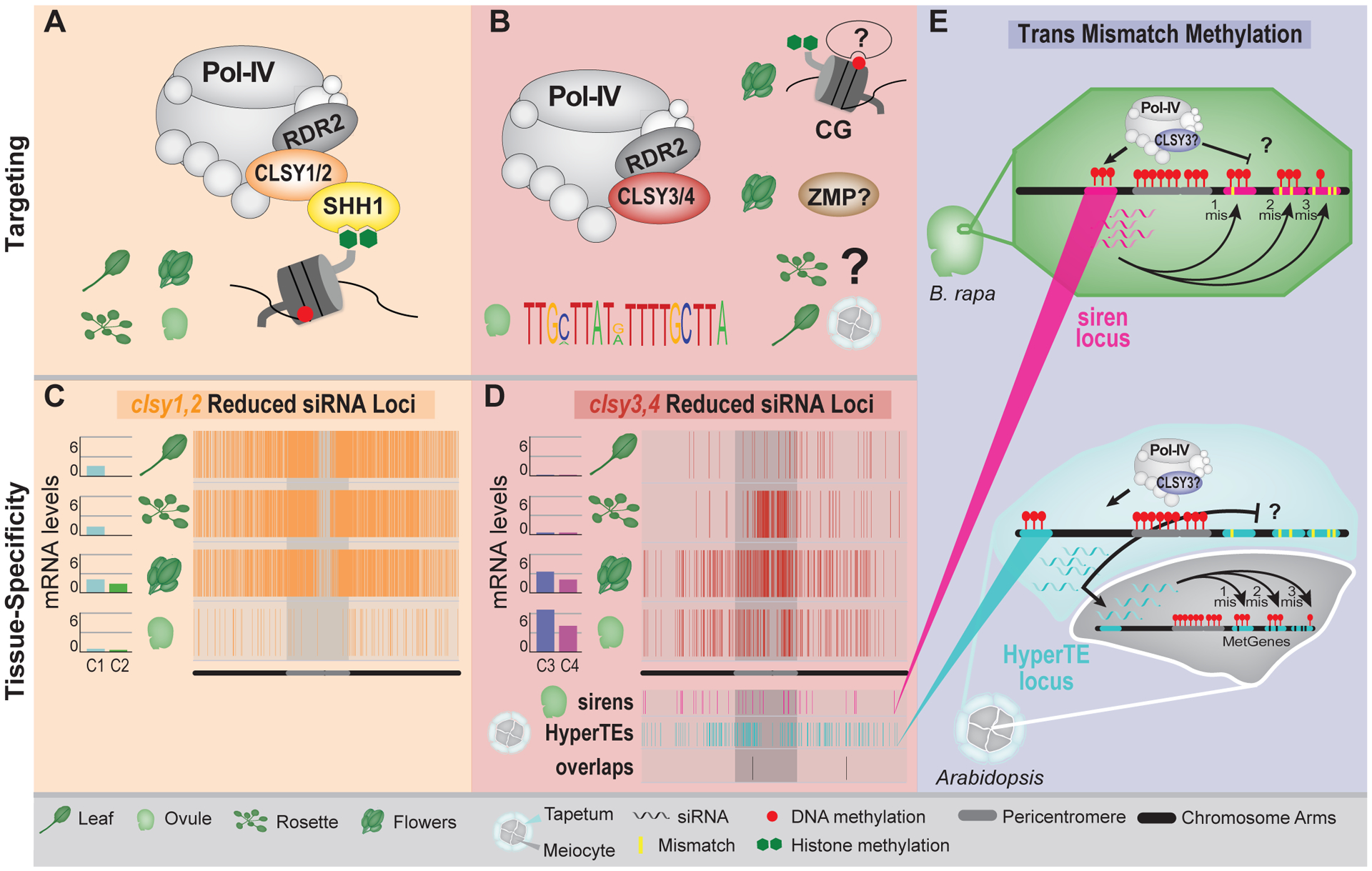
Mechanisms employed by Pol-IV targeting factors to generate epigenetic diversity within and between tissues. (A-B) Cartoons depicting the targeting mechanisms employed by CLSY1 and CLSY2 (A), or CLSY3 and CLSY4 (B), in the indicated tissues. In B, the targeting mechanism(s) in several tissues remain open questions (?). (C and D) CLSY expression levels [log2 (fpkm+1)] and genomic distributions (Chr 1) of reduced siRNA clusters in clsy1,2 (C) or clsy3,4 (D) mutants in the indicated tissues[20]. In D, the distributions of siren loci in ovules and HyperTEs in tapetum cells, as well as the overlaps of these loci, are also included. (E) Cartoons portraying the ability of siRNAs from siren loci to target methylation at genes with imperfect homology in ovule cells (Upper) and the ability of siRNAs from HyperTE loci to move from tapetum cells into neighboring meiocytes to target genes with imperfect homology (Lower). In the former, what blocks Pol-IV from generating perfectly matching siRNAs at the targeted genes remains an open question (?) and in the latter, what blocks the targeting of methylation at imperfectly matching targets in tapetum cells remains an open question (?). The background colors in A, B and E correspond to the Pol-IV targeting factors being discussed.
Besides the above-mentioned roles for the CLSYs in establishing DNA methylation, all four clsy single mutants, as wells as several triple mutant combinations, are also associated with locus-specific increases in DNA methylation[16], as are mutations in another Pol-IV-interacting protein, ZMP[7]. In these clsy mutants, the hypermethylated sites overlap loci regulated by demethylases, suggesting dual roles in the RdDM and demethylation pathways (Fig. 2B; inlay)[16]. However, as this hypermethylation depends on Pol-IV and the other CLSYs[16], it could instead result from hyperactive and/or redistributed RdDM (Fig. 2B; inlay). Indeed, such a hypothesis was proposed for the zmp mutant as both the hypomethylated loci in pericentromeric heterochromatin and the hypermethylated loci in more gene-rich regions are Pol-IV-dependent (Fig. 2B). In this case, Pol-IV targeting to heterochromatin might involve CLSY3 and/or CLSY4 (Fig. 3B) while restricting Pol-IV activity near genes (Fig. 2B) could rely on the preference of ZMP’s plant homeodomain (PHD) for unmodified H3K4 histones[7]. Although additional experimentation will be required to better understand the mechanisms underlying the hypermethylation phenotypes in the clsy and zmp mutants, these studies demonstrate that mutants that affect Pol-IV targeting can give rise to locus-specific losses and gains of DNA methylation, emphasizing the importance of a proper balance of Pol-IV targeting in shaping the epigenome during plant development.
In summary, these discoveries have uncovered Pol-IV targeting factors capable of exerting fine-tuned control over the RdDM pathway. They act at discrete loci, numbering in the 10s to 1000s depending on the factor, and display strong preferences for controlling DNA methylation, mainly in the non-CG context, in gene-rich (chromosome arms) or TE-rich (pericentromeric heterochromatin) regions of the genome. Attesting the importance of SHH1, the CLSYs, and ZMP beyond their roles in TE silencing, they regulated a number of protein-coding genes[7,8], a small subset of which are associated with important plant developmental traits. For example, CLSY1 is required to silence newly introduced copies of FLOWERING WAGENINGEN (FWA) and prevent a late-flowering phenotype (Fig. 4)[8,19]. On the other hand, hypermethylated loci in the zmp mutant are enriched for defense genes and this mutant is more susceptible to a virulent oomycete[7], suggesting an unknown gene important for pathogen defense may be aberrantly silenced by hypermethylation in zmp (Fig. 4). As more is learned about genes regulated by the RdDM pathway, additional examples are likely to emerge, expanding or understanding of how the locus-specific modulation of this pathway contributes to a variety of plant phenotypes.
Figure 4.
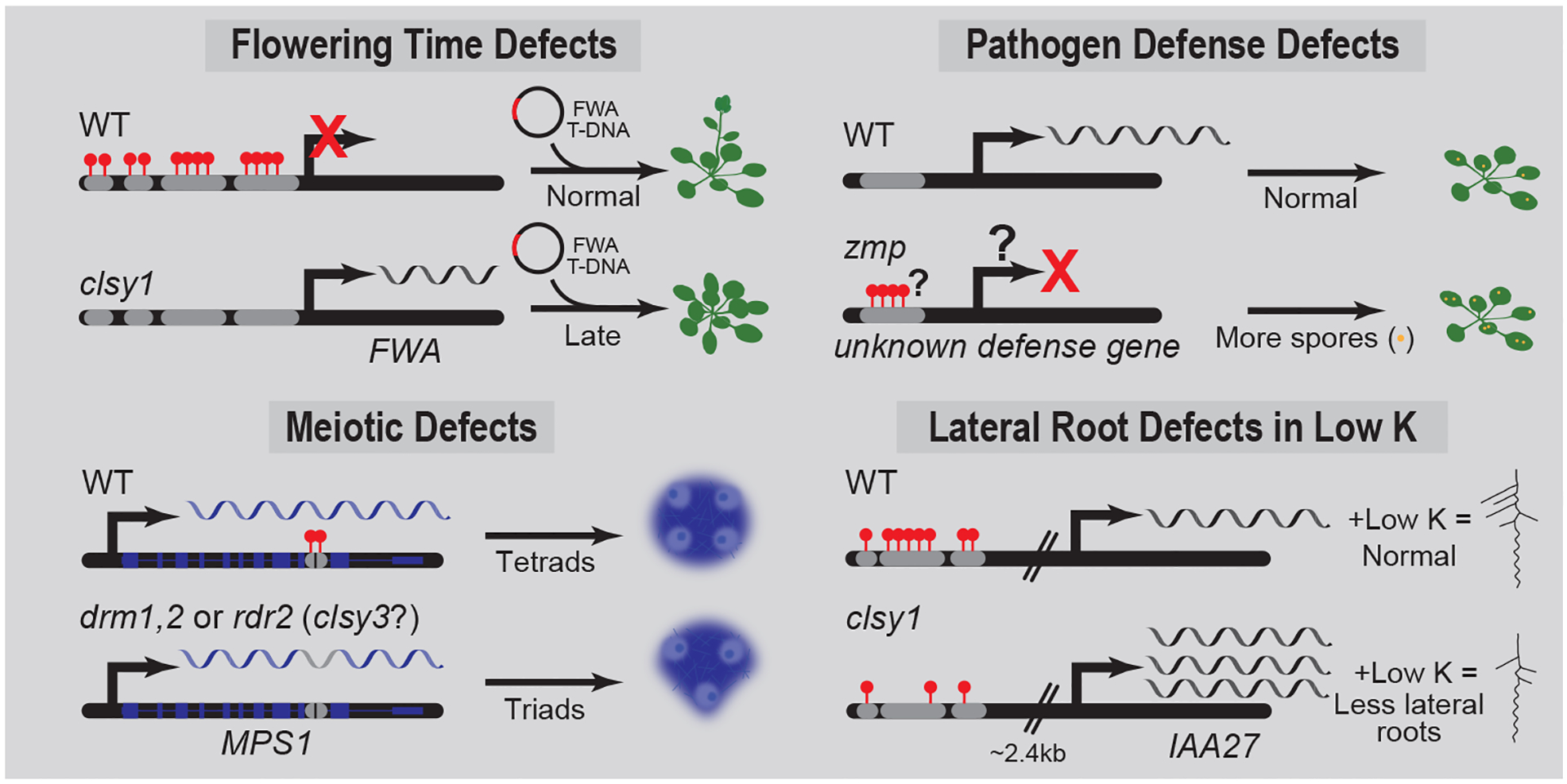
Examples of genes mis-regulated in RdDM mutants. Cartoons showing the locus-specific misregulation of the indicated genes in RdDM mutants along with their phenotypic consequences. For zmp, the target gene and mechanism of gene silencing remain to be determined as indicated by question marks (?). Root images were adapted from[40].
Combining tissue-specific expression with locus-specific targeting to generate methylation diversity
In addition to their distinct, locus-specific targeting mechanisms, the expression patterns of the CLSYs vary during plant development, suggesting they may also play roles in regulating tissue-specific DNA methylation patterns. In this section, we feature several recent studies demonstrating such roles for the CLSYs in flowers[8,20], ovules[20], leaves[20], rosettes[20], seedlings[16], tapetum cells and meiocytes[21]. In each case, the global methylation patterns differed, though some were more distinct than others, and relied to different extents on each of the four CLSY proteins. Furthermore, for many of these examples[20] the vast majority of methylation differences were directly attributed to the RdDM pathway[20]. Thus, while other pathways capable of altering DNA methylation patterns (i.e., those controlling the maintenance or removal of methylation) are certainly also important[2–4], they play minor roles in comparison to the RdDM pathway in generating DNA methylation diversity in the aforementioned tissues[20].
While the current studies are just the beginning in terms of understanding how the CLSYs modulate DNA methylation patterns in a tissue-specific manner, they highlight the different genetic requirements for, and relationships between, CLSY family members. For example, in contrast to the initial findings in flowers[8], there is minimal redundancy between CLSY1 and CLSY2 and very few loci regulated by CLSY3 and CLSY4 in seedlings, leaves, and rosettes, demonstrating CLSY1 is the main regulator of global siRNA levels in these tissues (Fig. 2A)[16,20]. In ovules, the opposite genetic requirements were revealed as in this tissue, CLSY3 plays the major role with some redundancy with CLSY4 (Fig. 2A)[20]. Notably, as the genetic requirements observed for the CLSYs correspond well with their expression levels (Fig. 3C, D)[20,21], transcriptome data alone may prove a useful proxy for predicting the relative contributions of the CLSYs in specific tissues, cell-types or environmental conditions.
Studies of the CLSYs are also uncovering some shared and some tissue-specific behaviors for these proteins. For example, in all tissues tested, CLSY1 and CLSY2 regulate loci that are enriched in the more gene-rich chromosome arms (Fig. 3C) and are also regulated by SHH1 [8,18,20]. Indeed, although a smaller subset of regions are targeted in ovules, similar loci are targeted in leaves, rosettes, and flowers[20], consistent with a common mode of targeting via SHH1 across tissues for these two CLSYs (Fig. 3A). In contrast, tissues that rely mainly on CLSY3 and CLSY4, including ovules[20], tapetum cells[21], and male meiocytes[22], show more variation in their epigenetic landscapes. This variation includes both the levels of expression observed at individual siRNA loci between tissues as well as the overall patterns of siRNA production (Fig. 3D); Instead of a large number of more lowly expressed siRNA loci, most of the siRNAs are generated from a small number of highly expressed loci, leading to distinctive global DNA methylation patterns. This is especially true in ovules where ~80–90% of siRNAs arise from 128[23]–133[20] loci designated as “siren loci”, but is also true in tapetum cells where ~95% of siRNAs arise from 797 loci designated as “HyperTE loci”[21] (Fig. 3D). What allows these loci to produce such high levels of siRNAs remains unclear. However this phenomenon, sans the mechanistic details, appears to be conserved as similar patterns of siRNA expression were observed in rice[24] and B. rapa[23] ovules (reviewed in[25]) and highly expressed siRNA loci were previously identified in maize tapetum cells and designated reproductive 24nt-phasiRNAs (reviewed in[25,26]). As detailed in the next section, another particularity of siRNAs from some of these highly expressed loci that is just beginning to be explored is their ability to target methylation at loci with imperfect sequence matches and to be produced in one cell-type, but act in another.
Mechanistically, the production of siRNAs at siren loci in ovules, and at HyperTE loci in tapetum cells, depends almost exclusively on CLSY3[20,21]. However, these loci show minimal overlaps, with only 22 loci in common genome-wide and just 2 overlapping on chromosome 1 (Fig. 3D), indicative of differential targeting modes for CLSY3. While the parameters controlling CLSY3 targeting in tapetum cells remain unknown, some insights into CLSY3 targeting in ovules were inferred based on its chromatin association profile in flowers[20]. These assays revealed that CLSY3 is strongly enriched at the majority of siren loci (94/133), and many of these binding sites (66/133) contained a highly conserved DNA motif (Fig. 3B)[20]. Interestingly, while the function of this motif remains unclear, it may be conserved as somewhat similar sequence motifs were identified in two TE families associated with siren loci in B. rapa[27]. It is also notable that none of the CLSY3 target loci that contain this motif overlap with the tapetum HyperTE loci. Therefore, unlike for CLSY1 and CLSY2, there appear to be tissue-specific factors and/or chromatin features shaping CLSY3 targeting, resulting in more unique siRNA and DNA methylation landscapes in tissues that rely on this CLSY (Fig. 3D).
In summary, these recent discoveries demonstrate that the proper combinations and expression patterns of CLSY proteins are necessary to generate the appropriate tissue-specific pattern of siRNAs and DNA methylation in diverse somatic tissues. These patterns are dictated by the different targeting mechanisms employed by the CLSYs, which vary in their genetic and epigenetic requirements. In moving forward, it will be especially critical to gain additional insights into the mechanisms controlling CLSY3 and CLSY4 targeting and to assess whether ectopic expression of the CLSYs is sufficient to alter siRNA and DNA methylation patterns in a predictable manner. Such advances would add a much-needed level of precision to ongoing efforts aimed and epigenetic engineering.
Expanding the purview of RdDM via trans-mapping of siRNAs to imperfectly matched targets
While it has long been appreciated that micro RNAs (miRNAs) can target and facilitate post-transcriptional suppression of imperfectly matching mRNA targets[28], whether siRNAs are able to target DNA methylation and facilitate transcriptional gene silencing at imperfectly matching loci has remained an open question until very recently. This lag in our understanding of the homology requirements for siRNAs stems largely from the self-reinforcing nature of the RdDM pathway wherein methylation at a locus potentiates the recruitment of RNA Pol-IV and Pol-V to generate more siRNAs and target more methylation, respectively[13]. Based on this model, once an imperfectly matched locus is sufficiently methylated, it would recruit Pol-IV and generate perfectly matched siRNAs, making it difficult to determine if the matched or mismatched siRNAs targeted methylation. In this section, we highlight several recent studies that have either supplied exogenous sources of mismatched siRNAs[29] or taken advantage of rare RdDM loci not subject to the aforementioned reinforcing loops[21,22,27] to study the impact of mismatches in targeting DNA methylation. They demonstrate that siRNAs can target methylation at loci with imperfect homology[21,27], expanding the set of genomic loci subject to regulation by the RdDM pathway and exposing the critical role of CLSY3 in this aspect of RdDM.
To directly test the ability of siRNAs to target methylation at loci with imperfect homology, Fei et al.[29] supplied exogenous sources of siRNAs using Virus Induced Gene Silencing (VIGS). Specifically, tobacco rattle virus (TRV) was engineered to encode an RNA fragment that, once processed by the host antiviral machinery, would generate siRNAs of all size classes (21–24nts) that perfectly match their host DNA target or contain one, two, or four mismatches. When delivered into tobacco, targeting a 35S promoter driving GFP, or into Arabidopsis, targeting FWA, constructs generating siRNAs with zero, one, or two mismatches were able to target methylation and suppress gene expression at essentially equal efficiency[29]. For the tobacco VIGS with 2 mismatches, small RNA sequencing revealed no perfectly matched siRNAs were generated from the genomically encoded 35S promoter[29], demonstrating that perfect matching is not required for targeting DNA methylation. However, a high degree of homology remains crucial, as constructs with four mismatches targeted lower methylation levels and displayed weaker gene silencing phenotypes[29]. Notably, in both tobacco and Arabidopsis, methylation targeted by siRNAs with imperfect homology was heritable, as silencing was observed in the generation after transient exposure to the virus[29].
The use of endogenous siRNAs to target methylation at loci with imperfect homology, hereafter referred to as trans-mismatch-methylation, was recently shown during both anther and ovule development (Fig. 3E)[21,27]. In each case, this phenomenon was illustrated by characterizing loci that behave outside the norm for RdDM loci. For anthers, a subset of 469 “MetGenes”, which are methylated in an RdDM-dependent manner in meiocytes but overlap with very few perfectly matched siRNAs, were identified[22]. As these genes share homology with HyperTE loci that produce high levels of siRNAs in tapetum cells, which surround the developing meiocytes, it was hypothesized that siRNAs produced from transposon loci in tapetum cells could target methylation at imperfectly matched loci in meiocytes (Fig. 3E). Several lines of investigation validate this hypothesis. First, siRNAs mapping to ~1/2 of the MetGenes were identified when up to three mismatches were allowed[21]. Second, CRISPR-mediated deletion of two individual HyperTE loci resulted in locus-specific decreases in methylation at the three homologous MetGenes[21]. Third, lines that are only active for siRNA production in tapetum cells are sufficient for trans-mismatch methylation of MetGenes in meiocytes, illustrative of siRNA movement between cell-types[21]. Fourth, clsy3 mutant lines, which are defective in RdDM at HyperTE loci, fail to induce methylation at MetGenes[21]. Thus, while it remains unclear why MetGenes fail to recruit Pol-IV to generate perfectly matched siRNAs, and likewise, how MetGenes in tapetum cells avoid being trans-methylated, studying this set of loci confirms that siRNAs do not require perfect matches to target methylation and suppress gene expression. In addition, this study adds another layer of complexity by revealing trans-mismatch-methylation can occur in a cell non-autonomous manner[21].
For ovules, evidence for trans-mismatch-methylation came from work in Brassica rapa studying siren loci, which are distinct in that they produce exceptionally high levels of siRNAs but, unlike most RdDM targets, they show low overall overlaps with transposons[23,27]. Instead, ~1/3 of siren loci in B. rapa were found to contain gene fragments, and in most cases, these fragments overlapped with the peaks of siRNA expression[27]. This observation led to the hypothesis that siren siRNAs might have roles outside of transposon silencing, by targeting and regulating genes with partial homology elsewhere in the genome[27]. Indeed, remapping of siren-derived 23–24nt siRNAs to the B. rapa genome allowing one, two, or three mismatches identified thousands of putative trans-mismatch-methylation targets with ≥1 reads per million (RPM) of mismatched siRNAs and <10% perfectly matched siRNAs (Fig. 3E)[27]. Vetting of these putative targets demonstrated that, as a group (n= 6,678), targets with increasing numbers of siRNAs harboring one or two mismatches displayed increasing levels of non-CG methylation and were also more likely to be upregulated in mutants that lack siRNAs[27]. Moreover, they showed that mismatched siRNAs alone appear to be capable of targeting the aforementioned methylation, as a similar correlation between siRNA levels and DNA methylation was seen in genomic windows where only mismatched siRNAs were observed[27]. However, for targets identified allowing three mismatches, these correlations were much lower, indicating those siRNAs are less effective at targeting methylation[27].
Genetically, the observed trans-mismatch methylation was shown to depend on the RdDM pathway and a role for B. rapa CLSY3 was hypothesized based on its expression profile[27] and its role in siren siRNA production in Arabidopsis[20]; Of all the B. rapa CLSYs, CLSY3 is the highest expressed in tissues where siren siRNAs are abundant and trans-mismatch methylation was observed (i.e., ovules, seedcoats, and endosperm, but not embryos)[27]. As siren loci in Arabidopsis and B. rapa share many features, including the presence of gene fragments from some common families, a targeted search was conducted and one putative target for mismatched siRNAs was identified in Arabidopsis[27]. Thus, while additional experimentation is required to vet this candidate and to comprehensively search for additional targets, the epigenetic regulation of genes with imperfect homology stemming from siren loci appears to be a conserved feature across species.
In summary, the ability of siRNAs to target methylation at loci with imperfect homology has now been demonstrated in three independent scenarios involving multiple different tissues and organisms[21,27,29]. This trans-mismatch-methylation can occur at hundreds to thousands of loci and can act within or between cell types (Fig. 3E)[21,27]. In many, though not all cases, it can affect gene expression and influence plant development (e.g., MULTIOLAR SPINDLE 1 (MPS1)[22] (Fig. 4)), and be trans-generationally inherited[21,27,29]. While much remains to learn about the rules governing the efficiency of targeting DNA methylation with mismatched siRNAs (e.g., siRNA size, position, type of mismatch, etc.), the level of homology is certainly important, with one or two mismatches being more effective than three or four[27,29]. Furthermore, it remains unclear what limits this activity to prevent the targeting of methylation and silencing of genes on a more global scale. Regardless, the ability of siRNAs to target methylation at imperfectly matched loci represents a previously unappreciated mechanism through which the RdDM pathway in general, and CLSY3, in particular, controls DNA methylation patterns in both somatic and reproductive tissues.
Natural variation within RdDM players modulates DNA methylation and influences plant development
Arabidopsis plants have adapted to diverse environments across broad geographic ranges resulting in natural accessions that differ in their physiology. To accelerate the discovery of genetic and epigenetic alterations underlying these differences, a global effort was launched to sequence >1001 Arabidopsis accessions[30] and to pair this data with methylomes and transcriptomes[31–34]. While the discovery of key DNA methylation players (e.g., NRPE1[31,35], CMT2[31,35–37], CMT3[36], and MET1[31]) from genome-wide association studies (GWAS) using variation in methylation patterns as a trait clearly demonstrates that natural variation in specific genes can shape global methylation patterns in trans, linking these methylation differences to specific traits and/or the regulation of specific causal genes has remained a challenge (reviewed in[38,39]). However, recent work has revealed the mechanism through which natural variation in CLSY1 promotes lateral root development under low potassium (K) conditions (Fig. 4)[40], marking a key advance in our understanding of how natural variation within RdDM machinery affects methylation in a locus-specific manner to facilitate normal plant development.
Specifically, Shahzad et al.[40] found a SNP in the coding region of Arabidopsis CLSY1, resulting in a D538E amino acid change (E allele), which is associated with a reduction in lateral root numbers, but only under low K conditions. To understand the nature of this cryptic variation, the authors first demonstrated that the CLSY1 gene, in general, and the E allele, in particular, is causal to the low K phenotype via several independent genetic experiments[40]. Available methylome data was then mined to find potential target genes regulated by CLSY1 with links to auxin signaling, which they found was central to the defects in lateral root development in the low K condition[40]. This identified INDOLE-3-ACETIC ACID INDUCIBLE 27 (IAA27)[40], a suppressor of auxin signaling[41]. Reductions in methylation at several transposons upstream of IAA27 were confirmed in roots from clsy1, nrpd1, and rdr2 mutants, as well as in 10 E allele accessions compared to 10 D allele accessions. Furthermore, these methylation decreases were shown to correlate with increased IAA27 expression, confirming the epigenetic regulation of this gene by the RdDM pathway (Fig. 4)[40]. In addition, overexpression of IAA27 recapitulated the low K lateral root defects, supporting are direct role for defects in IAA27 silencing in the observed phenotype[40]. However, the methylation and expression changes observed in CLSY1-defective lines were not low K dependent, demonstrating that the RdDM pathway is not directly regulated by low K[40]. Thus, in this case, the accumulation of IAA27 protein under low K, in combination with the release of IAA27 silencing by CLSY1, was proposed to exacerbate the suppression of auxin signaling and result in the formation of fewer lateral roots[40].
This work demonstrates the impact of natural variation in regulating DNA methylation and controlling agronomically important plant traits. As CLSY1 was recently associated with rice responses to anaerobic germination[42], additional examples are likely to follow. Furthermore, as nearly all the methylation data for the Arabidopsis accessions are from leaf tissue[31], expanding this resource to include data from other tissues may uncover traits regulated by the other CLSYs. Together such studies will expand our understanding of how natural variation with the RdDM machinery can impact methylation patterns in trans to enable plant adaptation and impart robustness to plastic traits, like root development and seed germination.
Conclusion
As we explore the epigenetic landscapes of different tissues, cell-types, and developmental stages, it is becoming increasingly clear that DNA methylation patterns in plants are more diverse than initially appreciated. Herein, we highlighted the ability of siRNAs to target methylation in trans and for even a small number of Pol-IV-targeting factors (i.e., SHH1, ZMP, and the CLSYs) to generate distinct methylation landscapes by leveraging tissue-specific expression patterns and locus-specific preferences for different chromatin features. Despite these seminal advances many open questions remain. Among the Pol-IV targeting factors, CLSY3 appears to be the most versatile, yet least well-understood; It is expressed in tissues and cell-types with the most divergent methylation patterns and its locus-specific functions are associated with a number of genetic and epigenetic features, but the mechanisms underlying these connections remain unknown. For CLSY1, natural variation is emerging as an additional avenue to tune the activity of the RdDM pathway and control important plant traits. However, it remains an open question how wide-spread such effects are for CLSY1 targets specifically, and for other RdDM factors, in general. Finally, the revelation that siRNAs can target methylation at loci with imperfect homology represents a new, largely unexplored frontier in RdDM, and raises important questions about how this ability is kept in check to protect the genome from aberrant and ever-increasing levels of genic DNA methylation. In moving forward, gaining additional insights into these aspects of RdDM regulation and continuing to identify and connect different DNA methylation patterns to the responsible cellular machinery will be critical. Such discoveries will not only expand our understanding of how specific methylation patterns are established during normal development, but will also provide key insights for utilizing these factors to manipulate the epigenome in a predictable manner in both model and crop species.
Highlights.
Pol-IV-targeting mutants cause locus-specific gains and losses of DNA methylation
The specificities of Pol-IV-targeting factors dictate tissue-specific methylation patterns
siRNAs can target methylation in trans at loci with imperfect homology
Natural variation within a Pol-IV-targeting factor affects root development
Acknowledgments
We thank colleagues and lab members for their helpful comments and discussions. For Fig. 2A, the scaled Venn diagrams adapted from[20].
Funding:
This work was supported by the National Institutes of Health [grant number GM112966].
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rymen B, Ferrafiat L, Blevins T: Non-coding RNA polymerases that silence transposable elements and reprogram gene expression in plants. Transcription 2020, 11:172–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Law JA, Jacobsen SE: Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat Rev Genet 2010, 11:204–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Matzke MA, Mosher RA: RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nature reviews. Genetics 2014, 15:394–408. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Gong Z, Zhu JK: Active DNA demethylation in plants: 20 years of discovery and beyond. J Integr Plant Biol 2022, 64:2217–2239. [DOI] [PubMed] [Google Scholar]
- 5.Kawashima T, Berger F: Epigenetic reprogramming in plant sexual reproduction. Nat Rev Genet 2014, 15:613–624. [DOI] [PubMed] [Google Scholar]
- 6.Law JA, Vashisht AA, Wohlschlegel JA, Jacobsen SE: SHH1, a homeodomain protein required for DNA methylation, as well as RDR2, RDM4, and chromatin remodeling factors, associate with RNA polymerase IV. PLoS Genet 2011, 7:e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Le BH, Wang J, You C, Zhao Y, Galli M, Xu Y, Gallavotti A, Eulgem T, Mo B, et al. : ZMP recruits and excludes Pol IV-mediated DNA methylation in a site-specific manner. Sci Adv 2022, 8:eadc9454. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Here, the authors report the identification of ZMP and demonstrate this Pol-IV-targeting protein promotes siRNA production in pericentromeric heterochromatin, but represses it in euchromatic regions. Mechanistically, they demonstrate that ZMP binds histone 3 tails, with a preference for low levels of lysine 4 methylation, and propose a model wherein this preference protects genes from aberrant silencing.
- 8.Zhou M, Palanca AMS, Law JA: Locus-specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nat Genet 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang K, Wu XX, Fang CL, Xu ZG, Zhang HW, Gao J, Zhou CM, You LL, Gu ZX, Mu WH, et al. : Pol IV and RDR2: A two-RNA-polymerase machine that produces double-stranded RNA. Science 2021, 374:1579–1586. [DOI] [PubMed] [Google Scholar]
- 10.Fukudome A, Singh J, Mishra V, Reddem E, Martinez-Marquez F, Wenzel S, Yan R, Shiozaki M, Yu Z, Wang JC, et al. : Structure and RNA template requirements of Arabidopsis RNA-DEPENDENT RNA POLYMERASE 2. Proc Natl Acad Sci U S A 2021, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh J, Pikaard CS: Reconstitution of siRNA Biogenesis In Vitro: Novel Reaction Mechanisms and RNA Channeling in the RNA-Directed DNA Methylation Pathway. Cold Spring Harb Symp Quant Biol 2019, 84:195–201. [DOI] [PubMed] [Google Scholar]
- 12.Loffer A, Singh J, Fukudome A, Mishra V, Wang F, Pikaard CS: A DCL3 dicing code within Pol IV-RDR2 transcripts diversifies the siRNA pool guiding RNA-directed DNA methylation. Elife 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou M, Law JA: RNA Pol IV and V in gene silencing: Rebel polymerases evolving away from Pol II’s rules. Curr Opin Plant Biol 2015, 27:154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang F, Huang HY, Huang J, Singh J, Pikaard CS: Enzymatic reactions of AGO4 in RNA-directed DNA methylation: siRNA duplex loading, passenger strand elimination, target RNA slicing, and sliced target retention. Genes Dev 2023, 37:103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smith LM, Pontes O, Searle I, Yelina N, Yousafzai FK, Herr AJ, Pikaard CS, Baulcombe DC: An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis. Plant Cell 2007, 19:1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang DL, Zhang G, Wang L, Li J, Xu D, Di C, Tang K, Yang L, Zeng L, Miki D, et al. : Four putative SWI2/SNF2 chromatin remodelers have dual roles in regulating DNA methylation in Arabidopsis. Cell Discov 2018, 4:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Law JA, Du J, Hale CJ, Feng S, Krajewski K, Palanca AM, Strahl BD, Patel DJ, Jacobsen SE: Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature 2013, 498:385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Ma ZY, Zeng L, Tanaka K, Zhang CJ, Ma J, Bai G, Wang P, Zhang SW, Liu ZW, et al. : DTF1 is a core component of RNA-directed DNA methylation and may assist in the recruitment of Pol IV. Proc Natl Acad Sci U S A 2013, 110:8290–8295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenberg MV, Ausin I, Chan SW, Cokus SJ, Cuperus JT, Feng S, Law JA, Chu C, Pellegrini M, Carrington JC, et al. : Identification of genes required for de novo DNA methylation in Arabidopsis. Epigenetics 2011, 6:344–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou M, Coruh C, Xu G, Martins LM, Bourbousse C, Lambolez A, Law JA: The CLASSY family controls tissue-specific DNA methylation patterns in Arabidopsis. Nat Commun 2022, 13:244. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This work demonstrates that patterns of DNA methylation differ significantly between somatic tissues and attributes these changes to the locus-specificities and expression patterns of the CLSYs. It also revealed that CLSY3 controls the distinct epigenetic landscape observed in ovules and connected this regulation to a highly conserved DNA motif, suggesting novel, tissue-specific, links between genetic elements and RdDM.
- 21.Long J, Walker J, She W, Aldridge B, Gao H, Deans S, Vickers M, Feng X: Nurse cell--derived small RNAs define paternal epigenetic inheritance in Arabidopsis. Science 2021, 373. [DOI] [PubMed] [Google Scholar]; **This study demonstrates that CLSY3-dependent siRNAs, which are generated from transposons in tapetum cells, are required to target methylation in meiocytes at genes with imperfect sequence homology. They also show complementation of RdDM defects specifically in tapetum cells restores methylation patterns to wild-type levels in sperm cells, revealing an important role for tapetum siRNAs in controlling methylation throughout germ cell development.
- 22.Walker J, Gao H, Zhang J, Aldridge B, Vickers M, Higgins JD, Feng X: Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat Genet 2018, 50:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grover JW, Burgess D, Kendall T, Baten A, Pokhrel S, King GJ, Meyers BC, Freeling M, Mosher RA: Abundant expression of maternal siRNAs is a conserved feature of seed development. Proc Natl Acad Sci U S A 2020, 117:15305–15315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, Xu H, Fu FF, Russell SD, Sundaresan V, Gent JI: Genome-wide redistribution of 24-nt siRNAs in rice gametes. Genome Res 2020, 30:173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chow HT, Mosher RA: Small RNA-mediated DNA methylation during plant reproduction. Plant Cell 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu Y, Teng C, Xia R, Meyers BC: PhasiRNAs in Plants: Their Biogenesis, Genic Sources, and Roles in Stress Responses, Development, and Reproduction. Plant Cell 2020, 32:3059–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Burgess D, Chow HT, Grover JW, Freeling M, Mosher RA: Ovule siRNAs methylate protein-coding genes in trans. Plant Cell 2022, 34:3647–3664. [DOI] [PMC free article] [PubMed] [Google Scholar]; *This publication reveals the presence of gene fragments under the peaks of siRNA expression within many siren loci in B. rapa ovules. It further demonstrates that these siRNAs are correlated with the targeting of methylation and the suppression of gene expression at homologous genic loci that contain up to two mismatches.
- 28.Carthew RW, Sontheimer EJ: Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136:642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fei Y, Nyiko T, Molnar A: Non-perfectly matching small RNAs can induce stable and heritable epigenetic modifications and can be used as molecular markers to trace the origin and fate of silencing RNAs. Nucleic Acids Res 2021, 49:1900–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]; *Using a Virus Induced Gene Silencing system, this paper demonstrates that small RNAs can target methylation and silence the expression of genes with imperfect homology in both tobacco and Arabidopsis. They further show that the amount of homology is important, as small RNAs with only one or two mismatches are more efficient at targeting methylation than those with four mismatches.
- 30.Genomes Consortium. Electronic address mngoaa, Genomes C: 1,135 Genomes Reveal the Global Pattern of Polymorphism in Arabidopsis thaliana. Cell 2016, 166:481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawakatsu T, Huang SC, Jupe F, Sasaki E, Schmitz RJ, Urich MA, Castanon R, Nery JR, Barragan C, He Y, et al. : Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166:492–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dubin MJ, Zhang P, Meng D, Remigereau MS, Osborne EJ, Paolo Casale F, Drewe P, Kahles A, Jean G, Vilhjalmsson B, et al. : DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 2015, 4:e05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagmann J, Becker C, Muller J, Stegle O, Meyer RC, Wang G, Schneeberger K, Fitz J, Altmann T, Bergelson J, et al. : Century-scale methylome stability in a recently diverged Arabidopsis thaliana lineage. PLoS Genet 2015, 11:e1004920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, et al. : Patterns of population epigenomic diversity. Nature 2013, 495:193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sasaki E, Kawakatsu T, Ecker JR, Nordborg M: Common alleles of CMT2 and NRPE1 are major determinants of CHH methylation variation in Arabidopsis thaliana. PLoS Genet 2019, 15:e1008492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sasaki E, Gunis J, Reichardt-Gomez I, Nizhynska V, Nordborg M: Conditional GWAS of non-CG transposon methylation in Arabidopsis thaliana reveals major polymorphisms in five genes. PLoS Genet 2022, 18:e1010345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shen X, De Jonge J, Forsberg SK, Pettersson ME, Sheng Z, Hennig L, Carlborg O: Natural CMT2 variation is associated with genome-wide methylation changes and temperature seasonality. PLoS Genet 2014, 10:e1004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seymour DK, Becker C: The causes and consequences of DNA methylome variation in plants. Curr Opin Plant Biol 2017, 36:56–63. [DOI] [PubMed] [Google Scholar]
- 39.Huther P, Hagmann J, Nunn A, Kakoulidou I, Pisupati R, Langenberger D, Weigel D, Johannes F, Schultheiss SJ, Becker C: MethylScore, a pipeline for accurate and context-aware identification of differentially methylated regions from population-scale plant whole-genome bisulfite sequencing data. Quant Plant Biol 2022, 3:e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shahzad Z, Eaglesfield R, Carr C, Amtmann A: Cryptic variation in RNA-directed DNA-methylation controls lateral root development when auxin signalling is perturbed. Nat Commun 2020, 11:218. [DOI] [PMC free article] [PubMed] [Google Scholar]; **The paper identified CLSY1 from a genome wide association study assessing lateral root development under low potassium conditions. They also identified a specific gene, IAA27, that is causal to the phenotype and demonstrated that DNA methylation and expression levels at this locus are regulated by CLSY1, directly linking natural variation in CLSY1 to a locus-specific difference in methylation that regulates an agronomically important root trait.
- 41.Cance C, Martin-Arevalillo R, Boubekeur K, Dumas R: Auxin response factors are keys to the many auxin doors. New Phytol 2022, 235:402–419. [DOI] [PubMed] [Google Scholar]
- 42.Castano-Duque L, Ghosal S, Quilloy FA, Mitchell-Olds T, Dixit S: An epigenetic pathway in rice connects genetic variation to anaerobic germination and seedling establishment. Plant Physiol 2021, 186:1042–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cuerda-Gil D, Slotkin RK: Non-canonical RNA-directed DNA methylation. Nat Plants 2016, 2:16163. [DOI] [PubMed] [Google Scholar]