

Abstract
Molnupiravir is an orally administered, small‐molecule ribonucleoside prodrug of β‐D‐N4‐hydroxycytidine (NHC) that has demonstrated potent, broad‐spectrum preclinical activity against RNA viruses and has a high barrier to the development of resistance. A double‐blind, placebo‐controlled, phase I trial was conducted to evaluate the pharmacokinetics (PKs), safety, and tolerability of 10.5‐day administration of multiple doses of molnupiravir and its metabolites in healthy, adult participants. Participants were randomly assigned (3:1) to receive molnupiravir (400 mg [n = 6], 600 mg [n = 6], and 800 mg [n = 12]) or matching placebo (n = 8) every 12 h (q12h) for 10.5 days. Blood was collected to evaluate the PKs of NHC in plasma and of its active metabolite, NHC‐triphosphate (NHC‐TP), in peripheral blood mononuclear cells (PBMCs). Molnupiravir was generally well‐tolerated. All adverse events were mild or moderate in severity and none led to treatment discontinuation. No clinically meaningful dose‐related safety findings were observed. Mean time to maximal concentration was ~1.50 to 1.98 h for plasma NHC and ~4.00 to 8.06 h for PBMC NHC‐TP. Accumulation was minimal (<1.2) for NHC and ~2‐ to 2.5‐fold for NHC‐TP. Plasma NHC PKs was generally dose proportional, and PBMC NHC‐TP PKs was less than dose proportional over the dose range studied. NHC and NHC‐TP PK support twice‐daily administration. Overall, molnupiravir administered at up to 800 mg q12h for 10.5 days was generally well‐tolerated in healthy participants with dose‐linear PKs, supporting the evaluation of longer molnupiravir dosing up to 10 days in future clinical trials.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Molnupiravir is an orally administered, small‐molecule ribonucleoside prodrug of β‐D‐N4‐hydroxycytidine (NHC) with potent, broad‐spectrum preclinical activity against coronaviruses and other RNA viruses and a high barrier to the development of resistance. Molnupiravir administration at 800 mg twice daily for 5.5 days is generally well‐tolerated and has been shown to reduce the risk of hospitalization or death in nonhospitalized, unvaccinated adults with mild to moderate coronavirus disease 2019 at risk of progression to severe disease. Safety and pharmacokinetic (PK) data to date are limited to ~5 days of molnupiravir administration.
WHAT QUESTION DID THIS STUDY ADDRESS?
This trial was designed to evaluate the PKs, safety, and tolerability of multiple doses (400, 600, and 800 mg) of molnupiravir and its metabolites following longer administration twice daily for 10.5 days in healthy participants.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Molnupiravir administration for 10.5 days within the studied dose range was generally well‐tolerated in healthy participants; all adverse events were mild or moderate in severity, and none resulted in trial discontinuation. Plasma NHC exhibited dose‐proportional PKs over the dose range studied, consistent with published PK data. The PKs of NHC‐triphosphate (NHC‐TP) in peripheral blood mononuclear cells exhibited less than dose‐proportional behavior. Minimal accumulation of NHC was exhibited and accumulation of ~2‐ to 2.5‐fold was seen for NHC‐TP following 10.5 days of molnupiravir administration.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The results from this trial support future clinical studies that may require longer‐term dosing of molnupiravir for up to 10 days.
INTRODUCTION
Coronavirus disease 2019 (COVID‐19), a disease resulting from severe acute respiratory syndrome‐coronavirus 2 (SARS‐CoV‐2) infection, was declared a global pandemic by the World Health Organization (WHO) in March 2020. As of February 2023, over 757 million confirmed cases of COVID‐19 and 6.8 million COVID‐19–related deaths have been reported worldwide. 1 COVID‐19 vaccinations and the availability of antiviral agents have reduced severe disease burden; 2 however, SARS‐CoV‐2 infections remain problematic 2 , 3 due to the emergence of SARS‐CoV‐2 variants conferring resistance to available therapy 4 , 5 and the ability to evade vaccine‐ and infection‐induced immunity. 4 , 6 , 7 , 8 There is still an unmet need for easily and conveniently administered outpatient interventions for the treatment and prevention of COVID‐19.
Molnupiravir is an orally administered, small‐molecule ribonucleoside prodrug of β‐D‐N4‐hydroxycytidine (NHC) that has demonstrated potent, broad‐spectrum preclinical activity against coronaviruses (including SARS‐CoV‐2 and its variants) and other RNA viruses and has a high barrier to the development of resistance. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 Clinically, early treatment with molnupiravir administered at 800 mg every 12 h (q12h) for 5 days was shown to reduce the risk of hospitalization or death in nonhospitalized, unvaccinated adults with mild to moderate COVID‐19 at risk of progression to severe disease. 17
Following oral administration, molnupiravir is rapidly absorbed and metabolized to NHC, which is widely distributed to tissues and then phosphorylated intracellularly to its pharmacologically active triphosphate form (NHC‐TP). Viral RNA polymerase incorporates NHC‐TP into the viral genome, resulting in viral error induction, ultimately leading to nonviable and noninfectious virus. 12 , 16 , 18 , 19 Molnupiravir has previously been evaluated in healthy participants following oral single (50–1600 mg) dose administration and multiple (50–800 mg) dose administration up to 5.5 days and in patients with COVID‐19 at 800 mg q12h for 5 days. 20 , 21 , 22 , 23 Due to rapid conversion, NHC is the primary circulating analyte, and molnupiravir is detected only at very low levels in plasma. Following oral administration, the time to peak NHC plasma concentration (T max) is attained at ~1.5 h, followed by elimination characterized by an effective terminal half‐life (t 1/2) of ~3.3 h and a slower terminal phase. 24 With multiple‐dose q12h administration, there is no clinically meaningful accumulation of NHC in plasma. NHC is eliminated primarily by metabolism through pathways involved in the metabolism of endogenous pyrimidines. 20 , 23 , 24
Administration of molnupiravir up to 5 days has been found to be generally well‐tolerated. 17 , 20 , 21 , 22 , 23 There are, however, limited clinical data for molnupiravir administration longer than 5 days. A molnupiravir dosing schedule exceeding 5 days may be needed for effective treatment or prevention of infections caused by RNA viruses. To obtain additional clinical pharmacokinetic (PK) and safety data in support of future clinical studies of molnupiravir, including pre‐ and post‐exposure prophylaxis studies, in which dosing of up to 10 days may be required, the current trial was conducted.
METHODS
The trial was conducted in accordance with principles of Good Clinical Practice and was approved by the appropriate institutional review boards and regulatory agencies. All participants provided written informed consent prior to enrollment.
Study design
A randomized, placebo‐controlled, multiple‐dose, double‐blind, phase I trial of molnupiravir (protocol MK‐4482‐012) was conducted from May 20, 2021, to October 18, 2021 (EudraCT, 2021‐000860‐30). Eligible participants (4 panels of 8 participants each) were randomly assigned to receive oral doses of molnupiravir (400 mg [panel A], 600 mg [panel B], and 800 mg [panels C and D]) or placebo q12h for 10.5 days (total of 21 doses). The final dose allowed for full PK sampling on day 11. Within each panel, six participants were randomly assigned to receive molnupiravir and two to receive placebo. Participants in panel A were dosed first at the lowest dose of molnupiravir (400 mg). Review of available safety data was conducted up to 24 h following the last dose of molnupiravir/placebo prior to escalating to the next dose level in panels B (600 mg) and C (800 mg). Panel D was conducted to expand the number of participants at the 800 mg dose level. All doses were given with ~250 mL of water at approximately the same times on each dosing day. On full PK sampling days (days 1 and 11), the morning dose was administered in the fasted state. All other doses were administered without regard to food.
Trial population
Healthy male or nonpregnant and not breastfeeding female participants aged 25–60 years (inclusive) with a body mass index (BMI) between 18 and 35 kg/m2 (inclusive) were eligible for inclusion. Due to the potential effect of molnupiravir on bone and cartilage growth, the lower age limit was set to 25 years to assure skeletal maturity. Women of childbearing potential and men were required to adhere to highly effective methods of contraception throughout the trial and for 28 and 90 days thereafter, timeframes corresponding to the duration of one menstrual or spermatogenesis cycle, respectively. Key exclusion criteria included history of any clinically significant medical or psychiatric condition or significant multiple and/or severe allergies or history of drug hypersensitivity.
Pharmacokinetic measurements
To determine NHC concentrations in plasma and intracellular NHC‐TP concentrations in peripheral blood mononuclear cells (PBMCs), blood samples were drawn predose and at selected timepoints up to 12 h postdose on day 1, prior to the morning dose on day 3 (plasma only), day 5, and day 7 (plasma only), and at selected timepoints up to 24 h (PBMC) and 72 h (plasma) postdose on day 11. Plasma NHC concentrations were determined using a validated high‐performance liquid chromatography tandem mass spectrometry method. 20 The analytical range was 1.00–1000 ng/mL (0.00386–3.86 μM). PBMC NHC‐TP concentrations were determined using a validated liquid chromatography tandem mass spectrometry method. 20 The analytical range was 5–2000 ng/mL (0.01–4.0 μM). PBMC NHC‐TP concentrations in nM were corrected for cell count using the following formula: 25
This value was then converted back to molar units, assuming 0.2 μL = 106 cells. Values below the lower limit of quantitation were treated as 0.
Key PK parameters of interest included the following: maximum concentration (C max), T max, area under the concentration‐time curve from time 0 to 12 h (AUC0–12), AUC to the last quantifiable concentration (AUC0–last), AUC extrapolated to infinity (AUC0–inf), accumulation ratio (AR), and apparent t 1/2; the t 1/2 and AUC0–inf were determined following the last dose only. PK parameter values were determined using noncompartmental modeling methods in Phoenix WinNonlin version 8.1 (Certara). AUCs were calculated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations (linear‐up, log‐down calculation method option in Phoenix WinNonlin). The apparent t 1/2 was calculated as the quotient of the natural log (ln) of 2 and λz (ln[2]/λz), where λz is the apparent first‐order terminal elimination rate constant calculated from the slope of the linear regression of the terminal log‐linear portion of the plasma concentration‐time profile. The AR was calculated based on C max and AUC0–12 on day 1 versus day 11.
Statistical analysis
For each PK parameter, values were natural‐log transformed and evaluated with separate linear mixed‐effects models containing a fixed effect for treatment, day (day 1 or day 11), and treatment by day interaction and a random effect for participant. Ninety‐five percent confidence intervals (CIs) for the least‐squares means (LSMs) by treatment and day were constructed on the natural log scale and referenced the t‐distribution. Exponentiating the LSMs and lower and upper limits of these CIs yielded estimates for the population geometric means and CIs about the geometric means on the original scale for each treatment and day. Similarly, the LSM differences (day 11 – day 1) and 90% CIs from this model were back‐transformed to obtain the geometric mean AR (day 11/day 1) and 90% CI for AUC0–12 and C max.
RESULTS
Trial participants
A total of 32 participants were enrolled and randomly assigned, of whom 24 were assigned to molnupiravir and eight were assigned to placebo. The median age (range) was 45 years (27–59), and the mean BMI was 24.9 kg/m2 (SD, 3.8); half of the participants were men (n = 16, 50%; Table S1). All 32 participants were White; none were of Hispanic or Latino ethnicity. All participants were 100% compliant with the protocol‐defined regimen of trial intervention.
Safety
In total, 27 participants (84.4%) reported at least one adverse event (AE), 21 participants (87.5%) who received molnupiravir, with a similar frequency across all three dose levels, and six participants (75.0%) who received placebo. A total of five participants (15.6%) did not report an AE. The most frequently reported AEs (in 2 or more participants) following molnupiravir administration (across all dose levels) were headache (n = 10, 41.7%), malaise (n = 4, 16.7%), and nausea, vomiting, fatigue, puncture site hematoma (following blood sampling for PK or safety analyses), myalgia, dizziness, presyncope, and skin irritation (n = 2 each, 8.3%). There was no apparent relationship of individual AEs with dose. Two participants who received molnupiravir reported AEs considered to be related to trial intervention by the investigator (2 episodes of dizziness reported by 1 participant who received molnupiravir 600 mg and 5 episodes of headache reported by 1 participant who received molnupiravir 800 mg). The most frequently reported AEs (in 2 or more participants) following placebo administration were headache and skin irritation (n = 2 each, 25.0%). All AEs in this trial were categorized as mild or moderate in severity by the investigator. No serious AEs, deaths, or discontinuations due to AEs were reported. No clinically meaningful dose‐related changes were observed for laboratory parameter values, vital signs, physical examination findings, or electrocardiograms.
Pharmacokinetics of NHC in plasma
The arithmetic mean plasma concentration‐time profiles of NHC and the summary PK statistics of NHC in plasma are presented in Figure 1 and Table 1, respectively. Plasma NHC was detectable at the first measured time point, 0.5 h after the first dose (Figure 1), with a median T max ranging from 1.50 to 1.98 h (Table 1). NHC was detectable up to 72 h (the last measured plasma concentration) following the last dose on day 11 (Figure 1). Biphasic elimination of NHC was apparent after the last dose, where it exhibited a rapid distributional phase followed by an extended terminal elimination phase (Figure 1). The geometric mean apparent t 1/2 of NHC on day 11 ranged from 16.41 to 20.65 h. The AR of day 11/day 1 for AUC0–12 and C max ranged from 0.98 to 1.15 and 0.81 to 1.16, respectively (Table 1). This indicated minimal to no accumulation of NHC exposure. A dose‐proportionality analysis from 400 to 800 mg of molnupiravir using AUC0–12 and C max, measured on day 11, was performed (Figure S1) with resulting least‐squares estimates (95% CI) of the slope of 1.18 (0.77–1.59) for AUC0–12 and 0.99 (0.48–1.50) for C max. These results suggest that AUC0–12 and C max of plasma NHC increased in a dose‐proportional manner over the dose range studied.
FIGURE 1.
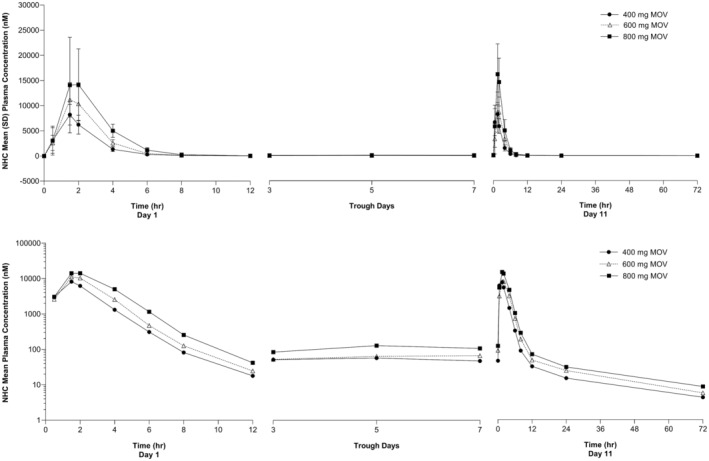
Arithmetic mean (±SD) concentration versus time profiles of NHC in plasma following administration of multiple oral doses of molnupiravir 400, 600, and 800 mg every 12 h for 10.5 days in healthy participants (n = 6 for 400 and 600 mg, n = 12 for 800 mg). Upper panel: linear scale; Lower panel: semi‐log scale. MOV, molnupiravir; NHC, β‐D‐N4‐hydroxycytidine; SD, standard deviation.
TABLE 1.
Summary statistics of plasma NHC pharmacokinetics following administration of multiple oral doses of molnupiravir 400, 600, and 800 mg every 12 h for 10.5 days in healthy participants.
| Pharmacokinetic parameter | Molnupiravir 400 mg (n = 6) | Molnupiravir 600 mg (n = 6) | Molnupiravir 800 mg (n = 12) |
|---|---|---|---|
| Day 1 (first dose) | |||
| AUC0–12 (h nmol/L) a | 18,000 (14,200–22,900) | 26,700 (21,000–33,900) | 38,100 (32,200–45,200) |
| AUC0–last (h nmol/L) a | 18,000 (14,200–22,900) | 26,700 (21,000–33,900) | 38,100 (32,200–45,200) |
| C max (nmol/L) a | 8320 (6150–11,300) | 11,000 (8120–14,900) | 14,000 (11,300–17,300) |
| T max (h) b | 1.50 (0.50, 1.55) | 1.50 (1.50, 2.00) | 1.98 (1.50, 4.00) |
| Day 11 (last dose) | |||
| AUC0–inf (h nmol/L) a | 20,500 (16,300–25,700) | 27,300 (21,800–34,300) | 45,500 (38,700–53,500) |
| AUC0–12 (h nmol/L) a | 19,700 (15,500–25,000) | 26,100 (20,500–33,200) | 43,900 (37,000–52,000) |
| AUC0–last (h nmol/L) a | 20,300 (16,000–25,800) | 27,100 (21,300–34,500) | 45,300 (38,200–53,600) |
| C max (nmol/L) a | 8400 (6210–11,400) | 8920 (6600–12,100) | 16,200 (13,100–20,100) |
| T max (h) b | 1.50 (0.57, 2.00) | 1.75 (1.50, 2.00) | 1.50 (1.50, 2.02) |
| Apparent t 1/2 (h) c | 16.41 (117.05) | 20.65 (22.67) | 20.64 (10.99) |
| Accumulation ratio day 11:day 1 | |||
| AUC0–12 (h nmol/L) d | 1.09 (0.97–1.23) | 0.98 (0.87–1.10) | 1.15 (1.06–1.25) |
| C max (nmol/L) d | 1.01 (0.81–1.26) | 0.81 (0.65–1.02) | 1.16 (0.99–1.36) |
Abbreviations: AUC0–12, area under the concentration‐time curve from time 0 to 12 h; AUC0–inf, AUC extrapolated to infinity; AUC0–last, AUC to the last quantifiable concentration; C max, maximum plasma concentration; NHC, β‐D‐N4‐hydroxycytidine; t 1/2, terminal half‐life; T max, time to maximum plasma concentration.
Back‐transformed least‐squares mean and 95% confidence interval from linear mixed‐effects model performed on natural‐log–transformed values.
Median (minimum, maximum) reported for T max (h).
Geometric mean and percent geometric coefficient of variation reported for apparent t 1/2 (h).
Back‐transformed least‐squares mean difference and 90% confidence interval from linear mixed‐effects model performed on natural‐log–transformed values.
Pharmacokinetics of NHC‐TP in PBMCs
The arithmetic mean PBMC concentration‐time profiles of NHC‐TP and the summary PK statistics of NHC‐TP in PBMCs are presented in Figure 2 and Table 2, respectively. NHC‐TP was detectable at the first measured timepoint, 4 h after the first dose (Figure 2), with a median T max ranging from 4.00 to 8.06 h (Table 2). NHC‐TP was detectable up to 24 h (the last measured PBMC concentration) following the last dose on day 11 in all but two molnupiravir‐treated participants from the 800‐mg dose group. The geometric mean apparent t 1/2 of NHC‐TP on day 11 ranged from 13.60 to 18.04 h over the dose range of 400–800 mg. The AR of AUC0–12 and C max ranged from 2.10 to 2.65 and 1.92 to 2.49, respectively (Table 2). A dose‐proportionality analysis of NHC‐TP in PBMCs for 400–800 mg of molnupiravir using AUC0–12 and C max day 11 data was performed (Figure S2), yielding least‐squares estimates (95% CI) of the slope of 0.60 (0.17–1.03) for AUC0–12 and 0.57 (0.10–1.04) for C max. These results suggest that AUC0–12 and C max of PBMC NHC‐TP increased in a less than dose‐proportional manner over the dose range studied.
FIGURE 2.
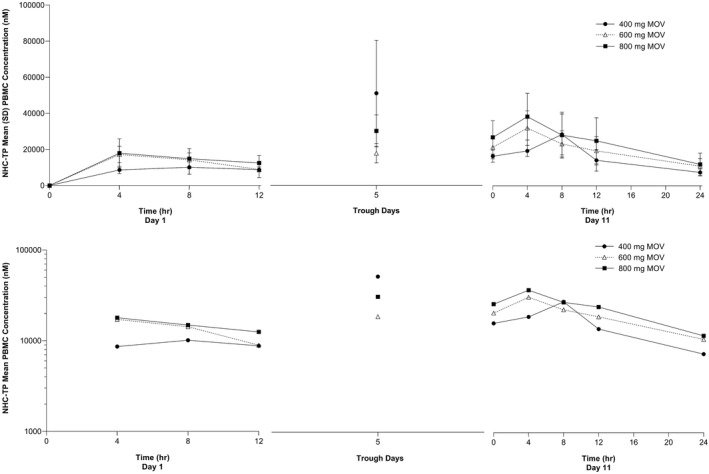
Arithmetic mean (±SD) concentration versus time profiles of NHC‐TP in PBMCs following administration of multiple oral doses of molnupiravir 400, 600, and 800 mg every 12 h for 10.5 days in healthy participants (n = 6 for 400 mg and 600 mg, n = 12 for 800 mg). Upper panel: linear scale; Lower panel: semi‐log scale. MOV, molnupiravir; NHC‐TP, β‐D‐N4‐hydroxycytidine‐triphosphate; PBMCs, peripheral blood mononuclear cells; SD, standard deviation.
TABLE 2.
Summary statistics of PBMC NHC‐TP pharmacokinetics following administration of multiple oral doses of molnupiravir 400, 600, and 800 mg every 12 h for 10.5 days in healthy participants.
| Pharmacokinetic parameter | Molnupiravir 400 mg (n = 6) | Molnupiravir 600 mg (n = 6) | Molnupiravir 800 mg (n = 12) |
|---|---|---|---|
| Day 1 (first dose) | |||
| AUC0–12 (h nmol/L) a | 88,400 (69,100–113,000) | 139,000 (109,000–178,000) | 144,000 (120,000–172,000) |
| AUC0–last (h nmol/L) a | 88,400 (68,100–115,000) | 139,000 (107,000–181,000) | 148,000 (123,000–178,000) |
| C max (nmol/L) a | 10,500 (7920–13,800) | 16,700 (12,700–22,000) | 17,100 (14,000–20,800) |
| T max (h) b | 8.06 (4.00, 12.00) | 4.00 (4.00, 4.00) | 4.00 (3.98, 8.15) |
| Day 11 (last dose) | |||
| AUC0–inf (h nmol/L) a | 334,000 (158,000–707,000) | 651,000 (466,000–909,000) | 978,000 (751,000–1,270,000) |
| AUC0–12 (h nmol/L) a | 234,000 (183,000–299,000) | 292,000 (229,000–374,000) | 354,000 (298,000–421,000) |
| AUC0–last (h nmol/L) a | 356,000 (274,000–461,000) | 459,000 (353,000–595,000) | 532,000 (442,000–640,000) |
| C max (nmol/L) a | 26,000 (19,700–34,300) | 32,100 (24,300–42,300) | 38,500 (31,700–46,900) |
| T max (h) b | 8.00 (3.92, 8.03) | 4.00 (4.00, 12.03) | 4.00 (0.00, 8.08) |
| Apparent t 1/2 (h) c | 13.60 e | 14.06 (28.52) | 18.04 (46.49) |
| Accumulation ratio day 11:day 1 | |||
| AUC0–12 (h nmol/L) d | 2.65 (2.29–3.07) | 2.10 (1.81–2.44) | 2.46 (2.20–2.76) |
| C max (nmol/L) d | 2.49 (2.05–3.02) | 1.92 (1.58–2.33) | 2.26 (1.97–2.59) |
Abbreviations: AUC0–12, area under the concentration‐time curve from time 0 to 12 h; AUC0–inf, AUC extrapolated to infinity; AUC0–last, AUC to the last quantifiable concentration; C max, maximum intracellular concentration; NHC‐TP, β‐D‐N4‐hydroxycytidine triphosphate; PBMC, peripheral blood mononuclear cell; t 1/2, terminal half‐life; T max, time to maximum intracellular concentration.
Back‐transformed least‐squares mean and 95% confidence interval from linear mixed‐effects model performed on natural‐log–transformed values.
Median (min, max) reported for T max (h).
Geometric mean and percent geometric coefficient of variation reported for apparent t 1/2 (h).
Back‐transformed least‐squares mean difference and 90% confidence interval from linear mixed‐effects model performed on natural‐log–transformed values.
Coefficient of variation not calculated due to insufficient concentration data during the terminal phase for five of the participants.
Plasma NHC versus PBMC NHC‐TP
The relationship of NHC exposures in plasma to NHC‐TP exposures in PBMCs was explored (Figure 3) by plotting the AUC0–12 of plasma NHC versus PBMC NHC‐TP for each individual participant and fitting with a linear regression line for day 1 and day 11. The adjusted coefficient of determination (R 2 ≥ 0.95) showed that exposures of NHC‐TP in PBMCs correlated well with plasma NHC exposures. The slope on day 11 was estimated to be approximately twice that of day 1, which is consistent with the observed AR of NHC‐TP in PBMCs.
FIGURE 3.
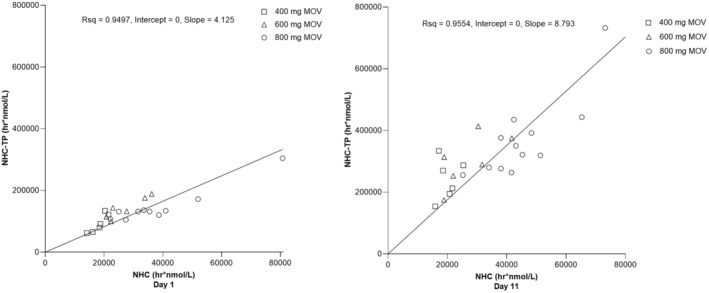
Individual AUC0–12 of NHC in plasma versus NHC‐TP in PBMCs following administration of multiple oral doses of molnupiravir 400 mg, 600 mg, and 800 mg every 12 h for 10.5 days to healthy participants (n = 6 for 400 and 600 mg, n = 12 for 800 mg). Left panel: day 1; Right panel: day 11. AUC0‐12, area under the concentration‐time curve from time 0 to 12 h; MOV, molnupiravir; NHC, β‐D‐N4‐hydroxycytidine; NHC‐TP, β‐D‐N4‐hydroxycytidine‐triphosphate; PBMCs, peripheral blood mononuclear cells; Rsq, coefficient of determination.
DISCUSSION
Molnupiravir is an orally administered prodrug with antiviral activity against RNA viruses, including SARS‐CoV‐2 and its variants of concern. Molnupiravir has been shown to reduce the risk of hospitalization or death in nonhospitalized patients with mild to moderate COVID‐19 following early treatment. 17 The oral route of administration of molnupiravir makes it convenient for administration to outpatients compared with COVID‐19 therapies that require intravenous administration (e.g., remdesivir). Additionally, because molnupiravir has no known drug–drug interactions (DDIs) and can be co‐administered without need for dose adjustments, 24 it is an important treatment option for high‐risk patients for whom other treatments may not be clinically appropriate due to potential DDIs with medications used to treat common underlying medical conditions. Administration of 800 mg molnupiravir q12h for up to 5 days was previously found to be generally well‐tolerated in healthy participants and patients with COVID‐19; 17 , 20 , 21 , 22 , 23 however, a safety and PK evaluation of molnupiravir administered for more than 5 days has not previously been published.
This trial evaluated the PKs, safety, and tolerability of multiple doses of molnupiravir up to 800 mg q12h for 10.5 days in healthy adult participants. Molnupiravir was found to be generally well‐tolerated with an acceptable safety profile over the 10.5‐day dosing period. Headache, malaise, and gastrointestinal symptoms were among the most frequently reported AEs following molnupiravir treatment. All AEs were mild or moderate in severity and none led to treatment discontinuation, consistent with results from previous phase I trials. 20 , 22 , 23 Furthermore, there was no apparent relationship between the incidence and intensity of AEs and molnupiravir dose, and no clinically relevant treatment‐ or dose‐related effects on vital signs, laboratory safety tests, physical examination findings, or electrocardiograms. Together, these results indicate that molnupiravir was generally well‐tolerated at the dose levels and duration tested supporting future clinical trials of molnupiravir that may require longer‐term dosing beyond 5.5 days (e.g., studies assessing the efficacy of molnupiravir as either pre‐ or post‐exposure prophylaxis). Another oral antiviral authorized for the treatment of COVID‐19, ritonavir‐boosted nirmatrelvir, has also been found to be generally well‐tolerated in healthy participants when administered q12h over 10 days. 26 However, ritonavir‐boosted nirmatrelvir requires a dose reduction in patients with moderate renal impairment, is not recommended for use in patients with severe renal or hepatic impairment, and may have significant DDIs with commonly used medications, 27 thereby restricting its use and complicating its prescribing. Molnupiravir offers an important treatment option for these vulnerable patient populations with no evident safety concerns.
The PKs of molnupiravir administered for 10.5 days was assessed by evaluating NHC in plasma and NHC‐TP in PBMCs, and findings were generally consistent with previously reported studies assessing 5.5 days of molnupiravir administration in healthy Japanese and White participants. 20 , 23 In this study, plasma NHC T max was ~1.5 h, corresponding to previous studies in both healthy participants and patients with COVID‐19 reporting a median T max between 1 and 2 h. 20 , 21 , 22 , 23 As described in previous phase I PK studies, 20 , 23 concentrations of NHC declined in a biphasic manner, where a rapid distributional phase followed by an extended terminal elimination phase was exhibited. The apparent terminal elimination t 1/2 of NHC was ~16 to 21 h. Despite this, the AR of AUC0–12 and C max was minimal (<1.2). This minimal accumulation indicates that steady‐state NHC levels were likely achieved within approximately one dosing interval (i.e., ~12 h). This trial was not designed to provide a definitive assessment of dose proportionality; however, the resulting least‐squares estimates of the slopes imply that AUC0–12 and C max of NHC increased in a dose‐proportional manner over the dose range studied.
Regarding the active metabolite of molnupiravir, NHC‐TP, T max was later than that of plasma NHC, with a median NHC‐TP T max of ~4 to 8 h. The apparent terminal elimination t 1/2 of NHC‐TP was estimated to be ~14 to 18 h. There are currently limited PK data available for NHC‐TP. However, in a recently published phase I trial in healthy Japanese participants who received molnupiravir for 5.5 days, NHC‐TP PMBC concentrations reached C max with a median T max between 3 and 5 h, and NHC‐TP was eliminated with an apparent t 1/2 of ~16 to 19 h on day 6. 20 In this study, the sampling duration of 24 h for NHC‐TP was less than two‐fold the estimated t 1/2; however, the coefficient of variation of the estimates is less than 50%, suggesting the estimates to be reasonably projected. NHC‐TP exposures in PBMCs after dosing q12h for 10.5 days were higher than after the first dose, as indicated by ARs of ~2 to 2.5. A similar AR of ~2.5 (day 6/day 1) was observed in healthy Japanese participants following 5.5 days of molnupiravir administration. 20 The AR and visual inspection of the concentration curves indicate that NHC‐TP levels likely achieved steady‐state by day 11. In an assessment of dose proportionality, the resulting least‐squares estimates of the slopes suggest that AUC0–12 and C max of PBMC NHC‐TP increased in a less than dose‐proportional manner over the dose range studied. Although it is difficult to compare exposures of NHC in relation to NHC‐TP, an exploratory evaluation of AUC0–12 demonstrated that PBMC NHC‐TP exposures generally correlated with plasma NHC exposures, with varying correlation on day 1 versus day 11.
This trial provides initial PK, safety, and tolerability results with molnupiravir q12h dosing up to 800 mg for 10.5 days but is limited by the small number of participants and the use of healthy participants versus patients with SARS‐CoV‐2 infection or other viral RNA infections. Another important limitation of the study is the lack of a diverse study population, with all participants being White; however, past data have indicated that race and ethnicity do not meaningfully influence the PKs of NHC and there are no known race‐based metabolism concerns. 24 Additional studies will be required in a broader population to assess the safety profile of molnupiravir more fully for dosing up to 10 days. Regarding PK evaluation, PBMC PK sampling was limited and did not adequately span the concentration tail; therefore, the determination of NHC‐TP half‐life is limited.
In summary, molnupiravir was generally well‐tolerated at up to 800 mg q12h for 10.5 days in healthy participants with no clinically meaningful dose‐related safety findings. Plasma NHC PK and PBMC NHC‐TP PK exhibited dose‐linear behavior over the dose range studied with dose‐proportional behavior for NHC and less than dose‐proportional behavior for NHC‐TP. The PK profile is supportive of twice‐daily administration. These results support future clinical studies of molnupiravir, including pre‐ or postexposure prophylaxis studies, for which dosing for up to 10 days may be needed for effective treatment or prevention of infection due to RNA viruses.
AUTHOR CONTRIBUTIONS
M.I. wrote the manuscript. M.I., G.G., K.E.D., B.M.M., and S.A.S. designed the research. M.I., K.E.D., P.K.W., T.Z., M.V.L., L.L., T.D., and S.R. performed the research. M.I., K.E.D., P.K.W., T.Z., M.V.L., L.L., T.D., S.R., B.M.M., G.G., and S.A.S. analyzed the data.
FUNDING INFORMATION
Funding for this research was provided by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
CONFLICT OF INTEREST STATEMENT
M.I., K.E.D., P.K.W., T.Z., M.V.L., L.L., T.D., B.M.M., G.G., and S.A.S. are employees of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and may own and/or hold stock options in Merck & Co., Inc., Rahway, NJ, USA. S.R. received research support from Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Supporting information
Figure S1
Figure S2
Table S1
ACKNOWLEDGMENTS
The authors wish to thank the study investigators and the participants for their participation in the trial. The authors additionally acknowledge Griet Van Lancker for contributions to this trial. Medical writing assistance was provided by Christina Balle, PhD, of ApotheCom, London, UK. This assistance was funded by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA.
Iwamoto M, Duncan KE, Wickremasingha PK, et al. Assessment of pharmacokinetics, safety, and tolerability following twice‐daily administration of molnupiravir for 10 days in healthy participants. Clin Transl Sci. 2023;16:1947‐1956. doi: 10.1111/cts.13602
DATA AVAILABILITY STATEMENT
The data sharing policy, including restrictions, of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA is available at https://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the Engage Zone site or via email to dataaccess@merck.com.
REFERENCES
- 1. World Health Organization . WHO COVID‐19 Dashboard. 2020. https://covid19.who.int/. Accessed February 24, 2023.
- 2. World Health Organization . Weekly epidemiological update on COVID‐19. https://www.who.int/publications/m/item/weekly‐epidemiological‐update‐on‐covid‐19‐‐‐11‐january‐2023. Accessed January 11, 2023.
- 3. Fiolet T, Kherabi Y, MacDonald CJ, Ghosn J, Peiffer‐Smadja N. Comparing COVID‐19 vaccines for their characteristics, efficacy and effectiveness against SARS‐CoV‐2 and variants of concern: a narrative review. Clin Microbiol Infect. 2022;28(2):202‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liu L, Iketani S, Guo Y, et al. Striking antibody evasion manifested by the Omicron variant of SARS‐CoV‐2. Nature. 2022;602(7898):676‐681. [DOI] [PubMed] [Google Scholar]
- 5. Cao Y, Jian F, Wang J, et al. Imprinted SARS‐CoV‐2 humoral immunity induces convergent Omicron RBD evolution. Nature. 2023;614(7948):521‐529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Evans JP, Zeng C, Qu P, et al. Neutralization of SARS‐CoV‐2 Omicron sub‐lineages BA.1, BA.1.1, and BA.2. Cell Host Microbe. 2022;30(8):1093‐1102.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qu P, Evans JP, Faraone JN, et al. Enhanced neutralization resistance of SARS‐CoV‐2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe. 2023;31(1):9‐17.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qu P, Faraone J, Evans JP, et al. Neutralization of the SARS‐CoV‐2 Omicron BA.4/5 and BA.2.12.1 subvariants. N Engl J Med. 2022;386(26):2526‐2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoon JJ, Toots M, Lee S, et al. Orally efficacious broad‐spectrum ribonucleoside analog inhibitor of influenza and respiratory syncytial viruses. Antimicrob Agents Chemother. 2018;62(8):e00766‐e00718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cox RM, Wolf JD, Plemper RK. Therapeutically administered ribonucleoside analogue MK‐4482/EIDD‐2801 blocks SARS‐CoV‐2 transmission in ferrets. Nat Microbiol. 2021;6(1):11‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Agostini ML, Pruijssers AJ, Chappell JD, et al. Small‐molecule antiviral β‐d‐N4‐hydroxycytidine inhibits a proofreading‐intact coronavirus with a high genetic barrier to resistance. J Virol. 2019;93(24):e01348‐e01319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sheahan TP, Sims AC, Zhou S, et al. An orally bioavailable broad‐spectrum antiviral inhibits SARS‐CoV‐2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med. 2020;12(541):eabb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wahl A, Gralinski LE, Johnson CE, et al. SARS‐CoV‐2 infection is effectively treated and prevented by EIDD‐2801. Nature. 2021;591(7850):451‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grobler J, Strizki J, Murgolo N, et al. Molnupiravir maintains antiviral activity against SARS‐CoV‐2 variants in vitro and in early clinical studies. Open Forum Infect Dis. 2021;8(Suppl 1):S373. [Google Scholar]
- 15. Abdelnabi R, Foo CS, De Jonghe S, Maes P, Weynand B, Neyts J. Molnupiravir inhibits replication of the emerging SARS‐CoV‐2 variants of concern in a hamster infection model. J Infect Dis. 2021;224(5):749‐753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Urakova N, Kuznetsova V, Crossman DK, et al. β‐d‐N4‐hydroxycytidine is a potent anti‐alphavirus compound that induces a high level of mutations in the viral genome. J Virol. 2018;92(3):e01965‐e01917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jayk Bernal A, Gomes da Silva MM, Musungaie DB, et al. Molnupiravir for oral treatment of Covid‐19 in nonhospitalized patients. N Engl J Med. 2022;386(6):509‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kabinger F, Stiller C, Schmitzová J, et al. Mechanism of molnupiravir‐induced SARS‐CoV‐2 mutagenesis. Nat Struct Mol Biol. 2021;28(9):740‐746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gordon CJ, Tchesnokov EP, Schinazi RF, Götte M. Molnupiravir promotes SARS‐CoV‐2 mutagenesis via the RNA template. J Biol Chem. 2021;297(1):100770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nakamura K, Fujimoto K, Hasegawa C, et al. A phase I, randomized, placebo‐controlled study of molnupiravir in healthy Japanese to support special approval in Japan to treat COVID‐19. Clin Transl Sci. 2022;15(11):2697‐2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. FitzGerald R, Dickinson L, Else L, et al. Pharmacokinetics of ß‐d‐N4‐hydroxycytidine, the parent nucleoside of prodrug molnupiravir, in nonplasma compartments of patients with severe acute respiratory syndrome coronavirus 2 infection. Clin Infect Dis. 2022;75(1):e525‐e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khoo SH, Fitzgerald R, Fletcher T, et al. Optimal dose and safety of molnupiravir in patients with early SARS‐CoV‐2: a phase I, open‐label, dose‐escalating, randomized controlled study. J Antimicrob Chemother. 2021;76(12):3286‐3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Painter WP, Holman W, Bush JA, et al. Human safety, tolerability, and pharmacokinetics of molnupiravir, a novel broad‐spectrum oral antiviral agent with activity against SARS‐CoV‐2. Antimicrob Agents Chemother. 2021;65(5):e02428‐e02420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Fact sheet for healthcare providers: emergency use authorization for Lagevrio™ (molnupiravir) capsules. https://www.fda.gov/media/155054/download. Accessed June 26, 2023.
- 25. Simiele M, D'Avolio A, Baietto L, et al. Evaluation of the mean corpuscular volume of peripheral blood mononuclear cells of HIV patients by a coulter counter to determine intracellular drug concentrations. Antimicrob Agents Chemother. 2011;55(6):2976‐2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh RSP, Toussi SS, Hackman F, et al. Innovative randomized phase I study and dosing regimen selection to accelerate and inform pivotal COVID‐19 trial of nirmatrelvir. Clin Pharmacol Ther. 2022;112(1):101‐111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. US Food and Drug Administration . Fact sheet for healthcare providers: emergency use authorization for Paxlovid™. https://www.fda.gov/media/155050/download. Accessed June 26, 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Table S1
Data Availability Statement
The data sharing policy, including restrictions, of Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA is available at https://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the Engage Zone site or via email to dataaccess@merck.com.