

Abstract
The amide is one of the most prevalent functional groups in all of pharmaceuticals and for this reason, reactions that introduce the amide moiety are of particular value. Intermolecular hydroamidation of alkenes remains an underexplored method for the synthesis of amide-containing compounds. The majority of hydroamidation procedures exhibit Markovnikov regioselectivity, while current methods for anti-Markovnikov hydroamidation are somewhat limited to activated alkene substrates or radical processes. Herein, we report a general method for the intermolecular anti-Markovnikov hydroamidation of unactivated alkenes under mild conditions, utilizing Rh(III) catalysis in conjunction with dioxazolone amidating reagents and isopropanol as an environmentally friendly hydride source. The reaction tolerates a wide range of functional groups, and efficiently converts electron-deficient alkenes, styrenes, and 1,1-disubstituted alkenes, in addition to unactivated alkenes, to their corresponding linear amides. Mechanistic studies reveal a reversible rhodium hydride migratory insertion step, leading to exquisite selectivity for the anti-Markovnikov product.
Graphical Abstract
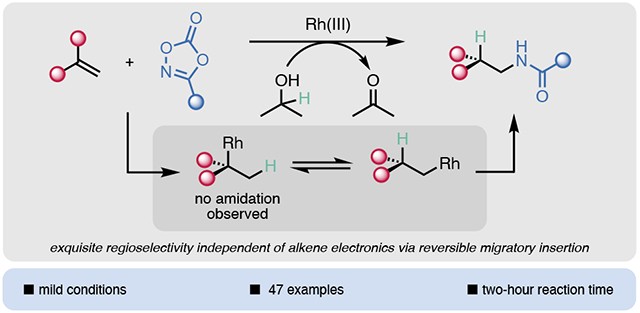
Due to the prevalence of nitrogen within bioactive molecules,1 C─N bond forming reactions are some of the most widely used in medicinal chemistry.2 Among these, the intermolecular hydroamination of alkenes is a valuable method of introducing nitrogen-containing moieties to widely available feedstock chemicals. Both Markovnikov and anti-Markovnikov hydroamination procedures have been developed with a variety of transition metal catalysts and nitrogen sources, including asymmetric methods for the synthesis of chiral amines.3,4,5,6,7,8,9,10 Recently, photoredox methodologies have also been leveraged to achieve hydroamination via the generation of radical species under mild conditions.11,12,13,14,15,16 Despite impressive recent advances in hydroamination, there has been limited success diversifying the nitrogen-containing moieties that can be introduced. Most hydroamination reactions result in aliphatic amine, aryl amine, or sulfonamide products. Due to the ubiquity of the amide in bioactive molecules, hydroamidation methods are of particular interest. According to a 2006 survey of leading pharmaceutical companies, 2/3 of drug candidates contained an amide bond.17 In 2020, a pharmaceutical company survey revealed that roughly one third of all reactions conducted internally were amide formation.18 The vast majority of these reactions are traditional couplings of acid derivatives and amines. A complementary strategy is to unite intact amides or amide surrogates with alkenes, one of the most readily available functional groups, in a catalytic hydroamidation reaction. This latter strategy is largely unexplored, likely as a consequence of the reduced nucleophilicity of amides compared to aliphatic amines (although there are a few examples of anti-Markovnikov hydroamidation to install the specialized phthalimide moiety).13,19 Recent examples, including this work, circumvent this constraint by using dioxazolones as electrophilic amide sources via metal nitrenoid formation,20,21,22,23,24 or generation of an amidyl radical.25,26
Most previous work on hydroamidation has yielded Markovnikov products (Scheme 1A). Early examples from Widenhoefer and Hartwig achieve hydroamidation using amide nucleophiles and unactivated alkenes, with platinum,27,28 gold,29 or iridium30 catalysts, but high reaction temperatures and a large excess of the alkene are generally required. More recently, Buchwald demonstrated an elegant copper-catalyzed Markovnikov hydroamidation of vinylarenes, using dioxazolones and a silane hydride source.21 Nickel and cobalt catalysis have also been leveraged for Markovnikov hydroamidation by S. Zhu22, Yu31, and R. Zhu.32
Scheme 1. Previous work on hydroamidation of alkenes.

Although even less explored than Markovnikov-selective methods, the anti-Markovnikov hydroamidation of alkenes has also been reported (Scheme 1B). Verma disclosed a metal-free hydroamidation of styrenes with aryl amides,25 and Xiao and C. Wang employed iron catalysis for the hydroamidation of allyl alcohol.33 While highly enabling, these methods are unable to engage unactivated alkenes as substrates. T. Wang used N-aminated dihydropyridines as redox-active amidyl radical precursors to effect anti-Markovnikov hydroamidation of unactivated alkenes under photocatalytic conditions.26 This work proceeds via a radical addition mechanism, leaving room for a complementary transition metal-catalyzed anti-Markovnikov hydroamidation procedure. Herein we report the anti-Markovnikov hydroamidation of unactivated alkenes, via a reversible and unselective Rh(III) hydride migratory insertion step, using dioxazolones as amide surrogates that are readily made in one or two steps from carboxylic acids, and isopropanol as an environmentally benign hydride source (Scheme 1C).
Our previous work has revealed that Rh(III) catalysts engage even simple alkenes in a variety of reactions.34,35 Extensive precedent by Krische and others36,37,38,39,40,41 has established that transfer hydrogenation can occur from isopropanol. We speculated that we could merge these technologies to leverage rhodium hydride species for alkene functionalization. Specifically, rhodium hydride insertion into an alkene, followed by trapping with a dioxazolone would lead to anti-Markovnikov hydroamidation products.
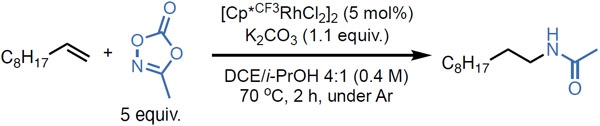
Through reaction optimization, we obtained high yield of the desired hydroamidation product with the conditions shown in Table 1 entry 1 (detailed optimization tables are available in the Supporting Information). Of particular note is the selection of the electron-deficient 1,2,3,4-tetramethyl-5-trifluoromethylcyclopenta-1,3-dienyl (Cp*CF3), which outperforms the prototypical pentamethylcyclopentadienyl (Cp*) ligand (Table 1, entry 2). Increased electron deficiency of the catalyst may aid migratory insertion into the alkene, resulting in higher reactivity.42,43 Control reactions demonstrated that rhodium, i-PrOH, and K2CO3 are all necessary components (Table 1, entries 4-6). We were pleased to observe that the hydroamidation product is still formed in synthetically useful yields when the equivalents of dioxazolone are reduced (2 equiv., 51%, Table 1, entry 7), and when the reaction is conducted without heating (55%, Table 1, entries 8). Additionally, the reaction exhibits no sensitivity to air (Table 1, entry 9).
Table 1.
Optimized reaction conditions and controls
| ||
|---|---|---|
| Entry | Deviation from Standard Conditions | Yield (%)a |
| 1 | none | 79 |
| 2 | [Cp*RhCl2]2 | 37 |
| 3 | no Rh | — |
| 4 | no i-PrOH | — |
| 5 | no K2CO3 | — |
| 6 | 2 equiv. dioxazolone | 51 |
| 7 | 22 °C | 55 |
| 8 | under air | 80 |
| ||
Optimization was performed on 0.1 mmol scale, using 1-decene and methyl dioxazolone (5 equiv.) in DCE/i-PrOH. aYields were determined by 1H NMR of the unpurified reaction mixture, with mesitylene as internal standard.
Having established optimized conditions for the anti-Markovnikov hydroamidation reaction, we next examined its scope (Scheme 2). Alkenes containing a wide range of functional groups are compatible with the reaction conditions, including free and protected alcohols (3c, 3h, 3j, 3m, 3o, 3p, 3q, 3aa, 3ac), protected amines (3b, 3g, 3i, 4e), and potentially sensitive leaving groups, including acetate, tosylate, and bromide (3j, 3o, and 3u respectively). Other functional groups that are tolerated include a nitrile (3l), Weinreb amide (3n), sulfone (3r), and epoxide (3ad). It is also notable that ketones (3s, 4l), are preserved, even under reducing metal-hydride conditions. More complex natural products linalool (3p) and sclareol (3q) undergo hydroamidation in good yields, and, notably, the trisubstituted alkene of linalool remains untouched. Likely, the more sterically hindered alkene region is unable to undergo migratory insertion, thereby enabling selective hydroamidation of the terminal alkene. Radical hydroamidation methods exhibit opposite chemoselectivity with analogous substrates.20 In addition to the aliphatic alkenes examined, the reaction also proceeds smoothly with styrenes (3v-3x) and electron deficient alkenes such as acrylates and vinyl sulfones (3s, 3y). We are also able to isolate terminally amidated products 3z and 3aa from internal alkene starting materials ethyl crotonate and trans 3-penten-1-ol, albeit in lower yields. These products most likely result from an alkene chain walk mechanism that first isomerizes the double bond to the terminal position, where it then undergoes hydroamidation. Similar chain walking has been previously described for Rh(I) hydride species by Shi.44
Scheme 2. Hydroamidation scope.

Unless otherwise noted, we report isolated yields of reactions run on 0.1 mmol scale using five equivalents of dioxazolone. a The NMR yield is reported. b The free alcohol was TMS-protected in the starting material. c With 2.5 equivalents of dioxazolone. d 0.05 mmol scale. Ac = acetyl, Bn = benzyl, Phth = phthaloyl, Ms = methanesulfonyl, TBDPS = tert-butyldiphenylsilyl, Ts = para-toluenesulfonyl.
Furthermore, we were excited to see that 1,1-disubstituted alkenes are also amenable to the hydroamidation procedure. Methacrylate derivatives (3ab-3ae) perform well in the method. α-substituted styrenes (3af, 3ag) undergo hydroamidation in moderate yields as well. Substituted methylenecyclohexanes (3ah-3al) also afford the desired products efficiently with moderate to excellent diasteroselectivity, but yield was found to decrease as the steric bulk of the substituents is increased. For example, the yield and diastereomeric ratio are minorly impacted moving from phenyl to tert-butyl (3ai, 3aj), but using significantly bulkier substrates β-pinene (3ak and (+)-longifolene (3al, results in a large drop in yield and significantly higher diastereomeric ratios of 4:1 and >20:1 respectively.
We also expanded the hydroamidation scope beyond the addition of an acetamide group by making use of different dioxazolones. Amides with longer linear aliphatic chains and rings of varying size were all installed efficiently (4a, 4b, 4c, 4d, 4f, 4g, 4h). N-phthaloyl glycine and dehydrocholic acid-derived dioxazolones (4e, 4i) afford the hydroamidated products in synthetically useful yields, despite using reduced equivalents of the dioxazolone due to solubility constraints.
We next turned our attention to elucidating the mechanism (Scheme 3). A rhodium hydride species was observed by stirring [Cp*CF3RhCl2]2, isopropanol, and K2CO3 in DCM-d2 at room temperature for one minute and taking a 1H NMR spectrum, where we observed the hydride as a triplet (−11.18 ppm, J = 20 Hz).45,46,47 Although further experiments suggest that the observed species is not catalytically active (see SI for details), this demonstrates the formation of a rhodium hydride under these conditions. In order to test whether a rhodium hydride is mechanistically active, we performed stoichiometric experiments with Et3SiH as a hydride source, which generate a small amount of active rhodium monohydride, as well as predominant formation of inactive rhodium dihydride species (see SI for details). Subjecting 1-decene to the standard conditions using Et3SiH instead of isopropanol, we observe the hydroamidation product, albeit in just 6% yield. On the basis of these experiments, as well as literature on rhodium transfer hydrogenation,48 we propose that rhodium hydride formation is likely the first step of the mechanism.
Scheme 3. Mechanistic studies.

We then sought to confirm that isopropanol is the hydride source by using isopropanol-d8 in the standard reaction conditions, and we indeed saw deuterium incorporation in the product (Scheme 3A). A second interesting observation from this experiment was that deuterium is incorporated equally at both C1 and C2, leading us to hypothesize that migratory insertion of the rhodium hydride is completely unselective. Since amidation occurs exclusively at the terminal position, we posited that migratory insertion is also reversible (supported by our prior observation that chain walking may occur to form products 3z and 3aa). 44,49 To test this, we subjected terminally-deuterated 1-undecene-d2 to the standard conditions (Scheme 3B). In the product we observed that some of the deuterium incorporation at C1 is transferred to C2, which further supports an unselective and reversible migratory insertion step. The exquisite terminal selectivity for amidation therefore suggests a higher barrier to amidation for the secondary alkylrhodium species.
To probe the reversibility of the alkene coordination step, we subjected 1-decene to the standard conditions with isopropanol-d8, but stopped the reaction after 30 minutes so that full conversion was not achieved (Scheme 3C). If alkene coordination is reversible, we should observe deuteration of the alkene due to migratory insertion of rhodium deuteride to form a secondary alkylrhodium species and subsequent β-hydride elimination (this pathway is implied by the deuteration of both C1 and C2 in Scheme 3A). Upon isolating the unreacted alkene, we did not observe deuterium incorporation, suggesting that alkene coordination is irreversible.
Subjecting 1-decene to the reaction conditions with a 1:1 mixture of isopropanol and isopropanol-d8 (Scheme 3D) results in no detectable deuterium incorporation in the product, implying a large primary KIE on the rhodium hydride-forming step and potentially on subsequent steps.50
Since the above experiments suggest that amidation is turnover-limiting, we used a competition experiment between fluoromethyl dioxazolone and methyl dioxazolone to determine whether dioxazolone coordination or nitrenoid formation is the turnover-limiting step (Scheme 3E). While methyl dioxazolone is more strongly coordinating, fluoromethyl dioxazolone is more easily activated to undergo nitrenoid formation. The product ratio of the reaction was 2.3:1 in favor of the fluorinated product, showing that the more weakly coordinating fluoromethyl dioxazolone outcompetes methyl dioxazolone. We thus conclude that dioxazolone coordination cannot be turnover limiting and that this outcompetition is a result of more facile dioxazolone activation, implying that nitrenoid formation is turnover-limiting.
On the basis of the above experiments, we propose the mechanism shown in Scheme 4. First, K2CO3 promotes the formation of rhodium alkoxide I from isopropanol. This species then undergoes β-hydride elimination to release acetone and form rhodium hydride II. Irreversible coordination of an alkene results in species III, which can undergo reversible migratory insertion to form either alkylrhodium species IV or IVa. Since the branched regioisomer of the product is not detected, we conclude that only IV is amidated, while IVa undergoes β-hydride elimination back to III. Upon dioxazolone coordination to IV to form V, turnover-limiting N─O bond cleavage and CO2 extrusion occur, forming rhodium nitrenoid VI. Migratory insertion and protodemetalation of the resultant N─Rh bond release the hydroamidated product and regenerate the catalyst.
Scheme 4. Proposed mechanism.

In summary, we have developed a novel method for highly selective Rh(III)-catalyzed anti-Markovnikov hydroamidation of unactivated alkenes, proceeding via reversible migratory insertion. The reaction exhibits a broad substrate scope under mild reaction conditions. In addition to tolerating a multitude of functional groups, the reaction is amenable to different classes of olefin, including electron deficient alkenes, styrenes, and 1,1-disubstituted alkenes, with no change in regioselectivity. Modulation of the dioxazolone also allows for the introduction of a variety of amide groups. Further advantages of the method include short (two-hour) reaction times, insensitivity to air, and the use of the ubiquitous solvent isopropanol as the hydride source. It is our hope that this reaction will provide a convenient way to append the prevalent amide functionality to molecules, and will enable new and streamlined synthetic pathways.
Supplementary Material
ACKNOWLEDGMENT
We thank NIGMS (GM80442) for support.
REFERENCES
- (1).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57 (24), 10257–10274. 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- (2).Roughley SD; Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 2011, 54 (10), 3451–3479. 10.1021/jm200187y. [DOI] [PubMed] [Google Scholar]
- (3).Huang L; Arndt M; Gooßen K; Heydt H; Gooßen LJ Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev 2015, 115 (7), 2596–2697. 10.1021/cr300389u. [DOI] [PubMed] [Google Scholar]
- (4).Hirano K; Miura M Hydroamination, Aminoboration, and Carboamination with Electrophilic Amination Reagents: Umpolung-Enabled Regio-and Stereoselective Synthesis of N-Containing Molecules from Alkenes and Alkynes. J. Am. Chem. Soc 2022, 144 (2), 648–661. 10.1021/jacs.1c12663. [DOI] [PubMed] [Google Scholar]
- (5).Huo J; He G; Chen W; Hu X; Deng Q; Chen D A Minireview of Hydroamination Catalysis: Alkene and Alkyne Substrate Selective, Metal Complex Design. BMC Chem. 2019, 13 (1), 89. 10.1186/s13065-019-0606-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Xi Y; Ma S; Hartwig JF Catalytic Asymmetric Addition of an Amine N─H Bond across Internal Alkenes. Nature 2020, 588 (7837), 254–260. 10.1038/s41586-020-2919-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ma S; Hill CK; Olen CL; Hartwig JF Ruthenium-Catalyzed Hydroamination of Unactivated Terminal Alkenes with Stoichiometric Amounts of Alkene and an Ammonia Surrogate by Sequential Oxidation and Reduction. J. Am. Chem. Soc 2021, 143 (1), 359–368. 10.1021/jacs.0c11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Ma S; Xi Y; Fan H; Roediger S; Hartwig JF Enantioselective Hydroamination of Unactivated Terminal Alkenes. Chem 2022, 8 (2), 532–542. 10.1016/j.chempr.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Jia S-M; Huang Y-H; Wang Z-L; Fan F-X; Fan B-H; Sun H-X; Wang H; Wang F Hydroamination of Unactivated Alkenes with Aliphatic Azides. J. Am. Chem. Soc 2022, 144 (36), 16316–16324. 10.1021/jacs.2c07643. [DOI] [PubMed] [Google Scholar]
- (10).Zhang X-G; He Z-X; Guo P; Chen Z; Ye K-Y Cobalt-Catalyzed Divergent Markovnikov and Anti-Markovnikov Hydroamination. Org. Lett 2022, 24 (1), 22–26. 10.1021/acs.orglett.1c03511. [DOI] [PubMed] [Google Scholar]
- (11).Musacchio AJ; Nguyen LQ; Beard GH; Knowles RR Catalytic Olefin Hydroamination with Aminium Radical Cations: A Photoredox Method for Direct C─N Bond Formation. J. Am. Chem. Soc 2014, 136 (35), 12217–12220. 10.1021/ja5056774. [DOI] [PubMed] [Google Scholar]
- (12).Nguyen TM; Manohar N; Nicewicz DA Anti-Markovnikov Hydroamination of Alkenes Catalyzed by a Two-Component Organic Photoredox System: Direct Access to Phenethylamine Derivatives. Angew. Chem. Int. Ed 2014, 53 (24), 6198–6201. 10.1002/anie.201402443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Lardy SW; Schmidt VA Intermolecular Radical Mediated Anti-Markovnikov Alkene Hydroamination Using N-Hydroxyphthalimide. J. Am. Chem. Soc 2018, 140 (39), 12318–12322. 10.1021/jacs.8b06881. [DOI] [PubMed] [Google Scholar]
- (14).Zhu Q; Graff DE; Knowles RR Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with Sulfonamides Enabled by Proton-Coupled Electron Transfer. J. Am. Chem. Soc 2018, 140 (2), 741–747. 10.1021/jacs.7b11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Miller DC; Ganley JM; Musacchio AJ; Sherwood TC; Ewing WR; Knowles RR Anti-Markovnikov Hydroamination of Unactivated Alkenes with Primary Alkyl Amines. J. Am. Chem. Soc 2019, 141 (42), 16590–16594. 10.1021/jacs.9b08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chinn AJ; Sedillo K; Doyle AG Phosphine/Photoredox Catalyzed Anti-Markovnikov Hydroamination of Olefins with Primary Sulfonamides via α-Scission from Phosphoranyl Radicals. J. Am. Chem. Soc 2021, 143 (43), 18331–18338. 10.1021/jacs.1c09484. [DOI] [PubMed] [Google Scholar]
- (17).Lundberg H; Tinnis F; Selander N; Adolfsson H Catalytic Amide Formation from Non-Activated Carboxylic Acids and Amines. Chem. Soc. Rev 2014, 43 (8), 2714–2742. 10.1039/C3CS60345H. [DOI] [PubMed] [Google Scholar]
- (18).Tomberg A; Boström J Can Easy Chemistry Produce Complex, Diverse, and Novel Molecules? Drug Discov. Today 2020, 25 (12), 2174–2181. 10.1016/j.drudis.2020.09.027. [DOI] [PubMed] [Google Scholar]
- (19).Ye Z-P; Hu Y-Z; Xia P-J; Xiang H-Y; Chen K; Yang H Photocatalytic Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes with N-Hydroxyphthalimide. Org. Chem. Front 2021, 8 (2), 273–277. 10.1039/D0QO01321H. [DOI] [Google Scholar]
- (20).Park Y; Park KT; Kim JG; Chang S Mechanistic Studies on the Rh(III)-Mediated Amido Transfer Process Leading to Robust C─H Amination with a New Type of Amidating Reagent. J. Am. Chem. Soc 2015, 137 (13), 4534–4542. 10.1021/jacs.5b01324. [DOI] [PubMed] [Google Scholar]
- (21).Zhou Y; Engl OD; Bandar JS; Chant ED; Buchwald SL CuH-Catalyzed Asymmetric Hydroamidation of Vinylarenes. Angew. Chem. Int. Ed 2018, 57 (22), 6672–6675. 10.1002/anie.201802797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Meng L; Yang J; Duan M; Wang Y; Zhu S Facile Synthesis of Chiral Arylamines, Alkylamines and Amides by Enantioselective NiH-Catalyzed Hydroamination. Angew. Chem. Int. Ed 2021, 60 (44), 23584–23589. 10.1002/anie.202109881. [DOI] [PubMed] [Google Scholar]
- (23).At the time of submission, Zhu and coworkers disclosed an asymmetric nickel-catalyzed hydroamidation of alkenyl boronates (see ref. 24).
- (24).Zhang Y; Qiao D; Duan M; Wang Y; Zhu S Enantioselective Synthesis of α-Aminoboronates by NiH-Catalysed Asymmetric Hydroamidation of Alkenyl Boronates. Nat. Commun 2022, 13 (1), 5630. 10.1038/s41467-022-33411-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ayushee; Patel M; Meena P; Jahan K; Bharatam PV; Verma AK Base-Mediated Anti-Markovnikov Hydroamidation of Vinyl Arenes with Arylamides. Org. Lett 2021, 23 (2), 565–570. 10.1021/acs.orglett.0c04084. [DOI] [PubMed] [Google Scholar]
- (26).Zhao G; Li J; Wang T Metal-Free Photocatalytic Intermolecular Anti-Markovnikov Hydroamination of Unactivated Alkenes. Eur. J. Org. Chem 2021, 2021 (18), 2650–2654. 10.1002/ejoc.202100049. [DOI] [Google Scholar]
- (27).Wang X; Widenhoefer RA Platinum-Catalyzed Intermolecular Hydroamination of Unactivated Olefins with Carboxamides. Organometallics 2004, 23 (8), 1649–1651. 10.1021/om0498549. [DOI] [Google Scholar]
- (28).Qian H; Widenhoefer RA Platinum-Catalyzed Intermolecular Hydroamination of Vinyl Arenes with Carboxamides. Org. Lett 2005, 7 (13), 2635–2638. 10.1021/ol050745f. [DOI] [PubMed] [Google Scholar]
- (29).Zhang Z; Lee SD; Widenhoefer RA Intermolecular Hydroamination of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes. J. Am. Chem. Soc 2009, 131 (15), 5372–5373. 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sevov CS; Zhou J; Hartwig JF Iridium-Catalyzed Intermolecular Hydroamination of Unactivated Aliphatic Alkenes with Amides and Sulfonamides. J. Am. Chem. Soc 2012, 134 (29), 11960–11963. 10.1021/ja3052848. [DOI] [PubMed] [Google Scholar]
- (31).Du B; Ouyang Y; Chen Q; Yu W-Y Thioether-Directed NiH-Catalyzed Remote γ-C(Sp3)─H Hydroamidation of Alkenes by 1,4,2-Dioxazol-5-Ones. J. Am. Chem. Soc 2021, 143 (37), 14962–14968. 10.1021/jacs.1c05834. [DOI] [PubMed] [Google Scholar]
- (32).Yin Y-N; Ding R-Q; Ouyang D-C; Zhang Q; Zhu R Highly Chemoselective Synthesis of Hindered Amides via Cobalt-Catalyzed Intermolecular Oxidative Hydroamidation. Nat. Commun 2021, 12 (1), 2552. 10.1038/s41467-021-22373-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Ma W; Zhang X; Fan J; Liu Y; Tang W; Xue D; Li C; Xiao J; Wang C Iron-Catalyzed Anti-Markovnikov Hydroamination and Hydroamidation of Allylic Alcohols. J. Am. Chem. Soc 2019, 141 (34), 13506–13515. 10.1021/jacs.9b05221. [DOI] [PubMed] [Google Scholar]
- (34).Lee S; Lei H; Rovis T A Rh(III)-Catalyzed Formal [4+1] Approach to Pyrrolidines from Unactivated Terminal Alkenes and Nitrene Sources. J. Am. Chem. Soc 2019, 141 (32), 12536–12540. 10.1021/jacs.9b07012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lee S; Semakul N; Rovis T Direct Regio- and Diastereoselective Synthesis of δ-Lactams from Acrylamides and Unactivated Alkenes Initiated by RhIII-Catalyzed C─H Activation. Angew. Chem. Int. Ed 2020, 59 (12), 4965–4969. 10.1002/anie.201916332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Noyori R; Hashiguchi S Asymmetric Transfer Hydrogenation Catalyzed by Chiral Ruthenium Complexes. Acc. Chem. Res 1997, 30 (2), 97–102. 10.1021/ar9502341. [DOI] [Google Scholar]
- (37).Bower JF; Skucas E; Patman RL; Krische MJ Catalytic C─C Coupling via Transfer Hydrogenation: Reverse Prenylation, Crotylation, and Allylation from the Alcohol or Aldehyde Oxidation Level. J. Am. Chem. Soc 2007, 129 (49), 15134–15135. 10.1021/ja077389b. [DOI] [PubMed] [Google Scholar]
- (38).Kim IS; Ngai M-Y; Krische MJ Enantioselective Iridium-Catalyzed Carbonyl Allylation from the Alcohol or Aldehyde Oxidation Level via Transfer Hydrogenative Coupling of Allyl Acetate: Departure from Chirally Modified Allyl Metal Reagents in Carbonyl Addition. J. Am. Chem. Soc 2008, 130 (44), 14891–14899. 10.1021/ja805722e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Liang T; Woo SK; Krische MJ C-Propargylation Overrides O-Propargylation in Reactions of Propargyl Chloride with Primary Alcohols: Rhodium-Catalyzed Transfer Hydrogenation. Angew. Chem. Int. Ed 2016, 55 (32), 9207–9211. 10.1002/anie.201603575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Imai H; Nishiguchi T; Fukuzumi K Transfer Hydrogenation and Transfer Hydrogenolysis. II. Catalytic Activity of Some Soluble Complexes in Hydrogen Transfer from Alcohols to Olefins and the Mechanism of the Reaction Catalyzed by Hydridotetrakis(Triphenylphosphine)Rhodium(I). J. Org. Chem 1974, 39 (12), 1622–1627. 10.1021/jo00925a004. [DOI] [Google Scholar]
- (41).Ok F; Aydemir M; Durap F; Baysal A Novel Half-Sandwich H5-Cp *–Rhodium(III) and H5-Cp *–Ruthenium(II) Complexes Bearing Bis(Phosphino)Amine Ligands and Their Use in the Transfer Hydrogenation of Aromatic Ketones. Appl. Organomet. Chem 2014, 28 (1), 38–43. 10.1002/aoc.3068. [DOI] [Google Scholar]
- (42).Richers CP; Roediger S; Laserna V; Hartwig JF Effects of Ligands on the Migratory Insertion of Alkenes into Rhodium–Oxygen Bonds. Chem. Sci 11 (38), 10449–10456. 10.1039/d0sc04402d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Rix FC; Brookhart M; White PS Electronic Effects on the β-Alkyl Migratory Insertion Reaction of Para-Substituted Styrene Methyl Palladium Complexes. J. Am. Chem. Soc 1996, 118 (10), 2436–2448. 10.1021/ja9527434. [DOI] [Google Scholar]
- (44).Borah AJ; Shi Z Rhodium-Catalyzed, Remote Terminal Hydroarylation of Activated Olefins through a Long-Range Deconjugative Isomerization. J. Am. Chem. Soc 2018, 140 (19), 6062–6066. 10.1021/jacs.8b03560. [DOI] [PubMed] [Google Scholar]
- (45).We believe this signal to be consistent with a rhodium-dimer species with a bridging hydride ligand (see refs. 46 and 47, and SI for further details on the nature of this complex). .
- (46).Boyd EA; Lionetti D; Henke WC; Day VW; Blakemore JD Preparation, Characterization, and Electrochemical Activation of a Model [Cp*Rh] Hydride. Inorg. Chem 2019, 58 (6), 3606–3615. 10.1021/acs.inorgchem.8b02160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Colebrooke SA; Duckett SB; Lohman JAB; Eisenberg R Hydrogenation Studies Involving Halobis(Phosphine)–Rhodium(i) Dimers: Use of Parahydrogen Induced Polarisation To Detect Species Present at Low Concentration. Chem. – Eur. J 2004, 10 (10), 2459–2474. 10.1002/chem.200305466. [DOI] [PubMed] [Google Scholar]
- (48).Samec JSM; Bäckvall J-E; Andersson PG; Brandt P Mechanistic Aspects of Transition Metal-Catalyzed Hydrogen Transfer Reactions. Chem. Soc. Rev 2006, 35 (3), 237–248. 10.1039/B515269K. [DOI] [PubMed] [Google Scholar]
- (49).Maity S; Potter TJ; Ellman JA α-Branched Amines by Catalytic 1,1-Addition of C─H Bonds and Aminating Agents to Terminal Alkenes. Nat. Catal 2019, 2 (9), 756–762. 10.1038/s41929-019-0330-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Guiral V; Delbecq F; Sautet P Hydride Transfer Reduction of Carbonyls by a Rhodium(I) Complex: A Theoretical Study. 1. The Two-Step Mechanism. Organometallics 2000, 19 (8), 1589–1598. 10.1021/om990603n. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.