

Abstract
Pediatric acute myeloid leukemia expressing the ETO2∷GLIS2 fusion oncogene is associated with dismal prognosis. Previous studies have shown that ETO2∷GLIS2 can efficiently induce leukemia development associated with strong transcriptional changes but those amenable to pharmacological targeting remained to be identified. By studying an inducible ETO2∷GLIS2 cellular model, we uncovered that de novo ETO2∷GLIS2 expression in human cells led to increased CASP3 transcription, CASP3 activation, and cell death. Patient-derived ETO2∷GLIS2+ leukemic cells expressed both high CASP3 and high BCL2. While BCL2 inhibition partly inhibited ETO2∷GLIS2+ leukemic cell proliferation, BH3 profiling revealed that it also sensitized these cells to MCL1 inhibition indicating a functional redundancy between BCL2 and MCL1. We further show that combined inhibition of BCL2 and MCL1 is mandatory to abrogate disease progression using in vivo patient-derived xenograft models. These data reveal that a transcriptional consequence of ETO2∷GLIS2 expression includes a positive regulation of the pro-apoptotic CASP3 and associates with a vulnerability to combined targeting of two BCL2 family members providing a novel therapeutic perspective for this aggressive pediatric AML subgroup.
INTRODUCTION
Acute myeloid leukemia (AML) remains a therapeutic challenge both in children and adults with a long-term disease-free survival rate of <60% and <30%, respectively [1, 2]. Among the numerous genetic alterations identified to date in pediatric AML, some molecular subgroups, including MLL, NUP98, and ETO2∷GLIS2 fusions are associated with particularly poor prognosis resulting in frequent resistance to treatment and relapses [3].
The resistance to cell death is a hallmark of tumor cells. A major regulatory mechanism controlling survival vs. mitochondrial-mediated apoptosis involves a balanced activity within the large BH3-containing proteins [4], including pro-apoptotic effectors (e.g., BAX, BAK, BOK), pro-apoptotic BH3-only (e.g., BAD, BIM, HRK) and anti-apoptotic (e.g., BCL2, MCL1, BCL-xL, BCL-W) factors. In this apoptotic process, caspase 3 (CASP3) is a major effector. In human adult AML, this led to the identification of BCL2 dependencies [5] and the approval of venetoclax (ABT199), a BH3 mimetic BCL2 inhibitor for the treatment of elderly patients with AML and other hematological malignancies [6, 7]. For pediatric leukemia, preclinical models indicate the relevance of BCL2 inhibition in lymphoid leukemia [8] and illustrate the predictive value of ex vivo functional assessment of BCL2 dependence using BH3-profiling [9]. Venetoclax alone or in combination with chemotherapy is being explored in pediatric patients and has shown activity in combination for AML [10, 11] and ALL [12]. However, while venetoclax provides promising short-term effects and clinical benefits for leukemia patients, it is often associated with relapse [13]. TP53 activity, cell-type specificities and metabolic priming have been proposed as potential resistance mechanisms to venetoclax treatment [14–17]. Also, in adult AML, the redundant activity of other anti-apoptotic proteins led to the proposed usage of combined inhibition of several BCL2 family members, including MCL1 [17–20]. While these data support that targeting the BCL2 family is of clinical interest for AML patients, the molecular determinants controlling the relative sensitivity or resistance to BCL2 inhibition remain to be better characterized to identify both the molecular subgroups that would benefit the most from such therapy and the mechanisms to overcome the frequently observed resistance.
Pediatric AML associated with the ETO2∷GLIS2 fusion exhibits features of aggressive AML and poor prognosis [21]. At the molecular level, ETO2∷GLIS2 can bind DNA both through ETO2-interacting transcription factors or directly through GLIS2 DNA binding domain [22]. ETO2∷GLIS2 binding is observed at enhancer regions and results in a strong imbalance in master transcription factor activities including GATA and ETS factors [22, 23]. In a murine model, ETO2∷GLIS2 expression induces leukemia development in a cell context-dependent manner [24] suggesting the importance of cell-specific chromatin state for transformation. Together, like other pediatric AML oncogenes including MLL or NUP98 fusions, ETO2∷GLIS2 induces strong transcriptional alterations that warrant further characterizations. Of note, while Glis2 has been suggested to contribute to hematopoietic stem cell biology [25] and differentiation of MOZ∷TIF2 AML blasts [26], its molecular consequences have been mostly studied in the kidney [27] and are poorly understood in the hematopoietic system. In addition, the relevance of GLIS-dependent mechanisms extends outside hematopoietic cancers as GLIS2 is involved in chemoresistance and metastastic properties [28, 29] and chromosomal translocations with GLIS1/GLIS3 are present in other cancers [30].
Here, we found that strong transcriptional deregulations imposed by ETO2∷GLIS2, include both a positive regulation of the pro-apoptotic factor CASP3 and a high BCL2 expression in ETO2∷GLIS2+ AMKL patient cells. This provided a molecular rationale for targeting the BCL2 family members within this aggressive pediatric AML subtype.
METHODS
Patient-derived data and material
Patient blood and BM samples were obtained with the written informed consent of the patient or their family in accordance with the Declaration of Helsinki and the study was approved by the Gustave Roussy institutional review board. The study was also approved by the Ethics Committee of Saint-Antoine Paris University Hospital (Assistance Publique-Hôpitaux de Paris) and by the Institutional Review Board of the French Regulatory Agency.
Vectors
The ETO2∷GLIS2, ETO2∷GLIS2ΔNHR2, and ETO2∷GLIS2C265G cDNA fused to GFP in Ct were cloned into the doxycycline (DOX) inducible LT3-GEPIR lentiviral vector (gift from J. Zuber, Austria).
Cell culture
HEL 92.1.7 (thereafter named HEL), CHRF-288-11 (thereafter named CHRF), CMS and WSU-AML cells were cultured in RPMI-1640 supplemented with 10% FBS, penicillin (100 U/mL)-streptomycin (100 μg/mL), and L-glutamine (2 mmol/L) (Gibco). For M07e cells, medium was supplemented with human GM-CSF (5 ng/mL) (PeproTech). For patient-derived xenograft cells medium was with human SCF, IL3, IL6, GM-CSF, TPO, and FLT3L (10 ng/ml of each). Clones of HEL cells with doxycycline-inducible expression of ETO2∷GLIS2 (and mutants) were generated by lentiviral transduction followed by 4-day of puromycin selection, cell sorting of single cells into 96-well plates and selection of the clones through qRT-PCR and western blot for ETO2∷GLIS2 expression.
Cell viability and flow cytometry
For viability assays, cells were seeded at 1–2 × 105 cells/well in 96-well plates. The CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega) was used according to the manufacturer’s instructions. Combination index (CI) was calculated with CompuSyn software (Combosyn Inc., Paramus, NJ). CI < 1 indicates a synergistic combination effect.
For flow cytometry, cells were stained in PBS 1X supplemented with 2% FBS for 30 min at 4 °C in the dark and washed prior to FACS analysis. Antibodies are described in Supplementary Table 1. Sytox blue (Thermo-Fisher Scientific) exclusion is used to identify live cells. For annexin V staining, stainings were performed after 48–72 h of drug treatment following the manufacturer’s recommendation (BD Biosciences). Data were acquired on FACSCanto II or FACSCanto X flow cytometer (BD). Analyses were performed using FlowJo software.
BH3 profiling
For BH3 profiling, 2 × 105 cells were washed with DTEB buffer (135 mM Trehalose, 20 μM EDTA, 20 μM EGTA, 5 mM Succinic Acid, 0.1% BSA, 10 mM HEPES, 50 mM KCl, pH 7.5) and permeabilized with DTEB + 5 μg/mL digitonin. Loss of mitochondrial membrane potential (ψm) was assessed using TMRE (200 nM) after one hour of incubation at 30 °C with BH3 peptides as described previously [31] (Supplementary Table 2). The fluorescence was measured using a MACSQuant® Analyzers (Miltenyi, France).
Caspase activity measurement assay
After stimulation, cells were lysed for 30 min at 4 °C in lysis buffer (50 mM HEPES pH 8, 150 mM NaCl, 20 mM EDTA, 1 mM PMSF, 10 μg/mL leupeptin, 10 μg/mL aprotinin, and 0.2% Triton X-100) and lysates were cleared at 10.000 g for 15 min at 4 °C. Each assay (in triplicate) was performed with 10 μg of protein prepared from control or stimulated cells. Briefly, cellular extracts were incubated in a 96-well plate with 0.2 mM of Ac-DEVD-AMC (Caspase-3) or Ac-LEHD-AMC (Caspase-9) as substrates for various times at 37 °C. Caspase activity was measured by following emission at 460 nm (excitation at 390 nm) in the presence or absence of 10 μM of DEVD-CHO or LEHD-CHO. Enzyme activities were expressed in arbitrary units (A.U.) per min and per mg of proteins.
Western blot and immunoprecipitation
Western blot analyses were performed with the primary antibodies listed in Supplementary Table 1 and detailed methods to evaluate BAX activation are provided in the Supplementary Methods.
Transcriptome
RNA was isolated from the HEL GFP (CTRL), ETO2∷GLIS2, ETO2∷GLIS2C265G, and ETO2∷GLIS2ΔNHR2 following 24 h of induction with 500 ng/mL of DOX using the RNeasy Mini/Micro Kit (Qiagen) and quantified by NanoDrop (ThermoScientific). 50-bp paired-end sequencing was performed. Detailed information on the bioinformatics analyses is provided in the Supplementary Methods.
Murine preclinical models
Patient-derived xenograft protocols and the AMKL7Luc model have been described previously [22, 32]. Cells were injected into NSG immunodeficient mice (Jackson Laboratory, Bar Harbor, ME, USA, stock number: 005557) maintained in the Gustave Roussy preclinical facility and all experiments were approved by the French National Animal Care and Use Committee (CEEA 26: #201712180922196_v2 and #2017122111548235_v2). 3–5 × 104 AMKL7Luc cells were intra-venously injected into irradiated (1.5 Gy) 6–12 weeks old NSG females. After confirmation of engraftment, the mice were randomly assigned to groups without blinding and treated (4 weeks) with vehicle, ABT (100 mg/kg, oral gavage 5 times per week), S63845 (40 mg/kg, intra-venously injection once a week), or the combination (n ≥ 7 per group). Mice were monitored by bioluminescence imaging (IVIS Spectrum and Ami HT) for AMKL7 or bone marrow sampling for CONECT-110 and analyzed upon disease development.
Statistical analysis
Statistical significance was calculated using Prism (version 6.0a) and is indicated as p values. Except when otherwise specified, Student’s t test was used. *: p < 0.05, **: p < 0.01, ***: p < 0.001.
RESULTS
The ETO2∷GLIS2 fusion is associated with high CASP3 expression
To analyze the consequence of ETO2∷GLIS2 expression in human cells, we generated doxycycline-inducible expression of GFP-tagged ETO2∷GLIS2 and two mutants [ETO2∷GLIS2C265G (shown to impede GLIS2 DNA binding properties [22, 33]) and ETO2∷-GLIS2ΔNHR2 (deleted for the NHR2 domain of ETO2 involved in protein interactions and tetramerization)] in the HEL cell line (Fig. 1A, Supplementary Fig. 1A). Surprisingly, ETO2∷GLIS2 expression led to a strong decrease in HEL cell numbers (Fig. 1B) associated with an increased Annexin V staining (Fig. 1C, Supplementary Fig. 1B). ETO2∷GLIS2ΔNHR2 showed a similar trend but with a significantly lower effect. However, ETO2∷GLIS2C265G expression did not induce any significant effect indicating a requirement for the GLIS2 DNA binding activity in cell death induction.
Fig. 1. ETO2∷GLIS2 induces cell death with CASP3 upregulation mostly via GLIS2 moiety.
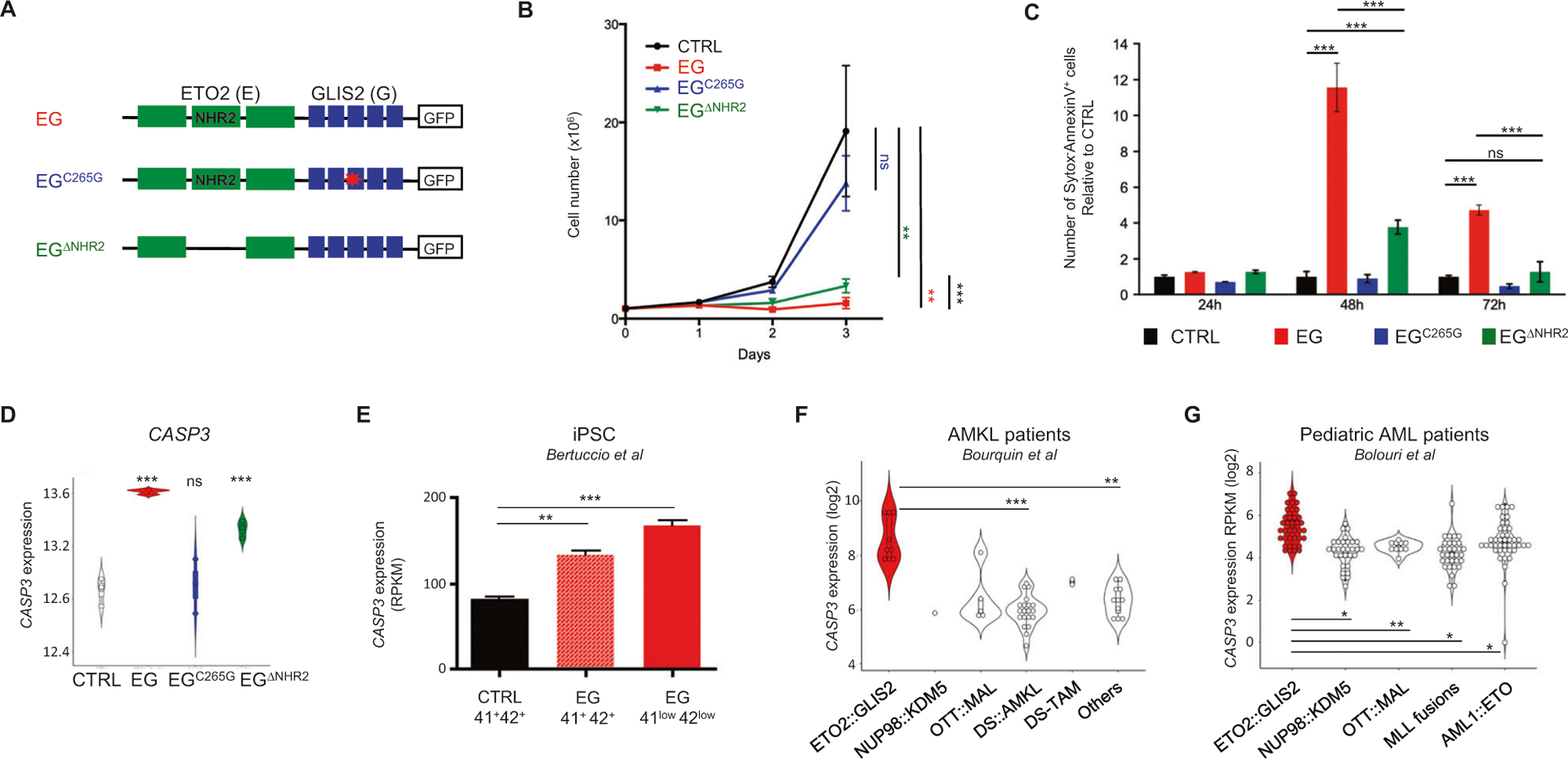
A HEL model of doxycycline-inducible expression of ETO2∷GLIS2 (EG), ETO2∷GLIS2ΔNHR2, and ETO2∷GLIS2C265G. Schematic representation of the constructs expressed through lentiviral delivery to generate HEL models. Empty box: GFP tag fused in Ct of the constructs. B Proliferation assay for HEL cells upon expression of ETO2∷GLIS2 and mutants. Mean ± SEM (n = 6) is shown. Statistical significance is indicated as p values (Student t-test). ns: p > 0.05, **p < 0.01, ***p < 0.001. C Histogram of the percentages of Sytox−AnnexinV+ apoptotic cells analyzed by flow cytometry. Mean ± SEM (n = 3) is represented. D CASP3 mRNA levels on HEL cells upon expression of ETO2∷GLIS2 and mutants (log2 RPKM). E CASP3 mRNA levels upon expression of ETO2∷GLIS2 in hematopoietic cells differentiated from wild-type (CTRL) and ETO2∷GLIS2 (EG) induced pluripotent stem cells [34]. 41+42+ indicates expression in maturing megakaryocytes. 41low42low indicates expression in EG-specific immature population presenting a molecular signature closer to ETO2∷GLIS2 patients. F CASP3 mRNA levels in pediatric AMKL subgroups [32, 54]. Wilcoxon test corrected for multiple comparisons was used. The low number of samples in some group precluded computation of statistical significance between some groups. G CASP3 mRNA levels in pediatric AML subgroups [3]. Wilcoxon test corrected for multiple comparisons (adjusted p-values computed with Benjamini-Hochberg method) was used. Statistical significance with the indicated test is shown: *<0.05, **<0.01, ***<0.001, ns >0.05.
To characterize the transcriptional consequences of ETO2∷GLIS2 expression in this model, we performed RNAseq shortly (24 h) after induction. Analyses by principal component analysis (Supplementary Fig. 1C) and computation of the number of differentially expressed genes with a twofold change threshold (Supplementary Fig. 1D) confirmed that ETO2∷GLIS2 induced important transcriptional changes. A single point mutation in the DNA binding domain of GLIS2 (ETO2∷GLIS2C265G mutant) strongly reduces the transcriptional changes imposed by ETO2∷GLIS2, while the deletion of the NHR2 domain of ETO2 had much less consequences, revealing the importance of the GLIS2 DNA binding properties in the ETO2∷GLIS2-imposed transcriptional signature. As expected, known ETO2∷GLIS2 targets including ERG and GATA1 [22], were upregulated and downregulated, respectively (Supplementary Fig. 1E). To search for the molecular bases of cell death, we first investigated expression of known pro-apoptotic regulators. CASP3, an essential effector of apoptosis, stood out as strongly upregulated upon ETO2∷GLIS2 and ETO2∷GLIS2ΔNHR2, but not upon ETO2∷GLIS2C265G expression (Fig. 1D). Previous expression data [22] also revealed that HEL transduction with GLIS2 induced CASP3 expression to a much higher extent than with ETO2 (Supplementary Fig. 1F). In another recent human cell model based on induced pluripotent stem cell hematopoietic differentiation, both maturing ETO2∷GLIS2 CD41+CD42+ cells and the more immature ETO2∷GLIS2 CD41lowC-D42low cell population, which showed the closest transcriptional signature to patients leukemic cells, expressed higher CASP3 compared to control cells (Fig. 1E) [34]. Importantly, two independent transcriptome datasets of pediatric AMKL (Fig. 1F) and pediatric AML (Fig. 1G) cohorts showed higher CASP3 expression in ETO2∷GLIS2+ samples compared to other molecular subgroups (including MLL, NUP98, or AML1∷ETO fusions).
These data indicate that ETO2∷GLIS2 ectopic expression induces cell death and that ETO2∷GLIS2 positively controls CASP3 transcription in part via the GLIS2 moiety.
Ectopic ETO2∷GLIS2 expression induces the intrinsic cell death pathway
We then investigated whether CASP3 was functionally activated upon ETO2∷GLIS2 expression. Both western blotting and flow cytometry analyses in HEL cells showed that ETO2∷GLIS2, but not ETO2∷GLIS2C265G, strongly induced expression of a cleaved CASP3 at the expected size for the apoptosis-inducing CASP3 form (~17–19 kDa) (Fig. 2A, Supplementary Fig. 2A). The ETO2∷-GLIS2ΔNHR2 mutant also induced a weak CASP3 activation. Consistently, the dosage of CASP3 activity using Ac-DEVD-AMC as a substrate showed a strong activation by ETO2∷GLIS2, a weaker activation by ETO2∷GLIS2ΔNHR2 and no significant activation by the ETO2∷GLIS2C265G (Fig. 2B).
Fig. 2. Ectopic ETO2∷GLIS2 expression induces CASP3 activation and the intrinsic apoptosis pathway.

A Western blot analysis of CASP3 expression in HEL cell 48 h post-induction. B Caspase cleavage activity using DEVD peptide. The AU/mg of protein ± SD (n = 3) is represented. C Representative TMRE staining in the absence or presence of doxycycline (DOX) for 48 h. D Quantification of results shown in C (n = 3). Statistical significance is indicated as p values (Student t test). ns: p > 0.05, **p < 0.01, ***p < 0.001. E Cell extracts were fractionated and analyzed for release of SMAC and CytoC in the cytoplasm. PDI and γ-Tubulin were used as markers of microsome and cytoplasm, respectively. F Detection of active BAX in ETO2∷GLIS2 vs. ETO2∷GLIS2C265G HEL cells. IP: immunoprecipitation. Ig: unspecific immunoglobulin. G Western blot analyses of the indicated proteins in HEL models and M07e cells. H CTRL and ETO2∷GLIS2 HEL cells were treated with 50 μM QVD and analyzed for Annexin V staining 48 h post-doxycycline induction. I Western blot analysis of CASP3 expression in cells analyzed in H. Statistical significance is indicated as adjusted p values (FDR): *<0.05, **<0.01, ***p < 0.001.
To define the mechanism upstream of CASP3 activation in ETO2∷GLIS2-expressing cells, we next investigated the involvement of the mitochondria. Indeed, in the intrinsic cell death pathway, apoptotic signals leading to the formation of BAX complexes induce mitochondrial depolarization and the release of Cytochrome C, then the activation of CASP9, which in turn cleaves CASP3 into its activated form [35]. Using tetramethylrhodamine ethyl ester (TMRE) to label active mitochondria, we observed that ETO2∷GLIS2 expression increased the number of depolarized mitochondria (52%) while this effect is reduced with ETO2∷GLIS2ΔNHR2 (28%) and abolished by the ETO2∷GLIS2C265G mutant (<5%)(Fig. 2C, D). The quantification of cytoplasmic and microsome contents of SMAC and Cytochrome C showed cytoplasmic released of SMAC/CytoC upon ETO2∷GLIS2 expression, which was abrogated upon ETO2∷-GLIS2C265G expression (Fig. 2E). Active BAX protein could be readily immunoprecipitated from ETO2∷GLIS2-, but not ETO2∷GLIS2C265G-expressing cells, and was barely detectable in ETO2∷GLIS2ΔNHR2-expressing cells (Fig. 2F). Also, pro-apoptotic BCL2 family members (e.g., BCL2, BCL-xL, MCL1) and anti-apoptotic factors (e.g., BIM, BMF, and NOXA) showed changes in expression upon ETO2∷GLIS2 induction (Supplementary Fig. 2B, C), suggesting that an imbalance in BAX-regulating factors may also contribute to ETO2∷GLIS2-induced mitochondrial depolarization. ETO2∷GLIS2-induced downstream caspase activation as shown by CASP9 and PARP cleavage (Fig. 2G), as well as Ac-LEHD-AMC cleavage assays revealing activation of CASP9 (Supplementary Fig. 2D). In addition, the pre-treatment with Q-VD-OPh (QVD), a pan-caspase inhibitor [36], abrogated ETO2∷GLIS2-induced cell death (Fig. 2H, I).
Together, these data indicate that ectopic ETO2∷GLIS2 expression results in a GLIS2-dependent induction of BAX activation, mitochondrial membrane depolarization and activation of a caspase-dependent apoptosis pathway. While involvement of the ETO2 moiety in this process is supported by the reduced induction of apoptosis features upon ETO2∷GLIS2ΔNHR2 expression, its significant effect on proliferation inhibition suggests the involvement of other mechanisms important for cell viability.
BH3-profiling indicates a dependence on BCL2 and MCL1 in ETO2∷GLIS2+ cells
To reconcile the observations that ectopic ETO2∷GLIS2 expression induces apoptosis with CASP3 activation, while ETO2∷GLIS2+ patient leukemic cells maintain high CASP3 expression without cell death, we then investigated anti-apoptotic factors including BCL2 that can antagonize mitochondria-induced apoptosis. BCL2 was highly expressed in three ETO2∷GLIS2+ patient cell lines (M07e, WSU-AML, CMS) and an ETO2∷GLIS2+ PDX-amplified patient sample (AMKL7) in comparison to two ETO2∷GLIS2− cells (CHRF, HEL) (Fig. 3A). Based on these data, we assessed the relevance of BCL2 activity in the survival of ETO2∷GLIS2+ cells by treating M07e cells with the BCL2 inhibitor ABT199 (a.k.a. venetoclax) [5]. ABT199 treatment led to a significant but moderate reduction in cell proliferation in a dose-dependent manner (Fig. 3B). The BCL2 family is composed of several anti-apoptotic regulators that present functional redundancy with BCL2 that may explain the moderate inhibitory effect observed in M07e cells. Western blot and transcriptome analysis showed BCL-xL, BCL-W and MCL1 were readily, but not specifically, expressed in ETO2∷GLIS2+ cells (M07e, WSU-AML, CMS, and AMKL7) (Fig. 3A, Supplementary Fig. 3). To test the sensibility of ETO2∷GLIS2 cells to BCL2 family proteins, we used seven well-characterized peptides [37] (Supplementary Table 2) to perform BH3-profiling. Response to the BAD peptide was consistent with a dependency on BCL2 in M07e (Supplementary Fig. 4A) and three other ETO2∷GLIS2+ cell lines (Supplementary Fig. 4B). Notably, expression of ETO2∷GLIS2, and to a lower extent ETO2∷GLIS2ΔNHR2, in HEL cells enhanced response to the BAD peptide (Supplementary Fig. 4C), confirming that expression of ETO2∷GLIS2 indeed provided an enhanced dependency on BCL2 activity.
Fig. 3. High BCL2 expression and redundancy between BCL2 and MCL1 in ETO2∷GLIS2+ leukemic cells.

A Western blot analysis of ETO2∷GLIS2− (CHRF, HEL) and ETO2∷GLIS2+ (M07e, WSU-AML, CMS) human leukemia cell lines and of cells from a PDX model of ETO2∷GLIS2+ AMKL (AMKL7). Red writing indicates ETO2∷GLIS2+ cells. B Relative cell number of M07e cells treated for 72 h with the indicated concentration of ABT199 using MTS assay. Counts were normalized to the DMSO (0) condition. Mean + /−SEM (n = 3) is indicated. C Representative BH3 profiling analysis in M07e cells. The percentages of TMRE-negative cells are indicated on the FACS plots. Lower raw: cells were pretreated with 50 nM of ABT199 for 16 h. D Histogram representation of the percentages of TMRE negative cells described in C. Mean ± SD of three independent analyses is shown. E Representative BH3 profiling analysis on cells from a PDX model (AML026). Lower raw: cells were pretreated with 5 nM of ABT199 for 16 h. F Histogram representation of the percentages of TMRE negative cells described in E. Mean ± SD of three independent PDX models (AML026, AMKL7, and CONECT-110) is shown. Statistical significance is indicated as p values (Student t test). *p < 0.05, **p < 0.01, ***p < 0.001.
As MCL1 was found redundant with BCL2 in adult AML and induces resistance to ABT199 treatment [18, 19], we then investigated the dependence of M07e cells toward MCL1 following pretreatment with a low concentration of ABT199 (50 nM) using BH3 profiling with increasing doses of the MS1 peptide specific for MCL1 activity. Mock-treated M07e cells presented a moderate response to the MS1 peptide, indicating that their mitochondrial potential is not significantly dependent on MCL1 at the basal level. However, a pretreatment with ABT199 strikingly decreased their mitochondrial potential (Fig. 3C, D). To confirm these results on other ETO2∷GLIS2+ cells, we performed additional ex vivo BH3 profiling using cells from 3 independent ETO2∷GLIS2+ PDX models (Fig. 3E, F and Supplementary Fig. 4D). As little as 5 nM of ABT199 pretreatment was sufficient to significantly sensitize these cells to the MS1 peptide. Of note, the basal loss of mitochondrial potential (visualized with the PUMA2A mutant peptide) was expectedly higher in PDX models as compared to the established M07e cell line.
Together, these results indicate that ETO2∷GLIS2+ cells mitochondrial potential is dependent in part on BCL2 with a functional redundancy between BCL2 and MCL1.
Combined BCL2 and MCL1 targeting inhibits ETO2∷GLIS2+ leukemia proliferation in vitro
The expression of BCL2 protein members and BH3 profiling results led us to test whether combined inhibition of both BCL2 and MCL1 would be required to achieve cell death in ETO2∷GLIS2+ cells. Therefore, we first compared the response of two ETO2∷GLIS2+ cell lines (M07e and WSU-AML) and an ETO2∷GLIS2-cell line (HEL) to various combinations of the BCL2 inhibitor (ABT199) and MCL1 inhibitor (S63845) [38] using proliferation assay after 72 h of treatment. While ABT199 or S63845 alone partly reduced M07e and WSU-AML cell viability in a dose-dependent manner, combined inhibition led to a complete abrogation of cell viability (Fig. 4A and Supplementary Fig. 5A). Similar combinations did not significantly affect HEL cells proliferation (Supplementary Fig. 5A). Western blotting for CASP3 (Fig. 4B) and quantification of its cleavage activity with the DEVD substrate (Fig. 4C) confirmed that combining ABT199 and S63845 resulted in increased CASP3 activation in M07e cells. The caspase activity of CASP9, an initiator caspase, was measured using Ac-LEHD-AMC as substrate and showed a strong increase in M07e upon combined treatment with ABT199 and S63845 as compared to single treatment, indicating increased effect of the combination on upstream caspase activation (Supplementary Fig. 5B). We then performed similar proliferation assays ex vivo on cells obtained from PDX models of pediatric AML presenting either ETO2∷GLIS2 (AML-026, AMKL7, CONECT-110) (Fig. 4D, E and Supplementary Fig. 5C) or other AMKL fusions (AMKL1: OTT∷MAL, AMKL4: NUP98∷KDM5A [32]) (Fig. 4E and Supplementary Fig. 5D) and an unrelated pediatric AML (CONECT-208) (Fig. 4E). ETO2∷GLIS2+ PDX-derived cells showed a significant inhibition of proliferation upon combined treatment. Notably, while AMKL samples with other fusions (AMKL1 and AMKL4) showed a lower response to ABT199 alone, they did exhibit a significant response to S63845 alone and to the combination. To assess whether the combined treatment presents a truly synergistic effect, we computed a combination index (Fig. 4F). For ETO2∷GLIS2+ cells, most analyses resulted in a combination index <1 supporting a synergistic effect between ABT119 and S63845.
Fig. 4. Combined BCL2 and MCL1 inhibition synergizes in ETO2∷GLIS2 AMKL in vitro.

A Relative cell number for the ETO2∷GLIS2+ M07e cell line following culture for 72 h with the indicated concentration of ABT199 and/or S63845 using MTS assay. Mean ± SEM (n = 3–6) is shown. B Western blot analysis of CASP3 expression in M07e cells after 48 h of treatment. C DEVD cleavage activity in M07e cells analyzed at 4 h following treatment with ABT199 (50 nM), S63845 (50 nM) or the combination of both. Mean ± SD of duplicates are shown. D Relative cell number cells following ex vivo treatment of cells from the AML026 PDX model for 48 h with the indicated concentration of ABT199 and/or S63845 using MTS assay. Mean ± SEM of three replicates is shown. E Heatmap representation of the relative cell number compared to controls following treatments with indicated doses as in C. Upper panel: human cell lines. Lower panel: ex vivo treatment of PDX models. Red writing indicates ETO2∷GLIS2+ cells. F Combination index computed with the Compusyn software using data generated in A, C, D. Each dot represents an independent replicate. Red dots indicate ETO2∷GLIS2+ samples. Statistical significance is indicated as p values (Student t test). *p < 0.05, **p < 0.01, ***p < 0.001.
These results demonstrate that ETO2∷GLIS2+ leukemic cells are vulnerable to the combined inhibition of BCL2 and MCL1, which led to a synergistic effect on CASP3 activation and induction of cell death in vitro.
Combined BCL2 and MCL1 inhibition abrogates disease development in PDX models
To ascertain the consequence of combining BCL2 and MCL1 inhibition on ETO2∷GLIS2+ leukemia in vivo, we performed preclinical assays on PDX models in which a lentivirally-delivered dual luciferase/mCherry expressing reporter allows to follow disease development through non-invasive bioluminescent imaging and flow cytometry, respectively. Treatment with ABT199 (100 mg/kg; daily oral gavage: 5 days ON, 2 days OFF) and S63845 (40 mg/kg; weekly i.v.) following a previously reported regimen [39] was performed for 4 weeks and animals were imaged during treatment and regularly during disease development (Fig. 5A). Treatment of the AMKL7 PDX model with ABT199 alone led to a significant but moderately delayed disease development while S63845 alone did not affect disease development compared to placebo-treated mice (Supplementary Fig. 5E). However, the combined treatment with ABT199 and S63845 abrogated disease development as assessed by bioluminescent imaging (Fig. 5B), flow cytometry analyses for the presence of leukemic cells at time of sacrifice (Fig. 5C) and overall survival (Fig. 5D). An independent preclinical assay on other ETO2∷GLIS2+ PDX model (CONECT-110) was also performed with full analyses of mice 6 weeks following the end of the treatment. As compared to placebo-treated, combined treatment also triggered an inhibition of disease development (Fig. 5E) and a lack of leukemic cells at the time of sacrifice (Fig. 5F).
Fig. 5. Combined BCL2 and MCL1 inhibition abrogates disease development in PDX models.

A Schematic representation of the 4-week treatment schedule for ABT199 alone, S63845 alone or the combination. Prior to injection into NSG immunodeficient murine recipients, cells from PDX models were transduced with dual mCherry/luciferase reporter. Arrow heads indicate timing of the imaging to quantify the luciferase (Luc) expressed in PDX leukemic cells. B Quantification of luciferase activity during and after treatment of the AMKL7 PDX model with vehicle only (placebo) (n = 9) or the combination of ABT199 and S63845 (n = 7). Left panel: mean ± SD of the indicated number of mice per group is shown. Right panel: representative images at 14 weeks. C Representative flow cytometry analysis of bone marrow cells to quantify mCherry+ leukemic cells. SSC-A: Side scatter area. D Kaplan–Meier survival plot for the mice described in B, C. Median survival: CTRL = 99.5 (n = 18), ABT199 = 130 (n = 10), S63845 = 98 (n = 10), ABT199 + S63845 = undefined (n = 7). Note that for the two animals that succumbed the ABT199 + S63845 group, flow cytometry could not be performed as the animals were found post-mortem but an analysis on the bone marrow aspirate two weeks prior to their death showed an absence of human leukemia similarly to the other mice in this group, leaving the cause of their death undetermined. E Quantification of luciferase activity during and after treatment of the CONECT-110 PDX model with vehicle only (placebo) or the combination of ABT199 and S63845. Mean ± SEM of the indicated number of mice per group in shown. Statistical significance is indicated as p values (Student t test). *p < 0.05, **p < 0.01, ***p < 0.001. F Representative flow cytometry analysis of bone marrow cells for the detection of KIT+CD41+ leukemic cells.
Altogether, these in vivo data show that combined treatment with BCL2 and MCL1 inhibitors abrogates disease development in these PDX models of ETO2∷GLIS2+ leukemia.
DISCUSSION
While resistance to cell-death is a hallmark of cancers and specific strategies are being actively developed to target this process in patients, a better knowledge of the molecular bases controlling the activity of pro/anti-apoptotic mechanisms is required to better predict patient’s response to novel BCL2-targeting therapies and overcome therapy-resistance. Here, we have established that the ETO2∷GLIS2 fusion oncogene associated with an aggressive subtype of pediatric AML generates an imbalance in the expression of pro and anti-apoptotic regulators with both mechanistic and clinical implications.
Firstly, this study provides novel mechanistic insights on the regulators of cell death in human cancer cells and identify the GLIS2 moiety of the ETO2∷GLIS2 fusion as important for induction of mitochondrial apoptosis. Indeed, ETO2∷GLIS2 fusion positively regulates transcription of CASP3, a major effector caspase. The transcription and activation of CASP3 was abrogated by a mutation in one of the GLIS2 zinc finger, indicating a clear dependence on GLIS2 DNA binding activity, but the relative contributions of the ETO2 and GLIS2 moiety within the fusion will require further investigations. As ETO2∷GLIS2 was associated with the deregulated expression of several cell survival/death pathway mediators, ETO2∷GLIS2-dependent activation of apoptosis likely depends on multiple effectors rather than solely on CASP3 transcriptional deregulation.
GLIS2 was reported to regulate apoptosis in a cell-context dependent manner, as an anti-apoptotic regulator in kidney [40, 41] and colorectal cancer through inhibition of the pro-apoptotic factor PUMA [42], and as a pro-apoptotic regulator in neural cells in which inactivation of Glis2 reduced the expression of active caspase 3 with concomitant upregulation of Bcl2 [43]. In hematopoiesis, GLIS2 has also been involved as a pro-apoptotic regulator in MOZ∷TIF2 AML blasts [26]. GLIS2 was also involved in the positive regulation of myeloid differentiation in acute myeloid leukemia with MOZ∷TIF2 fusion [26]. In the light of an essential non-apoptotic role of CASP3 activation for proper myeloid differentiation [44, 45], our data suggest that CASP3 activation by GLIS2 could be required for the differentiation effect observed in MOZ∷TIF2 blasts. Interestingly, CASP3 was also proposed to regulate hematopoietic stem cell differentiation [46], interfere with erythropoiesis [47] and present specific sub-cellular localization during megakaryocyte differentiation [48], supporting a non-apoptotic function in these cellular contexts. As ETO2∷GLIS2 is found in AML associated with either megakaryoblastic (AMKL) or myeloid features (AML-M2/M5) [24, 49], the high CASP3 activity found in ETO2∷GLIS2 cells may contribute to both the hematopoietic stem and progenitor cell survival and differentiation.
Secondly, as an important clinical perspective, our data provide a strong molecular rationale for the implementation of therapies combining BCL2 family targeting inhibitors in aggressive pediatric AML presenting the ETO2∷GLIS2 fusion. Indeed, we demonstrated that treatments with BCL2 or MCL1 inhibitor alone is not sufficient to inhibit disease progression in ETO2∷GLIS2+ PDX models. A recent report indicated a significant clinical response to a combination of venetoclax and 5-azacitidine in an ETO2∷GLIS2+ leukemia at relapse [50], supporting our finding that combination therapies, including venetoclax, present an interest for the treatment of aggressive ETO2∷GLIS2+ leukemia. However, as the response to this treatment was monitored for only 3 months, long-term response to such combination is unknown. Notably, in a large screening on various human cell lines, the wider-range ABT737 inhibitor (targeting both BCL2 and BCL-xL, but not MCL1, that led to the development of navitoclax) presented a significantly more potent 5-azacitidine-sensitizing property than the BCL2-specific ABT199 alone in ETO2∷GLIS2-expressing M07e cells [51], supporting that targeting at least two BCL2 family members could be mandatory even with 5-azacitidine treatment. As BCL-xL inhibition induces severe thrombocytopenia that is not observed upon MCL1 inhibition, the combination of BCL2 and MCL1 proposed here may represent a treatment strategy with less side effects.
Notably, our ex vivo treatments of cells from two additional PDX models, including OTT∷MAL+ and NUP98∷KDM5A+, also showed efficient responses upon combination of BCL2 and MCL1 inhibitors. While the molecular bases underlying the responses in other fusion-driven leukemia will need further investigation, our findings support a wider clinical relevance of the BCL2/MCL1 inhibitors association to several pediatric AMKL molecular subgroups, including another aggressive molecular subgroup (i.e., NUP98 fusions).
It is possible that the resistance mechanism to BCL2 inhibition in ETO2∷GLIS2+ cells may implicate other mechanisms. Recently, it was shown that CD44 cooperates with BCL2 to induce survival in AML [52]. As CD44 expression is high in ETO2∷GLIS2 [22] and as CD44 targeting therapies are being developed, a combined targeting of CD44 and BCL2 may therefore represent an alternate strategy in ETO2∷GLIS2+ leukemia. As the control of mitochondrial-mediated apoptosis is an important regulator of cell survival in ETO2∷GLIS2 cells, it is also likely that the mitochondrial integrity and structure [53] may contribute to leukemogenesis and the relative response to treatment.
Together, in-depth characterization of the transcriptional consequences of the ETO2∷GLIS2 fusion uncovered a vulnerability toward interference with anti-apoptotic BCL2 family activity and the need for a combined targeting of at least two members, BCL2 and MCL1 to efficiently eradicate disease in preclinical PDX models of pediatric AMKL. Beyond the strong molecular rationale to implement combined treatments for these poor prognosis patients, this study paves the way for further characterization of this novel association between GLIS2 function and leukemic cell survival. Indeed, the precise molecular transition from a cell in which the fusion arises and induces cell death pathways to a full-blown BCL2High leukemic cell remains unclear and may require both direct transcription deregulation by the fusion as well as epigenetic changes affecting other transcriptional regulators or signaling pathways.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Dr. Olivier Bernard and Dr. Jürg Schwaller for their scientific expertise and for providing essential feedback on this manuscript. We thank Dr. Nicola Salvatore Bertuccio for providing the WSU-AML cell line. This study was supported by Enfants Cancers Santé and clinical partner teams of the Société Française de lutte contre les Cancers et les leucémies de l’Enfant et de l’adolescent (SFCE), Sites de Recherche Intégrée sur le Cancer (SIRIC)-SOCRATE (INCa-DGOS-INSERM 12551), Fondation pour la Recherche Médicale (FRM-ING20150532273) and Carnot OPALE. TM is supported by PAIR-Pédiatrie/CONECT-AML (COllaborative Network for Children and Teenagers with Acute Myeloblastic Leukemia: INCa-ARC-LIGUE_11905 and Association Laurette Fugain), Institut National Du Cancer (PLBIO-2014-176 and PLBIO-2018-169), Ligue contre le cancer (Equipe labellisée, since 2016) and Gustave Roussy (PMS CRESCENDO). FP, PA, AP, and TM are members of the OPALE Carnot institute. PA team is also supported by grants from ARC Foundation (Equipe labellisée 2015-2020), the ALF association (2020-2021), INCA (PLBIO-2019-133). TM is a team of the PEDIAC consortium (INCA_15670, www.programme-pediac.com).
Footnotes
COMPETING INTERESTS
The authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41375-022-01800-0.
DATA AVAILABILITY
The RNAseq datasets generated in this study were deposited in Array-Express (E-MTAB-4332 and E-MTAB-12255). Previously published datasets were also used (GSE4119) [54] and (TARGET Data: https://ocg.cancer.gov/programs/target/data-matrix/) [3]. Other data are available from the authors upon reasonable request.
REFERENCES
- 1.Pession A, Masetti R, Rizzari C, Putti MC, Casale F, Fagioli F, et al. Results of the AIEOP AML 2002/01 multicenter prospective trial for the treatment of children with acute myeloid leukemia. Blood. 2013;122:170–8. [DOI] [PubMed] [Google Scholar]
- 2.Eisfeld A-K, Kohlschmidt J, Mrózek K, Blachly JS, Walker CJ, Nicolet D, et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: an analysis of Alliance studies. Leukemia. 2018;32:1338–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med. 2018;24:103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Delbridge ARD, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat Rev Cancer. 2016;16:99–109. [DOI] [PubMed] [Google Scholar]
- 5.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov. 2014;4:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richard-Carpentier G, DiNardo CD. Venetoclax for the treatment of newly diagnosed acute myeloid leukemia in patients who are ineligible for intensive chemotherapy. Ther Adv Hematol. 2019;10:2040620719882822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konopleva M, Letai A. BCL-2 inhibition in AML: an unexpected bonus? Blood. 2018;132:1007–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frismantas V, Dobay MP, Rinaldi A, Tchinda J, Dunn SH, Kunz J, et al. Ex vivo drug response profiling detects recurrent sensitivity patterns in drug-resistant acute lymphoblastic leukemia. Blood. 2017;129:e26–e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seyfried F, Demir S, Hörl RL, Stirnweiß FU, Ryan J, Scheffold A, et al. Prediction of venetoclax activity in precursor B-ALL by functional assessment of apoptosis signaling. Cell Death Dis. 2019;10:571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karol SE, Bittencourt H, Morgenstern DA, Macy ME, Khaw SL, Cooper TM, et al. Venetoclax alone or in combination with chemotherapy: responses in pediatric patients with relapsed/refractory acute myeloid leukemia with heterogeneous genomic profiles. Blood. 2020;136:30–31. [Google Scholar]
- 11.Karol SE, Alexander TB, Budhraja A, Pounds SB, Canavera K, Wang L, et al. Venetoclax in combination with cytarabine with or without idarubicin in children with relapsed or refractory acute myeloid leukaemia: a phase 1, dose-escalation study. Lancet Oncol. 2020;21:551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Place AE, Karol SE, Forlenza CJ, Gambart M, Cooper TM, Fraser C, et al. Pediatric patients with relapsed/refractory acute lymphoblastic leukemia harboring heterogeneous genomic profiles respond to venetoclax in combination with chemotherapy. Blood. 2020;136:37–38. [Google Scholar]
- 13.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax mono-therapy in patients with acute myelogenous leukemia. Cancer Disco. 2016;6:1106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pei S, Pollyea DA, Gustafson A, Stevens BM, Minhajuddin M, Fu R, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020;10:536–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bhatt S, Pioso MS, Olesinski EA, Yilma B, Ryan JA, Mashaka T, et al. Reduced mitochondrial apoptotic priming drives resistance to BH3 mimetics in acute myeloid leukemia. Cancer Cell. 2020;38:872–.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Short NJ, Konopleva M, Kadia TM, Borthakur G, Ravandi F, DiNardo CD, et al. Advances in the treatment of acute myeloid leukemia: new drugs and new challenges. Cancer Discov. 2020;10:506–25. [DOI] [PubMed] [Google Scholar]
- 17.Hashimoto M, Saito Y, Nakagawa R, Ogahara I, Takagi S, Takata S, et al. Combined inhibition of XIAP and BCL2 drives maximal therapeutic efficacy in genetically diverse aggressive acute myeloid leukemia. Nat Cancer. 2021;2:340–56. [DOI] [PubMed] [Google Scholar]
- 18.Lin KH, Winter PS, Xie A, Roth C, Martz CA, Stein EM, et al. Targeting MCL-1/BCL-XL forestalls the acquisition of resistance to ABT-199 in acute myeloid leukemia. Sci Rep. 2016;6:27696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov. 2018. 10.1158/2159-8290.CD-18-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thijssen R, Diepstraten ST, Moujalled D, Chew E, Flensburg C, Shi MX, et al. Intact TP-53 function is essential for sustaining durable responses to BH3-mimetic drugs in leukemias. Blood. 2021;137:2721–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Rooij JDE, Branstetter C, Ma J, Li Y, Walsh MP, Cheng J, et al. Pediatric non-Down syndrome acute megakaryoblastic leukemia is characterized by distinct genomic subsets with varying outcomes. Nat Genet. 2017;49:451–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thirant C, Ignacimouttou C, Lopez CK, Diop M, Le Mouël L, Thiollier C, et al. ETO2-GLIS2 hijacks transcriptional complexes to drive cellular identity and self-renewal in pediatric acute megakaryoblastic leukemia. Cancer Cell. 2017;31:452–65. [DOI] [PubMed] [Google Scholar]
- 23.Lopez CK, Malinge S, Gaudry M, Bernard OA, Mercher T. Pediatric acute megakaryoblastic leukemia: multitasking fusion proteins and oncogenic cooperations. Trends Cancer. 2017;3:631–42. [DOI] [PubMed] [Google Scholar]
- 24.Lopez CK, Noguera E, Stavropoulou V, Robert E, Aid Z, Ballerini P, et al. Ontogenic changes in hematopoietic hierarchy determine pediatric specificity and disease phenotype in fusion oncogene-driven myeloid leukemia. Cancer Discov. 2019;9:1736–53. [DOI] [PubMed] [Google Scholar]
- 25.Holmfeldt P, Ganuza M, Marathe H, He B, Hall T, Kang G, et al. Functional screen identifies regulators of murine hematopoietic stem cell repopulation. J Exp Med. 2016;213:433–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shima H, Takamatsu-Ichihara E, Shino M, Yamagata K, Katsumoto T, Aikawa Y, et al. Ring1A and Ring1B inhibit expression of Glis2 to maintain murine MOZ-TIF2 AML stem cells. Blood. 2018;131:1833–45. [DOI] [PubMed] [Google Scholar]
- 27.Scoville DW, Kang HS, Jetten AM. GLIS1–3: emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig. 2017;4:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guerra E, Trerotola M, Aloisi AL, Tripaldi R, Vacca G, La Sorda R, et al. The Trop-2 signalling network in cancer growth. Oncogene. 2013;32:1594–1600. [DOI] [PubMed] [Google Scholar]
- 29.Yuan J, Tan L, Yin Z, Tao K, Wang G, Shi W, et al. GLIS2 redundancy causes chemoresistance and poor prognosis of gastric cancer based on co‑expression network analysis. Oncol Rep. 2019;41:191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jetten AM. Emerging roles of GLI-similar Krüppel-like zinc finger transcription factors in leukemia and other cancers. Trends Cancer. 2019;5:547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan J, Letai A. BH3 profiling in whole cells by fluorimeter or FACS. Methods. 2013;61:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiollier C, Lopez CK, Gerby B, Ignacimouttou C, Poglio S, Duffourd Y, et al. Characterization of novel genomic alterations and therapeutic approaches using acute megakaryoblastic leukemia xenograft models. J Exp Med. 2012;209:2017–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasanth S, ZeRuth G, Kang HS, Jetten AM. Identification of nuclear localization, DNA binding, and transactivating mechanisms of Kruppel-like zinc finger protein Gli-similar 2 (Glis2). J Biol Chem. 2011;286:4749–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertuccio SN, Boudia F, Cambot M, Lopez CK, Lordier L, Donada A, et al. The pediatric acute leukemia fusion oncogene ETO2-GLIS2 increases self-renewal and alters differentiation in a human induced pluripotent stem cells-derived model. Hemasphere. 2020;4:e319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McArthur K, Kile BT. Apoptotic caspases: multiple or mistaken identities? Trends Cell Biol. 2018;28:475–93. [DOI] [PubMed] [Google Scholar]
- 36.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis. 2003;8:345–52. [DOI] [PubMed] [Google Scholar]
- 37.Montero J, Letai A. Why do BCL-2 inhibitors work and where should we use them in the clinic? Cell Death Differ. 2018;25:56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016;538:477–82. [DOI] [PubMed] [Google Scholar]
- 39.Mukherjee N, Skees J, Todd KJ, West DA, Lambert KA, Robinson WA, et al. MCL1 inhibitors S63845/MIK665 plus Navitoclax synergistically kill difficult-to-treat melanoma cells. Cell Death Dis. 2020;11:443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Attanasio M, Uhlenhaut NH, Sousa VH, O’Toole JF, Otto E, Anlag K, et al. Loss of GLIS2 causes nephronophthisis in humans and mice by increased apoptosis and fibrosis. Nat Genet. 2007;39:1018–24. [DOI] [PubMed] [Google Scholar]
- 41.Lu D, Rauhauser A, Li B, Ren C, McEnery K, Zhu J, et al. Loss of Glis2/NPHP7 causes kidney epithelial cell senescence and suppresses cyst growth in the Kif3a mouse model of cystic kidney disease. Kidney Int. 2016;89:1307–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao J, Lei P-J, Li Q-L, Chen J, Tang S-B, Xiao Q, et al. GLIS2 promotes colorectal cancer through repressing enhancer activation. Oncogenesis. 2020;9:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ke K, Song Y, Shen J, Niu M, Zhang H, Yuan D, et al. Up-regulation of Glis2 involves in neuronal apoptosis after intracerebral hemorrhage in adult rats. Cell Mol Neurobiol. 2015;35:345–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacquel A, Obba S, Solary E, Auberger P. Proper macrophagic differentiation requires both autophagy and caspase activation. Autophagy. 2012;8:1141–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pandey P, Nakazawa A, Ito Y, Datta R, Kharbanda S, Kufe D. Requirement for caspase activation in monocytic differentiation of myeloid leukemia cells. Oncogene. 2000;19:3941–7. [DOI] [PubMed] [Google Scholar]
- 46.Janzen V, Fleming HE, Riedt T, Karlsson G, Riese MJ, Lo Celso C, et al. Hematopoietic stem cell responsiveness to exogenous signals is limited by caspase-3. Cell Stem Cell. 2008;2:584–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ribeil J-A, Zermati Y, Vandekerckhove J, Cathelin S, Kersual J, Dussiot M, et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature. 2007;445:102–5. [DOI] [PubMed] [Google Scholar]
- 48.De Botton S, Sabri S, Daugas E, Zermati Y, Guidotti JE, Hermine O, et al. Platelet formation is the consequence of caspase activation within megakaryocytes. Blood. 2002;100:1310–7. [DOI] [PubMed] [Google Scholar]
- 49.Masetti R, Pigazzi M, Togni M, Astolfi A, Indio V, Manara E, et al. CBFA2T3-GLIS2 fusion transcript is a novel common feature in pediatric, cytogenetically normal AML, not restricted to FAB M7 subtype. Blood. 2013;121:3469–72. [DOI] [PubMed] [Google Scholar]
- 50.Mishra AK, Mullanfiroze K, Chiesa R, Vora A. Azacitidine and venetoclax for post-transplant relapse in a case of CBFA2T3/GLIS2 childhood acute myeloid leukaemia. Pediatr Blood Cancer. 2021;68:e29221. [DOI] [PubMed] [Google Scholar]
- 51.Bogenberger JM, Kornblau SM, Pierceall WE, Lena R, Chow D, Shi C-X, et al. BCL-2 family proteins as 5-Azacytidine-sensitizing targets and determinants of response in myeloid malignancies. Leukemia. 2014;28:1657–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yu X, Munoz-Sagredo L, Streule K, Muschong P, Bayer E, Walter RJ, et al. CD44 loss of function sensitizes AML cells to the BCL-2 inhibitor venetoclax by decreasing CXCL12-driven survival cues. Blood. 2021;138:1067–1080. [DOI] [PubMed] [Google Scholar]
- 53.Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. 2019;9:890–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bourquin J-P, Subramanian A, Langebrake C, Reinhardt D, Bernard O, Ballerini P, et al. Identification of distinct molecular phenotypes in acute megakaryoblastic leukemia by gene expression profiling. Proc Natl Acad Sci USA. 2006;103:3339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNAseq datasets generated in this study were deposited in Array-Express (E-MTAB-4332 and E-MTAB-12255). Previously published datasets were also used (GSE4119) [54] and (TARGET Data: https://ocg.cancer.gov/programs/target/data-matrix/) [3]. Other data are available from the authors upon reasonable request.