

Abstract
Spirochromanes incorporating Schiff's bases and semicarbazones 4a-e and 5a-j were synthesizedand analyzed for their potential antiproliferative activity using four human cancer cell lines (MCF-7, HCT-116, PC3, and A549). Compounds 5a, 5b and 5g possessed the highest antiproliferative activity among the tested compounds,with an IC50 range of 1.154–9.09 μM. Compound 5j selectively inhibited the PC3 cell proliferation (IC50 = 5.47 μM). Spirochromanes 5a, 5b and 5g exhibited high inhibitory activity against EGFR (IC50 = 0.116, 0.132, and 0.077 μM, respectively) and HER2 (IC50 = 0.055, 0.210 and 0.085 μM, respectively) compared with the references, erlotinib (IC50 = 0.090 and 0.038 μM, respectively) and gefitinib (IC50 = 0.052 and 0.072 μM, respectively). Cell cycle analysis and apoptosis results showed that compounds 5a, 5b and 5g arrested growth inthe S phase, and the programmed cell death induced by these compounds was an apoptotic mechanism rather than a necrotic pathway. Molecular docking studies of spirochromanes 5a, 5b and 5g to EGFR and HER2 binding sites were performed to explore the orientation mode and interaction.
Keywords: Spirochromane, Schiff's bases, Synthesis, Antiproliferative activity, Enzymatic assay, Apoptosis, Molecular docking
1. Introduction
Cancer is one of the leading public health problems worldwide,and many people are diagnosed with cancer annually (Torre et al., 2015, El-Husseiny et al., 2018, El-Azab et al., 2017, Al-Suwaidan et al., 2016, Hamdi et al., 2016). The use of chemotherapy in cancer treatment has become the traditional treatment; however, many cases exhibit no efficacy owing to poor selectivity and drug resistance, resulting in severe side effects (Holohan et al., 2013, Housman et al., 2014). Consequently, novel and effective anticancer agents are urgently required (El-Sherbeny et al., 2010, Alanazi et al., 2013, El-Azab et al., 2013, Mohamed et al., 2016). Moreover, the use of drug combinations in cancer treatment has several undesirable effects (Szakács et al., 2006, Bayat Mokhtari et al., 2017), which can be overcome by the preferred therapeutic strategy that involves using a single compound with multiple molecular mechanisms (Alkahtani et al., 2020, El-Azab et al., 2018, Fu et al., 2017, Antonello et al., 2006). Therefore, a single compound with multiple molecular mechanisms is used in a current trend therapy as multitargeted therapy (Xie and Bourne, 2015). In cell signalling pathways, survival of the cancer cell is enhanced through the activation of many kinases such as EGFR (epidermal growth factor receptor) and HER2 (human epidermal growth factor receptor 2) (Baker and Reddy, 2010, Christensen, 2007, Black et al., 2003, Abdelsalam et al., 2019). Targeting these kinases is a must to produce potent and efficient anticancer agents (Regad, 2015, Woodburn, 1999, Rusnak et al., 2001, El-Azab et al., 2020). In addition, apoptosis was found in various solid tumors due to the inhibition of the tyrosine kinases (Alkahtani et al., 2020, Gullick, 1991).
Sorafenib (Nexavar®), Gefitinib (Iressa®), Erlotinib (Tarceva®), Lapatinib (Tykerb®) and Vandetanib (Caprelsa®) are multikinase inhibitors that have been approved as chemotherapeutic agents with high selectivity and efficacy (Fig. 1) (Wang et al., 2017, Omar et al., 2016, Gundla et al., 2008, Madhusudan and Ganesan, 2004, Barker et al., 2001, Frampton, 2009, Takimoto and Awada, 2008, Dungo and Keating, 2013). On the other hand, spirochromanes and spirochromane piperidines I, II and III (Fig. 2) possess potent anticancer activity (Uto et al., 2010, Varasi et al., 2011). Also, compounds incorporating an aldimine or ketimine moiety (RHC = N-R) (Fig. 2), such as Schiff's bases, hydrazones and semicarbazones, have been characterized as efficient anticancer agents (compounds IV, V, and VI) (Xu et al., 2008, Abdel-Aziz et al., 2021, Krishnan et al., 2008, Peterson et al., 2009). Some of these compounds inhibit EGFR and HER2 and induce apoptosis (Şenkardeş et al., 2020, Vogel et al., 2008, George, 2018).
Fig. 1.

Approved multikinase inhibitors as chemotherapeutic agents.
Fig. 2.

Reported anticancer spirochromanes (I-III), Schiff's bases (IV, V and VI) and the designed compounds (4a-e and 5a-j).
Based on the aforementioned rationale, novel spirochromane piperidine derivatives 4a-e and 5a-j incorporating Schiff's bases (Fig. 2) were synthesized, and their antiproliferative activities were evaluated using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay. Structure-activity relationship (SAR) was also studied in diverse chemical spaces. In addition, the EGFR and HER2 inhibition assay for derivatives with promising antiproliferative activity was performed. Next, apoptosis and cell cycle status in tissue culture cell lines were determined by measuring the DNA content by flow cytometry. A docking protocol inside the binding sites of EGFR and HER2 tyrosine kinases was conducted to determine the binding modes of the promising compounds.
2. Material and methods
2.1. Chemistry
The melting points of the compounds (°C) were determined using a Stuart melting point apparatus (SMP 30). Infrared (IR) spectra were measured on an FT-IR 200 spectrophotometer (ύ cm−1) Faculty of Pharmacy, Mansoura University. The 1H NMR and 13C NMR spectra were recorded in CDCl3 or DMSO‑d6 at 1HNMR (400 MHz) and 13CNMR (100 MHz) on a Bruker NMR spectrometer (δ ppm) using tetramethylsilane(TMS) as an internal standard at the NMR Unit, Faculty of Pharmacy, Mansoura University. Mass spectra were obtained using the direct inlet of a mass analyzer in a Thermo Scientific GC/MS model ISQ at the Regional Center for Mycology and Biotechnology (RCMB), Al-Azhar University, Egypt. The cell cycle analysis was performed using a confirmatory diagnostic unit (VACSERA, Egypt). Compounds 2a and b were prepared as previously described (Battisti et al., 2014).
2.1.1. General procedure for preparation of hydrazone derivatives 3a,b
Hydrazine hydrate (3.6 mmol) in ethanol (20 ml) was added to a solution of spirochromanone derivatives 2a,b (3 mmol), and the mixture was left to stir at room temperature for 5 h.The reaction mixture was concentrated using a Rotavap, water was added, extracted with ethyl acetate (10 ml × 3) and evaporated under reduced pressure to generate yellow solids, which were used directly for the next step without any further purification.
2.1.1.1. (E)-4-Hydrazono-1′-methylspiro[chroman-2,4′-piperidine] (3a)
Yellow solid; (65%). M.p. 110 – 112 °C. IR (νmax/cm−1): 3345, 3400 (NH2), 3139, 2972, 2936 (CHs), 1615 (C = N), 1450, 1234, 1180. 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J = 7.9, 1.3 Hz, 1H), 7.24 – 7.19 (m, 1H), 6.97 – 6.89 (m, 2H), 5.31 (s, 2H), 2.60 (s, 2H), 2.53 (s, 2H), 2.40 (dd, J = 16.4, 6.7 Hz, 2H), 2.33 (s, 3H), 1.98 (d, J = 12.2 Hz, 2H), 1.70 (td, J = 13.7, 4.4 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 152.8, 141.0, 132.2, 129.9, 123.7, 122.1, 118.0, 72.6, 50.9, 45.8, 34.4, 34.1. MS m/z (%): 245.15 (M+, 17.88).
2.1.1.2. (E)-1′-Ethyl-4-hydrazonospiro[chroman-2,4′-piperidine] (3b)
Brown solid; (55%). M.p. 100−102 °C. IR (νmax/cm−1): 3338,3400 (NH2), 3134, 2971, 2936 (CHs), 1609 (C = N), 1454, 1234, 1181. 1H NMR (400 MHz, CDCl3) δ 7.87 (dd, J = 7.9, 1.3 Hz, 1H), 7.25 – 7.19 (m, 1H), 6.98 – 6.89 (m, 2H), 5.31 (s, 2H), 2.69 (d, J = 11.5 Hz, 2H), 2.53 (s, 2H), 2.48 (q, J = 7.2 Hz, 2H), 2.43 – 2.37 (m, 2H), 1.99 (d, J = 12.3 Hz, 2H), 1.76 – 1.67 (m, 2H), 1.12 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 153.0, 140.8, 132.0, 129.9, 123.7, 122.1, 118.5, 72.3, 50.2, 48.8, 34.4, 34.1, 12.9. MS m/z (%): 259.32 (M+, 26.03).
2.1.2. General procedure for preparation of hydrazone derivatives 4a-e
A mixture of hydrazone derivatives 3a,b (1.0 mmol), appropriate aldehyde (1.0 mmol) and catalytic drops of glacial acetic acid in ethanol (20 ml) was heated under reflux overnight. The reaction mixture was allowed to cool and then poured into icewater (60 ml). The resulting precipitate was collected, washed with water and recrystallized from ethanol.
2.1.2.1. (E)-4-[(E)-(4-Chlorobenzylidene)hydrazono]-1′-methylspiro(chroman-2,4′-piperidine) (4a)
White solid; (73%). M.p. 139−141 °C. IR (νmax/cm−1): 3062, 2938 (CHs), 1616 (C N), 1455, 1229, 1180. 1H NMR (400 MHz, CDCl3) δ 8.50 (s, 1H), 8.15 (d, J = 6.9 Hz, 1H), 7.79 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 8.4 Hz, 2H), 7.37 (t, J = 7.1 Hz, 1H), 7.02 – 6.96 (m, 2H), 3.14 (s, 2H), 2.61 (d, J = 11.4 Hz, 2H), 2.44 (t, J = 10.7 Hz, 2H), 2.34 (s, 3H), 2.01 (d, J = 13.1 Hz, 2H), 1.80 (d, J = 4.5 Hz, 2H). 13C NMR (100 MHz, DMSO‑d6) δ 159.1, 158.1, 155.81 (s), 136.1, 133.7, 133.3, 130.4, 129.4, 125.1, 121.2, 120.0, 118.5, 74.9, 51.0, 46.3, 35.8, 34.3. MS m/z (%): 367.69 (M+, 18.63).
2.1.2.2. 2.1.2.2.N,N-Dimethyl-4-((E)-{(E)-[1′-methylspiro(chroman-2,4′-piperidin)-4-ylidene]hydrazono}methyl)aniline (4b)
White solid; (60%). M.p. 155−157 °C. IR (νmax/cm−1): 3060, 2940 (CHs), 1618 (C N), 1450, 1220, 1189. 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.16 (d, J = 6.8 Hz, 1H), 7.73 (d, J = 7.7 Hz, 2H), 7.34 (t, J = 7.7 Hz, 1H), 7.03 – 6.93 (m, 2H), 6.75 (d, J = 7.7 Hz, 2H), 3.19 (s, 2H), 3.07 (s, 6H), 2.61 (d, J = 11.4 Hz, 2H), 2.44 (t, J = 10.5 Hz, 2H), 2.34 (s, 3H), 2.00 (d, J = 13.0 Hz, 2H), 1.81 (d, J = 4.2 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 160.1, 157.2, 155.2, 152.3, 132.0, 130.1, 125.0, 122.4, 120.9, 120.5, 118.0, 111.7, 74.0, 51.0, 45.9, 40.2, 36.1, 34.2.MS m/z (%): 376.99 (M+, 19.99).
2.1.2.3. 4-((E)-{(E)-[1′-Ethylspiro(chroman-2,4′-piperidin)-4-ylidene]hydrazono}methyl)-N,N-dimethylaniline (4c)
White solid; (58%). M.p. 110−112 °C. IR (νmax/cm−1): 3058, 2945 (CHs), 1620 (C N), 1450, 1225, 1110. 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.18 (d, J = 7.7 Hz, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.38 – 7.32 (m, 1H), 7.00 (t, J = 7.3 Hz, 1H), 6.94 (d, J = 8.1 Hz, 1H), 6.75 (d, J = 8.5 Hz, 2H), 3.23 (s, 2H), 3.07 (s, 6H), 2.97 – 2.90 (m, 2H), 2.72 – 2.64 (m, 4H), 2.06 (s, 4H), 1.25 (t, J = 7.2 Hz, 3H). MS m/z (%): 390.45 (M+, 26.70).
4-((E)-{(E)-[1′-Methylspiro(chroman-2,4′-piperidin)-4-ylidene]hydrazono}methyl)phenol (4d).
White solid; (52%). M.p. 166−168 °C. IR (νmax/cm−1): 3538 (OH), 3064, 2947 (CHs), 1609 (C N), 1513, 1457, 1160. 1H NMR (400 MHz, CDCl3) δ 8.48 (s, 1H), 8.16 (d, J = 7.4 Hz, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.36 (t, J = 7.2 Hz, 1H), 7.04 – 6.93 (m, 2H), 6.86 (d, J = 8.4 Hz, 2H), 3.15 (s, 2H), 3.00 (s, 1H), 2.77 (d, J = 11.1 Hz, 2H), 2.55 (t, J = 11.4 Hz, 2H), 2.42 (s, 3H), 2.03 (d, J = 13.3 Hz, 2H), 1.82 (t, J = 11.2 Hz, 2H). MS m/z (%): 349.34 (M+, 25.21).
4-((E)-{(E)-[1′-Ethylspiro(chroman-2,4′-piperidin)-4-ylidene]hydrazon}methyl)phenol (4e).
White solid; (50%). M.p. 135−137 °C. 3540 (OH), 3064, 2940 (CHs), 1612 (C N), 1513, 1457, 1180.1H NMR (400 MHz, CDCl3) δ 8.46 (s, 1H), 8.19 – 8.12 (m, 1H), 7.71 (d, J = 8.4 Hz, 2H), 7.38 – 7.32 (m, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.87 (d, J = 8.4 Hz, 2H), 5.02 (s, 1H), 3.15 (s, 2H), 2.92 (d, J = 11.3 Hz, 2H), 2.69 – 2.52 (m, 4H), 2.09 – 2.00 (m, 2H), 1.87 (dd, J = 19.4, 6.8 Hz, 2H), 1.21 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 160.4, 159.5, 157.9, 155.2, 132.3, 130.5, 126.1, 125.2, 121.1, 120.2, 117.9, 116.4, 74.2, 52.2, 48.1, 36.3, 33.4, 11.3.MS m/z (%): 363.80 (M+, 15.63).
2.1.3. General procedure for preparation of hydrazone derivatives 5a-j
The appropriate phenyl isocyanate derivative (2.0 mmol) was added to a solution of the hydrazone derivatives 3a,b (2.0 mmol) in THF (20 ml). The reaction mixture was stirred overnight at room temperature. After reaction completion was confirmed by thin layer chromatography, the reaction mixture was poured onto ice. The resulting solids were separated and recrystallized from ethanol.
2.1.3.1. (E)-2-(1′-Methylspirochroman-2,4′-piperidin-4-ylidene)-N-phenyl hydrazine carboxamide (5a)
White solid; (73%). M.p. 233−235 °C. IR (νmax/cm−1): 3368, 3203 (NHs), 3104, 2936 (CHs), 1679 (C O), 1597, 1538, 1128. 1H NMR (400 MHz, DMSO‑d6) δ 9.93 (s, 1H), 8.95 (s, 1H), 8.28 (d, J = 6.9 Hz, 1H), 7.66 (d, J = 7.9 Hz, 2H), 7.35 – 7.26 (m, 3H), 7.04 (t, J = 7.4 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 2.85 (s, 2H), 2.47 (d, J = 11.4 Hz, 2H), 2.32 (t, J = 9.5 Hz, 2H), 2.21 (s, 3H), 1.79 (d, J = 13.6 Hz, 2H), 1.64 (t, J = 9.7 Hz, 2H). 13C NMR (100 MHz, DMSO‑d6) δ 154.2, 154.0, 140.3, 139.5, 131.3, 128.9, 125.4, 123.1, 121.2, 120.6, 120.5, 118.2, 74.2, 50.9, 46.2, 34.6, 34.5.MS m/z (%): 364.25 (M+, 20.73).
(E)-2-(1′-Ethylspirochroman-2,4′-piperidin-4-ylidene)-N-phenyl hydrazine carboxamide (5b).
White solid; (69%). M.p. 228−230 °C. IR (νmax/cm−1): 3365, 3220 (NHs), 3110, 2930 (CHs), 1680 (C O), 1597, 1538, 1120. 1H NMR (400 MHz, DMSO‑d6) δ 9.93 (s, 1H), 8.95 (s, 1H), 8.28 (d, J = 7.8 Hz, 1H), 7.66 (d, J = 7.9 Hz, 2H), 7.37 – 7.27 (m, 3H), 7.05 (t, J = 7.3 Hz, 1H), 6.98 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.1 Hz, 1H), 2.85 (s, 2H), 2.56 (s, 2H), 2.37 (q, J = 7.1 Hz, 2H), 2.34 – 2.29 (m, 2H), 1.79 (d, J = 13.8 Hz, 2H), 1.63 (t, J = 9.9 Hz, 2H), 1.02 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO‑d6) δ 154.3, 154.0, 140.4, 139.5, 131.3, 128.9, 125.3, 123.1, 121.2, 120.7, 120.5, 118.2, 74.8, 51.9, 48.5, 34.7, 34.6, 12.7.MS m/z (%): 378.60 (M+, 13.65).
2.1.3.2. (E)-N-(4-Chlorophenyl)-2-(1′-methylspirochroman-2,4′-piperidin)-4-ylidene hydrazine carboxamide (5c)
White solid; (78%). M.p. 250−252 °C. IR (νmax/cm−1): 3368, 3205 (NHs), 3110, 2938 (CHs), 1678 (C O), 1590, 1200, 1128. 1H NMR (400 MHz, DMSO‑d6) δ 10.01 (s, 1H), 9.09 (s, 1H), 8.30 (d, J = 7.0 Hz, 1H), 7.73 (d, J = 8.8 Hz, 2H), 7.37 (d, J = 8.8 Hz, 2H), 7.29 (t, J = 7.0 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 6.90 (d, J = 8.2 Hz, 1H), 2.85 (s, 2H), 2.47 (d, J = 11.6 Hz, 2H), 2.32 (t, J = 9.4 Hz, 2H), 2.21 (s, 3H), 1.81 – 1.74 (m, 2H), 1.64 (t, J = 9.7 Hz, 2H). 13C NMR (100 MHz, DMSO‑d6) δ 154.3, 154.0, 140.7, 138.6, 131.5, 128.8, 126.6, 125.5, 122.1, 121.2, 120.5, 118.2, 74.2, 50.9, 46.2, 34.5, 34.4.MS m/z (%): 398.82 (M+, 25.63).
2.1.3.3. (E)-N-(4-Chlorophenyl)-2-(1′-ethylspirochroman-2,4′-piperidin-4-ylidene)hydrazine carboxamide (5d)
White solid; (74%). M.p. 242−244 °C. IR (νmax/cm−1): 3360, 3203 (NHs), 3104, 2940 (CHs), 1679 (C O), 1597, 1530, 1128. 1H NMR (400 MHz, DMSO‑d6) δ 10.01 (s, 1H), 9.09 (s, 1H), 8.29 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 8.7 Hz, 2H), 7.37 (d, J = 8.7 Hz, 2H), 7.29 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.0 Hz, 1H), 2.85 (s, 2H), 2.53 (s, 2H), 2.37 (q, J = 7.1 Hz, 2H), 2.34 – 2.29 (m, 2H), 1.78 (d, J = 12.9 Hz, 2H), 1.63 (t, J = 7.1 Hz, 2H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, DMSO‑d6) δ 154.2, 153.9, 140.7, 138.6, 131.4, 128.8, 126.6, 125.6, 122.2, 121.1, 120.6, 118.2, 74.8, 52.0, 48.5, 34.6, 34.5, 12.8.MS m/z (%): 412.75 (M+, 27.73).
2.1.3.4. (E)-2-(1′-Methylspirochroman-2,4′-piperidin-4-ylidene)-N-(4-nitrophenyl)hydrazine carboxamide (5e)
White solid; (61%). M.p. 270−272 °C. IR (νmax/cm−1): 3370, 3203 (NHs), 3109, 2930 (CHs), 1679 (C O), 1597, 1500, 1128. 1H NMR (400 MHz, DMSO‑d6) δ 10.27 (s, 1H), 9.58 (s, 1H), 8.30 (d, J = 7.1 Hz, 1H), 8.24 (d, J = 9.2 Hz, 2H), 8.01 (d, J = 9.2 Hz, 2H), 7.37 – 7.27 (m, 1H), 7.00 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 8.1 Hz, 1H), 2.87 (s, 2H), 2.47 (d, J = 11.3 Hz, 2H), 2.32 (t, J = 9.5 Hz, 2H), 2.21 (s, 3H), 1.79 (d, J = 14.4 Hz, 2H), 1.65 (t, J = 7.1 Hz, 2H). 13C NMR (100 MHz, DMSO‑d6) δ 154.4, 153.6, 146.2, 142.1, 141.7, 131.7, 125.6, 125.2, 121.2, 120.4, 119.6, 118.2, 74.2, 50.9, 46.2, 34.5, 29.5.MS m/z (%): 409.23 (M+, 15.69).
2.1.3.5. (E)-2-(1′-Ethylspirochroman-2,4′-piperidin-4-ylidene)-N-(4-nitrophenyl)hydrazine carboxamide (5f)
White solid; (63%). M.p. 247−249 °C. IR (νmax/cm−1): 3346, 3201 (NHs), 3108, 2939 (CHs), 1693 (C O), 1603, 1543, 1117. 1H NMR (400 MHz, DMSO‑d6) δ 10.26 (s, 1H), 9.58 (s, 1H), 8.30 (d, J = 7.8 Hz, 1H), 8.24 (d, J = 8.9 Hz, 2H), 8.01 (d, J = 8.9 Hz, 2H), 7.31 (t, J = 7.4 Hz, 1H), 7.00 (t, J = 7.4 Hz, 1H), 6.90 (d, J = 8.1 Hz, 1H), 2.87 (s, 2H), 2.53 (s, 2H), 2.37 (q, J = 7.1 Hz, 2H), 2.34 – 2.29 (m, 2H), 1.79 (d, J = 14.4 Hz, 2H), 1.63 (t, J = 9.7 Hz, 2H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO‑d6) δ 154.4, 153.6, 146.2, 142.1, 141.7, 131.6, 125.6, 125.2, 121.2, 120.4, 119.6, 118.2, 74.8, 52.0, 48.5, 34.6, 34.5, 12.8.MS m/z (%): 423.19 (M+, 22.65).
2.1.3.6. (E)-N-(4-Ethoxyphenyl)-2-(1′-methylspirochroman-2,4′-piperidin-4-ylidene)hydrazine carboxamide (5g)
White solid; (83%). M.p. 220−222 °C. IR (νmax/cm−1): 3375, 3210 (NHs), 3104, 2935 (CHs), 1679 (C O), 1597, 1210, 1130. 1H NMR (400 MHz, DMSO‑d6) δ 9.85 (s, 1H), 8.85 (s, 1H), 8.30 (dd, J = 7.9, 1.4 Hz, 1H), 7.53 (d, J = 9.0 Hz, 2H), 7.33 – 7.23 (m, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.92 – 6.88 (m, 3H), 3.75 (s, 3H), 2.84 (s, 2H), 2.50 – 2.40 (m, 2H), 2.32 (t, J = 9.3 Hz, 2H), 2.21 (s, 3H), 1.78 (d, J = 13.5 Hz, 2H), 1.65 (t, J = 9.3 Hz, 2H). 13C NMR (100 MHz, DMSO‑d6) δ 155.5, 154.3, 154.1, 139.9, 132.4, 131.2, 125.4, 122.7, 121.2, 120.7, 118.1, 114.1, 74.1, 55.7, 50.9, 46.2, 34.6, 34.5.MS m/z (%): 394.88 (M+, 34.49).
2.1.3.7. (E)-2-(1′-Ethylspirochroman-2,4′-piperidin-4-ylidene)-N-(4-methoxyphenyl)hydrazine carboxamide (5h)
White solid; (85%). M.p. 230−232 °C. IR (νmax/cm−1): 3375, 3203 (NHs), 3100, 2940 (CHs), 1678 (C O), 1597, 1540, 1108. 1H NMR (400 MHz, DMSO‑d6) δ 9.85 (s, 1H), 8.85 (s, 1H), 8.30 (d, J = 6.8 Hz, 1H), 7.53 (d, J = 8.9 Hz, 2H), 7.27 (dd, J = 11.1, 4.2 Hz, 1H), 6.96 (t, J = 7.6 Hz, 1H), 6.92 – 6.88 (m, 3H), 3.75 (s, 3H), 2.84 (s, 2H), 2.53 (s, 2H), 2.37 (q, J = 7.1 Hz, 2H), 2.34 – 2.29 (m, 2H), 1.79 (d, J = 13.5 Hz, 2H), 1.63 (t, J = 9.8 Hz, 2H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO‑d6) δ 155.5, 154.3, 154.2, 139.9, 132.4, 131.2, 125.4, 122.7, 121.1, 120.7, 118.1, 114.1, 74.7, 55.7, 52.0, 48.5, 34.6, 34.5, 12.8.MS m/z (%): 408.95 (M+, 13.06).
2.1.3.8. (E)-2-(1′-Methylspirochroman-2,4′-piperidin-4-ylidene)-N-(4-methyl phenyl)hydrazine carboxamide (5i)
White solid; (77%). M.p. 248−250 °C. IR (νmax/cm−1): 3365, 3213 (NHs), 3104, 2940 (CHs), 1675 (C O), 1597, 1530, 1128. 1H NMR (400 MHz, DMSO‑d6) δ 9.89 (s, 1H), 8.87 (s, 1H), 8.28 (d, J = 7.8 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.29 (t, J = 7.6 Hz, 1H), 7.13 (d, J = 8.2 Hz, 2H), 6.97 (t, J = 7.5 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 2.84 (s, 2H), 2.47 (d, J = 11.0 Hz, 2H), 2.33 (d, J = 9.9 Hz, 2H), 2.28 (s, 3H), 2.21 (s, 3H), 1.78 (d, J = 13.3 Hz, 2H), 1.64 (t, J = 9.9 Hz, 2H). 13C NMR (100 MHz, DMSO‑d6) δ 154.2, 154.0, 140.1, 136.9, 131.9, 131.3, 129.3, 125.4, 121.2, 120.7, 120.6, 118.2, 74.1, 50.9, 46.2, 34.5, 34.5, 20.9.MS m/z (%): 378.45 (M+, 19.55).
2.1.3.9. (E)-2-(1′-Ethylspirochroman-2,4′-piperidin-4-ylidene)-N-(4-methyl phenyl)hydrazine carboxamide (5j)
White solid; (75%). M.p. 223−225 °C. IR (νmax/cm−1): 3340, 3211 (NHs), 3104, 2900 (CHs), 1679 (C O), 1597, 1538, 1156. 1H NMR (400 MHz, DMSO‑d6) δ 9.90 (s, 1H), 8.88 (s, 1H), 8.28 (d, J = 6.9 Hz, 1H), 7.54 (d, J = 8.3 Hz, 2H), 7.28 (dd, J = 11.2, 4.2 Hz, 1H), 7.11 (d, J = 8.3 Hz, 2H), 6.97 (t, J = 7.4 Hz, 1H), 6.89 (d, J = 8.1 Hz, 1H), 2.84 (s, 2H), 2.53 (s, 2H), 2.37 (q, J = 7.1 Hz, 2H), 2.34 – 2.29 (m, 2H), 2.28 (s, 3H), 1.79 (d, J = 13.1 Hz, 2H), 1.64 (d, J = 9.8 Hz, 2H), 1.01 (t, J = 7.1 Hz, 3H). 13C NMR (100 MHz, DMSO‑d6) δ 154.2, 154.0, 140.1, 136.9, 131.9, 131.3, 129.3, 125.4, 121.2, 120.6, 118.7, 118.2, 74.7, 52.0, 48.5, 34.6, 34.5, 20.9, 12.8.MS m/z (%): 392.30 (M+, 26.05).
2.2. Biological evaluation
2.2.1. In vitro antiproliferative study against MCF-7, HCT-116, MCF-7, PC3 and A549 cell lines
The MTT assay was performed to evaluate the in vitro antiproliferative activity of the newly synthesized compounds according to the reported method using MCF-7, HCT-116, MCF-7, PC3 and A549 cancer cell lines (Denizot and Lang, 1986).
2.2.2. In vitro cytotoxic activity of all synthesized compounds against WI-38 cell line
The cytotoxicity of compounds 5a and 5 g was estimated according to the reported procedure (El-Azab et al., 2023, Al-Sanea et al., 2023).
2.2.3. In vitro enzyme inhibitory assays (against EGFR and Her-2)
Enzyme inhibitory assays for the most active compounds 5a,5b and 5 g were performed as described in the previous reports (El-Azab et al., 2020, Abdel-Aziz et al., 2021).
2.2.4. Flow cytometry analysis of the cell cycle distribution
Cell cycle analysis was performed using different cancer cell lines stained with propidium iodide (PI) and FACS Calibur flow cytometer as previously described (Ormerod, 2002, Mosmann, 1983, Othman et al., 2023).
2.2.5. Cellular apoptosis analysis
Apoptosis was induced using an annexin 5-FITC/PI detection kit, as previously reported (Vermes et al., 1995, Kumar et al., 2021, Hamdi et al., 2022a, Hamdi et al., 2022b, Al-Warhi et al., 2020).
2.3. Molecular docking and ADME methodology
Molecular docking methodology was performed using a well-established and reported protocols (Hamdi et al., 2022a, Hamdi et al., 2022b, Al-Suwaidan et al., 2015, Goda et al., 2005, El-Ayaan et al., 2007). ADME online tools were used to predict the pharmacokinetic, physicochemical, and drug-likeness properties of the target compounds (Lipinski et al., 2001, Hughes et al., 2008, Gleeson, 2008, Johnson et al., 2009).
3. Results
3.1. Chemistry
Scheme 1 outlines the synthesis of the targeted spirochromane compounds 4a-e and 5a-j. The starting materials, spirochromanones2a,b, were prepared using Kabbe's multicomponent reaction, which involved the thermal condensation of N-methylpiperidin-4-one or N-ethylpiperidin-4-onewith 2-hydroxyacetophenone, and pyrrolidine in methanol (Battisti et al., 2014, Abdelatef et al., 2018).Compounds 2a and 2b were reacted with hydrazine hydrate in ethanol at room temperature to afford the corresponding hydrazineylidene spirochromane intermediates 3a and 3b, which were used directly in the subsequent steps without further purification. The final Schiff's bases 4a-e were prepared by refluxing derivatives 3a and b with the appropriate aldehyde in ethanol, using glacial acetic acid as a catalyst. Finally, compounds 5a-j were obtained by coupling the properhydrazineylidenespirochromane 3a,b with various phenyl isocyanates to furnish them with reliable yields. All synthesized structures of the final novel spirochromane compounds were confirmed by NMR, IR and MS (Supplementary data). Overall, the 1HNMR spectra of all the final newderivatives revealed the disappearance of two amine protons related to the NH2 group of hydrazineylidenespirochromane derivatives 3a and 3b, which usually appeared as a singlet peak near 5.31 ppm. The 1HNMR spectra of Schiff's bases 4a-e showed a singlet proton around 8.48 ppm relative to the benzylidene proton (—CH N—). Also, derivatives 5a-j showed the appearance of two singlet protons near 8.95 and 9.93 ppm, assigned to the two protons of urea (HN-CO-NH) moiety.
Scheme 1.

Synthesis of the target spirochromanes 4a-e and 5a-j.
Moreover, the 13CNMRspectra of products 4a-e and 5a-j showed characteristic aliphatic peaks near 75.0, 51.0 and 36.0 ppm, corresponding to methylene groups of the spirochromane system. Besides, compounds 5a-j exhibited a distinct peak at approximately 154.0 ppmassigned to the carbonyl group of urea fragment. IR spectrum revealed characteristic peaks near 3300 cm−1 regions relative to the amino groups of urea moiety.
3.2. Biological activity
3.2.1. In vitro antiproliferative activity
The newly synthesized compounds 4a-e and 5a-j were screened for their antiproliferative activity against breast adenocarcinoma (MCF-7), human colon carcinoma (HCT-116), human prostate cancer (PC3) and human lung adenocarcinoma (A549) cells at various concentrations via the standard MTT assay method using Doxorubicin (DOX) and Afatinib as reference drugs (Table 1) (Denizot and Lang, 1986, Hamdi et al., 2022a, Hamdi et al., 2022b). Among the tested compounds, derivatives 4a and 5a-c showed the most potent antiproliferative activity against the four cancer cell lines with an IC50 range of 1.673–13.47 µM. Besides, compounds 4d and 5g showed strong antiproliferative activity against MCF-7, HCT-116, and PC3 (IC50 values range of 1.154–10.08 μM) and moderate activity against A549 with IC50 values of 23.20 and 29.36 μM, respectively.
Table 1.
In vitro antiproliferative study (IC50 values µM) of the final target compounds 4a-e and 5a-j against four cancer cell lines and WI38 normal cell line, in comparison with DOX and Afatinib.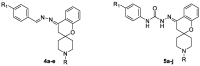
| Comp. No. | R | R1 |
In vitro Cytotoxicity IC50 (µM)a |
||||
|---|---|---|---|---|---|---|---|
| MCF7 | HCT-116 | PC3 | A549 | WI38 | |||
| 4a | CH3 | Cl | 8.74 ± 0.42 | 13.47 ± 0.57 | 12.71 ± 0.49 | 2.026 ± 0.09 | 75.71 ± 4.3 |
| 4b | CH3 | N(CH3)2 | 26.43 ± 1.27 | 31.02 ± 1.32 | 59.25 ± 2.28 | 42.54 ± 1.91 | 60.35 ± 3.7 |
| 4c | C2H5 | N(CH3)2 | 79.98 ± 3.83 | 56.20 ± 2.39 | 55.04 ± 2.12 | 48.53 ± 2.18 | 55.30 ± 3.3 |
| 4d | CH3 | OH | 10.08 ± 0.48 | 2.68 ± 0.11 | 2.371 ± 0.09 | 23.20 ± 1.04 | 33.26 ± 2.3 |
| 4e | C2H5 | OH | 35.55 ± 1.51 | 49.45 ± 2.1 | 22.2 ± 0.86 | 20.62 ± 0.93 | 47.53 ± 2.9 |
| 5a | CH3 | H | 1.673 ± 0.07 | 5.602 ± 0.24 | 12.33 ± 0.48 | 5.827 ± 0.26 | 49.62 ± 3.0 |
| 5b | C2H5 | H | 6.033 ± 0.27 | 2.991 ± 0.13 | 9.09 ± 0.35 | 1.904 ± 0.09 | 65.6 ± 2.7 |
| 5c | CH3 | Cl | 2.904 ± 0.11 | 2.575 ± 0.13 | 2.56 ± 0.13 | 8.029 ± 0.36 | 100 |
| 5d | C2H5 | Cl | 23.58 ± 0.92 | 21.74 ± 0.88 | 27.22 ± 1.05 | 47.67 ± 2.14 | 100 |
| 5e | CH3 | NO2 | 10.85 ± 0.42 | 20.74 ± 0.88 | 19.15 ± 0.74 | 71.46 ± 3.21 | 67.22 ± 3.9 |
| 5f | C2H5 | NO2 | 56.79 ± 2.21 | 62.74 ± 2.67 | 11.33 ± 0.44 | 21.73 ± 0.98 | 57.46 ± 2.4 |
| 5 g | CH3 | OCH3 | 6.674 ± 0.28 | 1.154 ± 0.05 | 3.302 ± 0.13 | 29.36 ± 1.32 | 72.93 ± 4.2 |
| 5 h | C2H5 | OCH3 | 38.95 ± 1.72 | 19.74 ± 0.84 | 15.51 ± 0.6 | 24.05 ± 1.08 | 83.47 ± 4.6 |
| 5i | CH3 | CH3 | 21.43 ± 0.91 | 7.75 ± 0.33 | 9.71 ± 0.37 | 5.129 ± 0.23 | 66.27 ± 3.7 |
| 5j | C2H5 | CH3 | 18.99 ± 0.84 | 21.31 ± 0.91 | 5.47 ± 0.21 | 11.91 ± 0.53 | 100 |
| Afatinib | – | – | 5.10 ± 0.31 | 10.01 ± 0.47 | 7.98 ± 0.37 | 8.09 ± 0.69 | 49.50 ± 2.5 |
| DOX | – | – | 4.50 ± 0.2 | 5.23 ± 0.3 | 4.17 ± 0.2 | 5.57 ± 0.4 | 6.72 ± 0.5 |
IC50 value is the concentration of compound that inhibits 50% of the cancer cell growth after 48 h of drug exposure as obtained from the MTT assay. Each value was shown as mean ± SD of three experiments. Bolded values represent the most potent antiproliferative derivatives.
In derivatives 4a-e, compound 4b exhibited moderate activity against MCF-7, HCT-116 and A549 with IC50 values of 26.43, 31.02, and 42.54 μM, respectively. However, a weak PC3 antiproliferative activity with IC50 of 59.25 μM was observed. In addition, compound 4e exerted moderate antiproliferative activity against the four cancer cell lines with an IC50 range of 20.62–49.45 µM. Derivative 4c possessed moderate A549 antiproliferative activity with an IC50 value of 48.53 μM and weak activity against all other cancer cell lines. Among the 5a-j series, compound 5i showedextreme activity against HCT-116, PC3 and A549 with IC50 values of 7.75, 9.71 and 5.12 μM, respectively, whereas it showed moderate effect against MCF-7 with IC50 values of 21.43 μM. Also, derivative 5j revealed extreme activity against PC3 with an IC50 value of 5.47 µM and potent activity against A549, MCF-7 and HCT-116 with IC50 values of 11.91, 18.99 and 21.31 µM in succession.Compound 5d exhibited moderate antiproliferative activity against the four cancer cell lines with an IC50 range of 21.74–47.67 µM, while compound 5e offered moderate activity against MCF-7, HCT-116 and PC3 (IC50 range of 10.85–20.74 µM) and weak inhibitory effect against A549. Besides, compound 5f exerted strong cytotoxic activity against PC3 (IC50 = 11.33 µM) and A549 (IC50 = 21.73 µM) but showed weak activity against MCF-7 (IC50 = 56.79 µM) and HCT-116 (IC50 = 62.74 µM) cells. Compound 5 h revealed vigorous activity against PC3 and HCT-116 cells with IC50 values of 15.51 and 19.74 µM in succession but showed moderate activity against A549 and MCF-7 with IC50 values of 24.05 and 38.95 µM, respectively.
3.2.2. In vitro cytotoxicity against normal human cell
A normal fibroblast-like fetal lung cell line (WI-38) was used further to investigate the therapeutic safety of the newly synthesized hybrids and evaluate their selective cytotoxicity displayed toward normal and tumour cells (El-Azab et al., 2023). DOX and Afatinib were used as the standard anticancer drugs for comparison (Table 1).
3.2.3. In vitro enzyme inhibition assays
The effect of the potent compounds 5a, 5b and 5 g was screened against two molecular targets, EGFR and HER2 (El-Azab et al., 2020). These compounds showed reasonable inhibitory activity against both EGFR and HER-2 compared with the reference drugs, as shown in Table 2.
Table 2.
In vitro EGFR-2, and Her-2 inhibitory effects of the synthesized compounds 5a, 5b and 5 g.
| Comp. No. | IC50 (µM)a |
|
|---|---|---|
| EGFR Inhibition |
Her2 Inhibition |
|
| 5a | 0.116 ± 0.005 | 0.055 ± 0.003 |
| 5b | 0.132 ± 0.006 | 0.21 ± 0.01 |
| 5 g | 0.077 ± 0.004 | 0.085 ± 0.004 |
| Erlotinib | 0.09 ± 1.65 | 0.038 ± 0.002 |
| Gefitinib | 0.052 ± 0.78 | 0.072 ± 0.98 |
IC50 value is the compound concentration required to produce 50% inhibition.
3.2.4. Cell cycle analysis
To obtain more detailed information regarding the mechanism of compounds 5a, 5b and 5g regarding cancer cell growth inhibition, cell cycle distribution analysis and apoptosis induction (Fig. 3 and Table 3) were assessed using a propidium iodide (PI) staining assay (Ormerod, 2002, Mosmann, 1983, Othman et al., 2023).
Fig. 3.

Flow cytometry analysis of DNA ploidy in different cancer cells after treatment with compounds 5a, 5b and 5g.
Table 3.
Effect of compounds 5a, 5b and 5 g on the cell cycle distribution in different cancer cell lines.
| Comp. No. |
Cell cycle distribution (%) |
||
|---|---|---|---|
| %G0-G1 | %S | %G2/M | |
| 5a/MCF7 | 46.35 | 43.25 | 10.4 |
| Cont.MCF7 | 55.49 | 25.94 | 18.57 |
| 5b/A549 | 56.19 | 37.26 | 6.55 |
| Cont. A549 | 61.06 | 28.44 | 10.5 |
| 5 g/HCT-116 | 47.83 | 42.66 | 9.51 |
| Cont. HCT-116 | 43.82 | 35.17 | 21.01 |
3.2.5. Apoptosis detection
The percentage of apoptosis induced by compounds 5a, 5b and 5 g was evaluated using an Annexin V-FITC/propidium iodide double-staining flow cytometry assay (Vermes et al., 1995, Kumar et al., 2021, Hamdi et al., 2022a, Hamdi et al., 2022b) (Fig. 4 and Table 4).
Fig. 4.

Effect of compounds 5a, 5b and 5g on the percentage of Annexin V-FITC-positive staining. The cells were treated with DMSO as a control, 5a, 5b and 5g for 24 h.
Table 4.
Total apoptosis (early, late) and necrosis induction analysis induced by compounds 5a, 5b and 5g, compared to control.
| Comp. No. |
Apoptosis |
Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 5a/MCF7 | 36.17 | 21.66 | 14.51 | 2.74 |
| Cont.MCF7 | 0.62 | 0.41 | 0.21 | 1.21 |
| 5b/A549 | 39.68 | 28.17 | 11.51 | 2.93 |
| Cont. A549 | 0.71 | 0.58 | 0.13 | 1.76 |
| 5g/HCT-116 | 47.31 | 31.02 | 16.29 | 3.72 |
| Cont. HCT-116 | 0.63 | 0.34 | 0.29 | 1.56 |
3.3. In silico study
Molecular modeling is an important method for studying the physicochemical, pharmacokinetic and biological activities of bioactive molecules (Goda et al., 2005, El-Ayaan et al., 2007). In addition, it is used to explore the interactionsof ligands within the receptor- or putative enzyme-binding sites (Al-Suwaidan et al., 2015, Alanazi et al., 2016). The selected compounds and co-crystallized inhibitors were subjected to molecular docking at the protein-binding sites to ensure docking accuracy and validation (Hamdi et al., 2022a, Hamdi et al., 2022b, Abuelizz et al., 2023).
3.3.1. Molecular docking of compounds 5a, 5b and 5 g with EGFR and HER2
Compounds 5a, 5b and 5g were dockedinto the binding siteof the EGFR, along with a co-crystallized bound inhibitor, to investigate the orientation and binding affinity of these compounds to the allosteric site of EGFR (Fig. 5, Fig. S1 and Table S1). The crystal 3D structure of EGFR and its bound inhibitor were obtained from PDB (PDB code: 7jxq) (Nicholson et al., 2001). Similarly, the 3D crystal structure of HER2 co-crystallized with its bound inhibitor was retrieved from PDB (PDB code: 7JXH) (Fig. 6, Fig. S2 and Table S2) (Son et al., 2022).
Fig. 5.

The 3D interaction profiles and best interaction poses of investigated compounds placed into the EGFR (7jxq) by docking study: co-crystallized inhibitor (upper left panel), 5a (upper right panel), 5b (lower left panel), and 5g (lower right panel).
Fig. 6.

The 3D interaction profiles and best interaction poses of investigated compounds placed into the Her2 (PDB ID: 7jxh) by docking study: co-crystallized inhibitor (upper left panel), 5a (upper right panel), 5b (lower left panel) and 5g (lower right panel).
3.3.2. Pharmacokinetic and physicochemical predictions
The pharmacokinetic and physicochemical properties of the most active compounds 5a, 5b and 5 g were predicted using the automated ADMETlab 2.0 web server (https://admetmesh.scbdd.com/) online calculation system (Table S3). Compounds 5a, 5b and 5 g demonstrated varying characteristics based on the drug-likeness and ADMET properties evaluation (Lipinski et al., 2001, Hughes et al., 2008, Gleeson, 2008, Johnson et al., 2009).
4. Discussion
Structure-activity relationship (SAR) analysis of the antiproliferative activity of the newly synthesized compounds against MCF-7, HCT-116, PC3 and A549 cells generally revealed that 1′-methyl-4-hydrazono spirochromane-2,4′-piperidines (4a, 4b, 4d, 5a, 5c, 5e, 5 g and 5i) showed improved activity compared to 1′-ethyl-4-hydrazono spirochromane-2,4′-piperidines (4c, 4e, 5b, 5d, 5f, 5 h and 5j). In addition, compounds 5a-j, with a hydrazine carboxamide moiety as a long linker, exhibited slightly better antiproliferative activity than arylidene incorporating derivatives 4a-e. Of the arylidene derivatives 4a-e, the best activity was from 4-chloro and 4-hydroxylbenzylidene derivatives 4a and 4d, which showed reliable antiproliferative activity against the four cancer cell lines with IC50 range of 2.02–23.20 μM. However, the less potent derivative among this series is ethyl piperidine derivative 4c, having 4-dimethylamino substituent with an IC50 range of 48.53–79.98 μM against all cancer cell lines.
As for the hydrazine carboxamide derivatives 5a-j, the results revealed that compounds containing unsubstituted phenyl moieties, such as compounds 5a and 5b, exhibited powerful antiproliferative activity against all tested cancer cell lines with IC50 range of 1.67–12.33 μM. Furthermore, the substitution of the phenyl moieties had a remarkable effect. Except for compound 5c, electron-donating groups notably enhanced the antiproliferative activity, as in compound 5 g substituted with a methoxy group and compound 5i substituted with amethyl group. These compounds exhibited activity ranging from very strong to moderate effect against all cancer cell lines (IC50 range of 1.154–29.36 μM). Besides, N-ethyl piperidine incorporating derivatives as compounds 5h and 5j, substituted with methoxy and methyl groups in success, showed remarkable antiproliferative activity against all tested cancer cell lines with IC50 values range of 5.47–38.95 μM.
In contrast, N-methylpiperidine derivative 5c with a 4-chloro phenyl fragment showed extreme antiproliferative activity against all tested cancer cell lines with an IC50 range of 2.575–8.029 μM. In contrast, N-ethylpiperidine derivative 5d with the same 4-chloro phenyl moiety displayed a moderate antiproliferative effect against the four cell lines with an IC50 range of 21.74–47.67 μM. Finally, N-methylpiperidine derivative 5e incorporating 4-nitro phenyl group exhibited a strong effect against MCF7, HCT-116, and PC3 with IC50 values of 10.85, 20.74 and 19.15 μM, respectively, while it showed a weak effect against A549 with IC50of 71.46 μM. Also, N-ethylpiperidine derivative 5f with nitrophenyl moiety exhibited strong activity against PC3 (IC50 = 11.33 μM) and A549 (IC50 = 21.73 μM), whereas it showed weak activity against the other two cell lines MCF7 (IC50 = 56.79 μM) and HCT-116 (IC50 = 62.74 μM).
As shown in Table 1, the investigated compounds exhibited lower cytotoxicity against normal fibroblast cells WI-38 as denoted from their IC50 values with the range of 33.26–100 μM, proving that they have substantially better results than DOX and Afatinib on normal cells. Notably, derivatives 4a, 4d, 5a, 5b, 5c, 5 g, 5i and 5j induced lower toxic effects on WI-38 cells, with IC50 values of 75.71, 33.26, 49.62, 65.6, 100, 72.93, 66.27 and 100 μM, respectively, compared with that by DOX (IC50 = 6.72 μM) and Afatinib (IC50 = 49.50 μM).
In enzyme inhibition assay, the newly synthesized spirochromane derivative 5a showed a vigorous inhibitory activity against HER2 (IC50 = 0.055 µM) compared with Gefitinib (IC50 = 0.072 µM). In addition, derivative 5 g showed more vigorous EGFR inhibitory activity (IC50 = 0.077 µM), compared to that of Erlotinib (IC50 = 0.09 µM) (Table 2).
On the other hand, cell cycle distribution results (Fig. 3 and Table 3) revealed that compound 5a induced apoptosis in the G0-G1 phase in 46.35% of MCF7 cells, whereas untreated cells showed 55.49% apoptosis. Also, derivative 5b induced apoptosis in the G0-G1 phase by 56.19 % in A549 cells, whereas untreated cells showed 61.06 % apoptosis. In addition, compound 5 g induced apoptosis in the G0-G1 phase in 47.83% of HCT-116 cells, whereas untreated cells showed 43.82% apoptosis. In the S phase, derivative 5a showed a high apoptotic effect (43.25%) compared to the untreated MCF7 cells (25.94%). Compound5b exhibited 37.26% apoptotic effect in comparison with untreated A549 cells (28.44%). Moreover, derivative 5 g showed a strong apoptotic effect (42.66%) compared to the untreated HCT-116 cells (35.17%). In the G2/M phase, compounds 5a, 5b and 5 g showed 10.4%, 6.55 % and9.51% apoptotic effect, respectively, whereas control cells showed 18.59%, 10.5% and 21.01% inhibition. Thus, compounds 5a, 5b and 5g arrested cell growth inthe S phase.
According to apoptosis detection, as shown in Fig. 4 and Table 4, compounds 5a, 5b and 5 g induced early apoptosis after 24 h incubation by 21.66% (MCF-7 cells), 28.17 % (A549cells) and 31.02% (HCT-116cells), respectively, whereas the untreated cells induced early apoptosis by approximately 0.34–0.58%.
In addition, the tested compounds 5a, 5b and 5 g enhanced the late apoptotic induction by 14.51%, 11.51 and 16.29%, in success, compared with that in the untreated control (0.13–0.29%). Furthermore, derivatives 5a, 5b and 5g promoted cell necrosis by 2.74%, 2.93% and 3.72%, respectively, compared with untreated cells, which showed approximately 1.21–1.76%. Cumulatively, compounds 5a, 5b and 5 g enhanced the total apoptosis by 36.17%, 39.68% and 47.31 %, respectively, compared with untreated cell (0.62–0.71%). In addition,the data support an apoptotic mechanism underlying the programmed cell death induced by compounds 5a, 5b and 5 g rather than a necrotic pathway.
Molecular docking of compounds 5a, 5b and 5g with EGFR revealed that EGFR allosteric inhibitors bound to EGFR at a site away from the tyrosine kinase domain, which passed the cysteine797 mediated resistance mechanism, demonstrating a promising strategy to overcome JBJ-09–063 resistance induced by cysteine797s. The docking of the high-potency compound 5 g with the EGFR resulted in a binding affinity of − 9.61 kcal/mol. Compound 5 g interacted with the allosteric site of EGFR via four H-bond donors of the crucial amino acid residues ASP855, LEU788 and MET790, along with two π-H bonds with PHE856 and ILE759 (Fig. 5, Table S1). Whereas compound 5a has a binding affinity of −8.99 kcal/mol and forms four strong H-bond donors with crucial amino acid residues MET790, LEU788 and ASP855, as well as two π-H bonds with LEU777 and ILE759 (Fig. 5, Table S1). Additionally, compound 5b possesses a binding affinity of −9.00 kcal/mol and interacts through three H-bond with main residue MET790 in the allosteric site of EGFR, besides five π-bond with amino acid residues PHE856, ILE759, LEU777 and ASP855 (Fig. 5, Table S1). Furthermore, Figure S1 shows the alignment of the co-crystalline ligand with the investigated compounds (5a, 5b and 5 g),which interact with identical residues as the co-crystalline ligand in the allosteric site.
Similarly, molecular docking of compounds 5a, 5b and 5g to the HER2-binding cavity revealed that compounds 5g and 5a demonstrated high binding affinities, whereas compound 5b showed moderate binding affinity. The docking simulation implied that the urea group in all investigated molecules (5g, 5a and 5b) was superimposed on the isatin group of the bound inhibitor within the hydrophobic binding pocket of HER2 (Fig. 6, Table S2). Moreover, the carbonyl group on the urea part of molecules 5a, 5b and 5g formed hydrogen bonds with the curial amino acid residue MET 801, which supported that these compounds may inhibit the same target enzyme asa co-crystallized inhibitor. In addition, compound 5g made two H-bond donors with THR798 and ASP863 and an H-bond acceptor with MET 801, as well as three π-bonds with LEU726 and VAL734, while compound 5a formed two H-bonds with curial residues MET 801 and ASN 850, beside π-bond with ASP 863 (Fig. 6, Table S2). Compound 5b interacted through two H-bonds with the curial residues MET801 and LYS753, in addition to three π-bonds with GLY804, MET801 and LEU726. The binding modes of compounds 5a, 5b and 5g were similar to those of the co-crystallized bound inhibitor with HER2 kinase, as shown in the overlay protocol (Figure S2).
The pharmacokinetic and physicochemical properties of compound 5a show good drug-likeness, according to Lipinski, Pfizer, GSK, and Golden Triangle. It has moderate medicinal chemistry properties, limited absorption, high distribution, moderate metabolism, and moderate toxicity. In contrast,derivative 5b exhibited good drug-likeness according to the Lipinski Rule and Golden Triangle but was rejected by the Pfizer and GSK Rules. It demonstrates moderate medicinal chemistry properties, limited absorption, limited distribution, moderate metabolism, and moderate toxicity. Also, derivative 5g possesses good drug-likeness based on the Lipinski Rule, Pfizer Rule, and Golden Triangle. It exhibited moderate medicinal chemistry properties, limited absorption, low distribution, moderate metabolism, and moderate toxicity. Considering these factors, spirochromane derivative5a emerged as the best candidate owing to its favourable drug-likeness profile and moderate ADMET properties.The ADMET lab 2.0 web tool calculations of compounds 5a, 5b and 5g predicted that they possessed appropriate pharmacokinetic and physicochemical properties (Table S3).
5. Conclusions
Two new series of spirochromanes 4a-e and 5a-j were synthesized and their antiproliferative activities were evaluated using four human cancer cell lines, i.e., MCF-7, HCT-116, PC3 and A549. Compounds 5b and 5c exhibited broad spectrum activity against the MCF-7 (IC50 of 6.03 and 2.90 µM, respectively), HCT-116 (IC50 of 2.99 and 2.575 µM, respectively), PC3 (IC50 of 9.09 and 2.56 µM, respectively) and A549 (IC50 of 1.90 and 8.029 µM, respectively) cell lines. Furthermore, derivatives 4d and 5 g showed broad spectrum activity against the MCF-7 (IC50 of 10.08 and 6.67 µM, respectively), HCT-116 (IC50 of 2.68 and 1.154 µM, respectively) and PC3 (IC50 of 2.37 and 3.30 µM, respectively) cell lines. Moreover, compound5j selectively inhibited the PC3 cell line with an IC50 value of 5.47 µM, while the MCF-7and A549 cell lines were selectively inhibited by derivative 4a (IC50 of 8.74 and 2.026 µM, respectively). HCT-116, PC3 and A549 cell lines were strongly inhibited by derivative 5i (IC50 of 7.75, 9.71, and 5.129 µM, respectively). Moreover, compound 5a possessed potential antiproliferative activity against MCF-7, HCT-116 and A549 cell lines (IC50 of 1.67, 5.60 and 5.827 µM, respectively). The IC50 values of spirochromanes 4a-e and 5a-j were compared with those of the reference drugs Doxorubicin (IC50 range of 4.17–5.57 µM) and Afatinib (IC50 range of 5.10–10.01 µM).
The most active antiproliferative agents, 5a, 5b and 5g, potently inhibited EGFR (IC50 = 0.116, 0.132, and 0.077 μM, respectively) and HER2 (IC50 = 0.055, 0.21 and 0.085 μM, respectively). The results were compared to the references Gefitinib (IC50 of 0.05 µM) and Erlotinib (IC50 of 0.038 µM). Cell cycle analysis and apoptosis detection data showed that compounds 5a, 5b and 5g arrested cell growth inthe S phase, and the apoptotic mechanism was the main mechanism rather than the necrotic pathway. Docking studies of derivatives 5a, 5b and 5 g into the putative binding sites of EGFR and HER2 were conducted to study the mode of interaction and requirements for antiproliferative activity. ADMETlab and drug-likeness predictions showed that compounds 5a, 5b and 5g exhibited goodpharmacokinetic, physicochemical, and drug-likeness properties.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors extend their appreciation to the Deputyship for Research & Innovation, “Ministry of Education” in Saudi Arabia, for funding this research (IFKSUOR3-435-1).
Footnotes
Peer review under responsibility of King Saud University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jsps.2023.101803.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Abdelatef S.A., El-Saadi M.T., Amin N.H., Abdelazeem A.H., Omar H.A., Abdellatif K.R. Design, synthesis and anticancer evaluation of novel spirobenzo[h]chromene and spirochromane derivatives with dual EGFR and B-RAF inhibitory activities. Eur. J. Med. Chem. 2018;150:567–578. doi: 10.1016/j.ejmech.2018.03.001. [DOI] [PubMed] [Google Scholar]
- Abdel-Aziz A.-A.-M., El-Azab A.S., AlSaif N.A., Obaidullah A.J., Al-Obaid A.M., Al-Suwaidan I.A. Synthesis, potential anti-tumor activity, cell cycle analysis, and multitarget mechanisms of novel hydrazones incorporating a 4-methylsulfonylbenzene scaffold: a molecular docking study. J. Enzyme Inhib. Med. Chem. 2021;36(1):1521–1539. doi: 10.1080/14756366.2021.1924698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelsalam E.A., Zaghary W.A., Amin K.M., Abou Taleb N.A., Mekawey A.A.I., Eldehna W.M., Abdel-Aziz H.A., Hammad S.F. Synthesis and in vitro anticancer evaluation of some fused indazoles, quinazolines and quinolines as potential EGFR inhibitors. Bioorg. Chem. 2019;89 doi: 10.1016/j.bioorg.2019.102985. [DOI] [PubMed] [Google Scholar]
- Abuelizz H.A., Bakheit A.H., Marzouk M., El-Senousy W.M., Abdellatif M.M., Mostafa G.A.E., Al-Salahi R. Evaluation of some Benzo[g]Quinazoline derivatives as antiviral agents against human rotavirus wa strain: biological screening and docking study. Curr. Issues Mol. Biol. 2023;45(3):2409–2421. doi: 10.3390/cimb45030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanazi A.M., El-Azab A.S., Al-Swaidan I.A., Maarouf A.R., El-Bendary E.R., El-Enin M.A.A., Abdel-Aziz A.-A.-M. Synthesis, single-crystal, in vitro anti-tumor evaluation and molecular docking of 3-substitued 5,5-diphenylimidazolidine-2,4-dione derivatives. Med. Chem. Res. 2013;22(12):6129–6142. [Google Scholar]
- Alanazi A.M., Abdel-Aziz A.-A.-M., Shawer T.Z., Ayyad R.R., Al-Obaid A.M., Al-Agamy M.H., Maarouf A.R., El-Azab A.S. Synthesis, anti-tumor and antimicrobial activity of some new 6-methyl-3-phenyl-4(3H)-quinazolinone analogues: in silico studies. J. Enzyme Inhib. Med. Chem. 2016;31(5):721–735. doi: 10.3109/14756366.2015.1060482. [DOI] [PubMed] [Google Scholar]
- Alkahtani H.M., Abdalla A.N., Obaidullah A.J., Alanazi M.M., Almehizia A.A., Alanazi M.G., Ahmed A.Y., Alwassil O.I., Darwish H.W., Abdel-Aziz A.-A.-M., El-Azab A.S. Synthesis, cytotoxic evaluation, and molecular docking studies of novel quinazoline derivatives with benzenesulfonamide and anilide tails: Dual inhibitors of EGFR/HER2. Bioorg. Chem. 2020;95 doi: 10.1016/j.bioorg.2019.103461. [DOI] [PubMed] [Google Scholar]
- Al-Sanea M.M., Hamdi A., Mohamed A.A., El-Shafey H.W., Moustafa M., Elgazar A.A., Eldehna W.M., Ur Rahman H., Parambi D.G., Elbargisy R.M., Selim S. New benzothiazole hybrids as potential VEGFR-2 inhibitors: design, synthesis, anticancer evaluation, and in silico study. J. Enzyme Inhib. Med. Chem. 2023;38(1):2166036. doi: 10.1080/14756366.2023.2166036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Suwaidan I.A., Abdel-Aziz N.I., El-Azab A.S., El-Sayed M.A., Alanazi A.M., El-Ashmawy M.B., Abdel-Aziz A.-A.-M. Anti-tumor evaluation and molecular docking study of substituted 2-benzylidenebutane-1,3-dione, 2-hydrazonobutane-1,3-dione and trifluoromethyl-1H-pyrazole analogues. J. Enzyme Inhib. Med. Chem. 2015;30(4):679–687. doi: 10.3109/14756366.2014.960863. [DOI] [PubMed] [Google Scholar]
- Al-Suwaidan I.A., Abdel-Aziz A.-A.-M., Shawer T.Z., Ayyad R.R., Alanazi A.M., El-Morsy A.M., Mohamed M.A., Abdel-Aziz N.I., El-Sayed M.-A.-A., El-Azab A.S. Synthesis, anti-tumor activity and molecular docking study of some novel 3-benzyl-4(3H)quinazolinone analogues. J. Enzyme Inhib. Med. Chem. 2016;31(1):78–89. doi: 10.3109/14756366.2015.1004059. [DOI] [PubMed] [Google Scholar]
- Al-Warhi T., Abo-Ashour M.F., Almahli H., Alotaibi O.J., Al-Sanea M.M., Al-Ansary G.H., Ahmed H.Y., Elaasser M.M., Eldehna W.M., Abdel-Aziz H.A. Novel [(N-alkyl-3-indolylmethylene)hydrazono]oxindoles arrest cell cycle and induce cell apoptosis by inhibiting CDK2 and Bcl-2: synthesis, biological evaluation and in silico studies. J. Enzyme Inhib. Med. Chem. 2020;35(1):1300–1309. doi: 10.1080/14756366.2020.1773814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonello A., Tarozzi A., Morroni F., Cavalli A., Rosini M., Hrelia P., Bolognesi M.L., Melchiorre C. Multitarget-directed drug design strategy: a novel molecule designed to block epidermal growth factor receptor (EGFR) and to exert proapoptotic effects. J. Med. Chem. 2006;49(23):6642–6645. doi: 10.1021/jm0608762. [DOI] [PubMed] [Google Scholar]
- Baker S.J., Reddy E.P. Targeted inhibition of kinases in cancer therapy. Mt Sinai J. Med. 2010;77(6):573–586. doi: 10.1002/msj.20220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker A.J., Gibson K.H., Grundy W., Godfrey A.A., Barlow J.J., Healy M.P., Woodburn J.R., Ashton S.E., Curry B.J., Scarlett L. Studies leading to the identification of ZD1839 (Iressa™): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001;11:1911–1914. doi: 10.1016/s0960-894x(01)00344-4. [DOI] [PubMed] [Google Scholar]
- Battisti U.M., Corrado S., Sorbi C., Cornia A., Tait A., Malfacini D., Cerlesi M.C., Calo G., Brasili L. Synthesis, enantiomeric separation and docking studies of spiropiperidine analogues as ligands of the nociceptin/orphanin FQ receptor. MedChemComm. 2014;5:973–983. [Google Scholar]
- Bayat Mokhtari R., Homayouni T.S., Baluch N., Morgatskaya E., Kumar S., Das B., Yeger H. Combination therapy in combating cancer. Oncotarget. 2017;8(23):38022–38043. doi: 10.18632/oncotarget.16723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black J.D., Brattain M.G., Krishnamurthi S.A., Dawson D.M., Willson J.K. ErbB family targeting. Curr. Opin. Invest. Drugs. 2003;4:1451–1454. [PubMed] [Google Scholar]
- Christensen J.G. A preclinical review of sunitinib, a multitargeted receptor tyrosine kinase inhibitor with anti-angiogenic and antitumour activities. Annal. Oncol.: Off. J. Eur. Soc. Medical Oncol. 2007;18(Suppl 10):x3–x. doi: 10.1093/annonc/mdm408. [DOI] [PubMed] [Google Scholar]
- Denizot F., Lang R. Rapid colorimetric assay for cell growth and survival: modifications to the tetrazolium dye procedure giving improved sensitivity and reliability. J. Immunol. Methods. 1986;89(2):271–277. doi: 10.1016/0022-1759(86)90368-6. [DOI] [PubMed] [Google Scholar]
- Dungo R.T., Keating G.M. Afatinib: first global approval. Drugs. 2013;73:1503–1515. doi: 10.1007/s40265-013-0111-6. [DOI] [PubMed] [Google Scholar]
- El-Ayaan U., Abdel-Aziz A.-A.-M., Al-Shihry S. Solvatochromism, DNA binding, anti-tumor activity and molecular modeling study of mixed-ligand copper(II) complexes containing the bulky ligand: bis[N-(p-tolyl)imino]acenaphthene. Eur. J. Med. Chem. 2007;42(11–12):1325–1333. doi: 10.1016/j.ejmech.2007.02.014. [DOI] [PubMed] [Google Scholar]
- El-Azab A.S., Alanazi A.M., Abdel-Aziz N.I., Al-Suwaidan I.A., El-Sayed M.A., El-Sherbeny M.A., Abdel-Aziz A.-A.-M. Synthesis, molecular modeling study, preliminary antibacterial, and anti-tumor evaluation of N-substituted naphthalimides and their structural analogues. Med. Chem. Res. 2013;22(5):2360–2375. [Google Scholar]
- El-Azab A.S., Al-Dhfyan A., Abdel-Aziz A.-A.-M., Abou-Zeid L.A., Alkahtani H.M., Al-Obaid A.M., Al-Gendy M.A. Synthesis, anticancer and apoptosis-inducing activities of quinazoline-isatin conjugates: epidermal growth factor receptor-tyrosine kinase assay and molecular docking studies. J. Enzyme Inhib. Med. Chem. 2017;32(1):935–944. doi: 10.1080/14756366.2017.1344981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A.S., Abdel-Aziz A.-A.-M., Abou-Zeid L.A., El-Husseiny W.M., El Morsy A.M., El-Gendy M.A., El-Sayed M.A. Synthesis, antitumour activities and molecular docking of thiocarboxylic acid ester-based NSAID scaffolds: COX-2 inhibition and mechanistic studies. J. Enzyme Inhib. Med. Chem. 2018;33(1):989–998. doi: 10.1080/14756366.2018.1474878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azab A.S., Abdel-Aziz A.A., AlSaif N.A., Alkahtani H.M., Alanazi M.M., Obaidullah A.J., Eskandrani R.O., Alharbi A. Anti-tumor activity, multitarget mechanisms, and molecular docking studies of quinazoline derivatives based on a benzenesulfonamide scaffold: Cell cycle analysis. Bioorg. Chem. 2020;104 doi: 10.1016/j.bioorg.2020.104345. [DOI] [PubMed] [Google Scholar]
- El-Azab A.S., Alkahtani H.M., AlSaif N.A., Al-Suwaidan I.A., Obaidullah A.J., Alanazi M.M., Al-Obaid A.M., Al-Agamy M.H.M., Abdel-Aziz A.-A.-M. Synthesis, antiproliferative and enzymatic inhibition activities of quinazolines incorporating benzenesulfonamide: Cell cycle analysis and molecular modeling study. J. Mol. Struct. 2023;127815 [Google Scholar]
- El-Husseiny W.M., El-Sayed M.A., Abdel-Aziz N.I., El-Azab A.S., Asiri Y.A., Abdel-Aziz A.-A.-M. Structural alterations based on naproxen scaffold: Synthesis, evaluation of anti-tumor activity and COX-2 inhibition, and molecular docking. Eur. J. Med. Chem. 2018;158:134–143. doi: 10.1016/j.ejmech.2018.09.007. [DOI] [PubMed] [Google Scholar]
- El-Sherbeny M.A., Abdel-Aziz A.-A.-M., Ahmed M.A. Synthesis and anti-tumor evaluation of novel diarylsulfonylurea derivatives: molecular modeling applications. Eur. J. Med. Chem. 2010;45(2):689–697. doi: 10.1016/j.ejmech.2009.11.014. [DOI] [PubMed] [Google Scholar]
- Frampton J.E. Lapatinib: a review of its use in the treatment of HER2-overexpressing, trastuzumab-refractory, advanced or metastatic breast cancer. Drugs. 2009;69:2125–2148. doi: 10.2165/11203240-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Fu R.G., Sun Y., Sheng W.B., Liao D.F. Designing multitargeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017;136:195–211. doi: 10.1016/j.ejmech.2017.05.016. [DOI] [PubMed] [Google Scholar]
- George R.F. Facile synthesis of simple 2-oxindole-based compounds with promising antiproliferative activity. Future Med. Chem. 2018;10(3):269–282. doi: 10.4155/fmc-2017-0148. [DOI] [PubMed] [Google Scholar]
- Gleeson M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008;51(4):817–834. doi: 10.1021/jm701122q. [DOI] [PubMed] [Google Scholar]
- Goda F.E., Abdel-Aziz A.-A.-M., Ghoneim H.A. Synthesis and biological evaluation of novel 6-nitro-5-substituted aminoquinolines as local anesthetic and anti-arrhythmic agents: molecular modeling study. Bioorg. Med. Chem. 2005;13(9):3175–3183. doi: 10.1016/j.bmc.2005.02.050. [DOI] [PubMed] [Google Scholar]
- Gullick W.J. Prevalence of aberrant expression of the epidermal growth factor receptor in human cancers. Br. Med. Bull. 1991;47(1):87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- Gundla R., Kazemi R., Sanam R., Muttineni R., Sarma J.A., Dayam R., Neamati N. Discovery of novel small-molecule inhibitors of human epidermal growth factor receptor-2: combined ligand and target-based approach. J. Med. Chem. 2008;51:3367–3377. doi: 10.1021/jm7013875. [DOI] [PubMed] [Google Scholar]
- Hamdi A., Said E., Farahat A., El-Bialy S., Massoud M. Synthesis and in vivo antifibrotic activity of novel leflunomide analogues. Lett. Drug Des. Discovery. 2016;13(9):912–920. [Google Scholar]
- Hamdi A., Elhusseiny W.M., Othman D.I., Haikal A., Bakheit A.H., El-Azab A.S., Al-Agamy M.H., Abdel-Aziz A.-A.-M. Synthesis, anti-tumor, and apoptosis-inducing activities of novel 5-arylidenethiazolidine-2,4-dione derivatives: Histone deacetylases inhibitory activity and molecular docking study. Eur. J. Med. Chem. 2022;244 doi: 10.1016/j.ejmech.2022.114827. [DOI] [PubMed] [Google Scholar]
- Hamdi A., El-Shafey H.W., Othman D.I., El-Azab A.S., AlSaif N.A., Abdel-Aziz A.-A.-M. Design, synthesis, anti-tumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: Molecular modeling studies. Bioorg. Chem. 2022;122 doi: 10.1016/j.bioorg.2022.105710. [DOI] [PubMed] [Google Scholar]
- Holohan C., Van Schaeybroeck S., Longley D.B., Johnston P.G. Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer. 2013;13(10):714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- Housman G., Byler S., Heerboth S., Lapinska K., Longacre M., Snyder N., Sarkar S. Drug resistance in cancer: an overview. Cancers. 2014;6(3):1769–1792. doi: 10.3390/cancers6031769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.D., Blagg J., Price D.A., Bailey S., Decrescenzo G.A., Devraj R.V., Ellsworth E., Fobian Y.M., Gibbs M.E., Gilles R.W., Greene N., Huang E., Krieger-Burke T., Loesel J., Wager T., Whiteley L., Zhang Y. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008;18(17):4872–4875. doi: 10.1016/j.bmcl.2008.07.071. [DOI] [PubMed] [Google Scholar]
- Johnson T.W., Dress K.R., Edwards M. Using the Golden Triangle to optimize clearance and oral absorption. Bioorg. Med. Chem. Lett. 2009;19(19):5560–5564. doi: 10.1016/j.bmcl.2009.08.045. [DOI] [PubMed] [Google Scholar]
- Krishnan K., Prathiba K., Jayaprakash V., Basu A., Mishra N., Zhou B., Hu S., Yen Y. Synthesis and ribonucleotide reductase inhibitory activity of thiosemicarbazones. Bioorg. Med. Chem. Lett. 2008;18(23):6248–6250. doi: 10.1016/j.bmcl.2008.09.097. [DOI] [PubMed] [Google Scholar]
- Kumar R., Saneja A., Panda A.K. An annexin V-FITC—propidium iodide-based method for detecting apoptosis in a non-small cell lung cancer cell line. Lung Cancer: Methods Protocols. 2021;2279:213–223. doi: 10.1007/978-1-0716-1278-1_17. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46(1–3):3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Madhusudan S., Ganesan T.S. Tyrosine kinase inhibitors in cancer therapy. Clin. Biochem. 2004;37:618–635. doi: 10.1016/j.clinbiochem.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Mohamed M.A., Ayyad R.R., Shawer T.Z., Abdel-Aziz A.-A.-M., El-Azab A.S. Synthesis and anti-tumor evaluation of trimethoxyanilides based on 4(3H)-quinazolinone scaffolds. Eur. J. Med. Chem. 2016;112:106–113. doi: 10.1016/j.ejmech.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65(1–2):55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Nicholson R.I., Gee J.M.W., Harper M.E. EGFR and cancer prognosis. Eur. J. Cancer. 2001;37:9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- Omar H.A., Tolba M.F., Hung J.-H., Al-Tel T.H. OSU-2S/Sorafenib synergistic anti-tumor combination against hepatocellular carcinoma: the role of PKCδ/p53. Front. Pharmacol. 2016;7:463. doi: 10.3389/fphar.2016.00463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormerod M.G. Investigating the relationship between the cell cycle and apoptosis using flow cytometry. J. Immunol. Methods. 2002;265:73–80. doi: 10.1016/s0022-1759(02)00071-6. [DOI] [PubMed] [Google Scholar]
- Othman D.I., Hamdi A., Tawfik S.S., Elgazar A.A., Mostafa A.S. Identification of new benzimidazole-triazole hybrids as anticancer agents: multi-target recognition, in vitro and in silico studies. J. Enzyme Inhib. Med. Chem. 2023;38(1):2166037. doi: 10.1080/14756366.2023.2166037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson Q.P., Hsu D.C., Goode D.R., Novotny C.J., Totten R.K., Hergenrother P.J. Procaspase-3 activation as an anticancer strategy: Structure− activity relationship of procaspase-activating compound 1 (PAC-1) and Its cellular co-localization with caspase-3. J. Med. Chem. 2009;52:5721–5731. doi: 10.1021/jm900722z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regad T. Targeting RTK Signaling Pathways in Cancer. Cancers. 2015;7(3):1758–1784. doi: 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusnak D.W., Lackey K., Affleck K., Wood E.R., Alligood K.J., Rhodes N., Keith B.R., Murray D.M., Knight W.B., Mullin R.J., Gilmer T.M. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001;1(2):85–94. [PubMed] [Google Scholar]
- Şenkardeş S., Han M., Kulabaş N., Abbak M., Çevik Ö., Küçükgüzel İ., Küçükgüzel Ş.G. Synthesis, molecular docking and evaluation of novel sulfonyl hydrazones as anticancer agents and COX-2 inhibitors. Mol. Divers. 2020;24(3):673–689. doi: 10.1007/s11030-019-09974-z. [DOI] [PubMed] [Google Scholar]
- Son J., Jang J., Beyett T.S., Eum Y., Haikala H.M., Verano A., Lin M., Hatcher J.M., Kwiatkowski N.P., Eser P.Ö., Poitras M.J., Wang S., Xu M., Gokhale P.C., Cameron M.D., Eck M.J., Gray N.S., Jänne P.A. A Novel HER2-Selective Kinase Inhibitor Is Effective in HER2 Mutant and Amplified Non-Small Cell Lung Cancer. Cancer Res. 2022;82(8):1633–1645. doi: 10.1158/0008-5472.CAN-21-2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács G., Paterson J.K., Ludwig J.A., Booth-Genthe C., Gottesman M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006;5(3):219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- Takimoto C.H., Awada A. Safety and anti-tumor activity of sorafenib (Nexavar) in combination with other anticancer agents: a review of clinical trials. Cancer Chemother. Pharmacol. 2008;61:535–548. doi: 10.1007/s00280-007-0639-9. [DOI] [PubMed] [Google Scholar]
- Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Uto Y., Ueno Y., Kiyotsuka Y., Miyazawa Y., Kurata H., Ogata T., Yamada M., Deguchi T., Konishi M., Takagi T. Synthesis and evaluation of novel stearoyl-CoA desaturase 1 inhibitors: 1'-{6-[5-(pyridin-3-ylmethyl)-1,3,4-oxadiazol-2-yl]pyridazin-3-yl}-3,4-dihydrospiro[chromene-2,4'-piperidine] analogs. Eur. J. Med. Chem. 2010;45(11):4788–4796. doi: 10.1016/j.ejmech.2010.07.044. [DOI] [PubMed] [Google Scholar]
- Varasi M., Thaler F., Abate A., Bigogno C., Boggio R., Carenzi G., Cataudella T., Dal Zuffo M.C., Fulco M.G.R. Discovery, synthesis, and pharmacological evaluation of spiropiperidine hydroxamic acid based derivatives as structurally novel histone deacetylase (HDAC) inhibitors. J. Med. Chem. 2011;54(8):3051–3064. doi: 10.1021/jm200146u. [DOI] [PubMed] [Google Scholar]
- Vermes I., Haanen C., Steffens-Nakken H., Reutellingsperger C. A novel assay for apoptosis flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J. Immunol. Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- Vogel S., Kaufmann D., Pojarová M., Müller C., Pfaller T., Kühne S., Bednarski P.J., von Angerer E. Aroyl hydrazones of 2-phenylindole-3-carbaldehydes as novel antimitotic agents. Bioorg. Med. Chem. 2008;16(12):6436–6447. doi: 10.1016/j.bmc.2008.04.071. [DOI] [PubMed] [Google Scholar]
- Wang S., Song Y., Liu D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017;385:51–54. doi: 10.1016/j.canlet.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Woodburn J.R. The epidermal growth factor receptor and its inhibition in cancer therapy. Pharmacol. Ther. 1999;82(2–3):241–250. doi: 10.1016/s0163-7258(98)00045-x. [DOI] [PubMed] [Google Scholar]
- Xie L., Bourne P.E. Developing multi-target therapeutics to fine-tune the evolutionary dynamics of the cancer ecosystem. Front. Pharmacol. 2015;6:209. doi: 10.3389/fphar.2015.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G., Abad M.C., Connolly P.J., Neeper M.P., Struble G.T., Springer B.A., Emanuel S.L., Pandey N., Gruninger R.H., Adams M., Moreno-Mazza S., Fuentes-Pesquera A.R., Middleton S.A. 4-Amino-6-arylamino-pyrimidine-5-carbaldehyde hydrazones as potent ErbB-2/EGFR dual kinase inhibitors. Bioorg. Med. Chem. Lett. 2008;18(16):4615–4619. doi: 10.1016/j.bmcl.2008.07.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.