

Abstract
Objective:
Calcific aortic valve disease (CAVD) is a progressive condition that shares some common pathogenic features with atherosclerosis. Transforming growth factor (TGF)-β1 is a recognised mediator of atherosclerosis and is expressed in aortic valve lesions. TGF-β1 stimulates glycosaminoglycan (GAG) elongation of proteoglycans that is associated with increased lipid binding. We investigated the presence of TGF-β1 and downstream signaling intermediates in diseased human aortic valves and the effects of activated TGF-β1 receptor signaling on aortic valve interstitial cell (VIC) proteoglycan synthesis and lipid binding as a possible mechanism for initiation of the early lesion of CAVD.
Methods and Results:
Diseased human aortic valve leaflets demonstrated strong immunohistochemical staining for TGF-β1 and phosphorylated Smad2/3. In primary porcine aortic VICs Western blots showed TGF-β1 stimulated phosphorylation in both the carboxy and linker regions of Smad2/3 which was inhibited by the TGF-β1 receptor inhibitor SB431542. Gel electrophoresis and size exclusion chromatography demonstrated that SB431542 decreased TGF-β1-mediated [35S]-sulfate incorporation into proteoglycans in a dose-dependent manner. Further, in proteoglycans derived from TGF-β1 treated VICs, gel mobility shift assays demonstrated that inhibition of TGF-β1 receptor signaling resulted in decreased lipid binding.
Conclusions:
Classic TGF-β1 signaling is present in human aortic valves in vivo, and contributes to the modification of proteoglycans expressed by VICs in vitro. These findings suggest that TGF-β1 may promote increased LDL binding in the early phases of CAVD.
Keywords: heart valves, Smad, transforming growth factor-β, lipids, glycosaminoglycans
Calcific aortic valve disease (CAVD), which includes aortic sclerosis and stenosis, is a progressive condition associated with substantial morbidity and mortality 1-2. The prevalence of aortic sclerosis increases with age, 20% in patients 65-75 years and 48% in patients older than 85 years 3. Advanced CAVD is the second most common indication for cardiac surgery in the Western world 4. Originally considered a degenerative disease CAVD is now recognized as the result of active pathological processes.5-6 There are indications that CAVD shares pathogenic features with atherosclerosis 5, 7-10. Risk factors including age, male gender, hypertension, hyperlipidemia and active inflammation have been associated with both CAVD and atherosclerosis 2, 11-14 however the disease processes are not identical. Laboratory and clinical studies have suggested that the progression of aortic stenosis might be retarded by the use of statins 15-18 and ACE inhibitors 19-22. However the results of controlled randomized intervention studies with statins in late-stage CAVD (i.e. aortic stenosis) have been negative 23-25. With the unmet clinical need for effective CAVD treatments it is recognized that the pathological consequences of early aortic valve changes in CAVD need to be elucidated at a molecular level to identify suitable new targets and develop rational therapeutic interventions.5-6, 26.
A hallmark of early CAVD is the structural and compositional change in the aortic valve extracellular matrix. Early valve thickening is characterized by disruption of the trilayer structure with disorganised collagen deposition in the fibrosa 27, accumulation of proteoglycans versican, biglycan and decorin 28-29, lipoproteins LDL, Lp(a), and ApoE as well as calcium 30 and inflammatory cell infiltrate 7. Recently it has been highlighted that despite the abundance of proteoglycans and glycosaminoglycans in sclerotic and stenotic aortic valves their role in CAVD is almost completely unexplored 31. Transforming growth factor (TGF)-β1 is a recognised mediator of atherosclerosis and is also expressed in aortic valve lesions 32. However the role of TGF-β1 in the initiation and progression of CAVD is not clear with context-dependent effects of TGF-β1 described with in vitro and in vivo models of CAVD. In atherosclerosis, TGF-β1 leads to the activation of proteoglycan synthesis in vascular smooth muscle cells and induces elongation of the glycosaminoglycan (GAG) chains 33-34. Elongated GAG chains are capable of increased binding of lipid 34-35 and this interaction underlies the “response to retention” hypothesis of Williams and Tabas regarding the origin of atherosclerosis 36. In this study we investigated TβR signaling in diseased human aortic valves and the effects of TGF-β1 on porcine aortic valve interstitial cell proteoglycan synthesis and lipid binding to determine whether the proteoglycans produced by aortic valve interstitial cells could have a role in the initiation of the early sclerotic stage of CAVD.
Methods
Immunohistochemical analysis
Valve leaflets were fixed in 4% paraformaldehyde in 0.1M phosphate buffer overnight prior to processing. Radial cross-sections from intact leaflets were cut from annulus to free edge, embedded in paraffin, and cryosectioned in 5 - 10 micron thick sections according to routine procedures. Immunohistochemistry was performed to evaluate distribution and expression of extracellular matrix components. Primary antibodies were to TGF-β1 (R&D Systems, 1:20), and phosphorylated Smad2/3 (Chemicon, 1:1000). A universal streptavidin/biotin (Vector Laboratories, Burlingame, CA) and diaminobenzidine detection system (DAKO, Denmark) was used for colorimetric detection. Negative controls for all markers were performed in the absence of primary antibodies. Sections were counterstained with hematoxylin and images recorded using a Zeiss microscope with a Zeiss Axiocam digital camera (Germany). A total of seven tricuspid aortic valves were studied. Stenotic valves (three female 74, 75 and 82 years and two male 62 and 76 years) were obtained at valve replacement surgery and control valves (female 54 and male 73 years) were collected from hearts undergoing non-valve disease-related heart surgery as approved by the Alfred Hospital Ethics Committee (Prahran, Australia).
Isolation of aortic valve interstitial cells
Aortic valve interstitial cells (VICs) were isolated from porcine aortic valves collected from three pig hearts according to previously published methods 37. Briefly, valve leaflets were dissected from the aortic root and rinsed in sterile PBS. To assist with removal of endothelial cells, partial digestion of the leaflets was performed with collagenase type II (2 mg/mL, Sigma-Aldrich, USA) in serum free DMEM (Invitrogen) for 20 minutes at 37°C in an incubated shaker. Leaflet surfaces were then wiped with sterile swabs, cut into 1 mm pieces and placed into collagenase type III (2 mg/mL in DMEM) for 2-3 hours at 37°C with shaking. The digest was strained through a 100 micron filter and cells were pelleted. Cells were then dispersed in fresh medium (DMEM containing 5 mmol/L glucose, 10% FBS, 1% antibiotics), and plated in 75 cm2 flasks. For experimentation, cells between passages one to four were used. Confluent cultures were serum-deprived by incubation in DMEM (5 mmol/L glucose, 0.1% FBS) for 48 hours prior to treatment.
Analysis of proteoglycans and glycosaminoglycans
Quiescent cells were treated in DMEM containing 5 mmol/L glucose, 0.1% FBS, 0.1% DMSO with SB431542 (0-10 μmol/L) an inhibitor of ALK5 or SIS3 (0.1-3 μM) an inhibitor of Smad3 phosphorylation and exposed to [35S]sulfate (1.85 MBq/mL) or [3H]glucosamine (0.37 MBq/mL) under basal conditions or in the presence of TGF-β1 (0-5 ng/mL) for 24 hours, unless otherwise stated. To synthesize xyloside-initiated GAG chains (an independent measure of GAG synthesis) treatment media was supplemented with methyl β-D-xylopyranoside (0.5 mmol/l, Sigma). Secreted proteoglycans were isolated and concentrated from the conditioned medium as described previously34. Radiolabel incorporation into proteoglycans was quantified using the CPC precipitation assay 38. The sizes of the proteoglycans and GAG chains (cleaved chemically from the proteoglycan core proteins via a β-elimination reaction 39) were analysed by gradient SDS-PAGE 38. GAG chain lengths also were analysed by size exclusion chromatography as described previously 40 and the data standardised by calculation of Kav values.
Western blotting
Total cell lysates from VIC cultures treated with TGF-β1 (2 ng/ml) and/or SB431542 (3 μM) for 24 hours were resolved on 10% SDS-PAGE and transferred onto PVDF membranes. Membranes were blocked with 5% skim milk powder for one hour at room temperature. Membranes were probed with primary antibodies as listed overnight at 4°C and then incubated with species specific secondary HRP-IgG (1:5000, 1 hour, RT) followed by enhanced chemiluminescent detection of proteins (ECL, Amersham). To assess protein loading the membrane was reprobed with anti-smooth muscle α-actin mouse monoclonal antibody (Dako, 1:1000, 1 hour, RT) or anti-GAPDH rabbit monoclonal antibody (Cell Signaling, USA) followed by incubation with anti-mouse IgG (1:5000, 1 hour, RT) or anti-rabbit IgG (1:5000, 1 hour, RT) respectively. Antibodies to phosphorylated-Smad2,-Smad2C and –Smad2L were obtained from Cell Signaling (USA).
Gel mobility shift assay
Quiescent VICs were treated with SB431542 (3 μmol/L) and TGF-β1 (2 ng/mL) for 24 hours in the presence of [35S]-Met/Cys (1.85 MBq/mL) to radiolabel the proteoglycan core proteins. Increasing concentrations of LDL (0-0.5 mg/mL) purified by ultracentrifugation from human blood41 were incubated with equal counts (1250 cpm) of proteoglycans. Bound and free proteoglycans were separated on flat bed agarose gels as described previously.40 Dried agarose gels were exposed to phosphorscreens and calculation of bound proteoglycans was performed using MacBas software (v1, Fuji Photo Film Co., Japan).
Data analyses
Western blots were in triplicate and density analysis performed using Image Lab (BioRad). All data for proteoglycan experiments were collected in triplicate from at least two separate experiments. Data were calculated as means and standard errors and were compared using one-way ANOVA, two-way ANOVA, or Student’s t-tests as indicated, with significance accepted at p<0.05.
RESULTS
Expression of TGF-β1 and phosphorylated Smad2/3 in human calcified and non-calcified aortic valve leaflets
Immunohistochemical staining was used to determine whether the TβR - Smad2/3 pathway is activated in CAVD. Human calcified and normal non-calcified aortic valve leaflets were examined for expression of the TβR ligand, TGF-β1 and downstream signaling intermediate phosphorylated Smad2/3 (pSmad2/3) and representative sections are shown (Figure 1). TGF-β1 staining was seen throughout the markedly thickened layers of diseased valves and the strongest expression was found in close proximity to calcific nodules (Figure 1B). The calcific remnants were stained dark purple by haematoxylin. A similar distribution of pSmad2/3 staining was visible in CAVD valves and prominent nuclear staining was also evident (Figure 1D). In contrast non-diseased valve leaflets had much lower levels of TGF-β1 (Figure 1A) and less intense pSmad2/3 staining with less prominent nuclear staining (Figure 1C). Thus in the calcific stages of CAVD we observed staining that indicates an activated TβR - Smad2/3 pathway in the valves.
Figure 1. Expression of TGF-β and phosphorylated Smad2/3 is increased in CAVD.
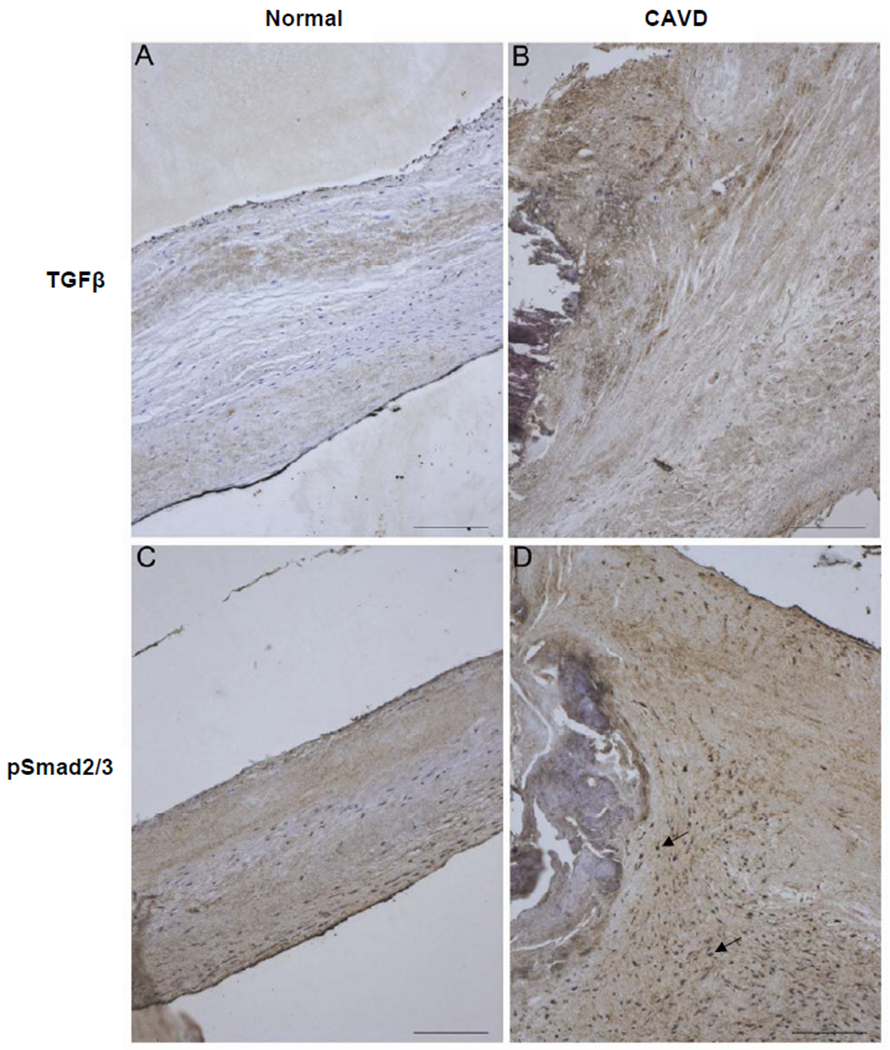
Immunohistochemistry on paraformaldehyde-fixed, paraffin-embedded human aortic valve leaflets, non-diseased (Normal) or late-stage CAVD (CAVD), was performed using antibodies against TGF-β (A and B) and phosphorylated Smad2/3 (C and D). Arrows indicate nuclear staining. Scale bars 100 μm.
TGF-β1 mediates phosphorylation of Smad2/3 in aortic valve interstitial cells
As it remains technically difficult to demonstrate the activity of the TβR - Smad2/3 pathway in situ at the initiation stages of CAVD due to the lack of availability of suitable valves we instead utilised isolated porcine aortic valve interstitial cells (VICs). Western blots demonstrated VICs expressed smooth muscle α-actin to the same extent under basal and TGFβ-stimulated conditions (Figure 2A) demonstrating an activated myofibroblast-like phenotype that is associated with aortic valve pathology and remodelling 42-43. After 4 hours stimulation with TGF-β1 (Figure 2A) VICs showed a nine-fold increase in serine phosphorylation of Smad2/3 at the carboxy terminus (pSmad2/3C) above basal and a five-fold increase in serine phosphorylation of the linker region of Smad2 (pSmad2L). Inhibition of TβR1 serine kinase activity with its highly specific inhibitor SB431542 (3 μM) reduced phosphorylation of Smad2/3 back to basal levels (Figure 2A). Total Smad2/3 expression levels remained constant under all treatments. Clearly in VICs TβR1 can be activated to phosphorylate multiple serine residues in different regions of Smad2/3.
Figure 2. TGF-β activates carboxy and linker region phosphorylation of Smad2/3 in VICs.
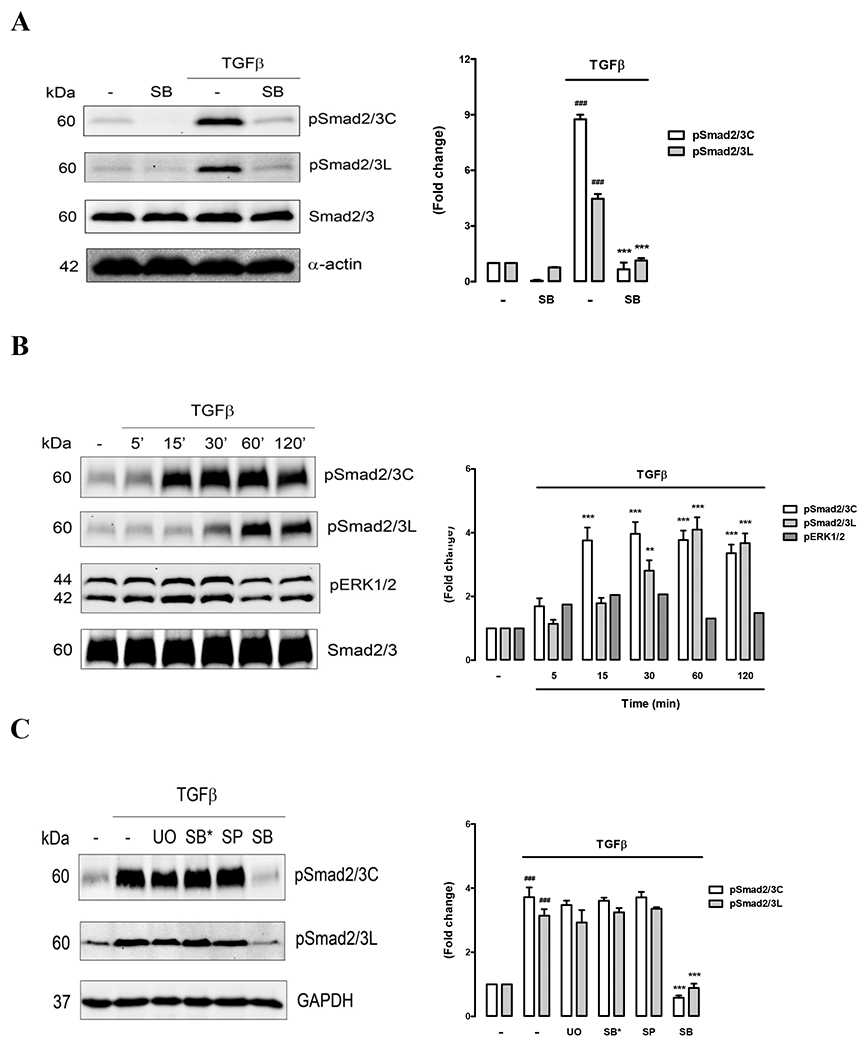
(A) Cells were pre-treated with medium (−) or the TβR1 inhibitor SB431542 (SB, 3 μM) for 30 min prior to the addition of TGF-β (2 ng/ml) for 4h. Western blots of whole cell lysates were probed for carboxy phosphorylated Smad2/3 (pSmad2/3C) or linker region phosphorylated Smad2/3 (pSmad2/3L) and reprobed for total Smad2 and smooth muscle α-actin. Density analysis of phospho-blots is plotted in the adjacent histogram. (B) Time-course of TGF-β-mediated Smad2/3 phosphorylation. Cells were treated with TGF-β (0 – 120 min) and Western blots of whole cell lysates probed for pSmad2/3C, pSmad2/3L and phosphorylated Erk1/2 (pErk1/2). (C) MAP kinase inhibition does not block Smad2/3 phosphorylation in VICs. Cells were pre-treated with either medium (−), Erk1/2 blocker U0126 (UO, 3 μM), p38 inhibitor SB202190 (SB*, 3 μM), Jnk inhibitor SP600125 (SP, 1 μM) or TβR1 inhibitor SB431542 (SB, 3 μM) for 30 min prior to the addition of TGF-β (2 ng/ml) for 4h. Western blots of whole cell lysates were probed for pSmad2/3C, pSmad2/3L and GAPDH as a loading control. ### P<0.001 versus control, *** P<0.001 versus TGF and ** P<0.01 versus TGF using a 1-way ANOVA.
We next performed an early time-course study of TβR1-mediated Smad2/3 phosphorylation. VICs were treated with TGF-β1 for 5, 15, 30, 60 and 120 minutes and Western blots of whole cell lysates probed for pSmad2/3C and pSmad2L (Figure 2B). Carboxy terminal phosphorylation was significantly above basal within 15 minutes (p<0.001) and was sustained up to 120 minutes. Linker region phosphorylation was slower peaking at 60 minutes. Smad2/3 linker region phosphorylation in some contexts is known to be a consequence of TβR1-mediated activation of the MAP kinase Erk1/2. The blots were reprobed to look for activation of Erk1/2 in VICs and we found a two-fold increase in pErk1/2 within 15 - 30 minutes of TGF-β1 stimulation (Figure 2B) however this did not reach statistical significance.
To clarify the extent of MAP kinase involvement in Smad2/3 linker phosphorylation we pre-incubated VICs with each of the specific MAP kinase inhibitors UO126 (MEK1/2 inhibitor that blocks Erk1/2), SP600125 (Jnk inhibitor) and SB202190 (p38 inhibitor) and then stimulated with TGF-β1 for 4 hours to ensure sustained Smad2/3 activation (Figure 2C). None of the MAP kinase inhibitors blocked carboxy or linker region phosphorylation of Smad2/3 significantly while TβR1 inhibitor SB431542 completely blocked phosphorylation as expected. These results suggest that in VICs at 4h post- TGF-β1 treatment Smad2/3 linker phosphorylation is not dependent on MAP kinase activation. However further studies would be useful to clarify the role of MAP kinases in Smad2/3 linker phosphorylation at 30 -60 minutes following TβR1 activation.
Antagonism of TβR1 inhibits TGF-β1 mediated proteoglycan synthesis and GAG elongation
In previous studies we and others have shown that activation of the TβR - Smad2/3 pathway increases proteoglycan synthesis in vascular smooth muscle cells and induces elongation of the glycosaminoglycan (GAG) chains and consequently greater lipid binding capacity33-34. From our findings of TβR - Smad2/3 pathway activation in VICs we hypothesized that a similar upregulation of proteoglycan synthesis and GAG elongation may occur in these cells and potentially contribute to the thickening of valve cusps in CAVD.
To evaluate the involvement of the classic TGF-β1-mediated cell signalling pathway in proteoglycan synthesis VICs were stimulated with TGF-β1 (2 ng/mL) in the presence of SB431542 (0 - 10 μmol/L). SB431542 decreased TGF-β1-mediated [35S]-sulfate incorporation into proteoglycans in a dose-dependent manner, with data normalised to 0% and 100% inhibition at 0 μmol/L and 0.03-10 μmol/L SB431542, respectively (Figure 3A). The IC50 for SB431542 was approximately 0.7 μmol/L. At 3 μmol/L SB431542, TGF-β1-mediated [35S]-sulfate incorporation was inhibited by 77%, with no observed decrease in cell number. This concentration of SB431542 was chosen for subsequent experiments. SB431542 treatment resulted in a dose-dependent increase in electrophoretic mobility of complete proteoglycans compared to TGF-β1 treatment alone, indicating a decrease in proteoglycan size (Figure 3B). An alternate radiolabel, [3H]-glucosamine was employed to confirm these findings. Under basal conditions, SB431542 decreased [3H]-glucosamine incorporation by approximately 20%, (Figure 3C). TGF-β1 stimulated an 87% increase in [3H]-glucosamine incorporation, p<0.01, which was 100% inhibited in the presence of SB431542, p<0.01 (Figure 3C). TGF-β1 stimulated a 12% increase in proteoglycan core protein synthesis compared to control (p<0.01), which was decreased by 19% in the presence of SB431542 (data not shown). To determine whether both Smad2 and Smad3 are involved in TGF-β1-mediated proteoglycan synthesis we employed SIS3, a compound which inhibits TGF-β1-mediated phosphorylation of Smad3. In VICs TGF-β1 stimulated a 70% increase in [35S]-sulfate incorporation into proteoglycans that was not significantly affected by the presence of 0.1-3 μM SIS3 (Figure 3D). These data indicate that in this context phosphorylation of Smad2 rather than Smad3 mediates TGF-β1-induced proteoglycan synthesis.
Figure 3. TβR1 mediates glycosaminoglycan elongation of VIC secreted proteoglycans.
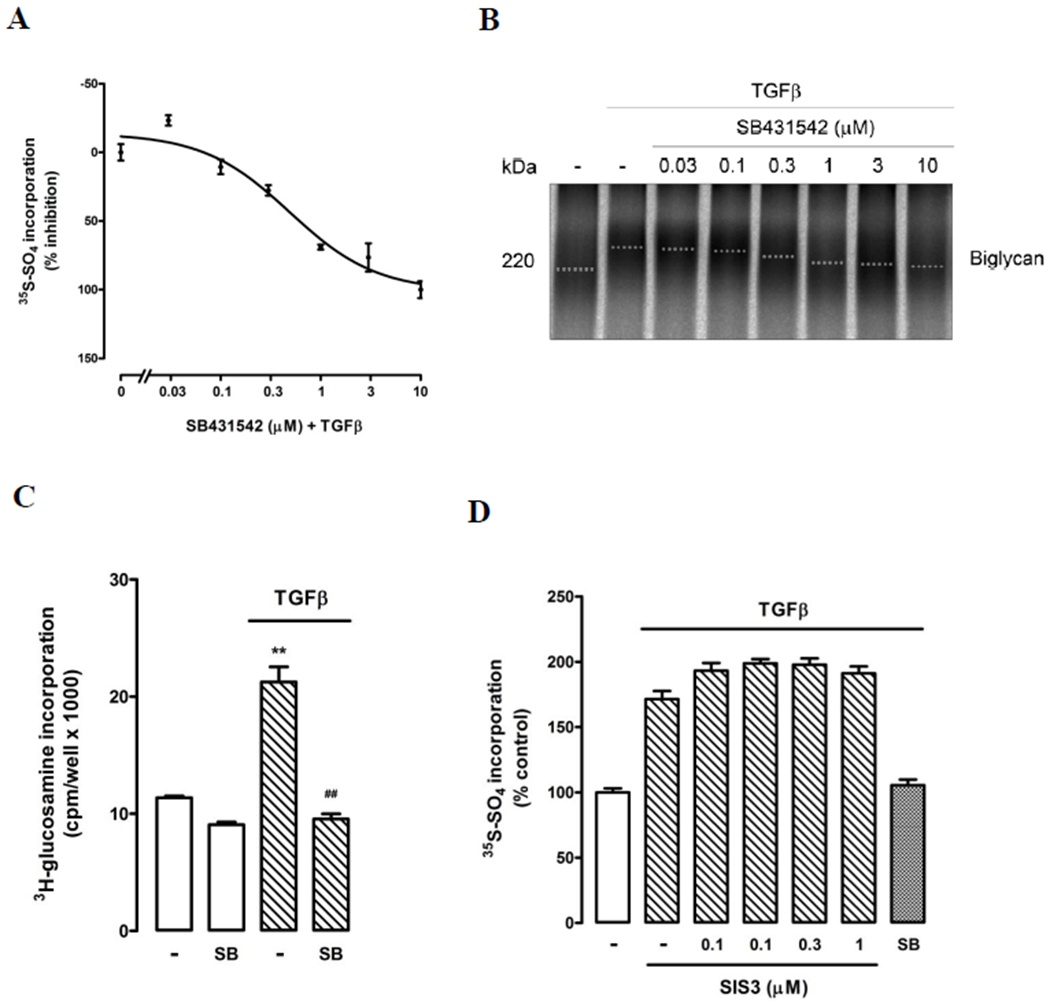
(A) Inhibition of TβR1 by SB431542 dose-dependently decreases TGF-β mediated [35S]-sulfate incorporation into proteoglycans secreted by VICs. Data was normalised to 0% and 100% inhibition at 0 μM and 10 μM SB431542, respectively. Normalised data are shown as mean ± SEM. (B). SB431542 treatment (0 – 10 μM) resulted in a concentration dependent increase in electrophoretic mobility of secreted proteoglycans compared to TGF-β (2 ng/ml) treatment alone (−), indicating a decrease in proteoglycan size. A representative gradient SDS-PAGE (4-13%) is shown, the dotted line indicates estimated mid-line of biglycan band size. (C) Confirmation of TGF-β and SB431542 effects seen in (A) using alternate radiolabel [3H]-glucosamine. Normalised data in each case are shown as mean ± SEM from at least two experiments in triplicate, **p<0.01 vs control and ##p<0.01 vs TGF-β using a 1-way ANOVA. (D) Specific inhibition of Smad3 phosphorylation using chemical inhibitor SIS3 (0-3 μM) does not block TGF-β mediated [35S]-sulfate incorporation into proteoglycans secreted by VICs.
To analyse whether the observed proteoglycan size changes were due to changes in GAG chain length, size exclusion chromatography was employed. Initially, GAG chains were chemically cleaved from proteoglycan core proteins via a β-elimination reaction and both complete proteoglycans and chemically cleaved GAG chains were separated by SDS-PAGE (Figure 4A). Under basal conditions, SB431542 treatment of VICs had negligible effect on the electrophoretic mobilities of either complete proteoglycans or cleaved GAG chains compared to control (Figure 4A). TGF-β1 treatment resulted in a decrease in electrophoretic mobility of both complete proteoglycans and cleaved GAG chains relative to control, which was attenuated in both cases in the presence of SB431542 (Figure 4A). Analysis of cleaved GAG chain length by size exclusion chromatography confirmed that the increase in GAG size demonstrated by SDS-PAGE was due to increased chain length. One representative analysis is shown in Figure 4B with the vertical line indicating mean Kav of control. Under basal conditions, cleaved GAG chains derived from SB431542 treated VICs had a mean Kav of 0.42±0.01 compared to 0.41±0.01 for control (Table 1). In the presence of TGF-β1, SB431542 treatment resulted in GAG chains with a mean Kav of 0.41±0.01 compared to 0.36±0.01 for TGF-β1 treatment alone, p<0.02 (Table 1) indicating a stimulation of GAG elongation in the presence of TGF-β1 that was inhibited by SB431542.
Figure 4. Size analysis of proteoglycans produced in VICs treated with TGF-β.
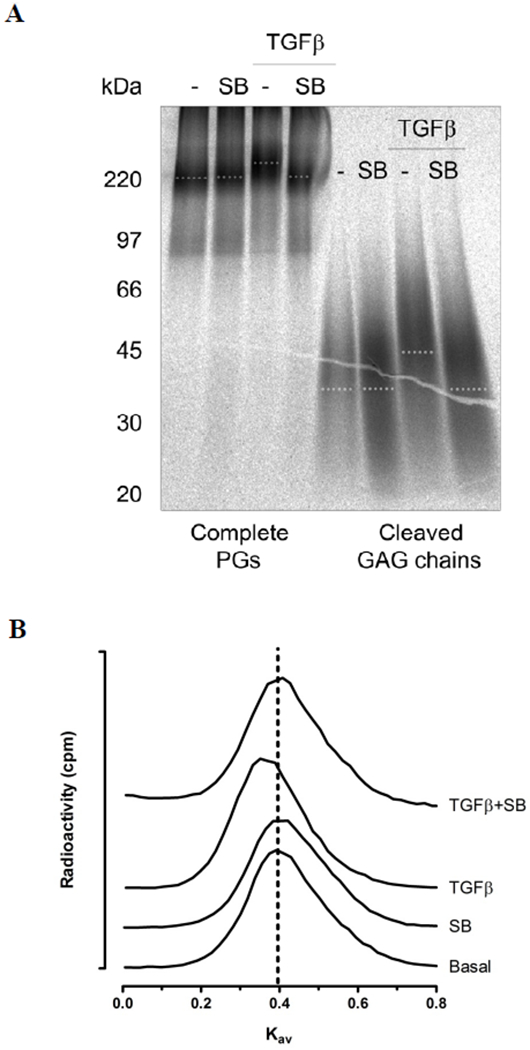
(A) VICs were pre-treated with medium (−) or SB431542 (SB, 3 μM) for 30 min prior to the addition of TGF-β (2 ng/ml) for 24h in the presence of [35S]-sulfate to metabolically label proteoglycans. Secreted proteoglycans were isolated and purified by Sepharose anion exchange chromatography and subjected to SDS-PAGE (Complete PGs). Replicate samples were treated with sodium cyanoborohydride and alkali to cleave the GAG chains from the proteoglycan core protein. Free GAG chains were also analysed by SDS-PAGE (Cleaved GAG chains). A representative gradient gel (4-20%) is shown. The dotted line indicates estimated mid-line of biglycan band size for complete PGs and mid-line of cleaved GAG chain sizes. (B) Size exclusion chromatography (Sepharose CL-6B) of radiolabeled cleaved GAG chains isolated from secreted proteoglycans from treatments given in (A) medium (Con), SB431542 (SB), TGF-β (TGF), SB431542 and TGF-β (SB + TGF). Vertical dotted line indicates the calculated apparent Kav of control GAG chains. Three measurements were performed from three separate experiments with a representative shown. Note that TGF-β shifts the peak to the left, indicating larger size.
Table 1.
Mean Kav values for GAG chains measured by size exclusion chromatography
| Control | SB | TGF-β | SB + TGF-β | |
|---|---|---|---|---|
| Cleaved chains | ||||
| Kav | 0.41±0.01 | 0.42±0.01 | 0.36±0.01# | 0.41±0.01* |
| Xyloside GAGs | ||||
| Kav | 0.53±0.01 | 0.52±0.01 | 0.48±0.01θ | 0.54±0.01κ |
SB SB154132, Kav values are expressed as mean ± SEM from three separate size exclusion experiments,
p<0.05 vs control,
p<0.02 vs TGF-β,
p<0.02 vs control,
p<0.05 vs TGF-β using a paired Student’s t-test.
Synthesis of GAG chains on a xyloside primer is a technique that allows evaluation of GAG synthesis independent of the proteoglycan core protein and was used to confirm TGF-β1-mediated effects on GAG chains. VICs were treated with SB431542 under basal conditions and in the presence of TGF-β1 in culture media supplemented with β-xyloside. TGF-β1 treatment increased [35S]-sulfate incorporation by 136%, p<0.01 (Figure 5A). SB431542 treatment had little effect under basal conditions but inhibited the TGF-β1-mediated increase in [35S]-sulfate incorporation by 88%, p<0.01 (Figure 5A). Xyloside-initiated GAG chains were separated by SDS-PAGE (Figure 4B). TGF-β1 treatment resulted in xyloside initiated GAG chains with decreased electrophoretic mobility, indicative of an increase in GAG size (Figure 5B). SB431542 inhibited this TGF-β1-mediated change in electrophoretic mobility. By size exclusion chromatography, TGF-β1 treatment decreased the mean Kav of xyloside-initiated GAG chains from 0.53±0.01 for control to 0.48±0.01, p<0.02, (Table 1, Figure 5C) indicating an increase in GAG chain size. SB431542 treatment under basal conditions and in the presence of TGF-β1 resulted in mean Kav values of 0.52±0.01 and 0.54±0.01 (p<0.05 vs TGF-β1), respectively (Table 1), indicating that SB431542 inhibited TGF-β1-mediated increases in xyloside-initiated GAG elongation.
Figure 5. TGF-β increases the size of GAG chains initiated on xyloside by VICs.
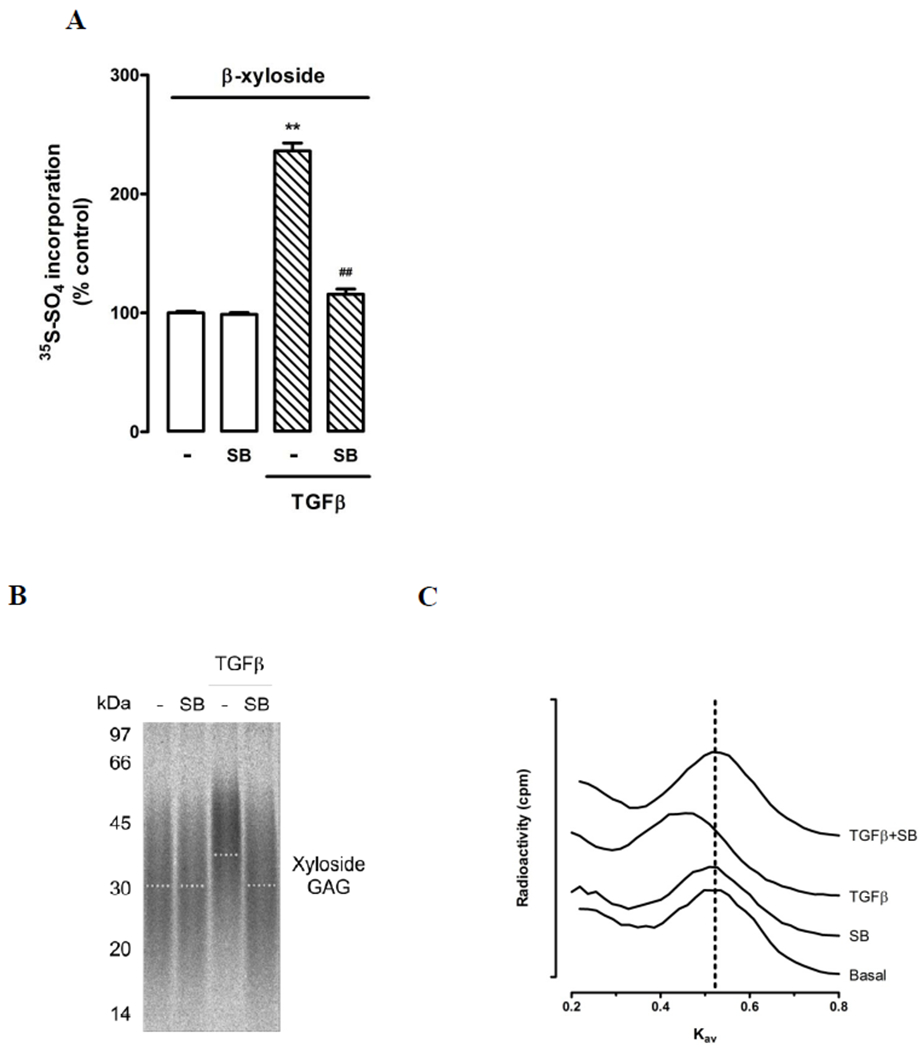
(A) TGF-β increased [35S]-sulfate incorporation into xyloside-GAGs and inhibition of TβR1. VICs were pretreated with medium (−) or SB431542 (SB 3 μM) for 30 min followed by TGF-β (2 ng/ml) for 24h in the presence of [35S]-sulfate and β-xyloside (0.5 mM). Data show mean ± SEM from two experiments in triplicate, **p<0.01 vs control and ##p<0.01 vs TGF-β using a 1-way ANOVA. (B) Xyloside-GAGs from above listed treatments were purified on DEAE-Sepharose ion-exchange columns and then run on gradient SDS-PAGE gels (4 – 20%). A representative gel from three separate experiments is shown and the dotted line indicates estimated mid-line of xyloside-GAGs size. (C) Size exclusion chromatography (Sepharose CL-6B) of radiolabeled xyloside-GAG chains isolated from VICs. Treatments as given above medium (Con), SB431542 (SB), TGF-β (TGF), SB431542 and TGF-β (SB + TGF). Vertical dotted line indicates the calculated apparent Kav of control GAG chains. Three measurements were performed from three separate experiments with a representative shown. Note that TGF-β shifts the peak to the left, indicating larger size.
TβR1 inhibition decreases VIC proteoglycan/low density lipoprotein binding
To assess the role of TβR1 in the LDL binding to secreted proteoglycans from TGF-β1-stimulated VICs, gel mobility shift assays were used (Figure 6). The migration of proteoglycans, metabolically radiolabelled with [35S]-Met/Cys, through agarose gel is reduced in the presence of increasing amounts of bound LDL. Proteoglycans from cells treated with SB431542 + TGF-β1 had decreased LDL binding compared to TGF-β1 treatment alone, p<0.001 (Figure 6A). The half maximal saturation value for proteoglycans from SB431542 treated cells in the presence of TGF-β1 was approximately four-fold higher at 0.13 mg/mL compared to 0.03 mg/mL for TGF-β1 treatment alone, indicating a substantial decrease in LDL binding affinity with SB431542 treatment. The LDL binding capacity of proteoglycans from SB431542 treated cells in the presence of TGF-β1 treated cells was also decreased compared to proteoglycans from TGF-β1 treated cells (Figure 6A). Thus inhibition of TβR1-mediated changes in VIC proteoglycans is sufficient to reduce the ability of the proteoglycans to bind LDL.
Figure 6. Inhibition of TβR1 reduces LDL binding to proteoglycans produced by TGF-β-treated VICs.
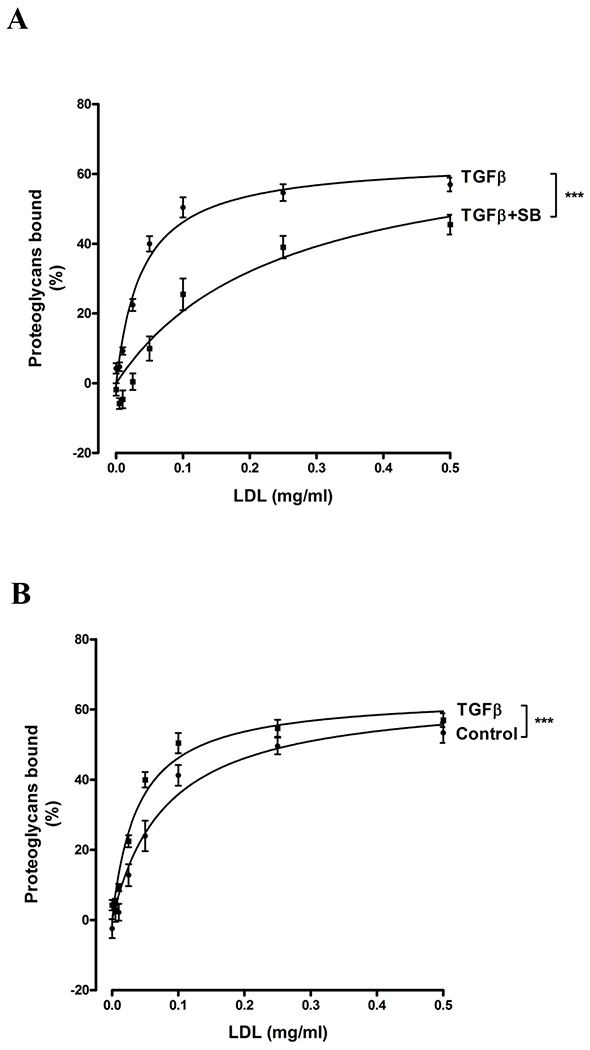
Gel mobility shift assay for the analysis of LDL binding to proteoglycans (A and B) VICs were treated with medium (Control), TGF-β (2 ng/ml), TGF-β + SB431542 (TGF-β+ SB) for 24h and proteoglycan core proteins were labelled with [35S]-Met/Cys (50 μCi/ml). Equal counts of core protein-radiolabeled proteoglycans were combined with increasing concentrations of LDL and separated using the gel shift mobility assay. Three separate experiments were performed with data shown as mean ± SEM. ***p<0.001 using a 2-way interaction ANOVA comparing the two data sets. Earlier experiments using proteoglycans isolated from vascular smooth muscle cells showed the effect of SB431542 treatment alone on LDL binding is negligible (unpublished data).
DISCUSSION
The pathological and actively regulated disease processes of CAVD have been well recognised recently and understanding basic valve biology and signaling pathways of VICs is acknowledged as a high priority for progress in CAVD research 44. Diseased valves have been shown previously to express the pleiotropic growth factor, TGF-β130, 32, 45 and a mouse model of CAVD has demonstrated elevated levels of phosphorylated Smad transcription factors in aortic valves 46. This study demonstrates that in human aortic valve lesions expression of both TGFβ1 and phosphorylated Smad2/3 are elevated throughout the thickened valve leaflets. Because TGF-β1 may be present in tissues in both active and latent forms, demonstrating the presence of TGF-β1 cannot determine whether the molecule is biologically active. However, canonical signaling by active TGF-β1 involves phosphorylation of Smad2/3 by TβR1 and subsequent translocation of pSmad2/3 to the nucleus. Thus, the demonstration that, in regions with immunostaining for TGF-β1, positive staining was also detected for both cytoplasmic and nuclear localization of pSmad2/3 provides supportive evidence of active TGF-β1 signalling in human aortic valve lesions. TGF-β1 and pSmad2/3 colocalization is also reported to occur in early lesions of the fibrosa in aortic valve leaflets from hypercholesterolemic pigs 47. Significantly elevated levels of pSmad2/3 are also observed in mouse models of early stage CAVD 46 and after lipid lowering treatment in late stage CAVD mouse models 48 and underlines the key role Smad2/3 in CAVD processes.
We investigated the signaling pathways through which TGF-β1 mediates its effects in VICs and demonstrated the involvement of the canonical Smad phosphorylation pathway. In porcine VICs TGF-β1 stimulated a significant increase in the level of both pSmad2/3C and pSmad2/3L. This specific response was blocked by SB431542, an inhibitor of the kinase activity of TβR1, for which both Smad2 and 3 are targets. Phosphorylation of Smad2/3 carboxy terminus was immediate and rapid, within 5 min, while linker region phosphorylation was elevated significantly after 30 minutes of TGF-β1 stimulation (Figure 2). A similar differential in regional Smad2/3 phosphorylation is also seen in vascular smooth muscle cells 49. Blocking pSmad2/3 phosphorylation inhibited proteoglycan GAG elongation however further studies are necessary to determine whether these downstream processes have a requirement for phosphorylation of both carboxy terminal and linker regions. Our results indicate that at 4h after TGF-β1 stimulation the linker phosphorylation is not conditional on MAP kinase activity. Further investigations are necessary to determine if MAP kinases are important immediately following TGF-β1 stimulation or whether alternate linker region phosphorylation kinases possibly cyclin-dependent kinase, glycogen synthase 3-β, calcium-calmodulin-dependent protein kinase II or G protein-coupled receptor kinase-2 are involved. Our findings correlate with recent reports of activated Smad2/3 signaling with TGF-β1 treated porcine VICs grown in Wnt3A-conditioned media demonstrating induction of pSmad2/3 nuclear translocation 47. The phosphorylation of Smad2/3 in aortic valves appears cell and context dependent since pSmad2 levels are not elevated in human aortic valve endothelium both in non-calcified and calcified valves 50.
Recently it was highlighted that despite an abundance of proteglycans and GAGs in valves in CAVD the role of proteoglycans and GAGs has not been investigated 31. Our study demonstrates that in porcine VICs TGF-β1 weakly induces proteoglycan core protein synthesis, and strongly stimulates proteoglycan GAG chain elongation. In addition we have clearly demonstrated that this is mediated by the classical TGF-β1 signaling pathway and by application of a pharmacological approach using SIS3 showed that Smad3 was not involved in proteoglycan synthesis, thereby demonstrating that the pathway specifically involves phosphorylation of Smad2. Previous in vitro studies using smooth muscle cells (SMCs) have demonstrated that TGF-β1 stimulates elongation of GAG chains on SMC-secreted proteoglycans 33, specifically on the chondroitin sulfate (CS) /dermatan sulfate (DS) proteoglycans, biglycan and decorin 33 and that these TGF-β1-induced changes in SMC-produced proteoglycans mediates increased lipoprotein binding 34. In the present study we show that TGF-β1 has similar effects on proteoglycans produced by aortic VICs. In VICs, TGF-β1 stimulated GAG chain elongation on CS/DS proteoglycans, as well as elongation of the GAGs initiated on xyloside. Moreover proteoglycans synthesized in response to TGF-β1 demonstrate enhanced LDL binding, an effect that is blunted by inhibition of classical TGF-β1 signalling via Smad2. This finding demonstrates that TGF-β1 is acting on proteoglycans primarily at the level of the GAG synthesizing machinery in the Golgi apparatus, an effect that is independent of the presence of core proteins. TGF-β1 also appears to stimulate a small increase in total proteoglycan core proteins by aortic VICs; we speculate that this is most likely an increase in biglycan and/or decorin. Although in this study we did not formally evaluate which individual core proteins were increased because the apparent effect on core protein synthesis was small and because our major interest was in the more robust effect on GAGs and their binding to LDL. Specific proteoglycan identification may be useful considering the impact of extracellular matrix components on myofibroblast differentiation and behaviour in CAVD 51. Biglycan and decorin are observed in early calcific nodule formation in human calcified aortic valves 29 and in late-stage CAVD there is increased accumulation of biglycan and with it a co-localization of oxidised-LDL, phospholipid transfer protein and Apo A1 52. By implicating biglycan and TGF-β1 influencing proteoglycan-mediated retention of lipoproteins within CAVD lesions, these findings suggest that inhibition of TGF-β1 and/or manipulation of proteoglycan/lipoprotein interactions may represent novel therapeutic targets in CAVD.
In conclusion, our data confirm that TGF-β1 is present in diseased human aortic valves, extends this observation to demonstrate the presence of important components of the canonical TGF-β1 signalling pathway in these same valves, and elucidates how stimulation of specific TGF-β1 signaling pathways affects proteoglycan synthesis and lipoprotein binding in cultured porcine VICs. Together, these findings provide strong support to the proposal that CAVD is a complex, mechanism based disease that shares some similar pathogenic features with atherosclerosis. Tentative evidence is provided that lipid trapping by modified GAG chains on proteoglycans may also represent an early step in CAVD, as has been hypothesized53 and strongly implicated by recent data54 for atherosclerosis. Although we have demonstrated that the canonical Smad phosphorylation pathway is present in diseased human aortic valves and cultured porcine VICs, studies of TGF-β1 signaling pathways in emerging animal models of CAVD would be of great interest. Moreover, TGF-β1 is critically involved in the immune system, most likely through Smad-dependent pathways. Thus, notwithstanding the involvement of TGF-β1 in CAVD, blocking those pathways may not be an option for treating CAVD and atherosclerosis. However, recent evidence has shown that TGF-β1 signalling pathways are much more complex and can involve tyrosine and serine/threonine phosphorylation and activation of MAP kinases , particularly ERK and p38 55-57. In addition to Smad,Erk1/2 and p38 MAP kinases are involved in the regulation of GAG synthesis in vascular smooth muscle cells.49, 57. Therefore, further investigations may provide an opportunity to discover Smad-independent TGF-β1 mediated signaling pathways that are associated with diseases processes and not immune functions, and that may well serve as more therapeutically useful targets for CAVD prevention and provides further strong impetus for studying the potential positive and/or negative effects of inhibiting these pathways in animal models of CAVD. Alternatively, strategies that avoid interference with TGF-β1 signaling by directly targeting proteoglycan/lipoprotein interactions may also represent useful therapeutic goals in CAVD.
ACKNOWLEDGEMENTS
This study was supported by National Health and Medical Research Fellowship (PJL) and a Diabetes Australia Research Trust grant (NO) and National Institutes of Health grant HL071739 (KDO). The work commenced with the support of a Human Frontier Science Program Short-Term Fellowship for JGA to spend a period of time in the laboratory of PJL.
DISCLOSURES
MLB – none; KJGA – none; RG – none; NO – none; SM – none; KDO - Speaking Honoraria from Merck; PJL – speaking honoraria from GSK; student grant from GSK.
ABBREVIATIONS
- CAVD
calcific aortic valve disease
- GAG
glycosaminoglycan
- LDL
low density lipoprotein
- pSmad2/3
phosphorylated Smad2/3
- TGF-β1
transforming growth factor
- VIC
valve interstitial cell
REFERENCES
- 1.Otto CM, Burwash IG, Legget ME, Munt BI, Fujioka M, Healy NL, Kraft CD, Miyake-Hull CY, Schwaegler RG. Prospective study of asymptomatic valvular aortic stenosis. Clinical, echocardiographic, and exercise predictors of outcome. Circulation. 1997;95:2262–2270 [DOI] [PubMed] [Google Scholar]
- 2.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147 [DOI] [PubMed] [Google Scholar]
- 3.Supino PG, Borer JS, Preibisz J, Bornstein A. The epidemiology of valvular heart disease: A growing public health problem. Heart Fail Clin. 2006;2:379–393 [DOI] [PubMed] [Google Scholar]
- 4.Roberts WC, Ko JM. Frequency by decades of unicuspid, bicuspid, and tricuspid aortic valves in adults having isolated aortic valve replacement for aortic stenosis, with or without associated aortic regurgitation. Circulation. 2005;111:920–925 [DOI] [PubMed] [Google Scholar]
- 5.O’Brien KD. Pathogenesis of calcific aortic valve disease: A disease process comes of age (and a good deal more). Arterioscler Thromb Vasc Biol. 2006;26:1721–1728 [DOI] [PubMed] [Google Scholar]
- 6.Grande-Allen KJ, Osman N, Ballinger ML, Dadlani H, Marasco S, Little PJ. Glycosaminoglycan synthesis and structure as targets for the prevention of calcific aortic valve disease. Cardiovasc Res. 2007;76:19–28 [DOI] [PubMed] [Google Scholar]
- 7.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O’Brien KD. Characterization of the early lesion of ‘degenerative’ valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. 1994;90:844–853 [DOI] [PubMed] [Google Scholar]
- 8.O’Brien KD, Olin KL, Alpers CE, Chiu W, Ferguson M, Hudkins K, Wight TN, Chait A. Comparison of apolipoprotein and proteoglycan deposits in human coronary atherosclerotic plaques: Colocalization of biglycan with apolipoproteins. Circulation. 1998;98:519–527 [DOI] [PubMed] [Google Scholar]
- 9.Mohler ER 3rd. Mechanisms of aortic valve calcification. Am J Cardiol. 2004;94:1396–1402, A1396 [DOI] [PubMed] [Google Scholar]
- 10.Rajamannan NM. Calcific aortic stenosis. Lessons learned from experimental and clinical studies. Arterioscler Thromb Vasc Biol. 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aronow WS, Ahn C, Kronzon I, Goldman ME. Association of coronary risk factors and use of statins with progression of mild valvular aortic stenosis in older persons. Am J Cardiol. 2001;88:693–695 [DOI] [PubMed] [Google Scholar]
- 12.Raggi P, Bommer J, Chertow GM. Valvular calcification in hemodialysis patients randomized to calcium-based phosphorus binders or sevelamer. J Heart Valve Dis. 2004;13:134–141 [PubMed] [Google Scholar]
- 13.Stewart BF, Siscovick D, Lind BK, Gardin JM, Gottdiener JS, Smith VE, Kitzman DW, Otto CM. Clinical factors associated with calcific aortic valve disease. Cardiovascular health study. J Am Coll Cardiol. 1997;29:630–634 [DOI] [PubMed] [Google Scholar]
- 14.Lindroos M, Kupari M, Heikkila J, Tilvis R. Prevalence of aortic valve abnormalities in the elderly: An echocardiographic study of a random population sample. J Am Coll Cardiol. 1993;21:1220–1225 [DOI] [PubMed] [Google Scholar]
- 15.Rajamannan NM, Subramaniam M, Springett M, Sebo TC, Niekrasz M, McConnell JP, Singh RJ, Stone NJ, Bonow RO, Spelsberg TC. Atorvastatin inhibits hypercholesterolemia-induced cellular proliferation and bone matrix production in the rabbit aortic valve. Circulation. 2002;105:2660–2665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu B, Elmariah S, Kaplan FS, Cheng G, Mohler ER 3rd. Paradoxical effects of statins on aortic valve myofibroblasts and osteoblasts: Implications for end-stage valvular heart disease. Arterioscler Thromb Vasc Biol. 2005;25:592–597 [DOI] [PubMed] [Google Scholar]
- 17.Pohle K, Maffert R, Ropers D, Moshage W, Stilianakis N, Daniel WG, Achenbach S. Progression of aortic valve calcification: Association with coronary atherosclerosis and cardiovascular risk factors. Circulation. 2001;104:1927–1932 [DOI] [PubMed] [Google Scholar]
- 18.Shavelle DM, Takasu J, Budoff MJ, Mao S, Zhao XQ, O’Brien KD. Hmg coa reductase inhibitor (statin) and aortic valve calcium. Lancet. 2002;359:1125–1126 [DOI] [PubMed] [Google Scholar]
- 19.O’Brien KD, Probstfield JL, Caulfield MT, Nasir K, Takasu J, Shavelle DM, Wu AH, Zhao XQ, Budoff MJ. Angiotensin-converting enzyme inhibitors and change in aortic valve calcium. Arch Intern Med. 2005;165:858–862 [DOI] [PubMed] [Google Scholar]
- 20.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–1295 [DOI] [PubMed] [Google Scholar]
- 21.O’Brien KD, Shavelle DM, Caulfield MT, McDonald TO, Olin-Lewis K, Otto CM, Probstfield JL. Association of angiotensin-converting enzyme with low-density lipoprotein in aortic valvular lesions and in human plasma. Circulation. 2002;106:2224–2230 [DOI] [PubMed] [Google Scholar]
- 22.Helske S, Lindstedt KA, Laine M, Mayranpaa M, Werkkala K, Lommi J, Turto H, Kupari M, Kovanen PT. Induction of local angiotensin ii-producing systems in stenotic aortic valves. J Am Coll Cardiol. 2004;44:1859–1866 [DOI] [PubMed] [Google Scholar]
- 23.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397 [DOI] [PubMed] [Google Scholar]
- 24.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356 [DOI] [PubMed] [Google Scholar]
- 25.Chan KL, Teo K, Dumesnil JG, Ni A, Tam J. Effect of lipid lowering with rosuvastatin on progression of aortic stenosis: Results of the aortic stenosis progression observation: Measuring effects of rosuvastatin (astronomer) trial. Circulation. 2010;121:306–314 [DOI] [PubMed] [Google Scholar]
- 26.Aikawa E, Nahrendorf M, Figueiredo JL, Swirski FK, Shtatland T, Kohler RH, Jaffer FA, Aikawa M, Weissleder R. Osteogenesis associates with inflammation in early-stage atherosclerosis evaluated by molecular imaging in vivo. Circulation. 2007;116:2841–2850 [DOI] [PubMed] [Google Scholar]
- 27.Cimini M, Boughner DR, Ronald JA, Aldington L, Rogers KA. Development of aortic valve sclerosis in a rabbit model of atherosclerosis: An immunohistochemical and histological study. J Heart Valve Dis. 2005;14:365–375 [PubMed] [Google Scholar]
- 28.O’Brien KD, Otto CM, Reichenbach DD, Alpers CE, Wight TW. Regional accumulation of proteoglycans in lesions of “Degenerative” Valvular aortic stenosis and their relationship to apolipoproteins. Circulation. 1995;92:612 [Google Scholar]
- 29.Stephens EH, Saltarrelli JG, Baggett LS, Nandi I, Kuo JJ, Davis AR, Olmsted-Davis EA, Reardon MJ, Morrisett JD, Grande-Allen KJ. Differential proteoglycan and hyaluronan distribution in calcified aortic valves. Cardiovasc Pathol. 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins b, (a), and e accumulate in the morphologically early lesion of ‘degenerative’ valvular aortic stenosis. Arterioscler Thromb Vasc Biol. 1996;16:523–532 [DOI] [PubMed] [Google Scholar]
- 31.Chen JH, Simmons CA. Cell-matrix interactions in the pathobiology of calcific aortic valve disease: Critical roles for matricellular, matricrine, and matrix mechanics cues. Circ Res. 2011;108:1510–1524 [DOI] [PubMed] [Google Scholar]
- 32.Jian B, Narula N, Li QY, Mohler ER 3rd, Levy RJ. Progression of aortic valve stenosis: Tgf-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465 [DOI] [PubMed] [Google Scholar]
- 33.Schonherr E, Jarvelainen HT, Kinsella MG, Sandell LJ, Wight TN. Platelet-derived growth factor and transforming growth factor-beta 1 differentially affect the synthesis of biglycan and decorin by monkey arterial smooth muscle cells. Arterioscler Thromb. 1993;13:1026–1036 [DOI] [PubMed] [Google Scholar]
- 34.Little PJ, Tannock L, Olin KL, Chait A, Wight TN. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to ldls. Arterioscler Thromb Vasc Biol. 2002;22:55–60 [DOI] [PubMed] [Google Scholar]
- 35.Ballinger ML, Nigro J, Frontanilla KV, Dart AM, Little PJ. Regulation of glycosaminoglycan structure and atherogenesis. Cell Mol Life Sci. 2004;61:1296–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. 1995;15:551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Blevins TL, Carroll JL, Raza AM, Grande-Allen KJ. Phenotypic characterization of isolated valvular interstitial cell subpopulations. J Heart Valve Dis. 2006;15:815–822 [PubMed] [Google Scholar]
- 38.Nigro J, Dilley RJ, Little PJ. Differential effects of gemfibrozil on migration, proliferation and proteoglycan production in human vascular smooth muscle cells. Atherosclerosis. 2002;162:119–129 [DOI] [PubMed] [Google Scholar]
- 39.Tannock LR, Little PJ, Tsoi C, Barrett PHR, Wight TN, Chait A. Thiazolidinediones reduce the ldl binding affinity of non-human primate vascular cell proteoglycans. Diabetologia. 2004;47:837–843 [DOI] [PubMed] [Google Scholar]
- 40.Nigro J, Ballinger M, Dilley R, Jennings G, Wight T, Little P. Fenofibrate modifies human vascular smooth muscle proteoglycans and reduces ldl binding. Diabetologia. 2004;47:2105–2113 [DOI] [PubMed] [Google Scholar]
- 41.Heinecke JW, Baker L, Rosen H, Chait A. Superoxide-mediated modification of low density lipoprotein by arterial smooth muscle cells. J Clin Invest. 1986;77:757–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taylor PM, Batten P, Brand NJ, Thomas PS, Yacoub MH. The cardiac valve interstitial cell. Int J Biochem Cell Biol. 2003;35:113–118 [DOI] [PubMed] [Google Scholar]
- 43.Rabkin-Aikawa E, Farber M, Aikawa M, Schoen FJ. Dynamic and reversible changes of interstitial cell phenotype during remodeling of cardiac valves. J Heart Valve Dis. 2004;13:841–847 [PubMed] [Google Scholar]
- 44.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O’Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: Not simply a degenerative process: A review and agenda for research from the national heart and lung and blood institute aortic stenosis working group * executive summary: Calcific aortic valve disease - 2011 update. Circulation. 2011;124:1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clark-Greuel JN, Connolly JM, Sorichillo E, Narula NR, Rapoport HS, Mohler ER 3rd, Gorman JH 3rd, Gorman RC, Levy RJ. Transforming growth factor-beta1 mechanisms in aortic valve calcification: Increased alkaline phosphatase and related events. Ann Thorac Surg. 2007;83:946–953 [DOI] [PubMed] [Google Scholar]
- 46.Miller JD, Weiss RM, Serrano KM, Brooks RM 2nd, Berry CJ, Zimmerman K, Young SG, Heistad DD. Lowering plasma cholesterol levels halts progression of aortic valve disease in mice. Circulation. 2009;119:2693–2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen JH, Chen WL, Sider KL, Yip CY, Simmons CA. Beta-catenin mediates mechanically regulated, transforming growth factor-beta1-induced myofibroblast differentiation of aortic valve interstitial cells. Arterioscler Thromb Vasc Biol. 2011;31:590–597 [DOI] [PubMed] [Google Scholar]
- 48.Miller JD, Weiss RM, Serrano KM, Castaneda LE, Brooks RM, Zimmerman K, Heistad DD. Evidence for active regulation of pro-osteogenic signaling in advanced aortic valve disease. Arterioscler Thromb Vasc Biol. 2010;30:2482–2486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burch ML, Yang SN, Ballinger ML, Getachew R, Osman N, Little PJ. Tgf-beta stimulates biglycan synthesis via p38 and erk phosphorylation of the linker region of smad2. Cell Mol Life Sci. 2010;67:2077–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ankeny RF, Thourani VH, Weiss D, Vega JD, Taylor WR, Nerem RM, Jo H. Preferential activation of smad1/5/8 on the fibrosa endothelium in calcified human aortic valves--association with low bmp antagonists and smad6. PLoS One. 2011;6:e20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller JD, Weiss RM, Heistad DD. Calcific aortic valve stenosis: Methods, models, and mechanisms. Circ Res. 2011;108:1392–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Derbali H, Bosse Y, Cote N, Pibarot P, Audet A, Pepin A, Arsenault B, Couture C, Despres JP, Mathieu P. Increased biglycan in aortic valve stenosis leads to the overexpression of phospholipid transfer protein via toll-like receptor 2. Am J Pathol. 2010;176:2638–2645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation. 2007;116:1832–1844 [DOI] [PubMed] [Google Scholar]
- 54.Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K. Early human atherosclerosis: Accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol. 2007;27:1159–1165 [DOI] [PubMed] [Google Scholar]
- 55.Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R. Tgf-beta activates erk map kinase signalling through direct phosphorylation of shca. Embo J. 2007;26:3957–3967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ungefroren H, Groth S, Ruhnke M, Kalthoff H, Fandrich F. Transforming growth factor-beta (tgf-beta) type i receptor/alk5-dependent activation of the gadd45beta gene mediates the induction of biglycan expression by tgf-beta. J Biol Chem. 2005;280:2644–2652 [DOI] [PubMed] [Google Scholar]
- 57.Dadlani H, Ballinger ML, Osman N, Getachew R, Little PJ. Smad and p38 map kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J Biol Chem. 2008;283:7844–7852 [DOI] [PubMed] [Google Scholar]