

Summary
ALDH1A1 is one of the classical stem cell markers for bladder cancer. Lysine 2-hydroxyisobutyrylation (Khib) is a newfound modification to modulate the protein expression, and the underlying mechanisms of how ALDH1A1 was regulated by Khib modification in bladder cancer remains unknown. Here, ALDH1A1 showed a decreased K260hib modification, as identified by protein modification omics in bladder cancer. Decreasing ALDH1A1 expression significantly suppressed the proliferation, migration and invasion of bladder cancer cells. Moreover, K260hib modification is responsible for the activity of ALDH1A1 in bladder cancer, which is regulated by HDAC2/3. Higher K260hib modification on ALDH1A1 promotes protein degradation through chaperone-mediated autophagy (CMA), and ALDH1A1 K260hib could sensitize bladder cancer cells to chemotherapeutic drugs. Higher ALDH1A1 expression with a lower K260hib modification indicates a poor prognosis in patients with bladder cancer. Overall, we demonstrated that K260hib of ALDH1A1 can be used as a potential therapeutic target for bladder cancer treatment.
Subject areas: Biochemistry, Cancer
Graphical abstract
Highlights
-
•
High expression of ALDH1A1 in bladder cancer promotes tumor progression
-
•
Higher K260hib modification of ALDH1A1 promotes protein degradation through CMA
-
•
K260hib of ALDH1A1 could decrease tumor growth and chemoresistance in bladder cancer
-
•
Higher ALDH1A1 expression with a lower K260hib indicates a poor prognosis
Biochemistry; Cancer
Introduction
Bladder cancer is the most common malignant tumor of the urinary system,1 and its incidence is increasing across the world. Most of the patients with bladder cancer were diagnosed with non-muscle-invasive cancer, and the recurrence rate of non-muscle-invasive bladder cancer (NMIBC) including local and treatable tumors by the transurethral resection of bladder tumor (TUR-BT) is relatively high; moreover, it is prone to progress to muscle-invasive bladder cancer (MIBC),2 accounting for approximately 20–25% of all cases. The 5-year survival rate of MIBC is extremely low due to the high heterogeneity of bladder cancer, despite the availability of multidisciplinary therapies such as radical surgery, chemotherapy, and immunotherapy.3
According to recent reports, post-translational modification (PTM) is closely related to tumor progression and recurrence.4 In the past two decades, new PTMs have been discovered, such as propionylation, malonylation, butyrylation, succinylation, crotonylation, β-hydroxybutyrylation, and 2-hydroxyisobutyrylation,5,6,7,8,9,10,11 which are structurally and functionally distinct from the widely studied lysine acetylation and play a significant role in the metabolic regulation of gene expression.11 However, the molecular mechanisms underlying these PTMs in bladder cancer have rarely been explored. Khib, a recently discovered post-translational modification, was closely associated with the processes of transcription, translation, protein degradation, and glucose metabolism,12,13 and it has been identified to play an ultimate role in metastasis of esophageal cancer by affecting the stability of NOTCH3.14 However, the effect of Khib modification on bladder cancer remains unclear.
Cancer stem cells (CSCs) are a group of cells that can self-new and multi-differentiate. The existence of CSCs have been proven to be closely associated with tumor recurrence, metastasis, and chemo-resistance.15,16,17,18 ALDH1A1 is one of the classical markers for CSC identification. The aberrant expression of ALDH1A1 can promote the catalytic NAD(P)+-dependent oxidation of retinaldehyde (RALD) to the corresponding retinoic acid (RA). RA then enters the nucleus and binds to retinoic acid receptors (RARs) or retinoid X receptors (RXRs) so as to induce the expression of its downstream target genes, which are especially focused on the cell differentiation and proliferation.19 In the past studies, ALDH1A1 has been recognized as a diagnostic target in bladder cancer,20,21,22,23,24,25 and ALDH1A1-induced drug resistance has been commonly reported in cancers. Moreover, specific ALDH1A1-targeted drugs developed by researchers have been used to treat cancers.26,27,28,29,30 However, no ALDH1A1-targeting drug has yet been applied to clinical treatment until date. We therefore attempted to explore multiple ways to block ALDH1A1, including the targeting of ALDH1A1-Khib.
In this work, we first detected the ubiquitination, lysine acetylation, lysine succinylation, lysine malonylation, and Khib modification by pan-antibody between bladder cancer and adjacent normal tissues. A significant difference was noted in Khib between them. Therefore, we conducted further research to explore the role of Khib modification in bladder cancer. Additionally, ALDH1A1 with a significantly lower level of Khib modification was identified in cases of bladder cancer. And a CMA pathway of ALDH1A1 degradation promoted by high K260hib modification was uncovered, and it could decrease tumor proliferation and metastasis in bladder cancer. Furthermore, ALDH1A1 K260hib was correlated with the clinical stage and prognosis of patients with bladder cancer. Moreover, K260hib of ALDH1A1 could sensitize bladder cancer cells to chemotherapeutic drugs. Altogether, these findings could not only enrich the research on Khib modification in cancer but also help explore the relationship between the Khib modification of ALDH1A1 and bladder cancer progression, thereby providing a promising therapy target for bladder cancer.
Results
Identification of ALDH1A1 with Khib modification in bladder cancer by MS
First, we identified 493 differential Khib modification sites in 287 proteins from 10 pairs of bladder cancer and the adjacent normal tissues by high pH reverse-phase HPLC/LC-MS/MS. Among these, the Khib level of 203 proteins with 391 modification sites was increased, and 84 proteins with 102 modification sites were decreased (Figure 1A). To further confirm the targeted Khib sequence preferences, motif analysis of all Khib sites revealed the flanking sequence motif of Khib proteins enriched with alanine, aspartic acid, and glutamic acid at −1, −2, −3, −4, +1, +2, +3, and +4 position, and histidine, isoleucine, methionine, and asparagine were the most depleted (Figure 1B). Generally, the subcellular localization of a protein is closely associated with its functions, and approximately half of these proteins are located in the cytoplasm (Figure 1C). In the GO enrichment analysis of these differential proteins between cancer and adjacent tissues, we identified the top 20 GO categories with significant enrichment of proteins relevant to cell secretion, extracellular space, and cell adhesion (Figure 1D). Moreover, the results of KEGG analysis indicated that different Khib modification levels of proteins were mainly enriched in the pathways of focal adhesion, tight junction, and leukocyte transendothelial migration. These pathways were closely associated with epithelial–to–mesenchymal transition (EMT) (Figure 1E). EMT is responsible for the acquisition of CSCs phenotypes in tumors.31 Among the proteins with Khib modification, ALDH1A1 with a significantly different Khib modification caught our attention as a marker of CSCs in bladder cancer.
Figure 1.

Identification ALDH1A1 with lysine 2-hydroxyisobutyrylation by MS from bladder cancer
(A) Differential Khib modification sites and proteins respectively between bladder cancer tissues and adjacent normal tissues (>1.2 or <0.83) (n = 10 per group).
(B) Flanking sequence analysis of all Khib sites.
(C) The subcellular localization of proteins.
(D) GO enrichment of differentially expressed proteins.
(E) KEGG analysis of proteins with the different Khib modification levels.
(F) Increased expression of ALDH1A1 was associated with advanced tumor stage in TCGA-BLCA database.
(G) Kaplan-Meier survival curves showed that higher ALDH1A1expression was significantly related with worse OS in TCGA-BLCA database.
(H) Higher expression of ALDH1A1 was related with advanced tumor stage in 105 patients with bladder cancer from our hospital (I) Kaplan-Meier survival curves revealed that higher ALDH1A1 expression was significantly related with worse OS in 105 patients with bladder cancer from our hospital.
(J‒L) The correlation of ALDH1A1 with CD44, CD133, and CD47 in the TCGA-BLCA database.
In the TCGA-BLCA database, the clinical data of the T stage was closely correlated with the expression of ALDH1A1, and the advanced stage was observed in association with a higher expression of ALDH1A1 (Figure 1F). Furthermore, patients with higher ALDH1A1 expression tended to have a worse overall survival (OS) (Figure 1G). Meanwhile, the expression of ALDH1A1 was detected by immunohistochemical staining in bladder cancer tissues collected from 105 patients at our hospital, and it was found that the expression of ALDH1A1 in advanced stage bladder cancer was significantly higher than that in the lower stage (Figure 1H), while the patients with higher ALDH1A1 expression showed association with a worse OS (Figure 1I). Furthermore, the correlation of ALDH1A1with other CSCs markers (such as CD44, CD133, and CD47) were positively correlated in the TCGA-BLCA database (Figures 1J‒1L). We found that the expression of CD44 was altered by ALDH1A1 knockdown and overexpression in T24 and UMUC3 cells (Figures S1A and S1B).
ALDH1A1 promoting tumor progression through the retinoic acid pathway in bladder cancer
Tumor progression and chemotherapy resistance are usually caused by bladder CSCs.32,33 First, we found that ALDH1A1 promotes tumor progression through retinol metabolism by Gene Set Variation Analysis (GSVA) in the TCGA-BLCA database (Figure S1C), and the β-catenin and p-AKT pathways were identified as the main signal pathways with ALDH1A1 through Gene Set Enrichment Analysis (GSEA) (Figure S1D). Next, we found that the expression of ALDH1A1 was increased in T24 and UMUC3 cells after treatment with gemcitabine and cisplatin (Figures 2A and 2B), and the expression of ALDH1A1 detected by IHC in bladder cancer tissues was significantly higher than that in its adjacent tissues (Figure S2A). Next, we efficiently knocked down the expression of ALDH1A1 by ALDH1A1-siRNA in T24 and UMUC3 cells (Figure S2B); knocking down ALDH1A1 could suppress the cell viability by the MTT assay in T24 and UMUC3 cells (Figures 2C and S2C). Meanwhile, we observed that the knockdown of ALDH1A1 could dramatically decrease the ability of cell migration and invasion (Figures 2D and S2D). For further research, the shALDH1A1-T24 cell line was established via lentivirus infection (Figure S2E). To evaluate the stem ability of ALDH1A1, a sphere-forming assay was performed to examine the spheroid formation ability, which demonstrated that knocking down ALDH1A1 could suppress the formation and number of spheroids (Figure 2E) as well as reduce the colony formation ability (Figure 2F).
Figure 2.

ALDH1A1 promoting tumor progress through the RA pathway
(A and B) The protein expression of ALDH1A1 was increased with the treatment of gemcitabine and cisplatin in T24 and UMUC3 cells. The final concentration of gemcitabine and cisplatin were 0, 0.1 μg/mL, 0.3 μg/mL and 0.5 μg/mL respectively.
(C) The cell proliferation and growth measured by the MTT assay in T24 cells.
(D) The migration and cell invasive ability was reduced in T24 cell line by knocking down ALDH1A1. Scale bar = 200 μm.
(E) Two stable cells (NC-shRNA and ALDH1A1-shRNA) were analyzed by sphere-forming assay. Scale bar = 100 μm.
(F) Two stable cells (NC-shRNA and ALDH1A1-shRNA) were seeded into the 6-well plates to subject to colony formation analysis.
(G and H) Western blotting analysis of ALDH1A1, RXRα, p-AKT, and β-catenin in T24 cells with different treatments.
(I and J) shALDH1A1 cells were cultured with ATRA gained a stronger ability of proliferation, migration and invasion. Scale bar = 200 μm.
(K and L) p-AKT and β-catenin were upregulated by added ATRA in shALDH1A1 cells by Western blotting. For cell experiments, each experiment was performed at least three times. Data are represented as mean ± SEM, ∗∗p < 0.01, ∗∗∗p < 0.001 by two-sided Student’s t test.
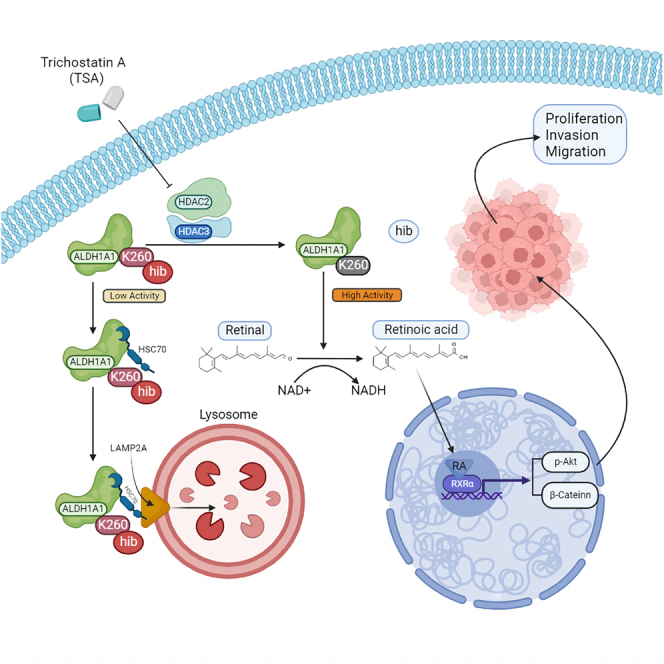
Next, we explored the molecular mechanisms underlying the oncogenic role of ALDH1A1 in bladder cancer. ALDH1A1 is a metabolic enzyme that specifically oxidizes the conversion of retinaldehyde (RALD) to retinoic acid. Therefore, we first evaluated the genes related to the RA pathway. RT-qPCR results revealed that RARα and RXRα were suppressed by the ALDH1A1 knockdown (Figure S2F). Furthermore, the detected protein expression of RXRα, p-AKT, and β-catenin varied consistently with the ALDH1A1 knockdown or overexpression in T24 and UMUC3 cells (Figures 2G, 2H, S2H, and S2I). Meanwhile, NCT-501, an inhibitor of ALDH1A1 (Figure S2G), was used to detect the expression of RXRα, p-AKT, and β-catenin, which displayed similar results (Figures 2G and S2H). For further analysis, the shALDH1A1-T24 cells were cultured with all-trans-retinoic acid (ATRA) or retinaldehyde (RALD), and the ATRA group demonstrated a stronger cell viability, migration and invasion abilities (Figures 2I and 2J). Meanwhile, the results showed that p-AKT and β-catenin were upregulated with the addition of ATRA instead of RALD (Figures 2K and 2L). In addition, the effect of ATRA on the ALDH1A1WT-T24 cells was consistent with that on the shALDH1A1-T24 cells (Figures S2J and S2K). All these results suggest that ALDH1A1 could promotes tumorigenesis by activating the RA signaling pathway in bladder cancer cells.
2-Hydroxyisobutyrylation at lysine 260 site of ALDH1A1
As a stem cell marker of bladder cancer, ALDH1A1 could promote tumorigenesis. Moreover, the 2-hydroxyisobutyrylation of ALDH1A1 in bladder cancer tissues was identified in our PTM proteomics results, and one 2-hydroxyisobutyrylation site was identified in ALDH1A1 K260 by MS (Figure 3A), after which we blasted the K260 site of ALDH1A1 among different species from rats to humans and noted that the K260 site was evolutionarily conserved among them, which further suggested the extreme importance of K260 for biological functions (Figure 3B). Furthermore, the lysine 2-hydroxyisobutyrylation modification level of ALDH1A1 was identified and heightened after treatment with trichostatin A (TSA), an inhibitor of histone deacetylase HDACs, rather than with another SIRT family of deacetylase inhibitor nicotinamide (NAM) in T24 bladder cancer cells (Figure 3C). A similar phenomenon was recorded in the 293T cells by transferring ALDH1A1 to detect the Khib modification level of ALDH1A1 (Figure 3D). These results suggested the presence of a Khib modification in ALDH1A1, which could be de-2-hydroxyisobutyrylated by HDACs. Meanwhile, we mutated the K260 site to threonine (T, mimicking the 2-hydroxyisobutyrylation status) or arginine (R, mimicking de-2-hydroxyisobutyrylation status) and found that K260 mutation decreased the 2-hydroxyisobutyrylation levels of ALDH1A1 with the ectopic ALDH1A1 overexpression (Figures 3E and S2L), which suggested that the K260 site was the main 2-hydroxyisobutyrylated site of ALDH1A1.
Figure 3.

Lowered ALDH1A1 enzyme activity and cell viability due to high levels of K260hib modification
(A) The 2-hydroxyisobutyrylation site of ALDH1A1was identified at K260 site by mass spectrometry.
(B) K260 site was evolutionarily conserved by T-coffee database.
(C) Treatment with NAM and TSA increases endogenous ALDH1A1 2-hydroxyisobutyrylation.
(D) ALDH1A1 was 2-hydroxyisobutyrylated, Flag-tagged ALDH1A1 was overexpressed in HEK 293T cells followed the treatment with the TSA and NAM, and then the level of 2-hydroxyisobutyrylation was analyzed by Western blotting with anti-pan 2-hydroxyisobutyrylation antibody.
(E) K260 mutation decreased the 2-hydroxyisobutyrylation levels of ALDH1A1 in 293T cells.
(F) Flag-tagged ALDH1A1WT, ALDH1A1K260T, and ALDH1A1K260R mutants were individually transfected into the 293T cells, and the dehydrogenase activity assay of ALDH1A1 was conducted with the NADH as the substrate after treatment of TSA, and changes of the absorbance at 340 nm were measured to reveal a decrease of NADH.
(G) The protein expression of ALDH1A1 in the four rescued stable cell lines were evaluated by Western blotting.
(H) MTT assay was performed to analyze the ability of proliferation for ALDH1A1 knockdown T24 re-expressed with vector, ALDH1A1WT, ALDH1A1K260T, and ALDH1A1K260R rescued cell lines.
(I) Four stable rescued cells (Vetor, ALDH1A1WT, ALDH1A1K260T, and ALDH1A1K260R) were seeded into the 6-well plates to subject to colony formation analysis.
(J) Indicated cells described as in (H, I) were analyzed by trans-well assay. Scale bar = 200 μm.
(K) ALDH1A1WT and ALDH1A1K260T were analyzed by sphere-forming assay. Scale bar = 100 μm. For cell experiments, each experiment was performed at least three times. Mean ± SEM, ∗p < 0.05, ∗∗∗p < 0.001 by two-sided Student’s t test or one-way ANOVA. ns: not significant.
As ALDH1A1 is a metabolic enzyme, it is speculated that the mutation of the conservative K260 site may induce a change in the enzyme activity. To investigate the role of ALDH1A1 K260 2-hydroxyisobutyrylation, the enzyme activity of ALDH1A1K260T and ALDH1A1K260R were compared with that of wild-type ALDH1A1. We found that the activity of ALDH1A1K260T demonstrated only 40% of the ALDH1A1WT (Figure S2M). Next, we examined the enzymatic activity of ALDH1A1 by TSA treatment to inhabit deacetylases, which decreased the enzyme activity by >60% relative to the control activity (Figure S2N). However, the activity of ALDH1A1K260T and ALDH1A1K260R showed only a slight effect after treatment of TSA (Figure 3F).
To explore the function of ALDH1A1 K260hib in the bladder cancer cells, we constructed four stable re-expression cell lines based on the shALDH1A1 cell line, namely, Vector, ALDH1A1(WT), ALDH1A1(K260T), and ALDH1A1(K260R) (Figure 3G). The re-expression of ALDH1A1WT cells could enhance cell growth when compared with that of the vector, with a significant decrease of ALDH1AK260T stable cells when compared with that of ALDH1A1WT cells, as assessed by MTT and colony formation assays (Figures 3H and 3I). Similarly, the re-expression of ALDH1A1WT cells, but not ALDH1A K260T cells increased the migration and invasion abilities (Figure 3J). Furthermore, the results of sphere-forming assay revealed that ALDH1A1 K260hib impaired the formation and number of spheroids (Figure 3K). Altogether, these results suggested that K260hib of ALDH1A1 could decrease the cell proliferation, spheroid formation, migration, and invasion abilities.
Lysine 2-hydroxyisobutyrylation and protein expression of ALDH1A1 regulated by HDAC2/3
As reported previously, HDAC2 and HDAC3 were identified as Khib deacetylases in mammal proteins.12 We found that the Khib level of ALDH1A1 decreased by HDAC2 and HDAC3 overexpression in our study (Figure 4A), meanwhile, knocking down HDAC2 or HDAC3 significantly enhanced the Khib level of ALDH1A1 (Figures 4B and 4C). In conclusion, HDAC2/3 could regulate the Khib level of ALDH1A1. Furthermore, the interaction of endogenous HDAC2/3 and ALDH1A1 was confirmed by co-IP in both T24 and UMUC3 bladder cancer cells (Figures 4D, 4E, S3A, and S3B), and the interaction between HDAC2/3 and ALDH1A1 was identified by their exogenous overexpression in 293T cells (Figures S3C and S3D). In addition to the observation of the interaction between HDAC2/3 and ALDH1A1, we explored whether ALDH1A1 could be regulated by HDAC2/3, followed by the determination of whether the regulation of ALDH1A1 by HDAC2/3 occurred at or after transcription. Quantitative RT-qPCR revealed that the overexpression of HDAC2/3 could not change the expression of ALDH1A1 at the mRNA level in both T24 and UMUC3, as was also noted after knocking down HDAC2/3 by RNA interference (Figures S3E and S3F). As HDAC2/3 did not regulate ALDH1A1 at the mRNA level, we speculated whether the protein level of ALDH1A1 could be altered by altering HDAC2/3. Western blotting results indicated that increasing and knocking down HDAC2/3 could induce a corresponding change at the protein levels in T24 and UMUC3 cells (Figures 4F, 4G, S3G, and S3H).
Figure 4.

Expression of ALDH1A1 with Khib modification regulated by HDAC2/3
(A) Flag-tagged ALDH1A1 was co-transfected with vector or HA-tagged HDAC2/HDAC3 respectively as indicated.
(B and C) Flag-tagged ALDH1A1 was overexpressed in T24 cells, and the Khib of ALDH1A1 was measured by knocking down HDAC2 and HDAC3.
(D and E) Endogenous ALDH1A1 was IPed from T24 cells. Rabbit IgG was used as a control.
(F and G) Western blotting analysis of HDAC2/3 and ALDH1A1 in HDAC2/3 overexpressed and knocking down T24 cells.
(H) T24 cells were either untreated or treated with TSA for different lengths of time as mentioned earlier, meanwhile, the cells were cultured with cycloheximide (CHX) and harvested at indicated time points. Then, immunoblotting against ALDH1A1 and α-actinin control was performed.
(I) MG132 was added into the culture medium without or with TSA in T24 cells, as indicated to determine the ubiquitination of ALDH1A1.For cell experiments, each experiment was performed at least three times.
Based on these results, TSA as an inhibitor of HDAC2/3 could increase the 2-hydroxyisobutylation modification of ALDH1A1. Next, we explored whether TSA could regulate ALDH1A1. Quantitative RT-qPCR was performed to measure the mRNA level of ALDH1A1 after treatment with TSA, and the results indicated no significant effect on the ALDH1A1 mRNA level (Figure S3I), suggesting the possible existence of the post-transcriptional regulation of ALDH1A1 protein by TSA. Next, we analyzed the protein levels of ALDH1A1 after treatment with TSA. Western blotting results demonstrated that the protein levels of ALDH1A1 gradually decreased with time (Figure S4A). To observe the effect of TSA on the half-life of ALDH1A1, we cultured T24 cells without or with TSA after treatment of cycloheximide, and the protein levels of ALDH1A1 were examined with respect to the corresponding time (Figure 4H). These results suggested that ALDH1A1 is a relatively stable protein with a half-life of >10 h and that TSA could shorten the half-life of ALDH1A1 protein within 8 h.
The ubiquitin-proteasome and autolysosome are two classical protein degradation pathways. We explored how to promote the ALDH1A1 degradation after using TSA. For this purpose, we first determined whether the inhibition of ubiquitin-proteasome by MG132 did not increase the amount of ALDH1A1 levels at the indicated time points in T24 and UMUC3 cells (Figures S4B and S4C). Next, the cell culture medium was supplemented with MG132 in T24 and UMUC3 cells after treatment with TSA, but it did not rescue the TSA-induced decrease in the AlDH1A1 protein level and the ubiquitination level of ALDH1A1 did not increase significantly after TSA treatment in T24 and UMUC3 cells (Figures 4I and S4D). To further examine how TSA degrades ALDH1A1, we treated the cells with the proteasome inhibitor MG132 or an inhibitor of lysosomal proteases chloroquine (CQ) to determine the protein levels of ALDH1A1. As expected, CQ could increase the protein levels of ALDH1A1 rather than MG132 (Figure S4E). Altogether, the abovementioned results implied that TSA may regulate the stability of ALDH1A1 through the lysosome pathway.
K260hib modification of ALDH1A1 promoting its degradation through chaperone-mediated autophagy
Autophagy is a major mechanism for regulating the catabolic process that is mainly involved in the degradation of cytoplasmic organelles and proteins.34 Macro-autophagy, micro-autophagy, and CMA are the three forms of autophagy in mammalian cells.35 The substrates of CMA are selectively specialized to certain proteins that exist with a relatively longer half-life.36 Since ALDH1A1 is a longevity protein, we speculated that it may be degraded through CMA. First, CQ could increase the protein levels of ALDH1A1at the indicated time (Figure 5A); meanwhile, the cells were cultured with MG132 or CQ after treatment with TSA, indicating that the ALDH1A1 protein evidently increased in the CQ cultured group by Western blotting (Figure 5B). As previously reported, prolonged serum starvation could activate CMA,37 and the ALDH1A1 protein level decreased continuously with the time of serum starvation (Figure S4F). Usually, CMA was defined as a process wherein the HSC70 chaperone first recognized the target proteins and then bind the LAMP2A receptor into the lysosome for degradation. To verify this notion, we found that knocking down LAMP2A could increase the ALDH1A1 protein level (Figure S4G). Furthermore, the ALDH1A1 protein level reduction induced via serum starvation could be blocked by LAMP2A knockdown (Figure 5C). Meanwhile, we observed that LAMP2A and ALDH1A1 not only interacted with each other in the T24 cell line (Figure 5D) but were also co-located in cells by IF (Figure 5E). In conclusion, the ALDH1A1 protein level was regulated by CMA in the bladder cells.
Figure 5.

2-Hydroxyisobutyrylation at lys-260 promoting ALDH1A1 degradation through chaperone-mediated autophagy
(A) T24 cells were treated with CQ, and the ALDH1A1 protein levels were detected by immunoblotting at the indicated time points.
(B) MG132 or CQ was cotreated with TSA in T24 cells to observe the protein levels of ALDH1A1 by Western blotting.
(C) LAMP2A was knocked down in T24 cells by siRNA, and the knockdown efficiency of LAMP2A and ALDH1A1 protein levels were examined by Western blotting.
(D) Endogenous LAMP2A was IPed from T24 cells. Rabbit IgG was used as a control.
(E) Colocalization of ALDH1A1 and LAMP2A in T24 cells was observed by confocal microscopy. Scale bar = 20 μm.
(F) TSA treatment decreased the level of ALDH1A1 wild-type, but not K260R mutant ALDH1A1 in T24 cells.
(G) TSA treatment decreased the level of ALDH1A1 wild-type, but not K260R mutant ALDH1A1 in 293T cells.
(H) HA-tagged HSC70 was co-transfected with vector, flag-tagged ALDH1A1WT, or flag-tagged ALDH1A1K260T into T24cells, the ALDH1A1-HSC70 binding was determined by immunoprecipitation-Western blotting analysis.
(I) The cell lysates were collected with ALDH1A1WT, ALDH1A1K260T, or ALDH1A1K260R overexpressed in T24 cells followed treatment with 1% glutaraldehyde, while the polymerizations of ALDH1A1 were examined by immunoblotting.
(J and K) The crystal structures of ALDH1A1WT and ALDH1A1K260T were analyzed by AlphaFold 2.0 software. For cell experiments, each experiment was performed at least three times.
We then overexpressed ALDH1A1WT, ALDH1A1K260T, and ALDH1A1K260R in the 293T and T24 cells and found that the protein level of ALDH1A1K260T decreased more than that of the ALDH1A1WT and that the protein level of ALDH1A1K260R did not reduce, rather increased, along with the protein expression of ALDH1A1 in the four stable cell lines mentioned above (Figures S5H and S5I). We found that the TSA only reduced the protein level of wild-type ALDH1A1, but had no decreasing effect on ALDH1A1K260R mutant in the T24 and 293T cells (Figures 5F and 5G). Based on these data, we hypothesized that the 2-hydroxyisobutyrylation of the K260 site could reduce ALDH1A1 protein stability. Accordingly, we further examined whether the 2-hydroxyisobutyrylation affects the stability of ALDH1A1 through CMA. For this purpose, we first co-expressed the ALDH1A1 and HSC70 in 293T and T24 cells and subjected them to co-IP. The results demonstrated that ALDH1A1K260T mutant had a much stronger interaction with HSC70 than the wild-type ALDH1A1 (Figures 5H and S5A); meanwhile, treatment with TSA could enhance the binding between wild-type ALDH1A1 and HSC70 (Figure S5B). This finding suggests that the 2-hydroxyisobutyrylation of ALDH1A1 at the K260 site may induce self-degradation through the CMA pathway.
ALDH1A1 can exist in monomeric, dimeric, or tetrameric forms,38 and ALDH1A1 exhibited a high enzyme activity only when it existed in a tetramer form.39 In our study, the K260 site of AlDH1A1 was modified by 2-hydroxyisobutylation; therefore, we investigated whether 2-hydroxyisobutylation at the K260 site affected the crystal structure and function of ALDH1A1. First, the crystal structure of the ALDH1A1 protein revealed that the K260 site was located on the external surface of the ALDH1A1 protein (Figure S5C). A previous study reported that this protein included an NAD+ binding domain (8–135 and 159–270 amino acids), a catalytic domain (271–470), and an oligomerization domain (140–158 and 486–495 amino acids).40 The crystal structure of ALDH1A1 demonstrated that the K260 site was far away from the catalytic active center (Figure S5D) and hence unlikely to affect its enzyme activity through domain change. Therefore, it is possible to reduce the enzyme activity by decreasing the protein stability by 2-hydroxyisobutylation. In light of this finding, the results of Western blotting suggested that ALDH1A1 mainly exists in the form of tetramer in bladder cancer and that ALDH1A1 decreased significantly after the treatment of TSA in T24 cells. This finding was consistent with the ALDH1A1 K260T mutant (Figure S5E). On the contrary, the ALDH1A1 K260R mutant evidently increased (Figure 5I). Then, we determined how 2-hydroxyisobutylation of K260 induced the protein reduction of ALDH1A1. Accordingly, we analyzed the ALDH1A1 monomer by AlphaFold 2.0 software, and the results revealed an electrostatic interaction between K260 and E256 at the interface (Figures 5J and 5K). Furthermore, salt bridges between K260 and E256 existed not only within the monomer but also between the monomers in tetrameric forms (Figures 5J and 5K). However, all the interactions were destroyed after the K260 mutation. This resulted in instability of the ALDH1A1 tetramer, which was then easily recognized by proteasomes or lysosomes, leading to protein degradation and a loss of enzyme activity.
Elevated K260hib of ALDH1A1 indicated a better prognosis in patients with bladder cancer
Next, we determined how Khib of ALDH1A1 altered in response to cellular stress. First, we treated cells in a medium with no serum to examine the level of Khib in ALDH1A1. The results showed that ALDH1A1 responded to no serum by reducing the level of Khib in 293T (Figure S5F) and T24 cells (Figure 6A). Moreover, we observed that Khib of ALDH1A1 decreased significantly in 293T (Figure S5G) and T24 cells with EBSS treatment (Figure 6B). Furthermore, Khib of ALDH1A1 was decreased significantly in T24 and UMUC3 cells after treatment with gemcitabine and cisplatin (Figures 6C and S5H). Subsequently, the results of MTT assay revealed that K260hib modification could sensitize the bladder cancer cells to gemcitabine and cisplatin treatment (Figure S6A). In addition, the apoptotic cells detected by flow cytometry were increased in the ALDH1A1 K260T group (Figures S6B and S6C). And the sphere-forming efficiency was also decreased in this ALDH1A1 K260T group (Figure S6D). Taken together, these results suggested that ALDH1A1 responded to an external stress by decreasing the level of Khib modification, and that the K260hib modification could reduce the chemotherapy resistance.
Figure 6.

2-Hydroxyisobutyrylation at lys-260 of ALDH1A1 affects tumor growth in vivo and was associated with prognosis in bladder cancer
(A and B) Flag-tagged ALDH1A1 was transfected into T24 cells, and the cells were treated with serum starvation and EBSS stresses for the indicated time to observe the level of 2-hydroxyisobutyrylation.
(C) T24 cells were treated with GC for the indicated time to observe the level of 2-hydroxyisobutyrylation. The final concentration of gemcitabine and cisplatin were 0, 0.1 μg/mL and 0.3 μg/mL respectively.
(D) Xenograft was performed in BALB/c nude mice injected with T24 ALDH1A1WT or ALDH1A1K260T rescued cells (n = 6 per group). The mice were sacrificed 2 weeks after injection, and the tumors were obtained and photographed. The volume of tumors were measured every 3 days after T24 ALDH1A1WT or ALDH1A1K260T rescued cells injection (p < 0.05).
(E) Representative pulmonary CT images acquired from mice with intravenous injection of ALDH1A1WT/ALDH1A1K260T at 5 weeks. Black arrows revealed the detection of the metastatic tumors in the lungs (n = 5 per group). Mice were sacrificed, the lungs were obtained and photographed. Red arrows revealed the detection of the metastatic nodesin the lungs (n = 5 per group).
(F) Then, the lungs were also fixed in 4% paraformaldehyde for HE staining. Up: Scale bar = 500 μm. Down: Scale bar = 50 μm.
(G) IHC analysis of the levels of ALDH1A1 and ALDH1A1-K260hib in bladder cancer tissues. Scale bar = 200 μm.
(H) Chi-square test was used to analyze the correlation between the expression levels of ALDH1A1 and ALDH1A1-K260hib in bladder cancer (p < 0.05).
(I) The lower expression of ALDH1A1K260hib was associated with an advanced tumor stage in 105 patients with bladder cancer from our hospital.
(J) Kaplan–Meier analysis of OS with 105 patients with bladder cancer was assessed between strong and weak staining of ALDH1A1-K260hib (log rank of p < 0.05).
(K) Kaplan–Meier analysis of RFS was evaluated according to the level of ALDH1A1 K260hib in 105 patients with bladder cancer. For cell experiments, each experiment was performed at least three times. For animal experiments, five to six per group were used for in vivo studies. Mean ± SEM, ∗p < 0.05, ∗∗∗p < 0.001 by two-sided Student’s t test.
Furthermore, we performed a xenograft assay to determine the physiological effect of ALDH1A1 K260hib on tumor growth in vivo. As expected, ALDH1A1K260T decreased the tumor growth in the xenograft models, The tumors obtained from the mice injected with the ALDH1A1K260T cells exhibited a 79.1% decrease in their volumes when compared with the tumors injected with the ALDH1A1WT cells (Figure 6D). Meanwhile, for the in vivo metastasis assay. First, CT scanning of the lungs from mice were performed in order to observe the metastatic lung nodules, and then, the lung samples from mice were obtained and photographed (Figure 6E). Then these lung specimen were analyzed by HE staining (Figure 6F). These results suggested that K260hib of ALDH1A1 could reduce the lung metastasis. Moreover, immunohistochemical staining were performed to assess the difference in ALDH1A1 between the two types of tumors (Figure S7A). Together, these results suggested that the 2-hydroxyisobutylation of ALDH1A1 at the K260 site decreased cell proliferation and metastasis in vivo.
Subsequently, to examine the K260hib level of ALDH1A1, a rabbit polyclonal antibody specifically against 2-hydroxyisobutylated-K260 of ALDH1A1 using the synthetic peptide (CIKEAAGK(2-Hydroxyisobutyric acid)SNLKR-NH2) was prepared. Next, this K260hib-ALDH1A1 antibody was evaluated by dot blotting, and the result demonstrated that the K260 hib-ALDH1A1 antibody specially recognized the K260 2-hydroxyisobutylated peptide rather than the unmodified peptide (Figure S7B). Next, Flag-ALDH1A1WT, Flag-ALDH1A1K260T, and Flag-ALDH1A1K260R were overexpressed in T24 cells, respectively, and the 2-hydroxyisobutylation of ALDH1A1 was evaluated by Western blotting (Figure S7C), suggesting that this K260hib-ALDH1A1 antibody specifically recognized the site of 2-hydroxyisobutylated K260.
To explore the association of ALDH1A1 K260hib with the prognosis of patients with bladder cancer, IHC was performed to evaluate the differences of ALDH1A1 K260hib expression between normal bladder tissues and the corresponding bladder cancer tissues with a specific K260hib antibody, indicating a relatively decreased expression of K260hib in the bladder cancer tissues than that in the normal tissues (Figure S7D). Meanwhile, we observed that the level of ALDH1A1-K260hib were opposite to its protein levels in bladder cancer tissues (Figures 6G and 6H). Moreover, the patients with bladder cancer were assigned to the higher and lower K260hib groups based on their IHC scores of ALDH1A1 K260hib. We next analyzed the correlation between K260hib and the clinical characteristics of the patients (Table 1), which showed a decrease along with the advancement of the bladder cancer stage (Figure 6I; Table 1). Furthermore, we assessed the overall survival rates of the expression of ALDH1A1 K260hib, and the results suggested that the patients with lower ALDH1A1 K260hib were associated with worse prognosis (Figure 6J). Meanwhile, the recurrence free survival rate of patients with lower ALDH1A1 K260hib had a relative poor prognosis (Figure 6K). Collectively, these findings indicated that ALDH1A1-K260hib may be a biomarker for bladder cancer diagnosis and a potential target for bladder cancer therapy.
Table 1.
Associations between K260hib-ALDH1A1 expression and clinical characteristics in patients with bladder cancer
| Characteristics | Low (K260hib) | High (K260hib) | p |
|---|---|---|---|
| Cases (n) | 55 (52.3%) | 50 (47.7%) | |
| Gender | 0.430 | ||
| Male | 50 (90.9%) | 43 (86%) | |
| Female | 5 (9.1%) | 7 (14%) | |
| Age (year) | 0.615 | ||
| <60 | 12 (21.8%) | 13 (26%) | |
| ≥60 | 43 (78.2%) | 37 (74%) | |
| Pathology grade | 0.448 | ||
| Low | 12 (21.8%) | 8 (16%) | |
| High | 43 (78.2%) | 42 (84%) | |
| AJCC | 0.009 | ||
| Ⅰ | 5 (9.1%) | 18 (36%) | |
| Ⅱ | 24 (43.6%) | 15 (30%) | |
| Ⅲ | 17 (30.9%) | 13 (26%) | |
| Ⅳ | 9 (16.4%) | 4 (8%) | |
| T stage | 0.202 | ||
| <T2 | 32 (58.2%) | 36 (72%) | |
| ≥T2 | 23 (41.8%) | 14 (28%) | |
| Lymph node metastasis | 0.322 | ||
| No | 47 (85.5%) | 39 (78%) | |
| Yes | 8 (14.5%) | 11 (22%) | |
| Distant metastasis | 0.862 | ||
| Negative | 49 (89.1%) | 46 (92%) | |
| Positive | 6 (10.9%) | 4 (8%) | |
| Molecular subtype | 0.075 | ||
| Luminal | 27 (49.1%) | 16 (32%) | |
| Basal | 28 (50.9%) | 34 (68%) |
Hib, 2-hydroxyisobutyrylation; AJCC, American Joint Committee on Cancer.
Discussion
In the past decades, several post-translational modifications (PTMs) have been identified recently, which mainly belong to the category of the lysine acylation of histones, including propionylation, β-hydroxybutyrylation, crotonylation, butyrylation, malonylation, glutarylation, and 2-hydroxyisobutyrylation. Recently, 2-hydroxyisobutyrylation was discovered in various organisms, displaying involvement in the regulation of biosynthesis, protein degradation, and energy metabolism.12 Moreover, it has been reported that 2-hydroxyisobutyrylation was distinguished from classical lysine methylation and acetylation owing to its unique genomic distribution9; hence, it may be of great significance in biological regulation as well as in classical lysine methylation and acetylation. However, no research has yet reported lysine 2-hydroxyisobutyrylation in bladder cancer. In this study, we discovered the lysine 2-hydroxyisobutyrylated-targeted proteins first by liquid chromatography-mass spectrometry-based quantitative proteomics in bladder cancer. ALDH1A1, which is one of the bladder CSCs markers, drew our attention to further study, which was observed with a low lysine 2-hydroxyisobutyrylation expression in our data. ALDH1A1 has been reported to drive cancer cell proliferation and metastasis in cancers.24,29,41,42,43,44 However, its role with lysine 2-hydroxyisobutyrylation has never been studied before. Therefore, we wonder whether lysine 2-hydroxyisobutyrylation of ALDH1A1 affects its function in bladder cancer.
The human ALDHs include 19 isoenzymes that oxidize aldehydes into the corresponding carboxylic acids.45 ALDH1A1 is one of the main members of the ALDH1 family (i.e., ALDH1A1, ALDH1A2, and ALDH1A3), which share 70% amino acid sequence homology, and ALDH1A1 showed a greater affinity for retinal than ALDH1A2 and ALDH1A3.19 ALDH1A2 play a crucial role in the occurrence and development of nervous system diseases, which, as a CSCs marker of neuroblastoma, was closely associated with chemotherapy resistance and tumor recurrence of nervous system neoplasms.46,47,48 While ALDH1A3 mainly contributes to CSC-mediated tumorigenicity of glioma,49 breast cancer,50 melanoma cancer,51 and gallbladder carcinoma.52 However, no study has yet focused on the ALDH1A2 and ALDH1A3 in bladder cancer. In the present study, we found that ALDH1A1 was closely related with CD44 and CD133 in the TCGA-BLCA database, while there was no strong correlation among ALDH1A2, ALDH1A3, CD44 and CD133 (Figures S8A and S8B). Moreover, the relationship among ALDH1A2, ALDH1A3 and ALDH1A1 was not significant (Figure S8C). Moreover, the expression of ALDH1A1, rather than ALDH1A2 and ALDH1A3, was increased in T24 and UMUC3 cells after treatment with different concentrations of gemcitabine and cisplatin (GC) (Figures S8D and S8E). In addition, the mRNA and protein expression of ALDH1A2 and ALDH1A3 were independent of the expression of ALDH1A1 change (Figures S8F‒S8I). These results together suggest that ALDH1A1, rather than ALDH1A2 and ALDH1A3, is a marker of bladder cancer stem cells.
ALDH1A1 plays an important role in oxidizing toxic retinaldehyde to RA. RA subsequently regulates targeted genes via RAR and RXR nuclear receptors to control tumor growth and aggression, which occurs in various tumors such as breast cancer, melanoma, hematological tumor, ovarian cancer and bladder cancer regarding its role of CSCs marker.1,20,43,53,54,55 It has been reported that PDC spheroids with high ALDH1A1 levels retained the tumorigenic capacity as a marker of CSCs in bladder cancer, whereas ALDH1A1 knockdown decreased tumor proliferation and spheroid formation.24 In our study, we observed that silencing ALDH1A1 could inhibit bladder cancer proliferation, migration, and invasion. Meanwhile, ALDH1A1 promoted tumor aggression depending on the RA signaling pathway, and the reduction ability of motility and invasiveness was recovered by RA addition. Furthermore, we found that RA increased tumor aggression via the RXRα-mediated p-AKT/β-catenin genes in the bladder cancer cells, which can be attributed to the role of ALDH1A1 as a marker of bladder CSCs.
Some studies have focused on the PTM of ALDH1A1, mainly because it is one of the CSCs markers, including lysine acetylation, phosphorylation, methylation, and ubiquitylation, as shown on the PhosphoSitePlus website. It has also been reported that the lysine acetylation of ALDH1A1 at the K353 site can inhibit its activity, consequently decreasing both the stem cell population and self-renewal properties of breast cancer cells.56 In addition, ALDH1A1, with phosphorylation at S75 and S274 sites, could inhibit its ubiquitylation, thereby correspondingly increasing its stability and protein levels. Simultaneously, phosphorylation increased its dehydrogenase activity by altering its oligomeric status.57 In our study, we found that ALDH1A1 with the 2-hydroxyisobutyrylation at the K260 site decreased its enzyme activity and protein stability in bladder cancer. Furthermore, we observed a change in the protein structure, which was altered by 2-hydroxyisobutyrylation at the K260 site. ALDH1A1 usually functions in tetramer,58 and we found that K260T mimicking 2-hydroxyisobutyrylation considerably reduced its tetrameric structure, thereby decreasing the ALDH1A1 levels. The results of Western blotting also confirmed that the 2-hydroxyisobutyrylation of ALDH1A1 at the K260 site decreased the protein levels, while simultaneously decreasing the occurrence of the tetrameric forms. Taken together, 2-hydroxyisobutyrylation of ALDH1A1 at the K260 site reduced its enzyme activity through the destruction of its tetramer structure.
To confirm the relationship between 2-hydroxyisobutyrylation of ALDH1A1 at the K260 site and protein stability, we undertook research of protein degradation via the ubiquitination–proteasome system and autophagy–lysosome system, which played an important role in regulating the protein homeostasis.59,60 Only a few studies have focused on the degradation pathway of ALDH1A1; we first found that 2-hydroxyisobutyrylation could promote ALDH1A1 degradation via the CMA pathway, a type of the autophagy–lysosome system. To further investigate the association of ALDH1A1 Khib with CMA, the Western blotting results suggested that HSC70 combined with ALDH1A1K260T more than ALDH1A1WT, similar to ALDH1A1WT treated with HDACs inhibitor TSA, which also reduced the half-life of ALDH1A. Furthermore, we found that TSA could decrease the protein of ALDH1A1 through the 2-hydroxyisobutyrylation at the K260 site. Some previous studies have shown that TSA could efficiently reduce the survival of pancreatic cancer cells,61,62 and a past study suggested that histone deacetylase inhibitors could render cancer cell more susceptible to chemotherapy.63 Hence, TSA can be potentially applied in the therapy of bladder cancer in the future.
ALDH1A1 could be recognized as a target in cancer therapy and chemoresistance owing to its role as a CSC marker.29,42,64 In our study, we detected the ALDH1A1 protein level and its 2-hydroxyisobutyrylation in bladder cancer cells after treatment of chemotherapeutic drugs, and we found that the level of ALDH1A1 was gradually increased after treatment of gemcitabine and cisplatin. Meanwhile, the 2-hydroxyisobutyrylation level of ALDH1A1 was constantly reduced with the indicated dosage of gemcitabine and cisplatin, implying the presence of a high protein level of ALDH1A1 with low 2-hydroxyisobutyrylation in the CSCs. Simultaneously, we observed the same change under the treatments without serum and EBSS stress. Hence, we concluded that ALDH1A1 is a tumor resistance factor against risk, which also explains its function as a CSC marker. Moreover, although the CSCs are relative rare in tumor, CSCs can promote the carcinoma growth and metastasis, which are responsible for tumor aggressiveness in bladder cancer.65,66,67,68,69 Therefore, we concluded that the expression of ALDH1A1 can be increased in response to the expansion of CSCs under adverse stimulation, such as chemotherapy, radiotherapy, and limotherapy.
Moreover, 2-hydroxyisobutyrylation of ALDH1A1 was observed in the bladder cancer tissues in this study, and the lower 2-hydroxyisobutyrylation with higher ALDH1A1 was markedly associated with an advanced stage in patients with bladder cancer. Furthermore, 2-hydroxyisobutyrylated-ALDH1A1 was closely associated with the prognosis of patients with bladder cancer, and higher 2-hydroxyisobutyrylation of ALDH1A1 demonstrated a better overall survival rate than the lower 2-hydroxyisobutyrylation of ALDH1A1. Moreover, in the present study, we found that K260hib of ALDH1A1 could reduce the resistance to conventional chemotherapy drugs. Altogether, 2-hydroxyisobutyrylation of ALDH1A1 was markedly associated with its protein level and tumor progression, which is likely to act as an indicator of treatment resistance and malignant progression.
Currently, past studies have increasingly identified protein PTMs as a target for anticancer drugs,70 and relevant studies about the targeted PTMs in bladder cancer has been reported.71,72,73,74 Tyrosine kinase inhibitors (TKIs) are the most widely studied PTMs inhibitors of proteins, which have been approved for the clinical treatment of cancers.75 Although TKIs are remarkably effective in the targeted therapy, their resistance is gradually increasing.76 Meanwhile, chemotherapy and immunotherapy usually incur serious adverse events. Recent studies have demonstrated that the protein PTMs may play a crucial role in regulating cancer stemness and tumor progression across various cancers.77 Moreover, abnormal protein PTMs are responsible for the therapeutic resistance by affecting the cancer stemness.78,79,80,81,82 Therefore, understanding the underlying mechanisms of CSCs for stemness properties acquirement through PTMs can promote specific targeted therapy of CSCs. Moreover, mounting evidence has demonstrated that protein PTMs of CSCs can serve as a potential therapeutic target for cancer treatment.83 These experiments together revealed that high ALDH1A1 K260hib modification can significantly weaken the proliferation, invasion, and metastasis of tumors, and that K260hib of ALDH1A1 could sensitize the bladder cancer cells to drugs treatment. Hence, targeted ALDH1A1 K260hib modification is a promising application in clinical practice for bladder cancer therapy in the future, as it can not only provides a potential therapeutic target for the treatment of bladder cancer, but also enrich the non-histone post-translational modification of tumors.
Altogether, our results uncovered a special lysosomal pathway of ALDH1A1 degradation promoted by K260-2-hydroxyisobutyrylation, that could decrease bladder cancer invasion and proliferation. Moreover, a higher level of K260-2-hydroxyisobutyrylated ALDH1A1 has been associated with good prognosis in patients with bladder cancer. Thus, these findings suggest K260hib of ALDH1A1 as a potential therapy target for bladder cancer.
Limitations of the study
Although HDAC2/3 can regulate the Khib modification of ALDH1A1, there is no specific inhibitor of HDAC2/3, warranting further exploration. The K260 site of ALDH1A1 has a hib modification, which necessitates the investigation of whether there are other modifications of this K260 site.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ALDH1A1 | Proteintech | Cat #: 15910; RRID:AB_2305276 |
| Mouse monoclonal anti-ALDH1A1 | Proteintech | Cat #: 60171; RRID:AB_10693634 |
| Rabbit polyclonal anti-AKT | Cell Signaling | Cat #: 9272; RRID:AB_329827 |
| Rabbit polyclonal anti-Phospho-Akt | Cell Signaling | Cat #:4060; RRID:AB_2315049 |
| Mouse polyclonal anti-CD44 | Cell Signaling | Cat#:5640; RRID:AB_AB_10547133 |
| Mouse monoclonal anti-HDAC3 | Cell Signaling | Cat #:3949; RRID:AB_2118371 |
| Rabbit polyclonal anti-HDAC3 | Proteintech | Cat #:10255; RRID:AB_2279733 |
| Rabbit polyclonal anti-HDAC2 | Proteintech | Cat #:12922; RRID:AB_2934877 |
| Mouse monoclonal anti-HDAC2 | Proteintech | Cat #:67165; RRID:AB_2920291 |
| Rabbit monoclonal anti-Flag-Tag | Cell Signaling | Cat #:14793; RRID:AB_2572291 |
| Rabbit monoclonal anti-HA-Tag | Cell Signaling | Cat #:3724; RRID:AB_1549585 |
| Mouse monoclonal anti-SQSTM1/p62 | Cell Signaling | Cat #:88588; RRID:AB_2800125 |
| Rabbit monoclonal anti-LC3 | Cell Signaling | Cat #:3868; RRID:AB_2137707 |
| Mouse monoclonal anti-β-catenin | Cell Signaling | Cat #:2698; RRID:AB_1030945 |
| Mouse monoclonal Anti-2-Hydroxyisobutyryllysine | PTMBIO | Cat #:PTM802; |
| Rabbit polyclonal anti-RXRα | Proteintech | Cat #:21218; RRID:AB_10693633 |
| Rabbit polyclonal anti-HSC70 | Proteintech | Cat #:10654; RRID:AB_2120153 |
| Rabbit monoclonal anti-LAMP2A | Abcam | Cat#:ab125068; RRID:AB_10971511 |
| Rabbit monoclonal anti-β-actin | Cell Signaling | Cat #:4970; RRID:AB_2223172 |
| Rabbit polyclonal anti-α-actinin | Proteintech | Cat #:11313; RRID:AB_2223815 |
| Rabbit polyclonal anti-GAPDH | Sigma | Cat #:67165; RRID:AB_796208 |
| Rabbit polyclonal anti-ALDH1A2 | Proteintech | Cat #: 13951; RRID:AB_2224033 |
| Rabbit polyclonal anti-ALDH1A3 | Proteintech | Cat #: 25167; RRID:AB_2879937 |
| Bacterial and virus strains | ||
| Sh-humanALDH1A1(5′ to 3’):GAACAGUGUGG GUGAAUUGTT |
GENEWIZ | N/A |
| Biological samples | ||
| Human bladder cancer tissues and adjacent tissues | The affiliated hospital of Qingdao university | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DMSO | Sigma-Aldrich | Cat #: D2650 |
| Nicotinamide | MCE | Cat #:HY-B0150 |
| MG132 | MCE | Cat #:HY-13259 |
| TSA | MCE | Cat #:HY-15144 |
| Gemcitabine | MCE | Cat #:HY-17026 |
| cisplatin | MCE | Cat #:HY-17394 |
| NCT501 | MCE | Cat #:HY-18768 |
| ATRA | MCE | Cat #:HY-14649 |
| RALD | MCE | Cat #:HY-W004500 |
| CQ | MCE | Cat #:HY-17589A |
| CHX | MCE | Cat #:HY-12320 |
| CIKEAAGK(2-Hydroxyisobutyric acid)SNLKR-NH2 | QYAOBIO | Cat #:4209-M |
| CIKEAAGKSNLKR-NH2 | QYAOBIO | Cat #:4209-C |
| Critical commercial assays | ||
| MTT reagent | R&D Systems | 4890-25-01 |
| Annexin V-FITC/PI detection kit | Yeasen | 40302ES50 |
| Acetaldehyde Dehydrogenase(ALDH) Activity Assay Kit | Solarbio | BC0755 |
| HiScript II One Step qRT-PCR SYBR Green Kit | Vazyme | Q221 |
| Deposited data | ||
| RNA-seq data | TCGA-BLCA | https://portal.gdc.cancer.gov/repository |
| Experimental models: Cell lines | ||
| T24 | the Chinese Academy of Sciences Cell Bank | SCSP-536 |
| UMUC3 | the Chinese Academy of Sciences Cell Bank | TCHu217 |
| HEK293T | the Chinese Academy of Sciences Cell Bank | GNHu17 |
| Experimental models: Organisms/strains | ||
| BALB/c nude | Vital River Laboratories, Beijing, China | N/A |
| Oligonucleotides | ||
| Si-humanALDH1A1(5′ to 3’):GAACAGUGUGGGU GAAUUGTT |
Genepharma | N/A |
| Si-humanHDAC2(5′ to 3’):UCCGUAAUGUUGCU CGAUGTT |
Genepharma | N/A |
| Si-humanHDAC31(5′ to 3’):AAUAUCCCUCUACUC GUGCUGA |
Genepharma | N/A |
| Si-humanLAMP2A(5′ to 3’):CUGCAAUCUGAUUGA UUAUUTT |
Genepharma | N/A |
| qPCR Primer sequences please see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| cDNA-Human ALDH1A1 | GENEWIZ | N/A |
| cDNA-Human ALDH1A1K260T | GENEWIZ | N/A |
| cDNA-Human ALDH1A1K260R | GENEWIZ | N/A |
| cDNA-Human HDAC2 | GENEWIZ | N/A |
| cDNA-Human HDAC3 | GENEWIZ | N/A |
| Software and algorithms | ||
| SPSS statistics 22.0 | IBM | https://www.ibm.com/spss |
| GraphPad Prism 8.0 | GraphPad | https://www.graphpad.com/scientifificsoftware/prism/ |
| R version 4.1.0 | The R Foundation | https://www.r-project.org |
| NovoExpress 1.6.1 | Agilent | https://www.agilent.com.cn/ |
| ImageJ | Schneider et al.84 | https://imagej.nih.gov/ij/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Haitao Niu (niuht0532@126.com).
Materials availability
This study did not generate any unique new reagent. All reagents used in this study are commercially available.
Experimental model and study participant details
Cell lines and cell culture
The T24 cell line (Female, 82Y), UMUC3 cell line (Male, age unspecified) and 293T cell line (Female, fetus) were supplied by the Cell Bank of the Chinese Academy of Sciences and cultured in Gibco Dulbecco’s Modified Eagle Medium (DMEM; China) supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin at 37°C and under 5% CO2 atmosphere.
Tissue samples and clinical data
A total of 35 pairs of bladder cancer tissues and the corresponding normal adjacent tissues were collected from the Department of Urology, the Affiliated Hospital of Qingdao University. Meanwhile, another 70 bladder cancer specimens with no normal adjacent tissues were obtained. These patients were all yellow races, and there was no influence of gender on this study. Furthermore, the clinicopathological characteristics of the patients were presented in Table 1. This study was approved by the hospital ethics committee of the affiliated hospital of Qingdao University.
Moreover, we explored the functions of ALDH1A1 in public databases, and the clinical information and levels of ALDH1A1 in bladder cancer were collected from The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and GEPIA database (http://gepia.cancer-pku.cn/). To evaluate the prognosis of ALDH1A1 in bladder cancer, the patients were assigned to either the high or low expression groups based on the median value of ALDH1A1 expression, while the association of ALDH1A1 with the overall survival (OS) was assessed by Kaplan–Meier assay. We also investigated the correlation between the ALDH1A1 expression and the tumor stage as well as explored the related mechanisms of ALDH1A1 in bladder cancer through GSVA and GSEA, respectively.
Xenograft model
BABL/c nude mice (age: 4 weeks, female, purchased from Vital River Laboratories, Beijing, China) were housed in a suitable environment. All operations on the animals were performed following the guidelines of the affiliated hospital of Qingdao University’s institutional animal care. T24 ALDH1A1 rescued WT or K260T cells were trypsinized and counted, and 5 × 106 cells were resuspended in PBS with Matrigel Matrix at 1:1 (v/v) and injected subcutaneously into the right flanks of mice. After the injection, the length (L) and width (W) of the tumor were measured with a caliper every 3 days. Tumor volume(mm3) was calculated by (L x W2)/2. Next, the mice were sacrificed after 2 weeks, and then the volume and weight of the tumor were analyzed. The tumor specimens obtained from mice were fixed in 4% formaldehyde, embedded in paraffin section, and these paraffin blocks were sliced into 4-μm-thick sections for the immunohistochemical and hematoxylin-eosin staining (HE) analyses.
For the metastasis assay in vivo, 5 × 105 cells were resuspended in 100 μL of PBS, which then were intravenously injected through the tail vein of mice. At 5 weeks after injection, mice were anesthetized with isoflurane mixed with oxygen and nitrous oxide (O2:N2O = 1:1) in order to observe the lung metastasis, which were performed by a spiral CT scanner (micro CT, PerkinElmer). During the CT scanning, mice breathed freely using a nose cone. The specific parameters were as follows: slice thickness, 0.1 mm; tube voltage, 90 kV; tube current, 88μA; Voxel Size, 72um; Dose 449mGy. Next, mice were sacrificed, the lungs were obtained and photographed. Relative number of metastatic lung nodules of mice was analyzed. Then, the lungs were also fixed in 4% paraformaldehyde for HE staining.
All experiments involving mice were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and approved by the hospital ethics committee of the affiliated hospital of the Qingdao University(Qingdao, Shandong; Registered number: QYFY WZLL 27206), which was also conducted in accordance with the principles of the Declaration of Helsinki.
Method details
Proteomic quantification of lysine 2-hydroxyisobutyrylation
The human bladder cancer tissues and adjacent normal tissues for the LC-MS/MS service were obtained from 10 patients with bladder cancer who had not received either chemotherapy or immunotherapy. The collected tissues were grounded into a powder form and then digested into peptides by trypsin, followed by labeling with TMT. The levels of lysine 2-hydroxyisobutyrylation were quantified through HPLC fractionation, affinity enrichment, and LC-MS/MS. Then, bioinformatic analysis was performed by GO annotation, domain annotation, KEGG pathway annotation, subcellular location, functional enrichment, and protein–protein interaction network.
Immunohistochemistry (IHC)
The tissue sections were deparaffinized in xylene and rehydrated in a graded series of ethanol (100%, 95%, 90%, 80%, and 70%), while antigen was retrieved in 10 mmol/L of sodium citrate buffer (pH 6.0) containing a sealing serum. These sections were incubated with primary antibodies (anti-ALDH1A1; Proteintech, Cat No: 15910-1-AP) and anti-ALDH1A1-K260hib antibody (ChinaPeptides Co., Ltd.) at 4°C overnight. The next day, the slides were stained with a biotinylated secondary antibody. Finally, a peroxidase kit (DAB kit, Servicebio) was used to detect the signal, and the resultant intensity was scored from 1 to 3, with “1” indicating no staining or weak staining, “2” indicating moderate staining, and “3” indicating strong staining, while the positive proportion was scored as 1 (0–25%), 2 (25–50%), 3 (50–75%), and 4 (75–100%), and the final score of the sample was obtained from the multiplication of two scores.
Plasmid construction and lentivirus transduction
cDNA of full-length WT-ALDH1A1 and its point mutations were synthesized (GENEWIZ, Suzhou, China) and cloned into a pcDNA3.1(+) vector. Similarly, HDAC2, HDAC3, and HSC70 cDNA plasmids were purchased from the same company (GENEWIZ, Suzhou). Lipofectamine 3000, P3000, and PEI were used for cell transfection as per the manufacturer’s instructions.
Control and ALDH1A1 T24 stable knockdown cell lines were generated by lentiviruses, including NC-shRNA vector and shRNA against human ALDH1A1. The ALDH1A1 shRNA sequences were fused into the plvx plasmid, which was co-transfected with packaging plasmids (psPAX2 and pMD2.G) into 293T cells, and the viral supernatant was collected after 48 h and 72 h, respectively. The targeted cells were infected by viral supernatant and screened with 1 μg/mL puromycin for 2 weeks. The interference effect of ALDH1A1 was confirmed by Western blotting, and the ALDH1A1 shRNA-targeted sequence was obtained as follows: ALDH1A1: 5′-GAACAGUGUGGGUGAAUUGTT-3’.
For ALDH1A1-rescued cell lines: the ALDH1A1-WT/K260T/K260R sequences were cloned into the Plvx lentivirus vector, and these plasmids were transfected with packaging plasmids (psPAX2 and pMD2.G) into 293T cells. The viral supernatant was collected after 48 h and 72 h, respectively. ALDH1A1 knockdown stable cell lines were infected by viral supernatant and screened with 200 μg/mL hygromycin for about 2 weeks.
RT-qPCR
The total RNA was extracted with the Trizol reagent (Takara), which was transcribed into cDNA using a reverse transcription kit (Perfect Real Time; Takara) as per the kit’s instructions. All primers synthesized by Huada Gene (Beijing, China) are listed in Table S1. Real-time PCR was performed in triplicate using the Roche Light Cycler 480II Real-time PCR detection system (Roche, Basel, Switzerland). The amount of mRNA was quantified by the 2−(ΔΔCt) method and normalized to actin or GAPDH levels.
Western blotting
The total protein content was extracted by Cell Lysis Buffer (Cell Signaling Technology, #9803), and the proteins were analyzed by Western blotting following the standard methods, while the immune complexes were detected by using the Enhanced Chemiluminescence Kit (Millipore).
Immunoprecipitation (IP) and Co-IP
The total cells were lysed in a cell lysis buffer (cell signaling technology, 9803) with the addition of a protease inhibitor cocktail (Sigma-Aldrich, 4693116001), PMSF (Beyotime, ST5062), TSA (MCE, HY-15144), and NAM (MCE, HY-B0150) to the lysis buffer. IP was performed as follows: the lysed cells were first incubated with Protein A/G Magnetic Beads (MCE, HY-K0202) and anti-ALDH1A1 at 4°C overnight, after which the beads were thrice eluted in a cell lysis buffer. The beads were then treated with 1× loading buffer and boiled at 95°C for about 5 min. The resultant protein complex was analyzed by Western blotting and immunoblotted with a related antibody.
Similarly, the cells were first lysed and then incubated with anti-ALDH1A1or anti-HDAC2/3 and anti-LAMP2A at 4°C overnight, after which the combined proteins were denatured and eluted from the beads. Finally, the proteins were evaluated by Western blotting.
ALDH1A1 enzyme activity assay
Flag-ALDH1A1 and its mutations were overexpressed in 293T cells. After transfection for 36 h, the ALDH1A1 enzyme activity was assessed by using the ALDH Activity Assay Kit (Solarbio, Beijing, China) according to the manufacturer’s instructions. The absorbance of NADH at 340 nm was measured by a microplate reader (Thermo, Peking, China).
Immunofluorescence staining (IF) for colocalization
Appropriate quantities of suitable T24 bladder cells were seeded and cultured on glass slides (BD Biosciences) and treated without or with serum for 12 h, after which the slides were fixed with 4% paraformaldehyde for 15 min and washed thrice with PBS for 5 min, followed by permeabilization with 0.1% Triton X-100 for 15 min. We then blocked the slides with 5% BSA for 1 h at room temperature and incubated the cells with anti-ALDH1A1 and anti-LAMP2A. Subsequently, the corresponding antibodies were added for 1 h at room temperature following counterstaining with 40,6-diamidino-2-phenylindole (DAPI; P36931; Life Technologies) for 15 min. Finally, the cells were observed and photographed under fluorescence microscopy.
Cell proliferation and colony formation
MTT assay
About 1000 cells were seeded into a 96-well plate and cultured for 1, 2, 3, and 4 days, respectively, after which 150 μL of MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added to the cell medium and the absorbance was measured after 4 h at the optical density value of 490 nm at room temperature.
Clone formation assay
A total of 500 cells were seeded into a 6-well plate and cultured with a normal medium for approximately 14 days. Then, the cells were fixed with 4% paraformaldehyde for 15 min and then stained with 0.1% crystal violet for 3 min, followed by colonies’ enumeration.
Apoptosis rate detection
The apoptosis rate of cells was evaluated by flow cytometry using FITC Annexin V Apoptosis Detection Kit (Yeasen Biotechnology, 40302ES50).The FITC-Annexin V and propidium iodide staining were performed based on the manufacturer’s instructions, followed by flow cytometry (Agilent NovoCyte 2060R). The apoptosis rates were calculated by combining with early-apoptotic cells and late-apoptotic cells. Data was analyzed using agilent software (NovoExpress 1.6.1, USA).
Cell migration and invasion assay
The transwell assay was performed in a 24-well Transwell Chamber (24-well plate; 8-μm pore size, Corning). A total of 5 × 104 cells/200 μL were seeded into the upper chamber coated without or with Matrigel (Corning biocoat 356234), which were cultured with a medium containing 5% FBS, while 10% FBS (500 μL) was placed in the lower chamber. After 36 h of treatment, the invasive and migrated cells were fixed in 4% paraformaldehyde for 30 min and stained with 1%crystal violet for another 30 min at room temperature. Subsequently, these cells were photographed under a microscope.
Sphere-forming assay
A total of 3000 cells were cultured in a media containing serum-free DMEM/F12 medium supplemented with 20 ng/mL of human recombinant epidermal growth factor (Thermo Fisher Scientific) and 10 ng/mL of human recombinant basic fibroblast growth factor (Thermo Fisher Scientific), which were seeded in ultralow attachment plates (Corning). The sphere-forming efficiency was evaluated when the spheres reached a diameter of 100 μm.
Quantification and statistical analysis
All data were analyzed by SPSS (IBM CORP), ImageJ,84 the Prism software program (GraphPad Software), and R version 4.1.0 (https://www.r-project.org/). The unpaired two-tailed Student’s t test was performed to compare the two study groups, and multiple groups were analyzed by one-way analysis of variance (ANOVA). Pearson Chi-Square test was used to evaluate the associations between the two groups and clinicopathological characteristics of the patients by SPSS ver. 22.0. Kaplan-Meier assay was utilized to estimate the survival rate of overall survival and recurrence-free survival between the groups. p < 0.05 was considered to indicate statistical significance in all experiments.
Acknowledgments
Thanks to PTM BIO for providing mass spectrum services. Funding for this work were supported by the National Natural Science Foundation of China [82071750, 81772713, 81472411]; Taishan Scholar Program of Shandong Province [tsqn20161077]; Major Science and technology innovation project of Shandong Province [2019JZZY021002]; Key projects of Qingdao Science and Technology Program [18-6-1-64-nsh]; Research and Development Program of Shandong Province [2018GSF118197]; Project ZR2021QH234 supported by Shandong Provincial Natural Science Foundation.
Author contributions
H.N. and Y.L. designed the experiments. Z.Z. and Y.W. wrote the article. Z.Z., Zhiqiang Li, and Zhijuan Liang performed the research and bioinformatics. Mingxin Zhang, Zhilei Zhang, Zhaoyuan Meng, Xiangyan Zhang, Guofeng Ma, Yuanbin Chen, and Yinjie Su analyzed the data. Zhilei Zhang and Xiangyan Zhang contributed patient samples and clinical data. All the authors read and approved the final article.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: October 5, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108142.
Contributor Information
Ye Liang, Email: liangye82812@163.com.
Haitao Niu, Email: niuht0532@126.com.
Supplemental information
Data and code availability
-
•
Data: The RNA-Seq data were collected from the online public database (The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and GEPIA database (http://gepia.cancer-pku.cn/). The proteomics data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP000478.
-
•
Code: This paper does not report the original code.
-
•
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021;71:7–33. doi: 10.3322/caac.21654. [DOI] [PubMed] [Google Scholar]
- 2.Rastinehad A.R., Andonian S., Smith A.D., Siegel D.N. Management of hemorrhagic complications associated with percutaneous nephrolithotomy. J. Endourol. 2009;23:1763–1767. doi: 10.1089/end.2009.1548. [DOI] [PubMed] [Google Scholar]
- 3.Moschini M., Sharma V., Dell'oglio P., Cucchiara V., Gandaglia G., Cantiello F., Zattoni F., Pellucchi F., Briganti A., Damiano R., et al. Comparing long-term outcomes of primary and progressive carcinoma invading bladder muscle after radical cystectomy. BJU Int. 2016;117:604–610. doi: 10.1111/bju.13146. [DOI] [PubMed] [Google Scholar]
- 4.Chen N., Zheng Q., Wan G., Guo F., Zeng X., Shi P. Impact of posttranslational modifications in pancreatic carcinogenesis and treatments. Cancer Metastasis Rev. 2021;40:739–759. doi: 10.1007/s10555-021-09980-4. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S.C., Falck J.R., Peng J., Gu W., Zhao Y. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007;6:812–819. doi: 10.1074/mcp.M700021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng C., Lu Z., Xie Z., Cheng Z., Chen Y., Tan M., Luo H., Zhang Y., He W., Yang K., et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.012658. M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N., et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Z., Tan M., Xie Z., Dai L., Chen Y., Zhao Y. Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol. 2011;7:58–63. doi: 10.1038/nchembio.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai L., Peng C., Montellier E., Lu Z., Chen Y., Ishii H., Debernardi A., Buchou T., Rousseaux S., Jin F., et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 2014;10:365–370. doi: 10.1038/nchembio.1497. [DOI] [PubMed] [Google Scholar]
- 10.Tan M., Peng C., Anderson K.A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014;19:605–617. doi: 10.1016/j.cmet.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabari B.R., Zhang D., Allis C.D., Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H., Luo Z., Qi S., Huang J., Xu P., Wang X., Gao L., Li F., Wang J., Zhao W., et al. Landscape of the regulatory elements for lysine 2-hydroxyisobutyrylation pathway. Cell Res. 2018;28:111–125. doi: 10.1038/cr.2017.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang H., Tang S., Ji M., Tang Z., Shimada M., Liu X., Qi S., Locasale J.W., Roeder R.G., Zhao Y., Li X. p300-Mediated Lysine 2-Hydroxyisobutyrylation Regulates Glycolysis. Mol. Cell. 2018;70:663–678.e6. doi: 10.1016/j.molcel.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liao L., He Y., Li S.J., Yu X.M., Liu Z.C., Liang Y.Y., Yang H., Yang J., Zhang G.G., Deng C.M., et al. Lysine 2-hydroxyisobutyrylation of NAT10 promotes cancer metastasis in an ac4C-dependent manner. Cell Res. 2023;33:355–371. doi: 10.1038/s41422-023-00793-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reya T., Morrison S.J., Clarke M.F., Weissman I.L. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 16.Choudhury N.J., Campanile A., Antic T., Yap K.L., Fitzpatrick C.A., Wade J.L., 3rd, Karrison T., Stadler W.M., Nakamura Y., O'Donnell P.H. Afatinib Activity in Platinum-Refractory Metastatic Urothelial Carcinoma in Patients With ERBB Alterations. J. Clin. Oncol. 2016;34:2165–2171. doi: 10.1200/JCO.2015.66.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg J.E., Hoffman-Censits J., Powles T., van der Heijden M.S., Balar A.V., Necchi A., Dawson N., O'Donnell P.H., Balmanoukian A., Loriot Y., et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet. 2016;387:1909–1920. doi: 10.1016/S0140-6736(16)00561-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zibelman M., Ramamurthy C., Plimack E.R. Emerging role of immunotherapy in urothelial carcinoma—Advanced disease. Urol. Oncol. 2016;34:538–547. doi: 10.1016/j.urolonc.2016.10.017. [DOI] [PubMed] [Google Scholar]
- 19.Tomita H., Tanaka K., Tanaka T., Hara A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget. 2016;7:11018–11032. doi: 10.18632/oncotarget.6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Su Y., Qiu Q., Zhang X., Jiang Z., Leng Q., Liu Z., Stass S.A., Jiang F. Aldehyde dehydrogenase 1 A1-positive cell population is enriched in tumor-initiating cells and associated with progression of bladder cancer. Cancer Epidemiol. Biomarkers Prev. 2010;19:327–337. doi: 10.1158/1055-9965.EPI-09-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keymoosi H., Gheytanchi E., Asgari M., Shariftabrizi A., Madjd Z. ALDH1 in combination with CD44 as putative cancer stem cell markers are correlated with poor prognosis in urothelial carcinoma of the urinary bladder. Asian Pac. J. Cancer Prev. 2014;15:2013–2020. doi: 10.7314/apjcp.2014.15.5.2013. [DOI] [PubMed] [Google Scholar]
- 22.Xu N., Shao M.M., Zhang H.T., Jin M.S., Dong Y., Ou R.J., Wang H.M., Shi A.P. Aldehyde dehydrogenase 1 (ALDH1) expression is associated with a poor prognosis of bladder cancer. Cancer Epidemiol. 2015;39:375–381. doi: 10.1016/j.canep.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Zhao A.Y., Dai Y.J., Lian J.F., Huang Y., Lin J.G., Dai Y.B., Xu T.W. YAP regulates ALDH1A1 expression and stem cell property of bladder cancer cells. OncoTargets Ther. 2018;11:6657–6663. doi: 10.2147/OTT.S170858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Namekawa T., Ikeda K., Horie-Inoue K., Suzuki T., Okamoto K., Ichikawa T., Yano A., Kawakami S., Inoue S. ALDH1A1 in patient-derived bladder cancer spheroids activates retinoic acid signaling leading to TUBB3 overexpression and tumor progression. Int. J. Cancer. 2020;146:1099–1113. doi: 10.1002/ijc.32505. [DOI] [PubMed] [Google Scholar]
- 25.Shukla V., Chandrasekaran B., Tyagi A., Navin A.K., Saran U., Adam R.M., Damodaran C. A Comprehensive Transcriptomic Analysis of Arsenic-Induced Bladder Carcinogenesis. Cells. 2022;11:2435. doi: 10.3390/cells11152435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patlolla J.M.R., Qian L., Biddick L., Zhang Y., Desai D., Amin S., Lightfoot S., Rao C.V. β-Escin inhibits NNK-induced lung adenocarcinoma and ALDH1A1 and RhoA/Rock expression in A/J mice and growth of H460 human lung cancer cells. Cancer Prev. Res. 2013;6:1140–1149. doi: 10.1158/1940-6207.CAPR-13-0216. [DOI] [PubMed] [Google Scholar]
- 27.Kwiatkowska-Borowczyk E., Czerwińska P., Mackiewicz J., Gryska K., Kazimierczak U., Tomela K., Przybyła A., Kozłowska A.K., Galus Ł., Kwinta Ł., et al. Whole cell melanoma vaccine genetically modified to stem cells like phenotype generates specific immune responses to ALDH1A1 and long-term survival in advanced melanoma patients. OncoImmunology. 2018;7 doi: 10.1080/2162402X.2018.1509821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Y., Song H., Jiang L., Qiao Y., Yang D., Wang D., Li J. Silybin Prevents Prostate Cancer by Inhibited the ALDH1A1 Expression in the Retinol Metabolism Pathway. Front. Cell Dev. Biol. 2020;8 doi: 10.3389/fcell.2020.574394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L., Cai S., Han C., Banerjee A., Wu D., Cui T., Xie G., Zhang J., Zhang X., McLaughlin E., et al. ALDH1A1 Contributes to PARP Inhibitor Resistance via Enhancing DNA Repair in BRCA2(-/-) Ovarian Cancer Cells. Mol. Cancer Ther. 2020;19:199–210. doi: 10.1158/1535-7163.MCT-19-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Z., Jiang L., Li B., Liang D., Feng Y., Liu L., Jiang C. Discovery of benzimidazole derivatives as potent and selective aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitors with glucose consumption improving activity. Bioorg. Med. Chem. 2021;46 doi: 10.1016/j.bmc.2021.116352. [DOI] [PubMed] [Google Scholar]
- 31.Castellanos J.A., Merchant N.B., Nagathihalli N.S. Emerging targets in pancreatic cancer: epithelial-mesenchymal transition and cancer stem cells. OncoTargets Ther. 2013;6:1261–1267. doi: 10.2147/OTT.S34670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aghaalikhani N., Rashtchizadeh N., Shadpour P., Allameh A., Mahmoodi M. Cancer stem cells as a therapeutic target in bladder cancer. J. Cell. Physiol. 2019;234:3197–3206. doi: 10.1002/jcp.26916. [DOI] [PubMed] [Google Scholar]
- 33.Wang H., Mei Y., Luo C., Huang Q., Wang Z., Lu G.M., Qin L., Sun Z., Huang C.W., Yang Z.W., et al. Single-Cell Analyses Reveal Mechanisms of Cancer Stem Cell Maintenance and Epithelial-Mesenchymal Transition in Recurrent Bladder Cancer. Clin. Cancer Res. 2021;27:6265–6278. doi: 10.1158/1078-0432.CCR-20-4796. [DOI] [PubMed] [Google Scholar]
- 34.Kliosnky D. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (3rd edition) Autophagy. 2016;12:1. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Majeski A.E., Dice J.F. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 36.Cuervo A.M. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol. Metab. 2010;21:142–150. doi: 10.1016/j.tem.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuervo A.M., Knecht E., Terlecky S.R., Dice J.F. Activation of a selective pathway of lysosomal proteolysis in rat liver by prolonged starvation. Am. J. Physiol. 1995;269:C1200–C1208. doi: 10.1152/ajpcell.1995.269.5.C1200. [DOI] [PubMed] [Google Scholar]
- 38.Rodriguez-Zavala J.S., Weiner H. Structural aspects of aldehyde dehydrogenase that influence dimer-tetramer formation. Biochemistry. 2002;41:8229–8237. doi: 10.1021/bi012081x. [DOI] [PubMed] [Google Scholar]
- 39.Yoval-Sánchez B., Pardo J.P., Rodríguez-Zavala J.S. New insights into the half-of-the-sites reactivity of human aldehyde dehydrogenase 1A1. Proteins. 2013;81:1330–1339. doi: 10.1002/prot.24274. [DOI] [PubMed] [Google Scholar]
- 40.Wang J., Nikhil K., Viccaro K., Chang L., White J., Shah K. Phosphorylation-dependent regulation of ALDH1A1 by Aurora kinase A: insights on their synergistic relationship in pancreatic cancer. BMC Biol. 2017;15:10. doi: 10.1186/s12915-016-0335-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang L., Ren Y., Yu X., Qian F., Bian B.S.J., Xiao H.L., Wang W.G., Xu S.L., Yang J., Cui W., et al. ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod. Pathol. 2014;27:775–783. doi: 10.1038/modpathol.2013.189. [DOI] [PubMed] [Google Scholar]
- 42.Kulsum S., Sudheendra H.V., Pandian R., Ravindra D.R., Siddappa G., Chevour P., Ramachandran B., Sagar M., Jayaprakash A., et al. Cancer stem cell mediated acquired chemoresistance in head and neck cancer can be abrogated by aldehyde dehydrogenase 1 A1 inhibition. Mol. Carcinog. 2017;56:694–711. doi: 10.1002/mc.22526. [DOI] [PubMed] [Google Scholar]
- 43.Ciccone V., Terzuoli E., Donnini S., Giachetti A., Morbidelli L., Ziche M. Stemness marker ALDH1A1 promotes tumor angiogenesis via retinoic acid/HIF-1α/VEGF signalling in MCF-7 breast cancer cells. J. Exp. Clin. Cancer Res. 2018;37:311. doi: 10.1186/s13046-018-0975-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biswas A.K., Han S., Tai Y., Ma W., Coker C., Quinn S.A., Shakri A.R., Zhong T.J., Scholze H., Lagos G.G., et al. Targeting S100A9-ALDH1A1-Retinoic Acid Signaling to Suppress Brain Relapse in EGFR-Mutant Lung Cancer. Cancer Discov. 2022;12:1002–1021. doi: 10.1158/2159-8290.CD-21-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou F., Mu Y.D., Liang J., Liu Z.X., Zhou D., Ning W.L., Li Y.Z., Ding D., Zhang J.F. Aldehyde dehydrogenase 1: A specific cancer stem cell marker for human colorectal carcinoma. Mol. Med. Rep. 2015;11:3894–3899. doi: 10.3892/mmr.2015.3195. [DOI] [PubMed] [Google Scholar]
- 46.Visvader J.E., Lindeman G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 47.Fragoso Y.D., Shearer K.D., Sementilli A., de Carvalho L.V., McCaffery P.J. High expression of retinoic acid receptors and synthetic enzymes in the human hippocampus. Brain Struct. Funct. 2012;217:473–483. doi: 10.1007/s00429-011-0359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ou A., Ott M., Fang D., Heimberger A.B. The Role and Therapeutic Targeting of JAK/STAT Signaling in Glioblastoma. Cancers. 2021;13 doi: 10.3390/cancers13030437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ni W., Xia Y., Luo L., Wen F., Hu D., Bi Y., Qi J. High expression of ALDH1A3 might independently influence poor progression-free and overall survival in patients with glioma via maintaining glucose uptake and lactate production. Cell Biol. Int. 2020;44:569–582. doi: 10.1002/cbin.11257. [DOI] [PubMed] [Google Scholar]
- 50.Thomas M.L., de Antueno R., Coyle K.M., Sultan M., Cruickshank B.M., Giacomantonio M.A., Giacomantonio C.A., Duncan R., Marcato P. Citral reduces breast tumor growth by inhibiting the cancer stem cell marker ALDH1A3. Mol. Oncol. 2016;10:1485–1496. doi: 10.1016/j.molonc.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pérez-Alea M., McGrail K., Sánchez-Redondo S., Ferrer B., Fournet G., Cortés J., Muñoz E., Hernandez-Losa J., Tenbaum S., Martin G., et al. ALDH1A3 is epigenetically regulated during melanocyte transformation and is a target for melanoma treatment. Oncogene. 2017;36:5695–5708. doi: 10.1038/onc.2017.160. [DOI] [PubMed] [Google Scholar]
- 52.Chen M.H., Weng J.J., Cheng C.T., Wu R.C., Huang S.C., Wu C.E., Chung Y.H., Liu C.Y., Chang M.H., Chen M.H., et al. ALDH1A3, the Major Aldehyde Dehydrogenase Isoform in Human Cholangiocarcinoma Cells, Affects Prognosis and Gemcitabine Resistance in Cholangiocarcinoma Patients. Clin. Cancer Res. 2016;22:4225–4235. doi: 10.1158/1078-0432.CCR-15-1800. [DOI] [PubMed] [Google Scholar]
- 53.Luo Y., Dallaglio K., Chen Y., Robinson W.A., Robinson S.E., McCarter M.D., Wang J., Gonzalez R., Thompson D.C., Norris D.A., et al. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cell. 2012;30:2100–2113. doi: 10.1002/stem.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gasparetto M., Pei S., Minhajuddin M., Khan N., Pollyea D.A., Myers J.R., Ashton J.M., Becker M.W., Vasiliou V., Humphries K.R., et al. Targeted therapy for a subset of acute myeloid leukemias that lack expression of aldehyde dehydrogenase 1A1. Haematologica. 2017;102:1054–1065. doi: 10.3324/haematol.2016.159053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cui T., Srivastava A.K., Han C., Wu D., Wani N., Liu L., Gao Z., Qu M., Zou N., Zhang X., et al. DDB2 represses ovarian cancer cell dedifferentiation by suppressing ALDH1A1. Cell Death Dis. 2018;9:561. doi: 10.1038/s41419-018-0585-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhao D., Mo Y., Li M.T., Zou S.W., Cheng Z.L., Sun Y.P., Xiong Y., Guan K.L., Lei Q.Y. NOTCH-induced aldehyde dehydrogenase 1A1 deacetylation promotes breast cancer stem cells. J. Clin. Invest. 2014;124:5453–5465. doi: 10.1172/JCI76611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nikhil K., Viccaro K., Shah K. Multifaceted Regulation of ALDH1A1 by Cdk5 in Alzheimer's Disease Pathogenesis. Mol. Neurobiol. 2019;56:1366–1390. doi: 10.1007/s12035-018-1114-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morgan C.A., Hurley T.D. Development of a high-throughput in vitro assay to identify selective inhibitors for human ALDH1A1. Chem. Biol. Interact. 2015;234:29–37. doi: 10.1016/j.cbi.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Milan E., Perini T., Resnati M., Orfanelli U., Oliva L., Raimondi A., Cascio P., Bachi A., Marcatti M., Ciceri F., Cenci S. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy. 2015;11:1161–1178. doi: 10.1080/15548627.2015.1052928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Meyer-Schwesinger C. The ubiquitin-proteasome system in kidney physiology and disease. Nat. Rev. Nephrol. 2019;15:393–411. doi: 10.1038/s41581-019-0148-1. [DOI] [PubMed] [Google Scholar]
- 61.Gilardini Montani M.S., Granato M., Santoni C., Del Porto P., Merendino N., D'Orazi G., Faggioni A., Cirone M. Histone deacetylase inhibitors VPA and TSA induce apoptosis and autophagy in pancreatic cancer cells. Cell. Oncol. 2017;40:167–180. doi: 10.1007/s13402-017-0314-z. [DOI] [PubMed] [Google Scholar]
- 62.Gilardini Montani M.S., Benedetti R., Piconese S., Pulcinelli F.M., Timperio A.M., Romeo M.A., Masuelli L., Mattei M., Bei R., D'Orazi G., Cirone M. PGE2 Released by Pancreatic Cancer Cells Undergoing ER Stress Transfers the Stress to DCs Impairing Their Immune Function. Mol. Cancer Ther. 2021;20:934–945. doi: 10.1158/1535-7163.MCT-20-0699. [DOI] [PubMed] [Google Scholar]
- 63.Lee H.S., Park S.B., Kim S.A., Kwon S.K., Cha H., Lee D.Y., Ro S., Cho J.M., Song S.Y. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci. Rep. 2017;7 doi: 10.1038/srep41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang S.M., Yasgar A., Miller B., Lal-Nag M., Brimacombe K., Hu X., Sun H., Wang A., Xu X., Nguyen K., et al. Discovery of NCT-501, a Potent and Selective Theophylline-Based Inhibitor of Aldehyde Dehydrogenase 1A1 (ALDH1A1) J. Med. Chem. 2015;58:5967–5978. doi: 10.1021/acs.jmedchem.5b00577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ben-Porath I., Thomson M.W., Carey V.J., Ge R., Bell G.W., Regev A., Weinberg R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chang C.C., Shieh G.S., Wu P., Lin C.C., Shiau A.L., Wu C.L. Oct-3/4 expression reflects tumor progression and regulates motility of bladder cancer cells. Cancer Res. 2008;68:6281–6291. doi: 10.1158/0008-5472.CAN-08-0094. [DOI] [PubMed] [Google Scholar]
- 67.She J.J., Zhang P.G., Wang Z.M., Gan W.M., Che X.M. Identification of side population cells from bladder cancer cells by DyeCycle Violet staining. Cancer Biol. Ther. 2008;7:1663–1668. doi: 10.4161/cbt.7.10.6637. [DOI] [PubMed] [Google Scholar]
- 68.He X., Marchionni L., Hansel D.E., Yu W., Sood A., Yang J., Parmigiani G., Matsui W., Berman D.M. Differentiation of a highly tumorigenic basal cell compartment in urothelial carcinoma. Stem Cell. 2009;27:1487–1495. doi: 10.1002/stem.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan K.S., Espinosa I., Chao M., Wong D., Ailles L., Diehn M., Gill H., Presti J., Jr., Chang H.Y., van de Rijn M., et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc. Natl. Acad. Sci. USA. 2009;106:14016–14021. doi: 10.1073/pnas.0906549106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trédan O., Galmarini C.M., Patel K., Tannock I.F. Drug resistance and the solid tumor microenvironment. J. Natl. Cancer Inst. 2007;99:1441–1454. doi: 10.1016/j.ebiom.2018.10.069. [DOI] [PubMed] [Google Scholar]
- 71.Seiler R., Oo H.Z., Tortora D., Clausen T.M., Wang C.K., Kumar G., Pereira M.A., Ørum-Madsen M.S., Agerbæk M.Ø., Gustavsson T., et al. An Oncofetal Glycosaminoglycan Modification Provides Therapeutic Access to Cisplatin-resistant Bladder Cancer. Eur. Urol. 2017;72:142–150. doi: 10.1016/j.eururo.2017.03.021. [DOI] [PubMed] [Google Scholar]
- 72.Cucci M.A., Compagnone A., Daga M., Grattarola M., Ullio C., Roetto A., Palmieri A., Rosa A.C., Argenziano M., Cavalli R., et al. Post-translational inhibition of YAP oncogene expression by 4-hydroxynonenal in bladder cancer cells. Free Radic. Biol. Med. 2019;141:205–219. doi: 10.1016/j.freeradbiomed.2019.06.009. [DOI] [PubMed] [Google Scholar]
- 73.Chen C., Zheng H., Luo Y., Kong Y., An M., Li Y., He W., Gao B., Zhao Y., Huang H., et al. SUMOylation promotes extracellular vesicle-mediated transmission of lncRNA ELNAT1 and lymph node metastasis in bladder cancer. J. Clin. Invest. 2021;131 doi: 10.1172/JCI146431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu J., Tan Z., Li H., Lin M., Jiang Y., Liang L., Ma Q., Gou J., Ning L., Li X., Guan F. Melatonin reduces proliferation and promotes apoptosis of bladder cancer cells by suppressing O-GlcNAcylation of cyclin-dependent-like kinase 5. J. Pineal Res. 2021;71 doi: 10.1111/jpi.12765. [DOI] [PubMed] [Google Scholar]
- 75.Huang L., Jiang S., Shi Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001-2020) J. Hematol. Oncol. 2020;13:143. doi: 10.1186/s13045-020-00977-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yang Y., Li S., Wang Y., Zhao Y., Li Q. Protein tyrosine kinase inhibitor resistance in malignant tumors: molecular mechanisms and future perspective. Signal Transduct. Target. Ther. 2022;7:329. doi: 10.1038/s41392-022-01168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Barkeer S., Chugh S., Batra S.K., Ponnusamy M.P. Glycosylation of Cancer Stem Cells: Function in Stemness, Tumorigenesis, and Metastasis. Neoplasia. 2018;20:813–825. doi: 10.1016/j.neo.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Blanas A., Zaal A., van der Haar Àvila I., Kempers M., Kruijssen L., de Kok M., Popovic M.A., van der Horst J.C., J van Vliet S. FUT9-Driven Programming of Colon Cancer Cells towards a Stem Cell-Like State. Cancers. 2020;12 doi: 10.3390/cancers12092580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu Y., Zhuang H., Cao F., Li J., Guo Y., Zhang J., Zhao Q., Liu Y. Shc3 promotes hepatocellular carcinoma stemness and drug resistance by interacting with β-catenin to inhibit its ubiquitin degradation pathway. Cell Death Dis. 2021;12:278. doi: 10.1038/s41419-021-03560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loong J.H., Wong T.L., Tong M., Sharma R., Zhou L., Ng K.Y., Yu H.J., Li C.H., Man K., Lo C.M., et al. Glucose deprivation-induced aberrant FUT1-mediated fucosylation drives cancer stemness in hepatocellular carcinoma. J. Clin. Invest. 2021;131 doi: 10.1172/JCI143377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuan Y., Wang L., Ge D., Tan L., Cao B., Fan H., Xue L. Exosomal O-GlcNAc transferase from esophageal carcinoma stem cell promotes cancer immunosuppression through up-regulation of PD-1 in CD8(+) T cells. Cancer Lett. 2021;500:98–106. doi: 10.1016/j.canlet.2020.12.012. [DOI] [PubMed] [Google Scholar]
- 82.Zhou S., Peng J., Xiao L., Zhou C., Fang Y., Ou Q., Qin J., Liu M., Pan Z., Hou Z. TRIM25 regulates oxaliplatin resistance in colorectal cancer by promoting EZH2 stability. Cell Death Dis. 2021;12:463. doi: 10.1038/s41419-021-03734-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y., Tong M. Protein Posttranslational Modification in Stemness Remodeling and Its Emerging Role as a Novel Therapeutic Target in Gastrointestinal Cancers. Int. J. Mol. Sci. 2023;24 doi: 10.3390/ijms24119173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data: The RNA-Seq data were collected from the online public database (The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov/) and GEPIA database (http://gepia.cancer-pku.cn/). The proteomics data that support the findings of this study have been deposited into CNGB Sequence Archive (CNSA) of China National GeneBank DataBase (CNGBdb) with accession number CNP000478.
-
•
Code: This paper does not report the original code.
-
•
Other items: Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.