

Abstract
Background and Aims:
HCC is a highly heterogeneous disease that is caused largely by genomic copy number variations. Herein, the mechanistic and therapeutically targeted role of vacuolar protein sorting 72 homologue (VPS72), a novel copy number variation cis-driven gained gene identified by genome-wide copy number variation and transcriptome analyses in HCC, is not well understood.
Approach and Results:
First, overexpression of VPS72 enhanced the initiation and progression of HCC in vitro and in vivo. Mechanistically, VPS72 interacted with the oncoproteins MYC and actin-like 6A (ACTL6A) and promoted the formation of the ACTL6A/MYC complex. Furthermore, ACTL6A regulated VPS72 protein stability by weakening the interaction between tripartite motif containing 21 (TRIM21) and VPS72. Thus, the interaction between VPS72 and ACTL6A enhanced the affinity of MYC for its target gene promoters and promoted their transcription, thereby contributing to HCC progression, which was inhibited by adeno-associated virus serotype 8 (AAV8)-mediated short hairpin RNA (shRNA) against VPS72.
Conclusions:
This study reveals the molecular mechanism of ACTL6A/VPS72/MYC in HCC, providing a theoretical basis and therapeutic target for this malignancy.

INTRODUCTION
Although substantial progress has been made in the treatment of hepatocellular carcinoma (HCC) in recent years, the prognosis remains less than ideal, with the 5-year overall survival (OS) rate being <50%.1 This may be because of the complex pathogenesis of HCC, which involves epigenetic and genetic changes, including mutations and the accumulation of copy number variations (CNVs).2–4
CNVs are chromosomal aberrations that include copy number deep losses/homozygous deletions, shallow deletions/shallow losses (heterozygous deletions), gains (a few additional copies, often broad) and amplifications (more copies, often focal), and can lead to the abnormal regulation of the genes driving or suppressing cancer.5 Although some studies have reported CNV loci in HCC and some specific gene copy number alterations, including the gain or amplification of CCND1 (11q13),6 VEGFA (6p21),7 and MYC (8q24),8 as well as the loss or deletion of TP53 (17p13)9 and CDKN2A (9p21),10 research on genes driving CNVs in HCC is still lacking. CNVs in the 1q region are highly variable, and patients with 1q21 gain have a worse prognosis than those without this alteration.11,12 However, the CNV driver genes in 1q are still worthy of further study.
MYC, which is highly amplified in cancer, is a multifunctional transcription factor that regulates multiple target genes by recognizing E-box sequences (5′-CACGTG‐3′) in the genome and thus controls the hallmark pathological functions of tumors.13 MYC drives the formation of HCC in mice.14 However, because directly targeting MYC is difficult, identifying factors that interact with the protein is essential for inhibiting its function. Previous studies on the MYC protein structure revealed 2 functional regions that are required for its bioactivity,15 a basic helix-loop-helix leucine zipper (bHLH/LZ) at the C-terminus, which binds DNA functional regions through E-box-dependent pattern recognition, and an N-terminal transcriptional activation domain, which contains multiple MYC homology boxes (MBs). Among them, MBII region of MYC is essential for maintaining its functional activity through cofactors.16
As a cofactor of MYC, actin-like 6A (ACTL6A) can activate the carcinogenicity of MYC.17 ACTL6A is a subunit of the SWI/SNF family, which plays roles in chromatin remodeling, transcriptional regulation, and nuclear transformation18,19 and plays a vital role in cancer. Previous studies have shown that ACTL6A can inhibit WWC1 by binding TP63 and further activate the YAP signaling pathway20 or directly bind and regulate YAP protein expression to promote tumor occurrence.21 However, the mechanisms by which ACTL6A regulates MYC in HCC and mediates changes in MYC activity have not been fully elucidated.
Here, we identified a novel HCC driver gene, vacuolar protein sorting 72 homolog (VPS72), with high copy number gain or amplification on chromosome 1q21.3. We found that CNV-driven VPS72 is highly expressed in HCC. Moreover, VPS72 was shown to act as a cofactor by binding to the MBII and HLH regions of MYC to promote the interaction between ACTL6A and MYC to further enhance the transcriptional activity of MYC. Furthermore, ACTL6A stabilizes the VPS72 protein by inhibiting the ubiquitination-mediated proteasome degradation induced by tripartite motif containing 21 (TRIM21). Collectively, the results of our study highlight the role and mechanism of VPS72 gain on chromosome 1q21.3 in HCC, thus identifying a potential target for this malignancy.
PATIENTS AND METHODS
Patient samples
We used 3 primary HCC cohorts from Tongji Hospital of Huazhong University of Science and Technology (Wuhan, China), namely, cohort 1, cohort 2, and cohort 3, which included a total of 214 HCC patients. All samples were stored at −80 °C. Cohort 1 contained 51 pairs of HCC tissues (pairs refer to tumor tissue and adjacent tissue from the same patient) for the western blot detection of the VPS72 protein, and 32 of the samples were used for CNV testing. Cohort 2 included 123 pairs of HCC tissue samples from patients who underwent primary HCC resection at the Hepatic Surgery Center of Tongji Hospital (Wuhan, China) between February 2012 and March 2016. The patients were pathologically diagnosed with HCC and followed up for prognostic assessment. Samples from this cohort were used to construct microarrays for immunohistochemistry experiments. Cohort 3 contained 40 pairs of HCC samples for the western blot analysis of the VPS72 and ACTL6A proteins. All samples were pathologically confirmed to be HCC. All patients provided signed informed consent forms. The study was conducted in accordance with both the Declarations of Helsinki and Istanbul, and was approved by the Ethics Committee of Tongji Hospital (Wuhan, China).
Animal models
All animal care and studies were performed according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Ethics Committee of Tongji Hospital (Wuhan, China). Four-week-old male BALB/C nude mice (Beijing HFK Bioscience Co., Ltd.) were used in this study. In the s.c. tumor formation assay, 1×106 logarithmic growth-phase HCC cells in 100 μL of serum-free high-glucose DMEM (Gibco) were injected into the axillary regions of the mice. For intrahepatic tumor model, 1×106 cells in 30 μL of serum-free DMEM were injected into the left lobe of the mouse liver. Approximately 4 weeks later, the mice were sacrificed, and the tumor size was detected. For HCC induced by hydrodynamic tail vein injection, male C57BL/6J mice (Beijing HFK Bioscience Co., Ltd.) at 6–8 weeks were used. For each mouse, pT3-myr-AKT (20 μg)/pT3-myr-AKT-IRES-luciferase (20 μg)/pT3-ΔN90-β-catenin (20 μg)/pT3-VPS72 (20 μg)/pT3-vector (20 μg) and SB transposase plasmids (1/25 of the total plasmid mass) were dissolved in 2 mL saline. Then, 2 mL of the plasmid mixture was rapidly delivered (~7 s) through the tail vein to the liver in vivo.
Statistics
Statistical analysis was performed with R software (version 3.63). Normal data distribution was assessed by 2-tailed Student t test or 1-way ANOVA. The significance of clinicopathological features and VPS72 expression was assessed by Fisher exact test or the chi-square test, and the prognosis was assessed by the log-rank test and Cox regression analysis. For publicly available prognostic data, the Survminer R package was used to analyze the significance of the VPS72 expression value and the prognosis of HCC patients. A p-value of <0.05 was considered statistically significant.
Full details of the methods are described in the Supplemental Materials and Methods (http://links.lww.com/HEP/B32).
RESULTS
Identification of the CNV-driven tumor-associated gene VPS72 in HCC
To screen activated or inhibited genes driven by CNVs in HCC, we analyzed whole-genome CNV data of 368 HCC patient samples from The Cancer Genome Atlas (TCGA). Significant gain peaks were concentrated on chromosomes 1q and 8q, and significant deletion peaks were concentrated on chromosomes 8p and 17p (Figure S1A). We focused on the peaks with the most gains, where the top 5 gain loci were 1q21.3, 1q24.2, 1q24.3, 1q25.1, and 1q25.3. Further analysis of the differentially expressed genes between HCC and nontumor tissues in Gene Expression Omnibus data sets showed that among the 50 genes most significantly upregulated in the 5 data sets (Figure S1B), 7 genes (VPS72, PSMD4, RFX5, PSMB4, UBAP2L, CKS1B, and PIGC) in the top 5 gain peaks were identified as potential CNV-driven genes (Figure S1C). Compared with the diploid tumor samples of each gene, 37% (136/366) of the samples showed high expression of VPS72 (z-score >2) (Table S1, http://links.lww.com/HEP/B32), and the proportions of other genes were 33% for PIGC, 29% for UBAP2L, 28% for PSMB4, 21% for PSMD4, 11% for RFX, and 0% for CKS1B (Figure S1D). This result suggests that VPS72 is expressed at higher levels in 37% of samples than in diploid patients, and 96% of the samples with altered VPS72 mRNA expression exhibited VPS72 amplification or gain (Figure S2A, Table S1, http://links.lww.com/HEP/B32). Moreover, the patients with these mRNA alterations had worse OS and disease-free survival (Figure S2B). Finally, VPS72 was selected as the gene for further study.
VPS72, located on 1q21.3, was amplified in 10.9% of the samples and gained in 61.4% of the TCGA samples (Figure 1A). In Gene Expression Omnibus data sets, there were more VPS72 copies in HCC tissues than in normal or cirrhotic tissues (Figure 1B). In Tongji cohort 1, the copy numbers of VPS72 were higher in HCC tissues, which was consistent with the public database (Figure 1C). In addition, CNV analysis of 33 tumor types in TCGA showed that the VPS72 gene was amplified in a variety of tumors. Surprisingly, VPS72 had the highest proportion of amplification in HCC (Figure 1D). In addition, VPS72 mutations in HCC were rare (Figure S2C), thereby excluding the influence of VPS72 mutants in HCC. Furthermore, we verified whether CNV of the VPS72 gene affected the mRNA expression of VPS72. In the Gene Expression Omnibus data set, VPS72 mRNA was strongly correlated with CNVs (Figure 1E), consistent with the results found with the Cancer Cell Line Encyclopedia cell lines (Figure 1F). In TCGA tissue samples, VPS72 expression gradually increased together with the CNV types in the VPS72 gene (Figure 1G).
Figure 1.

Identification of the CNV-driven tumor-associated gene VPS72 in HCC. (A) Schematic diagram of the types of CNVs found in the VPS72 gene in HCC [data from TCGA-LIHC. Amplification: a high-level amplification (more copies, often focal, CNV value from GISTIC 2.0 equals 2); gain: a low-level gain (a few additional copies, often broad, CNV value from GISTIC 2.0 equals 1); diploid: no change (CNV value from GISTIC 2.0 equals 0); shallow deletion: a shallow loss, literally a heterozygous deletion (CNV value from GISTIC 2.0 equals -1)]. (B) Comparison of the CNV unit values of the VPS72 gene in HCC and paracancerous tissues from public databases [data from Gene Expression Omnibus (GEO) data sets]. (C) Relative CNV values of the VPS72 gene in 32 paired cancerous and adjacent cancer tissues from Tongji cohort 1. (D) The amplification frequencies of the VSP72 gene in pan-cancers from the TCGA. (E) Pearson correlation analyses of the mRNA and CNV levels of the VPS72 gene in HCC samples from a public database (data from GEO data sets). (F) Pearson’s correlation analyses of the mRNA and CNV levels of the VPS72 gene in pan-cancer cell lines from a public database (data from Cancer Cell Line Encyclopedia). (G) VPS72 mRNA expression in HCC samples with different types of CNVs from the TCGA. (H) Differential expression of the VPS72 protein in 51 paired HCC and adjacent tissue samples in cohort 1 as determined by western blot. (I) Statistical analysis of the intensity values of VPS72 normalized to that of GAPDH. (J) Pearson correlation analyses of the VPS72 protein and CNV values in cohort 1. (K) Representative immunohistochemistry images of the VPS72 expression in cancerous and adjacent tissues in cohort 2 and a statistical evaluation of the staining intensity. (L) Kaplan-Meier plots of the overall survival (OS) and disease-free survival (DFS) rates of groups with differential VPS72 expression in cohort 2. (M) Meta-analysis of the OS in multiple HCC data sets as determined by the Cox proportional hazards model. Data are represented as means±SEM in the bar graphs. *p<0.05, **p<0.01, ***p<0.001. Abbreviations: AKT, AKT Serine/Threonine Kinase 1; CCLE, Cancer Cell Line Encyclopedia; HA, hemagglutinin tag; LIHC, Liver Hepatocellular Carcinoma; ns, not significant; OD, optical density; TCGA, The Cancer Genome Atlas; VPS72, vacuolar protein sorting 72 homologue.
Next, we consistently found high VPS72 expression in another 10 HCC Gene Expression Omnibus data sets (Figure S2D). In addition, VPS72 was significantly overexpressed in diethylnitrosamine-driven and oncogene-induced (AKT/β-catenin) mouse HCC models (Figure S2E). We further studied the role and function of the VPS72 protein in HCC. Western blotting from cohort 1 and immunohistochemistry staining from cohort 2 showed that VPS72 was highly expressed in HCC (Figure 1H, I, K). In cohort 1, protein expression was correlated with copy number alterations (Figure 1J). These results indicated that the high expression of the VPS72 protein was driven by the high copy number to some extent. Assessment of the clinical information revealed that VPS72 expression was significantly correlated with the tumor size, TNM stage, and recurrence (Table S2 http://links.lww.com/HEP/B32). Further analysis showed that the group with high VPS72 expression had lower OS and disease-free survival (Figure 1L), and this result was verified in the TCGA cohort (Figure S3). At the same time, samples from public data sets and our cohort were combined to assess the effect of VPS72 expression on the OS of HCC patients with a Cox model. Analysis of the other cohort and an overall meta-analysis showed that patients with high VPS72 expression had worse outcomes (Figure 1M).
VPS72 promotes HCC tumorigenesis and progression in vitro and in vivo
Moreover, we detected the copy number and mRNA of VPS72 in normal and HCC cell lines. The mRNA and copy number of VPS72, which were higher in HCC cell lines (Figure S4A-B), were highly correlated (Figure 2A). 97H, Huh7, and HepG2 cells were selected for the following study for their different mRNA expression and copy number units. In vitro, CCK-8 and colony formation assays showed that VPS72 knockdown significantly inhibited the proliferation of Huh7 and 97H cells, whereas overexpression of VPS72 significantly enhanced the proliferation of HepG2 cells (Figure 2B-C, Figure S4C, D). Next, cell cycle analysis revealed that VPS72 knockdown increased the proportion of cells in the G0/G1 phase, whereas VPS72 overexpression had the opposite effect (Figure 2D, Figure S4E, F). Subsequently, to confirm the phenotype, the CRISPR/Cas9 system was used to knockout VPS72 (Figure S5A). VPS72 protein deletion significantly inhibited growth and DNA replication in HCC cells (97H and HLF) (Figure S5B–D). These results suggest that VPS72 promotes HCC proliferation.
Figure 2.

VPS72 promotes hepatocellular carcinoma progression in vitro and in vivo. (A) Correlation between VPS72 mRNA and copy number units in hepatocyte and HCC cells. (B) CCK-8 assays of Huh7, 97H, and HepG2 cells after VPS72 knockdown or overexpression. (C) Colony formation assays of Huh7, 97H, and HepG2 cells after VPS72 knockdown or overexpression. (D) Statistical analysis revealed the cell cycle changes after VPS72 knockdown or overexpression in Huh7, 97H and HepG2 cells. (E) s.c. tumor model showed that 97H cells were significantly restricted after VPS72 knockdown, while HepG2 cells grew faster after VPS72 overexpressed. (F) Representative tumor images and tumor volumes after VPS72 knockout in the orthotopic 97H cell model (upper panel); representative tumor images and tumor volumes after VPS72 overexpression in the orthotopic HepG2 cell model (bottom panel). (G) Schematic diagram of hydrodynamic tail vein injection (HTVi) of oncogenic plasmids. (H) Representative liver images and hematoxylin and eosin-staining images of AKT/β-catenin+vector and AKT/β-catenin+VPS72 group, as well as ratio of liver and body weight between 2 groups. Data are represented as means±SEM in the bar graphs. *p<0.05, **p<0.01, ***p<0.001. Abbreviations: AKT, AKT Serine/Threonine Kinase 1;CNV, genomic copy number variation; ns, not significant; VPS72, vacuolar protein sorting 72 homologue; WT, wild type.
Furthermore, we verified the oncogenicity of VPS72 in HCC cell lines in vivo. First, the s.c. tumor model revealed that VPS72 knockdown significantly reduced the sizes and weights of tumors (Figure 2E). Next, VPS72 knockdown/knockout 97H cells were intrahepatically injected into the livers of nude mice, and these mice showed a significant reduction in tumors compared with controls (Figure 2F, Figure S5E). The opposite results were observed for HepG2 cells overexpressing VPS72 in these 2 models (Figure 2E, F). In addition, the positive proportion of Ki67 in s.c. tumor tissues was lower in cells with VPS72 knockdown and higher in HepG2 cells overexpressing VPS72 (Figure S5F).
To simulate the role of VPS72 amplification in HCC formation and development, we constructed an oncogene-induced HCC model in C57BL/6 mice through hydrodynamic tail vein injection of activated AKT (myr-AKT) and β-catenin (ΔN90-β-catenin) plasmids22,23 (Figure 2G). After confirming VPS72 overexpression in murine Hepa1-6 cells (Figure S5G), we transfected VPS72 with myr-AKT/ΔN90-β-catenin into mouse liver. Surprisingly, at the 9th week, mice with VPS72 overexpression showed a significant tumor burden in their livers, while in the control group, they showed liver damage and adipose degeneration (Figure 2H, Figure S5H). These results suggest that VPS72 promotes the formation and progression of HCC in vitro and in vivo.
VPS72 interacts with MYC and promotes its transcriptional activity
To study the mechanism by which VPS72 promotes HCC progression, we performed co-immunoprecipitation to enrich the proteins interacting with the VPS72 protein. Moreover, gene set enrichment analysis of VPS72 in TCGA-HCC data set showed that high expression of VPS72 was associated with a variety of nuclear processes (Figure S6A), including E2F1 targets and MYC targets. High abundance expression of VPS72 was determined by silver staining (Figure 3A), and 209 VPS72-interacting proteins were recognized by mass spectrometry (Table S3, http://links.lww.com/HEP/B32). A specific peptide of the MYC protein was identified (Figure 3B), which was consistent with the gene set enrichment analysis results in TCGA. After exogenous and endogenous co-immunoprecipitation validation, we verified the binding of VPS72 to MYC protein (Figure 3C, Figure S6B). Moreover, confocal immunofluorescence revealed that both VPS72 and MYC were colocalized in the nucleus (Figure 3D). In the functional domain (ΔMB0, ΔMBI, ΔMBII, ΔMBIIIA, ΔMBIIIB, ΔMBIV, ΔBR, ΔHLH, and ΔLZ) of MYC, the interaction between VPS72 and MYC was significantly weakened after deleting the MBII and HLH domains (Figure 3E), indicating that the binding of VPS72 to MYC depended on the MBII and HLH regions, which are required for the transcriptional activity of MYC and recognition of E-box sequences in the promoters of downstream target genes, respectively. In the study of MYC by Kalkat et al.24, the interactions between the ΔMBII and VPS72 peptides were significantly reduced (Figure S6C). In addition, MYC mainly interacted with the C-terminus of VPS72 (Figure 3F).
Figure 3.

VPS72 interacts with MYC and promotes its transcriptional activity. (A) Verification of the co-immunoprecipitation products by silver stain. (B) The peptide fragment of MYC as determined by mass spectrometry. (C) The interaction between endogenous MYC and VPS72 was confirmed in wild-type HCC cell lines (97H, Huh7, and HepG2). (D) Confocal immunofluorescence showed that VPS72 and MYC were colocalized in the cell nucleus. (E) Schematic diagram of the MYC functional region and the interactions between VPS72 and functionally mutated MYC. (F) Schematic diagram of the truncated regions (N-terminal and C-terminal) of VPS72 and the interaction of VPS72 with MYC. (G) E-box activity after VPS72 knockdown in HCC cells or VPS72 overexpression in HEK-293T cells. Data are represented as means±SEM in the bar graphs. *p<0.05, **p<0.01, ***p<0.001. Abbreviations: IP, immunoprecipitation; HA, hemagglutinin tag; MB, MYC homology box; ns, not significant; VPS72, vacuolar protein sorting 72 homologue.
Next, we found that the protein and mRNA levels of MYC were not altered after the knockdown/overexpression of VPS72 (Figure S6D, E). On the basis of the binding of VPS72 to the MBII and HLH domains, we speculated that VPS72 regulates the transcriptional activation pattern of MYC. The double-luciferase reporter gene assay revealed that the MYC E-box activity was significantly decreased in VPS72 knockdown HCC cells (Figure 3G), but increased in VPS72 overexpression cells (Figure 3G, Figure S6F). Furthermore, as a high-copy gene, we also examined the correlation between MYC and VPS72 gene copy number and protein expression, and the results showed that there was no correlation between MYC and VPS72 on CNV in either TCGA or Tongji cohort (Figure S6G, H). However, immunohistochemistry showed a significant correlation between the 2 proteins (Figure S6I). These results suggest that VPS72 promotes the transcriptional activation of MYC for downstream target genes.
VPS72 promotes HCC progression by regulating MYC target genes
To further study the effects of VPS72 on the downstream target genes of MYC, RNA-seq was performed on 97H cells with VPS72 knockdown and control cells (Figure S7A). Kyoto Encyclopedia of Genes and Genomes pathway enrichment analysis revealed that cancer-related pathways, papillomavirus infection, MAPK signaling pathway, and other pathways were significantly enriched after VPS72 knockdown, suggesting that VPS72 is involved in the cancer-related signaling pathway (Figure S7B). Subsequently, gene set enrichment analysis showed MYC target gene sets were significantly enriched in the control group (Figure 4A, B, Figure S7C). The same results were found in the TCGA data set (Figure S7E). In addition, RNA-seq of MYC knockdown was also performed. There was a significant positive correlation between the fold changes of differential genes for the siVPS72 and siMYC group (Figure 4C), and the normalized enrichment score in the gene set enrichment analysis results showed a strong positive correlation between siVPS72 and siMYC group (Figure S7D).
Figure 4.

VPS72 promotes HCC progression by regulating MYC target genes. (A) Bubble plot showing the results of gene set enrichment analysis (GSEA) between the siNC and siVPS72 groups in 50 hallmark gene sets. (B) GSEA plot of 2 MYC target gene sets. (C) Correlation analysis between differential genes after VPS72 knockdown and MYC knockdown. (D) Changes in the mRNA levels of MYC targets after VPS72 and MYC knockdown in 97H and Huh7 cells. (E) Changes in the protein levels of MYC targets after VPS72 knockdown in 97H and Huh7 cells. (F) Changes in the binding of MYC to the promoter of the MYC target gene after VPS72 knockdown in 97H and Huh7 cells. (G) CCK-8 assays of HepG2 and 97H cells with MYC knockdown on the basis of VPS72 overexpression. (H) Colony formation assays of HepG2 and 97H cells with MYC knockdown on the basis of VPS72 overexpression. (MYC siRNA transfection reversed the upregulation of MYC target mRNA (I) and protein (J) expression induced by VPS72 overexpression in HepG2 and 97H cells. Data are represented as means±SEM in the bar graphs. ns: not significant, *p<0.05, **p<0.01, ***p<0.001. Abbreviations: NES, normalized enrichment score; VPS72, vacuolar protein sorting 72 homologue.
Furthermore, we identified 5 differentially expressed MYC target genes (CCND1, CDK4, CAD, NCL, and EIF4A1) based on downregulated genes in both the siVPS72 and siMYC groups, as well as the pattern of MYC binding gene promoter loci (CACGTG) (Figure S7F). First, the expression of these 5 target genes was significantly correlated with VPS72 in the public database (Figure S7G). In Huh7 and 97H cells, the mRNA levels of these 5 target genes were significantly downregulated after VPS72 and MYC knockdown (Figure 4D, Figure S7H), and their protein levels were also verified after VPS72 knockdown (Figure 4E). Chromatin immunoprecipitation assays revealed that the enrichment of MYC in the 5 target gene promoters was decreased in 97H and Huh7 cells with VPS72 knockdown (Figure 4F). Moreover, MYC knockdown significantly attenuated the HCC proliferation induced by VPS72 overexpression (Figure 4G, H). In addition, the MYC target gene mRNA and protein upregulation induced by VPS72 overexpression was significantly attenuated by MYC knockdown (Figure 4I, J). These results show that VPS72 promotes the transcription of MYC target genes, suggesting that it promotes HCC progression through the MYC pathway.
VPS72 knockdown by adeno-associated virus attenuates the formation and progression of HCC
Then, to test the therapeutic effects of VPS72 on HCC via MYC target genes in vivo, we used adeno-associated virus 8 (AAV8) to package the short hairpin RNA against VPS72 (human) or Vps72 (murine) in 2 mouse models. In the intrahepatic tumor model, self-complementary AAV8 (scAAV8)-U6-shVPS72 and scAAV8-U6-shNC were injected through the tail vein 4 days after 97H cells were inoculated into the livers of nude mice (Figure 5A). Compared with scAAV8-U6-shNC group, mice injected with scAAV8-U6-shVPS72 had a lower tumor burden (Figure 5B). Also, protein expression levels of VPS72, MYC target genes (CCND1, CDK4, EIF4A1, NCL, and CAD), and Ki67 were significantly decreased in tumor tissues after infection with scAAV8-U6-shVPS72 (Figure 5C, D, Figure S8A). Furthermore, we developed an expressed-luciferase hydrodynamic tail vein injection model in immunocompetent C57BL/6 mice (Figure 5E). Recombination AAV (rAAV8)-shVps72/shNC was injected through the tail vein. The luciferase value and liver/body weight ratio of mice in rAAV8-shVps72 group were significantly decreased (Figure 5F). Consistent with the results in nude mice, the expression levels of VPS72, MYC target gene, and Ki67 were significantly decreased in the rAAV8-shVps72 group. These results suggested that targeting VPS72 inhibits the MYC pathway and has a potential therapeutic effect on HCC.
Figure 5.

VPS72 knockdown by adeno-associated virus attenuates the formation and progression of HCC. (A) Schematic diagram of AAV8 administration in nude mice with orthotopic 97H-luciferase cells. (B) The bioluminescence and liver with tumor images of orthotopically injected HCC 97H-luciferase cells in nude mice, following self-complementary AAV8 (scAAV8)-U6-shNC/shVPS72 administration for 36 days. (C) Representative images of H&E and immunohistochemistry (IHC) staining of MYC target genes (CAD, CDK4, CCND1, EIF4A1, and NCL) and Ki67 in the tumor sections between the scAAV8-shNC and shVPS72 groups in the intrahepatic tumor model. (D) Schematic diagram of oncogenic plasmids with luciferase injected into mice (upper panel); schematic diagram of AAV8 administration in C57BL/6 mice with hydrodynamic tail vein injection (HTVi) of oncogenic plasmids (bottom panel). (E) The bioluminescence and representative gross images of livers of HTVi-induced HCC model with luciferase in C57BL/6 mice, following rAAV8-shNC/shVps72 administration for 11 weeks. (F) Representative images of H&E and immunohistochemistry (IHC) staining of MYC target genes (Cad, Cdk4, Ccnd1, Eif4a1, and Ncl) and Ki67 in the tumor sections between the rAAV8-shNC and shVps72 groups in the HTVi model. Data are represented as means±SEM in the bar graphs. *p<0.05, **p<0.01, ***p<0.001. Abbreviations: H&E, hematoxylin and eosin; ns, not significant.
VPS72 interacts with ACTL6A and promotes the formation of the ACTL6A/MYC complex
Mass spectrometry revealed that the ACTL6A protein may bind to VPS72 (Figure S9A, Table S3, http://links.lww.com/HEP/B32). Because ACTL6A promotes HCC progression and is a potential cofactor of MYC,17,25 the relationship between VPS72 and ACTL6A deserves further study. The interaction between ACTL6A and VPS72 was confirmed at both endogenous and exogenous levels (Figure S9B, Figure 6A), and both were colocalized in the nucleus (Figure 6B). ACTL6A was shown to interact with the MBII domain of MYC (Figure 6C, Figure S9C), and VPS72 interacted with the second and third segments of ACTL6A (Figure S9D). However, unlike MYC, ACTL6A mainly interacted with the N-terminus of VPS72 (Figure 6D). As ACTL6A and MYC bind to different regions of VPS72, and both ACTL6A and VPS72 have a common functional domain (MBII) that binds to MYC, we hypothesized that VPS72 may act as an intermediate to facilitate the binding of ACTL6A and MYC. In the exogenous and endogenous co-immunoprecipitation assays, the interaction of ACTL6A with MYC was enhanced when VPS72 was overexpressed and attenuated when VPS72 was silenced (Figure S10A, B, Figure 6E). These results suggest that VPS72 enhances the interaction between ACTL6A and MYC.
Figure 6.
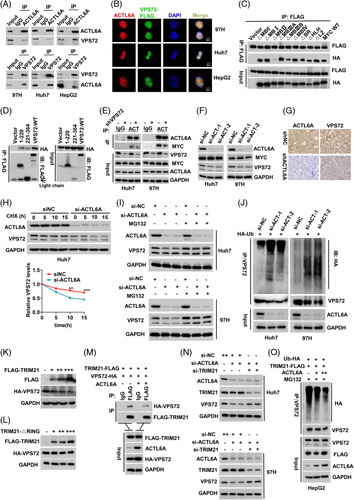
VPS72 interacts with ACTL6A, and the 2 regulates each other to enhance MYC function. (A) The interaction between endogenous ACTL6A and VPS72 was confirmed in wild-type HCC cell lines (97H, Huh7, and HepG2). (B) Confocal immunofluorescence analysis revealed the colocalization of VPS72 and ACTL6A in the cell nucleus. (C) The interactions between ACTL6A and different functional region mutants of MYC. (D) The interaction of the truncated VPS72 regions (N-terminal and C-terminal) with ACTL6A. (E) The interaction between ACTL6A and MYC was changed after VPS72 knockdown in HCC cell lines (97H and Huh7). (F) Expression of the VPS72 and MYC proteins after ACTL6A knockdown in HCC cells. (G) Immunohistochemistry staining of ACTL6A and VPS72 in s.c. tumor tissues comprised of 97H cells with ACTL6A knockdown. (H) Half-life of the VPS72 protein in Huh7 cells treated with cycloheximide (CHX) after ACTL6A knockdown. (I) Changes in the VPS72 protein expression in 97H and Huh7 cells treated with MG132 after ACTL6A knockdown. (J) After ACTL6A knockdown, cells were treated with 10-μM MG132 for 20 hours, and the ubiquitination level of VPS72 was detected by co-IP. (K) VPS72 protein changes after the gradient transfection of TRIM21 plasmids into HEK-293T cells. (L) The interaction between TRIM21 and VPS72 was weakened by the addition of ACTL6A plasmids. (M) VPS72 protein changes after the gradient transfection of TRIM21-ΔRing plasmids into HEK-293T cells. (N) TRIM21 knockdown abolished the downregulation of the VPS72 protein induced by ACTL6A knockdown in 97H and Huh7 cells. (O) Gradient transfection of the ACTL6A plasmids inhibited the ubiquitination of VPS72 protein induced by TRIM21 in HepG2 cells in a dose-dependent manner. Data are represented as means±SEM in the bar graphs. *p<0.05, **p<0.01, ***p<0.001. Abbreviations: ACTL6A, actin-like 6A; ns, not significant; VPS72, vacuolar protein sorting 72 homologue.
ACTL6A stabilizes VPS72 protein expression by inhibiting the interaction between TRIM21 and VPS72
To further understand the interaction between ACTL6A and VPS72, we silenced and overexpressed ACTL6A in HCC cells, and the VPS72 protein was downregulated and upregulated, respectively (Figure 6F, G, Figure S10C). In contrast, the expression of the MYC protein was not significantly altered. However, VPS72 mRNA expression was not significantly altered by the knockdown or overexpression of ACTL6A (Figure S10D), suggesting a posttranslational modification pathway. First, cells were treated with cycloheximide (CHX), showing that the half-life of the VPS72 protein was shorter after ACTL6A knockdown, whereas ACTL6A overexpression significantly extended the half-life of the VPS72 protein (Figure 6H, Figure S10E). In a previous study, ACTL6A was found to be deubiquitinated to stabilize the YAP protein.21 We investigated whether the interaction could remove the ubiquitinated VPS72 protein. The treatment of cells with MG132, a proteasome inhibitor, reversed the reduction in VPS72 protein expression induced by ACTL6A knockdown (Figure 6I), suggesting that ACTL6A affects the protein expression of VPS72 through the proteasome pathway.
Furthermore, co-immunoprecipitation analysis showed that the ubiquitination of VPS72 was increased after silencing ACTL6A but decreased after overexpressing ACTL6A (Figure 6J, Figure S10F). Also, we found that ACTL6A inhibited the polyubiquitination of the K48 linkage but not that of the K63 linkage (Figure S10G).
In addition, because the K48 linkage is ubiquitinated by E3 ligase, we screened 3 E3 ligases, TRIM21, TRIM27, and TRIM31, by mass spectrometry. First, we verified that all 3 E3 ligases interacted with VPS72 (Figure S10H). Further transfection showed that only VPS72 protein expression decreased dose dependently as TRIM21 expression increased (Figure 6K, Figure S10I). TRIM21 with a deleted ring domain lost the ability to inhibit VPS72 expression (Figure 6L). There was no interaction between ACTL6A and TRIM21 (Figure S10J). However, the interaction between TRIM21 and VPS72 was attenuated by ACTL6A overexpression (Figure 6M, Figure S10K). Furthermore, TRIM21 knockdown reversed the downregulation of VPS72 induced by ACTL6A knockdown in HCC cell lines (Figure 6N), and ACTL6A inhibited the TRIM21-mediated ubiquitination of VPS72 in a dose-dependent manner (Figure 6O). These results suggest that ACTL6A attenuated the ubiquitination of the VPS72 protein by inhibiting the interaction between TRIM21 and VPS72.
VPS72 directs ACTL6A to regulate MYC target gene transcription and promote HCC progression
Because of the mutual regulation between ACTL6A and VPS72, we further investigated the relationship among VPS72, ACTL6A, and MYC in the regulation of HCC progression. In vitro experiments with HCC cell lines, ACTL6A knockdown inhibited HCC proliferation (Figure S11A, B), whereas the overexpression of ACTL6A enhanced HCC proliferation (Figure S11A, B). Similar results were obtained with s.c. tumors and in situ liver tumor models (Figure S11C, D). Also, the 5 MYC target genes were downregulated at the mRNA level after ACTL6A knockdown (Figure S11E). Moreover, the knockdown of VPS72 in ACTL6A-overexpressing HCC cell lines limited MYC target gene mRNA and protein upregulation induced by ACTL6A overexpression (Figure 7A, B). Furthermore, the increased binding of MYC to the target gene promoters induced by ACTL6A overexpression was also abolished by VPS72 knockdown (Figure 7C). In addition, VPS72 knockdown significantly weakened the effect of ACTL6A overexpression on promoting HCC cell proliferation in vitro and in vivo (Figure 7D–F), and the overexpression of VPS72 reversed the inhibition of ACTL6A on HCC cell proliferation (Figure 7D, E). Therefore, these results suggest that VPS72 acts as a downstream mediator of ACTL6A to regulate the progression of HCC.
Figure 7.

VPS72 directs ACTL6A to regulate MYC target gene transcription and promote the progression of HCC. VPS72 knockdown reversed the mRNA (A) and protein (B) upregulation of MYC target genes induced by ACTL6A overexpression in HepG2 and 97H cells. (C) Changes in the sites at which MYC binds to the promoters of its target genes after VPS72 knockdown in HepG2 cells with ACTL6A overexpression. (D) CCK-8 assays of HepG2 and 97H cells with VPS72 knockdown on the basis of ACTL6A overexpression and of Huh7 cells with VPS72 overexpression on the basis of ACTL6A knockdown. (E) Colony formation assays of HepG2 and 97H cells with VPS72 knockdown on the basis of ACTL6A overexpression and of Huh7 cells with VPS72 overexpression on the basis of ACTL6A knockdown. (F) Tumor volumes and weights after VPS72 knockdown on the basis of ACTL6A overexpression in the s.c. tumor model comprised of HepG2 cells. Data are represented as means±SEM in the bar graphs. *p<0.05, **p<0.01, ***p<0.001. Abbreviations: ACTL6A, actin-like 6A; ns, not significant; VPS72, vacuolar protein sorting 72 homologue.
ACTL6A and VPS72 have similar expression characteristics in HCC
To determine the pathological features of ACTL6A and its relationship with VPS72 in HCC, Western blot and immunohistochemistry staining of HCC tissue samples were performed. In cohort 3, which included 40 pairs of tissues, ACTL6A was highly expressed in HCC and strongly correlated with VPS72 (Figure 8A–C), which was confirmed in the proteomic data of HCC cell lines from the Cancer Cell Line Encyclopedia (Figure 8D). Similarly, in cohort 2, the immunohistochemistry scores showed high expression of ACTL6A in HCC (Figure 8E). Moreover, HCC patients with high ACTL6A expression were more likely to have higher expression of VPS72 (Figure 8F). In addition, although the disease-free survival rates did not significantly differ between the ACTL6AHigh and ACTL6ALow patients, which was potentially related to the insufficient number of patients, the ACTL6AHigh patients had a worse OS (Figure 8G). Moreover, patients with high expression of both VPS72 and ACTL6A had the worst OS and disease-free survival (Figure 8H). These results indicate that both ACTL6A and VPS72 have similar expression characteristics in HCC. High expression of both ACTL6A and VPS72 suggests poor prognosis.
Figure 8.

ACTL6A and VPS72 have similar expression characteristics in HCC. (A) Differential expression of the VPS72 and ACTL6A proteins in 40 HCC and adjacent tissues from cohort 3 patients as determined by Western blot. (B) Statistical analysis of the intensity values of ACTL6A normalized to that of GAPDH in cohort 3. (C) Pearson correlation analysis of the ACTL6A and VPS72 protein levels in HCC cohort 3 as determined by Western blot. (D) Pearson correlation analysis of the ACTL6A and VPS72 protein levels in HCC cell lines from the Cancer Cell Line Encyclopedia proteome. (E) Representative immunohistochemistry images of the ACTL6A expression in cancerous and adjacent tissues from cohort 2 and a statistical evaluation of the staining intensity. (F) The immunohistochemistry scores demonstrated a high correlation between ACTL6A and VPS72 staining in HCC cohort 2 (chi-square test). (G) Kaplan-Meier curves of the overall survival (OS) and disease-free survival (DFS) rates between groups with differential ACTL6A expression in cohort 2. (H) Kaplan-Meier curves of the OS and DFS rates among groups with differential ACTL6A/VPS72 expression in cohort 2. (I) A schematic model of the mechanism by which VPS72 promotes HCC progression. Data are represented as means±SEM in the bar graphs. ns*p<0.05, **p<0.01, ***p<0.001. Abbreviations: ACTL6A, actin-like 6A; IHC, immunohistochemistry; ns, not significant; VPS72, vacuolar protein sorting 72 homologue.
Collectively, these results led to the hypothesis that genomic instability amplifies 1q21.3 and drives VPS72 expression in HCC. High expression of the VPS72 protein promotes the interaction of ACTL6A with MYC, and ACTL6A inhibits the TRIM21/VPS72 complex and stabilizes the VPS72 protein level. Therefore, the interaction between VPS72 and ACTL6A enhances the activity of MYC, promotes the transcription of MYC target genes and drives the progression of HCC (Figure 8I). Targeting VPS72 may be a potential treatment for HCC.
DISCUSSION
CNVs are very common in a variety of tumors due to genomic instability and can contribute to tumorigenesis and poor prognosis.26 Currently, targeted therapies for HCC are lacking, and the identification of CNV driver genes thus provides new opportunities for designing therapies targeting potential pathways.27 By analyzing the genome-wide CNVs in 368 HCC patients in TCGA, we identified the 1q genome-driven VPS72 as a key protein that regulates the ACTL6A/MYC complex in HCC.
Chromosome 1q gain or amplification is a crucial and highly frequent event occurring in HCC and other tumors.28–30 However, the extensive diffuse gain of 1q makes it difficult to identify functional targets for HCC. Although some typical 1q driver genes have been discovered successively, including the coding genes ARNT31 and BCL9,28 and the noncoding gene LINC00624,32 other driver genes on 1q may also be vital for the progression of HCC. Considering the cis-activated genes driven by CNVs, we described the coding gene VPS72 on 1q21.3 as a CNV-driven tumor-associated gene. In this study, CNV-driven VPS72 overexpression activated the malignant behavior of HCC and exerted a strong prognostic effect on HCC patients. A recent study revealed that VPS72 knockdown inhibited the growth and migration of HCC cells in vitro and inhibited the AKT signaling pathway.33 However, in our study, overexpression of VPS72 promoted the initiation and progression of an oncogene-induced (AKT/β-catenin) mouse HCC model, and targeting VPS72 inhibited the progression of this HCC model, suggesting that VPS72 is more likely to play a role in promoting HCC progression through pathways other than AKT.
Substantial amounts of evidence indicate that MYC plays a significant role in HCC, and targeting MYC may be a therapeutic target for HCC.34–36 Because targeting MYC directly is difficult, identifying its cofactors is a focus of research efforts.37 Evidence has indicated that the interactions of multiple cofactors with MYC enable it to work as an effective oncoprotein and that the function of MYC is based on its protein interactome.38 Targeting MYC cofactors or inhibiting their interactions with MYC is increasingly becoming a method for disrupting its function.38 Here, we reported that overexpressed VPS72 in HCC, as a novel MYC cofactor, interacted with the MBII and HLH domains of MYC. RNA-seq and gene set enrichment analysis enrichment analyses revealed that VPS72 can activate the binding of MYC to the promoters of target genes such as NCL, CAD, EIF4A1, CDK4. and CCND1. NCL, CAD, and EIF4A1 are highly correlated with the rapid development of HCC.39–41 CDK4 and CCND1 play key roles in regulating the G0/G1 phase of the cell cycle, which is consistent with the conclusion that VPS72 knockdown leads to G0/G1 blockade.42 In addition, MYC and VPS72 were independent of each other at the copy number level. However, their protein levels were significantly correlated in HCC. The correlation between these 2 proteins may be the result of the consistent expression profile of oncogenic proteins in tumors and the regulation of MYC expression by multiple pathways.
Furthermore, we identified an interaction between VPS72 and ACTL6A. Previous studies have demonstrated that ACTL6A regulates the Notch signaling pathway through SOX2 to promote HCC epithelial-mesenchymal transition.25 Interestingly, we revealed that VPS72 served as a platform to promote the interaction between MYC and ACTL6A, while ACTL6A stabilized the protein expression of VPS72 by attenuating the interaction between TRIM21 and VPS72. TRIM21 is a multisubstrate E3 ligase that interacts with different substrates in a variety of tumors. For example, TRIM21 promotes the degradation of fatty acid synthase ubiquitination and affects lipid metabolism in HCC.43 We showed that VSP72 served as a substrate for TRIM21 and that ACTL6A inhibited the TRIM21-mediated ubiquitination of VPS72. This result suggests that the regulation of the VPS72 protein in HCC is not only genomically driven by CNVs but also affected by high ACTL6A expression.
In summary, our study demonstrated that VPS72, a key tumor-associated gene gained on chromosome 1q, promotes the activity of the MYC transcription factor and influences the expression of multiple MYC target genes by interacting with ACTL6A in HCC. Considering that the 1q21 region accumulates the most gains in HCC, targeting VPS72 to thereby regulate MYC oncogenic activity may be a potential HCC treatment strategy.
Supplementary Material
Footnotes
Abbreviations: AAV8, adeno-associated virus serotype 8; ACTL6A, actin-like 6A; bHLH/LZ, basic helix-loop-helix leucine zipper; CCLE, Cancer Cell Line Encyclopedia; CHX, cycloheximide; CNV, genomic copy number variation; DEN, diethylnitrosamine; DFS, disease-free survival; GEO, gene expression omnibus; GSEA, gene set enrichment analysis; HA, hemagglutinin; H&E, hematoxylin and eosin; HTVi, hydrodynamic tail vein injection; IF, immunofluorescence; IHC, immunohistochemistry; MB, MYC homology box; NC, negative control; NES, normalized enrichment score; OS, overall survival; shRNA, short hairpin RNA; TCGA, The Cancer Genome Atlas; VPS72, vacuolar protein sorting 72 homologue; WT, wild type.
Furong Liu, Zhibin Liao, and Lu Qin contributed equally to this work.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com
Contributor Information
Furong Liu, Email: liufurong_tj@163.com.
Zhibin Liao, Email: zbliao@hust.edu.cn.
Lu Qin, Email: qinlumz@163.com.
Ze Zhang, Email: 2871634782@qq.com.
Qiaofeng Zhang, Email: 844065669@163.com.
Shenqi Han, Email: hanjj1103@outlook.com.
Weifeng Zeng, Email: laurentzeng@163.com.
Hongwei Zhang, Email: hwzhang636@126.com.
Yachong Liu, Email: liuyachongge@126.com.
Jia Song, Email: tjhsongjia@hust.edu.cn.
Wei Chen, Email: 438667390@qq.com.
He Zhu, Email: whtjzhuhe@163.com.
Huifang Liang, Email: lianghuifang1997@126.com.
Xiaoping Chen, Email: chenxpchenxp@163.com.
Bixiang Zhang, Email: bixiangzhang@163.com.
Zhanguo Zhang, Email: zhanguo_tjh@hust.edu.cn.
AUTHOR CONTRIBUTIONS
Furong Liu, Zhibin Liao, and Lu Qin contributed equally to this work. Ze Zhang, Bixiang Zhang, and Xiaoping Chen conceived this study. Ze Zhang, Bixiang Zhang, and Xiaoping Chen provided financial and administrative support. Furong Liu and Zhibin Liao conducted most experiments. Furong Liu and Lu Qin collected the public data and patients’ data of Tongji cohort. Shenqi Han, Hongwei Zhang, Yachong Liu, Jia Song, Huifang Liang, and Xiaoping Chen provided the advices of this study. Furong Liu, Lu Qin, and Ze Zhang wrote the manuscript.
FUNDING INFORMATION
This study was funded by the State Key Project on Infection Disease of China (No. 2018ZX10723204-003); the Chinese Ministry of Public Health for Key Clinical Project [No. (2010) 493-51]; National Natural Science Foundation of China (81502530, 81874189, 82172976, and 82103597).
CONFLICTS OF INTEREST
The authors have no conflicts to report.
DATA AVAILABILITY
The raw sequence data have been deposited in Genome Sequence Archive in National Genomics Data Center, Beijing Institute of Genomics (https://ngdc.cncb.ac.cn/gsa-human) with Project Accession No. PRJCA014662 and GSA Accession No. HRA003887.
REFERENCES
- 1.Villanueva A. Hepatocellular carcinoma. N Engl J Med. 2019;380:1450–62. [DOI] [PubMed] [Google Scholar]
- 2.Chen B, Garmire L, Calvisi DF, Chua MS, Kelley RK, Chen X. Harnessing big ‘omics’ data and AI for drug discovery in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol. 2020;17:238–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rebouissou S, Nault JC. Advances in molecular classification and precision oncology in hepatocellular carcinoma. J Hepatol. 2020;72:215–29. [DOI] [PubMed] [Google Scholar]
- 4.Xu LX, He MH, Dai ZH, Yu J, Wang JG, Li XC, et al. Genomic and transcriptional heterogeneity of multifocal hepatocellular carcinoma. Ann Oncol. 2019;30:990–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakamura Y. DNA variations in human and medical genetics: 25 years of my experience. J Hum Genet. 2009;54:1–08. [DOI] [PubMed] [Google Scholar]
- 6.Zhang YJ, Chen SY, Chen CJ, Santella RM. Polymorphisms in cyclin D1 gene and hepatocellular carcinoma. Mol Carcinog. 2002;33:125–29. [PubMed] [Google Scholar]
- 7.Llovet JM. Focal gains of VEGFA: candidate predictors of sorafenib response in hepatocellular carcinoma. Cancer Cell. 2014;25:560–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abou-Elella A, Gramlich T, Fritsch C, Gansler T. C-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod Pathol. 1996;9:95–98. [PubMed] [Google Scholar]
- 9.Liu F, Qin L, Liao Z, Song J, Yuan C, Liu Y, et al. Microenvironment characterization and multi-omics signatures related to prognosis and immunotherapy response of hepatocellular carcinoma. Exp Hematol Oncol. 2020;9:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liew CT, Li HM, Lo KW, Leow CK, Lau WY, Hin LY, et al. Frequent allelic loss on chromosome 9 in hepatocellular carcinoma. Int J Cancer. 1999;81:319–24. [DOI] [PubMed] [Google Scholar]
- 11.Chen L, Chan TH, Guan XY. Chromosome 1q21 amplification and oncogenes in hepatocellular carcinoma. Acta Pharmacol Sin. 2010;31:1165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen L, Yuan YF, Li Y, Chan TH, Zheng BJ, Huang J, et al. Clinical significance of CHD1L in hepatocellular carcinoma and therapeutic potentials of virus-mediated CHD1L depletion. Gut. 2011;60:534–43. [DOI] [PubMed] [Google Scholar]
- 13.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu Z, Xu M, Liu P, Zhang S, Shang R, Qiao Y, et al. The mTORC2-Akt1 cascade is crucial for c-Myc to promote hepatocarcinogenesis in mice and humans. Hepatology. 2019;70:1600–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4:a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tu WB, Shiah YJ, Lourenco C, Mullen PJ, Dingar D, Redel C, et al. MYC interacts with the g9a histone methyltransferase to drive transcriptional repression and tumorigenesis. Cancer Cell. 2018;34:579–95. [DOI] [PubMed] [Google Scholar]
- 17.Park J, Wood MA, Cole MD. BAF53 forms distinct nuclear complexes and functions as a critical c-Myc-interacting nuclear cofactor for oncogenic transformation. Mol Cell Biol. 2002;22:1307–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krasteva V, Buscarlet M, Diaz-Tellez A, Bernard MA, Crabtree GR, Lessard JA. The BAF53a subunit of SWI/SNF-like BAF complexes is essential for hemopoietic stem cell function. Blood. 2012;120:4720–32. [DOI] [PubMed] [Google Scholar]
- 19.Bao X, Tang J, Lopez-Pajares V, Tao S, Qu K, Crabtree GR, et al. ACTL6a enforces the epidermal progenitor state by suppressing SWI/SNF-dependent induction of KLF4. Cell Stem Cell. 2013;12:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saladi SV, Ross K, Karaayvaz M, Tata PR, Mou H, Rajagopal J, et al. ACTL6A is Co-Amplified with p63 in squamous cell carcinoma to drive YAP activation, regenerative proliferation, and poor prognosis. Cancer Cell. 2017;31:35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ji J, Xu R, Zhang X, Han M, Xu Y, Wei Y, et al. Actin like-6A promotes glioma progression through stabilization of transcriptional regulators YAP/TAZ. Cell Death Dis. 2018;9:517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184:912–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stauffer JK, Scarzello AJ, Andersen JB, De Kluyver RL, Back TC, Weiss JM, et al. Coactivation of AKT and beta-catenin in mice rapidly induces formation of lipogenic liver tumors. Cancer Res. 2011;71:2718–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ, et al. MYC protein interactome profiling reveals functionally distinct regions that cooperate to drive tumorigenesis. Mol Cell. 2018;72:836–48. [DOI] [PubMed] [Google Scholar]
- 25.Xiao S, Chang RM, Yang MY, Lei X, Liu X, Gao WB, et al. Actin-like 6A predicts poor prognosis of hepatocellular carcinoma and promotes metastasis and epithelial-mesenchymal transition. Hepatology. 2016;63:1256–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang L, Fang JY, Xu J. Gastric cancer and gene copy number variation: emerging cancer drivers for targeted therapy. Oncogene. 2016;35:1475–82. [DOI] [PubMed] [Google Scholar]
- 27.Guichard C, Amaddeo G, Imbeaud S, Ladeiro Y, Pelletier L, Maad IB, et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat Genet. 2012;44:694–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K, Lim HY, Shi S, Lee J, Deng S, Xie T, et al. Genomic landscape of copy number aberrations enables the identification of oncogenic drivers in hepatocellular carcinoma. Hepatology. 2013;58:706–17. [DOI] [PubMed] [Google Scholar]
- 29.Jia D, Wei L, Guo W, Zha R, Bao M, Chen Z, et al. Genome-wide copy number analyses identified novel cancer genes in hepatocellular carcinoma. Hepatology. 2011;54:1227–36. [DOI] [PubMed] [Google Scholar]
- 30.Cancer Genome Atlas Research Network. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim TM, Yim SH, Shin SH, Xu HD, Jung YC, Park CK, et al. Clinical implication of recurrent copy number alterations in hepatocellular carcinoma and putative oncogenes in recurrent gains on 1q. Int J Cancer. 2008;123:2808–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Z, Lu X, Liu Y, Zhao J, Ma S, Yin H, et al. Gain of LINC00624 enhances liver cancer progression by disrupting the histone deacetylase 6/Tripartite motif containing 28/Zinc finger protein 354C corepressor complex. Hepatology. 2021;73:1764–82. [DOI] [PubMed] [Google Scholar]
- 33.Chen T, Tu Y, Lv D, Lin K, Tang H, Huang W. Vacuolar protein sorting-associated protein 72 homolog (VPS72) binding to lysine acetyltransferase 5 (KAT5) promotes the proliferation, invasion and migration of hepatocellular carcinoma through regulating phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway. Bioengineered. 2022;13:9197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dang H, Takai A, Forgues M, Pomyen Y, Mou H, Xue W, et al. Oncogenic activation of the RNA binding protein NELFE and MYC signaling in hepatocellular carcinoma. Cancer Cell. 2017;32:101–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krishnan MS, Rajan KA, Park J, Arjunan V, Garcia MF, Bermudez A, et al. Genomic analysis of vascular invasion in HCC reveals molecular drivers and predictive biomarkers. Hepatology. 2021;73:2342–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Zhang S, Zhang Y, Jia J, Wang J, Liu X, et al. TAZ is indispensable for c-MYC-induced hepatocarcinogenesis. J Hepatol. 2021 10.1016/j.jhep.2021.08.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen H, Liu H, Qing G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct Target Ther. 2018;3:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lourenco C, Resetca D, Redel C, Lin P, MacDonald AS, Ciaccio R, et al. MYC protein interactors in gene transcription and cancer. Nat Rev Cancer. 2021;21:579–91. [DOI] [PubMed] [Google Scholar]
- 39.Guo L, Yi X, Chen L, Zhang T, Guo H, Chen Z, et al. Single cell DNA sequencing reveals punctuated and gradual clonal evolution in hepatocellular carcinoma. Gastroenterology. 2021 10.1053/j.gastro.2021.08.052 [DOI] [PubMed] [Google Scholar]
- 40.Burbano DLS, Tran D, Allister AB, Polenkowski M, Nashan B, Koch M, et al. C20orf204, a hepatocellular carcinoma-specific protein interacts with nucleolin and promotes cell proliferation. Oncogenesis. 2021;10:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen D, Zou J, Zhao Z, Tang X, Deng Z, Jia J, et al. TXNDC9 promotes hepatocellular carcinoma progression by positive regulation of MYC-mediated transcriptional network. Cell Death Dis. 2018;9:1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat Rev Cancer. 2011;11:558–72. [DOI] [PubMed] [Google Scholar]
- 43.Gu L, Zhu Y, Lin X, Tan X, Lu B, Li Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene. 2020;39:2437–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The raw sequence data have been deposited in Genome Sequence Archive in National Genomics Data Center, Beijing Institute of Genomics (https://ngdc.cncb.ac.cn/gsa-human) with Project Accession No. PRJCA014662 and GSA Accession No. HRA003887.