

Abstract
The serine hydrolases cytosolic phospholipase A2α (cPLA2α) and fatty acid amide hydrolase (FAAH) are interesting targets for the development of new anti-inflammatory and analgesic drugs. Structural modifications of a potent dual inhibitor with a propan-2-one substituted tetrazolylpropionic acid moiety led to compounds with also nanomolar activity against both enzymes but better physicochemical properties. The structure–activity relationships showed that the variations had partially divergent effects on the inhibitory activity of the compounds towards cPLA2α and FAAH reflecting differences in the binding mode to the enzymes. Furthermore, the metabolic stability of the target structures was investigated in vitro.
Structural variation of the potent dual cPLA2α/FAAH inhibitor 7 led to compounds with similar efficacy and improved physicochemical properties.
Introduction
Cytosolic phospholipase A2α (cPLA2α) and fatty acid amide hydrolase (FAAH) play an important role in several pathological processes. Thus, cPLA2α is considered a key enzyme in the synthesis of various pro-inflammatory and pain-causing lipid mediators such as prostaglandins, leukotrienes and platelet-activating factor by producing their precursors arachidonic acid and lysophospholipids through cleavage of membrane phospholipids at the sn-2 position.1,2 FAAH is a central enzyme in the endocannabinoid metabolism. It hydrolyses the endocannabinoid anandamide into arachidonic acid and ethanolamine, thereby terminating its anti-inflammatory and analgesic effects.3,4 Consequently, inhibitors of cPLA2α and FAAH could represent new treatment options for inflammatory conditions and pain.5–8
FAAH and cPLA2α have several features in common.9–11 Both enzymes are serine hydrolases that liberate arachidonic acid from their substrates. After formation of the enzyme–substrate complexes, the serines in the active site of the enzymes attack the arachidonoyl ester or amide, resulting in the formation of oxyanions. These tetrahedral transition states are stabilised by “oxyanion holes” formed by the amide groups of the two glycine residues of each enzyme. As the oxyanions collapse, serine–arachidonoyl intermediates are formed, which are finally hydrolysed by water. Various inhibitors of FAAH or cPLA2α inactivate these enzymes by covalent binding to the serines in their active centres.5–8 They are therefore called “serine-trap” inhibitors. The trifluoromethyl ketone 1 (AACOCF3) and the fluorophosphonate 2 (MAFP) (Fig. 1), which were developed as cPLA2α inhibitors, inhibit this enzyme via such a mechanism of action.12–14 Interestingly, these two compounds were later found to also affect FAAH activity accordingly.15–18 Such dual inhibitors could have additional advantages compared to selective inhibitors, as they would inhibit the formation of pro-inflammatory and the degradation of anti-inflammatory lipid mediators simultaneously.
Fig. 1. Structures of dual or selective cPLA2α and FAAH inhibitors.

Previously, we have discovered that AstraZeneca's potent cPLA2α inhibitor AR-C70484XX (3)19 and our structurally related propan-2-one derivative 420,21 are also dual inhibitors of cPLA2α and FAAH,22 although the cPLA2α inhibitory effect of 4 was significantly more pronounced than its FAAH inhibitory activity (Fig. 1). In the course of further investigations, we were able to find derivatives of 4 that either selectively or largely selectively inhibit only cPLA2α or FAAH like 5 and 6,22 or inhibit both enzymes to about the same extent like 7.23 An important structural element of these inhibitors is the central ketone group, which is activated by electron-withdrawing substituents. It is assumed that this reactive group can form covalent bonds with the catalytic serine residues in the active centres of cPLA2α or FAAH and thus contributes very strongly to the inhibitory effect, provided that the other functionalities of the molecules allow binding to the enzyme. This is evidenced by the fact that reduction of the ketone function to an alcohol, e.g., in compounds 3 and 4, results in metabolites that no longer inhibit cPLA2α.19,24 In addition, the alcohol derivative of the potent FAAH inhibitor 6 is not active any more at a test concentration of 10 μM.
Starting from our potent dual cPLA2α/FAAH inhibitor 7, it was now investigated whether structural variations in the region of the lipophilic octyl chain, the reactive ketone group as well as in the tetrazole ring system have the same effect on the inhibitory activity of the compound towards both enzymes or whether there are differences here. This should help to define the structural features of the respective pharmacophores required for activity against the enzymes.
Results and discussion
Chemistry
The preparation of the target compounds 8–23 bearing tetrazolylalkanoic acid and substituted phenoxy residues at the central ketone structure has already been decribed.25,26Scheme 1 gives an overview of the synthetic pathways used. First, ω-cyanopropionic- or -butyric acid methyl ester was reacted with trimethylsilyl azide in the presence of tetrabutylammonium fluoride to obtain the corresponding tetrazolyl substituted building blocks. These were treated with the appropriate (phenoxymethyl)oxirane27 under 1,4-diazabicyclo[2.2.2]octane (DABCO) catalysis. The attack on the tetrazole ring took place in the 1- as well as in the 2-position, whereby in each case the 2-substituted product was formed in larger amounts. The isomers could be well separated by silica gel chromatography. Structure assignment was possible by NMR spectroscopy as described recently.28 In most cases, only the main product was further reacted, sometimes also the side product. In the next step, the resulting secondary alcohol group was oxidised to the ketone with Dess–Martin periodinane. Finally, the ester moiety was hydrolysed under alkaline conditions at room temperature to obtain the desired carboxylic acid derivatives.
Scheme 1. Reagents and conditions: (a) trimethylsilyl azide, tetrabutylammonium fluoride hydrate, 100 °C, 8 h; (b) DABCO, 90–100 °C, 4–8 h; (c) Dess–Martin periodinane, dry CH2Cl2, room temperature, 4–24 h; (d) aqueous KOH solution, methanol, room temperature, 3–6 h.

For the synthesis of the two derivatives with triazolylpropionic acid residues 32 and 33, the oxirane ring of the starting compounds 24 or 2519,26 was opened with natrium azide to obtain the azido substituted secondary alcohols 26 and 27 (Scheme 2).26,29 The azide functionality of these substances was reacted in a click reaction with 4-pentynoic acid methyl ester to form the propionic acid ester substituted triazoles 28 and 29. Oxidation of their secondary alcohol functionalities to ketones with Dess–Martin periodinane followed by hydrolysis of the methyl ester groups with aqueous KOH in methanol provided the target compounds 32 and 33.25
Scheme 2. Reagents and conditions: (a) sodium azide, aqueous ammonium chloride solution, methanol, 70 °C, 4 h; (b) 4-pentynoic acid methyl ester, tert-butanol, water, aqueous sodium ascorbate solution, aqueous copper(ii) sulfate solution, room temperature, 24 h; (c) Dess–Martin periodinane, dry CH2Cl2, room temperature, 5 h; (d) aqueous KOH solution, methanol, room temperature, 3–5 h.

The derivative of compound 12, in which the 4-phenoxyphenoxymethylene residue attached to the reactive carbonyl group was replaced by a more rigid 6-phenylchroman-2-yl ring system, was prepared in a nine-step synthesis starting from 5-bromo-2-hydroxyacetophenone (34) (Scheme 3). First, its bromine atom was replaced by a phenyl ring in a Suzuki coupling reaction.30 Subsequently, the resulting compound 35 was reacted with diethyl oxalate in the presence of sodium ethanolate.31 The ethanolate deprotonates the CH-acidic methyl group of 35, thus forming a nucleophile that attacks the carbonyl moiety of diethyl oxalate leading to the formation of a new C–C bond. The appearance of two doublets in the 1H NMR spectrum of obtained compound 36 in the high field at 3.00 ppm and 3.42 ppm, each with an integral intensity of 1 and coupling constants of J = 16 Hz indicated that the substance was not present in the open-chain structure but in the form of the cyclic hemiacetal. Treatment of 36 with a mixture of acetic acid and concentrated HCl resulted in the elimination of water, giving the chromene derivative 37.31 In the next step, the double bond of the chromene was hydrogenated in acetic acid under catalysis with palladium on carbon to form the chromane 38. Then the ester group of this intermediate was reduced to an alcohol in diethyl ether with lithium aluminium hydride solution in THF. The primary alcohol functionality of 39 was oxidized with Dess–Martin reagent to an aldehyde (40), which was converted to an oxirane by reaction with sodium hydride and trimethylsulfonium iodide in DMSO. The obtained compound 41 was treated in acetonitrile with 3-(tetrazol-5-yl)propionic acid methyl ester in the presence of cesium carbonate to provide the substituted tetrazolyl derivative 42. The secondary alcohol group of this compound was then oxidized with Dess–Martin periodinane to a ketone group (43). Finally, the methyl ester group of 43 was saponified under alkaline conditions to give the desired propionic acid derivative 44.
Scheme 3. Reagents and conditions: (a) phenylboronic acid, K2CO3, bis(triphenylphosphine)palladium(ii)-dichloride, isopropanol, 85 °C, 2.5 h; (b) diethyl oxalate, sodium, ethanol, 100 °C, 2 h; (c) acetic acid, HCl conc. (30 : 1), 80 °C, 1.5 h; (d) acetic acid, Pd/C, H2, 1.5 bar, 80 °C, 1.5 h; (e) LiAlH4, diethyl ether, room temperature, 2 h; (f) Dess–Martin periodinane, dry CH2Cl2, room temperature, 4 h; (g) trimethylsulfonium iodide, NaH, DMSO, room temperature, 1 h; (h), Cs2CO3, acetonitrile, 80 °C, 5 h; (i) aqueous KOH, methanol, room temperature, 4 h.

Biological evaluation
For measuring the inhibition of cPLA2α the enzyme isolated from porcine platelets was used. The amount of arachidonic acid released from co-vesicles consisting of the substrate 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine and 1,2-dioleoyl-sn-glycerol was determined in dependence of the test compound concentration by reversed-phase HPLC and UV detection at 200 nm after online solid phase extraction.21 FAAH inhibition was evaluated using rat brain microsomes as the enzyme source and the fluorogenic substrate N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide. The product released by FAAH, 4-pyren-1-ylbutanoic acid, was determined by reversed-phase HPLC with fluorescence detection.32,33
Lead structure 7 has a relatively high lipophilicity (log P 5.5), especially due to the long octyl chain, which leads to unfavorable pharmacokinetic properties. Therefore, it should first be investigated how a reduction of lipophilicity by alkyl chain shortening or introduction of oxygen atoms affects the inhibitory effect against cPLA2α and FAAH. Replacing the octyl residue with a shorter hexyl or butyl chain did not change the efficacy against FAAH despite a significant decrease in lipophilicity (Table 1). The same was observed when the hexyl substituent was replaced by a hexyloxy or isohexyloxy residue. The IC50 value against this enzyme was about 0.030 μM in each case. However, with regard to the inhibition of cPLA2α, this structural variation led to significant changes in efficacy. A decrease in lipophilicity was associated here with a decrease in enzyme inhibition. Thus, the hexyl, hexyloxy, and isohexyloxy substituted compounds 8, 10 and 11, which have a log P of about 4, were about half as effective against cPLA2α as the lead compound 7. With an IC50 of 0.26 μM, the butyl substituted derivative 9, whose log P was only 3, was even more than ten-times less potent than this compound. A somewhat different picture emerges when the butyl residue is replaced by a phenyloxy substituent. Although the lipophilicity decreases further (log P of 12: 2.2), the cPLA2α inhibitory effect actually increases slightly (IC50 0.17 vs. 0.26 μM). Possibly, advantageous π–π interactions between the phenyl ring of the inhibitor 12 and the enzyme are responsible for this. In contrast, the inhibitory effect against FAAH decreases moderately due to this structural variation. Looking at the inhibition of both enzymes, it can be seen that the hexyloxy and isohexyloxy substituted compounds 10 and 11 are also potent dual cPLA2α/FAAH inhibitors, as is the case with lead 7, but with much more favorable log P values than this parent compound (3.7 and 3.6 vs. 5.5).
Inhibition of cPLA2α and FAAH.
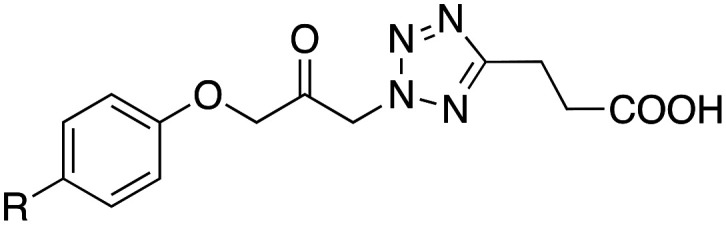
| ||||
|---|---|---|---|---|
| Comp. | R | Inhibition of cPLA2α IC50a (μM) | Inhibition of FAAH IC50a (μM) | log Pb |
| 7 | C8H17 | 0.019 | 0.030 | 5.5 |
| 8 | C6H13 | 0.036 | 0.023 | 4.2 |
| 9 | C4H9 | 0.260 | 0.027 | 3.0 |
| 10 | OC6H13 | 0.047 | 0.032 | 3.7 |
| 11 | O-isohexyl | 0.045 | 0.032 | 3.6 |
| 12 | O-phenyl | 0.170 | 0.054 | 2.2 |
Values are the means of at least two independent determinations, errors are within ± 20%; IC50 for cPLA2α reference inhibitor AR-C70484XX: 0.0065 ± 0.0011 μM (n = 3);21 IC50 for FAAH reference inhibitor URB597: 0.060 ± 0.0067 μM (mean ± standard deviation, n = 4).28
Determined by reversed-phase HPLC; log P of the references indomethacin and ibuprofen: 2.9 and 3.2, respectively.
Next, we investigated how the introduction of a methyl group at each of the carbon atoms adjacent to the carbonyl functionality and/or the replacement of the propionic acid with the longer butyric acid chain affected the activity towards the two enzymes. The starting compound was the hexyloxy substituted derivative 10. As the test data for compounds 13 and 14 show, the inhibitory potency against cPLA2α decreases by a factor of about 4 in each case by introducing one of these methyl substituents (Table 2). With regard to the inhibition of FAAH, these structural variations lead to much more pronounced effects. While the introduction of a methyl group at the carbon atom, which is located between the carbonyl group and the phenoxy residue, does not impair the efficacy of the compound (13), a methyl substituent at the methyl group between the carbonyl group and the tetrazole ring (14) causes an almost complete loss of inhibitory potency at the highest test concentration of 10 μM. This substituent apparently strongly impairs the attack of the catalytic serine residue of FAAH on the carbonyl group of the inhibitor and thus the formation of a covalent bond between the enzyme and the inhibitor. For both α-methyl substituted compounds, the extension of the acid chain by one carbon atom (15, 16) resulted in little or no loss of inhibitory activity towards cPLA2α. The inhibitory effect against FAAH is not affected by this variation for the potent inhibitor 13, as shown by the IC50 value of compound 15. For the other derivative with a methyl substituent on the methylene group attached to the tetrazole ring (14), the activity increases more than tenfold after this structural modification (16). The attack of the serine of the active site on the carbonyl group of the inhibitor is obviously facilitated again by the changed binding mode caused by the extended acid chain. Regarding the activity of the α-methylated compounds, it should be noted that they are racemic mixtures due to the chiral center they contain. The IC50 value of one of the two enantiomers could therefore be up to 50% lower if the other is less or not active.34
Inhibition of cPLA2α and FAAH.

| |||||
|---|---|---|---|---|---|
| Comp. | R1 | R2 | n | Inhibition of cPLA2α IC50a (μM) | Inhibition of FAAH IC50a (μM) |
| 10 | H | H | 2 | 0.047 | 0.032 |
| 13 | CH3 | H | 2 | 0.19 | 0.034 |
| 14 | H | CH3 | 2 | 0.18 | >10b |
| 15 | CH3 | H | 3 | 0.50 | 0.034 |
| 16 | H | CH3 | 3 | 0.25 | 0.75 |
Values are the means of at least two independent determinations, errors are within ±20%.
31% Inhibition at 10 μM.
For the four tetrazolylpropionic acid and -butyric acid derivatives with terminal phenoxy moiety 12, 17, 18 and 19, it was investigated how the potency changes when the acid chain and the residue bearing the ketone function are no longer in 1,3- but in 1,2-position to each other on the tetrazole residue. It was shown that the stronger angulation of the molecules (20–23) caused by this structural variation significantly reduced the inhibitory effect against both cPLA2α and FAAH in general (Table 3). The smallest loss of efficacy (by a factor of about 3–4) was observed with the two butyric acid derivatives 22 and 23 in terms of inhibition of FAAH.
Inhibition of cPLA2α and FAAH and metabolic stability in rat liver S9 fraction.

| |||||
|---|---|---|---|---|---|
| Comp. | R | n | Inhibition of cPLA2α IC50a (μM) | Inhibition of FAAH IC50a (μM) | Metabolic stabilityb (%) |
| 12 | H | 2 | 0.17 | 0.054 | <5 |
| 17 | CH3 | 2 | 0.54 | 0.044 | <5 |
| 18 | H | 3 | 0.28 | 0.054 | <5 |
| 19 | CH3 | 3 | 0.73 | 0.043 | <5 |
| 20 | H | 2 | 3.0 | 2.1 | 52 |
| 21 | CH3 | 2 | >10c | 0.88 | 86 |
| 22 | H | 3 | 3.3 | 0.17 | 36 |
| 23 | CH3 | 3 | 4.8 | 0.17 | 71 |
Values are the means of at least two independent determinations, errors are within ±20%.
Percent of parent remaining after incubation with rat liver S9 fraction for 30 min in presence of the co-factor NADPH; values are means of independent duplicates; metabolic stability of the reference daunorubicin: 28 ± 4% (n = 6).35
39% inhibition at 10 μM.
In addition, we replaced the tetrazole with a 1,2,3-triazole ring in the two phenoxy substituted tetrazolylpropionic acid derivatives 12 and 17. The compounds 32 and 33 obtained inhibited cPLA2α about sixfold and FAAH about threefold less strongly than the parent structures (Table 4).
Inhibition of cPLA2α and FAAH and metabolic stability in rat liver S9 fraction.

| ||||
|---|---|---|---|---|
| Comp. | R | Inhibition of cPLA2α IC50a (μM) | Inhibition of FAAH IC50a (μM) | Metabolic stabilityb (%) |
| 32 | H | 0.85 | 0.13 | <5 |
| 33 | CH3 | 3.1 | 0.17 | 29 |
| 44 | — | >10c | 0.094 | <5 |
Values are the means of at least two independent determinations, errors are within ±20%.
Percent of parent remaining after incubation with rat liver S9 fraction for 30 min in presence of the co-factor NADPH; values are means of independent duplicates.
34% inhibition at 10 μM.
Finally, we prepared a derivative of 17, namely compound 44, in which the relatively flexible lipophilic region of the molecule adjacent to the reactive carbonyl group was replaced by a rigid 6-phenyl-2,3-dihydrobenzopyran ring. This rigidification resulted in an almost complete loss of activity against cPLA2α (Table 4). In contrast, the inhibitory activity against FAAH decreased only by a factor of 2. Thus, with such a structural variation, selectivity can be improved in favor of FAAH. It should be noted that 44 does not contain a phenoxyphenoxy grouping like 17, but in principle a biphenyloxy substituent. Previous studies had shown that the replacement of a phenoxyphenoxy residue by a biphenyloxy residue in the investigated substance class did not lead to any significant change in activity against cPLA2α and FAAH.28,36,37 Accordingly, it is possible to evaluate the effect of rigidification by comparing the potency of 17 and 44.
An important pharmacophoric element of the compounds studied is the activated ketone group of their propan-2-one partial structure, which can apparently form covalent bonds with the serine of the active site of the serine hydrolases cPLA2α and FAAH. First, this was suggested in a model for binding of the cPLA2α inhibitor AR-C70484XX to the enzyme.19 Furthermore, we have recently found that inhibitory potency of propan-2-one inhibitors against FAAH in rat liver S9 homogenate increases when enzyme and inhibitor are preincubated before addition of the substrate, which indicates a covalent inhibition mechanism. However, when NADPH is added after preincubation, the inhibition strength is reduced due to a reduction of the ketones to inactive alcohols by carbonyl reductases present in the liver preparation,23 indicating the reversibility of the covalent inhibition. Due to the susceptibility of the ketone function to metabolic reduction by carbonyl reductases35,36 we have also examined all target structures for metabolic stability in rat liver S9 fraction in presence of NADPH.23,32 Under the experimental conditions, nearly all compounds were almost completely metabolised to the appropriate alcohols. The formation of hydroxylated products by cytochrome P450 enzymes was detected to a very low extent, if at all (see ESI†). The only exceptions were the triazole propionic acid 33 and the four compounds 20–23 in which the propan-2-one substituent and the acid chain on the tetrazole ring are in the 1,2-position relative to each other, but all have only a moderate inhibitory effect on the two target enzymes (Tables 3 and 4). Apparently, the angled structure of the molecules shields the reactive ketone group somewhat from the attack of carbonyl reductases and thus protects it from transformation. Moreover, the data show that a methyl group on the carbon atom between carbonyl group and the phenoxy residue, respectively, further hinders this enzymatic reduction.
Taken together, the highly active dual cPLA2α/FAAH inhibitors found in this work with a tetrazolylalkanoic acid structure such as 10 are obviously not suitable for systemic administration due to their metabolic instability. On the other hand, an application of 10 for example as an anti-inflammatory and analgesic skincare might be promising. In contrast to many other cPLA2α inhibitors, this substance has a surprisingly good water solubility.38 Thus, an amount of 500 μg39 was completely soluble in a volume of 1 mL phosphate buffer (pH 7.4). For comparison, a water solubility of about 250 μg mL−1 was determined for indomethacin under the same conditions. The development of specific dermal formulations with solvents, co-solvents or solubilized systems, which are essential for poorly soluble active substances, would therefore not be necessary at all for this compound.
Experimental
Chemistry
General
Column chromatography was performed on silica gel 60, particle size 0.040–0.063 mm, from Macherey & Nagel (Düren, Germany). Melting points were determined on a Büchi B-540 apparatus (Essen, Germany) and are uncorrected. 1H NMR spectra were measured on a DD2 spectrometer (400 MHz) from Agilent (Santa Clara, USA) or an Avance AV 300 spectrometer (300 MHz) from Bruker (Karlsruhe, Germany). Electron ionization (EI) mass spectra were obtained on a GCQ apparatus from Finnigan (Bremen, Germany). The high resolution mass spectra (HRMS) were recorded on a Bruker (Bremen, Germany) micrOTOF-Q II spectrometer applying electrospray ionization (ESI).
Purity of the target compounds was determined by reversed-phase HPLC with UV detection at 254 nm. The samples were prepared by diluting 20 μL of a 5 mM stock solution of the compound in DMSO with 180 μL acetonitrile. 5 μL of the solutions were injected into the HPLC-system using a Nucleosil EC 100-3 C18 3 μm column (3 mm inside diameter × 125 mm) (Macherey & Nagel, Düren, Germany) at a flow rate of 0.4 mL min−1 with a gradient consisting of acetonitrile/water/trifluoroacetic acid (42 : 58 : 0.1 to 86 : 14 : 0.1, v/v/v). The run-time was 35 min.
The syntheses of compounds 8–23 and 33 have already been described.25,26
1-Azido-3-(4-phenoxyphenoxy)propan-2-ol 26
To a solution of 2-[(4-phenoxyphenoxy)methyl]oxirane29 (785 mg, 3.24 mmol) in methanol (8 mL) was added sodium azide (320 mg, 4.92 mmol), ammonium chloride (256 mg, 4.79 mmol) and water (5 mL). The mixture was heated at 70 °C for 4 h. Then it was diluted with water and extracted three times with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated under reduced pressure. The residue was dissolved in a small volume of toluene and chromatographed on silica gel (hexane/ethyl acetate 8 : 2, 7 : 3) to give 26 as an oil (604 mg, 65%). C15H15N3O3 (285.3); 1H NMR (300 MHz, CDCl3) δ (ppm) 3.50–3.57 (m, 2H), 3.98–4.03 (m, 2H), 4.13–4.20 (m, 1H), 6.87–6.92 (m, 2H), 6.92–7.01 (m, 4H), 7.02–7.09 (m, 1H), 7.27–7.34 (m, 2H); MS (EI, 70 eV) m/z (%): 285 (55), 186 (100).
Methyl 3-{1-[2-hydroxy-3-(4-phenoxyphenoxy)propyl]-1H-1,2,3-triazol-4-yl}propionate 28
Compound 26 (593 mg, 2.08 mmol) and methyl pent-4-ynoate (277 mg, 2.47 mmol) were suspended in a mixture of equal parts of tert-butanol and water (8 mL). Then a solution of sodium ascorbate (59 mg) in water (0.3 mL) and a solution of copper(ii) sulfate hydrate (75 mg) in water (0.1 mL) were added successively. The mixture was stirred vigorously overnight at room temperature. After addition of water (30 mL), the reaction mixture was cooled to 0 °C and exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated under reduced pressure. Purification by chromatography on silica gel (hexane/ethyl acetate 6 : 4, 1 : 1, 4 : 6) yielded 28 as a solid (360 mg, 44%). C21H23N3O5 (397.4); 1H NMR (300 MHz, CDCl3) δ (ppm) 2.76 (t, J = 7.2 Hz, 2H), 3.07 (t, J = 7.2 Hz, 2H), 3.67 (s, 3H), 3.97 (dd, J = 5.4 and 1.3 Hz, 2H), 4.40–4.56 (m, 2H), 4.67–4.74 (m, 1H), 6.84–6.91 (m, 2H), 6.91–7.01 (m, 4H), 7.02–7.10 (m, 1H), 7.27–7.34 (m, 2H), 7.57 (s, 1H); MS (EI, 70 eV) m/z (%): 397 (6), 212 (100).
Methyl 3-{1-[2-oxo-3-(4-phenoxyphenoxy)propyl]-1H-1,2,3-triazol-4-yl}propionate 30
A solution of 28 (352 mg, 0.89 mmol) in dry dichloromethane (15 mL) was treated with Dess–Martin periodinane (750 mg, 1.77 mmol) and stirred at room temperature for 5 h. Then a 1 : 1 mixture of semi-concentrated aqueous sodium thiosulfate solution and saturated aqueous sodium hydrogen carbonate solution was added (10 mL) and stirring was continued for a further 10 min. The reaction mixture was exhaustively extracted with ethyl acetate and the combined organic phases were dried over sodium sulfate and concentrated under reduced pressure. The residue was dissolved in toluene (2 mL) and purified by chromatography on silica gel (hexane/ethyl acetate 6 : 4, 1 : 1) to give 30 as a solid (254 mg, 73%). C21H21N3O5 (395.4); 1H NMR (300 MHz, CDCl3) δ (ppm) 2.77 (t, J = 7.3 Hz, 2H), 3.09 (t, J = 7.3 Hz, 2H), 3.68 (s, 3H), 4.70 (s, 2H), 5.54 (s, 2H), 6.88–6.94 (m, 2H), 6.94–7.04 (m, 4H), 7.05–7.12 (m, 1H), 7.29–7.36 (m, 2H), 7.45 (s, 1H); MS (EI, 70 eV) m/z (%): 395 (16), 210 (100).
3-{1-[2-Oxo-3-(4-phenoxyphenoxy)propyl]-1H-1,2,3-triazol-4-yl}propionic acid 32
A solution of 30 (238 mg, 0.60 mmol) in methanol (15 mL) was treated with 10% aqueous KOH solution (10 mL) and stirred for 3 h at room temperature. After dilution with water, the reaction mixture was acidified with 1 M aqueous HCl and exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated under reduced pressure. The residue was chromatographed on silica gel (hexane/ethyl acetate/formic acid 3 : 7 : 0.1 followed by ethyl acetate/formic acid 10 : 0.1) to give 32 as a solid (199 mg, 87%). C20H19N3O5 (381.4); mp 164–166 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm) 2.59 (t, J = 7.4 Hz, 2H), 2.87 (t, J = 7.4 Hz, 2H), 5.00 (s, 2H), 5.60 (s, 2H), 6.91–6.96 (m, 2H), 6.97–7.04 (m, 4H), 7.05–7.10 (m, 1H), 7.31–7.38 (m, 2H), 7.78 (s, 1H), 12.20 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 20.6, 33.1, 55.4, 71.1, 116.0, 117.4, 120.6, 122.7, 123.5, 129.9, 145.7, 150.0, 153.9, 157.8, 173.6, 199.2; HRMS (APCI, direct probe) m/z [M + H]+ calc.: 382.1429, found: 382.1397.
1-(2-Hydroxy-5-phenylphenyl)ethan-1-one 35
5-Bromo-2-hydroxyacetophenone (34) (2.01 g, 9.35 mmol) and phenylboronic acid (1.14 g, 9.35 mmol) were treated with 2 M aqueous potassium carbonate solution (6 mL). After addition of isopropanol (20 mL) and bis(triphenylphosphine)palladium(ii)-dichloride (150 mg), the mixture was stirred under nitrogen atmosphere at room temperature for 20 min and at 85 °C for another 2 h. After cooling, the reaction mixture was diluted with water (20 mL) and exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. The residue was dissolved in a small volume of THF, treated with a small amount of silica gel and evaporated to dryness. The residue was placed on a silica gel column and eluted with hexane/ethyl acetate (19 : 1, 9 : 1, 8 : 2) to yield 3529 as a solid (1.70 g, 86%). C14H12O2 (212.2); 1H NMR (400 MHz, CDCl3) δ (ppm) 2.70 (s, 3H), 7.07 (d, J = 8.7 Hz, 1H), 7.32–7.39 (m, 1H), 7.43–7.49 (m, 2H), 7.52–7.58 (m, 2H), 7.72 (dd, J = 8.7 and 2.3 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), MS (EI, 70 eV) m/z (%): 212 (100), 197 (92).
Ethyl 2-hydroxy-4-oxo-6-phenylchromane-2-carboxylate 36
To solution of sodium ethoxide in ethanol prepared from sodium (367 mg, 16.0 mmol) and dry ethanol (15 mL) was added a solution of 35 (1.70 g, 8.01 mmol) in dry ethanol (10 mL). A yellow precipitate was formed which dissolved after addition of diethyl oxalate (2.2 mL, 16.2 mmol). The mixture was then heated under reflux for 2 h. After cooling to room temperature, the reaction mixture was diluted with water and exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated under reduced pressure. The residue was dissolved in THF (5 mL) and treated with silica gel (4 g). The mixture was evaporated to dryness and the residue was loaded on a silica gel column. After elution with hexane/ethyl acetate (8 : 2) followed by hexane/ethyl acetate/formic acid (7 : 3 : 0.5), 36 was obtained as a solid (2.06 g, 82%). C18H16O5 (312.3); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.37 (t, J = 7.1 Hz, 3H), 3.00 (d, J = 16.7 Hz, 1H), 3.42 (d, J = 16.7 Hz, 1H), 4.39 (q, J = 7.1 Hz, 2H), 4.55 (s, 1H), 7.07 (d, J = 8.5 Hz, 1H), 7.32–7.37 (m, 1H), 7.41–7.47 (m, 2H), 7.55–7.60 (m, 2H), 7.77 (dd, J = 8.6 and 2.4 Hz, 1H), 8.15 (d, J = 2.4 Hz, 1H); MS (EI, 70 eV) m/z (%): 312 (10), 197 (33), 239 (100).
Ethyl 4-oxo-6-phenyl-4H-chromene-2-carboxylate 37
A solution of 36 (2.00 g, 6.40 mmol) in a mixture of acetic acid (200 mL) and conc. HCl (7.5 mL) was heated at 80 °C for 90 min. The reaction mixture was cooled to room temperature, diluted with water (200 mL) and exhaustively extracted ethyl acetate. The combined organic phases were dried over sodium sulfate and the solvent removed under reduced pressure. The residue was dissolved in a small amount of THF and treated with a small amount of silica gel. The mixture was evaporated to dryness and the residue was loaded on a silica gel column. After elution hexane/ethyl acetate (9 : 1, 8 : 2, 7 : 3), 37 was yielded as a solid (1.60 g, 85%). C18H14O4 (294.3); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.45 (t, J = 7.1 Hz, 3H), 4.49 (q, J = 7.1 Hz, 2H), 7.15 (s, 1H), 7.37–7.43 (m, 1H), 7.45–7.52 (m, 2H), 7.65–7.68 (m, 2H), 7.70 (d, J = 8.7 Hz, 1H), 7.99 (dd, J = 8.8 and 2.4 Hz, 1H), 8.41 (d, J = 2.4 Hz, 1H); MS (EI, 70 eV) m/z (%): 294 (100), 266 (54).
Ethyl 6-phenylchromane-2-carboxylate 38
A solution of 37 (798 mg, 2.71 mmol) in acetic acid (15 mL) was treated with catalytic amounts (15 mg) of palladium on charcoal (10%) and heated under a hydrogen atmosphere at 1.5 bar and 90 °C for 2 h. The reaction mixture was cooled to room temperature and the catalyst was filtered off over Celite®545. The filter cake was washed several times with ethyl acetate and the combined filtrates were concentrated under reduced pressure to give 38 as an oil (762 mg, >99%). C18H18O3 (282.3); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.32 (t, J = 7.2 Hz, 3H), 2.20–2.36 (m, 2H), 2.70–2.95 (m, 2H), 4.28 (q, J = 7.2 Hz, 2H), 4.76 (dd, J = 7.4 and 3.7 Hz, 1H), 6.84–7.03 (m, 2H), 7.27–7.43 (m, 4H), 7.50–7.55 (m, 2H); MS (EI, 70 eV) m/z (%): 282 (100), 209 (96), 165 (55).
(6-Phenylchroman-2-yl)methanol 39
A solution of 38 (755 mg, 2.67 mmol) in dry diethyl ether (20 mL) was treated with a 2 M solution of lithium aluminium hydride in dry THF (2.5 mL) and stirred for 60 min at room temperature. After addition of ethyl acetate and water, a colourless solid precipitated, which was re-dissolved by addition of aqueous HCl. The mixture was exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. The residue was dissolved in a small amount of THF and treated with a small amount of silica gel. The mixture was evaporated to dryness and the residue was loaded on a silica gel column. After elution with hexane/ethyl acetate (7 : 3, 6 : 4), 39 was obtained as a solid (297 mg, 46%). C16H16O2 (240.3); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.83–2.04 (m, 2H), 2.78–3.01 (m, 2H), 3.79 (dd, J = 11.8 and 3.4 Hz, 1H), 3.87 (dd, J = 11.7 and 3.4 Hz, 1H), 4.13–4.20 (m, 1H), 6.91 (d, J = 8.4 Hz, 1H), 7.27–7.36 (m, 3H), 7.37–7.43 (m, 2H), 7.50–7.56 (m, 2H); MS (EI, 70 eV) m/z (%): 240 (100), 209 (80), 165 (55).
6-Phenylchromane-2-carbaldehyde 40
Compound 39 (238 mg, 0.99 mmol) was oxidized with Dess–Martin periodinane (778 mg, 1.83 mmol) in dry dichloromethane (7 mL) in a similar manner as described for the synthesis of 30 (reaction time 15 h). Chromatography on silica gel (hexane/ethyl acetate 8 : 2) gave 40 as a solid (185 mg, 78%). C16H14O2 (238.3); 1H NMR (400 MHz, CDCl3) δ (ppm) 2.06–2.15 (m, 1H), 2.22–2.31 (m, 1H), 2.79–2.98 (m, 2H), 4.53 (dd, J = 8.7 and 3.5 Hz, 1H), 7.03 (d, J = 8.5 Hz, 1H), 7.28–7.33 (m, 2H), 7.37–7.44 (m, 3H), 7.50–7.56 (m, 2H), 9.86 (s, 1H); MS (EI, 70 eV) m/z (%): 238 (100), 209 (97), 165 (72).
2-(Oxiran-2-yl)-6-phenylchromane 41
A mixture of sodium hydride (60% in mineral oil, 41 mg, 1.03 mmol), trimethylsulfonium iodide (210 mg, 1.03 mmol) and dry DMSO (4 mL) was stirred under nitrogen for 30 min. Then a solution of 40 (163 mg, 0.68 mmol) in dry DMSO (5 mL) was added dropwise, and stirring was continued for another 30 min. After addition of water and brine, the reaction mixture was exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated under reduced pressure to give 41 as a solid (172 mg, >99%). C17H16O2 (252.3); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.94–2.03 (m, 1H), 2.06–2.13 (m, 1H), 2.79–2.98 (m, 4H), 3.20–3.27 (m, 1H), 3.88–3.96 (m, 1H), 6.94 (d, J = 8.4 Hz, 1H), 7.27–7.43 (m, 5H), 7.49–7.57 (m, 2H); MS (EI, 70 eV) m/z (%): 252 (100), 207 (32).
Methyl 3-{2-[2-hydroxy-2-(6-phenylchroman-2-yl)ethyl]-2H-tetrazol-5-yl}propionate 42
To a solution of methyl tetrazol-5-ylpropionate (431 mg, 2.76 mmol) in dry acetonitrile (12 mL) was added dropwise a solution of 41 (172 mg, 0.68 mmol) in dry acetonitrile (8 mL). After addition of cesium carbonate (896 mg, 2.75 mmol), the mixture was heated at reflux for 5 h. Then water and brine were added successively, and the reaction mixture was exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated in vacuo. The residue was dissolved in a small volume of THF and treated with silica gel. The mixture was evaporated to dryness and the residue was placed on a silica gel column. After elution with hexane/ethyl acetate (9 : 1, 8 : 2, 6 : 4), 42 was obtained as a solid (25 mg, 9%). C22H24N4O4 (408.5); 1H NMR (400 MHz, CDCl3) δ (ppm) 1.83–1.98 (m, 1H), 2.04–2.15 (m, 1H), 2.82–2.98 (m, 4H), 3.25 (t, J = 7.4 Hz, 2H), 3.70 (s, 3H), 3.98–4.14 (m, 1H), 4.26–4.40 (m, 1H), 4.77–4.96 (m, 2H), 5.04 (dd, J = 14.0 and 2.9 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 7.28–7.44 (m, 5H), 7.49–7.56 (m, 2H); MS (EI, 70 eV) m/z (%): 408 (100), 209 (73), 141 (86).
Methyl 3-{2-[2-oxo-2-(6-phenylchroman-2-yl)ethyl]-2H-tetrazol-5-yl}propionate 43
Compound 42 (25 mg, 0.061 mmol) was oxidized with Dess–Martin periodinane (39 mg, 0.092 mmol) in dry dichloromethane (3 mL) in a similar manner as described for the synthesis of 30 (reaction time 15 h). Chromatography on silica gel (hexane/ethyl acetate 8 : 2) gave 43 as a solid (16 mg, 64%). C22H22N4O4 (406.4); 1H NMR (400 MHz, CDCl3) δ (ppm) 2.17–2.26 (m, 1H), 2.31–2.41 (m, 1H), 2.85–3.01 (m, 4H), 3.27 (t, J = 7.9 Hz, 2H), 3.70 (s, 3H), 4.77 (dd, J = 9.2 and 3.3 Hz, 1H), 5.73 (d, J = 18.4 Hz, 1H), 5.93 (d, J = 18.4 Hz, 1H), 7.02 (d, J = 8.5 Hz, 1H), 7.30–7.36 (m, 2H), 7.38–7.46 (m, 3H), 7.51–7.56 (m, 2H); MS (EI, 70 eV) m/z (%): 406 (52), 209 (64), 141 (100).
3-{2-[2-Oxo-2-(6-phenylchroman-2-yl)ethyl]-2H-tetrazol-5-yl}propionic acid 44
A solution of 43 (20 mg, 0.049 mmol) in methanol (5 mL) was treated with 10% aqueous KOH solution (1 mL) and stirred for 4 h at room temperature. After dilution with water, the reaction mixture was acidified with 6 M aqueous HCl, stirred for further 10 min and exhaustively extracted with ethyl acetate. The combined organic phases were dried over sodium sulfate and concentrated under reduced pressure. The residue was chromatographed on silica gel (hexane/ethyl acetate/formic acid 7 : 3 : 0.1, 6 : 4 : 0.1) to give 44 as a solid (14 mg, 73%). C21H20N4O4 (392.4); purity (HPLC) >99%; mp 175 °C; 1H NMR (400 MHz, DMSO-d6) δ (ppm) 2.14–2.32 (m, 2H), 2.70–2.82 (m, 3H), 2.88–2.99 (m, 1H), 3.09 (t, J = 7.2 Hz, 2H), 5.09 (dd, J = 8.2 and 3.6 Hz, 1H), 6.07 (d, J = 18.4 Hz, 1H), 6.21 (d, J = 18.4 Hz, 1H), 7.00 (d, J = 8.3 Hz, 1H), 7.28–7.35 (m, 1H), 7.39–7.47 (m, 4H), 7.58–7.65 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ (ppm) 21.0, 23.43, 23.54, 31.8, 58.1, 79.2, 117.6, 122.8, 126.4, 126.9, 127.4, 128.6, 129.5, 133.6, 140.5, 153.4, 166.0, 173.7, 201.2; HRMS (APCI, direct probe) m/z [M + H]+ calc.: 393.1557, found: 393.1564.
Biological and physicochemical evaluation
Inhibition of cytosolic phospholipase A2α (cPLA2α)
Inhibition of cPLA2α isolated from porcine platelets was measured by a recently published procedure.21 Briefly, co-vesicles consisting of the substrate 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (200 μM) and 1,2-dioleoyl-sn-glycerol (100 μM) were prepared in Tris buffer by sonication and incubated with the enzyme for 60 min. The enzyme reaction was terminated by adding a mixture of acetonitrile, methanol, and 0.1 M aqueous EDTA–Na2 solution containing 4-undecyloxybenzoic acid as an internal standard and nordihydroguaiaretic acid (NDGA) as an oxygen scavenger. cPLA2α activity was determined by measuring the arachidonic acid released by the enzyme in the absence and presence of a test compound using reversed-phase HPLC and UV detection at 200 nm after sample purification by online solid phase extraction.
Inhibition of fatty acid amide hydrolase (FAAH)
Inhibition of FAAH was determined as previously described.32 Briefly, the substrate N-(2-hydroxyethyl)-4-pyren-1-ylbutanamide33 dissolved in a solution of Triton X-100 (0.20%) in phosphate buffered saline was incubated with rat brain microsomes. The final substrate concentration was 100 μM. The enzyme reaction was terminated after 60 min by addition of a mixture of acetonitrile/methanol containing the internal standard 6-pyren-1-ylhexanoic acid. FAAH inhibition was determined by measuring the amount of 4-pyren-1-ylbutanoic acid released by the enzyme in the absence and presence of a test compound using reversed-phase HPLC with fluorescence detection.
Partition coefficient (log P)
log P values were determined by reversed-phase HPLC using a published OECD method as previously described.36 In this way, a log P value of 2.9 was determined for indomethacin.
Metabolic stability
The metabolic stability was tested using S9 fraction of rat liver homogenate as previously described.32 Briefly, the test compounds (final concentration: 20 μM) were incubated with the liver preparation under aerobic conditions in the absence and presence of the co-factor NADPH (final concentration: 1 mM). The metabolic reactions were terminated after 30 min. The extent of metabolism was evaluated by reversed-phase HPLC with UV or MS detection.23,32
Aqueous solubility
Thermodynamic solubility was determined analogously a previously described procedure.40 Briefly, to 1 mg of a test compound was added phosphate buffered saline (0.01 M, pH 7.4) (2 mL). The mixture was sonicated for 10 min in a bath sonifier and then shaken for 20 h at room temperature. After centrifugation, to an aliquot of the clear supernatant, acetonitrile was added, and the amount of the target compound present in the sample was determined by reversed-phase HPLC and UV detection at 240 nm. With this method, for the reference indomethacin an aqueous solubility of 246 ± 18 mg mL−1 (mean ± standard deviation, n = 4) was measured.
Conclusions
In summary, the structural modification of the dual cPLA2α/FAAH inhibitor 7 did not always affect the activity towards the two enzymes in the same direction. In particular, the introduction of methyl substituents on the carbon atoms adjacent to the ketone group led to markedly differing effects in terms of inhibitory potency. The most potent inhibitors found are relatively easily reduced at the ketone group, as shown in in vitro experiments with rat liver homogenate, resulting in a loss of their efficacy. Therefore, these substances are not suitable for systemic application. However, compound 10, for example, could be used topically for inflammatory diseases, as it has both a good inhibitory effect and favorable physicochemical properties.
Author contributions
Conceptualization: MEV, ML. Investigation: MEV, WH. Methodology: MEV, ML. Project administration: ML. Resources: ML. Supervision: ML. Validation: MEV, WH, ML. Visualization: MEV, ML. Writing – original draft: ML.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3md00224a
References
- Khan S. A. Ilies M. A. Int. J. Mol. Sci. 2023;24:1353. doi: 10.3390/ijms24021353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita Y. Shindou H. Shimizu T. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids. 2019;1864:838–845. doi: 10.1016/j.bbalip.2018.08.006. [DOI] [PubMed] [Google Scholar]
- Santoso A. D. De Ridder D. Cannabis Cannabinoid Res. 2023;8:56–76. doi: 10.1089/can.2021.0237. [DOI] [PubMed] [Google Scholar]
- Giacobbe J. Marrocu A. Di Benedetto M. G. Pariante C. M. Borsini A. Brain, Behav., Immun. 2021;93:353–367. doi: 10.1016/j.bbi.2020.12.024. [DOI] [PubMed] [Google Scholar]
- Batsika C. S. Gerogiannopoulou A. D. Mantzourani C. Vasilakaki S. Kokotos G. Expert Opin. Drug Discovery. 2021;16:1287–1305. doi: 10.1080/17460441.2021.1942835. [DOI] [PubMed] [Google Scholar]
- Soubhye J. van Antwerpen P. Dufrasne F. Curr. Med. Chem. 2018;25:2418–2447. doi: 10.2174/0929867325666180117103919. [DOI] [PubMed] [Google Scholar]
- Lodola A. Castelli R. Mor M. Rivara S. Expert Opin. Ther. Pat. 2015;25:1247–1266. doi: 10.1517/13543776.2015.1067683. [DOI] [PubMed] [Google Scholar]
- van Egmond N. Straub V. M. van der Stelt M. Annu. Rev. Pharmacol. Toxicol. 2021;61:441–463. doi: 10.1146/annurev-pharmtox-030220-112741. [DOI] [PubMed] [Google Scholar]
- Dessen A. Tang J. Schmidt H. Stahl M. Clark J. D. Seehra J. Somers W. S. Cell. 1999;97:349–360. doi: 10.1016/S0092-8674(00)80744-8. [DOI] [PubMed] [Google Scholar]
- Burke J. E. Babakhani A. Gorfe A. A. Kokotos G. Li S. Woods Jr. V. L. McCammon J. A. Dennis E. A. J. Am. Chem. Soc. 2009;13:8083–8091. doi: 10.1021/ja900098y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K. McKinneyn M. K. Cravatt B. F. Chem. Rev. 2008;108:1687–1707. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimble L. A. Street I. P. Perrier H. Tremblay N. M. Weech P. K. Bernstein M. A. Biochemistry. 1993;32:12560–12565. doi: 10.1021/bi00210a002. [DOI] [PubMed] [Google Scholar]
- Street I. P. Lin H. K. Laliberte F. Ghomashchi F. Wang Z. Perrier H. Tremblay N. M. Huang Z. Weech P. K. Gelb M. H. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- Huang Z. Liu S. Street I. Laliberte F. Abdullah K. Desmarais S. Wang Z. Kennedy B. Payette P. Riendeau D. Weech P. Gresser M. Mediators Inflammation. 1994;3:307. [Google Scholar]
- Deutsch D. Omeir R. Arreaza G. Salehani D. Prestwich G. D. Huang Z. Howlett A. Biochem. Pharmacol. 1997;53:255–260. doi: 10.1016/S0006-2952(96)00830-1. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L. Melck D. Ueda N. Maurelli S. Kurahashi Y. Yamamoto S. Marino G. Di Marzo V. Biochem. Biophys. Res. Commun. 1997;231:82–88. doi: 10.1006/bbrc.1997.6000. [DOI] [PubMed] [Google Scholar]
- Koutek B. Prestwich G. D. Howlett A. C. Chin S. A. Salehani D. Akhavan N. Deutsch D. G. J. Biol. Chem. 1994;269:22937–22940. doi: 10.1016/S0021-9258(17)31599-5. [DOI] [PubMed] [Google Scholar]
- Maurelli S. Bisogno T. De Petrocellis L. Di Luccia A. Marino G. Di Marzo V. FEBS Lett. 1995;377:82–86. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- Connolly S. Bennion C. Botterell S. Croshaw P. J. Hallam C. Hardy K. Hartopp P. Jackson C. G. King S. J. Lawrence L. Mete A. Murray D. Robinson D. H. Smith G. M. Stein L. Walters I. Wells E. Withnall W. J. J. Med. Chem. 2002;45:1348–1362. doi: 10.1021/jm011050x. [DOI] [PubMed] [Google Scholar]
- Ludwig J. Bovens S. Brauch C. Schulze Elfringhoff A. Lehr M. J. Med. Chem. 2006;49:2611–2620. doi: 10.1021/jm051243a. [DOI] [PubMed] [Google Scholar]
- Hanekamp W. Lehr M. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2012;900:79–84. doi: 10.1016/j.jchromb.2012.05.018. [DOI] [PubMed] [Google Scholar]
- Forster L. Ludwig J. Kaptur M. Bovens S. Schulze Elfringhoff A. Holtfrerich A. Lehr M. Bioorg. Med. Chem. 2010;18:945–952. doi: 10.1016/j.bmc.2009.11.028. [DOI] [PubMed] [Google Scholar]
- Garzinsky D. Zahov S. Ekodo Voundi M. Hanekamp W. Lehr M. Eur. J. Med. Chem. 2018;160:183–192. doi: 10.1016/j.ejmech.2018.10.021. [DOI] [PubMed] [Google Scholar]
- Fabian J. Lehr M. J. Pharm. Biomed. Anal. 2007;43:601–605. doi: 10.1016/j.jpba.2006.07.017. [DOI] [PubMed] [Google Scholar]
- Lehr M., Zahov S. and Ekodo Voundi M., DE102013016573, 2015
- Lehr M., Ekodo Voundi M. and Althaus J., WO2017093351, 2017
- Banville J., Remillard R., Balasubramanian N., Bouthillier G. and Martel A., US2002037875, 2002
- Zahov S. Garzinsky D. Hanekamp W. Lehr M. Bioorg. Med. Chem. 2017;25:825–837. doi: 10.1016/j.bmc.2016.11.025. [DOI] [PubMed] [Google Scholar]
- Sundermann T. Lehr M. Synth. Commun. 2014;44:1641–1648. doi: 10.1080/00397911.2013.868490. [DOI] [Google Scholar]
- Raju B. C. Rao R. N. Suman P. Yogeeswari P. Sriram D. Shaik T. B. Kalivendi S. V. Bioorg. Med. Chem. Lett. 2011;21:2855–2859. doi: 10.1016/j.bmcl.2011.03.079. [DOI] [PubMed] [Google Scholar]
- Witiak D. T. Heilman W. P. Sankarappa S. K. Cavestri R. C. Newman H. A. J. Med. Chem. 1975;18:935–942. doi: 10.1021/jm00243a015. [DOI] [PubMed] [Google Scholar]
- Holtfrerich A. Hanekamp W. Lehr M. Eur. J. Med. Chem. 2013;63:64–75. doi: 10.1016/j.ejmech.2013.01.050. [DOI] [PubMed] [Google Scholar]
- Forster L. Schulze Elfringhoff A. Lehr M. Anal. Bioanal. Chem. 2009;394:1679–1685. doi: 10.1007/s00216-009-2850-5. [DOI] [PubMed] [Google Scholar]
- Sundermann T. R. Lehr M. Tetrahedron: Asymmetry. 2017;28:447–453. doi: 10.1016/j.tetasy.2017.02.013. [DOI] [Google Scholar]
- Lehr M. Fabian J. Hanekamp W. Biopharm. Drug Dispos. 2015;36:398–404. doi: 10.1002/bdd.1946. [DOI] [PubMed] [Google Scholar]
- Drews A. Bovens S. Roebrock K. Sunderkötter C. Reinhardt D. Schäfers M. van der Velde A. Schulze Elfringhoff A. Fabian J. Lehr M. J. Med. Chem. 2010;53:5165–5178. doi: 10.1021/jm1001088. [DOI] [PubMed] [Google Scholar]
- Sundermann T. Fabian J. Hanekamp W. Lehr M. Bioorg. Med. Chem. 2015;23:2579–2592. doi: 10.1016/j.bmc.2015.03.033. [DOI] [PubMed] [Google Scholar]
- Chen L. Wang W. Lee K. L. Shen M. W. Murphy E. A. Zhang W. Xu X. Tam S. Nickerson-Nutter C. Goodwin D. G. Clark J. D. McKew J. C. J. Med. Chem. 2009;52:1156–1171. doi: 10.1021/jm8009876. [DOI] [PubMed] [Google Scholar]
- Valko K. Rexnolds D. P. Am. J. Drug Delivery. 2005;3:83–100. doi: 10.2165/00137696-200503020-00002. [DOI] [Google Scholar]
- Fritsche A. Schulze Elfringhoff A. Fabian J. Lehr M. Bioorg. Med. Chem. 2008;16:3489–3500. doi: 10.1016/j.bmc.2008.02.019. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.