

Abstract
The positive effect of levodopa in the treatment of Parkinson’s disease, although it is limited in time and has severe side effects, has encouraged the scientific community to look for new drugs that can stop the neurodegenerative process or even regenerate the neuromelanin-containing dopaminergic nigrostriatal neurons. Successful preclinical studies with coenzyme Q10, mitoquinone, isradipine, nilotinib, TCH346, neurturin, zonisamide, deferiprone, prasinezumab, and cinpanemab prompted clinical trials. However, these failed and after more than 50 years levodopa continues to be the key drug in the treatment of the disease, despite its severe side effects after 4–6 years of chronic treatment. The lack of translated successful results obtained in preclinical investigations based on the use of neurotoxins that do not exist in the human body as new drugs for Parkinson’s disease treatment is a big problem. In our opinion, the cause of these failures lies in the experimental animal models involving neurotoxins that do not exist in the human body, such as 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine and 6-hydroxydopamine, that induce a very fast, massive and expansive neurodegenerative process, which contrasts with the extremely slow one of neuromelanin-containing dopaminergic neurons. The exceedingly slow progress of the neurodegenerative process of the nigrostriatal neurons in idiopathic Parkinson’s patients is due to (i) a degenerative model in which the neurotoxic effect of an endogenous neurotoxin affects a single neuron, (ii) a neurotoxic event that is not expansive and (iii) the fact that the neurotoxin that triggers the neurodegenerative process is produced inside the neuromelanin-containing dopaminergic neurons. The endogenous neurotoxin that fits this degenerative model involving one single neuron at a time is aminochrome, since it (i) is generated within neuromelanin-containing dopaminergic neurons, (ii) does not cause an expansive neurotoxic effect and (iii) triggers all the mechanisms involved in the neurodegenerative process of the nigrostriatal neurons in idiopathic Parkinson’s disease. In conclusion, based on the hypothesis that the neurodegenerative process of idiopathic Parkinson’s disease corresponds to a single-neuron neurodegeneration model, we must search for molecules that increase the expression of the neuroprotective enzymes DT-diaphorase and glutathione transferase M2-2. It has been observed that the activation of the Kelch-like ECH-associated protein 1/nuclear factor (erythroid-derived 2)-like 2 pathway is associated with the transcriptional activation of the DT-diaphorase and glutathione transferase genes.
Keywords: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; 6-hydroxydopamine; aminochrome; dopaminergic neurons; DT-diaphorase; exogenous neurotoxins; glutathione transferase M2-2; neuromelanin; Parkinson’s disease; preclinical models
Parkinson’s Disease
The appearance of motor symptoms in Parkinson’s disease (PD) is preceded by a series of indications that are not related to the loss of dopaminergic (DA) nigrostriatal neurons. These premotor symptoms appear years before the motor ones. Different stages of PD have been proposed in which the premotor symptoms such as depression, hyposmia, anxiety, sleep disorders, and constipation present years before the motor ones. Lewy bodies and Lewy neurites appear in brain regions such as the medulla oblongata/pontine tegmentum, the olfactory bulb/anterior, and the olfactory nucleus (Braak et al., 2003). These are mainly composed of accumulations of alpha-synuclein (SNCA) fibrils. SNCA is a monomeric protein that under certain circumstances aggregates to form fibrils or alternatively oligomers (Mehra et al., 2019). The formation of these Lewy bodies and Lewy neurites in these regions spreads to other areas, affecting, after years, the substantia nigra, forebrain, and midbrain, which induces motor symptoms such as tremors at rest, muscular rigidity, global slowness, and instability. This model for the progression of idiopathic Parkinson’s disease (iPD) from one region of the brain to another is mediated by the propagation of Lewy bodies and Lewy neurites (Hijaz and Volpicelli-Daley, 2020).
The Lack of Advances in New Pharmacological Therapy in Parkinson’s Disease
In 1957, the association between dopamine depletion resulting from the loss of neuromelanin-containing (NM) DA neurons and motor symptoms in PD was discovered when it was observed that levodopa (L-dopa) decreased the immobilizing effect of reserpine (Carlsson, 1957). More than five decades have passed since L-dopa started to be used in the treatment of PD and this drug continues to be the key treatment in this disease. The effects of this pharmacological treatment are spectacular, as the patients recover their mobility and are able to return to daily life (Birkmayer and Hornykiewicz, 1998). However, severe side effects appear after four to six years of chronic treatment, such as dyskinesias, that change the patient’s life quality (Lane, 2019).
During the five decades since L-dopa was introduced into the treatment of PD, a series of clinical studies have been carried out on the basis of successful evidence obtained from experimental animal models based on neurotoxins that do not exist in the human body, such as 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). The problem is that these successful preclinical investigations have failed in the advanced phases of clinical ones. For example, the neuroprotective effect of coenzyme Q10 could not be verified in a phase 3 clinical study that included 67 different medical centers that found no clinical benefits (Parkinson Study Group QE3 Investigators et al., 2014). Another carried out in 45 medical centers with creatine for five years failed to demonstrate that creatine monohydrate produced neuroprotective effects (Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET-PD) Investigators et al., 2015). The protective effect of mitoquinone noted in preclinical research could not be confirmed in a clinical trial with 128 PD patients (Snow et al., 2010). The antioxidant effect of urate detected in an experimental animal model could not be validated in a phase 3 trial, as no difference with the placebo control group was observed (Schwarzschild et al., 2021).
Isradipine, an L-type calcium channel antagonist that had successful effects in experimental animal models, failed to produce beneficial clinical effects in an evaluation conducted at 57 medical centers (Simuni et al., 2020). The neuroprotection of DA neurons noted with the c-Abl inhibitor nilotinib in preclinical studies demonstrated no clinical benefit in a clinical one with 76 PD patients (Simuni et al., 2021). The protective effect of the antiapoptotic drug TCH346 in preclinical studies did not show any differences with the placebo in clinical ones (Warren Olanow et al., 2006). The results of glial cell-derived neurotrophic factor in preclinical studies were spectacular because the regeneration of DA neurons could be detected, which promised not only to halt the progress of the disease but also to treat patients with dyskinesias. However, a glial cell-derived neurotrophic factor analog, adeno-associated type-2 vector neurturin, indicated no clinical benefit in a 15- to 24-month multicenter investigation (Warren Olanow et al., 2015). The neuroprotective effect of the chelator agent deferiprone in preclinical studies could not be repeated in a clinical one with 367 PD patients who had never received L-dopa treatment. On the contrary, deferiprone worsens the symptoms of parkinsonism measured with the Movement Disorder Society-Sponsored Revision of the Unified PD Rating Scale (Devos et al., 2022). A clinical trial with zonisamide, an antiepileptic drug, did not decrease tremors in symptom-dominant patients more than the tremors that had been observed in preclinical ones (Pillai et al., 2022).
The possible role of SNCA aggregation in PD has motivated clinical studies with prasinezumab monoclonal antibodies. One phase 2 trial involved 316 participants who received these monoclonal antibodies for 52 weeks and were evaluated with the Movement Disorder Society-Sponsored Revision of the Unified PD Rating Scale and the measurement of dopamine transporter (DAT) levels measured with 123I-ioflupane single-photon emission computed tomography (SPECT). However, this was discontinued due to the lack of effect compared to the placebo group (Pagano et al., 2022). Another phase 2 trial with monoclonal antibodies derived from human SNCA (cinpanemab) showed no differences with the placebo group and the researchers decided to end these clinical studies (Lang et al., 2022).
Preclinical Models Based on Neurotoxins That Do Not Exist in the Human Body
The experimental animal models used in studying the mechanisms of PD and testing new molecules with therapeutic potential for the disease are based on neurotoxins that do not exist in the human body, including MPTP and 6-hydroxydopamine (Teixera et al., 2020; Capucciati et al., 2021; Qiu et al., 2022). These neurotoxins share an experimental animal model that, first, is of high specificity with respect to their neurotoxic effects on DA neurons. MPTP is easily transported through the blood-brain barrier and in the brain, it is taken up by astrocytes, where MPTP is converted to 1-methyl-4-phenylpyridinium (MPP+), which has a high affinity for the DAT, giving it its high specificity. Due to its high level of analogy with the dopamine structure, 6-hydroxydopamine itself has a high affinity for the DAT. In addition, 6-hydroxydopamine is highly unstable in the presence of dioxygen, which needs an injection of 6-hydroxydopamine in the presence of ascorbic acid to prevent its autoxidation before being injected into the nigrostriatal neurons. Second, these models also induce a massive, expansive, and extremely rapid neurodegeneration as all their molecules try to penetrate all the DA neurons to which they have access. The effect of these neurotoxins is the opposite of what happens in the disease, where the progress is very slow (Kostrzewa, 2022; Figure 1A). The difference between these two neurotoxins, in addition to their different structures, is that only MPTP has been used in humans, which occurred when several drug users with a knowledge of chemistry decided to synthesize their illicit drugs that were unfortunately contaminated with MPTP. The drug users who consumed these illicit drugs contaminated with MPTP developed severe parkinsonism in only three days (Williams, 1984; Figure 1C; Table 1).
Figure 1.
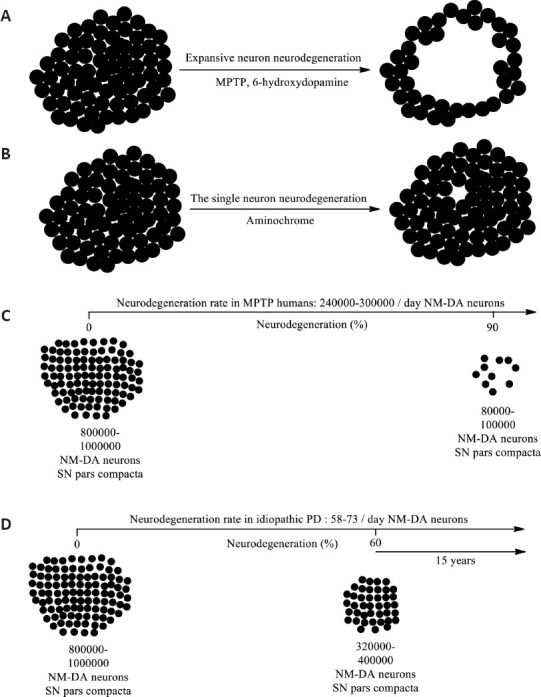
The single neuron degeneration model.
(A) MPTP or 6-hydroxydopamine induces a degenerative process of an expansive nature where the degenerative process does not stop at the time of the onset of the neurotoxic action of these neurotoxins but rather increases very rapidly day by day. (B) In the degenerative model of single neuron degeneration, an endogenous neurotoxin generated within the same affected neuron triggers a degenerative process that ends with the death of a single neuron, but unlike the expansive model, this degenerative process does not affect neighboring neurons. and it stops. In this single-neuron degenerative model, the death of a single neuron accumulates over time over the years. (C) MPTP-induced parkinsonism in humans in just 3 days 85–90% of NM DA neurons are lost. (D) It has been estimated that in the human substantia nigra pars compacta there are between 800,000 and 1,000,000 dopaminergic neurons, which implies that when motor symptoms appear, between 320,000 and 100,000 surviving neurons remain as a result of the loss of 60% of these neurons. If we consider that a patient survives 15 years after the onset of motor symptoms, this implies that the rate of loss of NM-DA neurons is 58–73 per day. Created with ChemDraw. DA: Dopaminergic; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NM: neuromelanin-containing; PD: Parkinson’s disease; SN: substantia nigra.
Table 1.
Limitations and advantages of preclinical models for Parkinson’s disease
| Preclinical models | Limitations | References |
|---|---|---|
| 6-Hydroxydopamine | Induces rapid, massive, and expansive neurotoxicity. It has high affinity with dopamine and norepinephrine transporters, which needs its use in the presence of a norepinephrine transporter inhibitor to ensure its specificity to the dopaminergic system. | Kostrzewa, 2022 |
| MPTP | It induces a massive and expansive neurotoxicity. In humans, it induces severe Parkinson’s in just three days. High specificity to the dopaminergic system but needs metabolism in astrocytes to convert MPTP to MPP+ which has a high affinity for the DAT. | Williams, 1984; Goloborshcheva et al., 2022 |
| Rotenone | It induces a massive, rapid and expansive neurotoxicity. Its effect is not specific and can affect other neuronal systems and glia cells. | Khalaf et al., 2023 |
| Paraquat | It induces a massive, rapid and expansive neurotoxicity. In humans, exposure to paraquat induces parkinsonism in young workers using this product. | Niso-Santano et al., 2010 |
| Tyrosinase transduction | It induces the formation of melanin which is different from neuromelanin. It produces an expansive neurotoxic effect that affects all types of neurons and glial cells. | Carballo-Carbajal et al., 2019 |
| 3,4-Hihydroxyphenylacetaldehyde (DOPAL) | The accumulation of DOPAL depends on a low expression of aldehyde dehydrogenase-1, which has been observed in postmortem material. The problem is that the postmortem tissue represents the neurons and glial cells that survived a degenerative process for years, since the tissue of the neurons that degenerated was eliminated by the microglia that phagocyted them years before; | Grünblatt et al., 2018; Goldstein, 2021 |
| Free neuromelanin | Free neuromelanin induces microglia activation and neurodegeneration. This model is expansive since the injection of free neuromelanin will affect all the neurons in the place where it is injected. | Zhang et al., 2011 |
| Aminochrome | Intracerebral injection in rats induces neuronal dysfunction and progressive degeneration. But this injection induces an expansive effect that affects all the neurons that are exposed to this endogenous neurotoxin. | Herrera et al., 2016 |
| One single-neuron degeneration | This model of neurodegeneration is not possible to implement in animals since it is impossible to inject a single dopaminergic neuron that contains neuromelanin. However, it is possible to study potential therapeutic agents to stop or slow down the degenerative process that aminochrome induces in a single neuron. One can search for compounds that activate the KEAP1/NRF2 pathway that is associated with the transcriptional activation of the DT-diaphorase and glutathione transferase genes. DT-diaphorase and glutathione transferase M2-2 prevent the neurotoxic effects of aminochrome. | Segura-Aguilar and Mannervik, 2023 |
DAT: Dopamine transporter; KEAP1/NRF2: Kelch-like ECH-associated protein 1/nuclear factor erythroid 2-related factor 2; MPP+: 1-methyl-4-phenylpyridinium ion; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.
Single-Neuron Neurodegeneration as a Degenerative Model for Idiopathic Parkinson’s Disease
To search for new pharmacological therapies in iPD, we must deeply analyze the characteristics of the disease. The neurodegenerative process in iPD is characterized by being an extremely slow one that takes years before motor symptoms appear. It has recently been estimated that the human substantia nigra pars compacta includes between 800,000 and 1,000,000 DA neurons, considering both hemispheres of the human brain (Ni and Ernst, 2022). This implies that an iPD patient has between 320,000 and 400,000 surviving DA neurons when motor symptoms appear after losing 60% of these neurons (Figure 1B). Generally, patients with PD live 15 to 20 years after the onset of motor symptoms, although there are individuals who live more than 20 years after diagnosis (Dorsey et al., 2018). Therefore, if we assume that a patient with iPD lives 15 years after the onset of motor symptoms, this implies that 58 to 73 NM DA neurons are lost every day (Segura-Aguilar and Mannervik, 2023). This is an estimate based on the years of survival of a patient with iPD from the onset of motor symptoms, at which point 60% of NM DA neurons have already been lost. However, if a greater number of neurons were lost, the years of survival would be fewer. Nor can it be ruled out that over a period of time, the neurodegeneration of these neurons is slower or faster. The important point is to keep in mind that the neurodegenerative process of these neurons is very slow and the UDPRS scale may not be sensitive enough to detect small changes. The single-neuron neurodegenerative process explains the extremely slow one in iPD since there is no expansive effect where the affected neurons secrete the neurotoxin to other neighboring ones. In one year of degeneration according to this single-neuron model, the loss of between 21,170 and 26,645 neurons would accumulate. The question is whether the methodology used in clinical studies to measure the effect of a new drug is sensitive enough to detect these small and slow changes in the neurodegenerative process of the disease. In phase 2 clinical trials with monoclonal antibodies against SNCA, no difference was observed with the placebo group after one year of SPECT measurements (Lang et al., 2022; Pagano et al., 2022).
The extremely rapid and massive neurodegenerative process that MPTP elicits in DA human nigrostriatal neurons (Williams, 1984) suggests that the neurodegenerative process in patients with iPD may not be triggered by neurotoxins that do not exist in the human body, but rather by an endogenous neurotoxin that is formed within NM DA neurons to cause the death of a single neuron. This implies that each degenerative event will be focused on and induce the death of a single NM DA neuron (Figure 1B). The single-neuron neurodegenerative process explains the extremely slow one in iPD, since there is no expansive effect where the affected neurons secrete the neurotoxin to other neighboring neurons (Figure 1D).
It is impossible to determine the therapeutic effect of a potential new drug in the treatment of iPD with an expansive, massive, and extremely rapid experimental animal model because it is entirely dissociated from the rate of the neurodegenerative process in the disease. In unsuccessful clinical studies with antioxidants, the therapeutic action of a daily dose neutralizes the oxidative stress produced in the 58–73 neurons that degenerate daily. However, this therapeutic action does not prevent other new neurons from degenerating the next day because this treatment does not inhibit the generator of oxidative stress. The number of NM DA neurons lost in the single-neuron neurodegenerative process in iPD increases at a very slow rate (58 to 73 neurons per day), and it takes many years to register the therapeutic effects that are observed in the aforementioned type of experimental animal models (Figure 1).
Possible Endogenous Neurotoxins for the Single-Neuron Neurodegenerative Model for Parkinson’s Disease
The extremely rapid, massive, and expansive effect of MPTP in humans strongly suggests that the neurotoxin that triggers all the mechanisms involved in the iPD process, such as mitochondrial impairment, oxidative and endoplasmic reticulum stress, and neuroinflammation, as well as the formation of neurotoxic SNCA oligomers and the dysfunction of both lysosomal and proteasomal protein degradation systems, must be of endogenous origin (Segura-Aguilar, 2021, 2022a). Following the logic of the single-neuron degenerative model, this endogenous neurotoxin must (i) be generated within NM DA nigrostriatal neurons, (ii) not have an expansive character that affects neighboring neurons and (iii) induce the mechanisms that are considered to be involved in the neurodegenerative process of iPD.
Three endogenous neurotoxins are known to exist that are formed in DA neurons containing neuromelanin:
SNCA is a protein that can aggregate to form fibrils that accumulate in Lewy bodies that have been observed in postmortem tissues that have survived the years-long neurodegenerative process in patients with PD. It has been postulated that Lewy bodies play a key role in PD, in which they are secreted into neighboring neurons and transferred from one region of the brain to another (Braak et al., 2003). This expansive character of Lewy bodies rules out the idea that this could be the neurotoxin that induces a single-neuron neurodegenerative process in iPD. In addition, it has been proposed that Lewy bodies have a protective role through accumulating SNCA fibrils that prevent the formation of the neurotoxic oligomers of this protein (Wakabayashi et al., 2013). SNCA can also aggregate into neurotoxic oligomers when there is a mutation that is associated with familial PD (Mehra et al., 2019). In the case of iPD, SNCA exists as a monomer and requires that the endogenous neurotoxin aminochrome (AM) causes the aggregation of SNCA to neurotoxic oligomers (Muñoz et al., 2015). This means that SNCA cannot be the endogenous neurotoxin that triggers the single-neuron neurodegenerative process, since it needs the action of another endogenous neurotoxin to form these neurotoxic oligomers. In addition, the lack of any effect of monoclonal antibodies against human SNCA in phase 2 clinical studies undertaken for one year questions the possible role of SNCA as an endogenous neurotoxin that triggers the loss of NM DA nigrostriatal neurons in iPD (Lang et al., 2022; Pagano et al., 2022).
Second, 3,4-dihydroxyphenylacetaldehyde (DOPAL) is a product of dopamine oxidative deamination catalyzed by monoamine oxidase (Goldstein, 2021). A study of enzyme expression in postmortem tissues of patients with PD found that the expression levels of the enzyme aldehyde dehydrogenase-1 were lower than those of tissues from control brains (Grünblatt et al., 2018), which implied that DOPAL should accumulate in DA neurons, generating neurotoxic effects. The problem is that the postmortem tissue represents the neurons and glial cells that have survived a neurodegenerative process for years, since the tissue of the neurons that degenerated was eliminated by the microglia that phagocyted them years before.
Free neuromelanin: the synthesis of neuromelanin needs the formation of neurotoxic ortho-quinones such as AM that are prevented by the enzymes DT-diaphorase (DT) and glutathione transferase M2-2 (GSTM2). Neuromelanin accumulates in isolated structures called NM organelles that prevent the oxidation of neuromelanin polymer catechol structures, which can be neurotoxic. The injection of neuromelanin into rodent substantia nigra triggers the activation of microglia, resulting in the loss of DA neurons. The rupture of NM organelles during the degradation of NM DA neurons would imply the exposure of free neuromelanin to all surrounding neurons that would cause an expansive neurotoxic effect (Zhang et al., 2011).
AM is generated during the synthesis of neuromelanin, which is a dark pigment that accumulates over the years in DA nigrostriatal neurons (Segura-Aguilar, 2021). At a physiological pH in the cytosol, dopamine can be oxidized to produce dopamine ortho-quinone, which is stable at a pH of less than 2.0. This transient metabolite of neuromelanin synthesis rapidly cyclizes at a rate of 0.1/second, finally forming AM. AM is also a transient metabolite that is formed during the synthesis of neuromelanin, but it is the most stable within the synthesis of this pigment since the rate of conversion of AM to 5,6-indolequinone is 0.06/minute and it is stable around 40 minutes in vitro (Zecca et al., 2002; Bisaglia et al., 2007; Figure 2). AM is neurotoxic in that it causes mitochondrial impairment by inhibiting complex I, leading to the dysfunction of both lysosomal and proteasomal protein degradation systems, as well as endoplasmic reticulum and oxidative stress, neuroinflammation, and the formation of neurotoxic oligomers of SNCA (Arriagada et al., 2004; Zafar et al., 2006; Paris et al., 2011; Aguirre et al., 2012; Muñoz et al., 2012a; Huenchuguala et al., 2014, 2017; Xiong et al., 2014; Santos et al., 2017). A unilateral injection of AM into the striatum of rats induces neuronal dysfunction with the progressive degeneration of tyrosine hydrolase-positive neurons in the substantia nigra, but the terminals of these neurons in the striatum remain intact. It also leads to neuronal dysfunction with significant decreases in denosine triphosphate production, the number of monoaminergic vesicles in the terminals, and dopamine release, as well as a significant increase in gamma-aminobutyric acid, a dramatic change in the morphology of tyrosine hydroxylase positive neurons called cell shrinkage, and contralateral behavior (Herrera et al., 2016).
Figure 2.
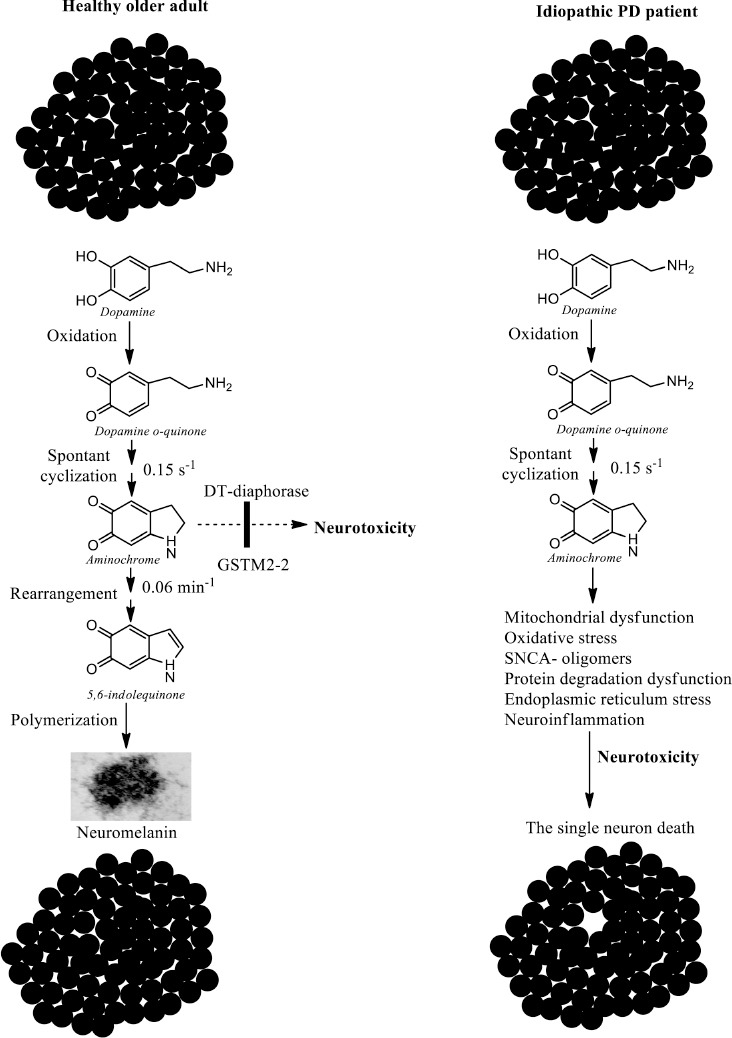
The single neuron death induced by aminochrome.
(A) Neuromelanin synthesis in healthy older adults needs the formation of the endogenous neurotoxin aminochrome, but the presence of the neuroprotective enzymes DT-diaphorase and glutathione transferase M2-2 (GSTM2-2) prevents the toxic effects of aminochrome, which explains why dopaminergic neurons containing neuromelanin are intact when they die. (B) In the patient with idiopathic Parkinson’s disease (PD), for some unknown reason, the enzymes DT-diaphorase and M2-2 glutathione transferase cannot prevent the neurotoxic action of aminochrome, which implies that aminochrome induces mitochondrial dysfunction, formation of neurotoxic oligomers of alpha-synuclein (SNCA), neuroinflammation, endoplasmic reticulum stress, dysfunction of both lysosomal and proteasomal protein degradation systems, and oxidative stress, which generates neurotoxicity that ultimately ends with the loss of a single neuron. Created with ChemDraw.
Why Neuromelanin Synthesis Is a Normal and Inoffensive Pathway in Healthy Seniors
Neuromelanin is a dark pigment that accumulates in double-membrane structures (Fuentes et al., 2007; Zucca et al., 2023) called NM organelles, which are composed of granules of neuromelanin and lipids with a size of 200-600 nm in the substantia nigra of the human brain (Biesemeier et al., 2016). It has been proposed that NM organelles are specialized autolysosomes that contain some membrane and matrix proteins of typical lysosomes, but are incapable of carrying out the degradation of proteins, lipids, and neuromelanin contained in NM organelles (Zucca et al., 2018) that accumulate with age (Zecca et al., 2002). The synthesis of neuromelanin needs the formation of three transient ortho-quinones (dopamine ortho-quinone, AM, and 5,6-indolequinone), which are potentially neurotoxic (Segura-Aguilar et al., 2014; Herrera et al., 2017). Neuromelanin accumulates in NM organelles and is inoffensive, but free neuromelanin activates microglia and induces the formation of inflammatory factors and consequent neurodegeneration (Zhang et al., 2011, 2013). However, the fact that NM DA neurons are intact in the substantia nigra pars compacta of healthy senior brains suggests that neuromelanin synthesis, although it needs the formation of ortho-quinones that are potentially neurotoxic, is a normal and inoffensive pathway. The explanation for this is that there are two enzymes that prevent the neurotoxic effects of AM: DT and GSTM2 (Segura-Aguilar et al., 2022a, b).
Using nicotinamide adenine dinucleotide + hydrogen or nicotinamide adenine dinucleotide phosphate as the electron donator, DT (NAD(P)H quinone oxidoreductase; NQO1) is the only flavoenzyme that catalyzes AM with two electrons to leukoaminochrome. DT is expressed in different organs, including in the substantia nigra, striatum, cerebral cortex, hypothalamus, hippocampus, and cerebellum in the brain. In the substantia nigra of rats, DT is responsible for 97% of the total quinone reductase activity. DT is present in DA neurons, astrocytes, Bergmann glia, and tanycytes (Schultzberg et al., 1988; Segura-Aguilar and Lind, 1989). DT prevents AM-induced cell death (Lozano et al., 2010), lysosomal dysfunction (Melendez et al., 2019), proteasome dysfunction (Zafar et al., 2006), autophagy dysfunction (Munoz et al., 2012), oxidative stress (Arriagada et al., 2004), mitochondrial impairment (Paris et al., 2011), cytoskeleton disruption (Paris et al., 2010) and the formation of SNCA neurotoxic oligomers (Muñoz et al., 2015).
GSTM2 expressed in the astrocytes catalyzes the glutathione conjugation of AM to 4-S-glutathionyl-5,6-dihydroxyindoline, which is resistant to biological agents such as hydrogen peroxide, superoxide, and dioxygen (Baez et al., 1997; Segura-Aguilar et al., 1997). This enzyme also causes the conjugation of dopamine ortho-quinone, a precursor of AM, to 5-glutathionyl dopamine (Dagnino-Subiabre et al., 2000). Glutathione is a tripeptide that is composed of the amino acid glutamate, cysteine, and glycine, and all glutathione conjugates are degraded by removing the amino acids glutamate and glycine. In addition, 5-glutathionyl dopamine breaks down to 5-cysteinyl dopamine, which has been detected in human neuromelanin and cerebrospinal fluid (Rosengren et al., 1985; Cheng et al., 1996). The presence of 5-cysteinyl dopamine in human neuromelanin and cerebrospinal fluid suggests that dopamine ortho-quinone conjugation is an end reaction, which supports the idea that GSTM2 is a neuroprotective enzyme. GSTM2 prevents AM-induced cell death, autophagic-lysosomal dysfunction, mitochondrial impairment (Huenchuguala et al., 2014; Segura-Aguilar and Huenchuguala, 2018), and the formation of neurotoxic SNCA oligomers (Huenchuguala et al., 2019). Human GSTM2 protects not only astrocytes from the neurotoxic effects of AM when the dopamine taken up is oxidized, but DA neurons as well. Interestingly, astrocytes also play a protective role for DA neurons by secreting exosomes loaded with GSTM2 that DA neurons take on, enhancing the neuroprotection of DT against the neurotoxic effects of AM in DA neurons (Cuevas et al., 2015; Valdes et al., 2021; Segura-Aguilar et al., 2022a, b; Figure 3).
Figure 3.
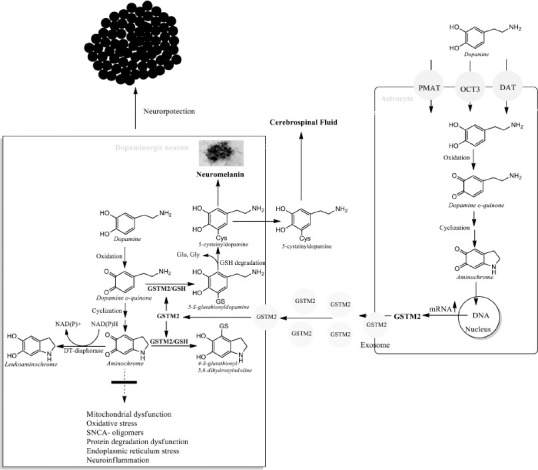
Astrocytes protect dopaminergic neurons.
DT-diaphorase and glutathione transferase M2-2 (GSTM2) prevent the neurotoxic effects of aminochrome. However, GSTM2 that is only expressed in human astrocytes also protects dopaminergic neurons, since astrocytes secrete GSTM2-laden exosomes that penetrate dopaminergic neurons and are released into the cytosol of dopaminergic neurons. Created with ChemDraw. GSH: reduced glutathione; GSTM2: glutathione transferase M2-2; SNCA: alpha-synuclein.
The Difficulties of Finding a Preclinical Model for Idiopathic Parkinson’s Disease
The lesson that can be drawn from the failed clinical studies is that the experimental animal models based on neurotoxins that do not exist in the human body, such as MPTP, 6-hydroxydopamine, and rotenone, do not represent what is happening in the neurodegenerative process in iPD. They induce a massive, expansive, and extremely rapid neurodegenerative process that is the complete opposite of what happens in iPD where, according to what we have calculated, between 58 and 73 DA neurons that contain neuromelanin degenerate per day. However, these experimental animal models are still being used to search for potential new drugs that can halt or slow down the progress of the disease (Alharthy et al., 2023; Frouni et al., 2023; Jalgaonkar et al., 2023).
During the last few years, several experimental animal models have been developed through the genetic manipulation of genes associated with familial PD that include knockout animals or the overexpression of some of these mutations (Chia et al., 2020). One such preclinical genetic model involved the expression of human SNCA A30P and A53T mutations in mice. However, these mutations did not induce Lewy body-like inclusions or the neurodegeneration of DA neurons in the substantia nigra (Matsuoka et al., 2001). The biggest problem with experimental animal models based on mutations associated with familial PD is that these mutations are not present in iPD. Such models may be useful for studying drugs to treat this form of PD, but not for the idiopathic form. The single-neuron degeneration model is not suitable for familial PD because this type of PD is caused by a mutation that leads to an early onset of the disease when the individual is between 21 and 40 years of age. The genes that trigger early-onset PD are recessive, such as those for deglycase 1, phosphatase and tensin homolog-induced kinase 1, and Parkin or the dominant SNCA gene (Post et al., 2020). Familial PD is not triggered by an excess of dopamine oxidation to AM in the synthesis of neuromelanin that exceeds the neuroprotective capacity of DT and M2-2 glutathione transferase in a single neuron that degenerates, but the mutation of a gene massively affects NM DA neurons, which explains its early onset.
It is important that the experimental animal models include the trigger for the mechanisms involved in the loss of NM DA neurons. The failure of phase 2 clinical studies with monoclonal antibodies against human SNCA (Lang et al., 2022; Pagano et al., 2022) can be partly explained by the fact that the neurodegenerative process is so slow that SPECT is possibly not capable of detecting differences in one year. On the other hand, the lack of any effect of these antibodies (Lang et al., 2022; Pagano et al., 2022) can be accounted for by the fact that the SNCA wild type by itself does not induce neurodegeneration, but needs a neurotoxic agent such as AM to cause the formation of neurotoxic oligomers (Muñoz et al., 2015). Therefore, it is critical that experimental animal models involve the neurotoxin that triggers mitochondrial impairment, oxidative and endoplasmic reticulum stress, and neuroinflammation, as well as the dysfunction of both proteasomal and lysosomal protein degradation systems and the formation of neurotoxic oligomers of alpha-synuclein.
One experimental animal model has recently been reported that uses tyrosinase, which plays a fundamental role in the generation of melanin in the skin, to stimulate the oxidation of dopamine to melanin in rat nigrostriatal neurons (Carballo-Carbajal et al., 2019). This model induces the progressive loss of neurons that are immunoreactive to tyrosine hydroxylase, hypokinesia, and the formation of Lewy bodies. It also causes the oxidation of dopamine to melanin, which includes the formation of AM. However, the genetic manipulation of the animal prevents this oxidative process from being similar to that which occurs in patients with iPD, since this model triggers the massive formation of AM in all neurons and glial cells that are transduced by the adeno-associated vector encoding the human tyrosinase gene. This massive transduction of tyrosinase contrasts with the single-neuron neurodegeneration model for iPD.
The unilateral injection of AM in rats induces contralateral behavior with the progressive degeneration of DA neurons in the substantia nigra, but intact DA terminals in the striatum, accompanied by mitochondrial impairment, which signifies decreased dopamine release and increased levels of gamma-aminobutyric acid and cell shrinkage (Herrera et al., 2016). Although the DA terminals of the striatum are intact, a significantly small number of monoaminergic vesicles in the terminals are observed. This unilateral injection of AM causes the neuronal dysfunction of the nigrostriatal neurons, but is not able to instigate single-neuron degeneration as proposed in the degenerative model for iPD.
With currently existing technology, it is practically impossible to implement a preclinical animal model for single-neuron neurodegeneration that replicates what happens in the nigrostriatal neurons of a patient with iPD. Therefore, if we agree that the single-neuron degeneration model represents what happens in the neurodegenerative process of iPD, preclinical studies should focus on the search for drugs that increase the expression of neuroprotective enzymes (DT and glutathione transferase M2-2) that prevent neurotoxic effects.
The activation of a transcription factor like nuclear factor (erythroid-derived 2)-like 2 (NRF2) results in the binding of NRF2 to the antioxidant-responsive element or electrophile-responsive element, which triggers the transition of several antioxidant genes including DT and glutathione transferases. Normally, NRF2 is bound to the Kelch-like ECH-associated protein 1 (KEAP1) which, in the presence of electrophilic or oxidizing molecules, releases the transcription factor NRF2 (Yamamoto et al., 2018; Yang et al., 2022; Segura-Aguilar and Mannervik, 2023). Future clinical studies based on drugs that increase the expression of DT and GSTM2 through the activation of the KEAP1/NRF2 pathway should take into account that the results should be evaluated after several years with a methodology that can quantify small changes in the number of NM DA neurons, such as SPECT. An analysis of phytocompounds that activate the NRF2 system published in 2022 identified possible activators of the KEAP1/NRF2 system that may have a potential neuroprotective effect by increasing the expression of DT and glutathione transferase M2-2. The phytochemicals identified were resveratrol, withametelin, ellagic acid, sesaminol, salidroside, naringenin, curcumin, myricetin, tetramethylpyrazine, ginsenoside Re, naringenin, tanshinone I, piperine, sulforaphane, and magnolol (Bai et al., 2023).
Search Strategy
All years were chosen in the search. These searches were performed between January and February 2023.
Conclusions
The extremely slow progress of the neurodegenerative process of the nigrostriatal neurons in iPD patients is the result of (i) a degenerative model in which the neurotoxic effect of an endogenous neurotoxin affects a single neuron, (ii) a neurotoxic event that is not expansive and (iii) the fact that the neurotoxin that triggers the neurodegenerative process is generated inside the NM DA neurons. The endogenous neurotoxin that fits this single-neuron degeneration model is AM since it (i) is produced within NM DA neurons, (ii) does not induce an expansive neurotoxic effect and (iii) triggers all the mechanisms involved in the neurodegenerative process of the nigrostriatal neurons in iPD. For this reason, the search for new molecules with potential therapeutic use in iPD should focus on ones that increase the expression of the neuroprotective enzymes DT and glutathione transferase M2-2, which prevent the neurotoxic effects of AM. It has been observed that the activation of the KEAP1/NRF2 pathway is associated with the transcriptional activation of the DT and glutathione transferase genes. It would be interesting to find out if the neuroprotective effects of the flavonoid rutin or nicotine against the neurotoxic effect of AM are connected to the activation of the KEAP1/NRF2 pathway (Muñoz et al., 2012b; Khan et al., 2020; Tizabi et al., 2021). Therefore, searching for molecules that activate the KEAP1/NRF2 pathway and inhibit the neurotoxic effects of AM could be useful in finding potential new molecules for future clinical studies that aim to halt or slow down the progress of iPD.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Data availability statement: The data are available from the corresponding author on reasonable request.
P-Reviewers: Kazanis I, Zecca L; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Aguirre P, Urrutia P, Tapia V, Villa M, Paris I, Segura-Aguilar J, Núñez MT. The dopamine metabolite aminochrome inhibits mitochondrial complex I and modifies the expression of iron transporters DMT1 and FPN1. Biometals. 2012;25:795–803. doi: 10.1007/s10534-012-9525-y. [DOI] [PubMed] [Google Scholar]
- 2.Alharthy KM, Althurwi HN, Albaqami FF, Altharawi A, Alzarea SI, Al-Abbasi FA, Nadeem MS, Kazmi I. Barbigerone potentially alleviates rotenone-activated Parkinson's disease in a rodent model by reducing oxidative stress and neuroinflammatory cytokines. ACS Omega. 2023;8:4608–4615. doi: 10.1021/acsomega.2c05837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arriagada C, Paris I, Sanchez de las Matas MJ, Martinez-Alvarado P, Cardenas S, Castañeda P, Graumann R, Perez-Pastene C, Olea-Azar C, Couve E, Herrero MT, Caviedes P, Segura-Aguilar J. On the neurotoxicity mechanism of leukoaminochrome o-semiquinone radical derived from dopamine oxidation:Mitochondria damage, necrosis, and hydroxyl radical formation. Neurobiol Dis. 2004;16:468–477. doi: 10.1016/j.nbd.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Baez S, Segura-Aguilar J, Widersten M, Johansson AS, Mannervik B. Glutathione transferases catalyse the detoxication of oxidized metabolites (o-quinones) of catecholamines and may serve as an antioxidant system preventing degenerative cellular processes. Biochem J. 1997;324:25–28. doi: 10.1042/bj3240025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bai X, Bian Z, Zhang M. Targeting the Nrf2 signaling pathway using phytochemical ingredients:a novel therapeutic road map to combat neurodegenerative diseases. Phytomedicine. 2023;109:154582. doi: 10.1016/j.phymed.2022.154582. [DOI] [PubMed] [Google Scholar]
- 6.Biesemeier A, Eibl O, Eswara S, Audinot JN, Wirtz T, Pezzoli G, Zucca FA, Zecca L, Schraermeyer U. Elemental mapping of neuromelanin organelles of human substantia nigra:correlative ultrastructural and chemical analysis by analytical transmission electron microscopy and nano-secondary ion mass spectrometry. J Neurochem. 2016;138:339–353. doi: 10.1111/jnc.13648. [DOI] [PubMed] [Google Scholar]
- 7.Birkmayer W, Hornykiewicz O. The effect of l-3,4-dihydroxyphenylalanine (=DOPA) on akinesia in parkinsonism. Parkinsonism Relat Disord. 1998;4:59–60. doi: 10.1016/s1353-8020(98)00013-3. [DOI] [PubMed] [Google Scholar]
- 8.Bisaglia M, Mammi S, Bubacco L. Kinetic and structural analysis of the early oxidation products of dopamine:analysis of the interactions with alpha-synuclein. J Biol Chem. 2007;282:15597–15605. doi: 10.1074/jbc.M610893200. [DOI] [PubMed] [Google Scholar]
- 9.Braak H, Del Tredici K, Rüb U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 10.Capucciati A, Zucca FA, Monzani E, Zecca L, Casella L, Hofer T. Interaction of neuromelanin with xenobiotics and consequences for neurodegeneration;promising experimental models. Antioxidants. 2021;10:824. doi: 10.3390/antiox10060824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carballo-Carbajal I, Laguna A, Romero-Giménez J, Cuadros T, Bové J, Martinez-Vicente M. Brain tyrosinase overexpression implicates age-dependent neuromelanin production in Parkinson's disease pathogenesis. Nat Commun. 2019;10:973. doi: 10.1038/s41467-019-08858-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlsson A, Linqvist M, Magnuson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200. doi: 10.1038/1801200a0. [DOI] [PubMed] [Google Scholar]
- 13.Cheng FC, Kuo JS, Chia LG, Dryhurst G. Elevated 5-S-cysteinyldopamine/homovanillic acid ratio and reduced homovanillic acid in cerebrospinal fluid:possible markers for and potential insights into the pathoetiology of Parkinson's disease. J Neural Transm. 1996;103:433–446. doi: 10.1007/BF01276419. [DOI] [PubMed] [Google Scholar]
- 14.Chia SJ, Tan EK, Chao YX. Historical perspective:models of Parkinson's disease. Int J Mol Sci. 2020;21:2464. doi: 10.3390/ijms21072464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuevas C, Huenchuguala S, Muñoz P, Villa M, Paris I, Mannervik B, Segura-Aguilar J. Glutathione transferase-M2-2 secreted from glioblastoma cell protects SH-SY5Y cells from aminochrome neurotoxicity. Neurotox Res. 2015;27:217–228. doi: 10.1007/s12640-014-9500-1. [DOI] [PubMed] [Google Scholar]
- 16.Dagnino-Subiabre A, Cassels BK, Baez S, Johansson AS, Mannervik B, Segura-Aguilar J. Glutathione transferase M2-2 catalyzes conjugation of dopamine and dopa o-quinones. Biochem Biophys Res Commun. 2000;274:32–36. doi: 10.1006/bbrc.2000.3087. [DOI] [PubMed] [Google Scholar]
- 17.Devos D, Labreuche J, Rascol O, Corvol JC, Duhamel A, Guyon Delannoy P, Poewe W, Compta Y, Pavese N, Růžička E, Dušek P, Post B, Bloem BR, Berg D, Maetzler W, Otto M, Habert MO, Lehericy S, Ferreira J, Dodel R, et al. FAIRPARK-II Study Group. Trial of deferiprone in Parkinson's disease. N Engl J Med. 2022;387:2045–2055. doi: 10.1056/NEJMoa2209254. [DOI] [PubMed] [Google Scholar]
- 18.Dorsey ER, Sherer T, Okun MS, Bloem BR. The emerging evidence of the Parkinson pandemic. J Parkinsons Dis. 2018;8:S3–8. doi: 10.3233/JPD-181474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frouni I, Kwan C, Belliveau S, Hamadjida A, Bédard D, Nuara SG, Gourdon JC, Huot P. Anti-parkinsonian effect of the mGlu2 positive allosteric modulator LY-487,379 as monotherapy and adjunct to a low L-DOPA dose in the MPTP-lesioned marmoset. Eur J Pharmacol. 2023;939:175429. doi: 10.1016/j.ejphar.2022.175429. [DOI] [PubMed] [Google Scholar]
- 20.Fuentes P, Paris I, Nassif M, Caviedes P, Segura-Aguilar J. Inhibition of VMAT-2 and DT-diaphorase induce cell death in a substantia nigra-derived cell line--an experimental cell model for dopamine toxicity studies. Chem Res Toxicol. 2007;20:776–783. doi: 10.1021/tx600325u. [DOI] [PubMed] [Google Scholar]
- 21.Goldstein DS. The catecholaldehyde hypothesis for the pathogenesis of catecholaminergic neurodegeneration:what we know and what we do not know. Int J Mol Sci. 2021;22:5999. doi: 10.3390/ijms22115999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goloborshcheva VV, Kucheryanu VG, Voronina NA, Teterina EV, Ustyugov AA, Morozov SG. Synuclein proteins in MPTP-induced death of substantia nigra pars compacta dopaminergic neurons. Biomedicines. 2022;10:2278. doi: 10.3390/biomedicines10092278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grünblatt E, Ruder J, Monoranu CM, Riederer P, Youdim MB, Mandel SA. Differential alterations in metabolism and proteolysis-related proteins in human Parkinson's disease substantia nigra. Neurotox Res. 2018;33:560–568. doi: 10.1007/s12640-017-9843-5. [DOI] [PubMed] [Google Scholar]
- 24.Herrera A, Muñoz P, Paris I, Díaz-Veliz G, Mora S, Inzunza J, Hultenby K, Cardenas C, Jaña F, Raisman-Vozari R, Gysling K, Abarca J, Steinbusch HW, Segura-Aguilar J. Aminochrome induces dopaminergic neuronal dysfunction:A new animal model for Parkinson's disease. Cell Mol Life Sci. 2016;73:3583–3597. doi: 10.1007/s00018-016-2182-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herrera A, Muñoz P, Steinbusch HWM, Segura-Aguilar J. Are dopamine oxidation metabolites involved in the loss of dopaminergic neurons in the nigrostriatal system in Parkinson's disease?ACS Chem Neurosci. 2017;8:702–711. doi: 10.1021/acschemneuro.7b00034. [DOI] [PubMed] [Google Scholar]
- 26.Hijaz BA, Volpicelli-Daley LA. Initiation and propagation of α-synuclein aggregation in the nervous system. Mol Neurodegener. 2020;15:19. doi: 10.1186/s13024-020-00368-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huenchuguala S, Muñoz P, Zavala P, Villa M, Cuevas C, Ahumada U, Graumann R, Nore BF, Couve E, Mannervik B, Paris I, Segura-Aguilar J. Glutathione transferase mu 2 protects glioblastoma cells against aminochrome toxicity by preventing autophagy and lysosome dysfunction. Autophagy. 2014;10:618–630. doi: 10.4161/auto.27720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huenchuguala S, Muñoz P, Segura-Aguilar J. The importance of mitophagy in maintaining mitochondrial function in U373MG cells. Bafilomycin A1 restores aminochrome-induced mitochondrial damage. ACS Chem Neurosci. 2017;8:2247–2253. doi: 10.1021/acschemneuro.7b00152. [DOI] [PubMed] [Google Scholar]
- 29.Huenchuguala S, Sjödin B, Mannervik B, Segura-Aguilar J. Novel alpha-synuclein oligomers formed with the aminochrome-glutathione conjugate are not neurotoxic. Neurotox Res. 2019;35:432–440. doi: 10.1007/s12640-018-9969-0. [DOI] [PubMed] [Google Scholar]
- 30.Jalgaonkar S, Gajbhiye S, Sayyed M, Tripathi R, Khatri N, Parmar U, Shankar A. S-adenosyl methionine improves motor co-ordination with reduced oxidative stress, dopaminergic neuronal loss, and DNA methylation in the brain striatum of 6-hydroxydopamine-induced neurodegeneration in rats. Anat Rec (Hoboken) 2023;306:820–830. doi: 10.1002/ar.24948. [DOI] [PubMed] [Google Scholar]
- 31.Khalaf MM, El-Sayed MM, Kandeil MA, Ahmed S. A novel protective modality against rotenone-induced Parkinson's disease:a pre-clinical study with dulaglutide. Int Immunopharmacol. 2023;119:110170. doi: 10.1016/j.intimp.2023.110170. [DOI] [PubMed] [Google Scholar]
- 32.Khan H, Tundis R, Ullah H, Aschner M, Belwal T, Mirzaei H, Akkol EK. Flavonoids targeting NRF2 in neurodegenerative disorders. Food Chem Toxicol. 2020;146:111817. doi: 10.1016/j.fct.2020.111817. [DOI] [PubMed] [Google Scholar]
- 33.Kostrzewa RM. Neonatal 6-hydroxydopamine lesioning of rats and dopaminergic neurotoxicity:proposed animal model of Parkinson's disease. J Neural Transm (Vienna) 2022;129:445–461. doi: 10.1007/s00702-022-02479-4. [DOI] [PubMed] [Google Scholar]
- 34.Lane EL. L-DOPA for Parkinson's disease-a bittersweet pill. Eur J Neurosci. 2019;49:384–398. doi: 10.1111/ejn.14119. [DOI] [PubMed] [Google Scholar]
- 35.Lang AE, Siderowf AD, Macklin EA, Poewe W, Brooks DJ, Fernandez HH, Rascol O, Giladi N, Stocchi F, Tanner CM, Postuma RB, Simon DK, Tolosa E, Mollenhauer B, Cedarbaum JM, Fraser K, Xiao J, Evans KC, Graham DL, Sapir I, et al. N Engl J Med. 2022;387:408–420. doi: 10.1056/NEJMoa2203395. [DOI] [PubMed] [Google Scholar]
- 36.Lozano J, Muñoz P, Nore BF, Ledoux S, Segura-Aguilar J. Stable expression of short interfering RNA for DT-diaphorase induces neurotoxicity. Chem Res Toxicol. 2010;23:1492–1496. doi: 10.1021/tx100182a. [DOI] [PubMed] [Google Scholar]
- 37.Matsuoka Y, Vila M, Lincoln S, McCormack A, Picciano M, LaFrancois J, Yu X, Dickson D, Langston WJ, McGowan E, Farrer M, Hardy J, Duff K, Przedborski S, Di Monte DA. Lack of nigral pathology in transgenic mice expressing human alpha-synuclein driven by the tyrosine hydroxylase promoter. Neurobiol Dis. 2001;8:535–539. doi: 10.1006/nbdi.2001.0392. [DOI] [PubMed] [Google Scholar]
- 38.Mehra S, Sahay S, Maji SK. α-Synuclein misfolding and aggregation:implications in Parkinson's disease pathogenesis. Biochim Biophys Acta Proteins Proteom. 2019;1867:890–908. doi: 10.1016/j.bbapap.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 39.Meléndez C, Muñoz P, Segura-Aguilar J. DT-diaphorase prevents aminochrome-induced lysosome dysfunction in SH-SY5Y cells. Neurotox Res. 2019;35:255–259. doi: 10.1007/s12640-018-9953-8. [DOI] [PubMed] [Google Scholar]
- 40.Muñoz P, Huenchuguala S, Paris I, Segura-Aguilar J. Dopamine oxidation and autophagy. Parkinsons Dis. 2012a;2012a:920953. doi: 10.1155/2012/920953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muñoz P, Huenchuguala S, Paris I, Cuevas C, Villa M, Caviedes P, Segura-Aguilar J, Tizabi Y. Protective effects of nicotine against aminochrome-induced toxicity in substantia nigra derived cells:implications for Parkinson's disease. Neurotox Res. 2012b;22:177–180. doi: 10.1007/s12640-012-9326-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muñoz P, Cardenas S, Huenchuguala S, Briceño A, Couve E, Paris I, Segura-Aguilar J. DT-diaphorase prevents aminochrome-induced alpha-synuclein oligomer formation and neurotoxicity. Toxicol Sci. 2015;145:37–47. doi: 10.1093/toxsci/kfv016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ni A, Ernst C. Evidence that substantia nigra pars compacta dopaminergic neurons are selectively vulnerable to oxidative stress because they are highly metabolically active. Front Cell Neurosci. 2022;16:826193. doi: 10.3389/fncel.2022.826193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niso-Santano M, Bravo-San Pedro JM, Gómez-Sánchez R, Climent V, Soler G, Fuentes JM, González-Polo RA. ASK1 overexpression accelerates paraquat-induced autophagy via endoplasmic reticulum stress. Toxicol Sci. 2011;119:156–168. doi: 10.1093/toxsci/kfq313. [DOI] [PubMed] [Google Scholar]
- 45.Olanow CW, Schapira AH, LeWitt PA, Kieburtz K, Sauer D, Olivieri G, Pohlmann H, Hubble J. TCH346 as a neuroprotective drug in Parkinson's disease:a double-blind, randomised, controlled trial. Lancet Neurol. 2006;5:1013–1020. doi: 10.1016/S1474-4422(06)70602-0. [DOI] [PubMed] [Google Scholar]
- 46.Olanow CW, Bartus RT, Volpicelli-Daley LA, Kordower JH. Trophic factors for Parkinson's disease:to live or let die. Mov Disord. 2015;30:1715–1724. doi: 10.1002/mds.26426. [DOI] [PubMed] [Google Scholar]
- 47.Pagano G, Taylor KI, Anzures-Cabrera J, Marchesi M, Simuni T, Marek K, Postuma RB, Pavese N, Stocchi F, Azulay JP, Mollenhauer B, López-Manzanares L, Russell DS, Boyd JT, Nicholas AP, Luquin MR, Hauser RA, Gasser T, Poewe W, Ricci B, et al. Trial of prasinezumab in early-stage Parkinson's disease. N Engl J Med. 2022;387:421–432. doi: 10.1056/NEJMoa2202867. [DOI] [PubMed] [Google Scholar]
- 48.Paris I, Perez-Pastene C, Cardenas S, Iturriaga-Vasquez P, Muñoz P, Couve E, Caviedes P, Segura-Aguilar J. Aminochrome induces disruption of actin, alpha-, and beta-tubulin cytoskeleton networks in substantia-nigra-derived cell line. Neurotox Res. 2010;18:82–92. doi: 10.1007/s12640-009-9148-4. [DOI] [PubMed] [Google Scholar]
- 49.Paris I, Muñoz P, Huenchuguala S, Couve E, Sanders LH, Greenamyre JT, Caviedes P, Segura-Aguilar J. Autophagy protects against aminochrome-induced cell death in substantia nigra-derived cell line. Toxicol Sci. 2011;121:376–388. doi: 10.1093/toxsci/kfr060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkinson Study Group QE3 Investigators. Beal MF, Oakes D, Shoulson I, Henchcliffe C, Galpern WR, Haas R, Juncos JL, Nutt JG, Voss TS, Ravina B, Shults CM, Helles K, Snively V, Lew MF, Griebner B, Watts A, Gao S, Pourcher E, Bond L, Kompoliti K, et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease:no evidence of benefit. JAMA Neurol. 2014;71:543–552. doi: 10.1001/jamaneurol.2014.131. [DOI] [PubMed] [Google Scholar]
- 51.Pillai KS, Bhat P, Srivastava AK, Rajan R, Radhakrishnan DM, Elavarasi A, Srivastava MP, Singh MB. Zonisamide add-on in tremor-dominant Parkinson's disease- A randomized controlled clinical trial. Parkinsonism Relat Disord. 2022;105:1–6. doi: 10.1016/j.parkreldis.2022.10.017. [DOI] [PubMed] [Google Scholar]
- 52.Post B, van den Heuvel L, van Prooije T, van Ruissen X, van de Warrenburg B, Nonnekes J. Young onset Parkinson's disease:a modern and tailored approach. J Parkinsons Dis. 2020;10:S29–36. doi: 10.3233/JPD-202135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiu J, Chen Y, Zhuo J, Zhang L, Liu J, Wang B, Sun D, Yu S, Lou H. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson's disease model. Neuropharmacology. 2022;207:108963. doi: 10.1016/j.neuropharm.2022.108963. [DOI] [PubMed] [Google Scholar]
- 54.Rosengren E, Linder-Eliasson E, Carlsson A. Detection of 5-S-cysteinyldopamine in human brain. J Neural Transm. 1985;63:247–253. doi: 10.1007/BF01252029. [DOI] [PubMed] [Google Scholar]
- 55.Santos CC, Araújo FM, Ferreira RS, Silva VB, Silva JHC, Grangeiro MS, Soares ÉN, Pereira ÉPL, Souza CS, Costa SL, Segura-Aguilar J, Silva VDA. Aminochrome induces microglia and astrocyte activation. Toxicol In Vitro. 2017;42:54–60. doi: 10.1016/j.tiv.2017.04.004. [DOI] [PubMed] [Google Scholar]
- 56.Schultzberg M, Segura-Aguilar J, Lind C. Distribution of DT diaphorase in the rat brain:biochemical and immunohistochemical studies. Neuroscience. 1988;27:763–776. doi: 10.1016/0306-4522(88)90181-9. [DOI] [PubMed] [Google Scholar]
- 57.Schwarzschild MA, Ascherio A, Casaceli C, Curhan GC, Fitzgerald R, Kamp C, Lungu C, Macklin EA. Effect of urate-elevating inosine on early parkinson disease progression:the SURE-PD3 randomized clinical trial. JAMA. 2021;326:926–939. doi: 10.1001/jama.2021.10207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Segura-Aguilar J, Lind C. On the mechanism of the Mn3(+)-induced neurotoxicity of dopamine:prevention of quinone-derived oxygen toxicity by DT diaphorase and superoxide dismutase. Chem Biol Interact. 1989;72:309–324. doi: 10.1016/0009-2797(89)90006-9. [DOI] [PubMed] [Google Scholar]
- 59.Segura-Aguilar J, Baez S, Widersten M, Welch CJ, Mannervik B. Human class Mu glutathione transferases, in particular isoenzyme M2-2, catalyze detoxication of the dopamine metabolite aminochrome. J Biol Chem. 1997;272:5727–5731. doi: 10.1074/jbc.272.9.5727. [DOI] [PubMed] [Google Scholar]
- 60.Segura-Aguilar J, Paris I, Muñoz P, Ferrari E, Zecca L, Zucca FA. Protective and toxic roles of dopamine in Parkinson's disease. J Neurochem. 2014;129:898–915. doi: 10.1111/jnc.12686. [DOI] [PubMed] [Google Scholar]
- 61.Segura-Aguilar J, Huenchuguala S. Aminochrome induces irreversible mitochondrial dysfunction by inducing autophagy dysfunction in Parkinson's disease. Front Neurosci. 2018;12:106. doi: 10.3389/fnins.2018.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Segura-Aguilar J. Preclinical models based on endogenous neurotoxins. In: Segura-Aguilar J, editor. Clinical studies and therapies in Parkinson's disease:translations from preclinical models. Cambridge: Elsevier; 2021. pp. 263–282. [Google Scholar]
- 63.Segura-Aguilar J, Muñoz P, Inzunza J, Varshney M, Nalvarte I, Mannervik B. Neuroprotection against aminochrome neurotoxicity:glutathione transferase M2-2 and DT-diaphorase. Antioxidants. 2022a;11:296. doi: 10.3390/antiox11020296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Segura-Aguilar J, Mannervik B, Inzunza J, Varshney M, Nalvarte I, Muñoz P. Astrocytes protect dopaminergic neurons against aminochrome neurotoxicity. Neural Regen Res. 2022b;17:1861–1866. doi: 10.4103/1673-5374.335690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segura-Aguilar J, Mannervik B. A preclinical model for Parkinson's disease based on transcriptional gene activation via KEAP1/NRF2 to develop new antioxidant therapies. Antioxidants. 2023;12:673. doi: 10.3390/antiox12030673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Simuni T, Oakes D, Biglan Kevin, Wendy R, Galpern WR. Isradipine versus placebo in early Parkinson disease:a randomized trial. Ann Intern Med. 2020;172:591–598. doi: 10.7326/M19-2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Simuni T, Fiske B, Merchant K, Coffey CS, Klingner E, Caspell-Garcia C, Lafontant DE, Matthews H, Wyse RK, Brundin P, Simon DK, Schwarzschild M, Weiner D, Adams J, Venuto C, Dawson TM, Baker L, Kostrzebski M, Ward T, Rafaloff G, et al. Efficacy of nilotinib in patients with moderately advanced Parkinson disease:a randomized clinical trial. JAMA Neurol. 2021;78:312–320. doi: 10.1001/jamaneurol.2020.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, Smith RA, Murphy MP, Taylor KM Protect Study Group. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 69.Teixeira FG, Vilaça-Faria H, Domingues AV, Campos J, Salgado AJ. Preclinical comparison of stem cells secretome and levodopa application in a 6-hydroxydopamine rat model of Parkinson's disease. Cells. 2020;9:315. doi: 10.3390/cells9020315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tizabi Y, Getachew B, Aschner M. Novel pharmacotherapies in Parkinson's disease. Neurotox Res. 2021;39:1381–1390. doi: 10.1007/s12640-021-00375-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Valdes R, Armijo A, Muñoz P, Hultenby K, Hagg A, Inzunza J, Nalvarte I, Varshney M, Mannervik B, Segura-Aguilar J. Cellular trafficking of glutathione transferase M2-2 between U373MG and SHSY-S7 cells is mediated by exosomes. Neurotox Res. 2021;39:182–190. doi: 10.1007/s12640-020-00327-5. [DOI] [PubMed] [Google Scholar]
- 72.Wakabayashi K, Tanji K, Odagiri S, Miki Y, Mori F, Takahashi H. The Lewy body in Parkinson's disease and related neurodegenerative disorders. Mol Neurobiol. 2013;47:495–508. doi: 10.1007/s12035-012-8280-y. [DOI] [PubMed] [Google Scholar]
- 73.Warren Olanow C, Bartus RT, Baumann TL, Factor S, Boulis N, Stacy M, Turner DA, Marks W, Larson P, Starr PA, Jankovic J, Simpson R, Watts R, Guthrie B, Poston K, Henderson JM, Stern M, Baltuch G, Goetz CG, Herzog C, et al. Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease:A double-blind, randomized, controlled trial. Ann Neurol. 2015;78:248–257. doi: 10.1002/ana.24436. [DOI] [PubMed] [Google Scholar]
- 74.Williams A. MPTP parkinsonism. Br Med J. 1984;289:1401–1402. doi: 10.1136/bmj.289.6456.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET-PD) Investigators. Kieburtz K, Tilley BC, Elm JJ, Babcock D, Hauser R, Ross GW, Augustine AH, Augustine EU, Aminoff MJ, Bodis-Wollner IG, Boyd J, Cambi F, Chou K, Christine CW, Cines M, Dahodwala N, Derwent L, Dewey RB, Jr, Hawthorne K, et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease:a randomized clinical trial. JAMA. 2015;313:584–593. doi: 10.1001/jama.2015.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xiong R, Siegel D, Ross D. Quinone-induced protein handling changes:implications for major protein handling systems in quinone-mediated toxicity. Toxicol Appl Pharmacol. 2014;280:285–295. doi: 10.1016/j.taap.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yamamoto M, Kensler TW, Motohashi H. The KEAP1-NRF2 system:a Thiol-based sensor-effector apparatus for main-taining redox homeostasis. Physiol Rev. 2018;98:1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yang XX, Yang R, Zhang F. Role of Nrf2 in Parkinson's disease:toward new perspectives. Front Pharmacol. 2022;13:919233. doi: 10.3389/fphar.2022.919233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zafar KS, Inayat-Hussain SH, Siegel D, Bao A, Shieh B, Ross D. Overexpression of NQO1 protects human SK-N-MC neuroblastoma cells against dopamine-induced cell death. Toxicol Lett. 2006;166:261–267. doi: 10.1016/j.toxlet.2006.07.340. [DOI] [PubMed] [Google Scholar]
- 80.Zecca L, Fariello R, Riederer P, Sulzer D, Gatti A, Tampellini D. The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson's disease. FEBS Lett. 2002;510:216–220. doi: 10.1016/s0014-5793(01)03269-0. [DOI] [PubMed] [Google Scholar]
- 81.Zhang W, Phillips K, Wielgus AR, Liu J, Albertini A, Zucca FA, Faust R, Qian SY, Miller DS, Chignell CF, Wilson B, Jackson-Lewis V, Przedborski S, Joset D, Loike J, Hong JS, Sulzer D, Zecca L. Neuromelanin activates microglia and induces degeneration of dopaminergic neurons:implications for progression of Parkinson's disease. Neurotox Res. 2011;19:63–72. doi: 10.1007/s12640-009-9140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang W, Zecca L, Wilson B, Ren HW, Wang YJ, Wang XM, Hong JS. Human neuromelanin:an endogenous microglial activator for dopaminergic neuron death. Elite, editor. Front Biosci. 2013;5:1–11. doi: 10.2741/e591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zucca FA, Vanna R, Cupaioli FA, Bellei C, De Palma A, Di Silvestre D, Mauri P, Grassi S, Prinetti A, Casella L, Sulzer D, Zecca L. Neuromelanin organelles are specialized autolysosomes that accumulate undegraded proteins and lipids in aging human brain and are likely involved in Parkinson's disease. NPJ Parkinsons Dis. 2018;4:17. doi: 10.1038/s41531-018-0050-8. [DOI] [PMC free article] [PubMed] [Google Scholar]