

Abstract
MEK1/2 are critical components of the RAS–RAF–MEK–ERK or MAPK signalling pathway that regulates a variety of cellular functions including proliferation, survival, and differentiation. In 1997, a lung cancer cell line was first found to have a MEK mutation (encoding MEK2P298L). MEK is involved in various human cancers such as non-small cell lung cancer (NSCLC), spurious melanoma, and pancreatic, colorectal, basal, breast, and liver cancer. To date, 4 MEK inhibitors i.e., trametinib, cobimetinib, selumetinib, and binimetinib have been approved by the FDA and several are under clinical trials. In this review, we have highlighted structural insights into the MEK1/2 proteins, such as the αC-helix, catalytic loop, P-loop, F-helix, hydrophobic pocket, and DFG motif. We have also discussed current issues with all FDA-approved MEK inhibitors or drugs under clinical trials and combination therapies to improve the efficacy of clinical drugs. Finally, this study addressed recent developments on synthetic MEK inhibitors (from their discovery in 1997 to 2022), their unique properties, and their relevance to MEK mutant inhibition.
MEK1/2 are critical components of RAS–RAF–MEK–ERK or MAPK signalling pathway.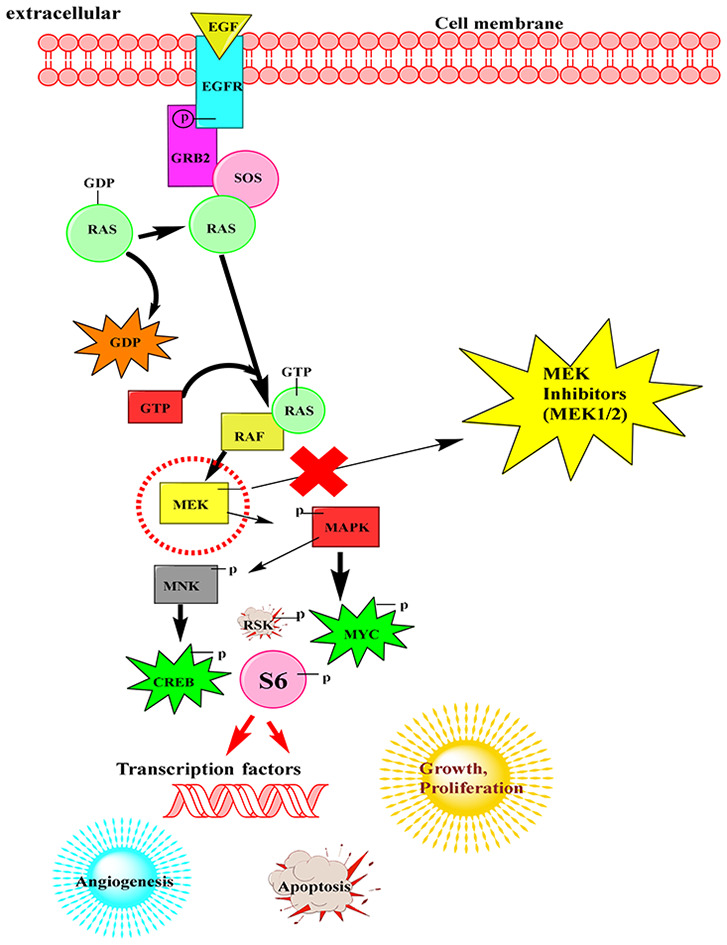
1. Introduction
Cancer incidence and mortality are rapidly increasing worldwide, and cancer is expected to be the major hindrance to improving life expectancy globally. Mitogen-activated protein kinases (MAPKs) are responsible for the development of various types of cancers. MAPKs are a family of conservative protein serine/threonine kinases that respond to a variety of extracellular stimuli and are involved in gene expression, cell metabolism, proliferation, differentiation, and apoptosis. The MAPK pathway includes three major kinases, MAPK kinase kinase (MAPKKK, MAP3K), MAPK kinase (MAPKK, MAP2K, MEK), and MAPK (ERK1/2), which activate and phosphorylate downstream proteins. The MAPK/ERK pathway is the binding of an external mitogen to a cell surface receptor. This allows a Ras protein (a small GTPase) to exchange a GDP molecule for a GTP molecule, thereby turning the signalling pathway on and off. The Ras protein can then stimulate MAP3K (e.g., Raf), which activates MAP2K, which in turn activates MAPK. MAPK (ERK1/2) regulates gene expression by directly phosphorylating transcription factors such as Ets, Elk and Myc. MAPK alters the level and activity of transcription factors, resulting in altered transcription of cell cycle-related genes. In the MAPK pathway, the MEK (MEK1/2) pathway is one of the most important.1–5 It involves a series of proteins in the cell that relay signals from a receptor on the cell surface to DNA in the nucleus. MEK1/2, also referred to as gatekeepers of ERK1/2, are responsible for the transduction of signals from a number of upstream kinases and are the only activators of downstream ERK1/2. At the same time, ERK1/2 are the only downstream MEK1/2 substrates.6 Nevertheless, cellular metamorphosis is a consequence of constitutive activation, and it is involved in the development of a variety of human cancers.7,8 The MEK pathway is one of the best-studied kinase cascades in cancer cell biology. Growth factors or activating mutations of the main oncogenic proteins in this pathway are the most common components of the MAPK pathway. Although mutations in MEK1 and MEK2 are rare in cancer, production of their mutant versions in constitutively active states (MEK1-DN3/S218E/S222D and MEK2-DN4/S222D/S226D, respectively) is sufficient to subject normal cells to oncogenic transformation.9 In 1997, a lung cancer cell line was first found to have a MEK mutation (encoding MEK2P298L), but the functional effects were not specified.10 In ovarian cancer cell lines, activating mutations in MEK1 or MEK2 were first detected in 2007.11 Since then, gain-of-function mutations in MEK1 (P124S, E203K, F53L, and N382H) or MEK2 (S154F) in melanoma, Y134C in MEK2 or Y130C in MEK1 in colorectal cancer (CRC), Q56P and K57N in MEK1 in lung cancer, TP53, CDKN2A, and SMAD4 in MEK1 in pancreatic cancer and D67N in MEK1 in ovarian cancer have been reported.12,13 Most of these mutations belong to mutations present in cardio-facio-cutaneous (CFC) syndrome, either in the N-terminal negative regulatory region or in the ATP-binding region of the N-terminal lobe. Since MEK activation represents a convergence point for the abnormal activation of other upstream signalling molecules, it may be a suitable molecular therapeutic target.14,15 Mutations in MEK occur at high frequency in numerous human malignancies, such as pancreatic cancer 70–90%, uveal melanoma 50%, liver cancer 20–40%, colorectal cancer 25–35%, melanoma 15–20%, NSCLC 10–20%, and basal-like breast cancer 1–5%. Statistics of new cases and deaths from all cancers worldwide for 2022 and details of specific cancers affected by the MEK mutation are shown in Fig. 1.13,16
Fig. 1. Global cancer statistics for new cases and deaths for 2022 and MEK (%) mutation in different cancers.

2. Regulation of the MEK signalling pathway
When a signalling molecule attaches to a cell surface receptor, signalling begins. It finishes when cellular DNA produces a protein, which results in cell growth. The excellent signal-regulated kinases (ERKs) or mitogen-activated protein kinases (MAPKs) interact by adding phosphate groups to neighbouring proteins (phosphorylation) and acting as “on” switches. When a protein in the signalling pathway is changed and stuck in the “on” or “off” state, it marks a critical stage in the growth of many malignancies. In fact, the MAPK/ERK pathway's components were initially discovered in cancer cells, and medications that turn the process on or off are currently being studied.1 EGF receptor (EGFR)-bound tyrosine kinases are activated by extracellular ligands like epidermal growth factor (EGF). EGF binds to an EGFR, which then causes the cytoplasmic domain of the protein to become active. The EGFR is phosphorylated as a result of tyrosine residues. The active receptor's phosphotyrosine residues interact with GRB2's SH2 domain.17 In order to connect to the guanine nucleotide exchange factor son of sevenless (SOS), the two SH3 domains of GRB2 engage with it. When the phosphorylated EGFR is bound by the GRB2–SOS complex, SOS is triggered.18 Activation of SOS then encourages the removal of guanosine diphosphate (GDP) from a Ras subfamily member. After binding of guanosine triphosphate (GTP), the Ras protein is subsequently activated. Along with fibroblast growth factor receptors (FGFRs), other cell surface receptors such as neurotrophin receptors (Trk A/B), FGFRs, and platelet derived growth factor receptors (PDGFRs) can activate this pathway via GRB2. As a result of the activated Ras's protein kinase activity, RAF kinase phosphorylates and activates a MAPK kinase (MEK). MEK is responsible for phosphorylating and activating a MAPK (ERK).19 Some of these phosphorylation events serve to enhance Raf activity (shown by a black P in a black circle) whereas others serve to inhibit Raf activity (shown by a black P in a red circle). Moreover, there are phosphatases, such as PP2A, which remove phosphates on certain regulatory residues. The downstream transcription factors regulated by this pathway are indicated by oval shaped outlines. Selective protein kinases for serine or threonine include RAF and MAPK/ERK. This mechanism has also been linked to apoptosis regulation. By post-translationally phosphorylating molecules like Bad, Mcl-1, Bcl-2, and Bim, RAF also activates mitochondrial localized proteins (Fig. 2).20
Fig. 2. Raf/MEK/ERK or MAPK pathway importance in cell proliferation and survival.

MEK is a kinase of serine, tyrosine, and threonine. MNK, RAF, MEK, and MAPK are technically referred to as mitogen-activated kinases. MAPKs were once known as microtubule-associated protein kinases (MAPKs) and extracellular signal-regulated kinases (ERKs). One of the earliest proteins shown to have been phosphorylated by ERK was a protein related to microtubules.
The translation of mRNA into proteins is one of the effects of MAPK activation. The S6 (RSK) kinase of the 40S ribosomal protein is phosphorylated by MAPK. This activates RSK which then phosphorylates the ribosomal protein S6.21 Several transcription factors are controlled by MAPK. C-MYC can be phosphorylated by MAPK. MNK is phosphorylated and activated by MAPK, which causes MNK to phosphorylate cyclic AMP (cAMP)-response element binding protein (CREB). Transcription of the C-FOS genes is also controlled by MAPK phosphorylated CREB. The transcription of C-FOS genes is also controlled by MAPK. By altering the levels and functions of transcription factors, MAPK affects the transcription of genes important for the cell cycle (Fig. 2).
3. Molecular structure of the MEK protein
The general features of protein kinases originally discovered for the cyclic AMP-dependent protein kinase are largely conserved and shared by kinases in the MAPK pathway.22 MEK proteins have two types, MEK1 and MEK2. MEK1 contains 393 amino acids and MEK2 contains 400 amino acids. They consist of two lobes, the N-lobe and the C-lobe. The N-lobe contains an αC-helix (L118/122) and a P-loop (74–82 in MEK1 and 78–86 in MEK2). The C-lobe has an activation segment (210–233 in MEK1 and 214–237 in MEK2), and an F-helix (D245/L253/M256 in MEK1 and 249/257/260 in MEK2). The catalytic loop (192–195 in MEK1 and 196–198 in MEK2), hydrophobic pocket (MET 143, MET 219, ALA 220, PHE 223) and DFG-motif (ASP 208, PHE 209, GLY 210 in MEK1 and ASP 212, PHE 213, GLY 214 in MEK2) are the critical binding pockets in the MEK1/2 proteins (Table 1). Both sides of MEK1 and MEK2's core catalytic domains are surrounded by proline-rich inserts and short amino and carboxy-terminal sections. These kinases are highly similar, sharing 90% of their kinase domain identities and exhibiting 80% total similarity. The flanking amino-terminal regions of MEK1 and MEK2, which are composed of amino acids 1–67 and 1–71, are 58% similar. Additionally, the first 32 and 36 residues of MEK1 and MEK2 exhibit a sequence similarity of 22%, lower than that of the next 28 and 24 residues (28% homology against 82% homology), respectively. This is significant because they are drawn to their ERK substrates at this location due to the lowest similarity. The first 10 residues of the D-domain (amino acids 1–32 in MEK1 and 1–36 in MEK2), commonly known as the docking site of ERK, are a short length of basic and hydrophobic amino acids.23 MEK must have a positively charged D-domain in order to attach to the associated acidic common docking domain in the carboxy termini of ERK1 and ERK2.24,25 The leucine-rich nuclear export signal (NES), which is made up of the amino acids 33–44 and 37–48 and is situated between the D-domain and the core catalytic domain, is essential for MEK's subcellular localization. MEKs are often seen in the cytoplasm. Catalytically inactive enzymes lacking NES convert lysine to alanine, and modify the distribution of MEKs in the cytoplasm and nucleus at steady state. This demonstrates that MEKs are quickly exported to the cytoplasm following activation and move to the nucleus in a NES-dependent manner.26,27 A negative regulatory area is positioned just downstream of the NES (NNR). When residues 44–51 from MEK1 and 48–55 from MEK2 are removed, the basal kinase activity increases by 60 and 9 times, respectively.28 These residues cause a disruption in the ATP-binding site, which inhibits MEK action. Changes in this area of the genome can affect MEK's catalytic activity.14 A proline-rich domain (amino acids 262–307 in MEK1 and 266–315 in MEK2) is located in the conserved core catalytic domain's carboxy-terminal region. It is hypothesised to facilitate certain protein–protein interactions that are crucial for the regulation of MEKs.29,30 MEK1 and MEK2 have 69% homology in the carboxy-terminal region, which includes the amino acids 74–82 of MEK1 and 78–86 of MEK2, the glycine-rich P-loop, the Mg2+ positioning loop, the ATP-binding site, and amino acids 143–146 of MEK1 and 147–150 of MEK2 (amino acids 362–393, 370–400 of C-terminal). This domain's specific purpose is unknown. However, according to Brunet et al., MEK1 is phosphorylated by ERK at T386 as part of a negative feedback loop that controls MEK1 inactivation. They featured a catalytic cleft to which Mg-ATP binds to allow phosphoryl transfer from the active site situated between the larger C-terminal lobe and the smaller N-terminal lobe. In addition to this, a conserved glycine-rich loop aids in positioning bound ATP for cleavage and phosphoryl transfer. The N-lobe of MEK has five β-sheets and also contains a conformation-dependent C-helix that is necessary for the activation state of the kinases.
Number of residues in MEK1 and MEK2.
| Structural insight | Residues in MEK1 | Residues in MEK2 |
|---|---|---|
| αC-helix (N-lobe) | L118 | 122 |
| P-loop/ADP (N-lobe) | 74–82 | 78–86 |
| ATP binding sites | 143–146 | 147–150 |
| Activation segment (C-lobe) | 210–233 | 214–237 |
| Catalytic loop | 191–195 | 196–198 |
| F-helix (C-lobe) | D245/L253/M256 | 249/257/260 |
| D-helix (C-lobe) | L151 | 155 |
| DFG motif | ASP208, PHE209, GLY212 | ASP12, PHE213, GLY214 |
| No. of residues | 393 | 400 |
| Molecular weight (kDa) | 43 kDa | 44 kDa |
Structural insights into the MEK protein are depicted in Fig. 3. The conserved valine (V81/85 in MEK1/2) interacts hydrophobically to the adenine of ATP after the glycine-rich loop. Six preserved helices are present along with four sheets in the C-terminal lobe. A motif called Ala-XXX-Lys may be found in the N-3 strand lobe (MEK1/2 residues 95–97/99–101). Kinase activation depends on forming a crucial salt bridge between the lysine of the third strand (residues 114/118 in MEK1/2) and the conserved glutamate of the C-helix to stabilize the active conformation. A conserved DFG motif appears at the beginning of the MEK activation domains, while a common APE motif appears at the end (SPE in MEK1). MEK has flexible hinge regions that allow the N and C lobes to rotate against one another. To form a closed active site and to bring distant active site remnants closer together, the N and C lobes must be turned inward towards one another. For ATP binding and ADP release to occur throughout the catalytic cycle, a very small rotation of the N and C lobes is needed. Aspartate's side chain must rotate inward in order to coordinate Mg2+ in the active site, making the D of the C-line of the DFG motif essential for adopting the active conformation. The aspartate marked “DFG out” is pointed outward in the inactive configuration. It is believed that the aspartate residue (residue 190/194 in MEK1/2) in the catalytic loop deprotonates the protein substrate and facilitates its nucleophilic assault on the ATP-phosphate.22 For an active site to operate, conformational changes must be caused by phosphorylation of the activation segment's residues. MEK1 and MEK2 both have two serine residues (at positions S218 and S222 and S222 and S226, respectively) in the activation region phosphorylated by RAF kinase. Although both residues are needed for activation, MEK is completely deactivated when the other one is dephosphorylated.31 Studying various protein kinases' inactive and active conformations resulted in the identification of numerous important residues that are a component of larger structural motifs called regulatory and catalytic spines, also known as R-spines and C-spines, respectively.32,33
Fig. 3. Structural insight into the MEK protein. (A) MEK linear models and binding pockets. The linear models of MEK1 and MEK2 show the mutations and functional regions. The linear models of both kinases, MEK1 and MEK2, are displayed in relation to their functional domain placements. Above each is a scale with hash marks at the locations of SNPs, somatic cancer mutations, syndrome-related mutations, experimentally discovered phosphorylation sites (blue hash with oval), and predicted phosphorylation sites (blue-green hash). The positions of the amino acids are indicated by numbers starting with the first Met residue. (B) Crystal structure of MEK protein. (C) The majority of MEK protein binding sites. Various binding locations are indicated by different colours. Burgundy, cyan, and blue (C-helix as well as the F-helix binding site). The P-loop/ADP binding site is cornflower blue, while the D-helix binding site is dark green. Green (catalytic loop); yellow and green (activation segment).

4. MEK inhibitors in cancer treatment
Many MEK inhibitors are presently undergoing testing in various clinical and preclinical stages. Contrary to RAF inhibitors, which are often ATP-competitive, a majority of MEK inhibitors are allosteric and not ATP-competitive.34 To date, four MEK inhibitors have achieved FDA approval, including cobimetinib for malignant melanomas with BRAFV600 mutations, selumetinib for NF1-related plexiform neurofibromas (PNs), and binimetinib and trametinib for BRAFV600E/K-mutated metastatic melanomas.35–37 Only moderate clinical success has been seen when MEK inhibitors are used as the sole therapy for patients with KRAS-mutated cancers. MEK inhibitors have demonstrated a strong anti-tumour effect and have been utilised as monotherapies for several types of RAS mutations; nevertheless, their performance is ultimately constrained by their dose-limiting toxicities and the possibility for resistance development.38,39 MEK inhibitors by themselves are less successful in treating tumours than BRAF plus MEK inhibitor combos, which have had tremendous success. For instance, in advanced BRAFV600E melanoma, the FDA-approved combination of trametinib and dabrafenib and cobimetinib and vemurafenib outperformed BRAF inhibitor monotherapy.40
4.1. FDA approved MEK inhibitors
The first MEK inhibitor, trametinib (GSK1120212, 73), received FDA approval in May 2013 for treating patients with metastatic or terminal BRAFV600E/K-mutated melanoma. In January 2014, the United States granted accelerated approval for trametinib and dabrafenib for the same indications. Trametinib was first developed by Japan Tobacco and then developed and introduced by GlaxoSmithKline under the trade name Mekinist as a DMSO solvate. It is an effective allosteric, non-competitive, and ATP-accessible inhibitor of the protein kinases MEK1 and MEK2.41 Trametinib can also inhibit MEK activation by decreasing phosphorylation at Ser-217. The typical dual phosphorylation of MEK would be disrupted, resulting in a primarily monophosphorylated protein at Ser-221.42 Trametinib in a phase 2 trial in combination with dabrafenib and pembrolizumab for the treatment of advanced melanoma was developed by Merck Sharp & Dohme Corp. and Novartis. A favourable toxicity profile was seen in the phase 1 research of patients with BRAFV600-mutated melanoma, and the continuing phase 2 inquiry will further assess the safety and effectiveness of this triple combination as a first-line therapy for BRAF-mutated melanoma.43
The second MEK inhibitor to receive approval was cobimetinib (GDC-0973, XL518), created by Exelixis and Genentech (Roche).44 Cobimetinib is an allosteric, non-ATP-competitive MEK inhibitor.45 In combination with vemurafenib, cobimetinib was licenced in Switzerland in August 2015 and in the United States and Europe in November 2015 to treat metastatic or unresectable melanoma that had the BRAFV600 mutation.44 Cobimetinib is a highly selective and effective reversible MEK inhibitor that prevents the phosphorylation of ERK1/2.46 Several clinical trials are now underway for cobimetinib in conjunction with several targeted medicines. For example, in the treatment of metastatic solid tumours, cobimetinib has been coupled with the PI3K or Erk1/2 inhibitor GDC-0941 or GDC-0994, and in the treatment of leukaemia with the p53 MDM2 inhibitor idasanutlin as well as the BCL-2 inhibitor venetoclax.47 Another contentious issue is the use of immunotherapy in conjunction with cobimetinib. A phase Ib dose-escalation and dose-extension study (NCT01988896) in melanoma patients revealed longer PFS with a median of 12.0 months in the combination therapy group compared to atezolizumab or cobimetinib alone.48 According to an updated phase Ib study, cobimetinib plus atezolizumab plus vemurafenib revealed an acceptable safety profile and possible antitumor effectiveness in BRAFV600-mutated metastatic melanoma.49 The combination of cobimetinib (Cobi) and atezolizumab (Atezo) is well tolerated at the highest dosages delivered. These findings support additional investigation into this therapy and suggest that individuals suffering from MSS CRC may benefit from the combination of Cobi and Atezo.50
Selumetinib (Koselugo; AZD6244; ARRY-142886) is an oral second-generation kinase inhibitor and a strong and selective non-ATP-competitive inhibitor of mitogen-activated protein kinase 1 and 2 (MEK1/2).36,51 Array BioPharma and AstraZeneca jointly developed selumetinib for clinical research in 2004, and it has subsequently undergone a number of phase I and phase II clinical trials for solid tumours as a monotherapy.52–54 Selumetinib received FDA approval in May 2016 to receive orphan drug designation for the treatment of people with stage III or IV differentiated thyroid cancer and as a therapy for neurofibromatosis type 1 (NF1) (in the US and EU).55 The FDA approved selumetinib (KOSELUGO, AstraZeneca) in April 2020 for the treatment of pediatric patients with neurofibromatosis type 1 (NF1) who had symptomatic and unresectable plexiform neurofibromas (PNs).56–58 The use of selumetinib in conjunction with sorafenib for advanced hepatocellular carcinoma (HCC) is also being studied (ClinicalTrials.gov identifier, NCT01029418).59 Although selumetinib plus docetaxel significantly improved the median PFS (5.3 vs. 2.1 months, p = 0.014) and objective response rate (37% vs. 0%, p = 0.0001) compared to docetaxel in a randomised phase II trial in KRAS-mutated NSCLC, the overall survival benefit (9.4 vs. 5.2 months) could not be replicated in the global phase III clinical trial. A total of 510 KRAS-mutated NSCLC patients were randomised to receive selumetinib plus docetaxel or a placebo plus docetaxel and the results indicated that selumetinib did not increase progression-free survival when compared to docetaxel alone (ClinicalTrials.gov number, NCT01933932).60
Binimetinib, 5-((4-bromo-2-fluorophenyl)amino)-4-fluoro-N-(2-hydroxyethoxy)-1-methyl-1H-benzo[d]imidazole-6-carboxamide (MEK162, ARRY-438162, Mektovi, also referred to as ARRY 162), is an anticancer small molecule developed by Array Biopharma to treat a range of tumours. With the potential to cure a number of malignancies, binimetinib is an orally available, highly selective, non-ATP-competitive MEK inhibitor. In June 2018, the FDA gave its approval for use in treating patients with metastatic or incurable BRAFV600E/V600K-positive melanoma when combined with encorafenib. In preclinical investigations using cell lines and animal models, binimetinib showed strong anticancer activity either alone or in conjunction with other medications. Combinations of the drug binimetinib with immunotherapies including pembrolizumab and encorafenib for the treatment of malignant melanoma and nivolumab, LGX818, and ipilimumab for the treatment of metastatic melanoma are used.61 FDA approved MEK inhibitors are depicted in Fig. 4 and Table 2 and their combination in different cancer treatment are shown in Table 3.
Fig. 4. FDA approved MEK inhibitors.

FDA approved MEK inhibitors.
| Sr. no. | FDA drugs | FDA approved | Target | Side effects | References |
|---|---|---|---|---|---|
| 01 | Trametinib | In 2013 | MEK1/2 | Fatigue rash, diarrhea, peripheral edema, and acneiform dermatitis | 62 |
| 02 | Selumetinib | In 2020 | MEK1 | Acneiform rash, gastro-intestinal effects, and asymptomatic creatine kinase elevation | 63 |
| 03 | Cobimetinib | In 2015 | MEK1/2 | Gastrointestinal disorders rash, pyrexia, increased blood CPK,12 and chorioretinopathy | 64 |
| 04 | Binimetinib | In 2018 | MEK1/2 | Rash, nausea, diarrhoea, peripheral oedema, and fatigue | 65 |
MEK inhibitors with combinations in different cancer treatments.
| Sr. no. | Drugs | Combination | FDA approved | Indication | References |
|---|---|---|---|---|---|
| 01 | Trametinib | GSK2141795 | In 2022 | Mutant melanoma | 66 |
| 02 | Trametinib | Dabrafenib | In 2014 | Malignant melanoma | 67 |
| 03 | Trametinib | Dabrafenib | In 2017 | Non-small cell lung cancer (NSCLC) | 68 |
| 04 | Trametinib | Dabrafenib | In 2018 | Anaplastic thyroid cancer (ATC) | 69, 70 |
| 05 | Selumetinib | Dacarbazine | — | Metastatic uveal melanoma | 71 |
| 06 | Cobimetinib | Vemurafenib | In 2015 | Metastatic melanoma | 72 |
| 07 | Cobimetinib | Atezolizumab | In 2020 | Metastatic colorectal cancer | 73 |
| 08 | Encorafenib | Binimetinib | In 2018 | Malignant melanoma | 74 |
| 09 | Pimasertib | Gemicitabine | In 2022 | Metastatic pancreatic adenocarcinoma | 75 |
4.2. MEK inhibitors under clinical trial
The efficacy, therapeutic indications, developers, and status of numerous MEK1/2 inhibitors have been developed and studied in clinical trials, which have been proven to be quite effective and selective.45 Under clinical trial MEK inhibitors is depicted in Fig. 5 and Table 4.
Fig. 5. Clinical trial MEK inhibitor drugs.

MEK inhibitors under clinical trials.
| Sr. no. | MEK inhibitors | Clinical trials | Mechanism of inhibition | Tumor types | Developer | References |
|---|---|---|---|---|---|---|
| 01 | CI-1040 (PD184352) | Phase 2 | Allosteric, non-ATP competitive inhibitor (MEK1/2) | Breast cancer, lung cancer, colon cancer and tumours of the pancreas | Pfizer | 106, 107 |
| 02 | Mirdametinib (PD-0325901) | Phase 2 | ATP competitive inhibitor (MEK1/2) | Colonic neoplasms, breast neoplasm carcinoma, melanoma skin cancer and NSCLC | Pfizer | 106, 108 |
| 03 | AZD-8330 | Phase 2 | Non-ATP competitive inhibitor (MEK1/2) | Advanced solid tumors | AstraZeneca | 109, 110 |
| 04 | TAK-733 | Phase 1 | Non-ATP competitive inhibitor (MEK1/2) | Advanced non-hematologic cancers and metastatic advanced melanoma | Millennium/Takeda | 82, 111 |
| 05 | GDC-0623 | Phase 1 | Allosteric, non-ATP competitive inhibitor (MEK1/2) | Solid metastatic tumors | Genentech | 112, 113 |
| 06 | Refametinib (RDEA-119, BYA-869766) | Phase 2 | Allosteric, non-ATP competitive inhibitor (MEK1/2) | Hepatocellular and colorectal cancer, and melanoma | Ardea Biosciences/Bayer | 45, 114 |
| 07 | Pimasertib (AS703026 or MSC1936369B) | Phase 2 | Non-ATP competitive inhibitor (MEK1/2) | Colorectal cancer and multiple myeloma | Merck and Co. | 45, 115 |
| 08 | RO4987655 (CH4987655) | Phase 1 | Non-ATP competitive inhibitor (MEK1/2) | Neoplasms | Hoffman-La Roche | 94, 107 |
| 09 | RO5126766 (Avutometinib) | Phase 1 | ATP competitive inhibitor (MEK1/2) | Neoplasms | Hoffman-La Roche | 94, 107 |
| 10 | EBI-1051 | Phase 3 | Non-ATP competitive inhibitor (MEK1/2) | Melanoma, and thyroid and colorectal cancer | Shanghai Hengrui Pharmaceutical Co. Ltd. | 116, 117 |
| 11 | DPS-2 | Phase 1 | Non-ATP competitive inhibitor (MEK1/2) | Colon cancer and melanoma | De Novo Pharmaceuticals | 99, 118 |
| 12 | KZ-001 | Preclinical trials | Non-ATP competitive inhibitor (MEK1/2) | Melanoma, and colon and non-small cell lung cancer | Innovent Biologics | 100, 110 |
| 13 | BI-847325 | Phase 1 trials (discontinued) | ATP-competitive inhibitor (MEK1/2) | Anaplastic thyroid carcinoma | Boehringer Ingelheim | 119, 120 |
| 14 | URML3881 | Phase 2 | Allosteric, non-ATP competitive inhibitor (MEK1/2) | Epithelial ovarian cancer | University of Rochester Medical Center (URMC) and Array BioPharma | 102 |
| 15 | WX-554 | Phase 2 | ATP-competitive inhibitor (MEK1/2) | Advanced solid tumor | Wilex AG, Germany | 103, 121 |
| 16 | KZ-02 | Phase 2 | Allosteric, non-ATP competitive inhibitor (MEK1/2) | Colorectal cancer | Kineta | 71 |
CI-1040, Pfizer/Warner-Lambert's MEK inhibitor, was the first to enter clinical trials as a highly potent and orally available small molecule inhibitor of MEK1/MEK2. It effectively blocked ERK phosphorylation and further signal transduction along this pathway. In preclinical models, this medication has demonstrated an anticancer effect, particularly against pancreatic, colorectal, and breast malignancies, which has been associated with its capacity to inhibit pERK. Phase II research on CI-1040 for the treatment of breast, colon, lung, and pancreatic malignancies revealed that it had poor solubility and quick elimination.76
Mirdametinib (PD-0325901) is an oral, highly selective small molecule inhibitor of MEK1 and MEK2 (MAPK/ERK kinase) and neurofibromatosis type 1-associated plexiform neurofibromas (NF1-PNs), which blocks the phosphorylation and subsequent activation of mitogen-activated protein kinase (MAPK). It has been obtained by optimization of the hydroxamate side chain of the MEK inhibitor CI-1040. Pfizer/Warner-Lambert's PD-0325901 caused dosage-dependent MEK inhibition and reduction in MAPK phosphorylation (pMAPK) in the liver and lungs following administration of PD-0325901 as an oral dose (PO) or as an intravenous injection (IV). Inhibition of pMAPK in the liver was usually equivalent among all routes of administration; however it remained longer in the lungs, which led to a higher maximum plasma concentration of PD-0325901 after IV dosing (Cmax).52,77 In phase II clinical studies for the treatment of non-small cell lung cancer with the KRAS mutation, PD-0325901 did not achieve its primary efficacy endpoint.78 Due to harm to the musculoskeletal system, the nervous system, and the eyes, a phase I/II trial for the treatment of breast, colon, and melanoma tumours was stopped in 2007.79 Research is going on regarding the use of PD-325901 in combination with palbociclib for the same indication. There are currently two more phase I or I/II investigations into KRAS-mutated cancers or colorectal cancer.
AZD8330, a MEK inhibitor with potential for anticancer activity, is a member of a different class of MEK inhibitors, has 6-oxo-1,6-dihydropyridazine as the basic structure38 and was developed as a non-ATP-competitive MEK1/2 inhibitor.80 The most frequently reported hazards associated with the use of AZD8330 as a single agent in treating solid tumours were lethargy, diarrhoea, vomiting, and acneiform dermatitis. Four participants experienced the following dose-limiting toxicities: rash (20 mg BID; twice daily; 1/9 patients) and mental status alterations including confusion and hallucinations (40 mg once daily; 2/9 patients and 60 mg once daily; 1/3 patients). Therefore, 20 mg twice daily was chosen as the highest dose that could be tolerated. AZD8330 exposure rose nearly proportionately with the dose in the dosing range of 0.5–60 mg once daily. ERK phosphorylation levels in peripheral blood mononuclear cells were used to confirm that the target was inhibited. AZD8330 displayed a manageable toxicity profile with fewer class impact adverse events (AEs) compared to other MEK inhibitors.81 No recent clinical studies have been published.
TAK-733 is an orally available, non-ATP-competitive small molecule MEK1/2 inhibitor with antitumor potential. With an EC50 (concentration for 50% of maximum effectiveness) of 0.19 nM against ERK phosphorylation in cells, TAK-733 is a highly effective and selective MEK inhibitor with allosteric targeting.82 The highest tolerable dose of TAK-733, which was produced by Millennium Pharmaceuticals Inc. in a phase I clinical trial, was determined to be 16 mg. A frequent drug-related side effect was dermatitis. The side effects of TAK-733 included acneiform rash, diarrhoea, and increased blood creatine phosphokinase levels. It had modest antitumor action.83 No new studies have been published recently.
GDC-0623 [(1-(5-((2-fluoro-4-iodophenyl)amino)imidazo[1,5-a]pyridin-6-yl)-2-(2-hydroxyethoxy)ethan-1-one)] is a strong, orally active, selective, non-ATP-competitive MEK inhibitor. It's a distinctive imidazo-pyridine structure invented by Genentech.84 In cell-based investigations, GDC-0623 has demonstrated great efficacy, particularly in cancer cell lines with KRAS and BRAF mutations as well as xenograft tumours.85 It is being researched for the treatment of patients with locally advanced or metastatic stable cancers. It serves as an EC 2.7.12.2 (MAPK kinase) inhibitor, antineoplastic, or an apoptosis inducer.86
Refametinib (RDEA-119, BYA-869766) is a powerful MEK inhibitor that is non-ATP-competitive, orally accessible, and has a low propensity to accumulate in the brain and other neural tissues.87 It was chosen for clinical research because of its effectiveness and great pharmacokinetic profile. The use of one or more agents has been studied in numerous phase I, I/II, and phase II clinical trials. In a phase I/II trial, refametinib plus gemcitabine showed a positive objective response rate and was well tolerated.88 Rafametinib and sorafenib combination therapy was tested in a phase II clinical trial for the first-line systemic treatment of RAS-mutated HCC due to the high prevalence of constitutive MAPK pathway activation in HCC. The outcomes showed that out of the 70 patients recruited, three showed partial remission and 25 had long-term stable illness.89 However, this combination exhibited some major side effects and toxicity which forced an alteration of the dosage for almost all patients.
Pimasertib, also known as AS703026 or MSC1936369B, is a selective, orally accessible, non-ATP competitive MEK1/2 inhibitor developed by Merck KGaA. In cell lines and xenograft models with constitutive MAPK pathway activation, it has demonstrated considerable anticancer efficacy. Its structure differs from that of other MEK inhibitors in that it includes a (2-fluoro-4-iodophenyl)amino group, a pyridine core structure, and an (S)-N-(2,3-dihydroxypropyl)acetamide side chain. An initial human trial on individuals with advanced solid tumours reported the pharmacokinetics (PK) and pharmacodynamics (PD) of pimasertib. Pimasertib showed a favourable PK profile in patients with solid tumours, and target action was shown by a decrease of phospho-ERK (pERK) in peripheral blood mononuclear cells (PBMCs).90,91 Clinical trials with pimasertib revealed a dose-dependent target-inhibitory impact. In melanomas with BRAF or NRAS mutations, sustained responses were primarily seen.92,93 Pimasertib is now being tested in phase I/II trials for advanced or metastatic solid tumours, including ovarian cancer, breast cancer, NRAS-mutated cutaneous melanoma, pancreatic cancer, NSCLC, hepatocellular carcinoma, and metastatic colorectal carcinoma.
The 3-oxo-oxazinane ring structure of RO4987655 (CH4987655), which is found at the 5-position of the benzamide core structure, is distinctive.94 The medication was made by Hoffman-La Roche. It was developed using the target enzyme's X-ray crystal structure as a starting point, and after that, it underwent multidimensional optimization, accounting for elements including metabolic stability, physicochemical qualities, and safety profiles. It maintained the desired metabolic stability, and only partially inhibited MEK in mouse brain, suggesting that RO4987655 will have few negative effects on the human central nervous system (CNS). Healthy participants in a phase I study had a favourable PK profile and evident target inhibition in PBMCs, while patients who had already received a number of therapies displayed favourable PK/PD profiles, moderate tolerability, and encouraging early anticancer activity.80,95,96
The allosteric inhibitor avutometinib (RO5126766), also known as CH5126766, binds to MEK directly and stops RAF from phosphorylating it by assembling a stable RAF–MEK complex. RO5126766 prevents ERK from being activated by MEK and the phosphorylation of MEK by RAF. Avutometinib efficiently inhibits a variety of human tumour cell lines, including KRAS/HRAS and BRAF mutant cell lines and KRAS/HRAS and BRAF wild-type cells.97
EBI-1051 is a safe and very efficient oral MEK inhibitor. A novel family of benzodihydrofuran compounds that function as potent MEK inhibitors has been developed by scaffold hopping using well-known medicines. Further SAR research and tuning led to the development of another benzofuran series with favourable oral absorption in rats. One of the substances, EBI-1051, demonstrated remarkable in vivo efficacy in mice Colo-205 tumour xenograft models and is appropriate for preclinical investigations for the treatment of melanoma and MEK-associated malignancies. EBI-1051 outperformed AZD6244 in treating a number of cancer cell lines, including Colo-205, A549, and MDA-MB-231.98
A recently developed small drug (DPS-2) shows potent anticancer activity in both cancer cells and animal models in CRC and melanoma and as a unique dual MEK–ERK and PI3K–AKT cell signalling pathway inhibitor. Notably, this drug has strong in vitro and in vivo apoptotic effectiveness against mutant KRAS and BRAF cancer cells and tumours, for which no effective therapeutics are present. To further explore its potential as an anticancer drug, the effects of the novel chemical DPS-2 on the MEK/ERK and PI3K/AKT signalling pathways (known to be involved in the growth of colon cancer and melanoma) need to be described and verified. Treatment of animal xenografts of Colo-205 colon cancer cells with DPS-2 significantly reduced tumour development, which further confirmed its antitumor efficacy in vivo. DPS-2 is highly effective against mouse xenografts of colorectal carcinoma cells in vivo.99
KZ-001 is a very potent and selective MEK1/2 inhibitor. Compared to selumetinib, the KZ-001 agent shows an estimated 30-fold higher suppression of BRAF and KRAS-mutated tumour cells. Additionally, in vivo xenograft models were used to illustrate these results. Furthermore, investigation of the pharmacokinetics of KZ-001 (PK) revealed that this chemical has high oral bioavailability (28%) and exposure (AUC0- = 337 169 ng h mL−1). The synergistic effect of KZ-001 with other drugs was studied in vitro and in vivo to determine its potential therapeutic benefit (xenograft models). In combination with the BRAF inhibitor vemurafenib and the microtubule-stabilising chemotherapeutic agent docetaxel, KZ-001 showed synergistic anti-cancer activity. KZ-001 also blocked the MAPK pathway like known MEK inhibitors.100
BI-847325 is a potent and ATP-competitive Aurora kinase and MEK inhibitor. It is orally accessible in therapeutic situations and models of drug-resistant BRAF-mutated melanoma. Cheng, Y. et al., demonstrated that BI-847325 is highly efficient in overcoming acquired BRAF resistance mediated by a variety of signalling pathways in both cell lines and mouse xenograft models of human melanoma. Further, BI-847325 was reported to have a novel mechanism of action that involves the downregulation of both Mcl-1 and MEK. In vivo and in vitro cancer models with BRAF and KRAS mutations responded favourably to BI-847325.100,101
An innovative MEK inhibitor, URML-3881, is being used to study the effects of MAPK inhibition in clear cell odontogenic carcinoma (CCOC). URML-3881 was found to inhibit apoptosis and proliferation but failed to induce regression of the in vivo tumour. Cisplatin alone also had little effect on tumour expansion, but surprisingly, the combination of cisplatin and MEK inhibition resulted in significant and long-lasting tumour shrinkage. These studies support the notion that URML-3881 and cisplatin in combination with MEK inhibition work better for CCOC than either drug alone.102
WX-554 is a MEK1/2 inhibitor that is currently undergoing preliminary human research. WX-554 was well tolerated, as demonstrated by pharmacokinetic and pharmacodynamic data for phase I investigation, and a phase II fixed dose of 75 mg twice weekly was advised.103 Unfortunately, for commercial reasons, two dose-escalation phase I/II studies in patients with advanced solid tumours were stopped.104
KZ-02 was developed for MEK inhibition, and causes the upregulation of Pim-1. Although KZ-02 increases the mRNA expression of Pim-1, it also promotes the proteasomal degradation of Pim-1. KZ-02 is a MEK inhibitor that exhibits unexpectedly high cytotoxicity. By targeting MEK and Pim-1 together, its anticancer activity was dramatically increased. KZ-02 is currently being tested in clinical trials for a variety of tumour types as a single agent or in combination with other cytotoxic chemotherapeutic agents or radiotherapy.71,105
5. Challenges with MEK inhibitors
Most cancers reactivate the MAPK pathway and ERK to overcome MEKi resistance and proliferate to maintain their growth. In RAS–RAF–MEK–ERK signaling pathway before signaling downstream to ERK, several signaling cascade such as NF1, MEK, RAS or RAF mutants.3 MEK may mutate on treatment with MEK inhibitors, which may lead to overactivation of MEK or make it difficult for inhibitors to bind to MEK. Literature reports demonstrated that the MYC-dependent transcriptional overexpression of ERBB3 may play a role in the resistance of KRAS-mutated lung and colorectal cancer. A patient's brain lesions had lower MYC levels than the lungs or colon, allowing ERBB3 to be produced at high levels, enabling adaptive MEKi resistance and rapid disease development in the brain only.122
Whenever the MAPK pathway is blocked (in order to get the signals needed to drive growth), cancer cells may switch to alternative signalling pathways leading to adaptive MEKi resistance. A well-known main resistance mechanism to MEK inhibition is the PI3K pathway. The development of various tumours has already been linked to this route, making it a viable target for treatment. Multiple studies in different malignancies have noted the stimulation of this pathway following the start of MEKi therapy.123 Multiple potential mutations that might lead to this system's dysfunction are a significant contributing factor to this pathway's high involvement in oncogenesis and MEKi resistance. Oncogenic RAS mutations can easily activate this pathway due to the stimulation provided by RAS, even when MEK is inhibited. Whenever MEK inhibition is active, alterations lower in the network, including active mutations in PIK3CA or deletion of PTEN, a tumor-suppressor gene, might overactivate this pathway.124
The capacity of tumour cells to change phenotype and rewire metabolic pathways is another possible route to resistance. A transcription factor and regulator of melanocyte formation called MITF was discovered to be more sensitive to MEK inhibition in melanoma cell lines with higher MITF expression than cell lines with lower MITF expression.125
6. Recent advancements in MEK inhibitors
Following the discovery of MEK mutations in 1997 and their importance in various cancers, many academic scientists/researchers started to work on them to solve problems related to different MEK mutations. In this context, various scaffolds such as 3-oxo-oxazinane, 2-aminopyrrole, indazole, sulfamide, 7-(pyrimidin-2-yloxy)-2H-chromen-2-one, bicyclic fused pyridine, imidazo[1,5-a]pyrazine, phenylsulfonylfuroxan and coumarin oxadiazole imidazole, pyrrole-3-carbonitriles, pyrimidine, benzofuran, 9-anilinoacridine phenyl-urea, carbazole, etc., and their hybrids were synthesized and their inhibitory activity against various cell lines such as A375, A375SM, C32 (melanoma), HCT116, Colo-205, HT-29 (colorectal wild type), A549 (lung adenocarcinoma), cancer cell lines, MCF-7, HeLa cells (breast cancer), etc., in enzyme (MEK) kinase assays were investigated and the results were published. A summary of the different scaffolds synthesized and their MEK inhibitory activity in relation to existing difficulties (from the discovery of the MEK mutant in 1997 to 2022) are shown in Fig. 6 and Table 5. Various derivatives of the parent scaffolds were synthesized, but only the most potent compounds were selected based on cell line activity as shown in Fig. 6 and Table 5.
Fig. 6. Various synthesized compounds with parent scaffolds as MEK mutant inhibitors.

Different scaffolds and their key aspects in MEK mutant inhibition.
| Cpd. no. | Derivatives | Cell line | Activity | Pin point | References |
|---|---|---|---|---|---|
| 01 | 3-Oxo-oxazinane | HT29 (CRC, Brafv600E) | IC50 1.7 nM | In vitro tests against a variety of tumour cells revealed strong antiproliferative action with little genotoxicity, hERG inhibition, or CYP inactivation | 94 |
| QG56 (NSCL, HrasQ61L) | IC50 9.5 nM | ||||
| MIA PaCa-2 panc. | IC50 3.3 nM | ||||
| KrasG12C C32 Brafv600E | IC50 8.4 nM | ||||
| Colo205 (CRC, Brafv600E) | IC50 0.86 nM | ||||
| 02 | 2-Aminopyrrole | Colo205 | EC50 0.012 (μM) | Important hydrogen bonds are formed between the oxygen of the acetyl group and the NH groups of Val211 and Ser212 in the backbone | 126 |
| A375 (melanoma) | EC50 0.014 (μM) | ||||
| 03 | Indazole | HCT116 | EC50 0.2 (nM) | Bidentate interaction with the Ser212 residue of MEK1 | 127 |
| A375 | EC50 0.4 (nM) | ||||
| 04 | Sulfamide | HCT116 cell line | IC50 8 (nM) | The compound's safety profiles and DMPK profiles (PK profiles in three animal species, CYP inhibition, and CYP induction) were unaffected by the sulfamide moiety (hERG and AMES assays). Powerful repression of HCT116 cell development | 128 |
| 05 | 7-(Pyrimidin-2-yloxy)-2H-chromen-2-one | HCT116 cell line | IC50 17 (nM) | No strong CYP inhibition activity | 129 |
| 06 | Sulfamide | A375 (B-Raf) | IC50 4 (nM) | Nanomolar cell potency of a very effective MEK inhibitor against B-RAF (V600E) and Ras-mutated cell lines | 130 |
| HCT116 (K-Ras) | IC50 180 (nM) | ||||
| 07 | Bicyclic fused pyridine | COLO-205 cell line | IC50 1.95 (nM) | Proven excellent in vitro Mek inhibitory activities | 131 |
| 08 | Imidazo[1,5-a]pyrazine | HCT116 cell line | IC50 0.107 (nM) | Rationalized by weaker interaction with the Ser-212 nitrogen due to the less basic, H-bond-accepting nitrogen | 132 |
| A375 cell line | IC50 0.007 (nM) | ||||
| 09 | Phenylsulfonylfuroxan and coumarin oxadiazole | A549 cell line | IC50 0.024 (μM) | The G2/M phase of the A2780 cell line's cell cycle was stopped | 133 |
| HeLa cell line | IC50 0.053 (μM) | ||||
| A2780 cell line | IC50 0.014 (μM) | ||||
| 10 | Pyrrole-3-carbonitriles | MCF-7 cells (breast) | IC50 1.35 ± 0.01 (μM) | It effectively inhibited proliferation in HT-29 (colon) and MCF-7 (breast) cells | 134 |
| HT-29 (colon) | IC50 1.47 ± 0.04 (μM) | ||||
| B16 cells | IC50 4.61 ± 0.01 (μM) | ||||
| 11 | Sulfamide | A375 (BRAF) | IC50 13 (nM) | BRAFV600E and Ras-mutant cell line resistance of cells (G13D) | 135 |
| HCT116 (K-Ras) | IC50 277 (nM) | ||||
| 12 | 9-Anilinoacridine phenyl-urea | K562 cell line | IC50 4.08 ± 0.14 (μM) | Against K562 and HepG-2 tumor cells | 136 |
| HepG-2 cell line | 9.41 ± 1.09 (μM) | ||||
| 13 | N-(Benzyloxy)-1,3-diphenyl-1H-pyrazole-4-carboxamide | HeLa cell line | GI50 1.18 ± 0.06 (μM) | This compound is most potent against the A549and Uo126 cancer cell lines | 137 |
| MCF-7 | GI50 2.11 ± 0.12 (μM) | ||||
| A549 cell line | GI50 0.26 ± 0.02 (μM) | ||||
| 293T cell line | CC50 20.57 ± 1.48 (μM) | ||||
| 14 | 2H-Chromen-2-one urea | MCF7 cell line | IC50 0.17 ± 0.07 (μM) | MCF7 breast cancer cell line and A549 lung cancer cell line activity, but no Hsp90 inhibitory activity | 138 |
| A549 cell line | IC50 0.15 ± 0.02 (μM) | ||||
| MRC-5 cell line | IC50 4.3 (μM) | ||||
| 15 | 1-(1H-Pyrazolo[4,3-c]pyridin-6-yl) urea | A375SM | IC50 43 (nM) | Strong tumor regression in BRAFV600E | 139 |
| 16 | Phenylsulfonylfuroxan and 3-benzyl coumarin | HeLa cells | IC50 2.8 (nM) | Hardly affected the cell cycle of A2780 | 140 |
| SKOV3 cells | IC50 8.3 (nM) | ||||
| A549 cell | IC50 3.7 (nM) | ||||
| OVCA429 cells | IC50 3.9 (nM) | ||||
| OVCA433 cells | IC50 3.3 (nM) | ||||
| A2780 cells | IC50 6.6 (nM) | ||||
| MDA-MB-231 | IC50 0.8 (nM) | ||||
| MCF-7 cells | IC50 2853 (nM) | ||||
| KB cells | IC50 3234 (nM) | ||||
| 17 | Benzofuran | Colo-205 cells | IC50 4.7 ± 1.5 (nM) | Excellent in vivo efficacy in mouse Colo-205 tumour xenograft models | 116 |
| 18 | Ursolic acid with hydrazide | MDA-MB-231 | IC50 0.12 ± 0.01 (μM) | HeLa cells undergo apoptosis, and the cell cycle is stopped in the G0/G1 phase | 141 |
| HeLa cells | IC50 0.08 ± 0.01 (μM) | ||||
| SMMC-7721 cells | IC50 0.34 ± 0.03 (μM) | ||||
| QSG-7701 cells | IC50 10.76 ± 0.72 (μM) | ||||
| 19 | Carbazole | HEK293 cells | IC50 8.9 ± 2.0 (μM) | Greatly reduced cytotoxicity to HEK293 cells | 142 |
| A549 cells | IC50 21.6 ± 6.1 (μM) | ||||
| A375 cells | IC50 7.7 ± 1.1 (μM) | ||||
| HL60 cells | IC50 17.2 ± 6.6 (μM) | ||||
| 20 | 1,2,5-Oxadiazole 2-oxide | MDA-MB-231 | IC50 0.034 ± 0.007 (μM) | Best cell growth inhibitory effect in MDA-MB-231 cells | 143 |
| HCT116 cells | IC50 0.64 ± 0.30 (μM) | ||||
| A549 cells | IC50 1.35 ± 0.94 (μM) | ||||
| Vero cells | IC50 21.07 ± 1.32 (μM) | ||||
| HL7702 cells | IC50 5.62 ± 0.82 | ||||
| 21 | 2H-Chromen-7-yl dimethylcarbamate | A549 cells | IG50 4.66 (μM) | Activity against the A549 and HCT116 cell lines | 144 |
| HCT116 cells | IG50 5.47 (μM) | ||||
| 22 | 5-Phenylamino-8-methylpyrido[2,3-d]pyrimidine-4,7(3H,8H)-dione | A375 cells | EC50 3.1 (nM) | Fluorine-containing diol compounds had the most potent enzyme and cell activity | 82 |
| Colo-205 cells | EC50 2.1 (nM) | ||||
| 23 | Bicyclic dihydroindolone pyrrole | Colo-205 cells | EC50 1.0 (nM) | Inhibition of HL-60 and regression of tumors in the promyelocytic leukemia xenograft model | 145 |
| A375 cells | EC50 2.0 (nM) | ||||
| 24 | Fused thiophene | HT-29 pERK | IC50 6 (nM) | Inhibition of pERK in the HT-29 tumor | 146 |
| HT-29 | IC50 79 (nM) | ||||
| 25 | 3-Benzylcoumarins | HEK293 cell line | EC50 19.38 (μM) | Inhibited virus (EV71) replication in HEK293 and RD cells | 147 |
| CC50 65.31 (μM) | |||||
| TI (CC50/IC50) 3.37 (μM) | |||||
| RD cell line | EC50 10 (μM) | ||||
| CC50 72.92 (μM) | |||||
| TI (CC50/IC50) 7.29 (μM) | |||||
| 26 | Propoxybenzamide | A375 cell line | IC50 0.0176 (μM) | Inhibited DNA replication of A375 cells | 148 |
| 27 | Benzothiazole-pyrrole | MCF-7 cells | GI50 0.92 ± 0.04 (μM) | Stopped G1 phase cells, signifying the G2/M cell cycle | 149 |
| MDA-MB231 cells | GI50 1.76 ± 0.352 (μM) | ||||
| 28 | Coumarin | HCT116 cells | IC50 40 (nM) | Inhibitory effect on HCT116 cell growth | 150 |
| 29 | 2-(1-Substituted benzyl-1H-tetrazol-5-yl)-3-phenylacrylonitrile | MCF-7 cells | IC50 ± 30 (μM) | The substituted group at the N1 position enhanced the antitumor activity of the parent compound | 151 |
| CaCo2 cells | IC50 ± 37 (μM) | ||||
| HeLa cells | IC50 ± 29 (μM) | ||||
| SkBr3 cell line | IC50 ± 35 (μM) | ||||
| 30 | 3-Benzyl-1,3-benzoxazine-2,4-dione | CPE cell line | EC50 1.02 ± 0.085 (μM) | Suppression of EV71 VPI expression, and an EV71 induced cytopathic effect | 152 |
| 31 | Thiosemicarbazone | MDA-MB-231 cells | IC50 1.9 ± 0.3 (μM ± SEM) | Demonstrated robust antitumor efficacy correlated with inhibition of MAPK kinase signal transduction | 153 |
| MDA-MB-468 cells | IC50 2.4 ± 0.2 (μM ± SEM) | ||||
| MCF-7 cells | IC50 2.1 ± 0.2 (μM ± SEM) | ||||
| BT-474 cells | IC50 2.6 ± 0.4 (μM ± SEM) | ||||
| SkBr3 cells | IC50 2.3 ± 0.4 (μM ± SEM) | ||||
| T-47D cell line | IC50 3.0 ± 0.4 (μM ± SEM) | ||||
| 32 | N-(Piperazin-1-yl)alkyl-1H-dibenzo[a,c]carbazole | SMMC-7721 | IC50 1.39 ± 0.13 (μM) | Damage the cell membrane's integrity, ultimately causing the HeG2 cells to undergo apoptosis and cancer | 154 |
| HepG2 cells | IC50 0.51 ± 0.09 (μM) | ||||
| Hep3B cells | IC50 0.73 ± 0.08 (μM) | ||||
| QSG-7701 cells | IC50 12.52 ± 0.58 (μM) | ||||
| 33 | Benzoxazole | A375 cells | IC50 4.3 ± 0.7 (nM) | G0/G1 phase cell cycle delay in A375 cells | 100 |
| Colo-205 cells | IC50 5.7 ± 0.3 (nM) | ||||
| HT-29 cells | IC50 2.9 ± 0.6 (nM) | ||||
| Calu-6 cells | IC50 169 ± 97.7 (nM) | ||||
| A431 cells | IC50 >3000 (nM) |
7. Future perspectives
MEKi resistance methods frequently include the activation of other cellular signalling pathways, like the PI3K/AKT/mTOR system or the STAT pathway. To avoid these procedures, several researchers have suggested and tried combination treatments that concurrently block different signalling pathways. The FDA has authorised the use of a BRAFi (encorafenib) and MEKi (binimetinib), a combination that has been demonstrated to be a more successful therapy than using either inhibitor alone for BRAF-mutated cutaneous melanoma.155,156 A different type of mechanism of resistance that frequently comes back following MEKi treatment is RTK production. The question of whether combination treatment, which inhibits these RTKs in addition to MEK, may overcome adaptive resistance mechanisms has been looked into. Stronger correlations between some RTKs and MEKi resistance have been found. Targeting resistance with epithelial-mesenchymal transition (EMT) is another strategy. As previously stated, lung cancer cells with the KRAS mutation which were resistant to MEKi expressed more ZEB1.122 These resistant cells can be made vulnerable to MEKi therapy by inhibiting ZEB1, which can be accomplished by upregulating miR-200 expression or with the HDAC inhibitor, mocetinostat. Combining MEKi with mocetinostat also had a synergistic impact in the reduction of the number and size of metastatic lung cancer malignant tumors. By screening synthetic lethal shRNA in cancer patients with KRAS mutations, BCL-XL was identified as a viable target for conjunction with MEKi. BCL-XL binds and inhibits the significant pro-apoptotic protein BIM, which MEKi has activated. BCL-2/BCL-XL inhibition enhances the effectiveness of MEK inhibition in lung and pancreatic malignant cell lines. In a patient-derived xenograft model of high-grade serous ovarian cancer, it has also been demonstrated that the combination of MEK and BCL-2/XL inhibitors is effective. Combination therapy is an option for dealing with the intricacy of the RAS–RAF–MEK–MAPK pathway linked to resistance to RAF and MEK inhibition and boosting the effectiveness of other anticancer medications by concurrently inhibiting the Ras–RAF–MEK–MAPK pathway. Clinical studies are testing a number of combination medicines based on MEK inhibitors; however, the toxicity of MEK inhibitors at significant doses limits the utility of these therapeutic approaches. MEK inhibitor-based therapy may be more effective when administered with other dose schedules, such as intermittent delivery, which might totally shut down the RAS–RAF–MEK–MAPK pathway while allowing normal tissue to recover. Combined inhibition of MEK and RAF kinases, which has advantages in terms of better efficacy and lower toxicity, is a prospective treatment strategy that focuses on the RAS–RAF–MEK–MAPK pathway.157–159
8. Conclusion
Several inhibitors that operate particularly on each of the many parts of the MEK pathway have been discovered. A few inhibitors have been FDA-approved for the treatment of different tumour types, while a majority are currently undergoing preclinical testing. Trametinib was the first MEK inhibitor that was approved by the FDA, followed by selumetinib, cobimetinib and binimetinib. A range of factors, such as paradoxical activation, toxicity and the evolution of resistance, might cause these inhibitors to lose their effectiveness. The RAF–MEK–ERK pathway's components frequently contain mutations that promote tumour heterogeneity and result in the establishment of resistance. When taken alone or in conjunction with other treatments, MEK inhibitors have been proven to have good antitumor activity against melanoma, lung cancer, and colorectal cancer. According to existing clinical evidence, combining a MEK inhibitor and a BRAF inhibitor may result in a more successful therapy. A MEK inhibitor in conjunction with a BRAF inhibitor or other targeted medications may change the immunisation process and boost immunological activation and improve efficacy. The ongoing ambiguity about toxicity is one of the difficulties in creating MEK inhibitors. The use of MEK inhibitors as a therapeutic strategy remains one of the most fascinating areas of Cancer Res. New small molecule inhibitors are anticipated to bring about a change in cancer treatment. Additionally, concurrent MEK kinase inhibition offers benefits in terms of increased effectiveness and lower toxicity, and it could be a future treatment strategy for the MAPK pathway.
Abbreviations
- ADP
Adenosine diphosphate
- ATP
Adenosine triphosphate
- CREB
cAMP response element binding protein
- ERK
Extracellular-signal regulated kinase
- EGFRs
Epidermal growth factor receptors
- FDA
Food and Drug Administration
- FGFRs
Fibroblast growth factor receptors
- GDP
Guanosine diphosphate
- GTP
Guanosine-5-triphosphate
- GRB2
Growth factor receptor bound protein 2
- MAPK
Mitogen-activated protein kinase
- MEK
Mitogen extracellular kinase
- NSCLC
Non-small cell lung cancer
- NF1
Neurofibromatosis type 1
- PBMCs
Peripheral blood mononuclear cells
- PD
Pharmacodynamics
- PDGFRs
Platelet derived growth factor receptors
- PK
Pharmacokinetics
- PNs
Plexiform neurofibromas
- RAF
Rapidly accelerated fibrosarcoma
- RSK
Ribosomal s6 kinase
- RAS
Rat sarcoma
- SOS
Son of sevenless
Author contributions
Conceptualization: Pradeep Kumar; data collection: Adarsh Kumar and Harshwardhan Singh; writing the manuscript: Teja Ram and Ankit Kumar Singh; sketching of figures and data interpretation: Prateek Pathak and Ankit Kumar Singh; writing, review and final editing of the manuscript: Habibullah Khalilullah, Mariusz Jaremko, Abdul-Hamid Emwas, Prateek Pathak, Amita Verma, Maria Grishina, Ankit Kumar Singh and Pradeep Kumar.
Conflicts of interest
The authors declare no competing interest.
Supplementary Material
Acknowledgments
The authors are thankful to DST-FIST, Central University of Punjab, Bathinda, for providing the necessary facilities to compose this manuscript. PP and MG acknowledge South Ural State University, Chelyabinsk, Russia under the Priority 2030 program.
References
- Orton R. J. Sturm O. E. Vyshemirsky V. Calder M. Gilbert D. R. Kolch W. Biochem. J. 2005;392:249–261. doi: 10.1042/BJ20050908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cargnello M. Roux P. P. Microbiol. Mol. Biol. Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt C. J. Sale M. J. Smith P. D. Cook S. J. Nat. Rev. Cancer. 2015;15:577–592. doi: 10.1038/nrc4000. [DOI] [PubMed] [Google Scholar]
- Singh A. K. Novak J. Kumar A. Singh H. Thareja S. Pathak P. Grishina M. Verma A. Yadav J. P. Khalilullah H. RSC Adv. 2022;12:30181–30200. doi: 10.1039/D2RA05751D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak P. Rimac H. Grishina M. Verma A. Potemkin V. ChemMedChem. 2021;16:822–838. doi: 10.1002/cmdc.202000646. [DOI] [PubMed] [Google Scholar]
- Sebolt-Leopold J. S. Oncogene. 2000;19:6594–6599. doi: 10.1038/sj.onc.1204083. [DOI] [PubMed] [Google Scholar]
- Hoshino R. Chatani Y. Yamori T. Tsuruo T. Oka H. Yoshida O. Shimada Y. Ari-i S. Wada H. Fujimoto J. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- Singh A. K. Kumar A. Thareja S. Kumar P. Anti-Cancer Agents Med. Chem. 2023;23(3):278–297. doi: 10.2174/1871520622666220624164152. [DOI] [PubMed] [Google Scholar]
- Favata M. F. Horiuchi K. Y. Manos E. J. Daulerio A. J. Stradley D. A. Feeser W. S. Van Dyk D. E. Pitts W. J. Earl R. A. Hobbs F. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Stuart K. A., PhD, University of Cambridge, 2019 [Google Scholar]
- Estep A. L. Palmer C. McCormick F. Rauen K. A. PLoS One. 2007;2:e1279. doi: 10.1371/journal.pone.0001279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks J. L. Gong Y. Chitale D. Golas B. McLellan M. D. Kasai Y. Ding L. Mardis E. R. Wilson R. K. Solit D. Cancer Res. 2008;68:5524–5528. doi: 10.1158/0008-5472.CAN-08-0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg-White J. L. Andersen N. J. Duesbery N. S. Briefings Funct. Genomics. 2012;11:300–310. doi: 10.1093/bfgp/els022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischmann T. O. Smith C. K. Mayhood T. W. Myers Jr J. E. Reichert P. Mannarino A. Carr D. Zhu H. Wong J. Yang R.-S. Biochemistry. 2009;48:2661–2674. doi: 10.1021/bi801898e. [DOI] [PubMed] [Google Scholar]
- Douillard J.-Y. Oliner K. S. Siena S. Tabernero J. Burkes R. Barugel M. Humblet Y. Bodoky G. Cunningham D. Jassem J. N. Engl. J. Med. 2013;369:1023–1034. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- Siegel R. L. Miller K. D. Fuchs H. E. Jemal A. Ca-Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- Oda K. Matsuoka Y. Funahashi A. Kitano H. Mol. Syst. Biol. 2005;1:2005.0010. doi: 10.1038/msb4100014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarich N. Oliva J. L. Martínez N. Jorge R. Ballester A. Gutiérrez-Eisman S. García-Vargas S. Rojas J. M. Mol. Biol. Cell. 2006;17:3591–3597. doi: 10.1091/mbc.e05-12-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avruch J. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog. Horm. Res. 2001;56:127–155. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- McCubrey J. A. Steelman L. S. Chappell W. H. Abrams S. L. Wong E. W. Chang F. Lehmann B. Terrian D. M. Milella M. Tafuri A. Biochim. Biophys. Acta, Mol. Cell Res. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pende M. Um S. H. Mieulet V. Sticker M. Goss V. L. Mestan J. Mueller M. Fumagalli S. Kozma S. C. Thomas G. Mol. Cell. Biol. 2004;24:3112–3124. doi: 10.1128/MCB.24.8.3112-3124.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knighton D. R. Zheng J. Ten Eyck L. F. Ashford V. A. Xuong N.-H. Taylor S. S. Sowadski J. M. Science. 1991;253:407–414. doi: 10.1126/science.1862342. [DOI] [PubMed] [Google Scholar]
- Xu B.-e. Wilsbacher J. L. Collisson T. Cobb M. H. J. Biol. Chem. 1999;274:34029–34035. doi: 10.1074/jbc.274.48.34029. [DOI] [PubMed] [Google Scholar]
- Yang S.-H. Whitmarsh A. J. Davis R. J. Sharrocks A. D. EMBO J. 1998;17:1740–1749. doi: 10.1093/emboj/17.6.1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinfeld H. Hanoch T. Seger R. J. Biol. Chem. 1999;274:30349–30352. doi: 10.1074/jbc.274.43.30349. [DOI] [PubMed] [Google Scholar]
- Jaaro H. Rubinfeld H. Hanoch T. Seger R. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3742–3747. doi: 10.1073/pnas.94.8.3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M. Gotoh I. Adachi M. Gotoh Y. Nishida E. J. Biol. Chem. 1997;272:32642–32648. doi: 10.1074/jbc.272.51.32642. [DOI] [PubMed] [Google Scholar]
- Mansour S. J. Candia J. M. Gloor K. K. Ahn N. Cell Growth Differ. 1996;7:243–250. [PubMed] [Google Scholar]
- Catling A. D. Schaeffer H.-J. Reuter C. Reddy G. R. Weber M. J. Mol. Cell. Biol. 1995;15:5214–5225. doi: 10.1128/MCB.15.10.5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer H. J. Catling A. D. Eblen S. T. Collier L. S. Krauss A. Weber M. J. Science. 1998;281:1668–1671. doi: 10.1126/science.281.5383.1668. [DOI] [PubMed] [Google Scholar]
- Alessi D. R. Saito Y. Campbell D. Cohen P. Sithanandam G. Rapp U. Ashworth A. Marshall C. Cowley S. EMBO J. 1994;13:1610–1619. doi: 10.1002/j.1460-2075.1994.tb06424.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A. P. Haste N. M. Taylor S. S. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. U. S. A. 2006:17783–17788. doi: 10.1073/pnas.0607656103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev A. P. Taylor S. S. Ten Eyck L. F. Proc. Natl. Acad. Sci. U. S. A. 2008;105:14377–14382. doi: 10.1073/pnas.0807988105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedüs L. Okumus Ö. Livingstone E. Baranyi M. Kovács I. Döme B. Tóvári J. Bánkfalvi Á. Schadendorf D. Aigner C. Cancers. 2021;13:829. doi: 10.3390/cancers13040829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P. A. Wallace E. Marlow A. Yeh T. Marsh V. Anderson D. Woessner R. Hurley B. Lyssikatos J. Poch G. Cancer Res. 2010;70:2515–2515. doi: 10.1158/1538-7445.AM10-2515. [DOI] [Google Scholar]
- Yeh T. C. Marsh V. Bernat B. A. Ballard J. Colwell H. Evans R. J. Parry J. Smith D. Brandhuber B. J. Gross S. Clin. Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- Musib L. Eppler S. Choo E. Deng A. Miles D. Hsu B. Rosen L. Sikic B. LoRusso P. Ma W. Cancer Res. 2011;71:1304. doi: 10.1158/1538-7445.AM2011-1304. [DOI] [Google Scholar]
- Cheng Y. Tian H. Molecules. 2017;22:1551. doi: 10.3390/molecules22101551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh S. J. Corrie P. G. Ther. Adv. Med. Oncol. 2015;7:122–136. doi: 10.1177/1758834014566428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J. Ascierto P. A. Dréno B. Atkinson V. Liszkay G. Maio M. Mandalà M. Demidov L. Stroyakovskiy D. Thomas L. N. Engl. J. Med. 2014;371:1867–1876. doi: 10.1056/NEJMoa1408868. [DOI] [PubMed] [Google Scholar]
- Salama A. K. Kim K. B. Curr. Oncol. Rep. 2013;15:473–482. doi: 10.1007/s11912-013-0336-2. [DOI] [PubMed] [Google Scholar]
- Salama A. K. Kim K. B. Expert Opin. Pharmacother. 2013;14:619–627. doi: 10.1517/14656566.2013.770475. [DOI] [PubMed] [Google Scholar]
- Simeone E. Grimaldi A. M. Festino L. Vanella V. Palla M. Ascierto P. A. BioDrugs. 2017;31:51–61. doi: 10.1007/s40259-016-0208-z. [DOI] [PubMed] [Google Scholar]
- Rosen L. S. LoRusso P. Ma W. W. Goldman J. W. Weise A. Colevas A. D. Adjei A. Yazji S. Shen A. Johnston S. Invest. New Drugs. 2016;34:604–613. doi: 10.1007/s10637-016-0374-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y. Adjei A. A. Nat. Rev. Clin. Oncol. 2014;11:385–400. doi: 10.1038/nrclinonc.2014.83. [DOI] [PubMed] [Google Scholar]
- Martini J.-F. Yu P. Li C. Rosario G. D. Choo E. F. Clin. Cancer Res. 2012;18:3090–3099. doi: 10.1158/1078-0432.CCR-12-0445. [DOI] [PubMed] [Google Scholar]
- Choo E. F. Ng C. M. Berry L. Belvin M. Lewin-Koh N. Merchant M. Salphati L. Cancer Chemother. Pharmacol. 2013;71:133–143. doi: 10.1007/s00280-012-1988-6. [DOI] [PubMed] [Google Scholar]
- Miller W. H. Kim T. M. Lee C. B. Flaherty K. T. Reddy S. Jamal R. Chow L. Q. Rooney I. A. Pitcher B. Cha E. J. Clin. Oncol. 2017:3057. [Google Scholar]
- Sullivan R. J. Gonzalez R. Lewis K. D. Hamid O. Infante J. R. Patel M. R. Hodi F. S. Wallin J. Pitcher B. Cha E. J. Clin. Oncol. 2017:3063. [Google Scholar]
- Bendell J. C. Kim T. W. Goh B. C. Wallin J. Oh D.-Y. Han S.-W. Lee C. B. Hellmann M. D. Desai J. Lewin J. H. J. Clin. Oncol. 2016:3502. [Google Scholar]
- Patel S. P. Lazar A. J. Papadopoulos N. E. Liu P. Infante J. R. Glass M. R. Vaughn C. S. LoRusso P. M. Cohen R. B. Davies M. A. Cancer. 2013;119:799–805. doi: 10.1002/cncr.27790. [DOI] [PubMed] [Google Scholar]
- El-Hoss J. Kolind M. Jackson M. Deo N. Mikulec K. McDonald M. Little C. Little D. Schindeler A. Bone. 2014;59:151–161. doi: 10.1016/j.bone.2013.11.013. [DOI] [PubMed] [Google Scholar]
- Jain N. Curran E. Iyengar N. M. Diaz-Flores E. Kunnavakkam R. Popplewell L. Kirschbaum M. H. Karrison T. Erba H. P. Green M. Clin. Cancer Res. 2014;20:490–498. doi: 10.1158/1078-0432.CCR-13-1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh H. Koong H. N. Poon D. Choo S. P. Toh H. C. Thng C. H. Chow P. Ong H. S. Chung A. Goh B. C. J. Hepatol. 2010;52:79–87. doi: 10.1016/j.jhep.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Banerji U. Camidge D. R. Verheul H. M. Agarwal R. Sarker D. Kaye S. B. Desar I. M. Timmer-Bonte J. N. Eckhardt S. G. Lewis K. D. Clin. Cancer Res. 2010;16:1613–1623. doi: 10.1158/1078-0432.CCR-09-2483. [DOI] [PubMed] [Google Scholar]
- Copley-Merriman C. Yang X. Juniper M. Amin S. Yoo H. K. Sen S. S. Adolesc. Health, Med. Ther. 2021;12:55. doi: 10.2147/AHMT.S303456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo F. Grasso A. G. Cortellazzo Wiel L. Maestro A. Trojniak M. P. Murru F. M. Basso L. Magnolato A. Bruno I. Barbi E. Pediatric Drugs. 2020;22:417–423. doi: 10.1007/s40272-020-00399-y. [DOI] [PubMed] [Google Scholar]
- Espírito Santo V. Passos J. Nzwalo H. Carvalho I. Santos F. Martins C. Salgado L. Vinhais S. Vilares M. Salgado D. J. Neuro-Oncol. 2020;147:459–463. doi: 10.1007/s11060-020-03443-6. [DOI] [PubMed] [Google Scholar]
- Tai W. Yong W. Lim C. Low L. Tham C. Koh T. Ng Q. Wang W. Wang L. Hartano S. Ann. Oncol. 2016;27:2210–2215. doi: 10.1093/annonc/mdw415. [DOI] [PubMed] [Google Scholar]
- Jänne P. A. Van Den Heuvel M. M. Barlesi F. Cobo M. Mazieres J. Crinò L. Orlov S. Blackhall F. Wolf J. Garrido P. JAMA, J. Am. Med. Assoc. 2017;317:1844–1853. doi: 10.1001/jama.2017.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran B. Cohen M. S. Expert Opin. Drug Discovery. 2020;15:745–754. doi: 10.1080/17460441.2020.1746265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolkind P. A. Campbell K. M. Lin T. Skidmore Z. Winkler A. Barnell E. Giri T. Adkins D. R. Griffith M. Dunn G. P. Clin. Cancer Res. 2017;23:PR01. doi: 10.1158/1557-3265.AACRAHNS17-PR01. [DOI] [Google Scholar]
- Coleman R. L. Sill M. W. Thaker P. H. Bender D. P. Street D. McGuire W. P. Johnston C. M. Rotmensch J. Gynecol. Oncol. 2015;138:30–35. doi: 10.1016/j.ygyno.2015.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice K. D. Aay N. Anand N. K. Blazey C. M. Bowles O. J. Bussenius J. Costanzo S. Curtis J. K. Defina S. C. Dubenko L. ACS Med. Chem. Lett. 2012;3:416–421. doi: 10.1021/ml300049d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso P. M. Sekulic A. Sosman J. A. Liang W. S. Carpten J. Craig D. W. Solit D. B. Bryce A. H. Kiefer J. A. Aldrich J. PLoS One. 2021;16:e0248097. doi: 10.1371/journal.pone.0248097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Algazi A. P. Esteve-Puig R. Nosrati A. Hinds B. Hobbs-Muthukumar A. Nandoskar P. Ortiz-Urda S. Chapman P. B. Daud A. Pigm. Cell Melanoma Res. 2018;31:110–114. doi: 10.1111/pcmr.12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies M. A. Saiag P. Robert C. Grob J.-J. Flaherty K. T. Arance A. Chiarion-Sileni V. Thomas L. Lesimple T. Mortier L. Lancet Oncol. 2017;18:863–873. doi: 10.1016/S1470-2045(17)30429-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall R. D. Kudchadkar R. R. Cancer Control. 2014;21:221–230. doi: 10.1177/107327481402100307. [DOI] [PubMed] [Google Scholar]
- Subbiah V. Kreitman R. J. Wainberg Z. A. Cho J. Y. Schellens J. H. Soria J. C. Wen P. Y. Zielinski C. Cabanillas M. E. Urbanowitz G. J. Clin. Oncol. 2018;36:7. doi: 10.1200/JCO.2017.73.6785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam B. Kreitman R. Wainberg Z. Cabanillas M. Cho D. Italiano A. Stein A. Cho J. Schellens J. Wen P. Ann. Oncol. 2018;29:viii645–viii646. doi: 10.1093/annonc/mdy302.002. [DOI] [Google Scholar]
- Carvajal R. D. Piperno-Neumann S. Kapiteijn E. Chapman P. B. Frank S. Joshua A. M. Piulats J. M. Wolter P. Cocquyt V. Chmielowski B. J. Clin. Oncol. 2018;36:1232–1239. doi: 10.1200/JCO.2017.74.1090. [DOI] [PubMed] [Google Scholar]
- Ascierto P. A. McArthur G. A. Dréno B. Atkinson V. Liszkay G. Di Giacomo A. M. Mandalà M. Demidov L. Stroyakovskiy D. Thomas L. Lancet Oncol. 2016;17:1248–1260. doi: 10.1016/S1470-2045(16)30122-X. [DOI] [PubMed] [Google Scholar]
- Eng C. Kim T. W. Bendell J. Argilés G. Tebbutt N. C. Di Bartolomeo M. Falcone A. Fakih M. Kozloff M. Segal N. H. Lancet Oncol. 2019;20:849–861. doi: 10.1016/S1470-2045(19)30027-0. [DOI] [PubMed] [Google Scholar]
- Koelblinger P. Thuerigen O. Dummer R. Curr. Opin. Oncol. 2018;30:125. doi: 10.1097/CCO.0000000000000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Cutsem E. Hidalgo M. Canon J. L. Macarulla T. Bazin I. Poddubskaya E. Manojlovic N. Radenkovic D. Verslype C. Raymond E. Int. J. Cancer. 2018;143:2053–2064. doi: 10.1002/ijc.31603. [DOI] [PubMed] [Google Scholar]
- Wabnitz P. A. Mitchell D. Wabnitz D. A. Pharm. Res. 2004;21:1670–1679. doi: 10.1023/B:PHAM.0000041464.27579.d0. [DOI] [PubMed] [Google Scholar]
- Brown A. P. Carlson T. C. Loi C.-M. Graziano M. J. Cancer Chemother. Pharmacol. 2007;59:671–679. doi: 10.1007/s00280-006-0323-5. [DOI] [PubMed] [Google Scholar]
- Haura E. B. Ricart A. D. Larson T. G. Stella P. J. Bazhenova L. Miller V. A. Cohen R. B. Eisenberg P. D. Selaru P. Wilner K. D. Clin. Cancer Res. 2010;16:2450–2457. doi: 10.1158/1078-0432.CCR-09-1920. [DOI] [PubMed] [Google Scholar]
- Boasberg P. D. Redfern C. H. Daniels G. A. Bodkin D. Garrett C. R. Ricart A. D. Cancer Chemother. Pharmacol. 2011;68:547–552. doi: 10.1007/s00280-011-1620-1. [DOI] [PubMed] [Google Scholar]
- Haasbach E. Hartmayer C. Planz O. Antiviral Res. 2013;98:319–324. doi: 10.1016/j.antiviral.2013.03.006. [DOI] [PubMed] [Google Scholar]
- Cohen R. B. Aamdal S. Nyakas M. Cavallin M. Green D. Learoyd M. Smith I. Kurzrock R. Eur. J. Cancer. 2013;49:1521–1529. doi: 10.1016/j.ejca.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Dong Q. Dougan D. R. Gong X. Halkowycz P. Jin B. Kanouni T. O'Connell S. M. Scorah N. Shi L. Wallace M. B. Bioorg. Med. Chem. Lett. 2011;21:1315–1319. doi: 10.1016/j.bmcl.2011.01.071. [DOI] [PubMed] [Google Scholar]
- Adjei A. A. LoRusso P. Ribas A. Sosman J. A. Pavlick A. Dy G. K. Zhou X. Gangolli E. Kneissl M. Faucette S. Invest. New Drugs. 2017;35:47–58. doi: 10.1007/s10637-016-0391-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G. Haling J. R. Chen H. Song K. Price S. Heald R. Hewitt J. F. Zak M. Peck A. Orr C. Nature. 2013;501:232–236. doi: 10.1038/nature12441. [DOI] [PubMed] [Google Scholar]
- Choo E. Belvin M. Merchant M. Chan E. Hollingshead P. Orr C. Boggs J. Plise E. Robarge K. Zak M. Eur. J. Cancer. 2012:155. doi: 10.1016/S0959-8049(12)72299-2. [DOI] [Google Scholar]
- Ait Tihyaty M., PhD, McGill University, 2014 [Google Scholar]
- Iverson C. Larson G. Lai C. Yeh L.-T. Dadson C. Weingarten P. Appleby T. Vo T. Maderna A. Vernier J.-M. Cancer Res. 2009;69:6839–6847. doi: 10.1158/0008-5472.CAN-09-0679. [DOI] [PubMed] [Google Scholar]
- Van Laethem J.-L. Riess H. Jassem J. Haas M. Martens U. M. Weekes C. Peeters M. Ross P. Bridgewater J. Melichar B. Target. Oncol. 2017;12:97–109. doi: 10.1007/s11523-016-0469-y. [DOI] [PubMed] [Google Scholar]
- Lim H. Y. Heo J. Choi H. J. Lin C.-Y. Yoon J.-H. Hsu C. Rau K.-M. Poon R. T. Yeo W. Park J.-W. Clin. Cancer Res. 2014;20:5976–5985. doi: 10.1158/1078-0432.CCR-13-3445. [DOI] [PubMed] [Google Scholar]
- Houédé N. Delord J. Awada A. Lebbe C. Lesimple T. Schellens J. Rottey S. Kefford R. von Richter O. Raymond E. Eur. J. Cancer. 2012:184. doi: 10.1016/S0959-8049(12)72397-3. [DOI] [Google Scholar]
- Naing A. Mita M. Komarnitsky P. Milner A. von Richter O. Ogden J. Piha-Paul S. Fu S. Asatiani E. Kurzrock R. Eur. J. Cancer. 2012:187. doi: 10.1016/S0959-8049(12)72405-X. [DOI] [Google Scholar]
- Awada A. Delord J. Houede N. Lebbe C. Lesimple T. Schellens J. Rottey S. Kefford R. Rejeb N. Raymond E. Eur. J. Cancer. 2012:185–186. doi: 10.1016/S0959-8049(12)72401-2. [DOI] [Google Scholar]
- Delord J. Houédé N. Awada A. Lebbe C. Lesimple T. Schellens J. Rottey S. Kefford R. Rejeb N. Raymond E. Eur. J. Cancer. 2012;48:190. doi: 10.1016/S0959-8049(12)72413-9. [DOI] [Google Scholar]
- Isshiki Y. Kohchi Y. Iikura H. Matsubara Y. Asoh K. Murata T. Kohchi M. Mizuguchi E. Tsujii S. Hattori K. Bioorg. Med. Chem. Lett. 2011;21:1795–1801. doi: 10.1016/j.bmcl.2011.01.062. [DOI] [PubMed] [Google Scholar]
- Kraeber-Bodéré F. Carlier T. Naegelen V. M. Shochat E. Lumbroso J. Trampal C. Nagarajah J. Chua S. Hugonnet F. Stokkel M. J. Nucl. Med. 2012;53:1836–1846. doi: 10.2967/jnumed.112.109421. [DOI] [PubMed] [Google Scholar]
- Leijen S. Middleton M. R. Tresca P. Kraeber-Bodéré F. Dieras V. Scheulen M. E. Gupta A. Lopez-Valverde V. Xu Z.-X. Rueger R. Clin. Cancer Res. 2012;18:4794–4805. doi: 10.1158/1078-0432.CCR-12-0868. [DOI] [PubMed] [Google Scholar]
- Martinez-Garcia M. Banerji U. Albanell J. Bahleda R. Dolly S. Kraeber-Bodéré F. Rojo F. Routier E. Guarin E. Xu Z.-X. Clin. Cancer Res. 2012;18:4806–4819. doi: 10.1158/1078-0432.CCR-12-0742. [DOI] [PubMed] [Google Scholar]
- González-Gómez J. C. Santana L. Uriarte E. Tetrahedron. 2005;61:4805–4810. doi: 10.1016/j.tet.2005.03.018. [DOI] [Google Scholar]
- Goulielmaki M. Assimomytis N. Rozanc J. Taki E. Christodoulou I. Alexopoulos L. G. Zoumpourlis V. Pintzas A. Papahatjis D. Transl. Oncol. 2019;12:932–950. doi: 10.1016/j.tranon.2019.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y. Wang X. Xia X. Zhang W. Tian H. Int. J. Cancer. 2019;145:586–596. doi: 10.1002/ijc.32119. [DOI] [PubMed] [Google Scholar]
- Liu Y. Hawkins O. E. Su Y. Vilgelm A. E. Sobolik T. Thu Y. M. Kantrow S. Splittgerber R. C. Short S. Amiri K. I. EMBO Mol. Med. 2013;5:149–166. doi: 10.1002/emmm.201201378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowswell-Turner R. B. Rutishauser J. A. Kim K. K. Khazan N. Sivagnanalingam U. Jones A. M. Singh R. K. Moore R. G. Transl. Oncol. 2019;12:917–924. doi: 10.1016/j.tranon.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson D. Griffin M. J. Sludden J. Drew Y. Cresti N. Swales K. Merriman M. Allen R. Bevan P. Buerkle M. Eur. J. Cancer. 2016;68:1–10. doi: 10.1016/j.ejca.2016.08.026. [DOI] [PubMed] [Google Scholar]
- Haagensen E. J. Thomas H. D. Schmalix W. A. Payne A. C. Kevorkian L. Allen R. A. Bevan P. Maxwell R. J. Newell D. R. Cancer Chemother. Pharmacol. 2016;78:1269–1281. doi: 10.1007/s00280-016-3186-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. Cheng Y. Zhang M. He X. Kong L. Zhou K. Zhou Y. Li L. Tian H. Song X. iScience. 2020;23:101254. doi: 10.1016/j.isci.2020.101254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett S. D. Bridges A. J. Dudley D. T. Saltiel A. R. Fergus J. H. Flamme C. M. Delaney A. M. Kaufman M. LePage S. Leopold W. R. Bioorg. Med. Chem. Lett. 2008;18:6501–6504. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]
- Wu P.-K. Park J.-I. Semin. Oncol. 2015;42:849–862. doi: 10.1053/j.seminoncol.2015.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Chi L. Yu F. Dai H. Si X. Gao C. Wang Z. Liu L. Zheng J. Ke Y. Bioorg. Med. Chem. 2022:116922. doi: 10.1016/j.bmc.2022.116922. [DOI] [PubMed] [Google Scholar]
- Planz O. Antiviral Res. 2013;98:457–468. doi: 10.1016/j.antiviral.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Cheng Y., PhD, Flinders University, College of eMedicine and Public Health, 2021 [Google Scholar]
- Miyake H. Tsuchiya S. Hoshino T. Hori A. Vincent P. W. Cancer Res. 2010;70:2516. doi: 10.1158/1538-7445.AM10-2516. [DOI] [Google Scholar]
- El-Khoueiry A. Kurkjian C. Semrad T. Musib L. Gates M. Eppler S. Chang I. Chan I. Rooney I. Bendell J. Mol. Cancer Ther. 2013;12:B75. doi: 10.1158/1535-7163.TARG-13-B75. [DOI] [Google Scholar]
- Ryan M. B. Der C. J. Wang-Gillam A. Cox A. D. Trends Cancer. 2015;1:183–198. doi: 10.1016/j.trecan.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanathan R. K. Von Hoff D. D. Eskens F. Blumenschein G. Richards D. Genvresse I. Reschke S. Granvil C. Skubala A. Peña C. Target. Oncol. 2020;15:163–174. doi: 10.1007/s11523-020-00714-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinelli E. Troiani T. D'Aiuto E. Morgillo F. Vitagliano D. Capasso A. Costantino S. Ciuffreda L. P. Merolla F. Vecchione L. Int. J. Cancer. 2013;133:2089–2101. doi: 10.1002/ijc.28236. [DOI] [PubMed] [Google Scholar]
- Lu B. Huang S. Cao J. Hu Q. Shen R. Wan H. Wang D. Yuan J. Zhang L. Zhang J. Bioorg. Med. Chem. 2018;26:581–589. doi: 10.1016/j.bmc.2017.12.019. [DOI] [PubMed] [Google Scholar]
- Zhang J. Lu B. Liu D. Shen R. Yan Y. Yang L. Zhang M. Zhang L. Cao G. Cao H. Cancer Biol. Ther. 2016;17:199–207. doi: 10.1080/15384047.2016.1139231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C. Wang H. Zheng C. Liu Z. Gao X. Xu F. Niu Y. Zhang L. Xu P. Eur. J. Med. Chem. 2021;218:113386. doi: 10.1016/j.ejmech.2021.113386. [DOI] [PubMed] [Google Scholar]
- Phadke M. S. Sini P. Smalley K. S. Mol. Cancer Ther. 2015;14:1354–1364. doi: 10.1158/1535-7163.MCT-14-0832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sini P. Gürtler U. Zahn S. K. Baumann C. Rudolph D. Baumgartinger R. Strauss E. Haslinger C. Tontsch-Grunt U. Waizenegger I. C. Mol. Cancer Ther. 2016;15:2388–2398. doi: 10.1158/1535-7163.MCT-16-0066. [DOI] [PubMed] [Google Scholar]
- Akinleye A. Furqan M. Mukhi N. Ravella P. Liu D. J. Hematol. Oncol. 2013;6:1–11. doi: 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo I. Garg S. Danesh A. Bruce J. Shaw P. Tan Q. Quevedo R. Braunstein M. Oza A. M. Pugh T. Mol. Case Stud. 2019;5:a004341. doi: 10.1101/mcs.a004341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine M. Stewart A. Pedersen B. Boyd S. Kefford R. Rizos H. Oncogenesis. 2018;7:1–11. doi: 10.1038/s41389-018-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H. Hugo W. Kong X. Hong A. Koya R. C. Moriceau G. Chodon T. Guo R. Johnson D. B. Dahlman K. B. Cancer Discovery. 2014;4:80–93. doi: 10.1158/2159-8290.CD-13-0642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper K. de Goeje P. L. Peeper D. S. van Amerongen R. Cancer Res. 2014;74:5937–5941. doi: 10.1158/0008-5472.CAN-14-1174. [DOI] [PubMed] [Google Scholar]
- Wallace M. B. Adams M. E. Kanouni T. Mol C. D. Dougan D. R. Feher V. A. O'Connell S. M. Shi L. Halkowycz P. Dong Q. Bioorg. Med. Chem. Lett. 2010;20:4156–4158. doi: 10.1016/j.bmcl.2010.05.058. [DOI] [PubMed] [Google Scholar]
- Heald R. A. Jackson P. Savy P. Jones M. Gancia E. Burton B. Newman R. Boggs J. Chan E. Chan J. J. Med. Chem. 2012;55:4594–4604. doi: 10.1021/jm2017094. [DOI] [PubMed] [Google Scholar]
- Aoki T. Hyohdoh I. Furuichi N. Ozawa S. Watanabe F. Matsushita M. Sakaitani M. Ori K. Takanashi K. Harada N. Bioorg. Med. Chem. Lett. 2013;23:6223–6227. doi: 10.1016/j.bmcl.2013.10.001. [DOI] [PubMed] [Google Scholar]
- Hyohdoh I. Furuichi N. Aoki T. Itezono Y. Shirai H. Ozawa S. Watanabe F. Matsushita M. Sakaitani M. Ho P.-S. ACS Med. Chem. Lett. 2013;4:1059–1063. doi: 10.1021/ml4002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung I. V. Hitchcock M. Pühler F. Neuhaus R. Scholz A. Hammer S. Petersen K. Siemeister G. Brittain D. Hillig R. C. Bioorg. Med. Chem. Lett. 2013;23:2384–2390. doi: 10.1016/j.bmcl.2013.02.028. [DOI] [PubMed] [Google Scholar]
- Lu H. Tu W. Fei H. Xu G. Hu Q. Zhang L. Lin B. Yuan J. Yin J. Gong A. Bioorg. Med. Chem. Lett. 2014;24:2555–2559. doi: 10.1016/j.bmcl.2014.03.086. [DOI] [PubMed] [Google Scholar]
- Robarge K. D. Lee W. Eigenbrot C. Ultsch M. Wiesmann C. Heald R. Price S. Hewitt J. Jackson P. Savy P. Bioorg. Med. Chem. Lett. 2014;24:4714–4723. doi: 10.1016/j.bmcl.2014.08.008. [DOI] [PubMed] [Google Scholar]
- Liu M.-M. Chen X.-Y. Huang Y.-Q. Feng P. Guo Y.-L. Yang G. Chen Y. J. Med. Chem. 2014;57:9343–9356. doi: 10.1021/jm500613m. [DOI] [PubMed] [Google Scholar]
- Pagadala R. Kommidi D. R. Kankala S. Maddila S. Singh P. Moodley B. Koorbanally N. Jonnalagadda S. B. Org. Biomol. Chem. 2015;13:1800–1806. doi: 10.1039/C4OB02229G. [DOI] [PubMed] [Google Scholar]
- Hartung I. V. Hammer S. Hitchcock M. Neuhaus R. Scholz A. Siemeister G. Bohlmann R. Hillig R. C. Pühler F. Bioorg. Med. Chem. Lett. 2016;26:186–193. doi: 10.1016/j.bmcl.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Cui Z. Li X. Li L. Zhang B. Gao C. Chen Y. Tan C. Liu H. Xie W. Yang T. Bioorg. Med. Chem. 2016;24:261–269. doi: 10.1016/j.bmc.2015.12.011. [DOI] [PubMed] [Google Scholar]
- Lv X.-H. Ren Z.-L. Zhou B.-G. Li Q.-S. Chu M.-J. Liu D.-H. Mo K. Zhang L.-S. Yao X.-K. Cao H.-Q. Bioorg. Med. Chem. 2016;24:4652–4659. doi: 10.1016/j.bmc.2016.08.002. [DOI] [PubMed] [Google Scholar]
- Hall J. A. Seedarala S. Zhao H. Garg G. Ghosh S. Blagg B. S. J. Med. Chem. 2016;59:925–933. doi: 10.1021/acs.jmedchem.5b01354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J. Kelley E. H. Methot J. L. Zhou H. Petrocchi A. Chen H. Hill S. E. Hinton M. C. Hruza A. Jung J. O. J. Med. Chem. 2016;59:6501–6511. doi: 10.1021/acs.jmedchem.6b00708. [DOI] [PubMed] [Google Scholar]
- Guo Y. Wang Y. Li H. Wang K. Wan Q. Li J. Zhou Y. Chen Y. ACS Med. Chem. Lett. 2018;9:502–506. doi: 10.1021/acsmedchemlett.8b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin X.-Y. Chen H. Li D.-D. Li A.-L. Wang W.-Y. Gu W. J. Enzyme Inhib. Med. Chem. 2019;34:955–972. doi: 10.1080/14756366.2019.1605364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi D. Niu Y. Li H. Noha S. M. Temml V. Schuster D. Wang C. Xu F. Xu P. Eur. J. Med. Chem. 2019;178:802–817. doi: 10.1016/j.ejmech.2019.06.027. [DOI] [PubMed] [Google Scholar]
- Wang C. Xi D. Wang H. Niu Y. Liang L. Xu F. Peng Y. Xu P. Eur. J. Med. Chem. 2020;196:112271. doi: 10.1016/j.ejmech.2020.112271. [DOI] [PubMed] [Google Scholar]
- Tao Q. Fang F. Li J. Wang Y. Zhao C. Liang J. Ma X. Wang H. Med. Chem. Res. 2020;29:519–527. doi: 10.1007/s00044-020-02502-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams M. E. Wallace M. B. Kanouni T. Scorah N. O'Connell S. M. Miyake H. Shi L. Halkowycz P. Zhang L. Dong Q. Bioorg. Med. Chem. Lett. 2012;22:2411–2414. doi: 10.1016/j.bmcl.2012.02.026. [DOI] [PubMed] [Google Scholar]
- Laing V. E. Brookings D. C. Carbery R. J. Simorte J. G. Hutchings M. C. Langham B. J. Lowe M. A. Allen R. A. Fetterman J. R. Turner J. Bioorg. Med. Chem. Lett. 2012;22:472–475. doi: 10.1016/j.bmcl.2011.10.105. [DOI] [PubMed] [Google Scholar]
- Wang C. Zhang H. Xu F. Niu Y. Wu Y. Wang X. Peng Y. Sun J. Liang L. Xu P. Molecules. 2013;18:6057–6091. doi: 10.3390/molecules18056057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. Hou J. Wang C. Liu X. He H. Xu P. Yang Z. Chen Z. Wu Y. Zhang L. Molecules. 2013;18:13957–13978. doi: 10.3390/molecules181113957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamal A. Faazil S. Ramaiah M. J. Ashraf M. Balakrishna M. Pushpavalli S. Patel N. Pal-Bhadra M. Bioorg. Med. Chem. Lett. 2013;23:5733–5739. doi: 10.1016/j.bmcl.2013.07.068. [DOI] [PubMed] [Google Scholar]
- Aoki T. Hyohdoh I. Furuichi N. Ozawa S. Watanabe F. Matsushita M. Sakaitani M. Morikami K. Takanashi K. Harada N. ACS Med. Chem. Lett. 2014;5:309–314. doi: 10.1021/ml400379x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddila S. Naicker K. Momin M. I. Rana S. Gorle S. Maddila S. Yalagala K. Singh M. Koorbanally N. A. Jonnalagadda S. B. Med. Chem. Res. 2016;25:283–291. doi: 10.1007/s00044-015-1482-x. [DOI] [Google Scholar]
- Sun J. Niu Y. Wang C. Zhang H. Xie B. Xu F. Jin H. Peng Y. Liang L. Xu P. Bioorg. Med. Chem. 2016;24:3472–3482. doi: 10.1016/j.bmc.2016.05.055. [DOI] [PubMed] [Google Scholar]
- Elsayed H. E. Ebrahim H. Y. Haggag E. G. Kamal A. M. El Sayed K. A. Bioorg. Med. Chem. 2017;25:6297–6312. doi: 10.1016/j.bmc.2017.09.033. [DOI] [PubMed] [Google Scholar]
- Chen H. Qiao C. Miao T.-T. Li A.-L. Wang W.-Y. Gu W. J. Enzyme Inhib. Med. Chem. 2019;34:1544–1561. doi: 10.1080/14756366.2019.1655407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozar I. Margue C. Rothengatter S. Haan C. Kreis S. Biochim. Biophys. Acta, Rev. Cancer. 2019;1871:313–322. doi: 10.1016/j.bbcan.2019.02.002. [DOI] [PubMed] [Google Scholar]
- Kakadia S. Yarlagadda N. Awad R. Kundranda M. Niu J. Naraev B. Mina L. Dragovich T. Gimbel M. Mahmoud F. OncoTargets Ther. 2018:7095–7107. doi: 10.2147/OTT.S182721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kun E. Tsang Y. Ng C. Gershenson D. Wong K. Cancer Treat. Rev. 2021;92:102137. doi: 10.1016/j.ctrv.2020.102137. [DOI] [PubMed] [Google Scholar]
- Meidhof S. Brabletz S. Lehmann W. Preca B. T. Mock K. Ruh M. Schüler J. Berthold M. Weber A. Burk U. EMBO Mol. Med. 2015;7:831–847. doi: 10.15252/emmm.201404396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K. Kumar A. Thareja S. Kumar P. Anti-Cancer Agents Med. Chem. 2023;23:278–297. doi: 10.2174/1871520622666220624164152. [DOI] [PubMed] [Google Scholar]