

Abstract
Neurodegenerative disorders of aging are characterized by the progressive accumulation of proteins such as α-synuclein (α-syn) and amyloid beta (Aβ). Misfolded and aggregated α-syn has been implicated in neurological disorders such as Parkinson’s disease, and Dementia with Lewy Bodies, but less so in Alzheimer’s Disease (AD), despite the fact that accumulation of α-syn has been confirmed in over 50% of postmortem brains neuropathologically diagnosed with AD. To date, no therapeutic strategy has effectively or consistently downregulated α-syn in AD. Here we tested the hypothesis that by using a systemically-delivered peptide (ApoB11) bound to a modified antisense oligonucleotide against α-syn (ASO-α-syn), we can downregulate α-syn expression in an AD mouse model and improve behavioral and neuropathologic phenotypes. Our results demonstrate that monthly systemic treatment with of ApoB11:ASO α-syn beginning at 6 months of age reduces expression of α-synuclein in the brains of 9-month-old AD mice. Downregulation of α-syn also led to reduction in Aβ plaque burden, prevented neuronal loss and astrogliosis. Furthermore, we found that AD mice treated with ApoB11:ASO α-syn had greatly improved hippocampal and spatial memory function in comparison to their control counterparts. Collectively, our data supports the reduction of α-syn through use of systemically-delivered ApoB11:ASO α-syn as a promising future disease-modifying therapeutic for AD.
Keywords: Amyloid beta, alpha synuclein, biomarkers, Alzheimer’s disease, antisense oligonucleotide, comorbidity
Graphical Abstract
Introduction
Alzheimer’s disease (AD) and AD related disorders (ADRD) are heterogeneous conditions characterized by the progressive accumulation of amyloid beta (Aβ) and tau neuropathology with neurodegeneration and inflammation in the CNS. In addition to Aβ and tau, recent studies from our group and others demonstrate that alpha-synuclein (α-syn) may also contribute to AD pathogenesis (Clinton et al., 2010; Jellinger, 2011; Mandal et al., 2006; Spencer et al., 2016a; Twohig and Nielsen, 2019).
Pathological comorbidity is common in neurodegenerative diseases. For example, co-occurrence of Aβ and α-syn neuropathology at autopsy is found in over 50% of brains of patients clinically diagnosed with AD antemortem (Hamilton, 2000; Rabinovici et al., 2017). Because comorbid cases often show increased rates of cognitive decline and shorter life span compared to AD without α-syn pathology (Matej et al., 2019; Spina et al., 2021; Visanji et al., 2019), our group and others have hypothesized that co-accumulation of these two proteins may synergistically increase neurotoxicity (Lashley et al., 2008; Spencer et al., 2016a). Data from cell models demonstrate that increased α-syn expression promotes amyloid precursor protein (APP) expression as well as β and γ secretase activity and increased Aβ (Roberts et al., 2017). The synergistic relationship underlying the Aβ and α-syn toxicity is not well understood but may involve hydrophobic domains in the Non-Amyloid Component (NAC) domain of α-syn and the core domain of Aβ42 (Jose et al., 2014). Inter-protein interactions through these hydrophobic domains have can promote cross-aggregation and toxicity in neuronal cells (Kazmierczak et al., 2008).
In addition to in vitro work and ex vivo human postmortem brain analyses, α-syn-Aβ neuropathological relationships have also been seen in vivo in AD models. For example, in AD transgenic models, α-syn interacts with both Aβ and tau and accumulates in selected brain regions (Ferman et al., 2018). Our recent mechanistic data suggests that enhanced expression of α-syn in APP mutant mice increases degeneration of cholinergic neurons in the nucleus basalis and hippocampal glutamatergic neurons (Spencer et al., 2016a). We also found that α-syn aggregates can target molecular components of the ESCRTIII/autophagy pathway and interfere with intracellular trafficking and degradation of toxic protein aggregates (Spencer et al., 2016b), a phenotype that could be blocked with treatment of α-syn antibodies (Spencer et al., 2014). In addition, previous work from our lab demonstrates that reduction of α-syn by short interfering RNA (siRNA) treatment or genetic ablation of α-syn prevents loss of cholinergic neurons and ameliorates the functional deficits in Lewy Body Dementia (Spencer et al., 2019) and AD mice (Spencer et al., 2016a). Taken together, data from our lab and others suggest that reducing α-syn may be a viable disease-modifying therapeutic strategy for not only synucleinopathies but also amyloidopathies such as AD.
Through use of anti-sense oligonucleotide (ASO) technology, there has been dramatic progress in developing strategies to decrease toxic proteins involved Spinal Muscular Atrophy, Amytrophic Lateral Sclerosis and Huntington’s disease (Dickey and La Spada, 2018; Keiser et al., 2016; McCampbell et al., 2018; Miller et al., 2013). Although these data have exciting implications and future therapeutic potential, delivery to the CNS with ASO therapy is challenging and invasive, as it relies upon repeated intrathecal and intracranial injections. Thus, there is great need for methodological advancement to develop systemic approaches to deliver ASOs into the CNS. Although strategies are being explored for synucleinopathies, AD and other tauopathies in mouse models and in vitro (Crunkhorn, 2017; DeVos and Hyman, 2017; Vossel et al., 2010), to our knowledge, few successful studies have shown delivery of ASOs to the CNS following systemic delivery, and none in AD by targeting α-syn. Other groups have published work on the use of other ASOs delivered peripherally to cross the blood-brain barrier. Examples of such studies are: the use of peptide nucleic acids against the brain receptor neurotensin, leading to the internalization into the CNS (Tyler et al., 1999); phosphorothioate ASOs which cross the BBB readily with various targets (Banks, 2016), including APP (Banks et al., 2001). Although encouraging from a conceptual standpoint, these studies showed minimal penetrance of ASO in the CNS.
To test the hypothesis that we can effectively administer ASO’s systemically and safely and non-invasively gain access to the brain to reduce/prevent Aβ toxicity, neuropathology and associated cognitive deficits in an AD mouse model through downregulation of α-syn expression, we leveraged published methodology using fragments of the Apolipoprotein B (ApoB) protein for systemic transport of oligonucleotides to the CNS (Spencer and Verma, 2007). We recently identified a 11-amino acid core domain that represents the minimal receptor binding domain of ApoB (i.e. ApoB11) (Spencer et al., 2019) and converted the siRNA sequence to an ASO 2’-O-Methyl-oligoribonucleotides (2’-OMe) for preclinical validation in AD mice.
We hypothesized that delivery of ApoB11:2’-OMe ASO targeted to α-syn by systemic administration would efficiently cross the BBB and reduces expression of α-syn in the AD brain, increases the survival of neurons and improves memory. We treated AD mice at early stages of disease (6 months of age), a timepoint at which Aβ plaques and cognitive deficits are beginning. We found that systemic delivery of ApoB11:ASO α-syn to these mice reduced expression of α-syn, which led to reduction of Aβ plaque burden, neuronal loss and astrogliosis by 9 months of age. Importantly, AD mice treated for three months with the ASO α-syn beginning at 6 months had greatly improved hippocampal and spatial memory function in comparison with their control counterparts.. Collectively, our data shown supports the use of ApoB11:ASO α-syn conjugates delivered systemically to downregulate α-syn as a promising future therapeutic strategy in AD.
Results
A α-syn antisense oligonucleotide conjugates with the ApoB11 peptide and crosses the blood brain barrier for delivery to neurons.
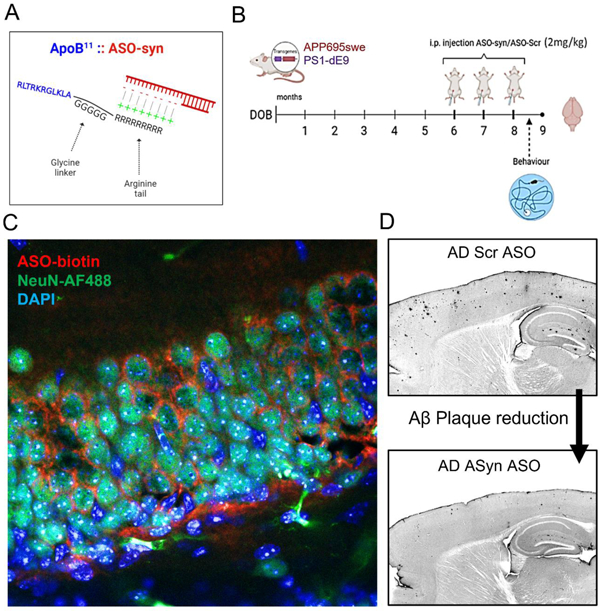
We converted the mouse/ human cross-reacting siRNA α-syn (as shown in (Spencer et al., 2019)) sequence to the 2’-OMe backbone and showed that this can bind to the ApoB11 peptide in vitro to form the peptide:oligonucleotide conjugate (Figure 1A,B). We first estimated the ratio of ApoB11:ASO α-syn that results in the maximum conjugation of both molecules. To that end we tested five different conjugation ratios in vitro and ran the resulting products on an ethidium bromide agarose gel. Unbound ASO is visible as a band of nucleotides whereas ASO bound to the ApoB11 peptide does not enter the gel and is not visible. At 1:10 ratio of ApoB11:ASO α-syn we did not see any band, in accordance with our previous results (Spencer et al., 2019), and this ratio was used for in vitro and in vivo experiments.
Figure 1.

Development of ApoB11:2’-OMe:α-syn ASO conjugates to cross the blood-brain-barrier in mice and downregulate translation of α-synuclein. (A) Schematical representation of the blood-brain-barrier and the distribution of the ApoB11:ASO α-syn conjugates, shown here to cross the BBB through RMT as described by other authors (Lajoie and Shusta, 2015). ASO (red), ApoB11 (blue). (B) Diagrammatic illustration of the 11 amino acid fragment coupled to a 5 Glycine linker and a 9 Arginine positively charged tail utilized to bind to negatively charged oligonucleotides and transport them into neuronal cells. (C) Agarose gel to test ratios of ApoB11:ASO i.e. the ratio where all ASO is conjugated with ApoB11 and thus is not visible in the gel – 1:10. (D) WT (C57BL6) mice received an IP injection with either ASO-syn (biotinylated) or PBS and sacrificed 24h later followed by immunofluorescence of brain slices – NeuN (AF-488, green), ASO-syn/Biotin (Tyr-Red), DAPI. Low-power magnification images were acquired with 10x objective (scale bar = 250μm). High-power magnification images were acquired with 63x objective, oil immersion (scale bar = 25 μm).
Next, we investigated whether we would be able to detect the ASO in the brain tissue of mice that were injected intraperitoneally. We injected wild-type (WT) mice (n=3) with a modified 5’-biotin labelled ASO-αsyn sequence. Male C57Bl/6 mice 8–10 weeks received either ApoB11:αsyn-ASO (2mg/kg) or PBS and were sacrificed 24 hours later. Brain sections were stained with Tyramide Red Signal Amplification Kit (to label the ASO-biotin in red dye) and anti-NeuN (AF-488, green). We found that mice injected with ASO-syn show profuse red signal throughout the brain, in between pyramidal cells of the hippocampus and cortex (Figure 1D and Figure S1), whereas mice injected with vehicle (PBS) did not.
Delivery of ApoB11:ASO α-syn reduced levels of α-syn protein in a neuronal cell line.
In order to determine whether ApoB11:ASO α-syn can reduce α-syn in a neuronal cell line, we infected differentiated SH-SY5Y human neuroblastoma cells with a lentivirus vector overexpressing human α-syn (LV-α-syn) or control vector (LV-Ctrl) to test ASO treatment under conditions of α-syn overexpression or normal levels, respectively. Cells were then treated with ApoB11:ASO α-syn or ApoB11:ASO-scrambled for 48 hours and then either fixed and immunostained for α-syn or lysed for immunoblot analysis of α-syn. We found that cells treated with scrambled ASO showed expected levels of endogenous α-syn and increased α-syn levels following infection with the LV-α-syn virus (Figure 2A,B). We found that treatment with ASO-α-syn reduced both endogenous and overexpression (LV-α-syn virus treated) levels of α-syn in SH-SY5Y cells (Figure 2B). We confirmed this by collecting cell lysates from each group and immunoblotting for total α-syn (Figure 2C,D). Collectively our data shown in Figure 2 and Figure S2 demonstrates that ASO α-syn reduces expression and accumulation of α-syn in an in vitro neuronal model.
Figure 2.

Delivery of ApoB11:ASO α-syn reduces the accumulation of α-syn in human neuronal cells in vitro. (A) Human neuroblastoma cells, SH-SY5Y, were plated in 12-well dishes, differentiated and infected with LV-α-syn or LV-Ctrl for 72 hours in differentiation media. Cells were then treated with 100pmol α-syn ASO (2’OMe) or ASO scrambled RNA conjugated to ApoB11 peptide. (B) Representative immunohistochemistry images of coverslips stained with an antibody against α-syn which recognizes total human α-syn (mouse anti- α-syn), counterstained with DAPI. Secondary blotting to detect α-syn was done using anti-mouse AF-488. Scale bars, 100μm in low-power images and 20μm in high-power images (C) Image analysis of SH-SY5Y α-syn stained cells represented as fluorescence intensity, corrected for background intensity (ImageJ). N=3 (3 different wells, one representative image per well). (D) Representative immunoblots from SH-SY5Y cells treated as described above and probed for α-syn and Actin. Secondary staining with anti-mouse-AF488 (green) for α-syn and anti-rabbit-AF594 (red) for actin. Densitometric analysis of α-syn immunoreactive band from immunoblot at 20kDa as a ratio to Actin (42 kDa). Blots for the LV-Asyn and the LV-Ctrl group are shown separately. N=2 (2 of the wells were used for each group). One-way ANOVA with post hoc Dunnett’s multiple comparisons test. ** p < 0.005, ****p<0.0001.
Delivery of ApoB11:ASO α-syn via intraperitoneal injection reduces the accumulation of phosphorylated α-syn in AD mice.
Thus far we have demonstrated that treatment of neuronal cells with ApoB11:ASO α-syn constructs is an effective strategy to reduce α-syn levels in vitro. We then investigated whether we could selectively reduce brain α-syn levels in in vivo in an mutant APP AD mouse model (Jankowsky et al., 2001). This AD model begins to accumulate Aβ at a young age and develop large numbers of fibrillar Aβ deposits in the cortex and hippocampus at about six months of age, considerably earlier than other AD Aβ models (Campbell et al., 2015; Holcomb et al., 1998; Zhang et al., 2016; Zhang et al., 2015).
We administered ApoB11:ASO to AD mice by intraperitoneal injections at 6, 7 and 8 months of age and sacrificed the animals at 9 months of age, a timepoint at which this model has mature cognitive deficits and Aβ plaques. Single intraperitoneal injections were done at each monthly timepoint using 2mg/kg of ApoB11:ASO α-syn or ASO-scrambled, as performed previously (Spencer et al., 2019). Spatial memory was tested by Morris water maze trials 2 weeks after the third and final injection. After sacrifice at 9 months, we immunostained brain sections by immunohistochemistry for α-syn using two antibodies: Syn1 , which reacts with both total human and mouse α-syn; and pSyn , which reacts with human and mouse α-syn that is phosphorylated at the S129 residue, a post-translational modification that is associated with pathological protein aggregation (Ma et al., 2016). Immunohistochemical experiments demonstrated a significant reduction in phosphorylated α-syn in all regions of the hippocampus of AD mice injected with ApoB11:ASO α-syn compared to the control ApoB11:ASO-scrambled (Figure 3). Differences were statistically significant in the three regions of the hippocampus analysed: CA1, CA3 and dentate gyrus (DG). Images for each brain region were selected based on Figure S3. No significant reduction was observed in other brain regions, including cortex, striatum and thalamus. Immunohistochemistry for total α-syn did not reveal any significant differences between ApoB11:ASO α-syn and ApoB11:ASO-scrambled (Figure S4).
Figure 3.

Downregulation of α-syn in vivo following the i.p. injection of ApoB11:ASO conjugates. (A) Mice from AD line were IP-injected with ApoB11:ASO α-syn or ApoB11:ASO-Scrambled at 6, 7 and 8 months of age. At 9 months of age, mice were sacrificed, brains were harvested for immunohistochemistry. (B) Immunohistochemistry using an antibody against phosphorylated synuclein (S129), highlighting cortex and hippocampal regions. (C) phosphorylated synuclein (S129) expression in different brain regions. Areas of section measured: Cortex 20um2, CA1 5um2, CA3 5um2, DG 5um2, (D) Quantification of staining (corrected optical density) for cortex, hippocampus (CA1, CA3 and dentate gyrus). Scale bars, 200μm in low-power images and 40μm in high-power images. N=5 mice per group (3 averaged sections from each brain). * p < 0.05, ** p < 0.005, *** p < 0.001. Comparisons between ASOs and vehicle via one-way ANOVA with post hoc Dunnett’s multiple comparisons test; comparisons between ASO-syn and ASO-scr via one-way ANOVA using Tukey’s test # p <0.05, ## p < 0.005, ### p < 0.001
Delivery of ApoB11:ASO α-syn to AD mice reduces accumulation of Aβ.
Having successfully established methodology for the knockdown of α-syn levels in vivo, we tested the hypothesis that reduction of α-syn would block Aβ accumulation in AD mice. As mentioned, these animals were injected intraperitoneally with either ApoB11:ASO α-syn or ApoB11:ASO-scrambled at 6, 7 and 8 months of age and sacrificed at 9 months. The number of Aβ plaques was counted in the cortex and hippocampus using previously published methods (Hampel et al., 2021; Palmqvist et al., 2017; Spencer et al., 2016a; Zhang et al., 2016). Immunohistochemistry using the anti-human monoclonal antibody 82E1 against the N-terminus of human Aβ showed a significant reduction in the number of Aβ plaques was observed in mice that were injected with the ApoB11:ASO α-syn and this reduction occurred both within the cortex and the hippocampus (Figure 4A,B).
Figure 4.

Downregulation of α-syn reveals significant reduction of Aβ plaques in AD mice. (A, B) Immunohistochemistry using 82E1 antibody that detects the APP transgene and quantification of plaques per volume. Plaques were counted in 200 μm2 for cortex and 150 μm2 for hippocampus. (i, ii) Detailed overview highlighting cortex, hippocampus and striatum regions of AD mouse brains, ASO-scrambled- and ASO-α-syn-treated. Plaques are seen in the cortex and hippocampus of mice treated with ASO-scrambled. (C, D). Labelling of brain sections using Thioflavin S and quantification of % area taken by plaques. Scale bars, 200μm in low-power images and 40μm in high-power images. N=5 mice per group (3 averaged sections from each brain). ** p < 0.005, **** p < 0.0001. One-way ANOVA with post-hoc Dunnett’s test for comparisons against vehicle control and Tukey’s test between ASOs, ## p<0.005.
We next confirmed our results with immunohistochemistry using Thioflavin S, a dye that binds selectively to the β-sheet structure, a method which detects the amyloid-core of aggregating proteins such as Aβ, α-syn or tau (Biancalana and Koide, 2010). We found that AD mice had significantly reduced numbers of Aβ plaques/aggregates when treated with ApoB11:ASO α-syn (Figure 4C,D).
ApoB11:ASO α-syn prevents neurodegeneration in AD mice
Previous studies have reported that α-syn can interact with and aggravate Aβ pathology leading to neurodegeneration (Colom-Cadena et al., 2013; Twohig and Nielsen, 2019). Thus, next we analyzed the effects of reducing α-syn levels on the degeneration of specific neuronal populations in the cortex, hippocampus, striatum and thalamus of AD mice. At 9 months of age, we found significant neuronal loss in the pre-frontal cortex and CA3 region of AD mouse brains injected with ApoB11:ASO-scrambled or PBS vehicle as compared with ApoB11:ASO-syn-treated mice, based on immunohistochemistry against pan-neuronal populations using NeuN antibody (Fig. 5A,B). In our hand, neuronal loss occurs earlier than what is described by other authors (22 months) (Sadowski et al., 2004), but we have previously reported neuronal and synaptic loss/changes at 12 months of age using the this AD mouse model (Zhang et al., 2016). Importantly, neuronal loss was rescued in AD mice that received ApoB11:ASO α-syn (Figure 5A,B), suggesting that this treatment can block early neurodegeneration in AD models. More generally, this suggests that the presence of α-syn may accelerate neurodegeneration in AD brains.
Figure 5.
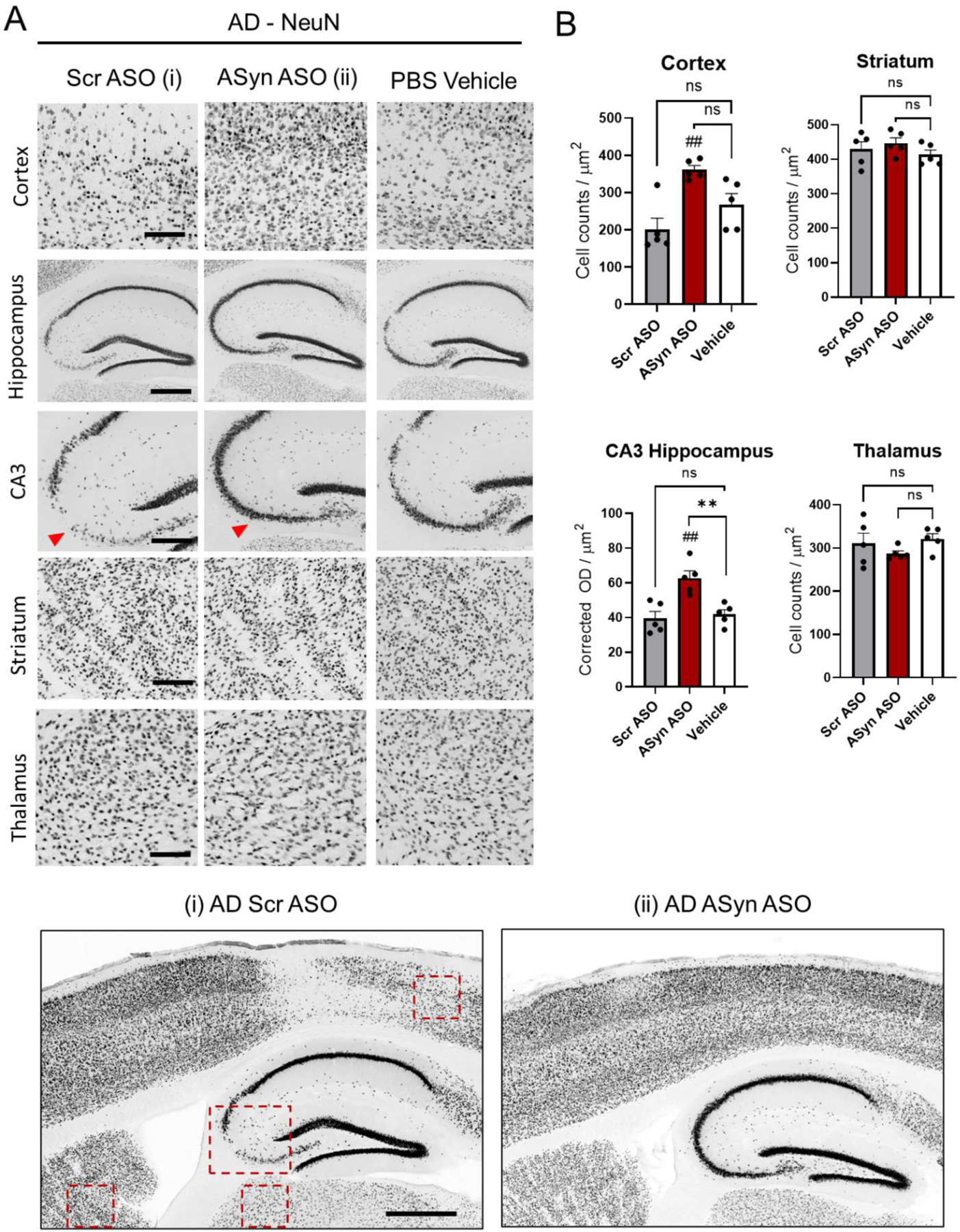
Neuronal loss is rescued in mice treated with ApoB11:ASO α-syn. (A) Vibratome sections were immunostained with an antibody against NeuN. Representative low- and high-magnification photomicrographs of the frontal cortex, CA3, striatum and thalamus from non-Tg and AD mice immunoreacted with anti-NeuN. Quantification: cortex 20um2, CA3 5um2, striatum 20um2, thalamus 20um2. (B) Quantification of cell counts per area and adjusted optical density in the cortex, hippocampus (CA3), striatum and thalamus. Patches without antibody coverage from folds were excluded from analysis. N=5 mice per group (3 averaged sections from each brain). Statistical analysis was conducted using one-way ANOVA post hoc Dunnett’s test against vehicle control ** p < 0.005 and Tukey’s test between ASOs, ##, p <0.005 Scale bars, 200μm in low-power images and 40μm in high-power images.
Downregulation of α-syn prevents glial-mediated neuroinflammatory phenotypes in AD Mice
The role of glial cells in the pathogenesis of several neurodegenerative diseases has been extensively studied (Bennett and Viaene, 2021; Brück et al., 2016; Gleichman and Carmichael, 2020; Mrak and Griffin, 2005). Astrocytes in particular have been demonstrated to associate closely with Aβ plaques, and reactive astrocytes are increased both in human AD brains and AD mouse models (Funato et al., 1998; Nagele et al., 2003). We hypothesized that alleviating Aβ burden via reduction of α-syn could also provide a means to reduce neuroinflammation in the brains of AD mice. Our results show in Figure 6 that GFAP staining is increased in the cortex and striatum of control (PBS and ASO-scrambled) AD mouse brains (up to 30 stained cells per volume of cortex stained, 20um3), when compared to mice treated with ApoB11:ASO α-syn. Differences in GFAP-stained cell counts were observed in the cortex and striatum only, due to the congregation of astrocytes around plaques to generate foci which were absent in these regions in the absence of plaques. On the other hand, in the hippocampus (specifically in the CA3), the presence of GFAP-stained cells is more ubiquitous and, even in the presence of plaques, we do not observe the formation of foci. Moreover, we performed immunohistochemistry experiments against Iba1 (activated microglia). Our data shows a reduction in activated microglial staining in treated mice, compared to untreated AD mice (Figure 6). These results demonstrate the potential for the downregulation of α-syn in preventing neuroimmune and inflammatory responses in AD mouse models.
Figure 6.

Astrogliosis is abrogated in mice treated systemically with ApoB11:ASO α-syn. Immunohistochemical analysis of changes in astrocytic and microglial activation (GFAP and Iba1 respectively) in AD mice treated with ApoB11:ASO α-syn as compared to ApoB11:ASO-scrambled. (A, C) Representative low- and high-magnification photomicrographs of the pre-frontal cortex, CA3, striatum and thalamus from AD mice immunoreacted with anti-GFAP and anti-Iba1. (i, ii) Detailed overview highlighting Cortex, Hippocampus and Striatum regions of AD ASO Scrambled and AD ASO ASyn mouse brains. In GFAP stains, astrogliosis foci are seen in the cortex and hippocampus of mice treated with ASO-Scrambled. (B, D) Quantification of cell counts per area in the cortex, hippocampus (CA3), striatum and thalamus respectively. Areas measured: cortex 20um2, CA3 5um2, striatum 50um2, thalamus 50um2. N=5 mice per group (3 averaged sections from each brain). Statistical analysis was conducted using one-way ANOVA post hoc Dunnett’s test ** p < 0.005 for comparisons against vehicle control and Tukey’s test between ASOs, ## p<0.005. Scale bars, 200μm in low-power images and 40μm in high-power images.
Downregulation of α-syn rescues spatial memory deficits in AD mice
We have thus far shown that the reduction of α-syn levels produces promising results in AD mice by reducing Aβ plaques, neuronal loss, and neuroinflammatory responses. AD mice develop spatial memory deficits as early as 3 to 6 months, even before accumulation of Aβ plaques (Campbell et al., 2015; Holcomb et al., 1998; Reiserer et al., 2007; Zhang et al., 2016). In order to understand whether reduction of α-syn would result in an amelioration of these behavioral deficits, we performed Morris water maze trials. We performed these experiments on 10 mice from each group, non-transgenic (non-tg) and AD (Figure 7).
Figure 7.

Morris water maze tests reveal amelioration of spatial memory deficits in AD mice treated with ApoB11:ASO α-syn. (A, B) Morris water maze test was performed in two phases. The first phase was the training portion conducted on days 1–3 and the second phase was with the platform hidden on days 4 –7. During the hidden platform test, AD mice performed worse in spatial learning portion of the test in comparison with non-tg mice, however this difference was abrogated upon treatment with ASO α-syn after days 6 and 7. (C, D, E) Probe test was performed at day 8, without the platform. The number of passes through target or amount of time spent in the quadrant that used to contain the platform were analyzed. AD mice spent significantly less time in the target quadrant compared with non-tg mice. AD mice treated with ASO α-syn show a trend to improve all parameters, albeit non-significant at day 8 without the platform. N=10 mice from each group. Statistical analysis was conducted using one-way ANOVA post hoc Dunnett’s test against non-tg control and students t test ASOscr vs ASOsyn (*p<0.05, **p<0.01, ***p<0.005, ****p<0.0001) and student’s t test test between ASO treatments (# p<0.05, ## p<0.005, #### p<0.0001). For analysis 10 mice of 9 months of age were used from each genotype.
During the first 3 days (learning task) all experiments were performed by adding a flag to the platform so it became visible to the animals. All mice successfully learned the location of the platform, as shown by a steep decrease in the escape latency (time to reach the visible platform) during the training section of the study. No statistically significant differences were found during the training section of the study between AD and the non-tg control group.
Between days 4 and 7, the flag was removed from the platform rendering it invisible to the animals, the memory portion of the test. All AD mice (both treated with ApoB11:ASO α-syn and ApoB11:ASO-scrambled) took a significantly longer time (by an order of magnitude of 3) to find the platform at days 4 and 5 as compared to the non-tg group (Figure 7A,B). Importantly, at day 6 behavioral deficits were partially rescued and at day 7 differences between ApoB11:ASO α-syn and the non-tg group became negligible, whereas AD mice injected with ASO scrambled were consistently unable to find the platform throughout all days of trials (Figure 7A,B).
On day 8 we conducted the probe portion of the test, where the platform was removed, and we measured the number of entrances as well as time the mice spent swimming inside the zone where the platform used to be. Results corroborated that the AD animals displayed deficits, as shown by lower number of entrances in correct zone, time spent in correct zone and passes through target quadrant. Importantly, we observed that the deficits in the probe trial were partially rescued by ApoB11:ASO α-syn treatment (Figure 7C–E). In Supplementary Figure 7 we characterize the statistical analysis to measure differences between animal groups.
We observed that the group of AD mice performed slower than the non-transgenic in the training portion of the test, although this difference was not significant at day 3. We also noticed different patterns in the swimming path of the mice belonging to AD versus non-tg (Figure 7 B). In order to characterize this we plotted thigmotaxis across 4 trial days (Supplementary figure 8). We observed no difference between AD mice treated with ASOsr or ASOsyn in thigmotaxis, however non-tg animals spent significantly less time on average in thigmotaxis as compared to AD mice.
In conclusion, our experiments demonstrate that monthly treatment of 2mg/kg ApoB11:ASO α-syn ASO to AD mice starting at 6 months of age results in the reduced accumulation of Aβ, rescue of neuronal loss, less cortical inflammation and improved spatial memory.
Materials and Methods
AD Transgenic Mice
In this study we used the mouse line PSAPP (or APP/PS1 MMRRC_034832-JAX) as a model of AD (Jankowsky et al., 2001). These AD mice are double transgenic, expressing a chimeric mouse/human amyloid precursor protein (Mo/HuAPP695swe) and a mutant human presenilin 1 (PS1-dE9), both directed to CNS neurons. These mice have increased Aβ42 production at 3-months of age and develop fibrillar Aβ deposits in the cerebral cortex and hippocampus at about six months of age. In parallel, there is a substantial increase in plaque-associated astrocytes and microglia, suggesting an overall increase in neuroinflammation between the ages of six and 16 months of age. In summary, these mice display early Aβ deposition, behavioral deficits, and neurodegeneration (Campbell et al., 2015; Holcomb et al., 1998; Zhang et al., 2016; Zhang et al., 2015). All experimental studies involving animals were approved by the Institutional Animal Care and Use Committee of University of California San Diego and performed in accordance with relevant guidelines and regulations established by NIH Guide for the Care and Use of Laboratory Animals under protocol #S02221 to R. Rissman.
Preparation of peptide vectors and antisense oligonucleotides and production of conjugates
For this study we utilized ApoB11, a novel vector derived from the ApoB38 peptide that we have previously shown to effectively transport antibodies, proteases and trophic factors across the BBB. ApoB11 is a smaller peptide, which includes the LDL-receptor binding domain of ApoB38 (NH2-RLTRKRGLKLAGGGGGRRRRRRRRR) (Spencer et al., 2019). The peptide was synthesized to 90% purity by Karebay Biochem, Inc. For delivery of oligonucleotides, we added a 5 Gly (G) linker and 9 Arg (R) amino acids to the C-terminus. The glycine residues act as flexible anchor and the 9 arginine residues are a positively charged tail for binding negatively charged nucleotides. The 2’-OMe modified ASO-syn (5’GAC UUU CAA AGG CCA AGG A) corresponding to nucleotides 168–186 of α-syn was chosen because we have previously shown targets both mouse and human α-syn. As control we used an ASO-scrambled (5′GGG CAU ACU GAG CUA ACA A). The antisense oligonucleotides were purchased from Microsynth, purified by HPLC and desalted. Oligonucleotides were re-suspended in RNAse free water (Life Technologies) at 100 mM and aliquoted. The ASO’s (si-sc and siα-syn) and peptides (ApoB11) were mixed at specified ratios in PBS (1:0, 0:1, 1;1, 1:10, 1:20, 1:40, 1:60) and incubated at room temperature for 30 min to allow RNA to bind to peptide. These were then run on an agarose gel to determine the optimal ratio at which no excess of (unconjugated) ASO is detected.
Treatment of SH-SY5Y cells with ApoB11:ASO conjugates and immunofluorescence
Our experimental design relied first on testing whether ApoB11:ASO α-syn could inhibit translation of α-syn in vitro. The human cell line SH-SY5Y was utilized for such in vitro experiments. Cells were plated at 1 × 105 cells/well of a 12 well dish on poly L-lysine coated glass coverslips in DMEM +1% FBS for 5 days to allow for differentiation. We infected differentiated SH-SY5Y human neuroblastoma cells with a lentivirus vector overexpressing human α-syn (LV-α-syn) or control vector (LV-Ctrl) to test the ASO treatment under conditions of α-syn overexpression or normal levels, respectively. Cells were then treated with 100 pmol ApoB11:ASO α-syn, ApoB11:ASO-scrambled for 48 hours and and then fixed in 4% paraformaldehyde and prepared for immunocytochemistry as follows. Cells in coverslips were immunolabelled with antibodies against α-syn (Syn-1, mouse monoclonal, 1:5000, BD Biosciences), α-syn was detected with AF-488 conjugated antibody (Invitrogen). Coverslips were imaged with a Leica Confocal microscope, image analysis was done using ImageJ to determine pixel intensity. We imaged a total of 3 wells confluent with cells, collected one representative image of each well for analysis, and collected cells from 2 of these wells for biochemistry, since we were unable to retrieve cells from all 3 wells successfully.
Immunoblotting of SH-SY5Y cell lysates
The levels of α-syn in the above neuronal cells were analyzed using lysates that were extracted using RIPA (Thermofisher) with the addition of protease inhibitor cocktails (Halt™ and PMSF – Thermofisher) followed by ultracentrifugation at 30,000rpm. Protein (20 μg/lane) was loaded onto 4–12% SDS/PAGE gels and blotted onto PVDF membranes and incubated with total α-syn antibody (1: 500, affinity-purified mouse monoclonal syn1 610787, BD Biosciences) followed by secondary staining with anti-mouse-AF488 (green, Invitrogen). As loading control we blotted with primary antibody against actin (rabbit polyclonal, Catalog number A2103, Sigma-Aldrich), followed by secondary blotting with anti-rabbit-AF594 (red, Invitrogen). Bands were imaged and analyzed with a quantitative Chemidoc MP imaging apparatus (Bio-Rad).
Treatment of mice with modified ASO for detection of ASO in brain tissue
WT mice (C57BL/6) (n=3) were injected with a modified version of the ASO-αsyn sequence mentioned above, so it included a biotin tag in its 5’ end. This was done in order to inquire whether we would be able to detect the presence of ASO in the brain tissue of mice that were injected intraperitoneally. Male C57BL/6 mice 8–10 weeks received either ApoB11:ASyn ASO (2mg/kg) or PBS and 24 hours later were sacrificed. Brain sections were stained with Tyramide Red Signal Amplification Kit (NEN Life Sciences) and with Anti-NeuN (Fisher) with AF-488 secondary. All sections were mounted with Anti-Fade with DAPI (nucleus). Sections were imaged with a Leica Confocal microscope, image analysis was done using ImageJ.
Treatments administered to mice – AD versus non-tg
In this study we used AD and non-transgenic (non-tg) littermates . Intraperitoneal injections were performed as three single injections (2 mg/kg) at 6, 7 and 8 months of age, based on protocols previously described for intraperitoneal injections of siRNAs (Spencer et al., 2019). Mice received intraperitoneal injections of 2mg/kg of ApoB11:ASO α-syn/scrambled at each time point. Animals were sacrificed 4 weeks after last intraperitoneal administration of ASO conjugates. We used 10 mice per condition, i.e. 10 non-tg and 10 AD mice for each treatment (ASO-syn and ASO-scrambled). For immunohistochemistry we used samples from a total of 5 mice from each group. For water-maze experiments we used a total of 10 mice per group. Immediately after sacrificing the animals, the tissue was collected and prepared as described below.
Tissue preparation
Following NIH guidelines for the humane treatment of animals, mice were anesthetized with isoflurane and flush-perfused transcardially with 0.9% saline. Brains were removed and divided in sagittal sections. The right hemibrain was postfixed in phosphate-buffered 4% PFA, pH 7.4, at 4°C for 48 h for neuropathological analysis and the left hemibrain was snap-frozen and stored at −70°C for subsequent protein analysis.
Morris water maze testing
To evaluate the functional effects of ablating synuclein, groups of male and female AD and non-transgenic littermates (non-tg) animals were tested in the water maze, as described previously. For this purpose, a pool (diameter 180 cm) was filled with opaque water (24°C) and mice were first trained to locate a visible platform (days 1–3 – platform marked with a flag) and then a submerged hidden platform (days 4 –7) in 3 daily trials 2–3 min apart. Mice that failed to find the hidden platform within 90 s were directed to the platform and allowed to remain there for 30 s. The same platform location was used for all sessions. The starting point at which each mouse was placed into the water was changed randomly between two alternative entry points located at a similar distance from the platform. Time to reach the platform (latency) and entrances into target quadrant were recorded with a Noldus Instruments EthoVision video tracking system set to analyze two samples per second. All experimental conditions were the same for the probe test (performed at day 8) except the platform was removed altogether and the amount of time spent in the quadrant that previously contained the platform was recorded. For these experiments, mice were 9 months old (n=10 mice/ group, half male and half female).
Immunohistochemical analysis
Analysis was performed using free floating, 40μM-thick, vibratome-cut, blind-coded sections, as described previously. Briefly, sections were incubated overnight at 4°C with antibodies against total α-syn (1: 500, affinity-purified mouse monoclonal syn211, Millipore), phosphorylated α-syn (1:500, phospho S129, affinity-purified rabbit monoclonal, EP1536Y), Aβ (82E1, mouse monoclonal; BioLegend), NeuN (1:500, affinity-purified monoclonal; Millipore), GFAP (1:500, affinity purified monoclonal; Millipore), followed by biotin-tagged anti-rabbit or anti-mouse IgG1 (1:100; Vector Laboratories) secondary antibodies, avidin D-HRP (1:200, ABC Elite; Vector Laboratories). Sections were scanned with a NanoZoomer S60 Digital slide scanner C13210–01 and images were analyzed using ImageJ for quantification of optical densities and cell counts, by correcting for background staining. For each group of animals, a total of five brains from 5 different mice were imaged, and one image from each subregion was analyzed. Supplementary Figure 3 represents the areas used for quantification.
The sections stained in this study correspond to 40 μm-thick sagittal sections taken between 1.5mm and 2mm from the midline, for a total of around 40 slices per mouse. We selected 3 sections randomly and performed immunohistochemistry. For histology we used the right hemisphere. For the analysis of the subregions of the hippocampus (i.e. CA1, CA3) for P-Syn and NeuN, the granular was measured for optical density and cell counts using the corpus callosum as background subtraction to normalize among sections. For each animal, 3 different sections were measured and averaged, for a total of 5 animals in each group of samples.
Measurements were analyzed in the following areas of brain sections (the values depicted in charts Figures 3 to 6 refer to these areas: 1) for PSyn cortex 20μm2, CA1 5μm2, CA3 5μm2, DG 5μm2, striatum 20μm2, thalamus 20μm2, 2) for NeuN cortex 20μm2, CA3 5μm2, striatum 20μm2, thalamus 20μm2, 3) for GFAP cortex 20μm2, CA3 5μm2, striatum 50μm2, thalamus 50μm2, 4) 82E1 cortex 300μm2, hippocampus 150μm2. Images collected for CA1, CA3 and DG included only granular layer and background was corrected against corpus callosum. Images for cortex were also corrected against the corpus callosum with cell counts and optical density being measured in the whole image, which included layers III and IV.
Statistical analysis
All analyses were performed using GraphPad Prism (version 9.0) software. Differences among means were assessed by one-way ANOVA with Dunnett’s post hoc test when compared against a common control (AD mice injected with PBS) or using Tukey-Kramer’s or student t test test when comparing between each treatment (ASO-syn vs ASO-scr).
Discussion
We and others have proposed that gene silencing strategies can modulate expression of aggregation-prone proteins and prevent/slow AD onset (Alves et al., 2016; Malm et al., 2011). Despite evidence demonstrating that α-syn is mechanistically involved in a large number of AD cases, to date no AD clinical trials have been conducted that target α-syn. In this study we took steps for developing and validating tools for future translation. We examined how reduction of α-syn could impact AD neuropathological and behavioral phenotypes, preclinically in an AD mouse model. Secondly, to increase translatability of our work, we developed methodology that would avoid intrathecal administration and allow transport of ASO from the periphery to the brain using the first class of BBB-penetrant conjugates.
Here we demonstrate that in vivo delivery of a modified ASO to silence α-syn successfully prevents formation of Aβ plaques in AD mice. This finding supports the notion that α-syn and Aβ are interrelated in their neuropathological accumulation. This is supported by the fact that treatment with ApoB11:ASO α-syn also led to a significantly less neuronal loss in the hippocampus, a neuropathological finding that has been shown to be the main correlating factor to cognitive and memory deficits in AD (Schmitz et al., 2004).
A central finding of our study is that reduction of α-syn levels leads to reduced accumulation of Aβ plaques in the cortical regions. Importantly, these results correlate with an amelioration of glial-mediated neuroinflammation, as shown by the reduction of immunostaining against astrocytes (GFAP).
Previous studies have shown that in AD, resident glial cells undergo proinflammatory activation, resulting in an increased capacity to convert resting astrocytes to reactive astrocytes (Halliday and Stevens, 2011; Park et al., 2021). Therefore, glial cells are a major therapeutic target for AD and blocking microglia/astrocyte activation has provided promising results in limiting neurodegeneration in AD (Park et al., 2021). Recent studies supporting this idea have shown that α-syn accumulation in microglia induces selective neuronal degeneration by promoting phagocytic exhaustion, an excessively toxic environment and the selective recruitment of peripheral immune cells. This inflammatory state creates a molecular feed-forward vicious cycle between glial cells and IFNγ-secreting immune cells infiltrating the brain parenchyma (Bido et al., 2021). Our results are promising insofar as we find that targeting α-syn could represent a proxy to an anti-neuroinflammatory treatment by blocking astrocyte activation in AD mice.
Whether Aβ plaques are predominantly inciters of neuroinflammation or instead arise from it is still an important topic of debate. Astrocytes are capable of accumulating substantial amounts of neuron-derived, Aβ as a consequence of their debris-clearing role during neurodegeneration. Astrocytes eventually undergo lysis and contribute to the dispersal of Aβ, leading to the deposition of GFAP-rich, astrocytic Aβ plaques, first appearing in the molecular layer of the cerebral cortex, as we observed in our study (Bido et al., 2021; Nagele et al., 2003; Nagele et al., 2004). Microglia are then recruited to plaques. However, studies have shown that phagocytosis and clearance of Aβ fibrils by microglia could be impaired in conditions where Aβ is prone to aggregate (Pan et al., 2011). Moreover, microglia could actually spread (more than clear) Aβ pathology, as demonstrated by a recent study (d’Errico et al., 2022). Fibril formation feeds the overburdening cycle, a cycle which α-syn could exacerbate, potentially explaining our findings.
Importantly, the neuropathological findings discussed in our study are also accompanied by promising results in the learning and memory tasks in AD mice. Our results indicate that reducing α-syn in vivo rescues spatial memory deficits to non-tg levels, after day 6 of the trial. Although this amelioration is only partial, as noted by the results of probe trial at day 8 (without any platform), an improvement is observed overall. This improvement is all the more intriguing if we consider the fact that α-syn is thought to not play any pathological role in AD dementias leading to loss of spatial memory. Our study challenges this notion and introduces the concept of α-syn as an accelerator and co-adjuvant in AD pathology.
Our spatial memory tests corroborate, to an extent, data from previous authors regarding differences between the memory and learning portions of the test in AD mice vs non-transgenic littermates. In a comprehensive study by (Huang et al., 2016) aged APP/PS1 mice showed memory deficits as well as anxiety, hyperactivity, and social interaction impairment. Consistently, there was deposition of amyloid plaques in the hippocampus with decreased expression of insulin-degrading enzyme, a proteolytic enzyme responsible for degradation of intracellular Aβ. The authors also observed decreases in hippocampal volum and neuronal number, as did we. However, here we report astrocyte accumulation around plaques, whereas at 24 months astrocyte atrophy has been described. Our study suggests that 9 months old APP(swe)/PS1(dE9) mice replicate cognitive and noncognitive behavioral abnormalities, hippocampal atrophy, and neuronal and astrocyte degeneration observed before in aged mice.
In AD, some initial feasibility work on genetic therapy has already been undertaken preclinically using a glycosylated “triple-interaction” stabilized polymeric siRNA nanomedicine compound to target BACE1 and found favorable safety and efficacy profiles (Zhou et al., 2020). Also, data from recent anti-Aβ immunotherapy trials have been very encouraging (e.g Aducanumab, Lecanumab), but it is uncertain whether these FDA approved approaches will be successful in AD participants who have comorbid α-syn pathology. The development of genetic approaches to α-syn reduction such as the one described here could be tested alone and/or used in primary prevention trials in combination with Aβ immunotherapy to fill this gap.
Limitations of the study
Although we were able to observe significant neuropathological effects when treating AD mice with the ASO, our study is not conclusive as to the degree and duration of the reduction in α-syn levels. Our measurements of optical density for Phosphorylated and total α-syn point to a significantly lower number of positive cells in the granular layers of the hippocampus, yet the magnitude of such reduction and the effect in subregional layers is not provided here. A more detailed characterization of sublayers in each hippocampal subregion for example, would clarify some of these points. Similarly, biochemistry data points to a reduction of α-synuclein but this reduction is modest and would benefit from follow-up studies using mouse ELISA kits.
Although our results are encouraging, it would be important to expand on this study and include a higher number of mice in future work. We restricted our histopathological analysis to 5 female mice from each group and variability in counts or optical density was generally high. Moreover, this study did not employ stereology methods, instead we selected three brain slices per mouse obtained through vibratome-cutting a region of the brain at a distance between 1.5 and 2mm from the midline. Because of this, our data provides mostly a reference to the number of cell counts or optical density for an approximate given area. Unbiased stereology methods should be employed to reveal the extent of differences between treatments and to be able to acquire data on the full number of cells or optical density of stained proteins per volume of a specific region.
Another limitation of our study is that behavior data was limited to spatial memory tests (Morris watermaze). Other more extensive parameters such as measurements of anxiety, social memory and/or novel object recognition would better inform on specific differences between each group.
This study was a first attempt at modulating α-syn levels in AD non-invasively. As such, it does not paint a complete picture of optimal therapeutical protocols nor do we show data regarding the biodistribution and half-life or toxicity of the ASO. We empirically used a dose of 2 mg/kg given to animals at 6,7 and 8 months of age based on previous approaches of systemic delivery of siRNAs in a mouse model of synucleinopathy (Spencer et al., 2019). Different dosages and protocols of application should be employed for further optimization purposes. Although the half-life of the ASO was not measured here, previous studies have shown the half-life of siRNA in the serum to be approximately 2 h (Bartlett and Davis, 2006; Javed et al., 2016; Layzer et al., 2004) and up to 3 weeks once it is internalized by neurons (Bartlett and Davis, 2006). We expect that introduction of the 2′-O’methyl sugar modification could increase endonuclease resistance as well the half-life compared to the ribonucleotide backbone, as shown extensively in literature (Layzer et al., 2004; Stein and Castanotto, 2017). For example, 2′-F modified siRNAs have an increased serum t1/2 of approximately 24 h (Stein and Castanotto, 2017). Thus, this modification could in theory allow for significantly fewer injections, lower dosages and increased effectiveness compared to conventional siRNA strategies. Other modifications that could be considered for future studies include morpholinos, peptide nucleic acids and phosphorothioate oligonucleotides.
Conclusion:
In conclusion, we presented here a strategy to therapeutically target α-syn in AD, a protein which is traditionally associated with other neurodegenerative diseases. Neurodegeneration is increasingly regarded as a multifactorial phenomenon, where several proteins co-accumulate in the brain and lead to functional deficits. Here we show that α-syn may be detrimental in the initiation and/or progression of AD. Its reduction leads to improvement in neuropathological and behavior phenotypes and should be considered at least as a co-adjuvant gene therapy strategy in the fight against AD.
Supplementary Material
Highlights.
Systemic delivery of a α-syn antisense oligonucleotide conjugated with a peptide derived from ApoB crosses the blood brain barrier for delivery to neurons.
Delivery of ApoB11:ASO α-syn to AD mice via three monthly intraperitoneal administrations starting at 6 months of age reduces the accumulation of phosphorylated α-syn in the hippocampus.
Downregulation of α-syn in AD mice via systemic administration of a α-syn ASO prevents the accumulation of Aβ plaques and glia-mediated responses.
Downregulation of α-syn rescues spatial memory deficits in 9 months-old AD mice.
Acknowledgments
The authors thank James Barlow, Floyd Sarsoza, Jazmin Florio, Michael Mante, Estefani Guzman, Sahar Salehi, Bao Quach and Carlos Arias from the Rissman lab for technical assistance with this project. This work was supported by NIH/NIA R01 grants AG018440 and AG073979 to RAR and NINDS grant number P30NS047101 to the Microscopy Core at UCSD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Section
Neurobiology of Disease
References
- Alves S, et al. 2016. Gene therapy strategies for Alzheimer’s disease: an overview. Human Gene Therapy 27, 100–107. [DOI] [PubMed] [Google Scholar]
- Banks WA, 2016. From blood–brain barrier to blood–brain interface: new opportunities for CNS drug delivery. Nature reviews Drug discovery 15, 275–292. [DOI] [PubMed] [Google Scholar]
- Banks WA, et al. 2001. Delivery across the blood-brain barrier of antisense directed against amyloid β: reversal of learning and memory deficits in mice overexpressing amyloid precursor protein. Journal of Pharmacology and Experimental Therapeutics 297, 1113–1121. [PubMed] [Google Scholar]
- Bartlett DW, Davis ME, 2006. Insights into the kinetics of siRNA-mediated gene silencing from live-cell and live-animal bioluminescent imaging. Nucleic acids research 34, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Viaene AN, 2021. What are activated and reactive glia and what is their role in neurodegeneration? Neurobiology of Disease 148, 105172. [DOI] [PubMed] [Google Scholar]
- Biancalana M, Koide S, 2010. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochimica et Biophysica Acta (BBA)-Proteins and Proteomics 1804, 1405–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bido S, et al. 2021. Microglia-specific overexpression of α-synuclein leads to severe dopaminergic neurodegeneration by phagocytic exhaustion and oxidative toxicity. Nature communications 12, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brück D, et al. 2016. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiology of disease 85, 262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell SN, et al. 2015. Impact of CRFR1 ablation on amyloid-β production and accumulation in a mouse model of Alzheimer’s disease. Journal of Alzheimer’s Disease 45, 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinton LK, et al. 2010. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 30, 7281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colom-Cadena M, et al. 2013. Confluence of α-synuclein, tau, and β-amyloid pathologies in dementia with Lewy bodies. Journal of Neuropathology & Experimental Neurology 72, 1203–1212. [DOI] [PubMed] [Google Scholar]
- Crunkhorn S, 2017. Antisense oligonucleotide reverses tau pathology. Nature Reviews Drug Discovery 16, 166–166. [DOI] [PubMed] [Google Scholar]
- d’Errico P, et al. 2022. Microglia contribute to the propagation of Aβ into unaffected brain tissue. Nature Neuroscience 25, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Hyman BT, 2017. Tau at the Crossroads between Neurotoxicity and Neuroprotection. Neuron 94, 703–704. [DOI] [PubMed] [Google Scholar]
- Dickey AS, La Spada AR, 2018. Therapy development in Huntington disease: from current strategies to emerging opportunities. American Journal of Medical Genetics Part A 176, 842–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferman TJ, et al. 2018. The limbic and neocortical contribution of α-synuclein, tau, and amyloid β to disease duration in dementia with Lewy bodies. Alzheimer’s & Dementia 14, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funato H, et al. 1998. Astrocytes containing amyloid beta-protein (Abeta)-positive granules are associated with Abeta40-positive diffuse plaques in the aged human brain. The American journal of pathology 152, 983. [PMC free article] [PubMed] [Google Scholar]
- Gleichman AJ, Carmichael ST, 2020. Glia in neurodegeneration: Drivers of disease or along for the ride? Neurobiology of Disease 142, 104957. [DOI] [PubMed] [Google Scholar]
- Halliday GM, Stevens CH, 2011. Glia: initiators and progressors of pathology in Parkinson’s disease. Movement Disorders 26, 6–17. [DOI] [PubMed] [Google Scholar]
- Hamilton RL, 2000. Lewy bodies in Alzheimer’s disease: a neuropathological review of 145 cases using α‐synuclein immunohistochemistry. Brain pathology 10, 378–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, et al. 2021. The amyloid-β pathway in Alzheimer’s disease. Molecular Psychiatry 26, 5481–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, et al. 1998. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nature medicine 4, 97–100. [DOI] [PubMed] [Google Scholar]
- Huang H, et al. 2016. Characterization of AD-like phenotype in aged APPSwe/PS1dE9 mice. Age 38, 303–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, et al. 2001. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomolecular engineering 17, 157–165. [DOI] [PubMed] [Google Scholar]
- Javed H, et al. 2016. Development of nonviral vectors targeting the brain as a therapeutic approach for Parkinson’s disease and other brain disorders. Molecular Therapy 24, 746–758. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Jellinger KA, 2011. Interaction between ?-Synuclein and Other Proteins in Neurodegenerative Disorders. TheScientificWorldJournal 11, 1893–1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jose JC, et al. 2014. Cross dimerization of amyloid-β and αSynuclein proteins in aqueous environment: a molecular dynamics simulations study. PloS one 9, e106883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmierczak A, et al. 2008. α-Synuclein enhances secretion and toxicity of amyloid beta peptides in PC12 cells. Neurochemistry international 53, 263–269. [DOI] [PubMed] [Google Scholar]
- Keiser MS, et al. 2016. Gene suppression strategies for dominantly inherited neurodegenerative diseases: lessons from Huntington’s disease and spinocerebellar ataxia. Human molecular genetics 25, R53–R64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lajoie JM, Shusta EV, 2015. Targeting receptor-mediated transport for delivery of biologics across the blood-brain barrier. Annual review of pharmacology and toxicology 55, 613–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashley T, et al. 2008. Cortical α-synuclein load is associated with amyloid-β plaque burden in a subset of Parkinson’s disease patients. Acta neuropathologica 115, 417–425. [DOI] [PubMed] [Google Scholar]
- Layzer JM, et al. 2004. In vivo activity of nuclease-resistant siRNAs. Rna 10, 766–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M-R, et al. 2016. Phosphorylation induces distinct alpha-synuclein strain formation. Scientific reports 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malm T, et al. 2011. Utilization of APPswe/PS1dE9 transgenic mice in research of Alzheimer’s disease: focus on gene therapy and cell-based therapy applications. International journal of Alzheimer’s disease 2011. [DOI] [PMC free article] [PubMed]
- Mandal PK, et al. 2006. Interaction between Aβ peptide and α synuclein: molecular mechanisms in overlapping pathology of Alzheimer’s and Parkinson’s in dementia with Lewy body disease. Neurochemical research 31, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Matej R, et al. 2019. Alzheimer’s disease and other neurodegenerative dementias in comorbidity: a clinical and neuropathological overview. Clinical biochemistry 73, 26–31. [DOI] [PubMed] [Google Scholar]
- McCampbell A, et al. 2018. Antisense oligonucleotides extend survival and reverse decrement in muscle response in ALS models. The Journal of clinical investigation 128, 3558–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TM, et al. 2013. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. The Lancet Neurology 12, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, Griffin WST, 2005. Glia and their cytokines in progression of neurodegeneration. Neurobiology of aging 26, 349–354. [DOI] [PubMed] [Google Scholar]
- Nagele RG, et al. 2003. Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain research 971, 197–209. [DOI] [PubMed] [Google Scholar]
- Nagele RG, et al. 2004. Contribution of glial cells to the development of amyloid plaques in Alzheimer’s disease. Neurobiology of aging 25, 663–674. [DOI] [PubMed] [Google Scholar]
- Palmqvist S, et al. 2017. Earliest accumulation of β-amyloid occurs within the default-mode network and concurrently affects brain connectivity. Nature communications 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X. d., et al. 2011. Microglial phagocytosis induced by fibrillar β-amyloid is attenuated by oligomeric β-amyloid: implications for Alzheimer’s disease. Molecular neurodegeneration 6, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J-S, et al. 2021. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta neuropathologica communications 9, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovici GD, et al. 2017. Multiple comorbid neuropathologies in the setting of Alzheimer’s disease neuropathology and implications for drug development. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 3, 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiserer R, et al. 2007. Impaired spatial learning in the APPSwe+ PSEN1ΔE9 bigenic mouse model of Alzheimer’s disease. Genes, Brain and Behavior 6, 54–65. [DOI] [PubMed] [Google Scholar]
- Roberts HL, et al. 2017. α-Synuclein increases β-amyloid secretion by promoting β-/γ-secretase processing of APP. PloS one 12, e0171925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M, et al. 2004. Amyloid-β deposition is associated with decreased hippocampal glucose metabolism and spatial memory impairment in APP/PS1 mice. Journal of neuropathology and experimental neurology 63, 418–428. [DOI] [PubMed] [Google Scholar]
- Schmitz C, et al. 2004. Hippocampal neuron loss exceeds amyloid plaque load in a transgenic mouse model of Alzheimer’s disease. The American journal of pathology 164, 1495–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, et al. 2016a. Reducing endogenous α-synuclein mitigates the degeneration of selective neuronal populations in an Alzheimer’s disease transgenic mouse model. Journal of Neuroscience 36, 7971–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, et al. 2014. ESCRT-mediated uptake and degradation of brain-targeted α-synuclein single chain antibody attenuates neuronal degeneration in vivo. Molecular Therapy 22, 1753–1767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Spencer B, et al. 2016b. α-Synuclein interferes with the ESCRT-III complex contributing to the pathogenesis of Lewy body disease. Human molecular genetics 25, 1100–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer B, et al. 2019. Systemic peptide mediated delivery of an siRNA targeting α-syn in the CNS ameliorates the neurodegenerative process in a transgenic model of Lewy body disease. Neurobiology of disease 127, 163–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer BJ, Verma IM, 2007. Targeted delivery of proteins across the blood–brain barrier. Proceedings of the National Academy of Sciences 104, 7594–7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spina S, et al. 2021. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain 144, 2186–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein CA, Castanotto D, 2017. FDA-approved oligonucleotide therapies in 2017. Molecular Therapy 25, 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twohig D, Nielsen HM, 2019. α-synuclein in the pathophysiology of Alzheimer’s disease. Molecular Neurodegeneration 14, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler BM, et al. 1999. Peptide nucleic acids targeted to the neurotensin receptor and administered ip cross the blood–brain barrier and specifically reduce gene expression. Proceedings of the National Academy of Sciences 96, 7053–7058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visanji NP, et al. 2019. Beyond the synucleinopathies: alpha synuclein as a driving force in neurodegenerative comorbidities. Translational neurodegeneration 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, et al. 2010. Tau reduction prevents Aβ-induced defects in axonal transport. Science 330, 198–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. 2016. Corticotropin-releasing factor receptor-1 antagonism mitigates beta amyloid pathology and cognitive and synaptic deficits in a mouse model of Alzheimer’s disease. Alzheimer’s & Dementia 12, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, et al. 2015. Corticotropin-Releasing factor Receptor-1 antagonism reduces oxidative damage in an Alzheimer’s disease transgenic mouse model. Journal of Alzheimer’s Disease 45, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, et al. 2020. Blood-brain barrier–penetrating siRNA nanomedicine for Alzheimer’s disease therapy. Science advances 6, eabc7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.