

This systematic review and meta-analysis assesses all reported genetic modifiers of sickle cell disease and evaluates the design and data reporting of each study to provide guidelines for future analyses to accelerate discovery and validation of genetic modifiers in this disease.
Key Points
Question
What genetic modifiers of sickle cell disease (SCD) are currently defined, and what are potential approaches to improve future studies?
Findings
In this systematic review and meta-analysis of 571 studies examining 29 670 individuals with SCD, 17 757 associations involving 1552 genes and 25 SCD phenotype categories were discovered; of these, only 173 associations met the study design, reporting, and phenotype or genotype harmonization required for meta-analysis. Gene variants regulating fetal hemoglobin and α-thalassemia were frequently identified, but other associations remained unconfirmed.
Meaning
While major genetic modifiers of SCD severity were identified, including some that are clinically relevant, validated genetic associations were lacking, in part due to suboptimal study design and data reporting.
Abstract
Importance
Sickle cell disease (SCD) is a monogenic disorder, yet clinical outcomes are influenced by additional genetic factors. Despite decades of research, the genetics of SCD remain poorly understood.
Objective
To assess all reported genetic modifiers of SCD, evaluate the design of associated studies, and provide guidelines for future analyses according to modern genetic study recommendations.
Data Sources
PubMed, Web of Science, and Scopus were searched through May 16, 2023, identifying 5290 publications.
Study Selection
At least 2 reviewers identified 571 original, peer-reviewed English-language publications reporting genetic modifiers of human SCD phenotypes, wherein the outcome was not treatment response, and the comparison was not between SCD subtypes or including healthy controls.
Data Extraction and Synthesis
Data relevant to all genetic modifiers of SCD were extracted, evaluated, and presented following STREGA and PRISMA guidelines. Weighted z score meta-analyses and pathway analyses were conducted.
Main Outcomes and Measures
Outcomes were aggregated into 25 categories, grouped as acute complications, chronic conditions, hematologic parameters or biomarkers, and general or mixed measures of SCD severity.
Results
The 571 included studies reported on 29 670 unique individuals (50% ≤ 18 years of age) from 43 countries. Of the 17 757 extracted results (4890 significant) in 1552 genes, 3675 results met the study criteria for meta-analysis: reported phenotype and genotype, association size and direction, variability measure, sample size, and statistical test. Only 173 results for 62 associations could be cross-study combined. The remaining associations could not be aggregated because they were only reported once or methods (eg, study design, reporting practice) and genotype or phenotype definitions were insufficiently harmonized. Gene variants regulating fetal hemoglobin and α-thalassemia (important markers for SCD severity) were frequently identified: 19 single-nucleotide variants in BCL11A, HBS1L-MYB, and HBG2 were significantly associated with fetal hemoglobin (absolute value of Z = 4.00 to 20.66; P = 8.63 × 10−95 to 6.19 × 10−5), and α-thalassemia deletions were significantly associated with increased hemoglobin level and reduced risk of albuminuria, abnormal transcranial Doppler velocity, and stroke (absolute value of Z = 3.43 to 5.16; P = 2.42 × 10−7 to 6.00 × 10−4). However, other associations remain unconfirmed. Pathway analyses of significant genes highlighted the importance of cellular adhesion, inflammation, oxidative and toxic stress, and blood vessel regulation in SCD (23 of the top 25 Gene Ontology pathways involve these processes) and suggested future research areas.
Conclusions and Relevance
The findings of this comprehensive systematic review and meta-analysis of all published genetic modifiers of SCD indicated that implementation of standardized phenotypes, statistical methods, and reporting practices should accelerate discovery and validation of genetic modifiers and development of clinically actionable genetic profiles.
Introduction
Sickle cell disease (SCD) is the most common monogenic disorder in the world due to the protection that heterozygosity affords against malaria.1 Although SCD most heavily impacts sub-Saharan Africa, population migration and relocation have resulted in 1 in 2000 infants born in the United States with SCD, and 1 in 67 infants will be heterozygous carriers.2,3,4 Demographic trends and widespread improvements in clinical care will result in an increase in the proportion of the world’s population affected by SCD.2 An improved understanding of the pathophysiology of SCD and the environmental and genetic drivers of disease severity is essential to improve the lives of individuals with this disease.
Most cases of SCD are caused by a homozygous variation in the HBB gene (p.Glu6Val) encoding the β-globin subunit of adult hemoglobin tetramer (α2β2).2 At low oxygen concentrations in venous capillaries, sickle hemoglobin (α2βS2) forms rigid polymers, causing circulating red blood cells to become stiff, sticky, and brittle, triggering a complex pathophysiology including hemolysis, vascular occlusion, and inflammation.2 Clinical manifestations include severe acute and chronic pain, immunodeficiency, multiorgan damage, and early mortality. Hemolysis-related cellular injury, partly mediated by circulating free heme released from red blood cells, is thought to drive progression of cerebrovascular disease, kidney disease, pulmonary hypertension, priapism, and leg ulcers,5 whereas vaso-occlusion is thought to precipitate acute pain episodes, acute chest syndrome, and avascular necrosis.2
Despite being a monogenic disorder, the symptoms of SCD vary between affected individuals. The influence of environment on SCD is illustrated by markedly different outcomes between sub-Saharan Africa, where approximately half of affected children die before 5 years of age,6 and high-income countries, where enhanced medical support extends patient lifespan, although most patients still suffer considerably and die prematurely.7
Manifestations of SCD are also influenced by genetic factors. For example, residual expression of fetal hemoglobin (HbF, α2γ2) in postnatal red blood cells, which reduces SCD severity by interfering with polymerization of sickle hemoglobin,8 is largely determined genetically. Coinherited hereditary persistence of fetal hemoglobin, caused by variants in the extended β-like globin locus, results in extremely high levels of HbF, eliminating many symptoms of SCD.9 Genome-wide association studies have shown that 20%-50% of the variation in HbF can be explained by single-nucleotide variants (SNVs) in 3 loci: BCL11A, HBS1L-MYB, and the extended β-like globin locus.10,11,12,13 The erythroid-specific enhancer BCL11A encodes a potent transcriptional repressor for the γ-globin genes (HBG1 and HBG2).10,11,14 This discovery led to gene therapy strategies aimed at reducing erythroid BCL11A expression, some of which are showing early signs of efficacy in clinical trials,15,16 illustrating how understanding the genetic modifiers of SCD can have profound therapeutic implications.
The genetic contributions to SCD-related complications are poorly defined, despite a preponderance of publications on this topic. As a motivating example, a recent polygenic score incorporating 21 SNVs in 9 genetic loci, including HbF modifiers, explained only 3.5% of the variation in acute pain episodes.17 A more complete understanding of how genetics influences pathophysiology could improve therapy by providing tools to predict outcomes and identifying new modes for therapeutic intervention. To assess current knowledge, we performed a systematic review of, to our knowledge, all publications reporting genetic modifiers of SCD, cataloged the findings by subdividing genotype-phenotype associations by quality of data analysis and reporting, and performed meta-analyses and pathway analyses. Based on our findings and current guidelines in the field of human genetics, we provide recommendations for analytical approaches and reporting to enhance scientific rigor, reduce spurious results, and facilitate cross-study data synthesis.
Methods
Article Search and Abstract Screening
This systematic review was prospectively registered with PROSPERO (No. CRD42021274466) and was reported following the Preferred Reporting Items for Systematic Reviews and Meta-analyses (PRISMA) reporting guideline and the Strengthening the Reporting of Genetic Association Studies (STREGA) reporting guideline. We searched PubMed, Web of Science, and Scopus for all studies reporting genetic modifiers of SCD, irrespective of SCD subtype, published before May 16, 2023 (search terms in eMethods in Supplement 1). A total of 8892 studies were identified (eFigure 1 in Supplement 1): 3132 from PubMed, 2443 from Web of Science, and 3317 from Scopus. After deduplication, 5290 unique manuscripts remained.
Abstracts were screened by 2 independent reviewers (J.K.K., S.R.R.), with a third reviewer (J.H.E.) blinded to other screening opinions resolving disagreements. To comprehensively aggregate all published mutations associated with SCD-related outcomes and to avoid excluding important genetic modifiers due to incorrect phenotypic or genotypic attribution, we included all reported phenotypes and genetic polymorphisms. Studies were excluded if the manuscript was unavailable in English, the research was not conducted in humans, individuals without SCD were included, the only analysis was nongenetic, the only comparison was between SCD subtypes, the outcome was treatment response only, the manuscript was not peer reviewed, or no original research was included. A total of 571 publications passed this screening (eFigure 1 in Supplement 1, eTable 1 in Supplement 2).8,11,12,13,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295,296,297,298,299,300,301,302,303,304,305,306,307,308,309,310,311,312,313,314,315,316,317,318,319,320,321,322,323,324,325,326,327,328,329,330,331,332,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358,359,360,361,362,363,364,365,366,367,368,369,370,371,372,373,374,375,376,377,378,379,380,381,382,383,384,385,386,387,388,389,390,391,392,393,394,395,396,397,398,399,400,401,402,403,404,405,406,407,408,409,410,411,412,413,414,415,416,417,418,419,420,421,422,423,424,425,426,427,428,429,430,431,432,433,434,435,436,437,438,439,440,441,442,443,444,445,446,447,448,449,450,451,452,453,454,455,456,457,458,459,460,461,462,463,464,465,466,467,468,469,470,471,472,473,474,475,476,477,478,479,480,481,482,483,484,485,486,487,488,489,490,491,492,493,494,495,496,497,498,499,500,501,502,503,504,505,506,507,508,509,510,511,512,513,514,515,516,517,518,519,520,521,522,523,524,525,526,527,528,529,530,531,532,533,534,535,536,537,538,539,540,541,542,543,544,545,546,547,548,549,550,551,552,553,554,555,556,557,558,559,560,561,562,563,564,565,566,567,568,569,570,571,572,573,574,575,576,577,578,579,580,581,582,583 Data from these studies were extracted (eMethods, eTable 2, and eTable 3 in Supplement 2). Following extraction, we standardized gene annotation and phenotype categories to facilitate results tabulation (eMethods, eTable 4 in Supplement 1). No individual-level participant data were used.
Risk of Bias
Evolving approaches to genetic studies and clinical care over time, combined with variability in study design, phenotype definitions, and reporting practices, resulted in highly heterogeneous data, even within a single publication. Rather than determining the risk of bias for each publication, we created 3 categories into which all results were assigned using the STREGA guidelines584 (eMethods in Supplement 1). Briefly, exploratory results were evaluated statistically but lacked information required for cross-study meta-analysis. Meta-suitable results contained the minimum information to allow for meta-analysis: clearly defined outcome and genetic variants, sample size, statistical test, association size, direction, and measure of variability. Contemporary results contained all requirements for the meta-suitable category plus further elements crucial for genetic association studies (ie, quality control checks, accounting for population stratification and relatedness, covariate adjustment, and external validation). We did not exclude results based on these categories; however, some sections only used meta-suitable and contemporary results (Figure 1; eMethods in Supplement 1).
Figure 1. Flowchart of Analysis.
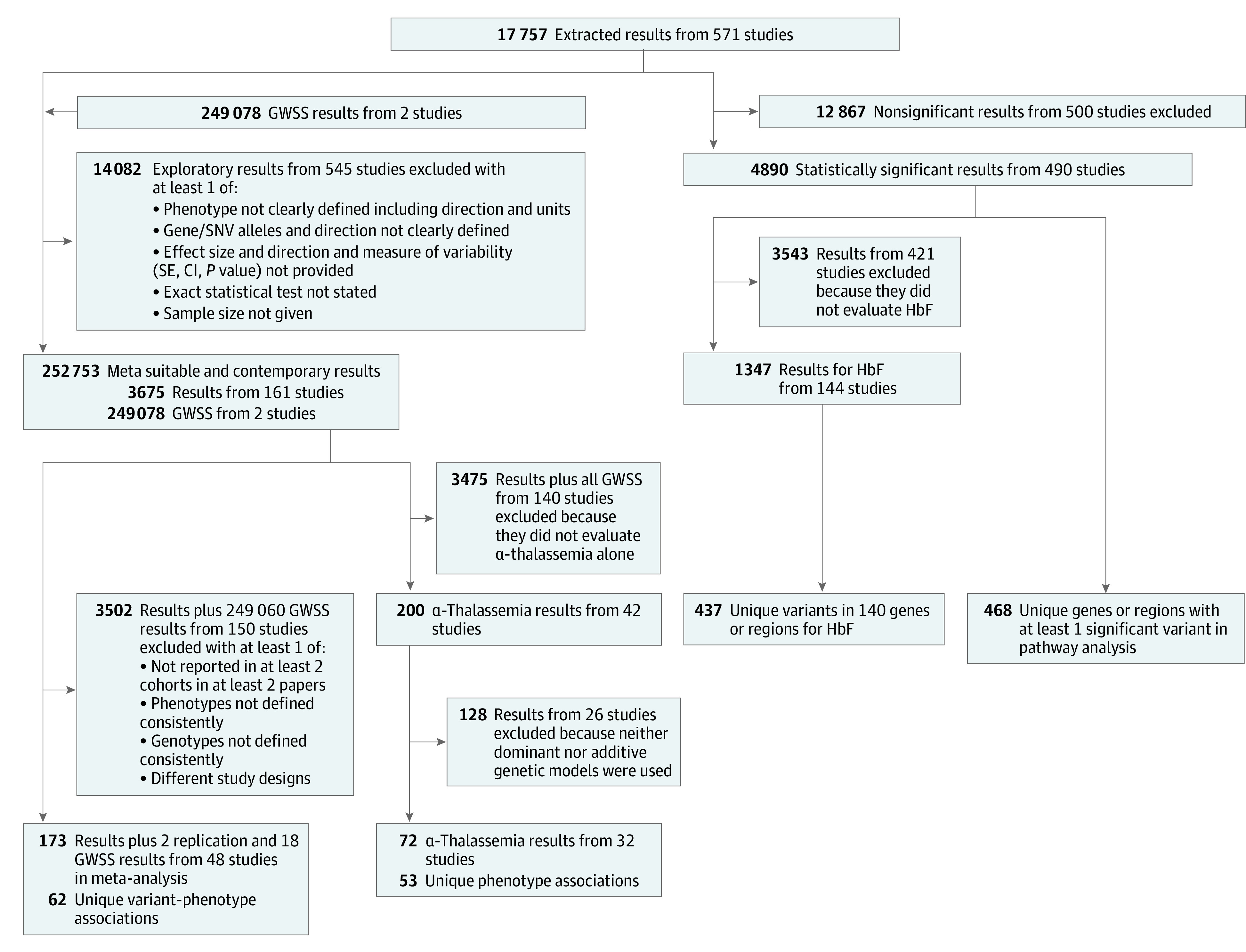
GWSS indicates genome-wide summary statistics; HbF, fetal hemoglobin.
Meta-Analysis
We conducted meta-analyses using a weighted z score–based approach (eMethods in Supplement 1) on all SNV-phenotype pairs with meta-suitable and contemporary results from at least 2 cohorts reported in at least 2 manuscripts in which the phenotype was the same, the same genotype comparison was done, and the same statistical test was performed. When a manuscript reported multiple results for the same cohort, we selected the one most similar to the other results being used in terms of adjustment for other covariates. If multiple studies reported results for the same cohort, we selected the result using the largest sample size.
Beyond Meta-Analysis
Our meta-analyses included only associations in which phenotypes and genotypes were defined consistently across studies. While statistically rigorous, this approach omitted biologically relevant associations established through repeated linking of loci to related phenotypes. As variability in study design and reporting prevented meta-analysis of most results, including well-established modifiers, we performed further data interrogations, as described here and in the eMethods in Supplement 1.
We examined genes with variants significantly associated with HbF in at least 3 manuscripts because HbF is a well-known disease modifier.8 Similarly, coinherited deletional α-thalassemia is common in SCD populations and modifies SCD pathophysiology,484 although the association with SCD varies across phenotypes and studies.2 To illustrate this comprehensively, we compared all meta-suitable and contemporary associations of SCD phenotypes with α-thalassemia deletions (eMethods in Supplement 1). Finally, we conducted pathway analyses to align significant findings with biological functions from the curated Gene Ontology (GO) and Reactome databases (eMethods in Supplement 1). This approach is used to analyze lists of important genes to facilitate interpretation and hypothesis generation.
Statistical Analysis
Unless stated otherwise, analyses were conducted with R, version 4.2.1 (CRAN). For the meta-analyses, a Bonferroni-corrected 2-sided P < 8.1 × 10−4 (.05/62) was considered statistically significant. For pathway analyses, an adjusted P < .05 was considered significant.
Results
We identified 571 manuscripts published before May 16, 2023, reporting genotype-phenotype associations across 29 670 unique individuals (50% ≤ 18 years of age) from 43 countries (eMethods, eFigures 1 and 2, and eTable 5 in Supplement 1; eTable 1 in Supplement 2).8,11,12,13,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157,158,159,160,161,162,163,164,165,166,167,168,169,170,171,172,173,174,175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201,202,203,204,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226,227,228,229,230,231,232,233,234,235,236,237,238,239,240,241,242,243,244,245,246,247,248,249,250,251,252,253,254,255,256,257,258,259,260,261,262,263,264,265,266,267,268,269,270,271,272,273,274,275,276,277,278,279,280,281,282,283,284,285,286,287,288,289,290,291,292,293,294,295,296,297,298,299,300,301,302,303,304,305,306,307,308,309,310,311,312,313,314,315,316,317,318,319,320,321,322,323,324,325,326,327,328,329,330,331,332,333,334,335,336,337,338,339,340,341,342,343,344,345,346,347,348,349,350,351,352,353,354,355,356,357,358,359,360,361,362,363,364,365,366,367,368,369,370,371,372,373,374,375,376,377,378,379,380,381,382,383,384,385,386,387,388,389,390,391,392,393,394,395,396,397,398,399,400,401,402,403,404,405,406,407,408,409,410,411,412,413,414,415,416,417,418,419,420,421,422,423,424,425,426,427,428,429,430,431,432,433,434,435,436,437,438,439,440,441,442,443,444,445,446,447,448,449,450,451,452,453,454,455,456,457,458,459,460,461,462,463,464,465,466,467,468,469,470,471,472,473,474,475,476,477,478,479,480,481,482,483,484,485,486,487,488,489,490,491,492,493,494,495,496,497,498,499,500,501,502,503,504,505,506,507,508,509,510,511,512,513,514,515,516,517,518,519,520,521,522,523,524,525,526,527,528,529,530,531,532,533,534,535,536,537,538,539,540,541,542,543,544,545,546,547,548,549,550,551,552,553,554,555,556,557,558,559,560,561,562,563,564,565,566,567,568,569,570,571,572,573,574,575,576,577,578,579,580,581,582,583 Approximately 52% of individuals resided in USA, Canada, France, or Brazil, while just 6628 individuals studied (22%) were from African cohorts (eFigure 2 and eTable 5 in Supplement 1). Fifty-five manuscripts (10%) assessed individuals from more than 1 country. At least 14 970 individuals were included in studies as children (eTable 1 in Supplement 2 and eTable 5 in Supplement 1), but some children were also included in studies as adults in longitudinal cohorts.
Risk of Bias
Of 17 757 reported associations, 3631 (20%) were meta-suitable, containing all elements required to perform a meta-analysis, and only 44 results from 2 studies met all contemporary criteria (eTable 2 in Supplement 2). The proportion of studies using a more rudimentary exploratory approach to a more rigorous meta-suitable approach did not appear to change over time (eFigure 3 in Supplement 1). For the rest of our analysis, we grouped the contemporary results with the meta-suitable results (3675 total results).
Genotype-Phenotype Associations
Across the 571 publications analyzed, 17 757 association results in 1552 unique genes were reported, along with 249 078 genome-wide association summary statistics (Figure 1, Table 1, and eTables 2 and 3 in Supplement 2). The number of genes interrogated varied by phenotype category (Table 1), ranging from 9 (retinopathy) to 452 (HbF). Of the 2399 unique gene- or polygene-phenotype category pairs (eTable 6 in Supplement 2), there was a median (IQR) of 2 (1-4) results, with 1976 (82%) limited to a single study. Overall, 4890 (28%) extracted results were statistically significant (eMethods in Supplement 1), but this does not account for varying significance thresholds (ie, 0.05, 5 × 10−8, etc).
Table 1. Number Of Studies, Total Results, and Unique Genes Reported for Each Phenotype Category.
| Phenotype categorya | Complication prevalence, %b | Total studies, No. | Total results, No. | Unique genes, No. |
|---|---|---|---|---|
| Acute SCD-related complications | 2316 | |||
| Acute pain episode | 100 | 140 | 1467 | 113 |
| ACS, pneumonia, or respiratory infection | 30 | 74 | 334 | 61 |
| Infection (excludes respiratory infection) | 10 | 42 | 290 | 35 |
| Priapism | 30c | 32 | 108 | 40 |
| Acute splenic sequestration | 15 | 19 | 51 | 11 |
| Other acute phenotype | NA | 24 | 66 | 17 |
| Chronic SCD-related complications | 6253 | |||
| Allo- or autoantibody or transfusion reaction | 20 | 14 | 1067 | 253 |
| Cerebrovascular disease | 50 | 106 | 1083 | 222 |
| Kidney dysfunction | 35 | 57 | 1174 | 220 |
| Cardiopulmonary dysfunction | 50 | 37 | 606 | 83 |
| Hyperbilirubinemia, cholelithiasis, cholecystitis, or cholecystectomy | 50 | 102 | 815 | 46 |
| Osteonecrosis | 30 | 59 | 227 | 45 |
| Leg ulcers | 15 | 42 | 163 | 43 |
| Iron overload | 30 | 30 | 81 | 29 |
| Chronic pain | 55 | 13 | 225 | 22 |
| Splenic dysfunction | 90 | 28 | 60 | 11 |
| Retinopathy | 50 | 13 | 52 | 9 |
| Other chronic phenotype | NA | 37 | 700 | 190 |
| Hematologic parameters and biomarkers of disease severity | 8582 | |||
| HbFd | NA | 240 | 3952 | 452 |
| Hemolysis | NA | 155 | 789 | 64 |
| Anemiad | 95 | 196 | 738 | 58 |
| Oxidative stress | NA | 23 | 196 | 22 |
| Other hematologic parameter | NA | 199 | 2345 | 113 |
| Other parameter or biomarker | NA | 63 | 562 | 78 |
| General or mixed measurement of SCD severity | NA | 89 | 606 | 171 |
| Totald | NA | 571 | 17 757 | 1552 |
Abbreviations: ACS, acute chest syndrome; HbF, fetal hemoglobin; NA, not applicable; SCD, sickle cell disease.
Within each subset, phenotype categories are ordered by decreasing number of total unique genes, excepting “other” categories, which are listed last.
Complication prevalence rates were obtained from published estimates among adults of all SCD subtypes within the United States, when available (eMethods in Supplement 1).
Among male participants.
Excludes 249 078 genome-wide summary statistics from 2 publications to avoid count distortion.
Meta-Analysis
While 3675 of the 17 757 total results (21%) were categorized as meta-suitable, due to differences in specific phenotypes, genotypes, or statistical methods, studies analyzing similar outcomes were often insufficiently harmonized for cross-comparison or were not replicated (Figure 1; eMethods in Supplement 1). Only 173 of 17 757 results (1%) plus 2 replication results and 18 genome-wide association summary statistics, representing 62 distinct genotype-phenotype associations, could be cross-study meta-analyzed (Figure 1, Table 2). Of these 193 results, 111 (58%) matched direction and significance with the meta-analysis results; of the remaining 82, 54 (66%) were in directional agreement but differed in statistical significance.
Table 2. Meta-Analysis Resultsa.
| Phenotype category | Specific outcome | Variant | Gene | EA | OA | No. of studiesb | Total sample size, No. | z Scorec | P valued | Directione | Significancef |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ACS, pneumonia, or respiratory infection189,363 | ACS (event occurrence) | rs2070744 | NOS3 | CC | TT or CT | 2 | 273 | 1.4 | .16 | +− | SN |
| Acute pain episode148,207,300 | VOC (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 3 | 435 | 2.18 | .03 | +++ | NNN |
| Acute pain episode185,333 | VOC (event occurrence) | rs5030737, rs1800450, rs1800451 | MBL2 | AA | AO or OO | 2 | 155 | −0.62 | .54 | +− | NS |
| Acute pain episode17,92 | VOC (event rate) | rs1042713 | ADRB2 | A | G | 2 | 463 | −1.16 | .25 | −− | NN |
| Acute pain episode17,92 | VOC (event rate) | rs1042713 | ADRB2 | AA or AG | GG | 2 | 463 | −2.55 | .01 | −− | SN |
| Acute pain episode17,113 | VOC (event rate) | rs10483639 | GCH1 | C | G | 2 | 458 | 1.96 | .05 | ++ | NN |
| Acute pain episode17,113 | VOC (event rate) | rs10483639 | GCH1 | CC or CG | GG | 2 | 458 | 1.61 | .11 | +− | NN |
| Acute pain episode17,159 | VOC (event rate) | rs1800587 | IL1A | A | G | 2 | 442 | 2.92 | .004 | +− | SN |
| Acute pain episode17,159 | VOC (event rate) | rs1800587 | IL1A | AA or AG | GG | 2 | 442 | 2.19 | .03 | +− | NN |
| Acute pain episode17,116 | VOC (event rate) | rs1947913 | TRPA1 | A | T | 2 | 459 | −1.67 | .10 | −+ | NN |
| Acute pain episode17,116 | VOC (event rate) | rs1947913 | TRPA1 | AA or AT | TT | 2 | 459 | 0.017 | .99 | −+ | NN |
| Acute pain episode17,112 | VOC (event rate) | rs2963155 | NR3C1 | A | G | 2 | 463 | 0.87 | .38 | +− | SS |
| Acute pain episode17,112 | VOC (event rate) | rs2963155 | NR3C1 | AA | GG or GA | 2 | 463 | 0.71 | .48 | +− | SS |
| Acute pain episode17,227 | VOC (event rate) | rs4680 | COMT | A | G | 2 | 457 | 3.3 | 9.60 × 10−4 | ++ | SN |
| Acute pain episode17,265 | VOC (event rate) | rs6858735 | TBC1D1 | T | C | 3 | 2228 | 4.57 | 4.81 × 10−6 | ++− | SNN |
| Acute pain episode17,265 | VOC (event rate) | rs7899453 | RPS24 | A | C | 3 | 2228 | 4.7 | 2.61 × 10−6 | +++ | NNN |
| Acute splenic sequestration148,300 | Acute splenic sequestration (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 239 | 1.59 | .11 | −+ | NS |
| Anemia148,207 | Blood transfusion (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 225 | −1.81 | .07 | −− | NN |
| Anemia118,436 | Hemoglobin (continuous) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 1193 | 5.12 | 3.11 × 10−7 | ++ | SS |
| Anemia115,223 | Hemoglobin (continuous) | rs66650371 | HBS1L-MYB | D | I | 2 | 986 | 2.96 | .003 | ++ | SN |
| Anemia115,223 | Hemoglobin (continuous) | rs7482144 | Extended β-globin locus | A | G | 2 | 986 | 2.4 | .02 | ++ | NS |
| Cerebrovascular disease25,389 | Abnormal TCD result (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 366 | −5.16 | 2.42 × 10−7 | −− | SS |
| Cerebrovascular disease21,118,148,300,311,445 | Stroke (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 6 | 3655 | −5.12 | 2.97 × 10−7 | −−−+−− | SSNNSS |
| Cerebrovascular disease382,400 | Stroke (event occurrence) | GT repeats | AGT | A3 and/or A4 | Other | 2 | 219 | −0.35 | 0.73 | −+ | NS |
| Cerebrovascular disease300,387 | Stroke (event occurrence) | rs1800629 | TNF | AA or GA | GG | 3 | 599 | −1.83 | .07 | −−− | NSN |
| Cerebrovascular disease196,224 | Stroke (time to event) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 595 | −3.43 | 6.00 × 10−4 | −− | SS |
| Hyperbilirubinemia, cholelithiasis, cholecystitis, or cholecystectomy148,289 | Cholelithiasis (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 242 | −2.48 | .01 | −− | NS |
| Hyperbilirubinemia, cholelithiasis, cholecystitis, or cholecystectomy344,380,205 | Cholelithiasis (event occurrence) | TA repeats | UGT1A locus | (6/6) | (7/7) | 4 | 821 | −5.09 | 3.57 × 10−7 | −−−− | NSSS |
| Hyperbilirubinemia, cholelithiasis, cholecystitis, or cholecystectomy344,380 | Cholelithiasis (event occurrence) | TA repeats | UGT1A locus | (6/6) | (6/7) | 3 | 719 | −1.92 | .06 | −−− | NNN |
| Hyperbilirubinemia, cholelithiasis, cholecystitis, or cholecystectomy380,205 | Cholelithiasis (event occurrence) | TA repeats | UGT1A locus | (6/6) | (7/8) | 3 | 668 | −4.27 | 1.93 × 10−5 | −−− | NNS |
| General or mixed measurement of SCD severity148,207 | Hospitalization (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 225 | −0.9 | .37 | −− | NN |
| HbF17,57,163,269,270,312 | HbF (continuous) | rs11886868 | BCL11A | T | C | 7 | 2339 | −15.3 | 7.08 × 10−53 | −−−−−−− | SSNNSSS |
| HbF17,115,122,163,202,214,269,270 | HbF (continuous) | rs1427407 | BCL11A | T | G | 10 | 3394 | 20.66 | 8.63 × 10−95 | ++++++++++ | SSSSSSSSNS |
| HbF17,228,312 | HbF (continuous) | rs28384513 | HBS1L-MYB | A | C | 4 | 1947 | 5.26 | 1.43 × 10−7 | ++−+ | NNNS |
| HbF17,210 | HbF (continuous) | rs35786788 | HBS1L-MYB | A | G | 2 | 1606 | 7.11 | 1.20 × 10−12 | ++ | NN |
| HbF17,163,228,269,270,312 | HbF (continuous) | rs4671393 | BCL11A | A | G | 7 | 2256 | 16.25 | 2.19 × 10−59 | +++++++ | SSSNSSS |
| HbF17,57,210,228,269,270 | HbF (continuous) | rs4895441 | HBS1L-MYB | A | G | 6 | 2221 | −7.55 | 4.24 × 10−14 | −−−−+− | SSNSNN |
| HbF115,122 | HbF (continuous) | rs6545816 | BCL11A | A | C | 2 | 841 | −4 | 6.19 × 10−5 | +− | NS |
| HbF17,115,122,210 | HbF (continuous) | rs66650371 | HBS1L-MYB | D | I | 4 | 2447 | 9.52 | 1.82 × 10−21 | ++++ | NSSS |
| HbF17,214 | HbF (continuous) | rs6706648 | BCL11A | T | C | 5 | 1728 | −12.57 | 3.01 × 10−36 | −−−−− | SNSNS |
| HbF269,270 | HbF (continuous) | rs6729815 | BCL11A | T | C | 2 | 198 | −0.12 | .91 | −+ | NN |
| HbF17,269,270 | HbF (continuous) | rs6732518 | BCL11A | T | C | 3 | 782 | −4.55 | 5.27 × 10−6 | −++ | SNN |
| HbF17,214 | HbF (continuous) | rs6738440 | BCL11A | A | G | 5 | 1728 | 9.7 | 3.10 × 10−22 | +++++ | SNSNS |
| HbF269,270 | HbF (continuous) | rs73555746 | HBS1L-MYB | A | C | 2 | 198 | 0.39 | .69 | +− | NN |
| HbF17,115,122,228,312 | HbF (continuous) | rs7482144 | Extended β-globin locus | A | G | 5 | 2637 | 7.14 | 9.27 × 10−13 | +++++ | NNSSS |
| HbF17,57 | HbF (continuous) | rs7557939 | BCL11A | A | G | 2 | 834 | −9.19 | 3.95 × 10−20 | −− | SN |
| HbF57,214 | HbF (continuous) | rs7599488 | BCL11A | T | C | 4 | 1298 | 0.47 | .63 | −++− | NNNN |
| HbF17,202,214 | HbF (continuous) | rs7606173 | BCL11A | C | G | 6 | 2354 | −13.68 | 1.41 × 10−42 | −−+−−− | SSNSNS |
| HbF17,269,270 | HbF (continuous) | rs766432 | BCL11A | A | C | 3 | 782 | −10.13 | 4.04 × 10−24 | −+− | SNS |
| HbF269,270 | HbF (continuous) | rs7775698 | HBS1L-MYB | A | G | 2 | 198 | 0.53 | 0.60 | +− | NN |
| HbF17,163,228,269,270,312 | HbF (continuous) | rs9399137 | HBS1L-MYB | T | C | 7 | 2256 | −7.24 | 4.64 × 10−13 | −−−−−−− | NSNNNSS |
| HbF17,115,163,269,270,312 | HbF (continuous) | rs9402686 | HBS1L-MYB | A | G | 7 | 2349 | 8.44 | 3.11 × 10−17 | +++−+++ | SSSNNSS |
| HbF17,312 | HbF (continuous) | rs9494142 | HBS1L-MYB | T | C | 3 | 1780 | −5.31 | 1.07 × 10−7 | −−− | SNN |
| HbF17,57,210 | HbF (continuous) | rs9494145 | HBS1L-MYB | T | C | 3 | 1856 | −7.88 | 3.33 × 10−15 | −−− | NSN |
| Leg ulcers148,118 | Leg ulcers (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 2 | 2336 | −2.05 | .04 | −− | NN |
| Other hematologic parameter115,223 | PLT (continuous) | rs66650371 | HBS1L-MYB | D | I | 2 | 986 | −0.96 | .34 | −+ | SN |
| Other hematologic parameter115,223 | PLT (continuous) | rs7482144 | Extended β-globin locus | A | G | 2 | 986 | −1.97 | .05 | −− | NN |
| Other hematologic parameter115,223 | RBC count (continuous) | rs66650371 | HBS1L-MYB | D | I | 2 | 986 | 1.89 | .06 | ++ | SN |
| Other hematologic parameter115,223 | RBC count (continuous) | rs7482144 | Extended β-globin locus | A | G | 2 | 986 | 1.66 | .10 | ++ | NN |
| Priapism21,118,148 | Priapism (event occurrence) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 3 | 1745 | −2.64 | .008 | −−+ | SNN |
| Kidney dysfunction20,320,514 | Albuminuria (time to event) | α-Thalassemia | α-Globin | 1 or 2 Deletions | No deletions | 3 | 978 | −3.54 | 4.10 × 10−4 | −−− | NSS |
| Kidney dysfunction170,543 | Albuminuria (event occurrence) | G1/G2 | APOL1 | Homozygous G1 or G2 or compound heterozygous | Other | 2 | 433 | 2.83 | .005 | ++ | NS |
Abbreviations: ACS, acute chest syndrome; EA, effect allele or genotype; HbF, fetal hemoglobin; OA, other allele or genotype; PLT, platelets; RBC, red blood cell; TCD, transcranial doppler; VOC, vaso-occlusive crisis.
Weighted z score meta-analyses were conducted for all single-nucleotide variant–phenotype pairs with meta-suitable or contemporary results from at least 2 cohorts that were reported in at least 2 manuscripts, where the phenotype was the same, the same genotype comparison was made, and the same statistical test was performed.
Represents the number of results combined in each meta-analysis, including replication results and genome-wide summary statistics.
Meta-analysis z score, indicating direction of association for EA.
Statistically significant at the Bonferroni-corrected threshold of P < 8.1 × 10−4 (.05/62).
Represents the association direction for the EA compared with the OA of each component study: + indicates increasing and −, decreasing.
Significance for each respective association is labeled S when it was reported as significant and N otherwise.
Meta-analysis results indicated that α-thalassemia deletions were significantly associated with increased hemoglobin level (Z = 5.12; P = 3.11 × 10−7) and reduced risk of albuminuria (Z = −3.54; P = 4.10 × 10−4), abnormal transcranial Doppler velocity (Z = −5.16; P = 2.42 × 10−7), and stroke (Z = −5.12; P = 2.97 × 10−7 for occurrence; Z = −3.43; P = 6.00 × 10−4 for time to event) (Table 2). Ten SNVs in BCL11A, 8 in HBS1L-MYB, and 1 in the γ-globin gene (rs7482144, the XmnI site of HBG2) were significantly associated with HbF (absolute value of Z = 4.00 to 20.66; P = 8.63 × 10−95 to 6.19 × 10−5). An increased number UGT1A1 promoter repeats was associated with increased risk for cholelithiasis (Z = 5.09, P = 3.57 × 10−7 for (TA)7/(TA)7; Z = 4.27, P = 1.93 × 10−5 for (TA)7/(TA)8; compared with (TA)6/(TA)6). Single-nucleotide variants in RPS24 (rs7899453-A, Z = 4.70; P = 2.61 × 10−6) and TBC1D1 (rs6858735-T, Z = 4.57; P = 4.81 × 10−6) were significantly associated with increased rate of vaso-occlusive crisis.
While high-risk G1/G2 APOL1 variants were frequently associated with numerous markers for kidney dysfunction (eTables 2 and 6 in Supplement 2), only 2 results could be meta-analyzed, showing nominal association with increased risk for albuminuria (Z = 2.83, P = .005) (Table 2). Similarly, 1 SNV in COMT (rs4680) was nominally associated with vaso-occlusive crisis in our meta-analyses (Z = 3.30, P = 9.60 × 10−4) (Table 2), but numerous SNVs and haplotypes within this gene have been associated with acute pain outcomes (eTables 2 and 6 in Supplement 2).
HbF
The genetics of HbF expression was more widely studied than any other SCD phenotype, with 240 of 571 studies (42%) reporting a total of 3952 associations involving 452 genes (Table 1; eTables 2 and 6 in Supplement 2). Significant associations with HbF were reported for 140 genes in 144 studies, yet 1220 of 1347 (91%) of these associations were exploratory (eFigure 4 in Supplement 1 and eTables 2 and 6 in Supplement 2). Most significant results identified the extended β-like globin locus, BCL11A, or HBS1L-MYB, with few repeated or meta-suitable results outside of these regions (eFigure 4 in Supplement 1). Numerous studies linked these same HbF modifier genes to specific complications of SCD, including acute pain, anemia, cerebrovascular disease, and hemolysis (eTables 2 and 6 in Supplement 2).
α-Thalassemia
Concurrent α-thalassemia was consistently associated with increased hemoglobin levels (eg, continuous β, 0.39; 95% CI, 0.24-0.53) and reduced risk for elevated markers of hemolysis (eg, hemolytic component β, −0.70; 95% CI, −1.26 to −0.14), hepatomegaly (≤4 cm vs >4 cm β, 1.82; 95% CI, 0.51-3.31), biliary dysfunction (eg, bilirubin levels as high vs low β, −1.32; 95% CI, −2.51 to −0.13), stroke (eg, occurrence β, −0.85; 95% CI, −1.18 to −0.52), and kidney dysfunction (eg, albuminuria occurrence β, −1.10; 95% CI, −1.74 to −0.47) (Figure 2). While less clear, there may be increased risk of acute pain crisis (eg, vaso-occlusive crisis events per year β, 0.29; 95% CI, −0.16 to 0.74), acute splenic sequestration (β, 0.72; 95% CI, −0.19 to 1.62), and osteonecrosis (β, 1.06; 95% CI, −0.22 to 2.36) and reduced risk of leg ulcers (β, −0.30; 95% CI, −0.59 to −0.01) and priapism (β, −0.42; 95% CI, −0.75 to −0.08).
Figure 2. Published α-Thalassemia Associations Across All Meta-Suitable Results.
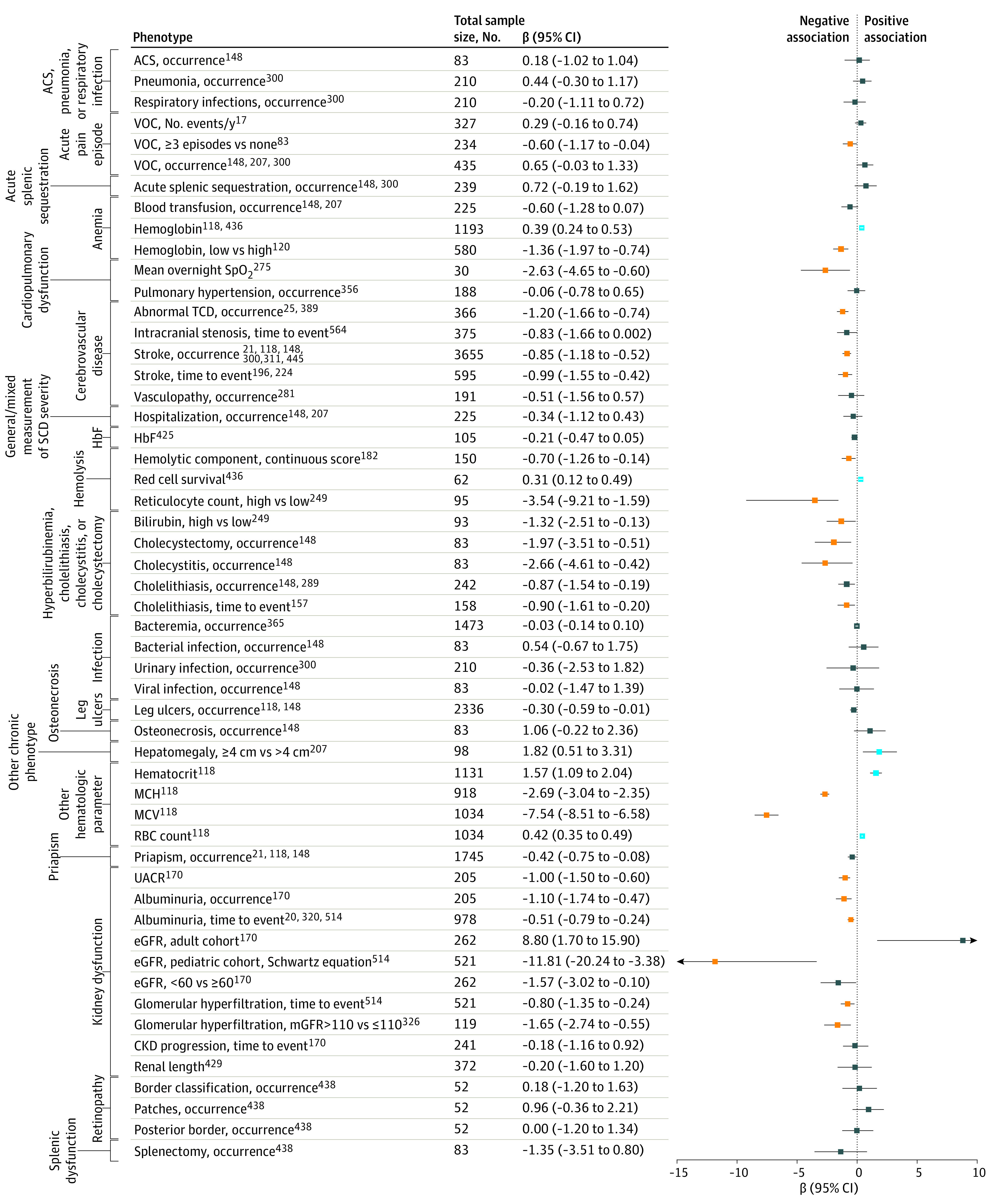
All associations are comparing 1 or 2 deletions vs no deletions, except fetal hemoglobin (HbF) and cholelithiasis (time to event), where only additive genotype coding was reported. Where appropriate, odds ratios and hazard ratios were transformed to β-scale (log odds ratio or log hazard ratio) for purposes of plotting. Results studied in more than 1 study were first combined via fixed-effects meta-analysis. Significant results as originally reported are shown in blue and gold for positive and negative association directions, respectively. For those meta-analyzed, significance was determined as P < 8.1 × 10−4 (.05/62). ACS indicates acute chest syndrome; CKD, chronic kidney disease; eGFR, estimated glomerular filtration rate; HbF, fetal hemoglobin; MCH, mean corpuscular hemoglobin; MCV, mean corpuscular volume; mGFR, measured glomerular filtration rate; RBC, red blood cell; SCD, sickle cell disease; SpO2, hemoglobin oxygen saturation; TCD, transcranial Doppler; UACR, urine albumin-to-creatinine ratio; and VOC, vaso-occlusive crisis.
Pathway Analysis
Consistent with the known pathophysiology of SCD, genes with at least 1 significant result for any outcome were enriched for cellular adhesion, oxidative and toxic stress, inflammation, and blood vessel regulation, with 23 of the 25 most enriched GO pathways representing these processes (Figure 3; eTables 7 and 8 in Supplement 2). Phenotype category-specific pathway analyses (eTables 7 and 8 in Supplement 2) identified numerous other pathway enrichments, including the flavonoid metabolic GO pathway, for which genes associated with hyperbilirubinemia or biliary dysfunction, hemolysis, and anemia were enriched.
Figure 3. Enriched Pathways for Genes Reported to be Significantly Associated With Any Sickle Cell Disease–Related Outcome.

Gene Ontology– and Reactome-curated pathways were examined for any pathways with significant enrichment among all genes with at least 1 reported significant result. The top 25 and all 22 with adjusted P < .05 are reported for Gene Ontology and Reactome, respectively. Circle size represents the number of genes in that pathway out of the total submitted genes (gene ratio); circle color, degree of significance after adjusting for multiple testing (adjusted P value, using the Benjamini-Hochberg method). ADME indicates absorption, distribution, metabolism, and excretion; BMP, bone morphogenetic proteins; CO2, carbon dioxide; O2, oxygen; PpS, Peters-plus syndrome; and TSR, thrombospondin type 1 repeat.
Discussion
This comprehensive systematic review and meta-analysis consolidates current knowledge of SCD genetic modifiers, incorporating data from 571 publications from 1981 to 2023 and describing at least 29 670 unique individuals residing in 43 countries. These 571 studies were assessed for quality of study design and reporting, according to STREGA guidelines. Remarkably, only 1% of results reported across the last 43 years of work met minimum standards for cross-study meta-analysis due to variability in study designs, reporting practices, and phenotype or genotype definitions.
The differing methodologies and reporting practices between screened studies limited our ability to include, aggregate, or analyze published data. Many manuscripts used individuals without SCD as controls and were excluded, as this does not assess the genetic modifiers of SCD severity. Similarly, studies using biochemical measures as surrogates for genotypes (ie, glucose-6-phosphate dehydrogenase levels) were excluded because biochemical measurements do not always align with genotype. Other limitations included use of rudimentary statistical tests, rather than regression-based techniques adjusting for confounders, and inconsistent phenotype or genotype definitions. Despite these methodological challenges, we resolved some previous discrepant results and provide guidelines for future studies.
Across 571 manuscripts, we, as in a recent review,585 identified several genes associated with SCD complications in at least 2 studies. However, among those validated in meta-analysis, most were related to HbF levels. Specifically, meta-analyses confirmed the polygenic regulation of HbF: 10 SNVs in BCL11A, 8 in HBS1L-MYB, and 1 HBG2 were significantly associated with HbF. Remarkably, 137 additional genes were reportedly associated with HbF but have not been confirmed due to lack of validation in a separate cohort or insufficient harmonization of phenotype (ie, dichotomous vs continuous, F cell percentage vs HbF percentage, unclear units) or genotype. Only 20%-50% of the genetic variability in HbF can be explained by currently validated variants (extended β-like globin locus, BCL11A, and HBS1L-MYB),10,11,12,13 which have relatively large association sizes. Most likely, many other loci with small association sizes or low frequency account for the remaining heritability. These additional modifier variants may be represented among the genes that lack validation.
This review also clarifies the role of α-thalassemia as a modifier of SCD severity by demonstrating that α-thalassemia was associated with reduced risk of clinically relevant SCD symptoms thought to be driven by hemolysis, including severe anemia, hyperbilirubinemia or gallstones, kidney dysfunction, and stroke. While this was a commonly accepted belief,232 our analysis consolidated the data to identify consistent trends among nonharmonized phenotypes with conflicting results.
To date, most validated studies of SCD modifiers have identified common variants with large association sizes (ie, “low-hanging fruit”) in relatively small cohorts. However, most genetic variation in health-related traits is driven by the interplay of many variants with small association sizes or low allele frequencies.586 Discovering such modifiers of SCD will require well-designed studies in larger cohorts using modern approaches to genetic analyses, including genome-wide association studies, adjustment for covariates, and, as discussed by Pincez, et al,585 multiomics. Future studies following best practices may also confirm candidate associations that have been reported in only 1 study. Moreover, studies in African cohorts could identify heretofore undiscovered variants with different frequencies in European and admixed cohorts. Most high-income countries, such as the US, have relatively few patients with SCD available for genetic studies. By contrast, while only 22% of individuals studied to date were from African cohorts, millions of individuals with SCD reside in sub-Saharan Africa, reflecting a fertile region for future research.
Advanced statistical and machine learning approaches, will also prove beneficial. For example, polygenic scores combining variants of small association sizes have been generated, including for HbF,17,81,104,122,544 pain,17,81,104 kidney,20,170 and cerebrovascular outcomes34,36,132 in SCD. However, those scores generally account for a small fraction of heritability and thus lack clinical utility. Improving polygenic scores for SCD phenotypes will require identifying more variants, validation, and rigorous testing, all of which would benefit from larger, more diverse cohorts and could be informed by analogous studies in non-SCD cohorts. Similarly, mendelian randomization, a method to explore causal relationships, has been used infrequently in SCD cohorts.544,576 In addition to increasing studies in Africa of both individuals with or without SCD, local ancestry inference in admixed individuals may help deconstruct associations driven by African ancestry.
Pathway analysis is another avenue for identifying potential candidate genes and generating clinically relevant hypotheses, even when traditional meta-analysis is not possible, as it can identify biologically meaningful pathways based on genes with significant associations and indicate other genes in these pathways that may contribute to disease risk. Among genes significantly associated with any SCD outcome in at least 1 study, we found enrichment in pathways controlling cellular adhesion, inflammation, response to toxic and oxidative stress, and blood vessel regulation, aligning with known disease pathophysiology. Other genes in those pathways are potential candidates for future investigation. We also identified potential therapeutic targets, such as flavonoid metabolic processes (of interest generally587,588 and in SCD589,590), which were enriched for genes associated with hemolysis, anemia, and hyperbilirubinemia or biliary complications.
Limitations
This study has limitations that may confound or reduce the generalizability of our results. Subtype of SCD, ancestry, and hydroxyurea treatment status were not often reported in detail or, in the case of SCD subtype and ancestry, determined genetically; thus, we made no attempt to assess the difference between SCD subtypes or ancestries or to examine treatment response. Because most studies used a candidate gene approach, our results may be biased toward genes or pathways that were historically of high interest. Similarly, our analysis could be affected by unreported negative or contradictory results arising from positive publication bias. There are some methods, such as bayesian approaches, that cannot be integrated into a meta-analysis, resulting in some high-quality results being classified as exploratory. Finally, while our analysis categories allowed for a measure of study design and reporting rigor, they did not constitute a formal risk of bias assessment.
Conclusions
Although this systematic review and meta-analysis assessed 571 manuscripts that collectively reported 17 757 genetic associations with outcomes related to SCD severity, those associations validated in cross-study meta-analysis were largely related to HbF. To accelerate the understanding of the genetic etiology of SCD, future genetic association studies should report sufficient information for results to be included in meta-analyses. Elements of contemporary study design and international collaborations will improve scientific rigor, reduce the risk of false positives, and expand generalizability of study results. To facilitate cross-study analysis, the use of consensus measures is recommended for phenotypes and exposures.591,592 Combined, these steps will generate the high quality results necessary to develop clinically actionable genetic tools.
eMethods.
eFigure 1. PRISMA Flow Diagram For Manuscript Identification, Screening, and Inclusion
eFigure 2. Map of Included Patient Cohort Locations by Number of Individuals
eFigure 3. Data Strength Categories by Time
eFigure 4. Number of Papers Reporting Significant Associations With Fetal Hemoglobin (HbF) and Number of Unique Variants for Genes With Significant Results Reported in at Least Three Manuscripts
eTable 4. Phenotype Categories With Major Constituent Phenotypes
eTable 5. Number of Unique Patients and Studies by Country
eReferences.
eTable 1. Manuscripts Included in This Systematic Review and Meta-Analysis
eTable 2. Data Extracted and Utilized in This Systematic Review and Meta-Analysis, Excluding Genome-Wide Summary Statistics
eTable 3. Genome-Wide Summary Statistics Extracted and Utilized in This Systematic Review and Meta-Analysis
eTable 6. Full List of Gene-Phenotype Category Associations
eTable 7. Gene Ontology Pathway Analysis Results
eTable 8. Reactome Pathway Analysis Results
Data Sharing Statement
References
- 1.Piel FB, Patil AP, Howes RE, et al. Global distribution of the sickle cell gene and geographical confirmation of the malaria hypothesis. Nat Commun. 2010;1:104. doi: 10.1038/ncomms1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi: 10.1038/nrdp.2018.10 [DOI] [PubMed] [Google Scholar]
- 3.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4)(suppl):S512-S521. doi: 10.1016/j.amepre.2009.12.022 [DOI] [PubMed] [Google Scholar]
- 4.Ojodu J, Hulihan MM, Pope SN, Grant AM; Centers for Disease Control and Prevention (CDC) . Incidence of sickle cell trait–United States, 2010. MMWR Morb Mortal Wkly Rep. 2014;63(49):1155-1158. [PMC free article] [PubMed] [Google Scholar]
- 5.Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37-47. doi: 10.1016/j.blre.2006.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grosse SD, Odame I, Atrash HK, Amendah DD, Piel FB, Williams TN. Sickle cell disease in Africa: a neglected cause of early childhood mortality. Am J Prev Med. 2011;41(6)(suppl 4):S398-S405. doi: 10.1016/j.amepre.2011.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lubeck D, Agodoa I, Bhakta N, et al. Estimated life expectancy and income of patients with sickle cell disease compared with those without sickle cell disease. JAMA Netw Open. 2019;2(11):e1915374. doi: 10.1001/jamanetworkopen.2019.15374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644. doi: 10.1056/NEJM199406093302303 [DOI] [PubMed] [Google Scholar]
- 9.Akinsheye I, Alsultan A, Solovieff N, et al. Fetal hemoglobin in sickle cell anemia. Blood. 2011;118(1):19-27. doi: 10.1182/blood-2011-03-325258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Menzel S, Garner C, Gut I, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007;39(10):1197-1199. doi: 10.1038/ng2108 [DOI] [PubMed] [Google Scholar]
- 11.Uda M, Galanello R, Sanna S, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc Natl Acad Sci U S A. 2008;105(5):1620-1625. doi: 10.1073/pnas.0711566105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lettre G, Sankaran VG, Bezerra MA, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and beta-globin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci U S A. 2008;105(33):11869-11874. doi: 10.1073/pnas.0804799105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Solovieff N, Milton JN, Hartley SW, et al. Fetal hemoglobin in sickle cell anemia: genome-wide association studies suggest a regulatory region in the 5′ olfactory receptor gene cluster. Blood. 2010;115(9):1815-1822. doi: 10.1182/blood-2009-08-239517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839-1842. doi: 10.1126/science.1165409 [DOI] [PubMed] [Google Scholar]
- 15.Esrick EB, Lehmann LE, Biffi A, et al. Post-transcriptional genetic silencing of bcl11a to treat sickle cell disease. N Engl J Med. 2021;384(3):205-215. doi: 10.1056/NEJMoa2029392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia. N Engl J Med. 2021;384(3):252-260. doi: 10.1056/NEJMoa2031054 [DOI] [PubMed] [Google Scholar]
- 17.Rampersaud E, Kang G, Palmer LE, et al. A polygenic score for acute vaso-occlusive pain in pediatric sickle cell disease. Blood Adv. 2021;5(14):2839-2851. doi: 10.1182/bloodadvances.2021004634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Akbulut-Jeradi N, Fernandez MJ, Al Khaldi R, Sukumaran J, Adekile A. Unique Polymorphisms at BCL11A, HBS1L-MYB and HBB loci associated with HbF in Kuwaiti patients with sickle cell disease. J Pers Med. 2021;11(6):567. doi: 10.3390/jpm11060567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adabale A, Makanjuola SBL, Akinbami A, et al. Frequency of beta S globin gene haplotypes among sickle cell patients in Nigeria. J Int Med Res. 2021;49(6):3000605211019918. doi: 10.1177/03000605211019918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rashkin SR, Rampersaud E, Kang G, et al. Generalization of a genetic risk score for time to first albuminuria in children with sickle cell anaemia: SCCRIP cohort study results. Br J Haematol. 2021;194(2):469-473. doi: 10.1111/bjh.17647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batista JVGF, Pereira-Martins DA, Falcão DA, et al. Association of KLOTHO polymorphisms with clinical complications of sickle cell anemia. Ann Hematol. 2021;100(8):1921-1927. doi: 10.1007/s00277-021-04532-w [DOI] [PubMed] [Google Scholar]
- 22.Delgadinho M, Ginete C, Santos B, Miranda A, Brito M. Genotypic diversity among Angolan children with sickle cell anemia. Int J Environ Res Public Health. 2021;18(10):5417. doi: 10.3390/ijerph18105417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alnafie AN, Alateeq SA, Al-Muhanna FA, et al. Exome sequencing in high and low fetal haemoglobin Arab-Indian haplotype sickle cell disease. Br J Haematol. 2021;194(2):e61-e64. doi: 10.1111/bjh.17542 [DOI] [PubMed] [Google Scholar]
- 24.Page GP, Kanias T, Guo YJ, et al. ; National Heart, Lung, and Blood Institute (NHLBI) Recipient Epidemiology Donor Evaluation Study–III (REDS-III) program . Multiple-ancestry genome-wide association study identifies 27 loci associated with measures of hemolysis following blood storage. J Clin Invest. 2021;131(13):e146077. doi: 10.1172/JCI146077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ojewunmi OO, Adeyemo TA, Oyetunji AI, Benn Y, Ekpo MG, Iwalokun BA. Association of alpha-thalassemia and glucose-6-phosphate dehydrogenase deficiency with transcranial Doppler ultrasonography in Nigerian children with sickle cell anemia. J Clin Lab Anal. 2021;35(6):e23802. doi: 10.1002/jcla.23802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brousse V, Pondarre C, Kossorotoff M, et al. Brain injury pathophysiology study by a multimodal approach in children with sickle cell anemia with no intra or extra cranial arteriopathy. Haematologica. 2022;107(4)958-965. doi: 10.3324/haematol.2020.278226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ngo-Bitoungui VJ, Belinga S, Mnika K, et al. Investigations of kidney dysfunction-related gene variants in sickle cell disease patients in Cameroon (sub-Saharan Africa). Front Genet. 2021;12:595702. doi: 10.3389/fgene.2021.595702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatzlhofer BLD, Pereira-Martins DA, de Farias Domingos I, et al. Alpha thalassemia, but not βS-globin haplotypes, influence sickle cell anemia clinical outcome in a large, single-center Brazilian cohort. Ann Hematol. 2021;100(4):921-931. doi: 10.1007/s00277-021-04450-x [DOI] [PubMed] [Google Scholar]
- 29.Romana M, Reminy K, Moeckesch B, et al. Loss of alpha globin genes is associated with improved microvascular function in patients with sickle cell anemia. Am J Hematol. 2021;96(5):E165-E168. doi: 10.1002/ajh.26126 [DOI] [PubMed] [Google Scholar]
- 30.Figueiredo CVB, Santiago RP, da Guarda CC, et al. Priapism in sickle cell disease: Associations between NOS3 and EDN1 genetic polymorphisms and laboratory biomarkers. PLoS One. 2021;16(2):e0246067. doi: 10.1371/journal.pone.0246067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Batista JVGF, Arcanjo GS, Batista THC, et al. Influence of UGT1A1 promoter polymorphism, α-thalassemia and βs haplotype in bilirubin levels and cholelithiasis in a large sickle cell anemia cohort. Ann Hematol. 2021;100(4):903-911. doi: 10.1007/s00277-021-04422-1 [DOI] [PubMed] [Google Scholar]
- 32.Al-Ali AK, Alsulaiman A, Alfarhan M, et al. Sickle cell disease in the eastern province of Saudi Arabia: clinical and laboratory features. Am J Hematol. 2021;96(4):E117-E121. doi: 10.1002/ajh.26096 [DOI] [PubMed] [Google Scholar]
- 33.Kumar R, Yadav R, Mishra S, et al. Krüppel-like factor 1 (KLF1) gene single nucleotide polymorphisms in sickle cell disease and its association with disease-related morbidities. Ann Hematol. 2021;100(2):365-373. doi: 10.1007/s00277-020-04381-z [DOI] [PubMed] [Google Scholar]
- 34.Brewin JN, Rooks H, Gardner K, et al. Genome wide association study of silent cerebral infarction in sickle cell disease (HbSS and HbSC). Haematologica. 2021;106(6):1770-1773. doi: 10.3324/haematol.2020.265827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saraf SL, Zhang X, Shah BN, et al. Engulfment and cell motility 1 (ELMO1) and apolipoprotein A1 (APOA1) as candidate genes for sickle cell nephropathy. Br J Haematol. 2021;193(3):628-632. doi: 10.1111/bjh.17224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Joly P, Bonello-Palot N, Badens C, et al. HbF-promoting polymorphisms may specifically reduce the residual risk of cerebral vasculopathy in SCA children with alpha-thalassemia. Clin Hemorheol Microcirc. 2021;77(3):267-272. doi: 10.3233/CH-200951 [DOI] [PubMed] [Google Scholar]
- 37.Santiago RP, Figueiredo CVB, Fiuza LM, et al. Transforming growth factor beta receptor 3 haplotypes in sickle cell disease are associated with lipid profile and clinical manifestations. Mediators Inflamm. 2020;2020:3185015. doi: 10.1155/2020/3185015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hariharan P, Chavan V, Nadkarni A. Significance of heme oxygenase-1(HMOX1) gene on fetal hemoglobin induction in sickle cell anemia patients. Sci Rep. 2020;10(1):18506. doi: 10.1038/s41598-020-75555-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Urio F, Nkya S, Rooks H, et al. F cell numbers are associated with an X-linked genetic polymorphism and correlate with haematological parameters in patients with sickle cell disease. Br J Haematol. 2020;191(5):888-896. doi: 10.1111/bjh.17102 [DOI] [PubMed] [Google Scholar]
- 40.Santiago RP, Figueiredo CVB, Fiuza LM, et al. TGFBR3 polymorphisms (rs1805110 and rs7526590) are associated with laboratory biomarkers and clinical manifestations in sickle cell anemia. Dis Markers. 2020;2020:8867986. doi: 10.1155/2020/8867986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tozatto-Maio K, Girot R, Ly ID, et al. Polymorphisms in inflammatory genes modulate clinical complications in patients with sickle cell disease. Front Immunol. 2020;11:2041. doi: 10.3389/fimmu.2020.02041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gueye Tall F, Martin C, Ndour EHM, et al. Influence of oxidative stress biomarkers and genetic polymorphisms on the clinical severity of hydroxyurea-free senegalese children with sickle cell anemia. Antioxidants (Basel). 2020;9(9):863. doi: 10.3390/antiox9090863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brewin JN, Smith AE, Cook R, et al. Genetic analysis of patients with sickle cell anemia and stroke before 4 years of age suggest an important role for apoliprotein E. Circ Genom Precis Med. 2020;13(5):531-540. doi: 10.1161/CIRCGEN.120.003025 [DOI] [PubMed] [Google Scholar]
- 44.Sinha S, Jit BP, Patro ARK, et al. Influence of rs1042713 and rs1042714 polymorphisms of β2-adrenergic receptor gene with erythrocyte cAMP in sickle cell disease patients from Odisha State, India. Ann Hematol. 2020;99(12):2737-2745. doi: 10.1007/s00277-020-04254-5 [DOI] [PubMed] [Google Scholar]
- 45.Elenga N, Cuadro-Alvarez E, Martin E, Njuieyon F, Defo A, Maniassom C. Influence of beta-cluster haplotypes, alpha-gene status and UGTA1 polymorphism on clinical and hematological data in sickle-cell disease children from French Guiana. PLoS One. 2020;15(9):e0238691. doi: 10.1371/journal.pone.0238691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khan J, Muhammad D, Ismail M, Khan I, Niaz S; Zia ur Rehman . Consanguinity, the driving force behind inheritance of HbS-β thalassemia in Southern Districts of KP. J Pak Med Assoc. 2020;70(6):978-983. [DOI] [PubMed] [Google Scholar]
- 47.El-Ghamrawy M, Yassa ME, Tousson AMS, et al. Association between BCL11A, HSB1L-MYB, and XmnI γG-158 (C/T) gene polymorphism and hemoglobin F level in Egyptian sickle cell disease patients. Ann Hematol. 2020;99(10):2279-2288. doi: 10.1007/s00277-020-04187-z [DOI] [PubMed] [Google Scholar]
- 48.Gordeuk VR, Shah BN, Zhang X, et al. The CYB5R3c. 350C>G and G6PD A alleles modify severity of anemia in malaria and sickle cell disease. Am J Hematol. 2020;95(11):1269-1279. doi: 10.1002/ajh.25941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costa Neto A, Santos F, Ribeiro I, et al. FcγR2B B2.4 haplotype predicts increased risk of red blood cell alloimmunization in sickle cell disease patients. Transfusion. 2020;60(7):1573-1578. doi: 10.1111/trf.15832 [DOI] [PubMed] [Google Scholar]
- 50.Pereira-Martins DA, Domingos IF, Belini-Junior E, et al. Association of HMIP1 C-893A polymorphism and disease severity in patients with sickle cell anemia. Hematol Transfus Cell Ther. 2021;43(3):243-248. doi: 10.1016/j.htct.2020.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santos B, Delgadinho M, Ferreira J, et al. Co-inheritance of alpha-thalassemia and sickle cell disease in a cohort of Angolan pediatric patients. Mol Biol Rep. 2020;47(7):5397-5402. doi: 10.1007/s11033-020-05628-8 [DOI] [PubMed] [Google Scholar]
- 52.Chamouine A, Saandi T, Muszlak M, et al. High fetal hemoglobin level is associated with increased risk of cerebral vasculopathy in children with sickle cell disease in Mayotte. BMC Pediatr. 2020;20(1):302. doi: 10.1186/s12887-020-02187-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Safwat NA, ELkhamisy MM, Abdel-Wahab SEA, Hamza MT, Boshnak NH, Kenny MA. Polymorphisms of the receptor for advanced glycation end products as vasculopathy predictor in sickle cell disease. Pediatr Res. 2021;89(1):185-190. doi: 10.1038/s41390-020-1014-3 [DOI] [PubMed] [Google Scholar]
- 54.Alaoui-Ismaili FZ, Laghmich A, Ghailani-Nourouti N, Barakat A, Bennani-Mechita M. XmnI Polymorphism in Sickle Cell Disease in North Morocco. Hemoglobin. 2020;44(3):190-194. doi: 10.1080/03630269.2020.1772284 [DOI] [PubMed] [Google Scholar]
- 55.Nkya S, Mwita L, Mgaya J, et al. Identifying genetic variants and pathways associated with extreme levels of fetal hemoglobin in sickle cell disease in Tanzania. BMC Med Genet. 2020;21(1):125. doi: 10.1186/s12881-020-01059-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chinedu O, Tonassé WV, Albuquerque DM, et al. Polymorphisms in the heme oxygenase-1 and bone morphogenetic protein receptor type 1b genes and estimated glomerular filtration rate in Brazilian sickle cell anemia patients. Hematol Transfus Cell Ther. 2021;43(2):165-170. doi: 10.1016/j.htct.2020.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sales RR, Belisário AR, Faria G, Mendes F, Luizon MR, Viana MB. Functional polymorphisms of BCL11A and HBS1L-MYB genes affect both fetal hemoglobin level and clinical outcomes in a cohort of children with sickle cell anemia. Ann Hematol. 2020;99(7):1453-1463. doi: 10.1007/s00277-020-04079-2 [DOI] [PubMed] [Google Scholar]
- 58.Silva M, Vargas S, Coelho A, et al. Biomarkers and genetic modulators of cerebral vasculopathy in sub-Saharan ancestry children with sickle cell anemia. Blood Cells Mol Dis. 2020;83:102436. doi: 10.1016/j.bcmd.2020.102436 [DOI] [PubMed] [Google Scholar]
- 59.Shaheen I, Khorshied M, Abdel-Raouf R, et al. L-selectin P213S and integrin alpha 2 C807T genetic polymorphisms in pediatric sickle cell disease patients. J Pediatr Hematol Oncol. 2020;42(8):e707-e711. doi: 10.1097/MPH.0000000000001839 [DOI] [PubMed] [Google Scholar]
- 60.Srisuwananukorn A, Raslan R, Zhang X, et al. Clinical, laboratory, and genetic risk factors for thrombosis in sickle cell disease. Blood Adv. 2020;4(9):1978-1986. doi: 10.1182/bloodadvances.2019001384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jhun EH, Sadhu N, He Y, et al. S100B single nucleotide polymorphisms exhibit sex-specific associations with chronic pain in sickle cell disease in a largely African-American cohort. PLoS One. 2020;15(5):e0232721. doi: 10.1371/journal.pone.0232721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Alves AC, da Silva VAL, Dos Santos A, et al. Sickle cell anemia in the state of Maranhão: a haplotype study. Ann Hematol. 2020;99(6):1225-1230. doi: 10.1007/s00277-020-04048-9 [DOI] [PubMed] [Google Scholar]
- 63.Alenzi FQ. New mutations of locus control region in Saudi sickle patients. Saudi J Biol Sci. 2020;27(5):1265-1270. doi: 10.1016/j.sjbs.2020.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Domingos IF, Pereira-Martins DA, Borges-Medeiros RL, et al. Evaluation of oxidative stress-related genetic variants for predicting stroke in patients with sickle cell anemia. J Neurol Sci. 2020;414:116839. doi: 10.1016/j.jns.2020.116839 [DOI] [PubMed] [Google Scholar]
- 65.Fong C, Mendoza Y, Barreto G. Genetic variants in the G gamma-globin promoter modulate fetal hemoglobin expression in the Colombian population. Genet Mol Biol. 2020;43(2):e20190076. doi: 10.1590/1678-4685-gmb-2019-0076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chenou F, Albuquerque DM, Leonardo DP, et al. Endothelial nitric oxide synthase (eNOS) gene polymorphisms and markers of hemolysis, inflammation and endothelial dysfunction in Brazilian sickle cell anemia patients. Biochem Genet. 2020;58(4):580-594. doi: 10.1007/s10528-020-09959-w [DOI] [PubMed] [Google Scholar]
- 67.Sadhu N, Jhun EH, Posen A, et al. Phenylethanolamine N-methyltransferase gene polymorphisms associate with crisis pain in sickle cell disease patients. Pharmacogenomics. 2020;21(4):269-278. doi: 10.2217/pgs-2019-0096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Willen SM, McNeil JB, Rodeghier M, et al. Haptoglobin genotype predicts severe acute vaso-occlusive pain episodes in children with sickle cell anemia. Am J Hematol. 2020;95(4):E92-E95. doi: 10.1002/ajh.25728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang X, Shah BN, Zhang W, et al. S100B has pleiotropic effects on vaso-occlusive manifestations in sickle cell disease. Am J Hematol. 2020;95(3):E62-E65. doi: 10.1002/ajh.25691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boulassel MR, Al-Zubaidi A, Al-Zadjali S, et al. Elevated levels of circulating invariant natural killer cell subsets are skewed toward Th2-like phenotype in children with sickle cell disease. Clin Immunol. 2020;210:108308. doi: 10.1016/j.clim.2019.108308 [DOI] [PubMed] [Google Scholar]
- 71.Pereira-Martins DA, Coelho-Silva JL, Domingos IF, et al. Association between ANXA2*5681 polymorphism (rs7170178) and osteonecrosis in haemoglobin SS-genotyped patients. Br J Haematol. 2020;188(3):e8-e11. doi: 10.1111/bjh.16267 [DOI] [PubMed] [Google Scholar]
- 72.Powell-Roach KL, Yao Y, Jhun EH, et al. Vasopressin SNP pain factors and stress in sickle cell disease. PLoS One. 2019;14(11):e0224886. doi: 10.1371/journal.pone.0224886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Valente-Frossard TNS, Cruz NRC, Ferreira FO, et al. Polymorphisms in genes that affect the variation of lipid levels in a Brazilian pediatric population with sickle cell disease: rs662799 APOA5 and rs964184 ZPR1. Blood Cells Mol Dis. 2020;80:102376. doi: 10.1016/j.bcmd.2019.102376 [DOI] [PubMed] [Google Scholar]
- 74.Cintho Ozahata M, Page GP, Guo Y, et al. ; International Component of the NHLBI Recipient Epidemiology and Donor Evaluation Study (REDS-III) . Clinical and genetic predictors of priapism in sickle cell disease: results from the Recipient Epidemiology And Donor Evaluation Study III Brazil cohort study. J Sex Med. 2019;16(12):1988-1999. doi: 10.1016/j.jsxm.2019.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ndidi US, Adanho CSA, Santiago RP, et al. Effect of N(Epsilon)-(carboxymethyl)lysine on laboratory parameters and its association with β S haplotype in children with sickle cell anemia. Dis Markers. 2019;2019:1580485. doi: 10.1155/2019/1580485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Olatunya OS, Albuquerque DM, Akanbi GO, et al. Uridine diphosphate glucuronosyl transferase 1A (UGT1A1) promoter polymorphism in young patients with sickle cell anaemia: report of the first cohort study from Nigeria. BMC Med Genet. 2019;20(1):160. doi: 10.1186/s12881-019-0899-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nicolau M, Vargas S, Silva M, et al. Genetic modulators of fetal hemoglobin expression and ischemic stroke occurrence in African descendant children with sickle cell anemia. Ann Hematol. 2019;98(12):2673-2681. doi: 10.1007/s00277-019-03783-y [DOI] [PubMed] [Google Scholar]
- 78.de Almeida E, Frantz SR, Cesar P, et al. Frequency of interleukins IL1β/IL18 and inflammasome NLRP1/NLRP3 polymorphisms in sickle cell anemia patients and their association with severity score. Curr Mol Med. 2019;19(10):776-783. doi: 10.2174/1566524019666190826143749 [DOI] [PubMed] [Google Scholar]
- 79.Meher S, Patel S, Das K, et al. Association of plasma homocysteine level with vaso-occlusive crisis in sickle cell anemia patients of Odisha, India. Ann Hematol. 2019;98(10):2257-2265. doi: 10.1007/s00277-019-03776-x [DOI] [PubMed] [Google Scholar]
- 80.Cruz PRS, Ananina G, Gil-da-Silva-Lopes VL, et al. Genetic comparison of sickle cell anaemia cohorts from Brazil and the United States reveals high levels of divergence. Sci Rep. 2019;9(1):10896. doi: 10.1038/s41598-019-47313-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gueye Tall F, Martin C, Ndour EHM, et al. Combined and differential effects of alpha-thalassemia and HbF-quantitative trait loci in Senegalese hydroxyurea-free children with sickle cell anemia. Pediatr Blood Cancer. 2019;66(10):e27934. doi: 10.1002/pbc.27934 [DOI] [PubMed] [Google Scholar]
- 82.Mikobi TM, Lukusa PT, Muamba JM, Rhama T. Homozygous deletion alpha-thalassemia and hereditary persistence of fetal hemoglobin, two genetic factors predictive the reduction of morbidity and mortality during pregnancy in sickle cell patients. a report from the Democratic Republic of Congo. Mediterr J Hematol Infect Dis. 2019;11(1):e2019039. doi: 10.4084/mjhid.2019.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jit BP, Mohanty PK, Pradhan A, et al. Erythrocyte cAMP in determining frequency of acute pain episodes in sickle cell disease patients from Odisha State, India. Hemoglobin. 2019;43(2):88-94. doi: 10.1080/03630269.2019.1623248 [DOI] [PubMed] [Google Scholar]
- 84.Afifi RAA, Sedky YM, Abd-ELKareem H, Botros SKA. IL-Iβ+3954 C/T polymorphism and its clinical associations in Egyptian sickle cell disease patients. Int J Hematol Oncol Stem Cell Res. 2019;13(1):35-41. [PMC free article] [PubMed] [Google Scholar]
- 85.Meinderts SM, Gerritsma JJ, Sins JWR, et al. Identification of genetic biomarkers for alloimmunization in sickle cell disease. Br J Haematol. 2019;186(6):887-899. doi: 10.1111/bjh.15998 [DOI] [PubMed] [Google Scholar]
- 86.Bhagat S, Thakur AS. Influence of β-globin haplotypes on oxidative stress, antioxidant capacity and inflammation in sickle cell patients of Chhattisgarh. Indian J Clin Biochem. 2019;34(2):201-206. doi: 10.1007/s12291-017-0729-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Carvalho-Siqueira GQ, Ananina G, de Souza BB, et al. Whole-exome sequencing indicates FLG2 variant associated with leg ulcers in Brazilian sickle cell anemia patients. Exp Biol Med (Maywood). 2019;244(11):932-939. doi: 10.1177/1535370219849592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bakr S, Khorshied M, Talha N, et al. Implication of HMOX1 and CCR5 genotypes on clinical phenotype of Egyptian patients with sickle cell anemia. Ann Hematol. 2019;98(8):1805-1812. doi: 10.1007/s00277-019-03697-9 [DOI] [PubMed] [Google Scholar]
- 89.Tozatto-Maio K, Girot R, Ly ID, et al. A Toll-like receptor 2 genetic variant modulates occurrence of bacterial infections in patients with sickle cell disease. Br J Haematol. 2019;185(5):918-924. doi: 10.1111/bjh.15875 [DOI] [PubMed] [Google Scholar]
- 90.Zahr RS, Rampersaud E, Kang G, et al. Children with sickle cell anemia and APOL1 genetic variants develop albuminuria early in life. Haematologica. 2019;104(9):e385-e387. doi: 10.3324/haematol.2018.212779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Milton JN, Shaikho EM, Steinberg MH. Haemolysis in sickle cell anaemia: effects of polymorphisms in α-globin gene regulatory elements. Br J Haematol. 2019;186(2):363-364. doi: 10.1111/bjh.15852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jhun EH, Sadhu N, Hu X, et al. Beta2-adrenergic receptor polymorphisms and haplotypes associate with chronic pain in sickle cell disease. Front Pharmacol. 2019;10:84. doi: 10.3389/fphar.2019.00084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Upadhye D, Jain D, Nadkarni A, Ghosh K, Colah R. Red cell indices and hemoglobin profile of newborn babies with both the sickle gene and alpha thalassaemia in central India. Indian J Hematol Blood Transfus. 2019;35(1):109-113. doi: 10.1007/s12288-018-0994-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rezende PV, Belisário AR, Oliveira EL, et al. Co-inheritance of a-thalassemia dramatically decreases the risk of acute splenic sequestration in a large cohort of newborns with hemoglobin SC. Haematologica. 2019;104(7):e281-e283. doi: 10.3324/haematol.2018.209221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Okumura JV, Silva DGH, Torres LS, et al. Atypical β-S haplotypes: classification and genetic modulation in patients with sickle cell anemia. J Hum Genet. 2019;64(3):239-248. doi: 10.1038/s10038-018-0554-4 [DOI] [PubMed] [Google Scholar]
- 96.Sabrie M, Cannas G, Tazarourte K, et al. Drepa-Opia: a pilot study to determine the predictive factors of morphine use and consumption in hospitalized adult patients with sickle cell disease. Hemoglobin. 2018;42(4):217-224. doi: 10.1080/03630269.2018.1529602 [DOI] [PubMed] [Google Scholar]
- 97.Jit BP, Mohanty PK, Purohit P, et al. Association of fetal hemoglobin level with frequency of acute pain episodes in sickle cell disease (HbS-only phenotype) patients. Blood Cells Mol Dis. 2019;75:30-34. doi: 10.1016/j.bcmd.2018.12.003 [DOI] [PubMed] [Google Scholar]
- 98.Williams LM, Qi Z, Batai K, et al. A locus on chromosome 5 shows African ancestry-limited association with alloimmunization in sickle cell disease. Blood Adv. 2018;2(24):3637-3647. doi: 10.1182/bloodadvances.2018020594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hassan FM, Al-Zahrani FM. BCL11A rs1427407 genotypes in sickle cell anemia patients undergo to stroke problems in Sudan. Korean J Fam Med. 2019;40(1):53-57. doi: 10.4082/kjfm.17.0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Dash PM, Sahu PK, Patel S, Mashon RS, Kharat KR, Mukherjee MB. Effect of assorted globin haplotypes and α-thalassemia on the clinical heterogeneity of Hb S-β-thalassemia. Hemoglobin. 2018;42(4):236-242. doi: 10.1080/03630269.2018.1536666 [DOI] [PubMed] [Google Scholar]
- 101.ElAlfy MS, Ebeid FSE, Kamal TM, Eissa DS, Ismail EAR, Mohamed SH. Angiotensinogen M235T gene polymorphism is a genetic determinant of cerebrovascular and cardiopulmonary morbidity in adolescents with sickle cell disease. J Stroke Cerebrovasc Dis. 2019;28(2):441-449. doi: 10.1016/j.jstrokecerebrovasdis.2018.10.019 [DOI] [PubMed] [Google Scholar]
- 102.Antwi-Boasiako C, Dzudzor B, Kudzi W, et al. Association between eNOS gene polymorphism (T786C and VNTR) and sickle cell disease patients in Ghana. Diseases. 2018;6(4):90. doi: 10.3390/diseases6040090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Rooks H, Brewin J, Gardner K, et al. A gain of function variant in PIEZO1 (E756del) and sickle cell disease. Haematologica. 2019;104(3):e91-e93. doi: 10.3324/haematol.2018.202697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Al-Allawi N, Qadir SMA, Puehringer H, Chui DHK, Farrell JJ, Oberkanins C. The association of HBG2, BCL11A, and HMIP polymorphisms with fetal hemoglobin and clinical phenotype in Iraqi Kurds with sickle cell disease. Int J Lab Hematol. 2019;41(1):87-93. doi: 10.1111/ijlh.12927 [DOI] [PubMed] [Google Scholar]
- 105.Ballas SK, Connes P; Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia . Rheological properties of sickle erythrocytes in patients with sickle-cell anemia: The effect of hydroxyurea, fetal hemoglobin, and α-thalassemia. Eur J Haematol. 2018;101(6):798-803. doi: 10.1111/ejh.13173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Alsultan A, Al-Suliman AM, Aleem A, AlGahtani FH, Alfadhel M. Utilizing Whole-exome sequencing to characterize the phenotypic variability of sickle cell disease. Genet Test Mol Biomarkers. 2018;22(9):561-567. doi: 10.1089/gtmb.2018.0058 [DOI] [PubMed] [Google Scholar]
- 107.Saraf SL, Viner M, Rischall A, et al. HMOX1 and acute kidney injury in sickle cell anemia. Blood. 2018;132(15):1621-1625. doi: 10.1182/blood-2018-05-853929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Olatunya OS, Albuquerque DM, Adekile A, Costa FF. Influence of alpha thalassemia on clinical and laboratory parameters among nigerian children with sickle cell anemia. J Clin Lab Anal. 2019;33(2):e22656. doi: 10.1002/jcla.22656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Marshall K, Howell S, Badaloo A, Reid M, McFarlane-Anderson N, McKenzie C. Exploring putative genetic determinants of inter-individual phenotypic heterogeneity in sickle cell disease: A cross-sectional Jamaican cohort-based study. Blood Cells Mol Dis. 2018;73:1-8. doi: 10.1016/j.bcmd.2018.08.001 [DOI] [PubMed] [Google Scholar]
- 110.Al-Ali ZA, Fallatah RK, Aljaffer EA, et al. ANTXR1 intronic variants are associated with fetal hemoglobin in the Arab-Indian haplotype of sickle cell disease. Acta Haematol. 2018;140(1):55-59. doi: 10.1159/000491688 [DOI] [PubMed] [Google Scholar]
- 111.Ilboudo Y, Bartolucci P, Garrett ME, et al. A common functional PIEZO1 deletion allele associates with red blood cell density in sickle cell disease patients. Am J Hematol. 2018;93(11):E362-E365. doi: 10.1002/ajh.25245 [DOI] [PubMed] [Google Scholar]
- 112.Jhun EH, Sadhu N, Yao Y, et al. Glucocorticoid receptor single nucleotide polymorphisms are associated with acute crisis pain in sickle cell disease. Pharmacogenomics. 2018;19(13):1003-1011. doi: 10.2217/pgs-2018-0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sadhu N, Jhun EH, Yao Y, et al. Genetic variants of GCH1 associate with chronic and acute crisis pain in African Americans with sickle cell disease. Exp Hematol. 2018;66:42-49. doi: 10.1016/j.exphem.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang Y, Paikari A, Sumazin P, et al. Metformin induces FOXO3-dependent fetal hemoglobin production in human primary erythroid cells. Blood. 2018;132(3):321-333. doi: 10.1182/blood-2017-11-814335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Adeyemo TA, Ojewunmi OO, Oyetunji IA, et al. A survey of genetic fetal-haemoglobin modifiers in Nigerian patients with sickle cell anaemia. PLoS One. 2018;13(6):e0197927. doi: 10.1371/journal.pone.0197927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Jhun EH, Hu X, Sadhu N, et al. Transient receptor potential polymorphism and haplotype associate with crisis pain in sickle cell disease. Pharmacogenomics. 2018;19(5):401-411. doi: 10.2217/pgs-2017-0198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yahouédéhou SCMA, Carvalho MOS, Oliveira RM, et al. Sickle cell anemia patients in use of hydroxyurea: association between polymorphisms in genes encoding metabolizing drug enzymes and laboratory parameters. Dis Markers. 2018;2018:6105691. doi: 10.1155/2018/6105691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Raffield LM, Ulirsch JC, Naik RP, et al. ; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium, Hematology & Hemostasis, Diabetes, and Structural Variation TOPMed Working Groups . Common α-globin variants modify hematologic and other clinical phenotypes in sickle cell trait and disease. PLoS Genet. 2018;14(3):e1007293. doi: 10.1371/journal.pgen.1007293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Y, Belfer I, Nouraie M, et al. Association of genetic variation in COMT gene with pain related to sickle cell disease in patients from the walk-PHaSST study. J Pain Res. 2018;11:537-543. doi: 10.2147/JPR.S149958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bernaudin F, Arnaud C, Kamdem A, et al. Biological impact of α genes, β haplotypes, and G6PD activity in sickle cell anemia at baseline and with hydroxyurea. Blood Adv. 2018;2(6):626-637. doi: 10.1182/bloodadvances.2017014555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Renoux C, Joly P, Faes C, et al. Association between oxidative stress, genetic factors, and clinical severity in children with sickle cell anemia. J Pediatr. 2018;195:228-235. doi: 10.1016/j.jpeds.2017.12.021 [DOI] [PubMed] [Google Scholar]
- 122.Gardner K, Fulford T, Silver N, et al. g(HbF): a genetic model of fetal hemoglobin in sickle cell disease. Blood Adv. 2018;2(3):235-239. doi: 10.1182/bloodadvances.2017009811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bhanushali AA, Himani K, Patra PK, Das BR. Hb F levels in Indian sickle cell patients and association with the HBB locus variant rs10128556 (C>T), and the HBG XmnI (Arab-Indian) variant. Hemoglobin. 2017;41(4-6):317-320. doi: 10.1080/03630269.2017.1414059 [DOI] [PubMed] [Google Scholar]
- 124.Mwesigwa S, Moulds JM, Chen A, et al. Whole-exome sequencing of sickle cell disease patients with hyperhemolysis syndrome suggests a role for rare variation in disease predisposition. Transfusion. 2018;58(3):726-735. doi: 10.1111/trf.14431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wonkam A, Mnika K, Ngo Bitoungui VJ, et al. Clinical and genetic factors are associated with pain and hospitalisation rates in sickle cell anaemia in Cameroon. Br J Haematol. 2018;180(1):134-146. doi: 10.1111/bjh.15011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Abou-Elew HH, Youssry I, Hefny S, Hashem RH, Fouad N, Zayed RA. βS globin gene haplotype and the stroke risk among Egyptian children with sickle cell disease. Hematology. 2018;23(6):362-367. doi: 10.1080/10245332.2017.1403736 [DOI] [PubMed] [Google Scholar]
- 127.Howell S, Marshall K, Reid M, McFarlane-Anderson N, McKenzie C. A cross-sectional clinic-based study exploring whether variants within the glutathione S-transferase, haptoglobin and uridine 5′-diphospho-glucuronosyltransferase 1A1 genes are associated with interindividual phenotypic variation in sickle cell anaemia in Jamaica. Eur J Haematol. 2018;100(2):147-153. doi: 10.1111/ejh.12993 [DOI] [PubMed] [Google Scholar]
- 128.Afifi RA, Kamal D, Sayed RE, et al. CD209-336A/G promotor polymorphism and its clinical associations in sickle cell disease Egyptian Pediatric patients. Hematol Oncol Stem Cell Ther. 2018;11(2):75-81. doi: 10.1016/j.hemonc.2017.09.002 [DOI] [PubMed] [Google Scholar]
- 129.Aleluia MM, Fonseca TCC, Souza RQ, et al. Comparative study of sickle cell anemia and hemoglobin SC disease: clinical characterization, laboratory biomarkers and genetic profiles. BMC Hematol. 2017;17:15. doi: 10.1186/s12878-017-0087-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Meinderts SM, Sins JWR, Fijnvandraat K, et al. Nonclassical FCGR2C haplotype is associated with protection from red blood cell alloimmunization in sickle cell disease. Blood. 2017;130(19):2121-2130. doi: 10.1182/blood-2017-05-784876 [DOI] [PubMed] [Google Scholar]
- 131.Rodrigues C, Sell AM, Guelsin GAS, et al. HLA polymorphisms and risk of red blood cell alloimmunisation in polytransfused patients with sickle cell anaemia. Transfus Med. 2017;27(6):437-443. doi: 10.1111/tme.12459 [DOI] [PubMed] [Google Scholar]
- 132.Saraf SL, Akingbola TS, Shah BN, et al. Associations of α-thalassemia and BCL11A with stroke in Nigerian, United States, and United Kingdom sickle cell anemia cohorts. Blood Adv. 2017;1(11):693-698. doi: 10.1182/bloodadvances.2017005231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Oliveira VB, Dezan MR, Gomes FCA, et al. -318C/T polymorphism of the CTLA-4 gene is an independent risk factor for RBC alloimmunization among sickle cell disease patients. Int J Immunogenet. 2017;44(5):219-224. doi: 10.1111/iji.12334 [DOI] [PubMed] [Google Scholar]
- 134.Santiago RP, Vieira C, Adanho CSA, et al. Laboratory and genetic biomarkers associated with cerebral blood flow velocity in hemoglobin SC disease. Dis Markers. 2017;2017:6359871. doi: 10.1155/2017/6359871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kormann R, Jannot AS, Narjoz C, et al. Roles of APOL1 G1 and G2 variants in sickle cell disease patients: kidney is the main target. Br J Haematol. 2017;179(2):323-335. doi: 10.1111/bjh.14842 [DOI] [PubMed] [Google Scholar]
- 136.David S, Aguiar P, Antunes L, et al. Variants in the non-coding region of the TLR2 gene associated with infectious subphenotypes in pediatric sickle cell anemia. Immunogenetics. 2018;70(1):37-51. doi: 10.1007/s00251-017-1013-7 [DOI] [PubMed] [Google Scholar]
- 137.Shaikho EM, Farrell JJ, Alsultan A, Sebastiani P, Steinberg MH. Genetic determinants of HbF in Saudi Arabian and African Benin haplotype sickle cell anemia. Am J Hematol. 2017;92(9):E555-E557. doi: 10.1002/ajh.24822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Belisário AR, Sales RR, Toledo NE, Velloso-Rodrigues C, Silva CM, Viana MB. Interleukin-10 haplotypes are not associated with acute cerebral ischemia or high-risk transcranial Doppler in a newborn cohort of 395 children with sickle cell anemia. Rev Bras Hematol Hemoter. 2017;39(2):108-114. doi: 10.1016/j.bjhh.2016.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.de Azevedo LA, Bonazzoni J, Wagner SC, et al. Do alpha thalassemia, fetal hemoglobin, and the UGT1A1 polymorphism have an influence on serum bilirubin levels and cholelithiasis in patients with sickle cell disease? Mol Diagn Ther. 2017;21(4):437-442. doi: 10.1007/s40291-017-0283-y [DOI] [PubMed] [Google Scholar]
- 140.Ilboudo Y, Bartolucci P, Rivera A, et al. Genome-wide association study of erythrocyte density in sickle cell disease patients. Blood Cells Mol Dis. 2017;65:60-65. doi: 10.1016/j.bcmd.2017.05.005 [DOI] [PubMed] [Google Scholar]
- 141.Khorshied MM, Mohamed NS, Hamza RS, Ali RM, El-Ghamrawy MK. Protein Z and endothelin-1 genetic polymorphisms in pediatric Egyptian sickle cell disease patients. J Clin Lab Anal. 2018;32(2):e22264. doi: 10.1002/jcla.22264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Domingos IF, Pereira-Martins DA, Coelho-Silva JL, et al. Interleukin-6 G-174C polymorphism predicts higher risk of stroke in sickle cell anaemia. Br J Haematol. 2018;182(2):294-297. doi: 10.1111/bjh.14773 [DOI] [PubMed] [Google Scholar]
- 143.Pule GD, Bitoungui VJN, Chemegni BC, Kengne AP, Wonkam A. SAR1a promoter polymorphisms are not associated with fetal hemoglobin in patients with sickle cell disease from Cameroon. BMC Res Notes. 2017;10(1):183. doi: 10.1186/s13104-017-2502-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Geard A, Pule GD, Chetcha Chemegni B, et al. Clinical and genetic predictors of renal dysfunctions in sickle cell anaemia in Cameroon. Br J Haematol. 2017;178(4):629-639. doi: 10.1111/bjh.14724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Aleluia MM, da Guarda CC, Santiago RP, et al. Association of classical markers and establishment of the dyslipidemic sub-phenotype of sickle cell anemia. Lipids Health Dis. 2017;16(1):74. doi: 10.1186/s12944-017-0454-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mikobi TM, Tshilobo Lukusa P, Aloni MN, et al. Protective BCL11A and HBS1L-MYB polymorphisms in a cohort of 102 Congolese patients suffering from sickle cell anemia. J Clin Lab Anal. 2018;32(1):e22207. doi: 10.1002/jcla.22207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Mikobi TM, Lukusa PT, Aloni MN, et al. Association between sickle cell anemia and alpha thalassemia reveals a high prevalence of the α3.7 triplication in congolese patients than in worldwide series. J Clin Lab Anal. 2018;32(1):e22186. doi: 10.1002/jcla.22186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Pontes RM, Costa ES, Siqueira PFR, et al. Protector effect of α-thalassaemia on cholecystitis and cholecystectomy in sickle cell disease. Hematology. 2017;22(7):444-449. doi: 10.1080/10245332.2017.1289325 [DOI] [PubMed] [Google Scholar]
- 149.Aleluia MM, Santiago RP, da Guarda CC, et al. Genetic modulation of fetal hemoglobin in hydroxyurea-treated sickle cell anemia. Am J Hematol. 2017;92(5):E70-E72. doi: 10.1002/ajh.24680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.da Silva DGH, Belini Junior E, de Souza Torres L, et al. Impact of genetic polymorphisms in key enzymes of homocysteine metabolism on the pathophysiology of sickle cell anemia. Free Radic Biol Med. 2017;106:53-61. doi: 10.1016/j.freeradbiomed.2017.02.019 [DOI] [PubMed] [Google Scholar]
- 151.Zhang X, Shah BN, Zhang W, et al. A genetic variation associated with plasma erythropoietin and a non-coding transcript of PRKAR1A in sickle cell disease. Hum Mol Genet. 2016;25(20):4601-4609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Renoux C, Connes P, Nader E, et al. Alpha-thalassaemia promotes frequent vaso-occlusive crises in children with sickle cell anaemia through haemorheological changes. Pediatr Blood Cancer. 2017;64(8). doi: 10.1002/pbc.26455 [DOI] [PubMed] [Google Scholar]
- 153.Elalfy MS, El Sherif NH, Kamal TM, Aly NH. Klf10 gene, a secondary modifier and a pharmacogenomic biomarker of hydroxyurea treatment among patients with hemoglobinopathies. J Pediatr Hematol Oncol. 2017;39(3):e155-e162. doi: 10.1097/MPH.0000000000000762 [DOI] [PubMed] [Google Scholar]
- 154.Adekile AD, Akbulut N, Azab AF, Al-Sharida S, Thomas D. The sickle β-thalassemia phenotype. J Pediatr Hematol Oncol. 2017;39(5):327-331. doi: 10.1097/MPH.0000000000000747 [DOI] [PubMed] [Google Scholar]
- 155.Jacob SA, Novelli EM, Isenberg JS, et al. Thrombospondin-1 gene polymorphism is associated with estimated pulmonary artery pressure in patients with sickle cell anemia. Am J Hematol. 2017;92(3):E31-E34. doi: 10.1002/ajh.24635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Abuamer S, Shome DK, Jaradat A, et al. Frequencies and phenotypic consequences of association of α- and β-thalassemia alleles with sickle-cell disease in Bahrain. Int J Lab Hematol. 2017;39(1):76-83. doi: 10.1111/ijlh.12577 [DOI] [PubMed] [Google Scholar]
- 157.Joly P, Renoux C, Lacan P, et al. UGT1A1 (TA)n genotype is not the major risk factor of cholelithiasis in sickle cell disease children. Eur J Haematol. 2017;98(3):296-301. doi: 10.1111/ejh.12838 [DOI] [PubMed] [Google Scholar]
- 158.Abu-Duhier F, Mir R. GSTT1 (rs4025935) null genotype is associated with increased risk of sickle cell disease in the populations of Tabuk-Northwestern region of Saudi Arabia. Hematology. 2017;22(3):172-177. doi: 10.1080/10245332.2016.1201631 [DOI] [PubMed] [Google Scholar]
- 159.Hu X, Jhun EH, Yao Y, et al. IL1A rs1800587 associates with chronic noncrisis pain in sickle cell disease. Pharmacogenomics. 2016;17(18):1999-2006. doi: 10.2217/pgs-2016-0085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Sippert EA, Visentainer JE, Alves HV, et al. Red blood cell alloimmunization in patients with sickle cell disease: correlation with HLA and cytokine gene polymorphisms. Transfusion. 2017;57(2):379-389. doi: 10.1111/trf.13920 [DOI] [PubMed] [Google Scholar]
- 161.Armenis I, Kalotychou V, Tzanetea R, et al. Prognostic value of T786C and G894T eNOS polymorphisms in sickle cell disease. Nitric Oxide. 2017;62:17-23. doi: 10.1016/j.niox.2016.11.002 [DOI] [PubMed] [Google Scholar]
- 162.Kalai M, Dridi M, Chaouch L, et al. The role of rs1984112_G at CD36 gene in increasing reticulocyte level among sickle cell disease patients. Hematology. 2017;22(3):178-182. doi: 10.1080/10245332.2016.1253253 [DOI] [PubMed] [Google Scholar]
- 163.Friedrisch JR, Sheehan V, Flanagan JM, et al. The role of BCL11A and HMIP-2 polymorphisms on endogenous and hydroxyurea induced levels of fetal hemoglobin in sickle cell anemia patients from southern Brazil. Blood Cells Mol Dis. 2016;62:32-37. doi: 10.1016/j.bcmd.2016.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Pule GD, Ngo Bitoungui VJ, Chetcha Chemegni B, Kengne AP, Wonkam A. Studies of novel variants associated with Hb F in Sardinians and Tanzanians in sickle cell disease patients from Cameroon. Hemoglobin. 2016;40(6):377-380. doi: 10.1080/03630269.2016.1251453 [DOI] [PubMed] [Google Scholar]
- 165.Aguiar L, Matos A, Gil Â, et al. Sickle cell anemia - nitric oxide related genetic modifiers of hematological and biochemical parameters. Clin Hemorheol Microcirc. 2016;64(4):957-963. doi: 10.3233/CH-168008 [DOI] [PubMed] [Google Scholar]
- 166.Vathipadiekal V, Farrell JJ, Wang S, et al. A candidate transacting modulator of fetal hemoglobin gene expression in the Arab-Indian haplotype of sickle cell anemia. Am J Hematol. 2016;91(11):1118-1122. doi: 10.1002/ajh.24527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Rodrigues DO, Ribeiro LC, Sudário LC, et al. Genetic determinants and stroke in children with sickle cell disease. J Pediatr (Rio J). 2016;92(6):602-608. doi: 10.1016/j.jped.2016.01.010 [DOI] [PubMed] [Google Scholar]
- 168.Schaefer BA, Flanagan JM, Alvarez OA, et al. Genetic modifiers of white blood cell count, albuminuria and glomerular filtration rate in children with sickle cell anemia. PLoS One. 2016;11(10):e0164364. doi: 10.1371/journal.pone.0164364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Martella M, Quaglia N, Frigo AC, Basso G, Colombatti R, Sainati L. Association between a combination of single nucleotide polymorphisms and large vessel cerebral vasculopathy in African children with sickle cell disease. Blood Cells Mol Dis. 2016;61:1-3. doi: 10.1016/j.bcmd.2016.07.005 [DOI] [PubMed] [Google Scholar]
- 170.Saraf SL, Shah BN, Zhang X, et al. APOL1, α-thalassemia, and BCL11A variants as a genetic risk profile for progression of chronic kidney disease in sickle cell anemia. Haematologica. 2017;102(1):e1-e6. doi: 10.3324/haematol.2016.154153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Duarte JD, Desai AA, Sysol JR, et al. Genome-wide analysis identifies IL-18 and FUCA2 as novel genes associated with diastolic function in African Americans with sickle cell disease. PLoS One. 2016;11(9):e0163013. doi: 10.1371/journal.pone.0163013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mendonça Belmont TF, do Ó KP, Soares da Silva A, et al. Single nucleotide polymorphisms at +191 and +292 of galectin-3 gene (LGALS3) related to lower GAL-3 serum levels are associated with frequent respiratory tract infection and vaso-occlusive crisis in children with sickle cell anemia. PLoS One. 2016;11(9):e0162297. doi: 10.1371/journal.pone.0162297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rezende PdoV, Costa KdaS, Domingues Junior JC, et al. Clinical, hematological and genetic data of a cohort of children with hemoglobin SD. Rev Bras Hematol Hemoter. 2016;38(3):240-246. doi: 10.1016/j.bjhh.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Belisário AR, Sales RR, Toledo NE, et al. Reticulocyte count is the most important predictor of acute cerebral ischemia and high-risk transcranial Doppler in a newborn cohort of 395 children with sickle cell anemia. Ann Hematol. 2016;95(11):1869-1880. doi: 10.1007/s00277-016-2789-5 [DOI] [PubMed] [Google Scholar]
- 175.Verma H, Mishra H, Khodiar PK, Patra PK, Bhaskar LV. NOS3 27-bp and IL4 70-bp VNTR polymorphisms do not contribute to the risk of sickle cell crisis. Turk J Haematol. 2016;33(4):365-366. doi: 10.4274/tjh.2016.0166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dadheech S, Madhulatha D, Jainc S, Joseph J, Jyothy A, Munshi A. Association of BCL11A genetic variant (rs11886868) with severity in β-thalassaemia major & sickle cell anaemia. Indian J Med Res. 2016;143(4):449-454. doi: 10.4103/0971-5916.184285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Shaikho EM, Habara AH, Alsultan A, et al. Variants of ZBTB7A (LRF) and its β-globin gene cluster binding motifs in sickle cell anemia. Blood Cells Mol Dis. 2016;59:49-51. doi: 10.1016/j.bcmd.2016.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Moreira JA, Machado RP, Laurentino MR, et al. Influence of βS-globin haplotypes and hydroxyurea on arginase I levels in sickle cell disease. Dis Markers. 2016;2016:9172726. doi: 10.1155/2016/9172726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Upadhye D, Jain D, Trivedi Y, Nadkarni A, Ghosh K, Colah R. Influence of single nucleotide polymorphisms in the BCL11A and HBS1L-MYB gene on the HbF levels and clinical severity of sickle cell anaemia patients. Ann Hematol. 2016;95(7):1201-1203. doi: 10.1007/s00277-016-2675-1 [DOI] [PubMed] [Google Scholar]
- 180.Terzi YK, Bulakbaşı Balcı T, Boğa C, et al. Effect of hereditary hemochromatosis gene H63D and C282Y mutations on iron overload in sickle cell disease patients. Turk J Haematol. 2016;33(4):320-325. doi: 10.4274/tjh.2015.0254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chaouch L, Moumni I, Ouragini H, et al. rs11886868 and rs4671393 of BCL11A associated with HbF level variation and modulate clinical events among sickle cell anemia patients. Hematology. 2016;21(7):425-429. doi: 10.1080/10245332.2015.1107275 [DOI] [PubMed] [Google Scholar]
- 182.Cita KC, Ferdinand S, Connes P, et al. Association of adenylyl cyclase 6 rs3730070 polymorphism and hemolytic level in patients with sickle cell anemia. Blood Cells Mol Dis. 2016;58:21-25. doi: 10.1016/j.bcmd.2016.02.006 [DOI] [PubMed] [Google Scholar]
- 183.Olenscki Gilli SC, Pericole FV, Benites BD, et al. Cytokine polymorphisms in sickle cell disease and the relationship with cytokine expression. Exp Hematol. 2016;44(7):583-589. doi: 10.1016/j.exphem.2016.03.008 [DOI] [PubMed] [Google Scholar]
- 184.Liu L, Pertsemlidis A, Ding LH, et al. Original research: a case-control genome-wide association study identifies genetic modifiers of fetal hemoglobin in sickle cell disease. Exp Biol Med (Maywood). 2016;241(7):706-718. doi: 10.1177/1535370216642047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zachariah M, Al Zadjali S, Bashir W, et al. Impact of mannose-binding protein gene polymorphisms in Omani sickle cell disease patients. Mediterr J Hematol Infect Dis. 2016;8(1):e2016013. doi: 10.4084/mjhid.2016.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Okumura JV, Silva DG, Torres LS, et al. Inheritance of the Bantu/Benin haplotype causes less severe hemolytic and oxidative stress in sickle cell anemia patients treated with hydroxycarbamide. J Hum Genet. 2016;61(7):605-611. doi: 10.1038/jhg.2016.16 [DOI] [PubMed] [Google Scholar]
- 187.Torres LdeS, Okumura JV, da Silva DG, et al. Plasma levels of TGF-β1 in homeostasis of the inflammation in sickle cell disease. Cytokine. 2016;80:18-25. doi: 10.1016/j.cyto.2016.02.012 [DOI] [PubMed] [Google Scholar]
- 188.Farawela HM, El-Ghamrawy M, Farhan MS, Soliman R, Yousry SM, AbdelRahman HA. Association between Duffy antigen receptor expression and disease severity in sickle cell disease patients. Hematology. 2016;21(8):474-479. doi: 10.1080/10245332.2015.1111643 [DOI] [PubMed] [Google Scholar]
- 189.Yousry SM, Ellithy HN, Shahin GH. Endothelial nitric oxide synthase gene polymorphisms and the risk of vasculopathy in sickle cell disease. Hematology. 2016;21(6):359-367. doi: 10.1080/10245332.2016.1142710 [DOI] [PubMed] [Google Scholar]
- 190.Leonardo FC, Brugnerotto AF, Domingos IF, et al. Reduced rate of sickle-related complications in Brazilian patients carrying HbF-promoting alleles at the BCL11A and HMIP-2 loci. Br J Haematol. 2016;173(3):456-460. doi: 10.1111/bjh.13961 [DOI] [PubMed] [Google Scholar]
- 191.Moumni I, Ben Mustapha M, Ben Mansour I, et al. Fetal hemoglobin in tunisian sickle cell disease patient: relationship with polymorphic sequences cis to the β-globin gene. Indian J Hematol Blood Transfus. 2016;32(1):114-119. doi: 10.1007/s12288-015-0504-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Fong C, Menzel S, Lizarralde MA, Barreto G. Genetic variants associated with fetal hemoglobin levels show the diverse ethnic origin in Colombian patients with sickle cell anemia. Biomedica. 2015;35(3):437-443. doi: 10.7705/biomedica.v35i3.2573 [DOI] [PubMed] [Google Scholar]
- 193.Belisário AR, Rodrigues Sales R, Evelin Toledo N, Velloso-Rodrigues C, Maria Silva C, Borato Viana M. Glucose-6-phosphate dehydrogenase deficiency in Brazilian children with sickle cell anemia is not associated with clinical ischemic stroke or high-risk transcranial Doppler. Pediatr Blood Cancer. 2016;63(6):1046-1049. doi: 10.1002/pbc.25924 [DOI] [PubMed] [Google Scholar]
- 194.Upadhye DS, Jain DL, Trivedi YL, Nadkarni AH, Ghosh K, Colah RB. Neonatal screening and the clinical outcome in children with sickle cell disease in central India. PLoS One. 2016;11(1):e0147081. doi: 10.1371/journal.pone.0147081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mousa SM, El-Ghamrawy MK, Gouda H, Khorshied M, El-Salam Ahmed DA, Shiba H. Prevalence of hepatitis c among Egyptian children with sickle cell disease and the role of IL28b gene polymorphisms in spontaneous viral clearance. Mediterr J Hematol Infect Dis. 2016;8(1):e2016007. doi: 10.4084/mjhid.2016.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sommet J, Alberti C, Couque N, et al. Clinical and haematological risk factors for cerebral macrovasculopathy in a sickle cell disease newborn cohort: a prospective study. Br J Haematol. 2016;172(6):966-977. doi: 10.1111/bjh.13916 [DOI] [PubMed] [Google Scholar]
- 197.Alkindi SY, Pathare A, Al Zadjali S, et al. Serum total bilirubin, not cholelithiasis, is influenced by UGT1A1 polymorphism, alpha thalassemia and β(s) haplotype: first report on comparison between Arab-Indian and African β(s) Genes. Mediterr J Hematol Infect Dis. 2015;7(1):e2015060. doi: 10.4084/mjhid.2015.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Damy T, Bodez D, Habibi A, et al. Haematological determinants of cardiac involvement in adults with sickle cell disease. Eur Heart J. 2016;37(14):1158-1167. doi: 10.1093/eurheartj/ehv555 [DOI] [PubMed] [Google Scholar]
- 199.Shimauti EL, Silva DG, de Souza EM, de Almeida EA, Leal FP, Bonini-Domingos CR. Prevalence of β(S)-globin gene haplotypes, α-thalassemia (3.7 kb deletion) and redox status in patients with sickle cell anemia in the state of Paraná, Brazil. Genet Mol Biol. 2015;38(3):316-323. doi: 10.1590/S1415-475738320140231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tatari-Calderone Z, Gordish-Dressman H, Fasano R, et al. Protective effect of HLA-DQB1 alleles against alloimmunization in patients with sickle cell disease. Hum Immunol. 2016;77(1):35-40. doi: 10.1016/j.humimm.2015.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.da Fonseca SF, Amorim T, Purificação A, Gonçalves M, Boa-Sorte N. Hemoglobin A2 values in sickle cell disease patients quantified by high performance liquid chromatography and the influence of alpha thalassemia. Rev Bras Hematol Hemoter. 2015;37(5):296-301. doi: 10.1016/j.bjhh.2015.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Pule GD, Ngo Bitoungui VJ, Chetcha Chemegni B, Kengne AP, Antonarakis S, Wonkam A. Association between variants at BCL11A erythroid-specific enhancer and fetal hemoglobin levels among sickle cell disease patients in Cameroon: implications for future therapeutic interventions. OMICS. 2015;19(10):627-631. doi: 10.1089/omi.2015.0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Belisário AR, Sales RR, Toledo NE, Velloso-Rodrigues C, Silva CM, Viana MB. Association between ENPP1 K173Q and stroke in a newborn cohort of 395 Brazilian children with sickle cell anemia. Blood. 2015;126(10):1259-1260. doi: 10.1182/blood-2015-05-645176 [DOI] [PubMed] [Google Scholar]
- 204.Saraf SL, Zhang X, Shah B, et al. Genetic variants and cell-free hemoglobin processing in sickle cell nephropathy. Haematologica. 2015;100(10):1275-1284. doi: 10.3324/haematol.2015.124875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Chaouch L, Kalai M I, Darragi I, et al. Genetic link with cholelithiasis among pediatric SCA Tunisian patients: examples of UGT1A1, SLCO1A2 and SLCO1B1. Hematology. 2016;21(2):121-125. [DOI] [PubMed] [Google Scholar]
- 206.Ferdinand S, Connes P, Brudey L, et al. Impact of eNOS polymorphisms on red blood cell aggregation in sickle cell disease. Blood Cells Mol Dis. 2015;55(2):151-153. doi: 10.1016/j.bcmd.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 207.Lubega I, Ndugwa CM, Mworozi EA, Tumwine JK. Alpha thalassemia among sickle cell anaemia patients in Kampala, Uganda. Afr Health Sci. 2015;15(2):682-689. doi: 10.4314/ahs.v15i2.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Joly P, Garnier N, Kebaili K, et al. G6PD deficiency and absence of α-thalassemia increase the risk for cerebral vasculopathy in children with sickle cell anemia. Eur J Haematol. 2016;96(4):404-408. doi: 10.1111/ejh.12607 [DOI] [PubMed] [Google Scholar]
- 209.Zhang X, Zhang W, Saraf SL, et al. Genetic polymorphism of APOB is associated with diabetes mellitus in sickle cell disease. Hum Genet. 2015;134(8):895-904. doi: 10.1007/s00439-015-1572-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Mtatiro SN, Mgaya J, Singh T, et al. Genetic association of fetal-hemoglobin levels in individuals with sickle cell disease in Tanzania maps to conserved regulatory elements within the MYB core enhancer. BMC Med Genet. 2015;16:4. doi: 10.1186/s12881-015-0148-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ellithy HN, Yousri S, Shahin GH. Relation between glutathione S-transferase genes (GSTM1, GSTT1, and GSTP1) polymorphisms and clinical manifestations of sickle cell disease in Egyptian patients. Hematology. 2015;20(10):598-606. doi: 10.1179/1607845415Y.0000000013 [DOI] [PubMed] [Google Scholar]
- 212.Adekile A, Menzel S, Gupta R, et al. Response to hydroxyurea among Kuwaiti patients with sickle cell disease and elevated baseline HbF levels. Am J Hematol. 2015;90(7):E138-E139. doi: 10.1002/ajh.24027 [DOI] [PubMed] [Google Scholar]
- 213.Muszlak M, Pissard S, Badens C, Chamouine A, Maillard O, Thuret I. Genetic modifiers of sickle cell disease: a genotype-phenotype relationship study in a cohort of 82 children on Mayotte Island. Hemoglobin. 2015;39(3):156-161. doi: 10.3109/03630269.2015.1023897 [DOI] [PubMed] [Google Scholar]
- 214.Sebastiani P, Farrell JJ, Alsultan A, et al. BCL11A enhancer haplotypes and fetal hemoglobin in sickle cell anemia. Blood Cells Mol Dis. 2015;54(3):224-230. doi: 10.1016/j.bcmd.2015.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hanchard NA, Moulds JM, Belmont JW, Chen A. A Genome-wide screen for large-effect alloimmunization susceptibility loci among red blood cell transfusion recipients with sickle cell disease. Transfus Med Hemother. 2014;41(6):453-461. doi: 10.1159/000369079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Bhanushali AA, Patra PK, Pradhan S, Khanka SS, Singh S, Das BR. Genetics of fetal hemoglobin in tribal Indian patients with sickle cell anemia. Transl Res. 2015;165(6):696-703. doi: 10.1016/j.trsl.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 217.Vicari P, Adegoke SA, Mazzotti DR, Cançado RD, Nogutti MA, Figueiredo MS. Interleukin-1β and interleukin-6 gene polymorphisms are associated with manifestations of sickle cell anemia. Blood Cells Mol Dis. 2015;54(3):244-249. doi: 10.1016/j.bcmd.2014.12.004 [DOI] [PubMed] [Google Scholar]
- 218.Bitoungui VJ, Ngogang J, Wonkam A. Polymorphism at BCL11A compared to HBS1L-MYB loci explains less of the variance in HbF in patients with sickle cell disease in Cameroon. Blood Cells Mol Dis. 2015;54(3):268-269. doi: 10.1016/j.bcmd.2014.11.010 [DOI] [PubMed] [Google Scholar]
- 219.Bhanushali AA, Patra PK, Nair D, Verma H, Das BR. Genetic variant in the BCL11A (rs1427407), but not HBS1-MYB (rs6934903) loci associate with fetal hemoglobin levels in Indian sickle cell disease patients. Blood Cells Mol Dis. 2015;54(1):4-8. doi: 10.1016/j.bcmd.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 220.Mtatiro SN, Singh T, Rooks H, et al. Genome wide association study of fetal hemoglobin in sickle cell anemia in Tanzania. PLoS One. 2014;9(11):e111464. doi: 10.1371/journal.pone.0111464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Camilo-Araújo RF, Amancio OM, Figueiredo MS, Cabanãs-Pedro AC, Braga JA. Molecular analysis and association with clinical and laboratory manifestations in children with sickle cell anemia. Rev Bras Hematol Hemoter. 2014;36(5):334-339. doi: 10.1016/j.bjhh.2014.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Tantawy AA, Adly AA, Ismail EA, Aly SH. Endothelial nitric oxide synthase gene intron 4 VNTR polymorphism in sickle cell disease: relation to vasculopathy and disease severity. Pediatr Blood Cancer. 2015;62(3):389-394. doi: 10.1002/pbc.25234 [DOI] [PubMed] [Google Scholar]
- 223.Mtatiro SN, Makani J, Mmbando B, Thein SL, Menzel S, Cox SE. Genetic variants at HbF-modifier loci moderate anemia and leukocytosis in sickle cell disease in Tanzania. Am J Hematol. 2015;90(1):E1-E4. doi: 10.1002/ajh.23859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Belisário AR, Nogueira FL, Rodrigues RS, et al. Association of alpha-thalassemia, TNF-alpha (−308G>A) and VCAM-1 (c.1238G>C) gene polymorphisms with cerebrovascular disease in a newborn cohort of 411 children with sickle cell anemia. Blood Cells Mol Dis. 2015;54(1):44-50. doi: 10.1016/j.bcmd.2014.08.001 [DOI] [PubMed] [Google Scholar]
- 225.Redha NA, Mahdi N, Al-Habboubi HH, Almawi WY. Impact of VEGFA −583C > T polymorphism on serum VEGF levels and the susceptibility to acute chest syndrome in pediatric patients with sickle cell disease. Pediatr Blood Cancer. 2014;61(12):2310-2312. doi: 10.1002/pbc.25158 [DOI] [PubMed] [Google Scholar]
- 226.Abdel Rahman HA, Abou-Elew HH, El-Shorbagy RM, Fawzy R, Youssry I. Influence of iron regulating genes mutations on iron status in Egyptian patients with sickle cell disease. Ann Clin Lab Sci. 2014;44(3):304-309. [PubMed] [Google Scholar]
- 227.Jhun E, He Y, Yao Y, Molokie RE, Wilkie DJ, Wang ZJ. Dopamine D3 receptor Ser9Gly and catechol-o-methyltransferase Val158Met polymorphisms and acute pain in sickle cell disease. Anesth Analg. 2014;119(5):1201-1207. doi: 10.1213/ANE.0000000000000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Cardoso GL, Diniz IG, Silva AN, et al. DNA polymorphisms at BCL11A, HBS1L-MYB and Xmn1-HBG2 site loci associated with fetal hemoglobin levels in sickle cell anemia patients from Northern Brazil. Blood Cells Mol Dis. 2014;53(4):176-179. doi: 10.1016/j.bcmd.2014.07.006 [DOI] [PubMed] [Google Scholar]
- 229.Griffin PJ, Sebastiani P, Edward H, et al. The genetics of hemoglobin A2 regulation in sickle cell anemia. Am J Hematol. 2014;89(11):1019-1023. doi: 10.1002/ajh.23811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Silva IV, Reis AF, Palaré MJ, Ferrão A, Rodrigues T, Morais A. Sickle cell disease in children: chronic complications and search of predictive factors for adverse outcomes. Eur J Haematol. 2015;94(2):157-161. doi: 10.1111/ejh.12411 [DOI] [PubMed] [Google Scholar]
- 231.Resende Cardoso PS, Lopes Pessoa de Aguiar RA, Viana MB. Clinical complications in pregnant women with sickle cell disease: prospective study of factors predicting maternal death or near miss. Rev Bras Hematol Hemoter. 2014;36(4):256-263. doi: 10.1016/j.bjhh.2014.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Rumaney MB, Ngo Bitoungui VJ, Vorster AA, et al. The co-inheritance of alpha-thalassemia and sickle cell anemia is associated with better hematological indices and lower consultations rate in Cameroonian patients and could improve their survival. PLoS One. 2014;9(6):e100516. doi: 10.1371/journal.pone.0100516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Shiba HF, El-Ghamrawy MK, Shaheen IA, Ali RA, Mousa SM. Glutathione S-transferase gene polymorphisms (GSTM1, GSTT1, and GSTP1) in Egyptian pediatric patients with sickle cell disease. Pediatr Dev Pathol. 2014;17(4):265-270. doi: 10.2350/14-03-1452-OA.1 [DOI] [PubMed] [Google Scholar]
- 234.Laurentino MR, Maia PAF, Barbosa MC, Bandeira IC, Rocha LB, Gonçalves RP. Influence of βS-globin haplotypes and hydroxyurea on tumor necrosis factor-alpha levels in sickle cell anemia. Rev Bras Hematol Hemoter. 2014;36(2):121-125. doi: 10.5581/1516-8484.20140028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Wonkam A, Ngo Bitoungui VJ, Vorster AA, et al. Association of variants at BCL11A and HBS1L-MYB with hemoglobin F and hospitalization rates among sickle cell patients in Cameroon. PLoS One. 2014;9(3):e92506. doi: 10.1371/journal.pone.0092506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Cox SE, Makani J, Soka D, et al. Haptoglobin, alpha-thalassaemia and glucose-6-phosphate dehydrogenase polymorphisms and risk of abnormal transcranial Doppler among patients with sickle cell anaemia in Tanzania. Br J Haematol. 2014;165(5):699-706. doi: 10.1111/bjh.12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Wonkam A, Rumaney MB, Ngo Bitoungui VJ, Vorster AA, Ramesar R, Ngogang J. Coinheritance of sickle cell anemia and α-thalassemia delays disease onset and could improve survival in Cameroonian’s patients (Sub-Saharan Africa). Am J Hematol. 2014;89(6):664-665. doi: 10.1002/ajh.23711 [DOI] [PubMed] [Google Scholar]
- 238.Connes P, Lamarre Y, Waltz X, et al. Haemolysis and abnormal haemorheology in sickle cell anaemia. Br J Haematol. 2014;165(4):564-572. doi: 10.1111/bjh.12786 [DOI] [PubMed] [Google Scholar]
- 239.Milton JN, Gordeuk VR, Taylor JG VI, Gladwin MT, Steinberg MH, Sebastiani P. Prediction of fetal hemoglobin in sickle cell anemia using an ensemble of genetic risk prediction models. Circ Cardiovasc Genet. 2014;7(2):110-115. doi: 10.1161/CIRCGENETICS.113.000387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Barbosa LC, Miranda-Vilela AL, Hiragi CdeO, et al. Haptoglobin and myeloperoxidase (- G463A) gene polymorphisms in Brazilian sickle cell patients with and without secondary iron overload. Blood Cells Mol Dis. 2014;52(2-3):95-107. doi: 10.1016/j.bcmd.2013.10.001 [DOI] [PubMed] [Google Scholar]
- 241.Zhang X, Zhang W, Ma SF, et al. Hypoxic response contributes to altered gene expression and precapillary pulmonary hypertension in patients with sickle cell disease. Circulation. 2014;129(16):1650-1658. doi: 10.1161/CIRCULATIONAHA.113.005296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Domingos IF, Falcão DA, Hatzlhofer BL, et al. Influence of the βs haplotype and α-thalassemia on stroke development in a Brazilian population with sickle cell anaemia. Ann Hematol. 2014;93(7):1123-1129. doi: 10.1007/s00277-014-2016-1 [DOI] [PubMed] [Google Scholar]
- 243.Dadheech S, Jain S, Madhulatha D, et al. Association of Xmn1 −158 γG variant with severity and HbF levels in β-thalassemia major and sickle cell anaemia. Mol Biol Rep. 2014;41(5):3331-3337. doi: 10.1007/s11033-014-3195-5 [DOI] [PubMed] [Google Scholar]
- 244.Pandey SK, Pandey S, Ranjan R, et al. Phenotypic effect of α-globin gene numbers on Indian sickle β-thalassemia patients. J Clin Lab Anal. 2014;28(2):110-113. doi: 10.1002/jcla.21652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Bandeira IC, Rocha LB, Barbosa MC, et al. Chronic inflammatory state in sickle cell anemia patients is associated with HBB(*)S haplotype. Cytokine. 2014;65(2):217-221. doi: 10.1016/j.cyto.2013.10.009 [DOI] [PubMed] [Google Scholar]
- 246.Chaouch L, Kalai M, Jbara MB, et al. Association between rs267196 and rs267201 of BMP6 gene and osteonecrosis among sickle cell aneamia patients. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2015;159(1):145-149. doi: 10.5507/bp.2013.080 [DOI] [PubMed] [Google Scholar]
- 247.Patel DK, Purohit P, Dehury S, et al. Fetal hemoglobin and alpha thalassemia modulate the phenotypic expression of HbSD-Punjab. Int J Lab Hematol. 2014;36(4):444-450. doi: 10.1111/ijlh.12165 [DOI] [PubMed] [Google Scholar]
- 248.AlFadhli S, Al-Jafer H, Hadi M, Al-Mutairi M, Nizam R. The effect of UGT1A1 promoter polymorphism in the development of hyperbilirubinemia and cholelithiasis in hemoglobinopathy patients. PLoS One. 2013;8(10):e77681. doi: 10.1371/journal.pone.0077681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Coelho A, Dias A, Morais A, et al. Genetic variation in CD36, HBA, NOS3 and VCAM1 is associated with chronic haemolysis level in sickle cell anaemia: a longitudinal study. Eur J Haematol. 2014;92(3):237-243. doi: 10.1111/ejh.12226 [DOI] [PubMed] [Google Scholar]
- 250.Chaouch L, Talbi E, Moumni I, et al. Early complication in sickle cell anemia children due to A(TA)nTAA polymorphism at the promoter of UGT1A1 gene. Dis Markers. 2013;35(2):67-72. doi: 10.1155/2013/173474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Belfer I, Youngblood V, Darbari DS, et al. A GCH1 haplotype confers sex-specific susceptibility to pain crises and altered endothelial function in adults with sickle cell anemia. Am J Hematol. 2014;89(2):187-193. doi: 10.1002/ajh.23613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Silva DG, Belini Junior E, Carrocini GC, et al. Genetic and biochemical markers of hydroxyurea therapeutic response in sickle cell anemia. BMC Med Genet. 2013;14:108. doi: 10.1186/1471-2350-14-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Nishank SS, Singh MP, Yadav R, Gupta RB, Gadge VS, Gwal A. Endothelial nitric oxide synthase gene polymorphism is associated with sickle cell disease patients in India. J Hum Genet. 2013;58(12):775-779. doi: 10.1038/jhg.2013.99 [DOI] [PubMed] [Google Scholar]
- 254.Hamad Z, Aljedai A, Halwani R, AlSultan A. UGT1A1 promoter polymorphism associated with serum bilirubin level in Saudi patients with sickle cell disease. Ann Saudi Med. 2013;33(4):372-376. doi: 10.5144/0256-4947.2013.372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Bhatnagar P, Barron-Casella E, Bean CJ, et al. Genome-wide meta-analysis of systolic blood pressure in children with sickle cell disease. PLoS One. 2013;8(9):e74193. doi: 10.1371/journal.pone.0074193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Lamarre Y, Romana M, Lemonne N, et al. Alpha thalassemia protects sickle cell anemia patients from macro-albuminuria through its effects on red blood cell rheological properties. Clin Hemorheol Microcirc. 2014;57(1):63-72. doi: 10.3233/CH-131772 [DOI] [PubMed] [Google Scholar]
- 257.Nishank SS, Singh MP, Yadav R. Clinical impact of factor V Leiden, prothrombin G20210A, and MTHFR C677T mutations among sickle cell disease patients of Central India. Eur J Haematol. 2013;91(5):462-466. doi: 10.1111/ejh.12190 [DOI] [PubMed] [Google Scholar]
- 258.Bean CJ, Boulet SL, Yang G, et al. Acute chest syndrome is associated with single nucleotide polymorphism-defined beta globin cluster haplotype in children with sickle cell anaemia. Br J Haematol. 2013;163(2):268-276. doi: 10.1111/bjh.12507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rocha LB, da Silva Jn GB, Daher EdeF, Rocha HA, Elias DB, Gonçalves RP. Kidney dysfunction and beta S-haplotypes in patients with sickle cell disease. Rev Bras Hematol Hemoter. 2013;35(3):171-173. doi: 10.5581/1516-8484.20130052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kalai M, Chaouch L, Mansour IB, Hafsia R, Ghanem A, Abbes S. Frequency of three polymorphisms of the CCL5 gene (rs2107538, rs2280788 and rs2280789) and their implications for the phenotypic expression of sickle cell anemia in Tunisia. Pol J Pathol. 2013;64(2):84-89. doi: 10.5114/pjp.2013.36012 [DOI] [PubMed] [Google Scholar]
- 261.Barbosa CG, Aleluia AC, Pacheco AP, et al. Genetic modulation of HbF in Brazilians with HbSC disease and sickle cell anemia. Am J Hematol. 2013;88(10):923-924. doi: 10.1002/ajh.23538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Nishank SS. Endothelial nitric oxide synthase (eNOS) gene polymorphism is associated with age onset of menarche in sickle cell disease females of India. Mediterr J Hematol Infect Dis. 2013;5(1):e2013036. doi: 10.4084/mjhid.2013.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Tatari-Calderone Z, Tamouza R, Le Bouder GP, et al. The association of CD81 polymorphisms with alloimmunization in sickle cell disease. Clin Dev Immunol. 2013;2013:937846. doi: 10.1155/2013/937846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Gil GP, Ananina G, Oliveira MB, et al. Polymorphism in the HMOX1 gene is associated with high levels of fetal hemoglobin in Brazilian patients with sickle cell anemia. Hemoglobin. 2013;37(4):315-324. doi: 10.3109/03630269.2013.789438 [DOI] [PubMed] [Google Scholar]
- 265.Galarneau G, Coady S, Garrett ME, et al. Gene-centric association study of acute chest syndrome and painful crisis in sickle cell disease patients. Blood. 2013;122(3):434-442. doi: 10.1182/blood-2013-01-478776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Cox SE, Makani J, Newton CR, Prentice AM, Kirkham FJ. Hematological and genetic predictors of daytime hemoglobin saturation in Tanzanian children with and without sickle cell anemia. ISRN Hematol. 2013;2013:472909. doi: 10.1155/2013/472909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Sheehan VA, Luo Z, Flanagan JM, et al. ; BABY HUG Investigators . Genetic modifiers of sickle cell anemia in the BABY HUG cohort: influence on laboratory and clinical phenotypes. Am J Hematol. 2013;88(7):571-576. doi: 10.1002/ajh.23457 [DOI] [PubMed] [Google Scholar]
- 268.de Oliveira Filho RA, Silva GJ, de Farias Domingos I, et al. Association between the genetic polymorphisms of glutathione S-transferase (GSTM1 and GSTT1) and the clinical manifestations in sickle cell anemia. Blood Cells Mol Dis. 2013;51(2):76-79. doi: 10.1016/j.bcmd.2013.03.003 [DOI] [PubMed] [Google Scholar]
- 269.Alsultan A, Ngo D, Bae H, et al. Genetic studies of fetal hemoglobin in the Arab-Indian haplotype sickle cell-β(0) thalassemia. Am J Hematol. 2013;88(6):531-532. doi: 10.1002/ajh.23434 [DOI] [PubMed] [Google Scholar]
- 270.Ngo D, Bae H, Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: genetic studies of the Arab-Indian haplotype. Blood Cells Mol Dis. 2013;51(1):22-26. doi: 10.1016/j.bcmd.2012.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Flanagan JM, Sheehan V, Linder H, et al. Genetic mapping and exome sequencing identify 2 mutations associated with stroke protection in pediatric patients with sickle cell anemia. Blood. 2013;121(16):3237-3245. doi: 10.1182/blood-2012-10-464156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Green NS, Ender KL, Pashankar F, et al. Candidate sequence variants and fetal hemoglobin in children with sickle cell disease treated with hydroxyurea. PLoS One. 2013;8(2):e55709. doi: 10.1371/journal.pone.0055709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Milton JN, Rooks H, Drasar E, et al. ; Walk-PHAAST Investigators . Genetic determinants of haemolysis in sickle cell anaemia. Br J Haematol. 2013;161(2):270-278. doi: 10.1111/bjh.12245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Carvalho-dos Santos BS, Dias-Elias DB, da Silva-Rocha LB, Cavalcante-Barbosa M, Pinheiro-Gonçalves R. Impact of β(S)-globin haplotypes on oxidative stress in patients with sickle cell anemia in steady state. Arch Med Res. 2012;43(7):536-540. doi: 10.1016/j.arcmed.2012.08.014 [DOI] [PubMed] [Google Scholar]
- 275.Cox SE, L’Esperance V, Makani J, et al. Sickle cell anemia: iron availability and nocturnal oximetry. J Clin Sleep Med. 2012;8(5):541-545. doi: 10.5664/jcsm.2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Nekhai S, Xu M, Foster A, et al. Reduced sensitivity of the ferroportin Q248H mutant to physiological concentrations of hepcidin. Haematologica. 2013;98(3):455-463. doi: 10.3324/haematol.2012.066530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.da Silva Filho IL, Ribeiro GS, Moura PG, Vechi ML, Cavalcante AC, de Andrada-Serpa MJ. Sickle cell disease: acute clinical manifestations in early childhood and molecular characteristics in a group of children in Rio de Janeiro. Rev Bras Hematol Hemoter. 2012;34(3):196-201. doi: 10.5581/1516-8484.20120049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Haghpanah S, Nasirabadi S, Kianmehr M, Afrasiabi A, Karimi M. Polymorphisms associated with sickle cell disease in Southern Iran. Genetika. 2012;48(7):890-893. [PubMed] [Google Scholar]
- 279.Nouraie M, Lee JS, Zhang Y, et al. ; Walk-PHASST Investigators and Patients . The relationship between the severity of hemolysis, clinical manifestations and risk of death in 415 patients with sickle cell anemia in the US and Europe. Haematologica. 2013;98(3):464-472. doi: 10.3324/haematol.2012.068965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Bean CJ, Boulet SL, Ellingsen D, et al. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood. 2012;120(18):3822-3828. doi: 10.1182/blood-2011-06-361642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Thangarajh M, Yang G, Fuchs D, et al. Magnetic resonance angiography-defined intracranial vasculopathy is associated with silent cerebral infarcts and glucose-6-phosphate dehydrogenase mutation in children with sickle cell anaemia. Br J Haematol. 2012;159(3):352-359. doi: 10.1111/bjh.12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Bae HT, Baldwin CT, Sebastiani P, et al. Meta-analysis of 2040 sickle cell anemia patients: BCL11A and HBS1L-MYB are the major modifiers of HbF in African Americans. Blood. 2012;120(9):1961-1962. doi: 10.1182/blood-2012-06-432849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Al-Habboubi HH, Mahdi N, Abu-Hijleh TM, Abu-Hijleh FM, Sater MS, Almawi WY. The relation of vascular endothelial growth factor (VEGF) gene polymorphisms on VEGF levels and the risk of vasoocclusive crisis in sickle cell disease. Eur J Haematol. 2012;89(5):403-409. doi: 10.1111/ejh.12003 [DOI] [PubMed] [Google Scholar]
- 284.Hatzlhofer BL, Bezerra MA, Santos MN, et al. MTHFR polymorphic variant C677T is associated to vascular complications in sickle-cell disease. Genet Test Mol Biomarkers. 2012;16(9):1038-1043. doi: 10.1089/gtmb.2011.0361 [DOI] [PubMed] [Google Scholar]
- 285.Bartolucci P, Brugnara C, Teixeira-Pinto A, et al. Erythrocyte density in sickle cell syndromes is associated with specific clinical manifestations and hemolysis. Blood. 2012;120(15):3136-3141. doi: 10.1182/blood-2012-04-424184 [DOI] [PubMed] [Google Scholar]
- 286.Desai AA, Zhou T, Ahmad H, et al. A novel molecular signature for elevated tricuspid regurgitation velocity in sickle cell disease. Am J Respir Crit Care Med. 2012;186(4):359-368. doi: 10.1164/rccm.201201-0057OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Mahdi N, Abu-Hijleh TM, Abu-Hijleh FM, Sater MS, Al-Ola K, Almawi WY. Protein Z polymorphisms associated with vaso-occlusive crisis in young sickle cell disease patients. Ann Hematol. 2012;91(8):1215-1220. doi: 10.1007/s00277-012-1474-6 [DOI] [PubMed] [Google Scholar]
- 288.Milton JN, Sebastiani P, Solovieff N, et al. A genome-wide association study of total bilirubin and cholelithiasis risk in sickle cell anemia. PLoS One. 2012;7(4):e34741. doi: 10.1371/journal.pone.0034741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Alsultan A, Aleem A, Ghabbour H, et al. Sickle cell disease subphenotypes in patients from Southwestern Province of Saudi Arabia. J Pediatr Hematol Oncol. 2012;34(2):79-84. doi: 10.1097/MPH.0b013e3182422844 [DOI] [PubMed] [Google Scholar]
- 290.Viana-Baracioli LM, Tukamoto Junior NC, Ricci Junior O, Mattos LC, Ângulo IL, Bonini-Domingos CR. Comparison of oxidative stress and the frequency of polymorphisms in the HFE gene between hemoglobin S trait blood donors and sickle cell disease patients. Genet Mol Res. 2011;10(4):3446-3454. doi: 10.4238/2011.December.8.4 [DOI] [PubMed] [Google Scholar]
- 291.Akinsheye I, Solovieff N, Ngo D, et al. Fetal hemoglobin in sickle cell anemia: molecular characterization of the unusually high fetal hemoglobin phenotype in African Americans. Am J Hematol. 2012;87(2):217-219. doi: 10.1002/ajh.22221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Vasavda N, Woodley C, Allman M, et al. Effects of co-existing α-thalassaemia in sickle cell disease on hydroxycarbamide therapy and circulating nucleic acids. Br J Haematol. 2012;157(2):249-252. doi: 10.1111/j.1365-2141.2011.08937.x [DOI] [PubMed] [Google Scholar]
- 293.Pandey S, Pandey S, Mishra RM, Sharma M, Saxena R. Genotypic influence of α-deletions on the phenotype of Indian sickle cell anemia patients. Korean J Hematol. 2011;46(3):192-195. doi: 10.5045/kjh.2011.46.3.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ngo DA, Aygun B, Akinsheye I, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. 2012;156(2):259-264. doi: 10.1111/j.1365-2141.2011.08916.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Day TG, Drasar ER, Fulford T, Sharpe CC, Thein SL. Association between hemolysis and albuminuria in adults with sickle cell anemia. Haematologica. 2012;97(2):201-205. doi: 10.3324/haematol.2011.050336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Joly P, Pondarré C, Bardel C, Francina A, Martin C. The alpha-globin genotype does not influence sickle cell disease severity in a retrospective cross-validation study of the pediatric severity score. Eur J Haematol. 2012;88(1):61-67. doi: 10.1111/j.1600-0609.2011.01705.x [DOI] [PubMed] [Google Scholar]
- 297.Ashley-Koch AE, Okocha EC, Garrett ME, et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. Br J Haematol. 2011;155(3):386-394. doi: 10.1111/j.1365-2141.2011.08832.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Darbari DS, Onyekwere O, Nouraie M, et al. Markers of severe vaso-occlusive painful episode frequency in children and adolescents with sickle cell anemia. J Pediatr. 2012;160(2):286-290. doi: 10.1016/j.jpeds.2011.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Ware RE, Despotovic JM, Mortier NA, et al. Pharmacokinetics, pharmacodynamics, and pharmacogenetics of hydroxyurea treatment for children with sickle cell anemia. Blood. 2011;118(18):4985-4991. doi: 10.1182/blood-2011-07-364190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Cajado C, Cerqueira BA, Couto FD, et al. TNF-alpha and IL-8: serum levels and gene polymorphisms (−308G>A and −251A>T) are associated with classical biomarkers and medical history in children with sickle cell anemia. Cytokine. 2011;56(2):312-317. doi: 10.1016/j.cyto.2011.07.002 [DOI] [PubMed] [Google Scholar]
- 301.Filho IL, Leite AC, Moura PG, et al. Genetic polymorphisms and cerebrovascular disease in children with sickle cell anemia from Rio de Janeiro, Brazil. Arq Neuropsiquiatr. 2011;69(3):431-435. doi: 10.1590/S0004-282X2011000400004 [DOI] [PubMed] [Google Scholar]
- 302.Kumar R, Panigrahi I, Dalal A, Agarwal S. Sickle cell anemia–molecular diagnosis and prenatal counseling: SGPGI experience. Indian J Pediatr. 2012;79(1):68-74. doi: 10.1007/s12098-011-0510-1 [DOI] [PubMed] [Google Scholar]
- 303.Alsultan A, Solovieff N, Aleem A, et al. Fetal hemoglobin in sickle cell anemia: Saudi patients from the Southwestern province have similar HBB haplotypes but higher HbF levels than African Americans. Am J Hematol. 2011;86(7):612-614. doi: 10.1002/ajh.22032 [DOI] [PubMed] [Google Scholar]
- 304.Flanagan JM, Frohlich DM, Howard TA, et al. Genetic predictors for stroke in children with sickle cell anemia. Blood. 2011;117(24):6681-6684. doi: 10.1182/blood-2011-01-332205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Silva DG, Belini Junior E, Torres LdeS, et al. Relationship between oxidative stress, glutathione S-transferase polymorphisms and hydroxyurea treatment in sickle cell anemia. Blood Cells Mol Dis. 2011;47(1):23-28. doi: 10.1016/j.bcmd.2011.03.004 [DOI] [PubMed] [Google Scholar]
- 306.Rusanova I, Cossio G, Moreno B, et al. β-globin gene cluster haplotypes in sickle cell patients from Panamá. Am J Hum Biol. 2011;23(3):377-380. doi: 10.1002/ajhb.21148 [DOI] [PubMed] [Google Scholar]
- 307.Bhatnagar P, Purvis S, Barron-Casella E, et al. Genome-wide association study identifies genetic variants influencing F-cell levels in sickle-cell patients. J Hum Genet. 2011;56(4):316-323. doi: 10.1038/jhg.2011.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Dworkis DA, Klings ES, Solovieff N, et al. Severe sickle cell anemia is associated with increased plasma levels of TNF-R1 and VCAM-1. Am J Hematol. 2011;86(2):220-223. doi: 10.1002/ajh.21928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Mukherjee MB, Nadkarni AH, Gorakshakar AC, Ghosh K, Mohanty D, Colah RB. Clinical, hematologic and molecular variability of sickle cell-β thalassemia in western India. Indian J Hum Genet. 2010;16(3):154-158. doi: 10.4103/0971-6866.73410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Vicari P, Silva GS, Nogutti MA, et al. Absence of association between TNF-α polymorphism and cerebral large-vessel abnormalities in adults with sickle cell anemia. Acta Haematol. 2011;125(3):141-144. doi: 10.1159/000321935 [DOI] [PubMed] [Google Scholar]
- 311.Belisário AR, Rodrigues CV, Martins ML, Silva CM, Viana MB. Coinheritance of α-thalassemia decreases the risk of cerebrovascular disease in a cohort of children with sickle cell anemia. Hemoglobin. 2010;34(6):516-529. doi: 10.3109/03630269.2010.526003 [DOI] [PubMed] [Google Scholar]
- 312.Makani J, Menzel S, Nkya S, et al. Genetics of fetal hemoglobin in Tanzanian and British patients with sickle cell anemia. Blood. 2011;117(4):1390-1392. doi: 10.1182/blood-2010-08-302703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42(12):1049-1051. doi: 10.1038/ng.707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Belisário AR, Martins ML, Brito AM, Rodrigues CV, Silva CM, Viana MB. β-globin gene cluster haplotypes in a cohort of 221 children with sickle cell anemia or Sβ0-thalassemia and their association with clinical and hematological features. Acta Haematol. 2010;124(3):162-170. doi: 10.1159/000320271 [DOI] [PubMed] [Google Scholar]
- 315.Adekile AD, Haider MZ. Haptoglobin gene polymorphisms in sickle cell disease patients with different βS-globin gene haplotypes. Med Princ Pract. 2010;19(6):447-450. doi: 10.1159/000320302 [DOI] [PubMed] [Google Scholar]
- 316.Rusanova I, Escames G, Cossio G, et al. Oxidative stress status, clinical outcome, and β-globin gene cluster haplotypes in pediatric patients with sickle cell disease. Eur J Haematol. 2010;85(6):529-537. doi: 10.1111/j.1600-0609.2010.01528.x [DOI] [PubMed] [Google Scholar]
- 317.Italia KY, Jijina FF, Jain D, et al. The effect of UGT1A1 promoter polymorphism on bilirubin response to hydroxyurea therapy in hemoglobinopathies. Clin Biochem. 2010;43(16-17):1329-1332. doi: 10.1016/j.clinbiochem.2010.08.006 [DOI] [PubMed] [Google Scholar]
- 318.Mecabo G, Hayashida DY, Azevedo-Shimmoto MM, et al. Duffy-negative is associated with hemolytic phenotype of sickle cell anemia. Clin Immunol. 2010;136(3):458-459. doi: 10.1016/j.clim.2010.06.006 [DOI] [PubMed] [Google Scholar]
- 319.Barbosa CG, Goncalves-Santos NJ, Souza-Ribeiro SB, et al. Promoter region sequence differences in the A and G gamma globin genes of Brazilian sickle cell anemia patients. Braz J Med Biol Res. 2010;43(8):705-711. doi: 10.1590/S0100-879X2010007500062 [DOI] [PubMed] [Google Scholar]
- 320.Nebor D, Broquere C, Brudey K, et al. Alpha-thalassemia is associated with a decreased occurrence and a delayed age-at-onset of albuminuria in sickle cell anemia patients. Blood Cells Mol Dis. 2010;45(2):154-158. doi: 10.1016/j.bcmd.2010.06.003 [DOI] [PubMed] [Google Scholar]
- 321.Nouraie M, Reading NS, Campbell A, et al. Association of G6PD with lower haemoglobin concentration but not increased haemolysis in patients with sickle cell anaemia. Br J Haematol. 2010;150(2):218-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Joannes MO, Loko G, Deloumeaux J, Chout R, Marianne-Pepin T. Association of the +874 T/A interferon gamma polymorphism with infections in sickle cell disease. Int J Immunogenet. 2010;37(4):219-223. doi: 10.1111/j.1744-313X.2010.00912.x [DOI] [PubMed] [Google Scholar]
- 323.Farra C, Zahed L, Nietert PJ, et al. Vascular at-risk genotypes and disease severity in Lebanese sickle cell disease patients. Am J Hematol. 2010;85(5):395-396. doi: 10.1002/ajh.21688 [DOI] [PubMed] [Google Scholar]
- 324.Nebor D, Durpes MC, Mougenel D, et al. Association between Duffy antigen receptor for chemokines expression and levels of inflammation markers in sickle cell anemia patients. Clin Immunol. 2010;136(1):116-122. doi: 10.1016/j.clim.2010.02.023 [DOI] [PubMed] [Google Scholar]
- 325.Al-Saqladi AW, Brabin BJ, Bin-Gadeem HA, Kanhai WA, Phylipsen M, Harteveld CL. Beta-globin gene cluster haplotypes in Yemeni children with sickle cell disease. Acta Haematol. 2010;123(3):182-185. doi: 10.1159/000294965 [DOI] [PubMed] [Google Scholar]
- 326.Haymann JP, Stankovic K, Levy P, et al. Glomerular hyperfiltration in adult sickle cell anemia: a frequent hemolysis associated feature. Clin J Am Soc Nephrol. 2010;5(5):756-761. doi: 10.2215/CJN.08511109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Mendonça TF, Oliveira MC, Vasconcelos LR, et al. Association of variant alleles of MBL2 gene with vasoocclusive crisis in children with sickle cell anemia. Blood Cells Mol Dis. 2010;44(4):224-228. doi: 10.1016/j.bcmd.2010.02.004 [DOI] [PubMed] [Google Scholar]
- 328.Al-Saqladi AW, Harper G, Delpisheh A, Fijnvandraat K, Bin-Gadeem HA, Brabin BJ. Frequency of the MTHFR C677T polymorphism in Yemeni children with sickle cell disease. Hemoglobin. 2010;34(1):67-77. doi: 10.3109/09687630903554111 [DOI] [PubMed] [Google Scholar]
- 329.Sebastiani P, Solovieff N, Hartley SW, et al. Genetic modifiers of the severity of sickle cell anemia identified through a genome-wide association study. Am J Hematol. 2010;85(1):29-35. doi: 10.1002/ajh.2157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Cordero EA, Veit TD, da Silva MA, Jacques SM, Silla LM, Chies JA. HLA-G polymorphism influences the susceptibility to HCV infection in sickle cell disease patients. Tissue Antigens. 2009;74(4):308-313. doi: 10.1111/j.1399-0039.2009.01331.x [DOI] [PubMed] [Google Scholar]
- 331.Al-Subaie AM, Fawaz NA, Mahdi N, et al. Human platelet alloantigens (HPA) 1, HPA2, HPA3, HPA4, and HPA5 polymorphisms in sickle cell anemia patients with vaso-occlusive crisis. Eur J Haematol. 2009;83(6):579-585. doi: 10.1111/j.1600-0609.2009.01339.x [DOI] [PubMed] [Google Scholar]
- 332.Hoppe C, Klitz W, Vichinsky E, Styles L. HLA type and risk of alloimmunization in sickle cell disease. Am J Hematol. 2009;84(7):462-464. doi: 10.1002/ajh.21442 [DOI] [PubMed] [Google Scholar]
- 333.Oliveira MC, Mendonça TF, Vasconcelos LR, et al. Association of the MBL2 gene EXON1 polymorphism and vasoocclusive crisis in patients with sickle cell anemia. Acta Haematol. 2009;121(4):212-215. doi: 10.1159/000220335 [DOI] [PubMed] [Google Scholar]
- 334.Mahdi N, Al-Subaie AM, Al-Ola K, et al. HLA DRB1*130101-DQB1*060101 haplotype is associated with acute chest syndrome in sickle cell anemia patients. Tissue Antigens. 2009;73(3):245-249. doi: 10.1111/j.1399-0039.2008.01189.x [DOI] [PubMed] [Google Scholar]
- 335.Tatari-Calderone Z, Minniti CP, Kratovil T, et al. rs660 polymorphism in Ro52 (SSA1; TRIM21) is a marker for age-dependent tolerance induction and efficiency of alloimmunization in sickle cell disease. Mol Immunol. 2009;47(1):64-70. doi: 10.1016/j.molimm.2008.12.027 [DOI] [PubMed] [Google Scholar]
- 336.Creary LE, Ulug P, Menzel S, et al. Genetic variation on chromosome 6 influences F cell levels in healthy individuals of African descent and HbF levels in sickle cell patients. PLoS One. 2009;4(1):e4218. doi: 10.1371/journal.pone.0004218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Ulug P, Vasavda N, Awogbade M, Cunningham J, Menzel S, Thein SL. Association of sickle avascular necrosis with bone morphogenic protein 6. Ann Hematol. 2009;88(8):803-805. doi: 10.1007/s00277-008-0659-5 [DOI] [PubMed] [Google Scholar]
- 338.Bernaudin F, Verlhac S, Chevret S, et al. G6PD deficiency, absence of alpha-thalassemia, and hemolytic rate at baseline are significant independent risk factors for abnormally high cerebral velocities in patients with sickle cell anemia. Blood. 2008;112(10):4314-4317. doi: 10.1182/blood-2008-03-143891 [DOI] [PubMed] [Google Scholar]
- 339.Carpenter SL, Lieff S, Howard TA, Eggleston B, Ware RE. UGT1A1 promoter polymorphisms and the development of hyperbilirubinemia and gallbladder disease in children with sickle cell anemia. Am J Hematol. 2008;83(10):800-803. doi: 10.1002/ajh.21264 [DOI] [PubMed] [Google Scholar]
- 340.Sedgewick AE, Timofeev N, Sebastiani P, et al. BCL11A is a major HbF quantitative trait locus in three different populations with beta-hemoglobinopathies. Blood Cells Mol Dis. 2008;41(3):255-258. doi: 10.1016/j.bcmd.2008.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Ugochukwu CC, Okpala I, Pantelidis P, Inusa B, Ibegbulam O, Onyekwere O. l-selectin gene polymorphisms and complications of sickle cell disease. Int J Lab Hematol. 2008;30(4):312-316. doi: 10.1111/j.1751-553X.2007.00961.x [DOI] [PubMed] [Google Scholar]
- 342.Ganesh A, Al-Zuhaibi S, Pathare A, et al. Orbital infarction in sickle cell disease. Am J Ophthalmol. 2008;146(4):595-601. doi: 10.1016/j.ajo.2008.05.041 [DOI] [PubMed] [Google Scholar]
- 343.Taylor JG VI, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT, Steinberg MH. Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with less frequent vasoocclusive pain. PLoS One. 2008;3(5):e2095. doi: 10.1371/journal.pone.0002095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Martins R, Morais A, Dias A, et al. Early modification of sickle cell disease clinical course by UDP-glucuronosyltransferase 1A1 gene promoter polymorphism. J Hum Genet. 2008;53(6):524-528. doi: 10.1007/s10038-008-0281-3 [DOI] [PubMed] [Google Scholar]
- 345.Eyler CE, Jackson T, Elliott LE, et al. beta(2)-Adrenergic receptor and adenylate cyclase gene polymorphisms affect sickle red cell adhesion. Br J Haematol. 2008;141(1):105-108. doi: 10.1111/j.1365-2141.2008.07008.x [DOI] [PubMed] [Google Scholar]
- 346.Kumkhaek C, Taylor JG VI, Zhu J, Hoppe C, Kato GJ, Rodgers GP. Fetal haemoglobin response to hydroxycarbamide treatment and sar1a promoter polymorphisms in sickle cell anaemia. Br J Haematol. 2008;141(2):254-259. doi: 10.1111/j.1365-2141.2008.07045.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Al-Ola K, Mahdi N, Al-Subaie AM, Ali ME, Al-Absi IK, Almawi WY. Evidence for HLA class II susceptible and protective haplotypes for osteomyelitis in pediatric patients with sickle cell anemia. Tissue Antigens. 2008;71(5):453-457. doi: 10.1111/j.1399-0039.2008.01012.x [DOI] [PubMed] [Google Scholar]
- 348.Mahdi N, Al-Ola K, Al-Subaie AM, et al. HLA class II haplotypes distinctly associated with vaso-occlusion in children with sickle cell disease. Clin Vaccine Immunol. 2008;15(4):729-731. doi: 10.1128/CVI.00425-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Afenyi-Annan A, Kail M, Combs MR, Orringer EP, Ashley-Koch A, Telen MJ. Lack of Duffy antigen expression is associated with organ damage in patients with sickle cell disease. Transfusion. 2008;48(5):917-924. doi: 10.1111/j.1537-2995.2007.01622.x [DOI] [PubMed] [Google Scholar]
- 350.Sebastiani P, Zhao Z, Abad-Grau MM, et al. A hierarchical and modular approach to the discovery of robust associations in genome-wide association studies from pooled DNA samples. BMC Genet. 2008;9:6. doi: 10.1186/1471-2156-9-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Ashley-Koch AE, Elliott L, Kail ME, et al. Identification of genetic polymorphisms associated with risk for pulmonary hypertension in sickle cell disease. Blood. 2008;111(12):5721-5726. doi: 10.1182/blood-2007-02-074849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Mourad H, Fadel W, El Batch M, Rowisha M. Heamostatic and genetic predisposing factors for stroke in children with sickle cell anemia. Egypt J Immunol. 2008;15(1):25-37. [PubMed] [Google Scholar]
- 353.Bagdasaryan R, Glasser L, Quillen K, Chaves F, Xu D. Effect of hydroxyurea on immature reticulocyte fraction in sickle cell anemia. Lab Hematol. 2007;13(3):93-97. doi: 10.1532/LH96.07008 [DOI] [PubMed] [Google Scholar]
- 354.Tamouza R, Busson M, Fortier C, et al. HLA-E*0101 allele in homozygous state favors severe bacterial infections in sickle cell anemia. Hum Immunol. 2007;68(10):849-853. doi: 10.1016/j.humimm.2007.08.260 [DOI] [PubMed] [Google Scholar]
- 355.Sebastiani P, Wang L, Nolan VG, et al. Fetal hemoglobin in sickle cell anemia: bayesian modeling of genetic associations. Am J Hematol. 2008;83(3):189-195. doi: 10.1002/ajh.21048 [DOI] [PubMed] [Google Scholar]
- 356.Taylor JG VI, Ackah D, Cobb C, et al. Mutations and polymorphisms in hemoglobin genes and the risk of pulmonary hypertension and death in sickle cell disease. Am J Hematol. 2008;83(1):6-14. doi: 10.1002/ajh.21035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Hoppe C, Klitz W, D’Harlingue K, et al. ; Stroke Prevention Trial in Sickle Cell Anemia (STOP) Investigators . Confirmation of an association between the TNF(−308) promoter polymorphism and stroke risk in children with sickle cell anemia. Stroke. 2007;38(8):2241-2246. doi: 10.1161/STROKEAHA.107.483115 [DOI] [PubMed] [Google Scholar]
- 358.Vasavda N, Menzel S, Kondaveeti S, et al. The linear effects of alpha-thalassaemia, the UGT1A1 and HMOX1 polymorphisms on cholelithiasis in sickle cell disease. Br J Haematol. 2007;138(2):263-270. doi: 10.1111/j.1365-2141.2007.06643.x [DOI] [PubMed] [Google Scholar]
- 359.Adekile A, Al-Kandari M, Haider M, Rajaa M, D’Souza M, Sukumaran J. Hemoglobin F concentration as a function of age in Kuwaiti sickle cell disease patients. Med Princ Pract. 2007;16(4):286-290. doi: 10.1159/000102151 [DOI] [PubMed] [Google Scholar]
- 360.Elliott L, Ashley-Koch AE, De Castro L, et al. Genetic polymorphisms associated with priapism in sickle cell disease. Br J Haematol. 2007;137(3):262-267. doi: 10.1111/j.1365-2141.2007.06560.x [DOI] [PubMed] [Google Scholar]
- 361.Duckworth L, Hsu L, Feng H, et al. Physician-diagnosed asthma and acute chest syndrome: associations with NOS polymorphisms. Pediatr Pulmonol. 2007;42(4):332-338. doi: 10.1002/ppul.20582 [DOI] [PubMed] [Google Scholar]
- 362.Nolan VG, Ma Q, Cohen HT, et al. Estimated glomerular filtration rate in sickle cell anemia is associated with polymorphisms of bone morphogenetic protein receptor 1B. Am J Hematol. 2007;82(3):179-184. doi: 10.1002/ajh.20800 [DOI] [PubMed] [Google Scholar]
- 363.Chaar V, Tarer V, Etienne-Julan M, Diara JP, Elion J, Romana M. ET-1 and ecNOS gene polymorphisms andsusceptibility to acute chest syndrome and painful vaso-occlusive crises in children with sickle cell anemia. Haematologica. 2006;91(9):1277-1278. [PubMed] [Google Scholar]
- 364.Moreira Neto F, Lourenço DM, Noguti MA, et al. The clinical impact of MTHFR polymorphism on the vascular complications of sickle cell disease. Braz J Med Biol Res. 2006;39(10):1291-1295. doi: 10.1590/S0100-879X2006001000004 [DOI] [PubMed] [Google Scholar]
- 365.Adewoye AH, Nolan VG, Ma Q, et al. Association of polymorphisms of IGF1R and genes in the transforming growth factor- beta /bone morphogenetic protein pathway with bacteremia in sickle cell anemia. Clin Infect Dis. 2006;43(5):593-598. doi: 10.1086/506356 [DOI] [PubMed] [Google Scholar]
- 366.Nolan VG, Adewoye A, Baldwin C, et al. Sickle cell leg ulcers: associations with haemolysis and SNPs in Klotho, TEK and genes of the TGF-beta/BMP pathway. Br J Haematol. 2006;133(5):570-578. doi: 10.1111/j.1365-2141.2006.06074.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Lima CS, Rocha EM, Silva NM, Sonatti MF, Costa FF, Saad ST. Risk factors for conjunctival and retinal vessel alterations in sickle cell disease. Acta Ophthalmol Scand. 2006;84(2):234-241. doi: 10.1111/j.1600-0420.2005.00604.x [DOI] [PubMed] [Google Scholar]
- 368.Chaar V, Kéclard L, Etienne-Julan M, et al. UGT1A1 polymorphism outweighs the modest effect of deletional (−3.7 kb) alpha-thalassemia on cholelithogenesis in sickle cell anemia. Am J Hematol. 2006;81(5):377-379. doi: 10.1002/ajh.20574 [DOI] [PubMed] [Google Scholar]
- 369.Tarer V, Etienne-Julan M, Diara JP, et al. Sickle cell anemia in Guadeloupean children: pattern and prevalence of acute clinical events. Eur J Haematol. 2006;76(3):193-199. doi: 10.1111/j.1600-0609.2005.00590.x [DOI] [PubMed] [Google Scholar]
- 370.Vargas AE, da Silva MA, Silla L, Chies JA. Polymorphisms of chemokine receptors and eNOS in Brazilian patients with sickle cell disease. Tissue Antigens. 2005;66(6):683-690. doi: 10.1111/j.1399-0039.2005.00506.x [DOI] [PubMed] [Google Scholar]
- 371.Adekile A, Kutlar F, McKie K, et al. The influence of uridine diphosphate glucuronosyl transferase 1A promoter polymorphisms, beta-globin gene haplotype, co-inherited alpha-thalassemia trait and Hb F on steady-state serum bilirubin levels in sickle cell anemia. Eur J Haematol. 2005;75(2):150-155. doi: 10.1111/j.1600-0609.2005.00477.x [DOI] [PubMed] [Google Scholar]
- 372.Costa RN, Conran N, Albuquerque DM, Soares PH, Saad ST, Costa FF. Association of the G-463A myeloperoxidase polymorphism with infection in sickle cell anemia. Haematologica. 2005;90(7):977-979. [PubMed] [Google Scholar]
- 373.Adekile A, Haider MZ, Marouf R, Adekile AD. HLA-DRB1 alleles in Hb SS patients with avascular necrosis of the femoral head. Am J Hematol. 2005;79(1):8-10. doi: 10.1002/ajh.20311 [DOI] [PubMed] [Google Scholar]
- 374.Baldwin C, Nolan VG, Wyszynski DF, et al. Association of klotho, bone morphogenic protein 6, and annexin A2 polymorphisms with sickle cell osteonecrosis. Blood. 2005;106(1):372-375. doi: 10.1182/blood-2005-02-0548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Sebastiani P, Ramoni MF, Nolan V, Baldwin CT, Steinberg MH. Genetic dissection and prognostic modeling of overt stroke in sickle cell anemia. Nat Genet. 2005;37(4):435-440. doi: 10.1038/ng1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Chaar V, Kéclard L, Diara JP, et al. Association of UGT1A1 polymorphism with prevalence and age at onset of cholelithiasis in sickle cell anemia. Haematologica. 2005;90(2):188-199. [PubMed] [Google Scholar]
- 377.Nolan VG, Baldwin C, Ma Q, et al. Association of single nucleotide polymorphisms in klotho with priapism in sickle cell anaemia. Br J Haematol. 2005;128(2):266-272. doi: 10.1111/j.1365-2141.2004.05295.x [DOI] [PubMed] [Google Scholar]
- 378.Adorno EV, Zanette A, Lyra I, et al. The beta-globin gene cluster haplotypes in sickle cell anemia patients from Northeast Brazil: a clinical and molecular view. Hemoglobin. 2004;28(3):267-271. doi: 10.1081/HEM-120040310 [DOI] [PubMed] [Google Scholar]
- 379.Bakanay SM, Dainer E, Clair B, et al. Mortality in sickle cell patients on hydroxyurea therapy. Blood. 2005;105(2):545-547. doi: 10.1182/blood-2004-01-0322 [DOI] [PubMed] [Google Scholar]
- 380.Haverfield EV, McKenzie CA, Forrester T, et al. UGT1A1 variation and gallstone formation in sickle cell disease. Blood. 2005;105(3):968-972. doi: 10.1182/blood-2004-02-0521 [DOI] [PubMed] [Google Scholar]
- 381.Castro V, Alberto FL, Costa RN, et al. Polymorphism of the human platelet antigen-5 system is a risk factor for occlusive vascular complications in patients with sickle cell anemia. Vox Sang. 2004;87(2):118-123. doi: 10.1111/j.1423-0410.2004.00536.x [DOI] [PubMed] [Google Scholar]
- 382.Romana M, Diara JP, Doumbo L, et al. Angiotensinogen gene associated polymorphisms and risk of stroke in sickle cell anemia: Additional data supporting an association. Am J Hematol. 2004;76(3):310-311. doi: 10.1002/ajh.20078 [DOI] [PubMed] [Google Scholar]
- 383.Ofori-Acquah SF, Lalloz MR, Serjeant G, Layton DM. Dominant influence of gamma-globin promoter polymorphisms on fetal haemoglobin expression in sickle cell disease. Cell Mol Biol (Noisy-le-grand). 2004;50(1):35-42. [PubMed] [Google Scholar]
- 384.Wyszynski DF, Baldwin CT, Cleves MA, et al. Polymorphisms near a chromosome 6q QTL area are associated with modulation of fetal hemoglobin levels in sickle cell anemia. Cell Mol Biol (Noisy-le-grand). 2004;50(1):23-33. [PubMed] [Google Scholar]
- 385.Sharan K, Surrey S, Ballas S, et al. Association of T-786C eNOS gene polymorphism with increased susceptibility to acute chest syndrome in females with sickle cell disease. Br J Haematol. 2004;124(2):240-243. doi: 10.1046/j.1365-2141.2003.04762.x [DOI] [PubMed] [Google Scholar]
- 386.Jeng MR, Adams-Graves P, Howard TA, Whorton MR, Li CS, Ware RE. Identification of hemochromatosis gene polymorphisms in chronically transfused patients with sickle cell disease. Am J Hematol. 2003;74(4):243-248. doi: 10.1002/ajh.10426 [DOI] [PubMed] [Google Scholar]
- 387.Hoppe C, Klitz W, Cheng S, et al. ; CSSCD Investigators . Gene interactions and stroke risk in children with sickle cell anemia. Blood. 2004;103(6):2391-2396. doi: 10.1182/blood-2003-09-3015 [DOI] [PubMed] [Google Scholar]
- 388.Gonçalves MS, Bomfim GC, Maciel E, et al. BetaS-haplotypes in sickle cell anemia patients from Salvador, Bahia, Northeastern Brazil. Braz J Med Biol Res. 2003;36(10):1283-1288. doi: 10.1590/S0100-879X2003001000001 [DOI] [PubMed] [Google Scholar]
- 389.Hsu LL, Miller ST, Wright E, et al. ; Stroke Prevention Trial (STOP) and the Cooperative Study of Sickle Cell Disease (CSSCD) . Alpha thalassemia is associated with decreased risk of abnormal transcranial Doppler ultrasonography in children with sickle cell anemia. J Pediatr Hematol Oncol. 2003;25(8):622-628. doi: 10.1097/00043426-200308000-00007 [DOI] [PubMed] [Google Scholar]
- 390.Fertrin KY, Melo MB, Assis AM, Saad ST, Costa FF. UDP-glucuronosyltransferase 1 gene promoter polymorphism is associated with increased serum bilirubin levels and cholecystectomy in patients with sickle cell anemia. Clin Genet. 2003;64(2):160-162. doi: 10.1034/j.1399-0004.2003.00113.x [DOI] [PubMed] [Google Scholar]
- 391.Inati A, Taher A, Bou Alawi W, et al. Beta-globin gene cluster haplotypes and HbF levels are not the only modulators of sickle cell disease in Lebanon. Eur J Haematol. 2003;70(2):79-83. doi: 10.1034/j.1600-0609.2003.00016.x [DOI] [PubMed] [Google Scholar]
- 392.Taylor JG VI, Tang DC, Savage SA, et al. Variants in the VCAM1 gene and risk for symptomatic stroke in sickle cell disease. Blood. 2002;100(13):4303-4309. doi: 10.1182/blood-2001-12-0306 [DOI] [PubMed] [Google Scholar]
- 393.Wali YA, Al-Lamki Z, Hussein SS, et al. Splenic function in Omani children with sickle cell disease: correlation with severity index, hemoglobin phenotype, iron status, and alpha-thalassemia trait. Pediatr Hematol Oncol. 2002;19(7):491-500. doi: 10.1080/08880010290097314 [DOI] [PubMed] [Google Scholar]
- 394.Powars DR, Hiti A, Ramicone E, Johnson C, Chan L. Outcome in hemoglobin SC disease: a four-decade observational study of clinical, hematologic, and genetic factors. Am J Hematol. 2002;70(3):206-215. doi: 10.1002/ajh.10140 [DOI] [PubMed] [Google Scholar]
- 395.el-Hazmi MA, Warsy AS. Pattern for alpha-thalassaemia in Yemeni sickle-cell-disease patients. East Mediterr Health J. 1999;5(6):1159-1164. doi: 10.26719/1999.5.6.1159 [DOI] [PubMed] [Google Scholar]
- 396.Passon RG, Howard TA, Zimmerman SA, Schultz WH, Ware RE. Influence of bilirubin uridine diphosphate-glucuronosyltransferase 1A promoter polymorphisms on serum bilirubin levels and cholelithiasis in children with sickle cell anemia. J Pediatr Hematol Oncol. 2001;23(7):448-451. doi: 10.1097/00043426-200110000-00011 [DOI] [PubMed] [Google Scholar]
- 397.Tamouza R, Neonato MG, Busson M, et al. Infectious complications in sickle cell disease are influenced by HLA class II alleles. Hum Immunol. 2002;63(3):194-199. doi: 10.1016/S0198-8859(01)00378-0 [DOI] [PubMed] [Google Scholar]
- 398.Romana M, Muralitharan S, Ramasawmy R, Nagel RL, Krishnamoorthy R. Thrombosis-associated gene variants in sickle cell anemia. Thromb Haemost. 2002;87(2):356-358. doi: 10.1055/s-0037-1613007 [DOI] [PubMed] [Google Scholar]
- 399.Taylor JG, Tang D, Foster CB, Serjeant GR, Rodgers GP, Chanock SJ. Patterns of low-affinity immunoglobulin receptor polymorphisms in stroke and homozygous sickle cell disease. Am J Hematol. 2002;69(2):109-114. doi: 10.1002/ajh.10048 [DOI] [PubMed] [Google Scholar]
- 400.Tang DC, Prauner R, Liu W, et al. Polymorphisms within the angiotensinogen gene (GT-repeat) and the risk of stroke in pediatric patients with sickle cell disease: a case-control study. Am J Hematol. 2001;68(3):164-169. doi: 10.1002/ajh.1173 [DOI] [PubMed] [Google Scholar]
- 401.Luporini SM, Bendit I, Manhani R, Bracco OL, Manzella L, Giannella-Neto D. Growth hormone and insulin-like growth factor I axis and growth of children with different sickle cell anemia haplotypes. J Pediatr Hematol Oncol. 2001;23(6):357-363. doi: 10.1097/00043426-200108000-00007 [DOI] [PubMed] [Google Scholar]
- 402.Kutlar A, Kutlar F, Turker I, Tural C. The methylene tetrahydrofolate reductase (C677T) mutation as a potential risk factor for avascular necrosis in sickle cell disease. Hemoglobin. 2001;25(2):213-217. doi: 10.1081/HEM-100104029 [DOI] [PubMed] [Google Scholar]
- 403.Olatunji PO, Davies SC. The predictive value of white cell count in assessing clinical severity of sickle cell anaemia in Afro-Caribbeans patients. Afr J Med Med Sci. 2000;29(1):27-30. [PubMed] [Google Scholar]
- 404.Adekile AD, Gupta R, Yacoub F, Sinan T, Al-Bloushi M, Haider MZ. Avascular necrosis of the hip in children with sickle cell disease and high Hb F: magnetic resonance imaging findings and influence of alpha-thalassemia trait. Acta Haematol. 2001;105(1):27-31. doi: 10.1159/000046529 [DOI] [PubMed] [Google Scholar]
- 405.Adekile AD, Kutlar F, Haider MZ, Kutlar A. Frequency of the 677 C→T mutation of the methylenetetrahydrofolate reductase gene among Kuwaiti sickle cell disease patients. Am J Hematol. 2001;66(4):263-266. doi: 10.1002/ajh.1055 [DOI] [PubMed] [Google Scholar]
- 406.Ballas SK, Marcolina MJ. Determinants of red cell survival and erythropoietic activity in patients with sickle cell anemia in the steady state. Hemoglobin. 2000;24(4):277-286. doi: 10.3109/03630260008993134 [DOI] [PubMed] [Google Scholar]
- 407.Raghupathy R, Haider MZ, Azizieh F, Abdelsalam R, D’Souza TM, Adekile AD. Th1 and Th2 cytokine profiles in sickle cell disease. Acta Haematol. 2000;103(4):197-202. doi: 10.1159/000041049 [DOI] [PubMed] [Google Scholar]
- 408.Neonato MG, Guilloud-Bataille M, Beauvais P, et al. ; French Study Group on Sickle Cell Disease . Acute clinical events in 299 homozygous sickle cell patients living in France. Eur J Haematol. 2000;65(3):155-164. doi: 10.1034/j.1600-0609.2000.90210.x [DOI] [PubMed] [Google Scholar]
- 409.Styles LA, Hoppe C, Klitz W, Vichinsky E, Lubin B, Trachtenberg E. Evidence for HLA-related susceptibility for stroke in children with sickle cell disease. Blood. 2000;95(11):3562-3567. doi: 10.1182/blood.V95.11.3562 [DOI] [PubMed] [Google Scholar]
- 410.Mouélé R, Pambou O, Feingold J, Galactéros F. Alpha-thalassemia in Bantu population from Congo-Brazzaville: its interaction with sickle cell anemia. Hum Hered. 2000;50(2):118-125. doi: 10.1159/000022899 [DOI] [PubMed] [Google Scholar]
- 411.el-Hazmi MA, Warsy AS. Alpha thalassaemia in Yemeni children with sickle cell disease. J Trop Pediatr. 1999;45(6):370-374. doi: 10.1093/tropej/45.6.370 [DOI] [PubMed] [Google Scholar]
- 412.Driscoll MC, Prauner R. The methylenetetrahydrofolate reductase gene C677T mutant and ischemic stroke in sickle cell disease. Thromb Haemost. 1999;82(6):1780-1781. [PubMed] [Google Scholar]
- 413.Cumming AM, Olujohungbe A, Keeney S, Singh H, Hay CR, Serjeant GR. The methylenetetrahydrofolate reductase gene C677T polymorphism in patients with homozygous sickle cell disease and stroke. Br J Haematol. 1999;107(3):569-571. doi: 10.1046/j.1365-2141.1999.01728.x [DOI] [PubMed] [Google Scholar]
- 414.Balasa VV, Gruppo RA, Gartside PS, Kalinyak KA. Correlation of the C677T MTHFR genotype with homocysteine levels in children with sickle cell disease. J Pediatr Hematol Oncol. 1999;21(5):397-400. doi: 10.1097/00043426-199909000-00011 [DOI] [PubMed] [Google Scholar]
- 415.Neonato MG, Lu CY, Guilloud-Bataille M, et al. Genetic polymorphism of the mannose-binding protein gene in children with sickle cell disease: identification of three new variant alleles and relationship to infections. Eur J Hum Genet. 1999;7(6):679-686. doi: 10.1038/sj.ejhg.5200360 [DOI] [PubMed] [Google Scholar]
- 416.Mouélé R, Boukila V, Fourcade V, Feingold J, Galactéros F. Sickle-cell disease in Brazzaville, Congo: genetical, hematological, biochemical and clinical aspects. Acta Haematol. 1999;101(4):178-184. doi: 10.1159/000040950 [DOI] [PubMed] [Google Scholar]
- 417.Guasch A, Zayas CF, Eckman JR, Muralidharan K, Zhang W, Elsas LJ. Evidence that microdeletions in the alpha globin gene protect against the development of sickle cell glomerulopathy in humans. J Am Soc Nephrol. 1999;10(5):1014-1019. doi: 10.1681/ASN.V1051014 [DOI] [PubMed] [Google Scholar]
- 418.Kinney TR, Sleeper LA, Wang WC, et al. ; The Cooperative Study of Sickle Cell Disease . Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. Pediatrics. 1999;103(3):640-645. doi: 10.1542/peds.103.3.640 [DOI] [PubMed] [Google Scholar]
- 419.Ofori-Acquah SF, Lalloz MR, Layton DM. Localisation of cis regulatory elements at the beta-globin locus: analysis of hybrid haplotype chromosomes. Biochem Biophys Res Commun. 1999;254(1):181-187. doi: 10.1006/bbrc.1998.9901 [DOI] [PubMed] [Google Scholar]
- 420.Haider MZ, Ashebu S, Aduh P, Adekile AD. Influence of alpha-thalassemia on cholelithiasis in SS patients with elevated Hb F. Acta Haematol. 1998;100(3):147-150. doi: 10.1159/000040890 [DOI] [PubMed] [Google Scholar]
- 421.Zimmerman SA, Ware RE. Inherited DNA mutations contributing to thrombotic complications in patients with sickle cell disease. Am J Hematol. 1998;59(4):267-272. doi: [DOI] [PubMed] [Google Scholar]
- 422.Steinberg MH, Lu ZH, Nagel RL, et al. Hematological effects of atypical and Cameroon beta-globin gene haplotypes in adult sickle cell anemia. Am J Hematol. 1998;59(2):121-126. doi: [DOI] [PubMed] [Google Scholar]
- 423.Lee K, Préhu C, Mérault G, et al. Genetic and hematological studies in a group of 114 adult patients with SC sickle cell disease. Am J Hematol. 1998;59(1):15-21. doi: [DOI] [PubMed] [Google Scholar]
- 424.Mukherjee MB, Surve R, Tamankar A, et al. The influence of alpha-thalassaemia on the haematological & clinical expression of sickle cell disease in western India. Indian J Med Res. 1998;107:178-181. [PubMed] [Google Scholar]
- 425.Chang YP, Maier-Redelsperger M, Smith KD, et al. The relative importance of the X-linked FCP locus and beta-globin haplotypes in determining haemoglobin F levels: a study of SS patients homozygous for beta S haplotypes. Br J Haematol. 1997;96(4):806-814. doi: 10.1046/j.1365-2141.1997.d01-2094.x [DOI] [PubMed] [Google Scholar]
- 426.Thomas PW, Higgs DR, Serjeant GR. Benign clinical course in homozygous sickle cell disease: a search for predictors. J Clin Epidemiol. 1997;50(2):121-126. doi: 10.1016/S0895-4356(96)00320-4 [DOI] [PubMed] [Google Scholar]
- 427.Steinberg MH, Lu ZH, Barton FB, Terrin ML, Charache S, Dover GJ; Multicenter Study of Hydroxyurea . Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Blood. 1997;89(3):1078-1088. doi: 10.1182/blood.V89.3.1078 [DOI] [PubMed] [Google Scholar]
- 428.Mukherjee MB, Colah RB, Ghosh K, Mohanty D, Krishnamoorthy R. Milder clinical course of sickle cell disease in patients with alpha thalassemia in the Indian subcontinent. Blood. 1997;89(2):732. doi: 10.1182/blood.V89.2.732a [DOI] [PubMed] [Google Scholar]
- 429.Walker TM, Beardsall K, Thomas PW, Serjeant GR. Renal length in sickle cell disease: observations from a cohort study. Clin Nephrol. 1996;46(6):384-388. [PubMed] [Google Scholar]
- 430.Figueirido MS, Steinberg MH. 5′ hypersensitive site-2 and fetal hemoglobin in Brazilians. Hemoglobin. 1996;20(4):435-438. doi: 10.3109/03630269609005847 [DOI] [PubMed] [Google Scholar]
- 431.Figueiredo MS, Kerbauy J, Gonçalves MS, et al. Effect of alpha-thalassemia and beta-globin gene cluster haplotypes on the hematological and clinical features of sickle-cell anemia in Brazil. Am J Hematol. 1996;53(2):72-76. doi: [DOI] [PubMed] [Google Scholar]
- 432.Adekile AD, Tuli M, Haider MZ, Al-Zaabi K, Mohannadi S, Owunwanne A. Influence of alpha-thalassemia trait on spleen function in sickle cell anemia patients with high HbF. Am J Hematol. 1996;53(1):1-5. doi: [DOI] [PubMed] [Google Scholar]
- 433.Steinberg MH, Nagel RL, Lawrence C, et al. Beta-globin gene haplotype in Hb SC disease. Am J Hematol. 1996;52(3):189-191. doi: [DOI] [PubMed] [Google Scholar]
- 434.Braden DS, Covitz W, Milner PF. Cardiovascular function during rest and exercise in patients with sickle-cell anemia and coexisting alpha thalassemia-2. Am J Hematol. 1996;52(2):96-102. doi: [DOI] [PubMed] [Google Scholar]
- 435.Kéclard L, Ollendorf V, Berchel C, Loret H, Mérault G. beta S haplotypes, alpha-globin gene status, and hematological data of sickle cell disease patients in Guadeloupe (F.W.I.). Hemoglobin. 1996;20(1):63-74. doi: 10.3109/03630269609027911 [DOI] [PubMed] [Google Scholar]
- 436.Serjeant G, Serjeant B, Stephens A, et al. Determinants of haemoglobin level in steady-state homozygous sickle cell disease. Br J Haematol. 1996;92(1):143-149. doi: 10.1046/j.1365-2141.1996.284816.x [DOI] [PubMed] [Google Scholar]
- 437.Gill FM, Sleeper LA, Weiner SJ, et al. ; Cooperative Study of Sickle Cell Disease . Clinical events in the first decade in a cohort of infants with sickle cell disease. Blood. 1995;86(2):776-783. doi: 10.1182/blood.V86.2.776.bloodjournal862776 [DOI] [PubMed] [Google Scholar]
- 438.al-Hazzaa S, Bird AC, Kulozik A, et al. Ocular findings in Saudi Arabian patients with sickle cell disease. Br J Ophthalmol. 1995;79(5):457-461. doi: 10.1136/bjo.79.5.457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 439.Steinberg MH, Hsu H, Nagel RL, et al. Gender and haplotype effects upon hematological manifestations of adult sickle cell anemia. Am J Hematol. 1995;48(3):175-181. doi: 10.1002/ajh.2830480307 [DOI] [PubMed] [Google Scholar]
- 440.Chang YC, Smith KD, Moore RD, Serjeant GR, Dover GJ. An analysis of fetal hemoglobin variation in sickle cell disease: the relative contributions of the X-linked factor, beta-globin haplotypes, alpha-globin gene number, gender, and age. Blood. 1995;85(4):1111-1117. doi: 10.1182/blood.V85.4.1111.bloodjournal8541111 [DOI] [PubMed] [Google Scholar]
- 441.Padmos A, Roberts G, Lindahl S, et al. Avascular necrosis of the femoral head in Saudi Arabians with homozygous sickle cell disease - risk factors. Ann Saudi Med. 1995;15(1):21-24. doi: 10.5144/0256-4947.1995.21 [DOI] [PubMed] [Google Scholar]
- 442.Brown AK, Sleeper LA, Miller ST, Pegelow CH, Gill FM, Waclawiw MA; Cooperative Study of Sickle Cell Disease . Reference values and hematologic changes from birth to 5 years in patients with sickle cell disease. Arch Pediatr Adolesc Med. 1994;148(8):796-804. doi: 10.1001/archpedi.1994.02170080026005 [DOI] [PubMed] [Google Scholar]
- 443.Castro O, Brambilla DJ, Thorington B, et al. ; The Cooperative Study of Sickle Cell Disease . The acute chest syndrome in sickle cell disease: incidence and risk factors. Blood. 1994;84(2):643-649. doi: 10.1182/blood.V84.2.643.643 [DOI] [PubMed] [Google Scholar]
- 444.el-Hazmi MA, Warsy AS, al-Swailem AR, al-Faleh FZ, al-Jabbar FA. Genetic compounds–Hb S, thalassaemias and enzymopathies: spectrum of interactions. J Trop Pediatr. 1994;40(3):149-156. doi: 10.1093/tropej/40.3.149 [DOI] [PubMed] [Google Scholar]
- 445.Adams RJ, Kutlar A, McKie V, et al. Alpha thalassemia and stroke risk in sickle cell anemia. Am J Hematol. 1994;45(4):279-282. doi: 10.1002/ajh.2830450402 [DOI] [PubMed] [Google Scholar]
- 446.Falusi AG, Olatunji PO. Effects of alpha thalassaemia and haemoglobin F (HbF) level on the clinical severity of sickle-cell anaemia. Eur J Haematol. 1994;52(1):13-15. doi: 10.1111/j.1600-0609.1994.tb01278.x [DOI] [PubMed] [Google Scholar]
- 447.de Montalembert M, Maier-Redelsperger M, Girot R, et al. Beta-globin gene cluster haplotype and alpha-thalassemia do not correlate with the acute clinical manifestations of sickle cell disease in children. Blood. 1993;82(8):2595-2596. doi: 10.1182/blood.V82.8.2595b.2595b [DOI] [PubMed] [Google Scholar]
- 448.Green NS, Fabry ME, Kaptue-Noche L, Nagel RL. Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia. Am J Hematol. 1993;44(2):145-146. doi: 10.1002/ajh.2830440214 [DOI] [PubMed] [Google Scholar]
- 449.Milner PF, Kraus AP, Sebes JI, et al. Osteonecrosis of the humeral head in sickle cell disease. Clin Orthop Relat Res. 1993;(289):136-143. doi: 10.1097/00003086-199304000-00018 [DOI] [PubMed] [Google Scholar]
- 450.Adekile AD, McKie KM, Adeodu OO, et al. Spleen in sickle cell anemia: comparative studies of Nigerian and U.S. patients. Am J Hematol. 1993;42(3):316-321. doi: 10.1002/ajh.2830420313 [DOI] [PubMed] [Google Scholar]
- 451.Fox PD, Higgs DR, Serjeant GR. Influence of alpha thalassaemia on the retinopathy of homozygous sickle cell disease. Br J Ophthalmol. 1993;77(2):89-90. doi: 10.1136/bjo.77.2.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.el-Hazmi MA. Heterogeneity and variation of clinical and haematological expression of haemoglobin S in Saudi Arabs. Acta Haematol. 1992;88(2-3):67-71. doi: 10.1159/000204654 [DOI] [PubMed] [Google Scholar]
- 453.el-Hazmi MA, Bahakim HM, Warsy AS. DNA polymorphism in the beta-globin gene cluster in Saudi Arabs: relation to severity of sickle cell anaemia. Acta Haematol. 1992;88(2-3):61-66. doi: 10.1159/000204653 [DOI] [PubMed] [Google Scholar]
- 454.Milner PF, Kraus AP, Sebes JI, et al. Sickle cell disease as a cause of osteonecrosis of the femoral head. N Engl J Med. 1991;325(21):1476-1481. doi: 10.1056/NEJM199111213252104 [DOI] [PubMed] [Google Scholar]
- 455.Powars DR, Elliott-Mills DD, Chan L, et al. Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality. Ann Intern Med. 1991;115(8):614-620. doi: 10.7326/0003-4819-115-8-614 [DOI] [PubMed] [Google Scholar]
- 456.Platt OS, Thorington BD, Brambilla DJ, et al. Pain in sickle cell disease: rates and risk factors. N Engl J Med. 1991;325(1):11-16. doi: 10.1056/NEJM199107043250103 [DOI] [PubMed] [Google Scholar]
- 457.Nagel RL, Erlingsson S, Fabry ME, et al. The Senegal DNA haplotype is associated with the amelioration of anemia in African-American sickle cell anemia patients. Blood. 1991;77(6):1371-1375. doi: 10.1182/blood.V77.6.1371.1371 [DOI] [PubMed] [Google Scholar]
- 458.Rieder RF, Safaya S, Gillette P, et al. Effect of beta-globin gene cluster haplotype on the hematological and clinical features of sickle cell anemia. Am J Hematol. 1991;36(3):184-189. doi: 10.1002/ajh.2830360305 [DOI] [PubMed] [Google Scholar]
- 459.Ballas SK, Talacki CA, Adachi K, Schwartz E, Surrey S, Rappaport E. The Xmn I site (−158, C—-T) 5′ to the G gamma gene: correlation with the Senegalese haplotype and G gamma globin expression. Hemoglobin. 1991;15(5):393-405. doi: 10.3109/03630269108998859 [DOI] [PubMed] [Google Scholar]
- 460.el-Hazmi MA. Beta-globin gene haplotypes in the Saudi sickle cell anaemia patients. Hum Hered. 1990;40(3):177-186. doi: 10.1159/000153927 [DOI] [PubMed] [Google Scholar]
- 461.Powars DR, Chan L, Schroeder WA. Beta S-gene-cluster haplotypes in sickle cell anemia: clinical implications. Am J Pediatr Hematol Oncol. 1990;12(3):367-374. doi: 10.1097/00043426-199023000-00022 [DOI] [PubMed] [Google Scholar]
- 462.Koshy M, Entsuah R, Koranda A, et al. Leg ulcers in patients with sickle cell disease. Blood. 1989;74(4):1403-1408. doi: 10.1182/blood.V74.4.1403.1403 [DOI] [PubMed] [Google Scholar]
- 463.Ballas SK, Talacki CA, Rao VM, Steiner RM. The prevalence of avascular necrosis in sickle cell anemia: correlation with alpha-thalassemia. Hemoglobin. 1989;13(7-8):649-655. doi: 10.3109/03630268908998842 [DOI] [PubMed] [Google Scholar]
- 464.Tejuca M, Martinez G, Ferreira R, et al. Alpha-thalassemia changes the cell density profile in sickle cell anaemia. Haematologia (Budap). 1989;22(3):175-180. [PubMed] [Google Scholar]
- 465.Schroeder WA, Powars DR, Kay LM, et al. Beta-cluster haplotypes, alpha-gene status, and hematological data from SS, SC, and S-beta-thalassemia patients in southern California. Hemoglobin. 1989;13(4):325-353. doi: 10.3109/03630268909003397 [DOI] [PubMed] [Google Scholar]
- 466.Ballas SK, Larner J, Smith ED, Surrey S, Schwartz E, Rappaport EF. Rheologic predictors of the severity of the painful sickle cell crisis. Blood. 1988;72(4):1216-1223. doi: 10.1182/blood.V72.4.1216.1216 [DOI] [PubMed] [Google Scholar]
- 467.Kulozik AE, Kar BC, Serjeant GR, Serjeant BE, Weatherall DJ. The molecular basis of alpha thalassemia in India. Its interaction with the sickle cell gene. Blood. 1988;71(2):467-472. doi: 10.1182/blood.V71.2.467.467 [DOI] [PubMed] [Google Scholar]
- 468.Pajot N, Maier-Redelsperger M, Dode C, Labie D, Girot R. Density distribution of red cells and prognostic significance in 50 patients with homozygous sickle-cell disease. Haematologia (Budap). 1988;21(4):189-197. [PubMed] [Google Scholar]
- 469.Felice AE, McKie KM, Cleek MP, Marino EM, Kutlar A, McKie VC. Effects of alpha-thalassemia-2 on the developmental changes of hematological values in children with sickle cell disease from Georgia. Am J Hematol. 1987;25(4):389-400. doi: 10.1002/ajh.2830250405 [DOI] [PubMed] [Google Scholar]
- 470.Falusi AG, Esan GJ, Ayyub H, Higgs DR. Alpha-thalassaemia in Nigeria: its interaction with sickle-cell disease. Eur J Haematol. 1987;38(4):370-375. doi: 10.1111/j.1600-0609.1987.tb00013.x [DOI] [PubMed] [Google Scholar]
- 471.Dover GJ, Chang VT, Boyer SH, Serjeant GR, Antonarakis S, Higgs DR. The cellular basis for different fetal hemoglobin levels among sickle cell individuals with two, three, and four alpha-globin genes. Blood. 1987;69(1):341-344. doi: 10.1182/blood.V69.1.341.341 [DOI] [PubMed] [Google Scholar]
- 472.Williams S, Maude GH, Serjeant GR. Clinical presentation of sickle cell-hemoglobin C disease. J Pediatr. 1986;109(4):586-589. doi: 10.1016/S0022-3476(86)80217-7 [DOI] [PubMed] [Google Scholar]
- 473.Billett HH, Kim K, Fabry ME, Nagel RL. The percentage of dense red cells does not predict incidence of sickle cell painful crisis. Blood. 1986;68(1):301-303. doi: 10.1182/blood.V68.1.301.301 [DOI] [PubMed] [Google Scholar]
- 474.Milner PF, Garbutt GJ, Nolan-Davis LV, Jonah F, Wilson LB, Wilson JT. The effect of Hb F and alpha-thalassemia on the red cell indices in sickle cell anemia. Am J Hematol. 1986;21(4):383-395. doi: 10.1002/ajh.2830210407 [DOI] [PubMed] [Google Scholar]
- 475.Stevens MC, Maude GH, Beckford M, et al. Alpha thalassemia and the hematology of homozygous sickle cell disease in childhood. Blood. 1986;67(2):411-414. doi: 10.1182/blood.V67.2.411.411 [DOI] [PubMed] [Google Scholar]
- 476.Bainbridge R, Higgs DR, Maude GH, Serjeant GR. Clinical presentation of homozygous sickle cell disease. J Pediatr. 1985;106(6):881-885. doi: 10.1016/S0022-3476(85)80230-4 [DOI] [PubMed] [Google Scholar]
- 477.Noguchi CT, Dover GJ, Rodgers GP, et al. Alpha thalassemia changes erythrocyte heterogeneity in sickle cell disease. J Clin Invest. 1985;75(5):1632-1637. doi: 10.1172/JCI111870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Nagel RL, Fabry ME, Pagnier J, et al. Hematologically and genetically distinct forms of sickle cell anemia in Africa: the Senegal type and the Benin type. N Engl J Med. 1985;312(14):880-884. doi: 10.1056/NEJM198504043121403 [DOI] [PubMed] [Google Scholar]
- 479.Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma-globin gene expression and high Hb F levels in beta-thalassemia and sickle cell anemia patients. Proc Natl Acad Sci U S A. 1985;82(7):2111-2114. doi: 10.1073/pnas.82.7.2111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.el-Hazmi MA. Clinical manifestation and laboratory findings of sickle cell anaemia in association with alpha-thalassaemia in Saudi Arabia. Acta Haematol. 1985;74(3):155-160. doi: 10.1159/000206194 [DOI] [PubMed] [Google Scholar]
- 481.Steinberg MH, Coleman MB, Adams JG, Rosenstock W. Interaction between HBS-beta-o-thalassemia and alpha-thalassemia. Am J Med Sci. 1984;288(5):195-199. doi: 10.1097/00000441-198412000-00001 [DOI] [PubMed] [Google Scholar]
- 482.Fabry ME, Mears JG, Patel P, et al. Dense cells in sickle cell anemia: the effects of gene interaction. Blood. 1984;64(5):1042-1046. doi: 10.1182/blood.V64.5.1042.1042 [DOI] [PubMed] [Google Scholar]
- 483.Steinberg MH, Rosenstock W, Coleman MB, et al. Effects of thalassemia and microcytosis on the hematologic and vasoocclusive severity of sickle cell anemia. Blood. 1984;63(6):1353-1360. doi: 10.1182/blood.V63.6.1353.1353 [DOI] [PubMed] [Google Scholar]
- 484.Embury SH, Clark MR, Monroy G, Mohandas N. Concurrent sickle cell anemia and alpha-thalassemia: effect on pathological properties of sickle erythrocytes. J Clin Invest. 1984;73(1):116-123. doi: 10.1172/JCI111181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Rucknagel D, Ferrucci S, Whitten CF, et al. Alpha-thalassemia and HbF concentration in sickle cell anemia. Prog Clin Biol Res. 1984;165:103-120. [PubMed] [Google Scholar]
- 486.Higgs DR, Aldridge BE, Lamb J, et al. The interaction of alpha-thalassemia and homozygous sickle-cell disease. N Engl J Med. 1982;306(24):1441-1446. doi: 10.1056/NEJM198206173062402 [DOI] [PubMed] [Google Scholar]
- 487.Embury SH, Dozy AM, Miller J, et al. Concurrent sickle-cell anemia and alpha-thalassemia: effect on severity of anemia. N Engl J Med. 1982;306(5):270-274. doi: 10.1056/NEJM198202043060504 [DOI] [PubMed] [Google Scholar]
- 488.Altay C, Gravely ME, Joseph BR, Williams DF. Alpha-thalassemia-2 and the variability of hematological values in children with sickle cell anemia. Pediatr Res. 1981;15(8):1093-1096. doi: 10.1203/00006450-198108000-00004 [DOI] [PubMed] [Google Scholar]
- 489.Bauer DE, Kamran SC, Lessard S, et al. An erythroid enhancer of BCL11A subject to genetic variation determines fetal hemoglobin level. Science. 2013;342(6155):253-257. doi: 10.1126/science.1242088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Chaturvedi S, Bhatnagar P, Bean CJ, et al. Genome-wide association study to identify variants associated with acute severe vaso-occlusive pain in sickle cell anemia. Blood. 2017;130(5):686-688. doi: 10.1182/blood-2017-02-769661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Lee N, Makani J, Tluway F, et al. Decreased hepcidin levels are associated with low steady-state hemoglobin in children with sickle cell disease in Tanzania. EBioMedicine. 2018;34:158-164. doi: 10.1016/j.ebiom.2018.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Adorno EV, Zanette Â, Lyra I, Seixas MO, Reis MG, Gonçalves MS. Clinical and molecular characteristics of sickle cell anemia in the northeast of Brazil. Genet Mol Biol. 2008;31(3):621-625. doi: 10.1590/S1415-47572008000400003 [DOI] [Google Scholar]
- 493.Marouf R, Gupta R, Haider MZ, Al-Wazzan H, Adekile AD. Avascular necrosis of the femoral head in adult Kuwaiti sickle cell disease patients. Acta Haematol. 2003;110(1):11-15. doi: 10.1159/000072406 [DOI] [PubMed] [Google Scholar]
- 494.Darshana T, Bandara D, Nawarathne U, et al. Sickle cell disease in Sri Lanka: clinical and molecular basis and the unanswered questions about disease severity. Orphanet J Rare Dis. 2020;15(1):177. doi: 10.1186/s13023-020-01458-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Costa FF, Tavella MH, Zago MA. Deletion type alpha-thalassemia among Brazilian patients with sickle-cell anemia. Rev Bras Genet. 1989;12(3):605-611. [Google Scholar]
- 496.Serjeant GR, Chin N, Asnani MR, et al. Causes of death and early life determinants of survival in homozygous sickle cell disease: The Jamaican cohort study from birth. PLoS One. 2018;13(3):e0192710. doi: 10.1371/journal.pone.0192710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Condon PI, Marsh RJ, Maude GH, Higgs DR, Weatherall DJ, Serjeant GR. Alpha thalassaemia and the macular vasculature in homozygous sickle cell disease. Br J Ophthalmol. 1983;67(11):779-781. doi: 10.1136/bjo.67.11.779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Serjeant BE, Mason KP, Kenny MW, et al. Effect of alpha thalassaemia on the rheology of homozygous sickle cell disease. Br J Haematol. 1983;55(3):479-486. doi: 10.1111/j.1365-2141.1983.tb02163.x [DOI] [PubMed] [Google Scholar]
- 499.Torres LS, Okumura JV, Silva DG, et al. Inflammation in sickle cell disease: differential and down-expressed plasma levels of annexin A1 protein. PLoS One. 2016;11(11):e0165833. doi: 10.1371/journal.pone.0165833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Adekile AD, Haider MZ, Serebour F, Al-Zaabi K, Tuli M. Serum immunoglobulins and IgG subclasses in SS patients with Saudi Arabia/India haplotype. Med Princ Pract. 1999;8(3):183-188. doi: 10.1159/000026090 [DOI] [Google Scholar]
- 501.Vyas P, Higgs DR, Weatherall DJ, Dunn D, Serjeant BE, Serjeant GR. The interaction of alpha thalassaemia and sickle cell-beta zero thalassaemia. Br J Haematol. 1988;70(4):449-454. doi: 10.1111/j.1365-2141.1988.tb02515.x [DOI] [PubMed] [Google Scholar]
- 502.Cumming V, King L, Fraser R, Serjeant G, Reid M. Venous incompetence, poverty and lactate dehydrogenase in Jamaica are important predictors of leg ulceration in sickle cell anaemia. Br J Haematol. 2008;142(1):119-125. doi: 10.1111/j.1365-2141.2008.07115.x [DOI] [PubMed] [Google Scholar]
- 503.Dossou-Yovo OP, Zaccaria I, Benkerrou M, et al. Effects of RANTES and MBL2 gene polymorphisms in sickle cell disease clinical outcomes: association of the g.In1.1T>C RANTES variant with protection against infections. Am J Hematol. 2009;84(6):378-380. doi: 10.1002/ajh.21411 [DOI] [PubMed] [Google Scholar]
- 504.Rahimi Z, Vaisi-Raygani A, Merat A, Haghshenass M, Rezaei M. Level of hemoglobin F and Gg gene expression in sickle cell disease and their association with haplotype and XmnI polymorphic site in south of Iran. Iran J Med Sci. 2007;32(4):234-239. [Google Scholar]
- 505.Adekile AD, Owunwanne A, Al-Za’abi K, Haider MZ, Tuli M, Al-Mohannadi S. Temporal sequence of splenic dysfunction in sickle cell disease. Am J Hematol. 2002;69(1):23-27. doi: 10.1002/ajh.10010 [DOI] [PubMed] [Google Scholar]
- 506.Hoppe C, Cheng S, Grow M, et al. A novel multilocus genotyping assay to identify genetic predictors of stroke in sickle cell anaemia. Br J Haematol. 2001;114(3):718-720. doi: 10.1046/j.1365-2141.2001.02997.x [DOI] [PubMed] [Google Scholar]
- 507.Al-Lamki Z, Wali YA, Shah W, Zachariah M, Rafique B, Ahmed S. Natural history of sickle hemoglobinopathies in Omani children. J Pediatr Hematol Oncol. 2000;7(2):101-107. [Google Scholar]
- 508.el-Hazmi MA, al-Swailem AR, Bahakim HM, al-Faleh FZ, Warsy AS. Effect of alpha thalassaemia, G-6-PD deficiency and Hb F on the nature of sickle cell anaemia in south-western Saudi Arabia. Trop Geogr Med. 1990;42(3):241-247. [PubMed] [Google Scholar]
- 509.el-Hazmi MA, Bahakim HM, al-Swailem AM, Warsy AS. The features of sickle cell disease in Saudi children. J Trop Pediatr. 1990;36(4):148-155. doi: 10.1093/tropej/36.4.148 [DOI] [PubMed] [Google Scholar]
- 510.Falusi AG, Esan GJ. Foetal haemoglobin levels in sickle cell anaemia in Nigerians. Afr J Med Med Sci. 1989;18(2):145-149. [PubMed] [Google Scholar]
- 511.de Ceulaer K, Higgs DR, Weatherall DJ, Hayes RJ, Serjeant BE, Serjeant GR. Alpha-thalassemia reduces the hemolytic rate in homozygous sickle-cell disease. N Engl J Med. 1983;309(3):189-190. doi: 10.1056/NEJM198307213090320 [DOI] [PubMed] [Google Scholar]
- 512.Steinberg MH, Coleman MB, Adams JG, Platica O, Gillette P, Rieder RF. The effects of alpha-thalassaemia in HbSC disease. Br J Haematol. 1983;55(3):487-492. doi: 10.1111/j.1365-2141.1983.tb02164.x [DOI] [PubMed] [Google Scholar]
- 513.Bernardo VS, Torres FF, Chaves NA, Okumura JV, da Silva DGH, Bonini-Domingos CR. Relationship of polymorphism rs3800231 in FOXO3 gene and clinical severity with oxidative stress markers in sickle cell disease. Meta Gene. 2020;24:100660. doi: 10.1016/j.mgene.2020.100660 [DOI] [Google Scholar]
- 514.Belisário AR, de Almeida JA, Mendes FG, et al. Prevalence and risk factors for albuminuria and glomerular hyperfiltration in a large cohort of children with sickle cell anemia. Am J Hematol. 2020;95(5):E125-E128. doi: 10.1002/ajh.25763 [DOI] [PubMed] [Google Scholar]
- 515.Patel R, Kang S, Valeshabad AK, et al. Kidney ultrasound findings according to kidney function in sickle cell anemia. Am J Hematol. 2019;94(11):E288-E291. doi: 10.1002/ajh.25602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 516.Uyoga S, Macharia AW, Mochamah G, et al. The epidemiology of sickle cell disease in children recruited in infancy in Kilifi, Kenya: a prospective cohort study. Lancet Glob Health. 2019;7(10):e1458-e1466. doi: 10.1016/S2214-109X(19)30328-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 517.Amle DB, Patnayak RL, Verma V, et al. VEGF Promoter region 18-bp insertion-deletion polymorphism in sickle cell disease patients with microalbuminuria: a pilot study. Indian J Hematol Blood Transfus. 2019;35(2):278-283. doi: 10.1007/s12288-018-1018-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Hassan FM, Alzahrani FM. Cytokines TNF-alpha and IL-8 gene polymorphisms in sickle cell anaemia patients under hydroxyurea treatment. J Clin Diagn Res. 2018;12(6):EC14-EC17. doi: 10.7860/JCDR/2018/35740.11681 [DOI] [Google Scholar]
- 519.Laurentino MR, Barbosa MC, Santos TEJ, Perdigão ACB, Araújo FMC, Lemes RPG. Analysis of BCL11A gene polymorphisms and hemolysis parameters in patients with: sickle-cell disease. Brazilian Journal of Pathology and Laboratory Medicine. 2018;54(3):132-137. doi: 10.5935/1676-2444.20180025 [DOI] [Google Scholar]
- 520.Vasseur C, Domingues-Hamdi E, Pakdaman S, et al. Elevated soluble α-hemoglobin pool in sickle cell anemia. Am J Hematol. 2017;92(10):E593-E595. doi: 10.1002/ajh.24835 [DOI] [PubMed] [Google Scholar]
- 521.Howell S, Marshall K, Reid M, McFarlane-Anderson N, McKenzie C. A cross-sectional clinic-based study exploring whether variants within genes coding for enzymes of the transmethylation and trans-sulphuration pathways are associated with inter-individual phenotypic variation in sickle cell anaemia in Jamaica. West Indian Med J. 2017;66(4):510-517. doi: 10.7727/wimj.2017.205 [DOI] [Google Scholar]
- 522.Medeiros FS, Mendonça TF, Lopes KAM, et al. Combined genotypes of the MBL2 gene related to low mannose-binding lectin levels are associated with vaso-occlusive events in children with sickle cell anemia. Genet Mol Biol. 2017;40(3):600-603. doi: 10.1590/1678-4685-gmb-2016-0161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Watanabe AM, Pianovski MAD, Lenzi L, Cat R. The frequency of βS-globin haplotypes in the state of Paraná, Brazil, and clinical: manifestations of sickle cell anemia. Brazilian Journal of Pathology and Laboratory Medicine. 2017;53(1):24-30. doi: 10.5935/1676-2444.20170007 [DOI] [Google Scholar]
- 524.Figueiredo CVB, Carvalho MOS, Santiago RP, et al. Leptin—2548 G > A gene polymorphism is associated with lipids metabolism and TGF-β alteration in sickle cell disease. Meta Gene. 2016;10:27-31. doi: 10.1016/j.mgene.2016.10.001 [DOI] [Google Scholar]
- 525.Ben Sassi M, Chaouch L, Kalai M, et al. Association of rs1319868, rs1567811 and rs8041224 of IGF1R gene with infection among sickle cell anemia Tunisian patients. Acta Haematol Pol. 2016;47(4):242-247. doi: 10.1016/j.achaem.2016.10.004 [DOI] [Google Scholar]
- 526.Purohit P, Patel S, Mohanty PK, Das P, Panigrahi J. Fetal hemoglobin modifies the disease manifestation of severe plasmodium falciparum malaria in adult patients with sickle cell anemia. Mediterr J Hematol Infect Dis. 2016;8(1):e2016055. doi: 10.4084/mjhid.2016.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Leal AS, Martins PRJ, Balarin MAS, Pereira GA, Resende GAD. Haplotypes βs-globin and its clinical-haematological correlation in patients with sickle-cell anemia in Triângulo Mineiro, Minas Gerais, Brazil. Brazilian Journal of Pathology and Laboratory Medicine. 2016;52(1):5-10. doi: 10.5935/1676-2444.20160001 [DOI] [Google Scholar]
- 528.Araujo NB, Domingos IF, Medeiros FS, et al. Lack of association between the Duffy antigen receptor for chemokines (DARC) expression and clinical outcome of children with sickle cell anemia. Immunol Lett. 2015;166(2):140-142. doi: 10.1016/j.imlet.2015.05.015 [DOI] [PubMed] [Google Scholar]
- 529.Chaouch L, Kalai M, Chaouachi D, et al. Gilbert syndrome acts as a risk factor of developing gallstone among β hemoglobinopathy Tunisian patients. Tunis Med. 2015;93(4):237-241. [PubMed] [Google Scholar]
- 530.Lustosa Souza CR, Azevedo Shimmoto MM, Vicari P, Mecabo G, Arruda MM, Figueiredo MS. Klotho gene polymorphisms and their association with sickle cell disease phenotypes. Rev Bras Hematol Hemoter. 2015;37(4):275-276. doi: 10.1016/j.bjhh.2015.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Rabab A. GSTM1 and GSTT1 polymorphism in Egyptian sickle cell anemia patients. Int J Hematol Oncol. 2013;23(4):269-275. doi: 10.4999/uhod.13025 [DOI] [Google Scholar]
- 532.Kangne HK, Jijina FF, Italia YM, et al. The Fc receptor polymorphisms and expression of neutrophil activation markers in patients with sickle cell disease from Western India. Biomed Res Int. 2013;2013:457656. doi: 10.1155/2013/457656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Drasar ER, Menzel S, Fulford T, Thein SL. The effect of Duffy antigen receptor for chemokines on severity in sickle cell disease. Haematologica. 2013;98(8):e87-e89. doi: 10.3324/haematol.2013.089243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Lemonne N, Lamarre Y, Romana M, et al. Does increased red blood cell deformability raise the risk for osteonecrosis in sickle cell anemia?. Blood. 2013;121(15):3054-3056. doi: 10.1182/blood-2013-01-480277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.El Deen MAK, Khorshied MM, El Sadani ZA, Amrousy YM, Galal NM. Mannose-binding lectin (MBL2) gene polymorphism in sickle cell anemia: an Egyptian study. Comparative Clinical Pathology. 2012;22(3):387-394. doi: 10.1007/s00580-012-1420-y [DOI] [Google Scholar]
- 536.Pandey S, Ranjan R, Firdos A, et al. Relation between the uridine diphosphate glucuronosyltransferase 1A1 polymorphism and the bilirubin levels in sickle cell disease. J Clin Diagn Res. 2012;6(5):821-824. [Google Scholar]
- 537.Pandey S, Pandey S, Mishra RM, Saxena R. Modulating effect of the −158 γ (C→T) Xmn1 polymorphism in Indian sickle cell patients. Mediterr J Hematol Infect Dis. 2012;4(1):e2012001. doi: 10.4084/mjhid.2012.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Heitzer AM, Longoria J, Rampersaud E, et al. Fetal hemoglobin modulates neurocognitive performance in sickle cell anemia✰,✰✰. Curr Res Transl Med. 2022;70(3):103335. doi: 10.1016/j.retram.2022.103335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.de Freitas Dutra V, Leal VNC, Fernandes FP, Souza CRL, Figueiredo MS, Pontillo A. Genetic contribution and functional impairment of inflammasome in sickle cell disease. Cytokine. 2022;149:155717. doi: 10.1016/j.cyto.2021.155717 [DOI] [PubMed] [Google Scholar]
- 540.Garrett ME, Soldano KL, Erwin KN, et al. Genome-wide meta-analysis identifies new candidate genes for sickle cell disease nephropathy. Blood Adv. 2023;7(17):4782-4793. doi: 10.1182/bloodadvances.2022007451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Nardo-Marino A, Glenthøj A, Brewin JN, et al. The significance of spleen size in children with sickle cell anemia. Am J Hematol. 2022;97(12):1520-1528. doi: 10.1002/ajh.26703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Wang X, Gardner K, Tegegn MB, et al. Genetic variants of PKLR are associated with acute pain in sickle cell disease. Blood Adv. 2022;6(11):3535-3540. doi: 10.1182/bloodadvances.2021006668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Adebayo OC, Betukumesu DK, Nkoy AB, et al. Clinical and genetic factors are associated with kidney complications in African children with sickle cell anaemia. Br J Haematol. 2022;196(1):204-214. doi: 10.1111/bjh.17832 [DOI] [PubMed] [Google Scholar]
- 544.Pincez T, Lo KS, D’Orengiani APHD, et al. Variation and impact of polygenic hematologic traits in monogenic sickle cell disease. Haematologica. 2023;108(3):870-881. doi: 10.3324/haematol.2022.281180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Walker TM, Hambleton IR, Mason KP, Serjeant G. Spleen size in homozygous sickle cell disease: trends in a birth cohort using ultrasound. Br J Radiol. 2022;95(1140):20220634. doi: 10.1259/bjr.20220634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Ibrahim NS, Makhlouf MM, Shahin GH, Elghamrawy MK, Hussein NM. The impact of MCP1-2518A/G and CCR2-V64I genetic polymorphisms in Egyptian sickle cell disease patients. Exp Mol Pathol. 2022;128:104834. doi: 10.1016/j.yexmp.2022.104834 [DOI] [PubMed] [Google Scholar]
- 547.Allard P, Alhaj N, Lobitz S, et al. Genetic modifiers of fetal hemoglobin affect the course of sickle cell disease in patients treated with hydroxyurea. Haematologica. 2022;107(7):1577-1588. doi: 10.3324/haematol.2021.278952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Liyanage JSS, Estepp JH, Srivastava K, Li Y, Mori M, Kang G. GMEPS: a fast and efficient likelihood approach for genome-wide mediation analysis under extreme phenotype sequencing. Stat Appl Genet Mol Biol. 2022;21(1). doi: 10.1515/sagmb-2021-0071 [DOI] [PubMed] [Google Scholar]
- 549.Rizo-de la Torre LC, Borrayo-López FJ, Perea-Díaz FJ, et al. Fetal hemoglobin regulating genetic variants identified in homozygous (HbSS) and heterozygous (HbSA) subjects from South Mexico. J Trop Pediatr. 2022;68(5):fmac073. doi: 10.1093/tropej/fmac073 [DOI] [PubMed] [Google Scholar]
- 550.Boisson C, Renoux C, Nader E, et al. Comparisons of oxygen gradient ektacytometry parameters between sickle cell patients with or without α-thalassaemia. Br J Haematol. 2021;195(4):629-633. doi: 10.1111/bjh.17777 [DOI] [PubMed] [Google Scholar]
- 551.Osunkalu VO, Ogbenna AA, Davies NO, et al. Assessment of MTR Rs1805087 SNP as possible modifier of sickle cell disease severity in a Nigerian population. West Afr J Med. 2022;39(11):1198-1204. [PubMed] [Google Scholar]
- 552.Belvitch P, Casanova N, Sun X, et al. A cortactin CTTN coding SNP contributes to lung vascular permeability and inflammatory disease severity in African descent subjects. Transl Res. 2022;244:56-74. doi: 10.1016/j.trsl.2022.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Germano I, Santos B, Delgadinho M, et al. Genetic modulation of anemia severity, hemolysis level, and hospitalization rate in Angolan children with sickle cell anemia. Mol Biol Rep. 2022;49(11):10347-10356. doi: 10.1007/s11033-022-07831-1 [DOI] [PubMed] [Google Scholar]
- 554.Hariharan P, Gorivale M, Sawant P, Mehta P, Nadkarni A. Significance of genetic modifiers of hemoglobinopathies leading towards precision medicine. Sci Rep. 2021;11(1):20906. doi: 10.1038/s41598-021-00169-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Prohaska CC, Zhang X, Schwantes-An TL, et al. RASA3 is a candidate gene in sickle cell disease-associated pulmonary hypertension and pulmonary arterial hypertension. Pulm Circ. 2023;13(2):e12227. doi: 10.1002/pul2.12227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 556.Powell-Roach KL, Yao Y, Wallace MR, et al. HUMAN STUDY COMT and DRD3 haplotype-associated pain intensity and acute care utilization in adult sickle cell disease. Exp Biol Med (Maywood). 2022;247(17):1601-1608. doi: 10.1177/15353702221080716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Ndour EHM, Mnika K, Guèye Tall F, et al. Effects of Senegal haplotype (Xmn1-rs7412844), alpha-thalassemia (3.7kb HBA1/HBA2 deletion), NPRL3-rs11248850 and BCL11A-rs4671393 variants on sickle cell nephropathy. Int J Biochem Mol Biol. 2022;13(2):5-16. [PMC free article] [PubMed] [Google Scholar]
- 558.Menezes JF, Carvalho MOS, Rocha LC, et al. Role of paraoxonase 1 activity and PON1 gene polymorphisms in sickle cell disease. Sci Rep. 2023;13(1):7215. doi: 10.1038/s41598-023-34396-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Sales RR, Nogueira BL, Belisário AR, et al. Fetal hemoglobin-boosting haplotypes of BCL11A gene and HBS1L-MYB intergenic region in the prediction of clinical and hematological outcomes in a cohort of children with sickle cell anemia. J Hum Genet. 2022;67(12):701-709. doi: 10.1038/s10038-022-01079-0 [DOI] [PubMed] [Google Scholar]
- 560.Manu GP, Segbefia C, N’guessan BB, Coffie SA, Adjei GO. Association between selected single nucleotide polymorphisms in globin and related genes and response to hydroxyurea therapy in Ghanaian children with sickle cell disease. Pharmgenomics Pers Med. 2022;15:205-214. doi: 10.2147/PGPM.S351599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Brewin JN, Nardo-Marino A, Stuart-Smith S, et al. The pleiotropic effects of α-thalassemia on HbSS and HbSC sickle cell disease: Reduced erythrocyte cation co-transport activity, serum erythropoietin, and transfusion burden, do not translate into increased survival. Am J Hematol. 2022;97(10):1275-1285. doi: 10.1002/ajh.26652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Powell-Roach KL, Yao Y, Cao X, et al. Analysis of AVPR1A, thermal and pressure pain thresholds, and stress in sickle cell disease. Front Pain Res (Lausanne). 2023;3:1060245. doi: 10.3389/fpain.2022.1060245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.Hamadi A, Mir R, Mahzari A, et al. Molecular determination of vascular endothelial growth factor, miRNA-423 gene abnormalities by utilizing ARMS-PCR and their association with fetal hemoglobin expression in the patients with sickle cell disease. Curr Issues Mol Biol. 2022;44(6):2569-2582. doi: 10.3390/cimb44060175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Bernaudin F, Arnaud C, Kamdem A, et al. Incidence, kinetics, and risk factors for intra- and extracranial cerebral arteriopathies in a newborn sickle cell disease cohort early assessed by transcranial and cervical color Doppler ultrasound. Front Neurol. 2022;13:846596. doi: 10.3389/fneur.2022.846596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Duru AN, Ocheni S, Ibegbulam O, Okpala I. Plasma concentration of 12-hydroxyeicosatetraenoic acid, single nucleotide polymorphisms of 12-lipooxygenase gene and vaso-occlusion in sickle cell disease. Front Genome Ed. 2021;3:722190. doi: 10.3389/fgeed.2021.722190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 566.Dosunmu-Ogunbi A, Yuan S, Reynolds M, et al. SOD2 V16A amplifies vascular dysfunction in sickle cell patients by curtailing mitochondria complex IV activity. Blood. 2022;139(11):1760-1765. doi: 10.1182/blood.2021013350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 567.Kumkhaek C, Kim C, Kurban G, et al. Single nucleotide polymorphisms in SAR1A coding regions in sickle cell disease and their potential miRNA binding sites. EJHaem. 2022;3(4):1438-1441. doi: 10.1002/jha2.542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Dehury S, Mohanty PK, Patel S, et al. Profiling of 35 cases of Hb S/Hb E (HBB: c.20A>T/HBB: c.79G>a), disease and association with α-thalassemia and β-globin gene cluster haplotypes from Odisha, India. Hemoglobin. 2021;45(6):380-386. doi: 10.1080/03630269.2021.1965618 [DOI] [PubMed] [Google Scholar]
- 569.Hassab H, Hanafi M, Elbeheiry A, Hassan M, Chazli YE. Does TGFBR3 polymorphism increase the risk of silent cerebral infarction in egyptian children with sickle cell disease? Indian J Pediatr. 2023;90(2):146-152. doi: 10.1007/s12098-022-04181-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Knisely MR, Yang Q, Stauffer N, et al. Evaluating associations between average pain intensity and genetic variation in people with sickle cell disease: an exploratory study. Pain Manag Nurs. 2023;24(1):12-18. doi: 10.1016/j.pmn.2022.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.do Kleyton Palmeira Ó, da Silva Freire AK, de Nóbrega DN, et al. Polymorphisms and gene expression of metalloproteinases and their inhibitors associated with cerebral ischemic stroke in young patients with sickle cell anemia. Mol Biol Rep. 2023;50(4):3341-3353. doi: 10.1007/s11033-023-08262-2 [DOI] [PubMed] [Google Scholar]
- 572.Purohit P, Mohanty PK, Panigrahi J, Das K, Patel S. Effect of α+ thalassemia on the severity of Plasmodium falciparum malaria in different sickle cell genotypes in Indian adults: a hospital-based study. Hemoglobin. 2023;47(1):11-18. doi: 10.1080/03630269.2023.2168201 [DOI] [PubMed] [Google Scholar]
- 573.Earley EJ, Kelly S, Fang F, et al. ; International Component of the NHLBI Recipient Epidemiology and Donor Evaluation Study (REDS-III) and the NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium . Genome-wide association study of early ischaemic stroke risk in Brazilian individuals with sickle cell disease implicates ADAMTS2 and CDK18 and uncovers novel loci. Br J Haematol. 2023;201(2):343-352. doi: 10.1111/bjh.18637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 574.Olupot-Olupot P, Tomlinson G, Williams TN, et al. Hydroxyurea treatment is associated with lower malaria incidence in children with sickle cell anemia in sub-Saharan Africa. Blood. 2023;141(12):1402-1410. doi: 10.1182/blood.2022017051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Cappelli B, Scigliuolo GM, Boukouaci W, et al. Impact of the human leucocyte antigen (HLA)-B leader peptide dimorphism and HLA-A expression on outcomes of stem cell transplantation for sickle cell disease. Br J Haematol. 2021;195(2):e128-e131. doi: 10.1111/bjh.17665 [DOI] [PubMed] [Google Scholar]
- 576.Liyanage JSS, Estepp JH, Srivastava K, et al. A versatile and efficient novel approach for mendelian randomization analysis with application to assess the causal effect of fetal hemoglobin on anemia in sickle cell anemia. Mathematics. 2022;10(20):3743. [Google Scholar]
- 577.Khorshied MM, Shaheen IA, Selim YMM, Elshahawy AO, Youssry I. Impact of superoxide dismutase genetic polymorphism (SOD2 Val16Ala) and superoxide dismutase level on disease severity in a cohort of Egyptian sickle cell disease patients. Mediterr J Hematol Infect Dis. 2022;14(1):e2022037. doi: 10.4084/MJHID.2022.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Nardo-Marino A, Petersen J, Brewin JN, et al. Oxygen gradient ektacytometry does not predict pain in children with sickle cell anaemia. Br J Haematol. 2022;197(5):609-617. doi: 10.1111/bjh.17975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Martins JO, Pagani F, Dezan MR, et al. Impact of HLA-G +3142C>G on the development of antibodies to blood group systems other than the Rh and Kell among sensitized patients with sickle cell disease. Transfus Apher Sci. 2022;61(5):103447. doi: 10.1016/j.transci.2022.103447 [DOI] [PubMed] [Google Scholar]
- 580.Costa-Júnior DAD, Santos APP, da Silva CM, Velloso-Rodrigues C. Growth hormone/insulin-like growth factor 1 axis associated with modifier factors in children with sickle cell anemia. Endocr Metab Immune Disord Drug Targets. 2022;22(9):954-962. doi: 10.2174/1871530322666220303164029 [DOI] [PubMed] [Google Scholar]
- 581.Kengne Fotsing CB, Pieme CA, Biapa Nya PC, et al. Relation between haptoglobin polymorphism and oxidative stress status, lipid profile, and cardiovascular risk in sickle cell anemia patients. Health Sci Rep. 2022;5(1):e465. doi: 10.1002/hsr2.465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Sokkar MF, Kamal L, Salama N, Hamdy M. Thrombophilic mutations and risk of vascular complications in sickle cell disease. Gene Rep. 2022;27:101595. doi: 10.1016/j.genrep.2022.101595 [DOI] [Google Scholar]
- 583.Lakkakula BVKS, Pattnaik S. HBB gene cluster haplotype diversity in sickle cell anemia patients of Chhattisgarh, India. J Appl Biol Biotechnol. 2021;9(5):64-69. [Google Scholar]
- 584.Little J, Higgins JP, Ioannidis JP, et al. ; STrengthening the REporting of Genetic Association Studies . STrengthening the REporting of Genetic Association Studies (STREGA): an extension of the STROBE statement. PLoS Med. 2009;6(2):e22. doi: 10.1371/journal.pmed.1000022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Pincez T, Ashley-Koch AE, Lettre G, Telen MJ. Genetic modifiers of sickle cell disease. Hematol Oncol Clin North Am. 2022;36(6):1097-1124. doi: 10.1016/j.hoc.2022.06.006 [DOI] [PubMed] [Google Scholar]
- 586.Manolio TA, Collins FS, Cox NJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461(7265):747-753. doi: 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 587.Moridani MY, Pourahmad J, Bui H, Siraki A, O’Brien PJ. Dietary flavonoid iron complexes as cytoprotective superoxide radical scavengers. Free Radic Biol Med. 2003;34(2):243-253. doi: 10.1016/S0891-5849(02)01241-8 [DOI] [PubMed] [Google Scholar]
- 588.Rees A, Dodd GF, Spencer JPE. The Effects of flavonoids on cardiovascular health: a review of human intervention trials and implications for cerebrovascular function. Nutrients. 2018;10(12):1852. doi: 10.3390/nu10121852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Henneberg R, Otuki MF, Furman AE, Hermann P, do Nascimento AJ, Leonart MS. Protective effect of flavonoids against reactive oxygen species production in sickle cell anemia patients treated with hydroxyurea. Rev Bras Hematol Hemoter. 2013;35(1):52-55. doi: 10.5581/1516-8484.20130015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 590.Lizarralde MA, Merriweather B, Conrey A, Saxena A, Shet AS. Effects of flavonoid quercetin on thrombo-inflammatory processes in patients with sickle cell disease. Blood. 2021;138(suppl 1):2020. doi: 10.1182/blood-2021-148601 [DOI] [Google Scholar]
- 591.Hamilton CM, Strader LC, Pratt JG, et al. The PhenX Toolkit: get the most from your measures. Am J Epidemiol. 2011;174(3):253-260. doi: 10.1093/aje/kwr193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 592.Hripcsak G, Duke JD, Shah NH, et al. Observational Health Data Sciences and Informatics (OHDSI): opportunities for observational researchers. Stud Health Technol Inform. 2015;216:574-578. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eMethods.
eFigure 1. PRISMA Flow Diagram For Manuscript Identification, Screening, and Inclusion
eFigure 2. Map of Included Patient Cohort Locations by Number of Individuals
eFigure 3. Data Strength Categories by Time
eFigure 4. Number of Papers Reporting Significant Associations With Fetal Hemoglobin (HbF) and Number of Unique Variants for Genes With Significant Results Reported in at Least Three Manuscripts
eTable 4. Phenotype Categories With Major Constituent Phenotypes
eTable 5. Number of Unique Patients and Studies by Country
eReferences.
eTable 1. Manuscripts Included in This Systematic Review and Meta-Analysis
eTable 2. Data Extracted and Utilized in This Systematic Review and Meta-Analysis, Excluding Genome-Wide Summary Statistics
eTable 3. Genome-Wide Summary Statistics Extracted and Utilized in This Systematic Review and Meta-Analysis
eTable 6. Full List of Gene-Phenotype Category Associations
eTable 7. Gene Ontology Pathway Analysis Results
eTable 8. Reactome Pathway Analysis Results
Data Sharing Statement