

Abstract
Current immunotherapeutic targets are often shared between neoplastic and normal hematopoietic stem and progenitor cells (HSPCs), leading to unwanted on-target, off-tumor toxicities. Deletion or modification of such targets to protect normal HSPCs is, therefore, of great interest. Although HSPC modifications commonly aim to mimic naturally occurring phenotypes, the long-term persistence and safety of gene-edited cells need to be evaluated. Here, we deleted the V-set domain of CD33, the immune-dominant domain targeted by most anti-CD33 antibodies used to treat CD33-positive malignancies, including acute myeloid leukemia, in the HSPCs of two rhesus macaques, performed autologous transplantation after myeloablative conditioning, and followed the animals for up to 3 years. CD33-edited HSPCs engrafted without any delay in recovery of neutrophils, the primary cell type expressing CD33. No impact on the blood composition, reconstitution of the bone marrow stem cell compartment, or myeloid differentiation potential was observed. Up to 20% long-term gene editing in HSPCs and blood cell lineages was seen with robust loss of CD33 detection on myeloid lineages. In conclusion, deletion of the V-set domain of CD33 on HSPCs, progenitors, and myeloid lineages did not show any adverse effects on their homing and engraftment potential or the differentiation and functionality of myeloid progenitors and lineages.
Keywords: hematopoietic stem cells, HSCs, gene therapy and editing, CRISPR, CD33, non-human primate, NHP, autologous transplantation
Graphical abstract
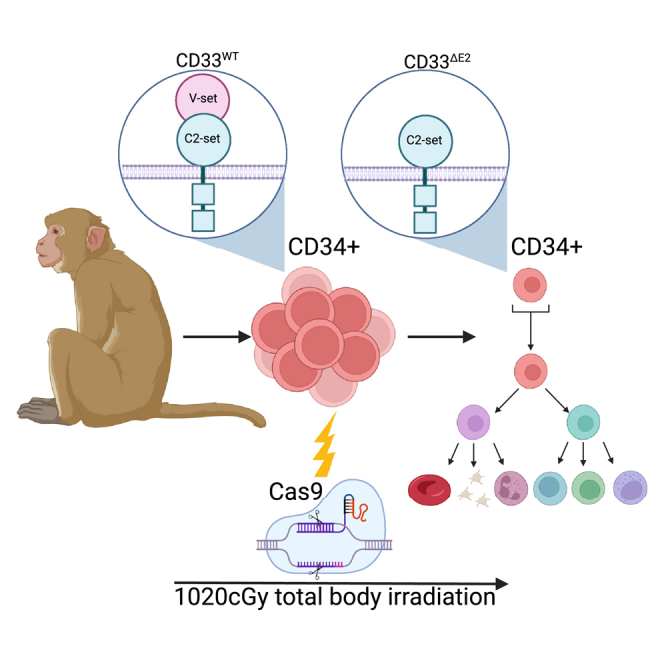
Petty and colleagues investigate the efficiency and long-term persistence of Cas9-mediated ablation of CD33 in non-human primate hematopoiesis. Gene-modified hematopoietic stem cells engrafted long term and maintained up to 20% editing across all blood lineages. These results pave the way for the clinical application of CD33 editing for selection and protection.
Introduction
Cancer cells often upregulate cell surface markers present on healthy tissues, leading to challenges for the design of antigen-specific treatment strategies. For example, immunotherapeutics such as the CD33 antibody-drug conjugate gemtuzumab ozogamicin (GO), used for the treatment of acute myeloid leukemia (AML), show off-leukemia toxicities on healthy hematopoietic cells because of the expression of CD33 on normal myeloid cells.1,2,3,4 To avoid such effects, multiple groups have used CRISPR to modify CD33 expression on the surface of human and nonhuman primate (NHP) hematopoietic stem and progenitor cells (HSPCs).5,6,7,8 While identical in goal, CD33 CRISPR-Cas9 editing has been reported either via gene ablation using a standard CRISPR-Cas9 approach to disrupt CD33 expression with insertions or deletions (indels)4 or by mimicking a naturally occurring phenotype using a dual guide excision approach removing exon 2, which encodes the extracellular V-set domain of CD33, the immune-dominant domain recognized by GO and most other current anti-CD33 targeting moieties.9 Successful non-homologous end-joining-mediated disruption of CD33 was achieved by Kim et al.6 in human and NHP HSPCs without any impairment of the engraftment or differentiation potential of edited cells in the mouse xenograft or autologous NHP models. Similarly, we have shown that deletion of exon 2 in human HSPCs does not impair cell proliferation, differentiation, or multilineage engraftment in the NSG mouse model.5 Here, we comprehensively evaluated the safety and long-term persistence of CD33 exon 2-deleted HSPCs in NHPs after autologous transplantation of ex vivo gene-edited HSPCs.
Results
Efficient CRISPR-mediated deletion of the V-set domain of CD33 in NHP HSPCs
To test the feasibility and efficiency of our CD33-deletion approach in NHP cells, we transplanted two rhesus macaques (A17039 and A18092) with V-set domain deletion of CD33 (CD33ΔE2) HSPCs. Briefly, CD34+ cells were isolated and CD33 deleted by excising exon 2 using CRISPR-Cas9 as previously described5,7 using two human/NHP cross-reactive guide RNAs (Figure 1A). Successful editing of the CD33 locus in NHP CD34+ HSPCs was confirmed by flow cytometry showing 53.4% editing efficiency in A17039 and 46.7% in A18092 (Figure 1B). To confirm loss of CD33 antibody binding, edited CD34+ cells were cultured for 7 days, showing highly efficient loss of CD33 expression as assessed by flow cytometry using the anti-CD33 antibody, clone AC104.3E4, which binds the V-set domain (Figure 1C). To ensure that CD33ΔE2 does not impact myeloid differentiation, CD33-edited bulk CD34+ cells, as well as fluorescence-activated cell sorting (FACS)-purified CD34-subsets enriched for HSCs (CD34+CD90+), erythro-myeloid progenitors (EMPs: CD34+CD90–), and lympho-myeloid progenitors (LMPs: CD34+CD45RA+) were introduced into colony-forming cell (CFC) assays. Edited cells were not impaired in their total capacity to form colonies (total height of bars) or in their ability to form erythroid, granulocytic, and monocytic lineages (Figure 1D). As previously described,10 granulocyte colonies were enriched in the HSC-containing subset, erythroid progenitors in the CD34+CD90– cell fraction, and granulocyte/monocyte progenitors in the CD34+CD45RA+ subset.
Figure 1.

Efficient deletion of CD33 in NHP CD34+ cells without impacting the in vitro differentiation potential
(A) Experimental workflow for the CRISPR-Cas9-mediated deletion of CD33. (B and C) Loss of CD33 signal on CD34+ cells 7 days post-editing measured by flow cytometry. (D) Colony-forming potential of gene-modified CD34+ cells and FACS-purified CD34-subsets enriched for HSCs (CD34+CD90+), EMPs (CD34+CD90–), and LMPs (CD34+CD45RA+). BFU, burst-forming unit; E, erythrocyte; G, granulocytes; GM, granulocyte-macrophage/monocyte; M, macrophages/monocytes; MIX, erythro-myeloid colony.
The CD33ΔE2 phenotype persists long-term in the peripheral blood
Edited CD34+ cells were transplanted into animals after total body irradiation (TBI) (1,020 cGy) to assess the long-term multilineage engraftment potential. A total of 26.2 × 106 and 20.5 × 106 CD34+ cells per kilogram bodyweight were infused into A17039 and A18092, respectively. In comparison with historic controls,10,11 no impact of CD33 editing was seen on the hematopoietic recovery as measured by the time to recovery for both neutrophils (A17039, day 10; A18092, day 13) and platelets (A17039, day 26; A18092, day 19) (Figures 2A and 2B). Total white blood cell and lymphocyte counts in both animals were in the normal range after 1–2 months and remained stable (Figures 2C and 2D). Detailed analysis of the blood composition by flow cytometry showed recovery of all immune compartments in both animals, with granulocytes rapidly recovering after transplantation, followed by T cells and B cells recovering by 2 months post-transplant (Figure 2E).
Figure 2.

Hematopoietic recovery of the PB after transplant
(A–D) Neutrophil, platelet, white blood cell (WBC), and lymphocyte counts measured by complete blood cell counts. Horizontal dotted lines indicate range of normal as determined by historical data as well as published values.12 (E) Phenotypic composition of the PB measured by flow cytometry.
Long-term persistence of editing was assessed by flow cytometric analysis of CD33 surface expression within the granulocyte compartment in the peripheral blood (PB) (Figure 3A). Immediately after transplant, both animals demonstrated near-total loss of CD33 signal on granulocytes. CD33+ granulocyte signals rebounded after transplantation to 60% in A17039 and 10% in A18092 and remained stable throughout the entire course of the long-term follow-up.
Figure 3.

Long-term persisting multi-lineage gene-editing in the PB
(A) Loss of CD33 signal on PB granulocytes measured with flow cytometry. (B) Genomic deletion of CD33 measured by ddPCR. (C) CD33 deletion efficiency in FACS-purified PB lineages measured by ddPCR at 1 year post transplant.
To verify CD33 editing across all blood cells, droplet digital PCR (ddPCR) was performed (Figure 3B). Peak editing of 50%–60% was seen immediately after transplant, declining over the course of 2 months, then stabilizing long term at 20% for A17039 and 10% for A18092. Next, we investigated whether the editing was equally present in all lineages, confirming successful introduction of the CD33ΔE2 edit in long-term persisting multipotent HSCs. T cell (CD3+), B cells (CD20+), natural killer cells (CD16+), as well as granulocytes (CD11b+CD14–SSChigh) and monocytes (CD11b+CD14+SSClow), were purified using FACS; DNA was analyzed by ddPCR. Editing in all lineages at 1 year and 3 years post-transplant closely tracked with the editing we observed in bulk white blood cells, averaging 20% in A17039 and 10%–15% in A18092 (Figure 3C).
Efficient engraftment of CD33ΔE2 HSPCs in the bone marrow stem cell compartment
Concurrent with our tracking in the PB, we obtained BM (BM) samples from both animals at 1 and 3 years post-transplant. Gene-edited CD34+ cells efficiently engrafted into the BM stem cell niche without any impairment caused by the deletion of CD33 (Figure 4A). All CD34 subsets including phenotypical HSCs (CD34+CD90+), EMPs (CD34+CD90–), and LMPs (CD34+CD45RA+) were re-established at normal frequencies.10 CD33 surface expression was inconsistent in between the CD34 subsets and did not reflect the downregulation seen on granulocytes in the PB (Figure 3A). The lowest expression was found on HSCs (A17039, 6.3%; A18092, 1.2%), whereas EMPs (A17039, 29.6%; A18092, 40.8%) and LMPs (A17039, 58.7%; A18092, 30.9%) showed heterogeneous presentation of CD33 at 3 years post-transplant. To verify successful genomic editing, bulk CD34+ cells as well as phenotypic subsets were FACS purified and CD33 deletion was quantified using ddPCR (Figure 4B). Genomic editing efficiency in both animals was highly consistent across the different cell populations and between the two BM draws at 1 and 3 years post-transplant. Consistent with the editing levels seen in PB lineages (Figure 3C), 15%–25% and 8%–12% CD33 editing was seen in A17039 and A18092, respectively.
Figure 4.

Long-term engraftment of CD33-edited HSPCs in the BM
(A) Flow-cytometric assessment of the recovery of the BM stem cell compartment and loss of CD33 on the surface of CD34+ cells 3 years post transplant. (B) Quantification of CD33 editing by ddPCR 1 year and 3 years post transplant.
Long-term engrafted CD33ΔE2 HSCs maintained erythro-myeloid differentiation potential
To verify that long-term engrafted CD33ΔE2 HSPCs retain their full erythro-myeloid differentiation potential, CD34+ cells and subsets were FACS purified and introduced into CFC assays at 1 and 3 years post-transplant. CD33 editing did not impact the total colony forming potential or the ability to generate erythroid, myeloid, or mixed colonies in A17039 (Figure 5A). However, in A18092, impaired CFC potential was seen in EMPs (CD90–) and LMPs (CD45RA+), with skewing toward granulocytes in year 1 that entirely resolved in year 3. PCR-based assessment of extracted single-cell-derived colonies from both animals at year 3 showed monoallelic as well as biallelic deletion of exon 2 (Figures 5B and 5C). Finally, indel formation of at both cut sites was assessed on wildtype and monoallelic CD33ΔE2 colonies from A18092. No indels were found in wildtype colonies, whereas in 5 out of 31 monoallelic CD33ΔE2 colonies indels causing a frameshift were found on the second allele (Table S2).
Figure 5.

CFC potential of CD33ΔE2 HSPCs in the BM stem cell compartment
(A) (top) Total CFC potential and (bottom) erythro-myeloid differentiation potential of HSPCs 1 year and 3 years post-transplant. BFU-E, burst-forming unit erythrocyte; G, granulocytes; GM, granulocytes-macrophage/monocytes; GEMM, granulocytes-erythrocytes-monocytes-megakaryocytes; M, macrophage/monocytes. (B) Representative PCR-based assessment of CD33 deletion in individual colonies. ΔE2, CD33ΔE2; WT, wildtype. (C) Quantification of colonies with WT CD33 or mono-/biallelic editing of CD33ΔE2.
Discussion
In this study, we demonstrate that CRISPR-mediated deletion of the CD33 V-set domain in NHP HSCs preserves their ability to stably engraft and persist long term without any measurable impact on their multilineage differentiation potential or the development of myeloid lineages. CD33 editing neither impacted homing of HSCs to the BM nor prolonged the neutrophil recovery after transplantation in comparison with historic control animals. No obvious impact was seen on the myeloid colony-forming potential of HSCs and generation of phenotypic neutrophils in the PB or BM. Most important, highly efficient editing was achieved with our two-guide CRISPR approach ex vivo on NHP HSPCs, with editing levels remaining long term in reach of the therapeutic threshold.
We here successfully used a previously reported two-guide CRISPR-based approach to delete the CD33ΔE2 in NHP HSPCs. We observed ex vivo an almost complete loss (80%–90% reduction) of CD33 detection by flow cytometry on the surface of CD34+ HSPCs using a V-set domain-directed anti-CD33 antibody, closely recapitulating our previous observations in human HSPCs using the exact same guide sequences.5 In contrast, 3%–5% editing of CD33 was achieved by Kim et al.6 using either a single- or dual-guide CRISPR approach introducing indels with the goal to create a full knockout of the protein. Similar to our studies in human HSPCs, our CD33 editing approach did not have any quantitative or qualitative impact on the differentiation potential of HSPCs in CFC assays realizing erythroid as well as myeloid lineages.10 Similarly, we did not appreciate any negative impacts of our editing strategy on cell viability or engraftment potential, despite the previously reported Cas9-induced p53 induction or the known occurrence of large scale genomic rearrangements caused by double-strand break formation.13,14
Importantly, ablation of exon 2 did not have any negative impact on the reconstitution potential after myeloablative irradiation and autologous transplantation of ex vivo gene-edited CD34+ cells. No delay in the neutrophil recovery was seen, with the only cell type in the PB expressing CD33 on the cell surface, indicating there was no functional impairment of the myeloid compartment due to exon 2 ablation. Similarly, all other blood lineages and cell types recovered to normal levels within 1–2 months without any delay in comparison with historic controls undergoing autologous HSPC gene therapy, suggesting that the genetic modification of CD33 does not trigger a delay in the maturation or differentiation of HSCs into progenitors and mature blood cells.
As previously reported by us and others in the autologous NHP gene therapy model,11,15 editing levels in the PB peaked shortly after transplant, reaching similar frequencies as observed ex vivo in the infusion product. Thereafter, the frequency of CD33– granulocytes by flow cytometry, as well as CD33-edited PB mononuclear cells by ddPCR, gradually decreased and plateaued at 10% in A18092 and 20% in A17039 because of the competition with remaining endogenous HSPCs after non-lethal TBI at 1,020 cGy. Observed frequencies of editing in our CD33 exon 2 excision approach are above the single-guide CRISPR study by Kim et al.6 that reported about 20% at peak and 3%–4% editing long term in steady-state. The frequency of editing in both animals remained stable throughout the 3 years of follow-up, demonstrating no selective advantage or disadvantage.
Last, the long-term frequency of editing in the PB closely matched the editing achieved in long-term persisting, BM-engrafted HSPCs, ensuring no disadvantage of CD33 ablation on the homing and reconstitution potential of ex vivo edited HSPCs. The BM stem cell compartment fully recovered, establishing a similar composition of phenotypic and functional subsets as previously reported for naive animals.11 CD33-edited HSPCs showed no change in the lineage output or bias toward distinct lineages in the PB or BM indicating that CD33 is not essential for the long-term persistence and maturation of HSCs. Like the study by Kim et al., monoallelic as well as biallelic edits were found in engrafted NHP HSPCs at 3 years post transplant, demonstrating no obvious selection against CD33 biallelic-edited HSCs or impairment in the long-term persistence.6
A major concern in the field of gene-edited HSCTs is the ability to reach and maintain sufficient editing to convey therapeutic benefit. It has been shown previously that, in the case of sickle cell disease, 20% editing is a critical threshold for therapeutic efficacy.16,17,18 Although the editing levels in the reported animals here remained below the therapeutic threshold, we demonstrate feasibility to introduce a non-therapeutic edit that allows targeted depletion of non-edited cells and provides the opportunity to increase the levels above the 20% threshold. Our group has previously explored overexpression of O6-methylguanine-DNA methyltransferase P140K to this effect.19,20 We and others are also investigating alternative non-genotoxic approaches using modifications to CD117 and CD45 to allow antibody-mediated selection of gene-modified cells; however, the editing efficiency required to effectively leverage these modalities for enrichment is subject to future studies.21,22,23
In regard to CD33, previous studies have explored selection modalities such as antibody-drug conjugates (ADCs) and chimeric antigen receptor (CAR) T cells in the context of CD33-overexpressing AMLs, both of which are currently US Food and Drug Administration-approved or undergoing phase Ib/II clinical trials, respectively. Recent work with CD33-directed CAR T cells as well as the ADC GO has demonstrated selective killing of CD33+ HSCs, potentially allowing these modalities to be paired with CD33 editing multiplexed with a therapeutic edit to facilitate enrichment of edited cells.3,24,25 However, prior work from our group and others has demonstrated that CRISPR-Cas9, particularly with multiple guide RNAs, can lead to large-scale recombination events such as inversions and translocations.26 The advent of base editing technology, which does not rely on double-strand breaks for editing, will allow for safer multiplex editing without concern for unwanted off-target effects.27
In conclusion, we demonstrate the long-term safety and feasibility of a two-guide CRISPR approach to CD33 editing in an NHP model. Deletion of exon 2 not only allowed us to attain higher editing than previously observed in NHP models, but also had no observable impact on reconstitution of the hematopoietic system after transplantation and did not alter the constitution of the PB. Even after 3 years of longitudinal follow-up, we maintained up to 20% CD33 editing and did not observe any significant adverse events associated with the transplantation of CD33-edited HSCs. This work supports the safety of CD33 editing in the context of anti-CD33 AML therapeutics, but the ramifications extend beyond this and toward the use of CD33 editing as an enrichment handle for therapeutic edits geared toward other hematologic diseases.
Materials and methods
Animals
Healthy juvenile rhesus macaques were housed at the University of Washington (UW) National Primate Research Center (WaNPRC) under conditions approved by the American Association for the Accreditation of Laboratory Animal Care. All experimental procedures performed were reviewed and approved by the Institutional Animal Care and Use Committee of the Fred Hutchinson Cancer Research Center (Fred Hutch) and UW (Protocol No. 3235–01). This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (The Guide), and monkeys were randomly assigned to the study. This study included at least twice daily observation by animal technicians for basic husbandry parameters (e.g., food intake, activity, stool consistency, and overall appearance), as well as daily observation by a veterinary technician and/or veterinarian. Animals were housed in cages approved by The Guide and in accordance with Animal Welfare Act regulations. Animals were fed twice daily and were fasted for up to 14 h before sedation. Environmental enrichment included grouping in compound, large activity, or run-through connected cages, perches, toys, food treats, and foraging activities. If a clinical abnormality was noted by WaNPRC personnel, standard WaNPRC procedures were followed to notify the veterinary staff for evaluation and determination for admission as a clinical case. Animals were sedated by administration of ketamine HCl and/or telazol and supportive agents for balanced anesthesia (such as diazepam and midazolam) before all procedures. After sedation, animals were monitored according to WaNPRC standard protocols. WaNPRC surgical support staff are trained and experienced in the administration of anesthetics and have monitoring equipment available to assist with electronic monitoring of heart rate, respiration, and blood oxygenation; audible alarms and digital readouts; and monitoring of blood pressure, temperature. For minor procedures, the presence or absence of deep pain was tested by the toe-pinch reflex, and the absence of response (leg flexion) to this test indicated adequate anesthesia. In cases of general anesthesia, similar monitoring parameters were used, and anesthesia was tested by the loss of palpebral reflexes (eye blink). Analgesics (generally buprenorphine with meloxicam or buprenorphine slow release) were provided as prescribed by the clinical veterinary staff for at least 48 h after the procedures and could be extended at the discretion of the clinical veterinarian based on clinical signs. Autologous NHP transplantation, priming (mobilization), collection of cells, and genetic engineering were conducted consistent with our previously published protocols.10,13,28 In parallel to cell processing, macaques were conditioned with myeloablative TBI of 1,020 cGy from a 6-MV X-ray beam of a single-source linear accelerator located at the Fred Hutch South Lake Union Facility (Seattle); irradiation was administered as a fractionated dose over the 2 days before cell infusion. During irradiation, animals were housed in a specially modified cage that provided unrestricted access for the irradiation while simultaneously minimizing excess movement. The dose was administered at a rate of 7 cGy/min delivered as a midline tissue dose. Granulocyte colony-stimulating factor was administered daily from the day of cell infusion until the animals began to show onset of neutrophil recovery. Supportive care, including antibiotics, electrolytes, fluids, and transfusions, was given as necessary, and blood counts were analyzed daily to monitor hematopoietic recovery. Cytomegalovirus (CMV) monitoring was performed by quantitative PCR as described previously.29 Briefly, DNA was isolated from whole blood using Qiagen DNA Blood Mini Kit (Qiagen) using the manufacturer’s instructions. Isolated DNA was then used for quantitative real-time PCR analysis on a QuantStudio Real-Time PCR system (12K Flex) (Applied Biosystems) using CMV-specific primers (forward: ATC CGC GTT CCA ATG CA; reverse: CGG AGG AGC ACC ATA GAA GGT) and a TaqMan Probe (6FAM CCT TCC CGG CTA TGG MGBNFQ). Each sample was run in triplicate for 40 cycles along with positive controls. Copy numbers were calculated by comparison to a standard curve, and the viral load was reported as CMV copies per milliliter of whole blood.
CD34 enrichment and in vitro cultures
Primed NHP BM was harvested, enriched, and cultured as previously described.28,30 Briefly, before enrichment of CD34+ cells, red cells were lysed in ammonium chloride lysis buffer, and white blood cells were incubated for 30 min with the 12.8 immunoglobulin M anti-CD34 antibody and then washed and incubated for another 30 min with magnetic-activated cell-sorting anti–immunoglobulin M microbeads (Miltenyi Biotec). The cell suspension was run through magnetic columns enriching for CD34+ cell fractions with a purity of 60%–80% confirmed by flow cytometry. Enriched CD34+ cells were cultured in StemSpan serum-free expansion medium II (SFEM II) (StemCell Technologies) supplemented with penicillin and streptomycin (100 U/mL; ThermoFisher Scientific), stem cell factor (PeproTech), thrombopoietin (PeproTech), and Fms-related tyrosine kinase 3 ligand (Miltenyi Biotec) (100 ng/mL for each cytokine).
CFC assay
For CFC assays, 1,000–1,200 sorted cells were seeded into 3.5-mL ColonyGEL 1402 (ReachBio). Hematopoietic colonies were scored after 12 to 14 days. Arising colonies were identified as colony-forming unit (CFU) granulocyte, CFU macrophage, CFU granulocyte-macrophage, and burst-forming unit-erythrocyte. Colonies consisting of erythroid and myeloid cells were scored as CFU-MIX. Colony forming potential was calculated following the equation: (Number of colonies formed/Number of cells plated) × 100.
Colony PCR
White blood cells
Genomic DNA was extracted from samples using the QIAamp DNA Blood Mini Kit (Qiagen). Excision of exon 2 was detected by PCR amplification on 40–60 ng genomic DNA using 20 μM forward and reverse primers as listed in Table S1. The target sequence was amplified running 35 cycles (assumed to be within the window of linearity), and the resulting amplicon was run on a 2% agarose gel (UltraPure Agarose, Thermo Fisher Scientific) with a 100-bp ladder (Thermo Fisher Scientific). Bands corresponding to 200 bp indicated the deletion of exon 2, while bands corresponding with 601 bp indicated the wildtype exon.
CFC colonies
Individual colonies were picked and stored in QuickExtract DNA Extraction Solution (Lucigen, QE09050). Colonies were heat lysed at 65°C for 20 min, 99°C for 10 min, and then cooled down. We used 5 μL of the lysed colony as a template for PCR amplification using the forward and reverse primers listed above. Resulting amplicons were run on a 2% agarose gel with 100-bp ladder. Bands corresponding with 200 bp indicated the deletion of exon 2, while bands corresponding to 600 bp indicate the wildtype exon.
CRISPR-Cas9 gene editing
Cas9 purified protein was obtained from Thermo Fisher Scientific (TrueCut Cas9 Protein v2, 5 g/L, A36499), and chemically modified guide RNA (gRNAs) (2′-O-methyl analogs and 3′ phosphorothioate internucleotide linkages at the first three 5′ and 3′ terminal RNA residues) were custom ordered from Synthego and are listed in Table S1. Lyophilized gRNAs were resuspended in nuclease-free water at a concentration of 200 pmol/L and stored as frozen aliquots at −80°C. For the editing of rhesus CD34+ cells, cells were first cultured overnight after enrichment and/or sorting. CRISPR-Cas9 RNPs were formed by combining 180 pmol of Cas9 protein with 1,800 pmol gRNA at room temperature for 10 min and used for the electroporation of 3 million cells in 2-mm cuvettes following the manufacturer’s instructions (Harvard Apparatus). Edited cells were recovered in medium overnight at 37°C before infusion in the animals.
Flow cytometry and FACS
Antibodies used for analysis and FACS sorting of NHP cells are listed in Tables S3 and S4. Dead cells and debris were excluded by FSC/SSC gating. Flow cytometric analysis was performed on an LSR Iiu, FACSAria Iiu, and Canto II (BD). Cells for in vitro assays as well as autologous NHP stem cell transplants were sorted right after CD34+ enrichment using a FACSAria Iiu cell sorter (BD) and sort purity assessed by recovery of sorted cells.
ddPCR
Two sets of primer and probe were selected based on three CD33 sequences provided (human, rhesus, and pseudogene), which share the same forward primer and probe. The reverse primer CD33Rev1 1210–1189 is specific to rhesus CD33. Duplex rhesus CD33 ddPCR is performed with CD33Fwd723-740, CD33Probe760-774, CD33Rev1 1210–1189 (Rhesus), and rhesus RPP30 primer and probe set. The primer and probe sequences are all listed in Table S1. The DNA ddPCR was performed on a BioRad QX200 instrument with ddPCR Supermix for probe (Biorad). The DNA droplets were generated with an Auto-Droplet Generator. The thermal condition of ddPCR were 95°C for 10 min, then 40 cycles at 94°C for 30 s, 60°C for 1 min, followed by 98°C for 10 min. The copy number of CD33 and RPP30 per reaction were analyzed and calculated with QuantaSoft analysis Pro software (BioRad).
Statistics
Statistical analysis of data was performed using GraphPad Prism Version 5. Details for the statistical analyses used can be found in the figure legends.
Acknowledgments
We thank Helen Crawford for help in preparing this manuscript and figures. This work was supported in part by grants to from the National Institutes of Health (R01 AI135953-01 [to H.P.K.], R01 HL136135 [to H.P.K.]), R01 CA266556 [to R.B.W.], and R21 CA245594 [to R.B.W.]). H.P.K. also received support as a Markey Molecular Medicine Investigator, as the inaugural recipient of the José Carreras/E. Donnall Thomas Endowed Chair for Cancer Research and as the Stephanus Family Endowed Chair for Cell and Gene Therapy.
Author contributions
S.R., O.H., R.B.W., and H.P.K. designed the study; E.F., M.L., H.Z., K.J., S.R., and O.H. performed longitudinal follow-up, CD34 enrichments, ddPCR, and FACS-sorts; S.R., N.E.P., and E.F. generated the figures; H.P.K. funded the study; N.E.P., S.R., G.S.L., R.B.W., and H.P.K. wrote the manuscript. All authors reviewed and edited the final manuscript.
Declaration of interests
S.R. is a consultant to Forty-Eight BIO Inc. and Ensoma Inc. R.B.W. received laboratory research grants and/or clinical trial support from Amgen, Aptevo, Celgene, ImmunoGen, Janssen, Jazz, Kura, MacroGenics, and Pfizer; has ownership interests in Amphivena; and is (or has been) a consultant to Abbvie, Adicet, Amphivena, BerGenBio, Bristol Myers Squibb, GlaxoSmithKline, ImmunoGen, Kura, and Orum. H.P.K is or was a consultant to and has or had ownership interests with Rocket Pharmaceuticals, Homology Medicines, V.O.R. Biopharma, and Ensoma Inc. H.P.K. has also been a consultant to CSL Behring and Magenta Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.101121.
Supplemental information
Data and code availability
Data presented in this manuscript are available upon request.
References
- 1.Laszlo G.S., Estey E.H., Walter R.B. The past and future of CD33 as therapeutic target in acute myeloid leukemia. Blood Rev. 2014;28:143–153. doi: 10.1016/j.blre.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 2.Appelbaum F.R., Bernstein I.D. Gemtuzumab ozogamicin for acute myeloid leukemia. Blood. 2017;130:2373–2376. doi: 10.1182/blood-2017-09-797712. [DOI] [PubMed] [Google Scholar]
- 3.Liu Y., Wang S., Schubert M.L., Lauk A., Yao H., Blank M.F., Cui C., Janssen M., Schmidt C., Göllner S., et al. CD33-directed immunotherapy with third-generation chimeric antigen receptor T cells and gemtuzumab ozogamicin in intact and CD33-edited acute myeloid leukemia and hematopoietic stem and progenitor cells. Int. J. Cancer. 2022;150:1141–1155. doi: 10.1002/ijc.33865. [DOI] [PubMed] [Google Scholar]
- 4.Jilani I., Estey E., Huh Y., Joe Y., Manshouri T., Yared M., Giles F., Kantarjian H., Cortes J., Thomas D., et al. Differences in CD33 intensity between various myeloid neoplasms. Am. J. Clin. Pathol. 2002;118:560–566. doi: 10.1309/1WMW-CMXX-4WN4-T55U. [DOI] [PubMed] [Google Scholar]
- 5.Humbert O., Laszlo G.S., Sichel S., Ironside C., Haworth K.G., Bates O.M., Beddoe M.E., Carrillo R.R., Kiem H.P., Walter R.B. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia. 2019;33:762–808. doi: 10.1038/s41375-018-0277-8. [DOI] [PubMed] [Google Scholar]
- 6.Kim M.Y., Yu K.R., Kenderian S.S., Ruella M., Chen S., Shin T.H., Aljanahi A.A., Schreeder D., Klichinsky M., Shestova O., et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. 2018;173:1439–1453.e19. doi: 10.1016/j.cell.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.El-Kharrag R., Berckmueller K.E., Madhu R., Cui M., Campoy G., Mack H.M., Wolf C.B., Perez A.M., Humbert O., Kiem H.P., Radtke S. Efficient polymer nanoparticle-mediated delivery of gene editing reagents into human hematopoietic stem and progenitor cells. Mol. Ther. 2022;30:2186–2198. doi: 10.1016/j.ymthe.2022.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borot F., Wang H., Ma Y., Jafarov T., Raza A., Ali A.M., Mukherjee S. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc. Natl. Acad. Sci. USA. 2019;116:11978–11987. doi: 10.1073/pnas.1819992116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw B.C., Estus S. Pseudogene-mediated gene conversion after CRISPR-Cas9 editing demonstrated by partial CD33 conversion with SIGLEC22P. CRISPR J. 2021;4:699–709. doi: 10.1089/crispr.2021.0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Radtke S., Adair J.E., Giese M.A., Chan Y.Y., Norgaard Z.K., Enstrom M., Haworth K.G., Schefter L.E., Kiem H.P. A distinct hematopoietic stem cell population for rapid multilineage engraftment in nonhuman primates. Sci. Transl. Med. 2017;9 doi: 10.1126/scitranslmed.aan1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Humbert O., Radtke S., Carrillo R.R., Reddy S.S., Perez A.M., Lux C., Peterson C.W., Schefter L.E., Negre O., Rawlings D.J., et al. Transplantation and persistence of CRISPR/Cas9-edited hematopoietic stem and progenitors cells for the reactivation of fetal hemoglobin in nonhuman primates (Abstract) Mol. Ther. 2018;26:296. [Google Scholar]
- 12.Bernacky B.J., Gibson S.V., Keeling M.E., Abee C.R. In: Laboratory Animal Medicine. 2nd edn. Fox J.G., Anderson L.C., Loew F.M., Quimby F.W., editors. Academic Press; 2002. Nonhuman Primates; pp. 675–791. [Google Scholar]
- 13.Humbert O., Radtke S., Samuelson C., Carrillo R.R., Perez A.M., Reddy S.S., Lux C., Pattabhi S., Schefter L.E., Negre O., et al. Therapeutically relevant engraftment of a CRISPR-Cas9-edited HSC-enriched population with HbF reactivation in nonhuman primates. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.AlJanahi A.A., Lazzarotto C.R., Chen S., Shin T.H., Cordes S., Fan X., Jabara I., Zhou Y., Young D.J., Lee B.C., et al. Prediction and validation of hematopoietic stem and progenitor cell off-target editing in transplanted rhesus macaques. Mol. Ther. 2022;30:209–222. doi: 10.1016/j.ymthe.2021.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson C.W., Haworth K.G., Burke B.P., Polacino P., Norman K.K., Adair J.E., Hu S.L., Bartlett J.S., Symonds G.P., Kiem H.P. Multilineage polyclonal engraftment of Cal-1 gene-modified cells and in vivo selection after SHIV infection in a nonhuman primate model of AIDS. Mol. Ther. Methods Clin. Dev. 2016;3 doi: 10.1038/mtm.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abraham A., Hsieh M., Eapen M., Fitzhugh C., Carreras J., Keesler D., Guilcher G., Kamani N., Walters M.C., Boelens J.J., et al. Relationship between mixed donor-recipient chimerism and disease recurrence after hematopoietic cell transplantation for sickle cell disease. Biol. Blood Marrow Transplant. 2017;23:2178–2183. doi: 10.1016/j.bbmt.2017.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fitzhugh C.D., Cordes S., Taylor T., Coles W., Roskom K., Link M., Hsieh M.M., Tisdale J.F. At least 20% donor myeloid chimerism is necessary to reverse the sickle phenotype after allogeneic HSCT. Blood. 2017;130:1946–1948. doi: 10.1182/blood-2017-03-772392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walters M.C., Patience M., Leisenring W., Rogers Z.R., Aquino V.M., Buchanan G.R., Roberts I.A., Yeager A.M., Hsu L., Adamkiewicz T., et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol. Blood Marrow Transplant. 2001;7:665–673. doi: 10.1053/bbmt.2001.v7.pm11787529. [DOI] [PubMed] [Google Scholar]
- 19.Beard B.C., Trobridge G.D., Ironside C., McCune J.S., Adair J.E., Kiem H.P. Efficient and stable MGMT-mediated selection of long-term repopulating stem cells in nonhuman primates. J. Clin. Invest. 2010;120:2345–2354. doi: 10.1172/JCI40767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Humbert O., Peterson C.W., Norgaard Z.K., Radtke S., Kiem H.P. A nonhuman primate transplantation model to evaluate hematopoietic stem cell gene editing strategies for beta-hemoglobinopathies. Mol. Ther. Methods Clin. Dev. 2018;8:75–86. doi: 10.1016/j.omtm.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Czechowicz A., Palchaudhuri R., Scheck A., Hu Y., Hoggatt J., Saez B., Pang W.W., Mansour M.K., Tate T.A., Chan Y.Y., et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat. Commun. 2019;10:617. doi: 10.1038/s41467-018-08201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Srikanthan M.A., Humbert O., Haworth K.G., Ironside C., Rajawat Y.S., Blazar B.R., Palchaudhuri R., Boitano A.E., Cooke M.P., Scadden D.T., Kiem H.P. Effective multi-lineage engraftment in a mouse model of Fanconi anemia using non-genotoxic antibody-based conditioning. Mol. Ther. Methods Clin. Dev. 2020;17:455–464. doi: 10.1016/j.omtm.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saha A., Hyzy S., Lamothe T., Hammond K., Clark N., Lanieri L., Bhattarai P., Palchaudhuri R., Gillard G.O., Proctor J., et al. A CD45-targeted antibody-drug conjugate successfully conditions for allogeneic hematopoietic stem cell transplantation in mice. Blood. 2022;139:1743–1759. doi: 10.1182/blood.2021012366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu B., Liu D. Gemtuzumab ozogamicin and novel antibody-drug conjugates in clinical trials for acute myeloid leukemia. Biomark. Res. 2019;7:24. doi: 10.1186/s40364-019-0175-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cummins K.D., Salas-McKee J., Kulikovskaya I., Gohil M., Nobles C., Bushman F., Lacey S., Gill S. Optimization of CRISPR-Cas9 knock-out of CD33 in human hematopoietic stem/progenitor cells for allogeneic transplantation in patients with acute myeloid leukemia. Cytotherapy. 2019;21:S16–S17. doi: 10.1016/j.jcyt.2019.03.585. [DOI] [Google Scholar]
- 26.Samuelson C., Radtke S., Zhu H., Llewellyn M., Fields E., Cook S., Huang M.L.W., Jerome K.R., Kiem H.P., Humbert O. Multiplex CRISPR/Cas9 genome editing in hematopoietic stem cells for fetal hemoglobin reinduction generates chromosomal translocations. Mol. Ther. Methods Clin. Dev. 2021;23:507–523. doi: 10.1016/j.omtm.2021.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gaudelli N.M., Komor A.C., Rees H.A., Packer M.S., Badran A.H., Bryson D.I., Liu D.R. Programmable base editing of A∗T to G∗C in genomic DNA without DNA cleavage. Nature. 2017;551:464–471. doi: 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trobridge G.D., Beard B.C., Gooch C., Wohlfahrt M., Olsen P., Fletcher J., Malik P., Kiem H.P. Efficient transduction of pigtailed macaque hematopoietic repopulating cells with HIV-based lentiviral vectors. Blood. 2008;111:5537–5543. doi: 10.1182/blood-2007-09-115022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng H.B., Watkins B., Tkachev V., Yu S., Tran D., Furlan S., Zeleski K., Singh K., Hamby K., Hotchkiss C., et al. The knife's edge of tolerance: Inducing stable multilineage mixed chimerism but with a significant risk of CMV reactivation and disease in rhesus macaques. Am. J. Transplant. 2017;17:657–670. doi: 10.1111/ajt.14006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adair J.E., Waters T., Haworth K.G., Kubek S.P., Trobridge G.D., Hocum J.D., Heimfeld S., Kiem H.P. Semi-automated closed system manufacturing of lentivirus gene-modified haematopoietic stem cells for gene therapy. Nat. Commun. 2016;7 doi: 10.1038/ncomms13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data presented in this manuscript are available upon request.